
Submitted:
08 July 2024
Posted:
09 July 2024
You are already at the latest version
Abstract
Chitosan is a biodegradable polymer derived from chitin, that is a versatile material for various biological applications for its attractive properties such as biocompatibility, biodegradability, and non-toxicity. Furthermore, chitosan possesses Second Harmonic Generation properties, that are useful for bio-sensing applications.
In this work we explored the possibility to exploit chitosan-based nanospheres as SHG-based bio-sensors, and also as carriers of 89Sr radionuclide, an Active Pharmaceutical Ingredient for radiopharmaceutical treatments in cancer therapy. To load opportunely the Sr ion on the nanospheres, we used a fructose-based Metal Organic Framework, of formula [Sr(C6H12O6)2(H2O)2]Cl2·H2O, because the sugar was able to drive the Sr ions on the chitosan matrix.
The Sr-loaded chitosan nanospheres were synthetized, characterized, and their SHG response was measured. The results encouraged us to propose the nanospheres for theranostic purposes, i.e. at the same time valuable for both therapy and diagnostic applications.
Keywords:
Metal Organic Frameworks
; Chitosan nanospheres
; theranostics
; radiopharmaceutical
; Second Harmonic Generation properties
1. Introduction
Chitosan is a biodegradable polymer derived from chitin, found in the shells of marine crustaceans. It consists of β (1-4)-linked 2-amino-2-deoxy-D-glucose (D-glucosamine) and 2-acetamido-2-deoxy-D-glucosamine (N-acetyl-D-glucosamine) units, sharing structural similarities with cellulose [1,2]. Being the second most diffuse polysaccharide on the Earth, it can be considered a very cheap material with attractive properties such as biocompatibility, biodegradability, non-toxicity, adhesion, and sorption, making it versatile for various biological applications [3,4,5]. Extensive in vitro studies evidenced positive cellular responses to chitosan in several tissues, including smooth muscle cells, macrophages, osteoblasts, chondrocytes, erythrocytes, and whole blood [6,7].
The application of chitosan as a carrier of Active Pharmaceutical Ingredient (API) has also been widely explored both in vitro and in vivo [2,8,9,10], especially in the form of nanospheres (NPs) that were proposed as novel non-toxic and efficient drug delivery systems [11,12,13]. Another noteworthy property of chitosan is related to its Non-Linear Optical (NLO) properties, particularly Second Harmonic Generation (SHG) properties [14], that in principle allows the exploitation of this material also for bio-imaging. Optical imaging techniques based on SHG properties are particularly advantageous for bio-sensing, since the SHG-based nanoprobes do not undergo bleaching or blinking, as observed for fluorescence probes, and the second-harmonic signal does not saturate with increasing illumination intensity [15,16].
In particular, we are interested in developing new biologically compatible radioisotope carriers. Radionuclide delivery bio-systems are the new frontiers of cancer therapy [17,18,19], because they allow the development of systemic personal therapies that can noticeably reduce the devastating side effects of conventional therapies. These carriers should be appropriately engineered, in order to drive the radioisotopes quickly toward the targeted tumoral tissue, reducing as much as possible their permanence in the health tissues.
In this work, we propose the use of chitosan NPs, loaded with 89Sr radionuclide, for radiopharmaceutical treatments in cancer therapy. The 89Sr radionuclide is a readily available and affordable beta-emitter. It is already extensively employed in radiotherapy for treating metastases and pain in bone cancer because of its low chemical toxicity and low half-life (50.57 d), which reduces post-treatment risks [20,21,22,23]. The possibility of carrying the 89Sr radioisotope through biocompatible nanoparticles, that can be concentrated to specific target tumor cells and also in tissue different from bones, can open new opportunities in the systemic cancer treatments with this radioisotope. For this purpose, chitosan is a promising carrier, since suitably functionalized chitosan can selectively permeate through tumor cells, increasing its concentration in targeted tumor tissue and avoiding damage in healthy tissue [24,25,26,27]. Furthermore, since chitosan possesses the SHG property [28], the NPs loaded with 89Sr can be considered excellent candidates for applications in theranostics, i.e., contemporary therapeutic and diagnostic purposes.
Thus, the aim of this work is to synthetize chitosan-based NPs with SHG properties loaded with Sr, that can also be enriched with 89Sr radionuclide. To promote the encapsulation of the metallic ions on the chitosan NPs, we used a D-fructose-based Metal Organic Framework (MOF) of formula [Sr(C6H12O6)2(H2O)2]Cl2·H2O (SrFRUCl) [29], since Sr ions surrounded by sugar could approach more easily the chitosan matrix. We characterized the chitosan-based NPs loaded with Sr (Sr-NPs) by X-ray Powder Diffraction (XRPD), Fourier Transform Infrared (FTIR) spectroscopy, Scanning Electron Microscopy (SEM) and energy dispersive X-Ray spectroscopy (EDS). Furthermore, in order to understand the mechanism of interaction between the chitosan and the Sr and Cl ions, theoretical calculations were performed using Born-Oppenheimer molecular dynamics (BOMD) at the density functional theory (DFT) level [30,31]. Finally, we tested the SHG properties of the NPs, before and after the Sr loading, using the SHG microscopy technique.
2. Results and Discussion
2.1. Synthesis of Chitosan-Based NPs Loaded with Sr (Sr-NPs)
The first step to synthesize the chitosan-based NPs suitable for our purposes was the formation of a hydrogel by mixing two polymeric aqueous solutions of chitosan (CS) and poly(vinyl alcohol) (PVA) at a 3:1 volume ratio, according to the procedure reported in the literature [32,33,34]. This CS:PVA volume ratio promotes the miscibility and formation of a polymeric network with strong intermolecular interactions, yielding CS/PVA hybrid hydrogels with the best chemical and physical properties in terms of porosity and swelling capability for their loading with an API [35].
The second step involved forming the polymeric hybrid nanospheres by slowly adding the CS/PVA hydrogel solution to an emulsion of polyisobutylene and sorbitan monooleate emulsion, using the oil-in-water emulsion method [36]. The chitosan-based NPs were formed and suspended in the emulsion after the addition of the cross-linker GA. The mixture was then centrifuged, and the chitosan-based NPs were collected from the bottom of the test tube. Subsequently, they were washed first with hexane and then with deionized water to remove any residual oil and surfactant. After washing with water, the NPs were kept wet to prevent cluster formation.
In order to promote the encapsulation of the metallic ions on the chitosan NPs, we used the SrFRUCl MOF, since each metal in the structure was strictly bonded with four fructose molecules (see Figure S1 in Supplementary Material), which could facilitate the approach to the chitosan matrix. Thus, an equal amount of wet NPs and finely ground SrFRUCl powder were suspended in ethanol and sonicated for 10 min, and the MOF and the chitosan-based NPs remained in contact for 48 h. The resulting sample appeared highly homogeneous and was further washed with deionized water to remove the excess of SrFRUCl MOF, resulting in Sr-NPs.
2.2. Characterization of Chitosan-Based NPs Loaded with Sr (Sr-NPs)
2.2.1. FTIR Spectroscopy
FTIR spectroscopy was conducted on both the pure components of the chitosan-based NPs (i.e., CS, PVA, and GA) and their corresponding blends (i.e., CS/PVA and CS/PVA/GA), in the form of films or nanospheres. The FTIR spectra obtained are presented in Figure 1.
For CS (Figure 1a), typical absorption bands were observed at 3348 and 3268 cm−1 (NH2 and O-H stretching vibration modes), 2925-2860 cm−1 (axial stretching of C-H bonds), and at 1645, 1549, 1375 and 1310 cm−1. These correspond to amide I (stretching of the C=O group), amine/amide II (NH2 deformation and mixed vibration of N-H bending and C-N stretching in secondary amides), CH-CH2 and CH-OH bending modes, respectively. The position of the amide II band at 1548-1560 cm−1 indicates the protonation of primary amino groups (-NH2 → -NH3+) [37,38]. Additionally, bands associated with C-O and C-N bond stretching modes were located at 1060-1030 cm−1, while bands at 1152 and 894 cm−1 correspond to saccharide rings. The FTIR spectrum of pure PVA (Figure 1b) exhibited characteristic bands, including hydroxyl groups at 3500-3000 cm−1, stretching vibrational modes for C-H bonds at 2935-2900 cm−1, CH-CH2 and O-H bending modes at 1411 and 1323 cm−1, respectively, stretching of C-O bonds at 1085 cm−1 and out-of-plane O-H deformation bonds at 833 cm−1.
As observed in Figure 1c, the FTIR spectrum of the CS/PVA hydrogel exhibits characteristic bands corresponding to its individual components. Compared to the PVA spectrum, CS/PVA shows a slightly wider band at 3500-3000 cm−1, indicating an increase in OH bonds due to interactions between NH2 and/or OH groups in CS with the OH groups in PVA polymeric chains. Additionally, the band associated with amide II of CS shifts to 1559 cm−1, resulting from ionizing of the free primary amino groups in the acid medium (-NH3+).
The FTIR spectrum of the chitosan-based NPs (CS/PVA/GA, Figure 1d) exhibited differences compared to that of CS/PVA hydrogels. An increase in the intensity of the NH2/O-H band was detected at 3500-3000 cm−1, attributed to the moisture present in the wet sample. The small shoulder observed at 1708 cm−1 corresponds to the deformation of C=O bonds in the terminal groups of GA. The cross-linking reaction between CS and GA was evidenced by the strong band a 1630 cm−1 (C=N, imine bonds in Schiff bases), which overlaps with the amide I band. Furthermore, a decrease in intensity of the amine/amide II band intensity at 1559 cm−1 was observed, indicating the formation of the Schiff bases at the expense of -NH2 groups. Additionally, the definition of bands at 1060-1030 cm−1 related to C-O bonds suggests the formation of acetal groups (O-C-O), indicating cross-linking of PVA chains by GA [39,40]. These results suggest that GA induced extensive cross-linking, resulting in a chemically and mechanically stable NPs over time and in diverse chemical environments.
New bands at 1108, 1000 and 970 cm−1 correspond to unreacted GA (Figure 1d, dashed lines marked with an asterisk). Note that these bands align with absorption bands in the fingerprint region of the GA spectrum (Figure 1e). Therefore, their presence indicates that additional washes are necessary to completely remove the unreacted cross-linking reagent from the chitosan-based NPs completely. Similar results have been reported by Ceylan et al. when the concentration of GA in the CS/PVA blend exceeds 5% w/w [41].
Figure 2 shows the FTIR spectrum of the chitosan-based NPs loaded with Sr (Sr-NPs, Figure 2a), alongside that of the pure chitosan-based NPs (CS/PVA/GA, Figure 2b) previously discussed. As can be observed, after loading the chitosan-based NPs with Sr (Sr-NPs), their FTIR spectrum (Figure 2a) exhibited noticeable changes compared to the chitosan-based NPs spectrum (Figure 2b), particularly in the region between 1750 and 750 cm−1. In this region, the band intensity was significantly reduced at 1710 cm−1, corresponding to the C=O bond of the GA dialdehyde terminal groups. Additionally, the bands associated with GA (1108, 1000, and 997 cm−1) observed in the chitosan-based NPs were absent in the Sr-NPs spectrum, which instead showed only the characteristic absorption band for C-O/C-N bonds (1060-1030 cm−1) of CS/PVA hydrogels. These findings indicate that the remnants of unreacted GA were removed entirely from the Sr-NPs during washing.
The appearance of a weak band at 856-812 cm−1 in the Sr-NPs spectrum (Figure 2a) could be attributable to the presence of Sr. According to several studies, bending vibrations of Sr-O bonds typically occur at this wavenumber [42,43,44]. The change in intensity and a slight shift to lower frequencies of the amide II band (from 1564 to 1552 cm−1) might be associated with the binding of Cl ions, which could interact with the protonated amines in chitosan through electrostatic attractions. Finally, the band between 3500-3000 cm−1 corresponds to the overlapping stretching vibrations of N-H and O-H bonds, showing a decrease in intensity compared to that of chitosan-based NPs, which is attributed to lower moisture content in the Sr-NPs sample.
2.2.2. XRPD Characterization
The XRPD patterns of polymers are typically characterized by wide bands due to a prominent low crystallinity of the material, and furthermore they can show preferred orientation. For this reason, to obtain definitely reproducible results, we collected the XRPD pattern with a diffractometer equipped with a 2D detector, that permits to collect the whole diffraction circles. Then, the final XRPD pattern was calculated by averaging the intensities of the points of each circle drawn around the zero value of 2θ. In this way, for each 2θ value any eventual lower intensity points were compensated by the higher intensity ones, and the XRPD patterns results much more reproducible.
In Figure 3 are reported the XRPD patterns of the chitosan-based NPs (blue), chitosan-based NPs filtered from the SrFRUCl solution (red), and the washed Sr-NPs (black). The chitosan-based NPs obtained from the procedure proposed in this work show a pattern typical of a polymer with low crystallinity: the pattern is characterized by two wide bands at 2θ values of ca. 15.5° and 19.7°. The chitosan-based NPs filtered from the SrFRUCl solution, instead, show the typical pattern of the crystalline material SrFRUCl (green), even if the relative intensities were not completely respected, since the sample was not ground for stability reasons. After washing with water, the XRPD pattern of Sr-NPs completely lost the crystalline SrFRUCl peaks, and the pattern of the low crystalline polymer resurfaces again, slightly modified: the broad band at 2θ ca. 15.5° almost disappeared, while the band at 2θ ca. 19.7° is maintained. Thus, the SrFRUCl MOF was not incorporated as crystalline fragments inside the chitosan-based NPs and, consequently, where dissolved by the washing with water, but the Sr-NPs XRPD pattern is different from the one of the pure chitosan-based NPs: clearly, the polymer structure is slightly altered by the treatment with the solution of the MOF. This agrees with the possibility of embedding of Sr and/or Cl ions inside the NPs.
2.2.3. SEM Characterization
Figure 4 reports the SEM characterization of the Sr-NPs. The nanoparticles show a spherical morphology in the nanoscale dimension (Figure 4a), with an average size of 0.39 microns and a standard deviation of 0.19 microns. The Map Sum Spectrum in Figure 4d reports the chemical composition of the NPs: besides the C, O, and N atoms expected from the polymer, there is also present a low quantity of Cl and Sr. Figure 4b and 4c depict, respectively, images of the distribution of Sr (purple) and Cl (blue) demonstrating a homogeneous distribution of these two ions in the sample.
To determine the role of the MOF in the Sr loading on the NPs, we prepared chitosan-based NPs also loaded with SrCl2, or a mixture of SrCl2 and fructose, instead of the SrFRUCl MOF powder, using the same procedure used for the MOF. Table 1 reports the relative abundances, averaged with respect to different points of the sample, of the Sr and Cl ions resulting from Energy Dispersive Spectroscopy (EDS) analysis for the Sr-NPs, chitosan-based NPs loaded with SrCl2 alone, and those loaded with SrCl2 and fructose.
From the results in Table 1, it is clear that chlorides were loaded on the NPs in similar amounts in all the sample, while Sr was loaded only in the presence of the SrFRUCl MOF, confirming the role of the MOF in the encapsulation of the Sr cation. Notably, a significant amount of chlorides was loaded on the chitosan NPs loaded with SrCl2. Thus, the chitosan NPs should possess high positive charges that are balanced by the negative Cl anions. This suggests that the chitosan NPs would refuse the Sr2+ ions, and only the presence of the negative Cl ions induced the encapsulation of the Sr cations.
2.3. Geometry Optimization and Born-Oppenheimer Molecular Dynamics (BOMD)
To elucidate the interaction between the chitosan-based NPs and the Sr2+ and Cl- ions, we performed ab initio calculations, starting from the simulation of a fragment of chitosan, linking two units of chitin, the constituent monomer of the chitosan, whose XRD structure was downloaded from the crystallographic CSD databank (CCDC 1425611, the image of the unit and some crystallographic data are reported in Figure S2 and Table S1 in Supplementary Materials). In a previous study on the CS/PVA/GA hydrogel [45], it was suggested that interactions through hydrogen bonding between chitosan and PVA and/or its chemical cross-linking with GA, induce modifications to the amine groups. Therefore, to simulate a fragment of chitosan NPs, we assembled two chitin units, and one amide group was modified to NH2 (FRAG 1, Figure 5).
In Table 2, the bond lengths of the FRAG1, calculated at the B3LYP/6-31G-d level of theory, are compared to the corresponding X-ray structural data of chitin downloaded from CSD. Computational optimized coordinates (Å) for FRAG1 are reported in Table 3S. The low differences (the largest is 0.09 Å) between the XRD and optimized B3LYP parameters, suggest that the chosen model adequately simulates the chitin unit.
Starting from the optimized geometry of FRAG1, we performed Born-Oppenheimer molecular dynamics (BOMD) calculations on fragments containing an Sr2+ ion, two chlorides, and a fructose unit (FRAG2, Figure 6a). To construct FRAG2, we were inspired by the original metal-organic framework (MOF) structure, a crystalline material with fully known atomic positions. As shown in Figure 6b, the oxygen atoms of the hydroxyl groups in the MOF coordinate the Sr2+ ion. A similar structure was considered for BOMD calculations.
This new fragment was optimized at the same level of theory used for FRAG1. Computational optimized coordinates (Å) for FRAG2 are reported in Table S4. In Table 3, the bond lengths of the chitin units in FRAG2 are compared to the corresponding X-ray structural data of chitin from the CSD. Again, the low differences (the largest being 0.08 Å) between the XRD and optimized B3LYP parameters suggest that the chosen model adequately simulates the chitin unit.
Part of the interest in conducting BOMD was to assess the stability of the system presented. The stability was evaluated based on changes in their coordinate configurations and energy levels. To measure variations in coordinate configurations, we employed pairwise Root Mean Square Deviation (RMSD), which considers all possible pairs of points between two structures, unlike standard RMSD. This method provides a more robust description of positional changes in the compounds.
Figure 7 displays the pairwise RMSD graph for FRAG2. The vertical and horizontal axes represent an ordered pair by comparing two frames. The color bar on the side indicates the RMSD values, where yellow represents the highest value and purple is the lowest.
There are only 12 frames representing the total simulation time. This is because the configuration represents a very unstable system, causing some atoms to separate so much during the simulation that the dynamics could not continue due to convergence issues.
Since FRAG2 is unstable, we modified the chitosan model, considering that the environment for forming the CS/PVA/GA hydrogel is acid. Thus, we can hypothesize that the NH2 group actually was present as NH3+. Therefore, the chitosan fragment should have positive charges on the surface, which should be responsible for the attraction of the chlorides in the chitosan structure. With these considerations, we created a new model of chitosan (FRAG3, Figure 8), in which one NH2 of a chitin unit is protonated. Table 4 shows the bond lengths of the chitin units of the FRAG3, optimized at the B3LYP/6-31G-d level of theory, compared to the corresponding X-ray structural data. Also in this case, we found low differences (the largest is 0.09 Å), so we performed the BOMBD calculations on this fragment (Figure 9).
Figure 9 reports the Pairwise RMSD graph for the configuration FRAG4. There are 3000 frames representing the total simulation time. On the contrary with respect to FRAG2, FRAG3 not only completed the total simulation time, but also exhibited very low RMSD variations: the highest RMSD recorded was 0.06, which is very low. These data indicate a very stable configuration.
The results of FRAG3 can explain the SEM results for the chitosan-based NPs treated with SrCl2, in which only chlorides were encapsulated on the polymeric matrix, and encouraged us to consider a new fragment composed of FRAG3 with Sr2+ and other two Cl- added (FRAG4) and connected as shown in Figure 10. Again, we optimized at B3LYP/6-31G(d) level, and we compared with the chitin structure (see Table 5), and finally we performed BOMD (Figure 11)
Figure 11 displays the pairwise RMSD graphs for FRAG4. There are only 12 frames representing the total simulation time. As for FRAG2, the configuration represents a very unstable system, causing some atoms to separate so much during the simulation that the dynamics could not continue due to convergence issues.
The graphs for the three systems are presented as follows in Figure 12. It can be seen that graph b) shows the slightest variation, whereas graphs a) and c) exhibit diverging energy values.
In summary, the simulations indicate that the protonation of the NH2 group to NH3+ contributes to the system’s stability and explains the experimentally observed encapsulation of chlorides. However, the overall interaction with Sr2+ remains unstable, requiring a more complex system.
2.4. SHG Measurements
The SHG properties of the chitosan-based NPs before and after the Sr loading were characterized using the SHG microscopy technique. Three SHG images were collected in different positions for each sample, and the average intensity generated from the samples was measured (Figure 13). All the images show similar SHG intensities, with a slightly higher brightness of the Sr-loaded chitosan-based NPs than the not-loaded ones (419 ±25 counts after Sr loading versus 397 ±49 counts before Sr loading).
It is noteworthy that chitosan-based NPs before and after Sr loading show a suitable SHG emission for the application as biosensors. This result encouraged us to propose Sr loaded chitosan-based NPs (Sr-NPs) as SHG biosensors that, if opportunely functionalized on the surface, can also carry 89Sr radioisotope for radiotherapy.
3. Materials and Methods
3.1. Synthesis of Chitosan/Poly(Vinyl Alcohol)/Glutaraldehyde Nanospheres Loaded with Sr (Sr- NPs)
The synthesis process of the chitosan/poly(vinyl alcohol) (CS/PVA) nanospheres chemically cross-linked with glutaraldehyde (GA) and loaded with Sr is described in this section. All chemicals utilized were purchased from Sigma-Aldrich and used as received.
3.1.1. Preparation of CS/PVA Hydrogel Blend
The CS/PVA hydrogel blend was obtained by dissolving chitosan (CS) powder in a 1% (w/v) acetic acid water solution, under magnetic stirring for 24 h at room temperature, to obtain a 2.5% w/v chitosan solution. The acetic acid was HPLC-grade, and the CS powder was of medium-molecular weight, with a 75–85% degree of deacetylation. An aqueous solution of poly(vinyl alcohol) (PVA, 10% w/v) was prepared by dissolving PVA, with a 99% degree of hydrolysis and a typical average molecular weight (Mw) of 89,000–98,000 g/mol, in deionized water under magnetic stirring at 80 °C for two hours. Both polymeric solutions were then mixed at a 3:1 (CS:PVA) volume ratio, under magnetic stirring at room temperature for about two hours, until a homogeneous polymeric blend was obtained.
3.1.2. Preparation of CS/PVA/GA Nanospheres
An emulsion was prepared by stirring a solution of polyisobutylene (Polysciences, CAS: 9003-27-4) and sorbitan monooleate (SPAN 80, Fluka, CAS: 1338- 43-8) with an Ultra-Turrax disperser at 12,000 rpm. The concentration of the SPAN-80 in the emulsion was 1% (w/v). Subsequently, the CS/PVA solution was added dropwise to the emulsion, followed by the addition of GA (analytical-grade, 25% w/v) to achieve a weight ratio of approximately 1:1 relative to the polymers in the emulsion. The emulsion was magnetically stirred for 40 min at room temperature, and then centrifuged at 4,000 rpm for 10 min. The formed CS/PVA/GA NPs, named as chitosan-based NPs henceforth, were collected from the bottom of the test tube and washed with two hexane aliquots followed by two washes of deionized water. The wet powder containing the NPs was used without drying in further assays.
3.1.3. Synthesis of the SrFRUCl MOF
To synthesize the SrFRUCl MOF, 0.66 g of strontium chloride hexahydrate and 0.46 g of D-fructose were dissolved in 12 mL of ethanol at 74.85 °C (348 K), at a 1:1 stoichiometric ratio. After a few minutes, both solids were utterly dissolved, precipitating a fine white powder of SrFRUCl. The obtained powder was subsequently washed with a few drops of ethanol, filtered, and air-dried.
Loading of chitosan-based NPs with Sr (Sr-NPs). To load the chitosan-based NPs with Sr, 51 mg of the wet CS/PVA/GA NPs were suspended in 1 mL of ethanol with an equal amount (51 mg) of SrFRUCl MOF fine powder. Subsequently, the suspension was sonicated for ten minutes. After sonication, the suspension appeared homogeneous and was left to rest for two days to favor absorption by the chitosan-based NPs. Finally, the nanospheres were filtered and washed with deionized water to obtain the Sr-NPs.
3.2. Nanosphere Characterization
3.2.1. Fourier Transform Infrared (FTIR) Spectroscopy
An FTIR spectrometer (Nicolet iS50R, Thermo Scientific) equipped with an attenuated total reflectance (ATR) module was used for this analysis. The FTIR spectra were acquired over a wavenumber range from 4000 to 650 cm−1, using 32 scans and a resolution of 2 cm−1. All FTIR spectra were normalized concerning the band at 2950-2850 cm−1, corresponding to the stretching of C-H bonds.
3.2.2. X-ray Powder Diffraction (XRPD) Characterization
The XRPD patterns were collected using the Atlas S2 Rigaku-Oxford Diffraction Gemini R-Ultra diffractometer, equipped with mirror monochromatized Cu-Kα (1.5418 Å) radiation. The wet powders of nanospheres were easily compacted and molded into balls of ca. 0.45 mm in diameter (smaller than the diameter of the X-ray beam). Each ball was glued to a glass capillary and mounted on the goniometer head of the instrument. Each powder pattern was collected by rotating 60 degrees, with an exposure time of 60 s. The CrysAlisPro package (CrysAlisPro 1.171.42.49, Rigaku Oxford Diffraction, 2022) was used for data collection and integration.
3.2.3. Scanning Electron Microscopy (SEM)/Energy Dispersive Spectroscopy (EDS)
Field-Emission Scanning Electron Microscopy (FESEM) and EDS were performed using a TESCAN S9000G FESEM 3010 microscope working at 30kV and equipped with a high-brightness Schottky emitter. For the microanalysis, the OXFORD Ultim Max-software Aztec was used. To average the measurements of the nanospheres, the program ImageJ was used [46].
3.3. Theoretical Simulations
Geometry Optimization. Theoretical calculations were performed using the GAUSSIAN16 program [47]. All the geometries in this work were optimized by gradient-based techniques [48,49,50] with no symmetry constraints at the DFT B3LYP level of theory [51,52], in conjunction with the 6–31G(d) basis set for the C, H, and N atoms [53]. For Sr, the LANL2DZ basis was used [54]. All critical points were characterized as energy minima by calculating their analytical frequencies.
3.3.1. Born-Oppenheimer Molecular Dynamics (BOMD)
BOMD was conducted using the NorthWest Chemistry (NWChem) modeling software [55]. To maintain consistency with other theoretical calculations, the B3LYP functional was used with the 6–31G (d, p) basis set, and a polarization function was added to enhance the description of quantum effects. The basis set used for the Sr ion was Def2-TZVP [56]. The dynamics spanned a total of 3 ps with a timestep of 1 fs.
3.4. Second Harmonic Generation (SHG) Measurements
An Yb-fiber-based pump laser (Emerald Engine HP Basic DUO, A.P.E. GmbH, Berlin, Germany) was used to provide an optimized pulsed laser source to pump an Optical Parametric Oscillator (Levante Emerald OPO) (APE, Berlin, Germany). The excitation source was tuned at about 892.5 nm with a pulse width of about 2 ps, and a repetition rate of 80 MHz.
The beam entered an upright microscope (BX51WI, Olympus, Tokyo, Japan) through the scanning unit (FluoView FV300, Olympus, Tokyo, Japan) for image acquisition. The total average power of the excitation pulses at the sample was set to about 30 mW. The excitation beams were focused on the powder using a microscopy objective (UPlanSApo 20x NA = 0.75, W.D.= 0.65 mm, Olympus), and a condenser objective (UPLSAPO 10× objective NA = 0.4, Olympus) was used to collect forward SHG signal at about 446 nm, that was optically filtered and then detected using a PMT (R3896, Hamamatsu, Japan).
The images were measured by Kalman, averaging five acquisitions, keeping the PMT voltage, gain, and offset constant for all the acquisitions, and using a pixel depth of 12 bits. The collected images were processed in a way to extract the average intensity of the foreground using the same threshold level of 300 counts.
4. Conclusions
In this work, we synthesized new chitosan-based NPs loaded with Sr and Cl ions, that can be applied as carriers of the radioisotope 89Sr for radio-cancer therapy. From SEM analyses of the NPs, we observed a good homogeneously dispersed amount of Sr and Cl ions in the NPs. However, to load the metal ion on the NPs, the role of the SrFRUCl MOF is fundamental, since the loading with SrCl2 produces NPs loaded only with chlorides. Evidently, the fructose that coordinates the Sr cation in the MOF has a role in the encapsulation of the metal ion.
In order to understand the interaction of the chitosan matrix with the Sr and Cl ions, BOMD calculations were performed on different fragments, that simulate small chitosan matrices with the ions encapsulated. The calculations evidenced that chitosan-based NPs, during their synthesis in an acidic environment, acquire positive charges on the surfaces, due to the acidification of some NH2 groups of chitosan. The positive charges are able to attract the chlorides, which explains the amount of chlorides encapsulated on NPs observed on EDS spectroscopy, more than the stoichiometric ratio Sr:Cl 1:2 expected for the charge balance. However, BOMD simulations of the fragments with Sr ion encapsulated were not stable, probably because the chitosan matrix simulated was too small, and unfortunately, we were not yet able to clearly understand the interaction of this ion with chitosan with our current simulations. However, the NPs loaded with the Sr ions, show a good SHG emission, which encouraged us to propose them also as biosensors. Thus, the chitosan NPs enriched with 89Sr can be proposed for theranostic purposes, i.e., at the same time for therapy and diagnostic purposes.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., (1) description of X-ray diffraction structure of the SrFruCl MOF, (2) details on X-ray molecular structure of chitin, (3) computational optimized coordinates of fragments.
Funding
this work was developed with financial support from PAPIIT DGAPA-UNAM program through grant IN101624, and from MIUR (Ministero dell’Istruzione, dell’Università e della Ricerca).
Data Availability Statement
The original contributions presented in this study are included in the article and further inquiries can be directed to the corresponding authors.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ravi Kumar, M.N.V. A review of chitin and chitosan applications. React. Funct. Polym. 2000, 46, 1–27. [Google Scholar] [CrossRef]
- Wong, T.W.; Chan, L.W.; Kho, S.B.; Sia Heng, P.W. Design of controlled-release solid dosage forms of alginate and chitosan using microwave. J. Control. Release 2002, 84, 99–114. [Google Scholar] [CrossRef]
- Abdeen, R.; Salahuddin, N. Modified chitosan-clay nanocomposite as a drug delivery system intercalation and in vitro release of ibuprofen. J. Chem. 2013, 9, 576370–576379. [Google Scholar]
- Wong, T.W.; Chan, V.S.; Kho, B.; Sia Heng, P.W. Design of controlled-release solid dosage forms of alginate and chitosan using microwave. J. Control. Release 2002, 84, 99–114. [Google Scholar] [CrossRef]
- Nanda, R.; Sasmal, A.; Nayak, P.L. Preparation and characterization of chitosan–polylactide composites blended with Cloisite 30B for control release of the anticancer drug paclitaxel. Carbohydr. Polym. 2011, 83, 988–994. [Google Scholar] [CrossRef]
- Chandy, T.; Sharma, C.P. Chitosan as a biomaterial. Biomater. Artif. Cell. 1990, 18, 1–24. [Google Scholar] [CrossRef]
- Pathania, D.; Gupta, D.; Agarwal, S.; Asif, M.; Gupta, V.K. Fabrication of chitosan-g-poly (acrylamide)/CuS nanocomposite for controlled drug delivery and antibacterial activity. Materials Science and Engineering C 2016, 64, 428–435. [Google Scholar] [CrossRef]
- Kato, Y.; Onishi, H.; Machida, Y. Application of chitin and chitosan derivatives in the pharmaceutical field. Curr. Pharm. Biotechnol. 2003, 4, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Singla, A.K.; Chawla, M. Chitosan: Some pharmaceutical and biological aspects—an update. J. Pharm. Pharmacol. 2001, 53, 1047–1067. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.A.; Syeda, J.T.M.; Wasan, K.M.; Wasan, E. K. An Overview of Chitosan Nanoparticles and Its Application in Non-Parenteral Drug Delivery. Pharmaceutics 2017, 9, 53–79. [Google Scholar] [CrossRef]
- Yanat, M.; Schroën, K. Preparation methods and applications of chitosan nanoparticles; with an outlook toward reinforcement of biodegradable packaging. React. Funct. Polym. 2021, 161, 104849–104861. [Google Scholar] [CrossRef]
- Ji, J.; Hao, S.; Wu, D.; Huang, R.; Xu, Y. Preparation, characterization and in vitro release of chitosan nanoparticles loaded with gentamicin and salicylic acid. Carbohydr. Polym. 2011, 85, 803–808. [Google Scholar] [CrossRef]
- Ghadi, A.; Mahjoub, S.; Tabandeh, F.; Talebnia, F. Synthesis and optimization of chitosan nanoparticles: Potential applications in nanomedicine and biomedical engineering. Caspian J Intern Med. 2014, 5, 156–161. [Google Scholar] [PubMed]
- Praveen, E.; Muruganb, S.; Jayakumar, K. Investigations on the existence of piezoelectric property of a bio-polymer – chitosan and its application in vibration sensors. RSC Adv. 2017, 7, 35490–35495. [Google Scholar] [CrossRef]
- Aghigh, A. , Bancelin, S.; Rivard, M., Pinsard, M., Ibrahim, H., Légaré, F. Second harmonic generation microscopy: a powerful tool for bio-imaging. Biophys. Rev. 2023, 15, 43–70. [Google Scholar] [CrossRef]
- Tran, R.J.; Sly, K.L.; Conboy, J.C. Applications of Surface Second Harmonic Generation in Biological Sensing. Annu. Rev. Anal. Chem. 2017, 10, 387–414. [Google Scholar] [CrossRef]
- Salih, S.; Alkatheeri, A.; Alomaim, W.; Elliyanti, A. Radiopharmaceutical Treatments for Cancer Therapy, Radionuclide Characteristic, Applications, and Challenges. Molecules 2022, 27, 5231–5246. [Google Scholar] [CrossRef] [PubMed]
- Asadian, S.; Mirzaei, H.; Kalantari, B.A.; Davarpanah, M.R.; Mohamadi, M.; Shpichka, A.; Nasehi, L.; Es, H.A.; Timashev, P.; Najimi, M.; Gheibi, N.; Hassan, M.; Vosough, M. β-radiating radionuclides in cancer treatment, novel insight into promising approach. Pharmacol. Res. 2020, 160, 105070–105081. [Google Scholar] [CrossRef]
- Widel, M.; Przybyszewski, W.M.; Cieslar-Pobpuda, A.; Saenko, Y.V.; Rzeszowska-Wolny, J. ; Bystander normal human fibroblasts reduce damage response in radiation targeted cancer cells through intercellular ROS level modulation. Muta. Res. 2012, 731, 117–124. [Google Scholar] [CrossRef]
- Yaneva, M.P.; Semerdjieva, M.; Radev, L.R.; Ivanova, E.K.; Geiman, M.; Vlaikova, M.I.; Mihova, L.S. Radionuclide therapy of cancer patients with bone metastases. Folia Med. (Plovdiv) 2005, 47, 63–69. [Google Scholar]
- Nightengale, B.; Brune, M.; Blizzard, S.P. , Ashley-Johnson, M.; Slan, S. Am. J. Health Syst. Pharm. 1995, 52, 2189–2195. [Google Scholar] [CrossRef] [PubMed]
- Dafermou, A.; Colamussi, P.; Giganti, M.; Cittanti, C.; Bestagno, M.; Piffanelliet, A. A multicentre observational study of radionuclide therapy in patients with painful bone metastases of prostate cancer. Eur. J. Nucl. Med. 2001, 28, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Nightengale, B.; Brune, M.; Blizzard, S.P.; Ashley-Johnson, M.; Slan, S. Strontium chloride Sr 89 for treating pain from metastatic bone disease. Am. J. Health-Syst. Ph. 1995, 52, 2189–2195. [Google Scholar] [CrossRef]
- Adhikari, H.S.; Yadav, P.N. Anticancer Activity of Chitosan, Chitosan Derivatives, and Their Mechanism of Action. Int. J. Biomater. 2018, 2952085–2952114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, F.; Li, Y.; Yang, X.; Huang, J.; Lv, T.; Zhang, Y.; Chen, J.; Chen, H.; Gao, Y.; Liu, G.; Jia, L. Chitosan-based nanoparticles for improved anticancer efficacy and bioavailability of mifepristone. Beilstein J. Nanotechnol. 2016, 7, 1861–1870. [Google Scholar] [CrossRef] [PubMed]
- Gavhane, Y.N.; Gurav, A.S.; Yadav, A.V. Chitosan and Its Applications: A Review of Literature. Int. J. Res. Pharm. Biomed. Sci. 2013, 4, 312–331. [Google Scholar]
- Thanou, M.; Verhoef, J.C.; Junginger, H.E. Chitosan and its derivatives as intestinal absorption enhancers. Adv. Drug Delivery Rev. 2001, 50, S91–S101. [Google Scholar] [CrossRef]
- Praveen, E.; Murugan, S.; Jayakumar, K. Investigations on the existence of piezoelectric property of a bio-polymer – chitosan and its application in vibration sensors. RSC Adv. 2017, 7, 35490–35495. [Google Scholar] [CrossRef]
- Marabello, D.; Antoniotti, P.; Benzi, P.; Canepa, C.; Mortati, L.; Sassi, M.P. Synthesis, structure and non-linear optical properties of new isostructural b-D-fructopyranose alkaline halide metal–organic frameworks: a theoretical and an experimental study. Acta Cryst. 2017, B73, 737–743. [Google Scholar] [CrossRef]
- Barnett, R.N.; Landman, U. Born-Oppenheimer molecular-dynamics simulations of finite systems: Structure and dynamics of (H2O)2. Physical Review B 1993, 48, 2081–2097. [Google Scholar] [CrossRef]
- Vásquez-Pérez, J.M.; Martínez, G.U.G.; Köster, A.M.; Calaminici, P. The discovery of unexpected isomers in sodium heptamers by Born–Oppenheimer molecular dynamics. J. Chem. Phys. 2009, 131, 124126–124136. [Google Scholar] [CrossRef] [PubMed]
- Naveen Kumar, H.M.P.; Prabhakar, M.N.; Venkata Prasad, C.; Madhusudhan Rao, K.; Ashok Kumar Reddy, T.V.; Chowdoji Rao, K.; Subha, M.C.S. Compatibility studies of chitosan/PVA blend in 2% aqueous acetic acid solution at 30°C. Carbohyd. Polym. 2010, 82, 251–255. [Google Scholar] [CrossRef]
- Zhuang, P.-Y.; Li, Y.-L.; Fan, L.; Lin, J.; Qiao-Ling, H. Modification of chitosan membrane with poly(vinyl alcohol) and biocompatibility evaluation. Int. J. Biol. Macromol. 2012, 50, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Pizano, M.D.; Vélaz, I.; Penas, F.J.; Zavala-Rivera, P.; Rosas-Durazo, A.J.; Maldonado-Arce, A.D.; Martínez-Barbosa, M.E. Effect of freeze-thawing conditions for preparation of chitosanpoly(vinyl alcohol) hydrogels and drug release studies. Carbohyd. Polym. 2018, 195, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Brunel, F.; Véron, L.; David, L.; Domard, A.; Delair, T. A Novel Synthesis of Chitosan Nanoparticles in Reverse Emulsion. Langmuir 2008, 24, 11370–11377. [Google Scholar] [CrossRef] [PubMed]
- Sinha, V.R.; Singla, A.K.; Wadhawan, S.; Kaushik, R.; Kumria, R.; Bansal, K.; Dhawan, S. Chitosan microspheres as a potential carrier for drugs. Int. J. Pharm. 2004, 274, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Costa-Júnior, E.S.; Barbosa-Stancioli, E.F.; Mansur, A.A.P.; Vasconcelos, W.L.; Mansur, H.S. Preparation and characterization of chitosan/poly(vinyl alcohol) chemically crosslinked blends for biomedical applications. Carbohyd. Polym. 2009, 76, 472–481. [Google Scholar] [CrossRef]
- He, Q.; Gong, K.; Ao, Q.; Ma, T.; Yan, Y.; Gong, Y.; Zhang, X. Positive charge of chitosan retards blood coagulation on chitosan films. J. Biomater. Appl. 2013, 27, 1032–1045. [Google Scholar] [CrossRef] [PubMed]
- Gonenc, I.; Us, F. Effect of glutaraldehyde crosslinking on degree of substitution, thermal, structural, and physicochemical properties of corn starch. Starch-Stärke 2019, 71, 1800046–1800056. [Google Scholar] [CrossRef]
- Mansur, H.S.; Sadahira, C.M.; Souza, A.N.; Mansur, A.A. FTIR spectroscopy characterization of poly (vinyl alcohol) hydrogel with different hydrolysis degree and chemically crosslinked with glutaraldehyde. Mat. Sci. Eng. C 2008, 28, 539–548. [Google Scholar] [CrossRef]
- Ceylan, S.; Alatepeli, B. Evaluation of PVA/Chitosan Cryogels as Potential Tissue Engineering Scaffolds; Synthesis, cytotoxicity and genotoxicity investigations. JOTCSA 2021, 8, 69–78. [Google Scholar] [CrossRef]
- Alimuddin, A.; Rafeeq, M. Synthesis and Characterization of Strontium Oxide Nano Particle by Sol-Gel Method. Orient. J. Chem. 2021, 37, 177–180. [Google Scholar] [CrossRef]
- Khalil, K.D.; Riyadh, S.M.; Alkayal, N.S.; Bashal, A.H.; Alharbi, K.H.; Alharbi, W. Chitosan-Strontium Oxide Nanocomposite: Preparation, Characterization, and Catalytic Potency in Thiadiazoles Synthesis. Polymers 2022, 14, 2827. [Google Scholar] [CrossRef] [PubMed]
- Apsana, G.; George, P.P.; Devanna, N.; Yuvasravana, R. Biomimetic Synthesis and Antibacterial Properties of Strontium Oxide Nanoparticles Using Ocimum Sanctum Leaf Extract. Asian J. Pharm. Clin. Res. 2018, 11, 384–389. [Google Scholar]
- Garnica-Palafox, I.M.; Sánchez-Arévalo, F.M. Influence of natural and synthetic crosslinking reagents on the structural and mechanical properties of chitosan-based hybrid hydrogels. Carbohyd. Polym. 2016, 151, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.J. ImageJ for microscopy. Biotechniques 2007, 43, S25–S30. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A. ; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A.V.; Bloino, J.; Janesko, B.G.; Gomperts, R.; Mennucci, B.; Hratchian, H.P.; Ortiz, J.V.; Izmaylov, A.F.; Sonnenberg, V.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V.G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery Jr., J.A.; Peralta, J.E.; Ogliaro, F.; Bearpark, M.J.; Heyd, J.J.; Brothers, E.N.; Kudin, K.N.; Staroverov, V.N.; Keith, T.A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.P.; Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; Millam, J.M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Farkas, O.; Foresman, J.B.; Fox, D.J., Gaussian 16, Revision C.01, Inc., Wallingford CT, 2016.
- Schlegel, H.B.; Daudel, C. Computational Theoretical Organic Chemistry. Reidel, Dordrecht, 1981, 129, ed. I. G. Csizsmadia and R. Daudel.
- Schlegel, H.B. An efficient algorithm for calculating ab initio energy gradients using s, p Cartesian Gaussians. J. Chem. Phys. 1982, 77, 3676–3681. [Google Scholar] [CrossRef]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A, 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Hehre, W.J.; Radom, L.; Vr Schleyer, P.; Pople, J.A. Ab Initio Molecular Orbital Theory. Wiley, New York, 1986.
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Aprà, E.; Bylaska, E.J.; de Jong, W.A.; Govind, N.; Kowalski, K.; Straatsma, T.P.; Valiev, M.; Van Dam, H.J.J.; Alexeev, Y.; Anchell, J.; Anisimov, V.; Aquino, F.W.; Atta-Fynn, R.; Autschbach, J.; Bauman, N.P.; Becca, J.C.; Bernholdt, D.E.; Bhaskaran-Nair, K.; Bogatko, S.; Borowski, P.; Boschen, J.; Brabec, J.; Bruner, A.; Cauët, E.; Chen, Y.; Chuev, G.N.; Cramer, C.J.; Daily, J.; Deegan, M.J.O.; Dunning Jr., T.H.; Dupuis, M.; Dyall, K.G.; Fann, G.I.; Fischer, S.A.; Fonari, A.; Früchtl, H.; Gagliardi, L.; Garza, J.; Gawande, N.; Ghosh, S.; Glaesemann, K.; Götz, A.W.; Hammond, J.; Helms, V.; Hermes, E.D.; Hirao, K.; Hirata, S.; Jacquelin, M.; Jensen, L.; Johnson, B.G.; Jónsson, H.; Kendall, R.A.; Klemm, M.; Kobayashi, R.; Konkov, V.; Krishnamoorthy, S.; Krishnan, M.; Lin, Z.; Lins, R.D.; Littlefield, R.J.; Logsdail, A.J.; Lopata, K.; Ma, W.; Marenich, A.V.; Martin del Campo, J.; Mejia-Rodriguez, D.; E. Moore, J.; Mullin, J.M.; Nakajima, T.; Nascimento, D.R.; Nichols, J.A.; Nichols, P.J.; Nieplocha, J.; Otero-de-la-Roza, A.; Palmer, B.; Panyala, A.; Pirojsirikul, T.; Peng, B.; Peverati, R.; Pittner, J.; Pollack, L.; Richard, R.M.; Sadayappan, P.; Schatz,G. C.; Shelton, W.A.; Silverstein, D.W.; Smith, D.M.A.; Soares, T.A.; Song, D.; Swart, M.; Taylor, H.L.; Thomas, G.S.; Tipparaju, V.; Truhlar, D.G.; Tsemekhman, K.; Van Voorhis, T.; Vázquez-Mayagoitia, Á.; Verma, P.; Villa, O.; Vishnu, A.; Vogiatzis, K.D.; Wang, D.; Weare, J.H.; Williamson, M.J.; Windus, T.L.; Woliński, K.; Wong, A.T.; Wu, Q.; Yang, C.; Yu, Q.; Zacharias, M.; Zhang, Z.; Zhao, Y.; Harrison, R.J. NWChem: Past, present, and future. J. Chem. Phys. 2020, 152, 184102–184128.
- Xu, X.; Truhlar, D.G. Performance of Effective Core Potentials for Density Functional Calculations on 3d Transition Metals. J. Chem. Theory Comput. 2012, 8, 80–90. [Google Scholar] [CrossRef]
Figure 1.
FTIR spectra of a) CS-film, b) PVA film, c) CS/PVA film, d) Chitosan-based NPs, and e) GA solution. The dotted lines indicate the assignment of the bands to the relevant functional groups.
Figure 1.
FTIR spectra of a) CS-film, b) PVA film, c) CS/PVA film, d) Chitosan-based NPs, and e) GA solution. The dotted lines indicate the assignment of the bands to the relevant functional groups.
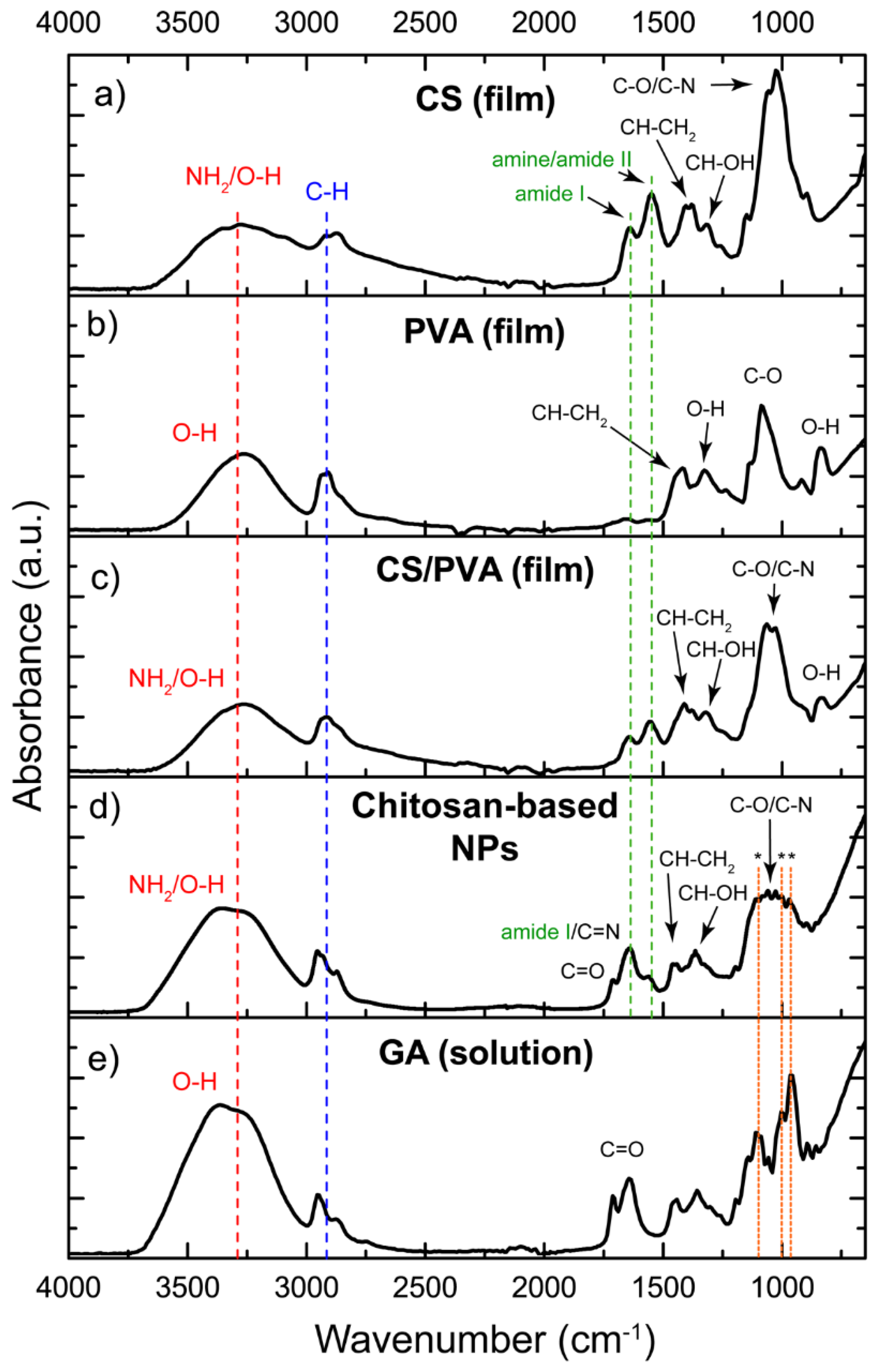
Figure 2.
FTIR spectra of a) Sr-NPs and b) chitosan-based NPs. The dotted lines indicate the assignment of the bands to the relevant functional groups.
Figure 2.
FTIR spectra of a) Sr-NPs and b) chitosan-based NPs. The dotted lines indicate the assignment of the bands to the relevant functional groups.
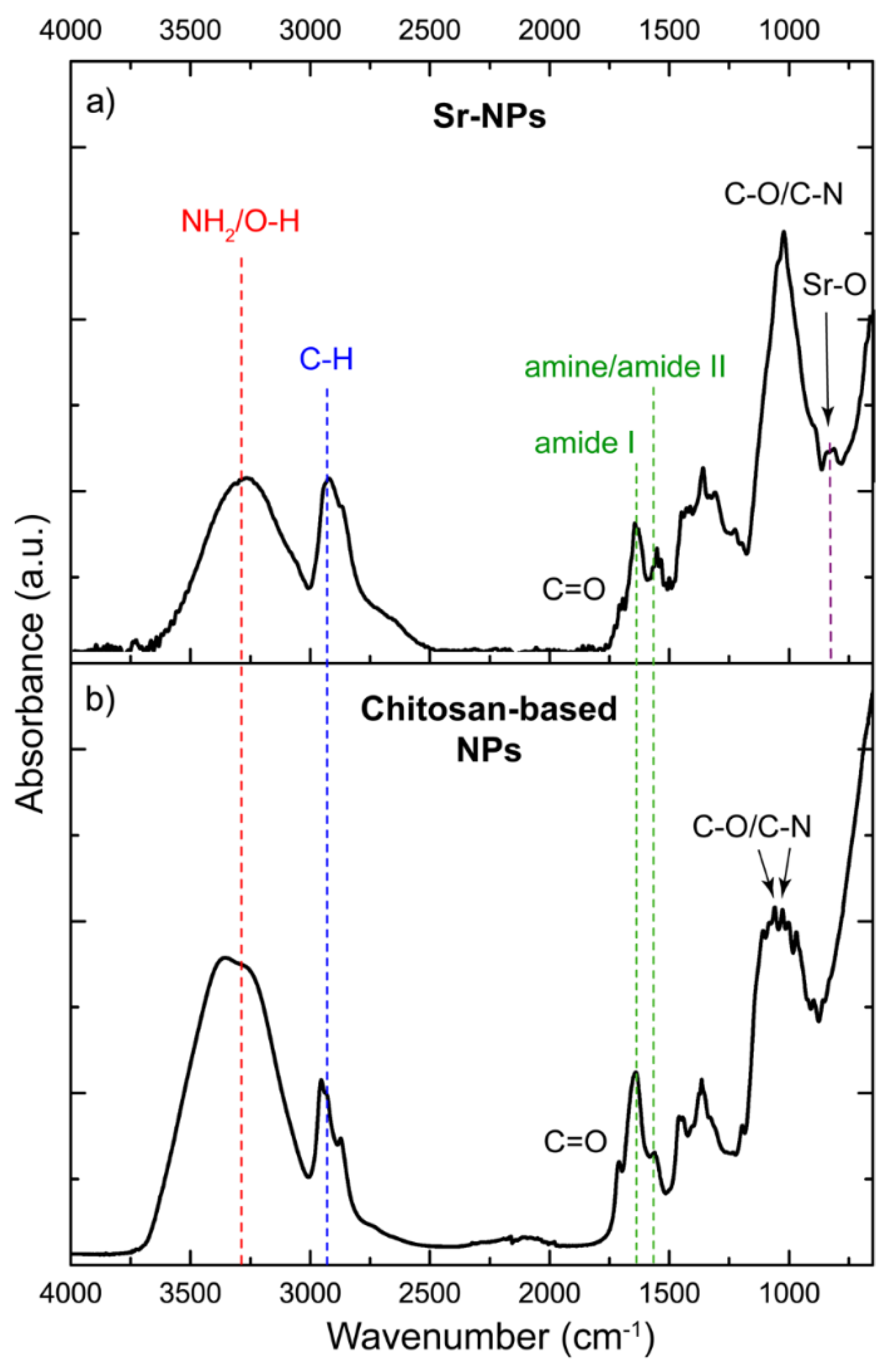
Figure 3.
Measured XRPD patterns of pure chitosan-based NPs (blue), chitosan-based NPs filtered from the SrFRUCl solution (red), Sr-NPs (black), and of the SrFRUCl MOF calculated from the XRD structure (green).
Figure 3.
Measured XRPD patterns of pure chitosan-based NPs (blue), chitosan-based NPs filtered from the SrFRUCl solution (red), Sr-NPs (black), and of the SrFRUCl MOF calculated from the XRD structure (green).
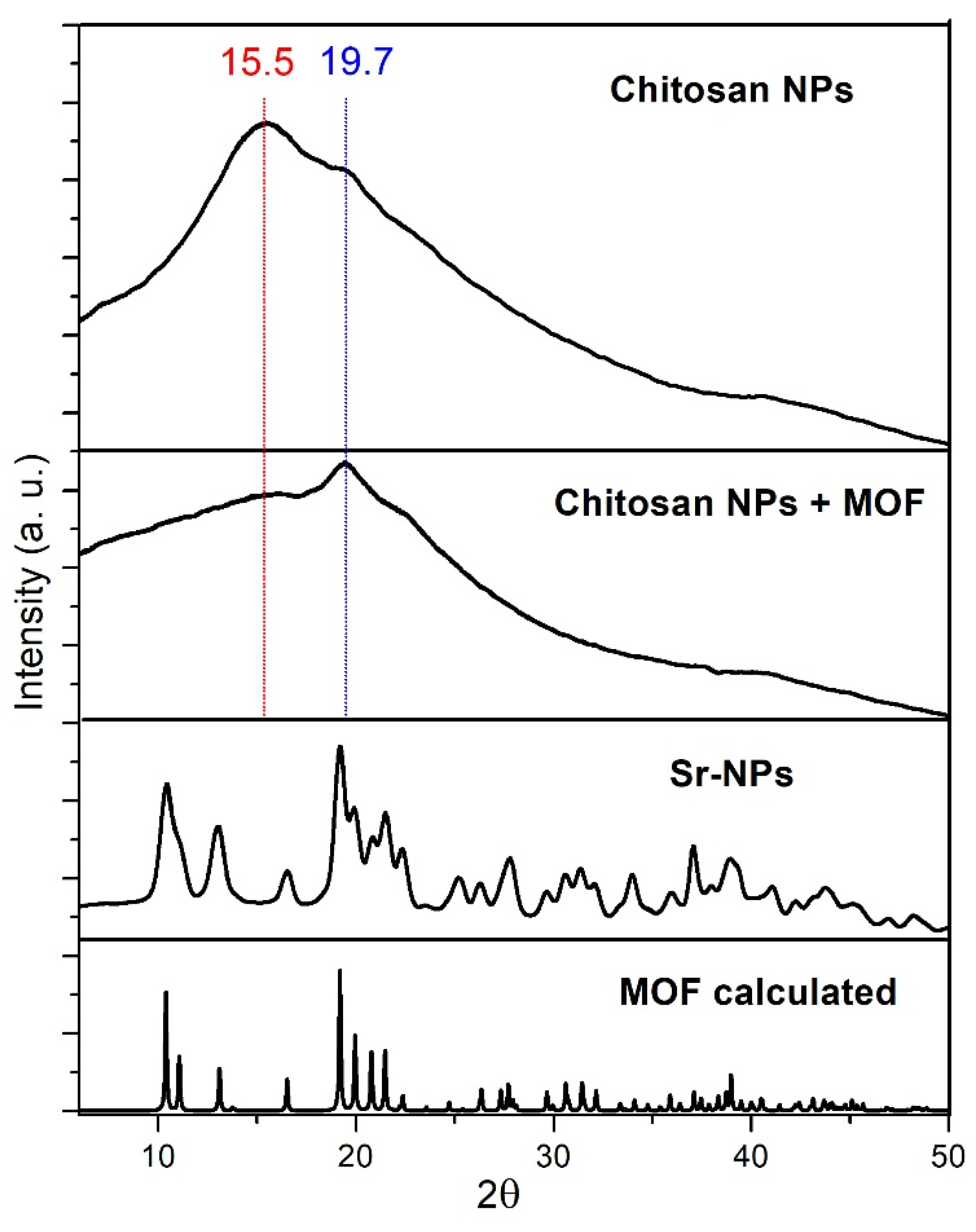
Figure 4.
FESEM and EDS characterization of Sr-NPs: a) SEM electron image of Sr-NPs, b)-c) distribution of Sr and Cl ions, and d) mappng the sum spectrum of Sr-NPs.
Figure 4.
FESEM and EDS characterization of Sr-NPs: a) SEM electron image of Sr-NPs, b)-c) distribution of Sr and Cl ions, and d) mappng the sum spectrum of Sr-NPs.
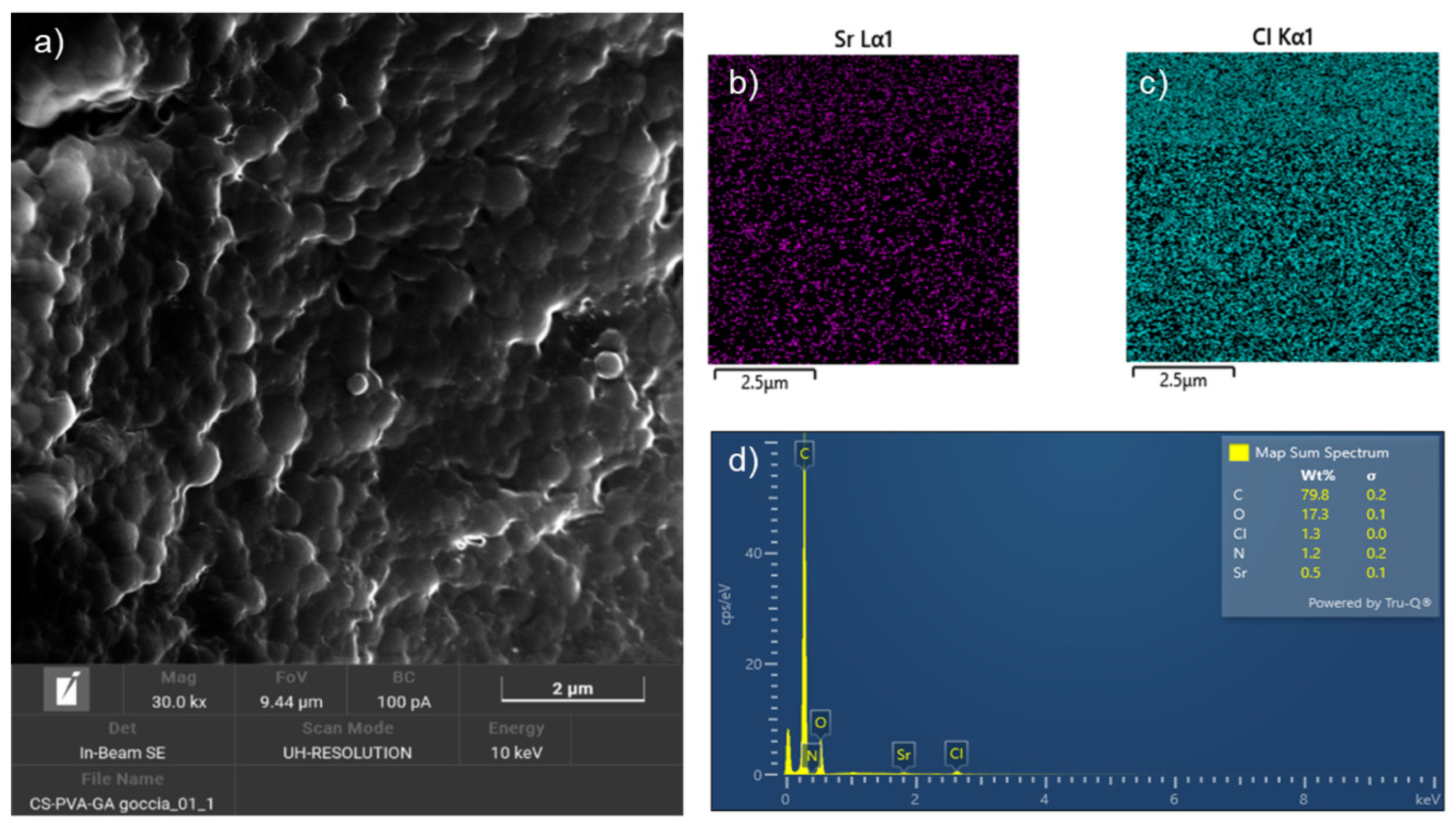
Figure 5.
View of the FRAG1. the fragment composed of two connected chitin units that simulate a fragment of chitosan. One amide group of a chitin unit was modified to NH2. This fragment (FRAG1) was the starting structure for the theoretical calculations. Red=oxygen, blue=nitrogen, grey=carbon, white=hydrogen.
Figure 5.
View of the FRAG1. the fragment composed of two connected chitin units that simulate a fragment of chitosan. One amide group of a chitin unit was modified to NH2. This fragment (FRAG1) was the starting structure for the theoretical calculations. Red=oxygen, blue=nitrogen, grey=carbon, white=hydrogen.
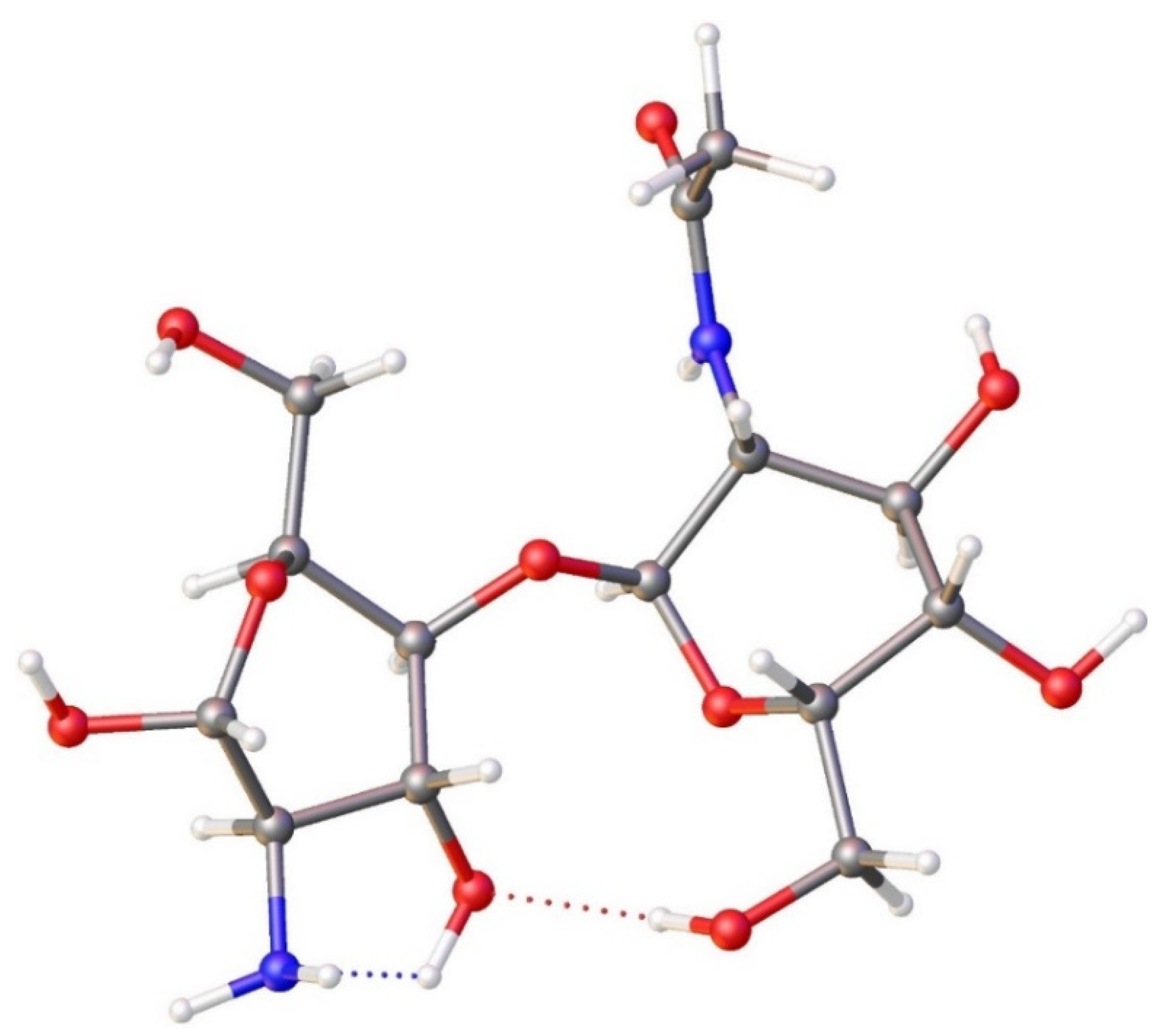
Figure 6.
Molecular fragments used in Born-Oppenheimer molecular dynamics (BOMD) calculations. a) View of FRAG2, composed of FRAG1 with Sr2+, two Cl- and a fructose molecule added. b) Fragment of the structure of MOF SrFRUCl. Red=oxygen, blue=nitrogen, grey=carbon, white=hydrogen.
Figure 6.
Molecular fragments used in Born-Oppenheimer molecular dynamics (BOMD) calculations. a) View of FRAG2, composed of FRAG1 with Sr2+, two Cl- and a fructose molecule added. b) Fragment of the structure of MOF SrFRUCl. Red=oxygen, blue=nitrogen, grey=carbon, white=hydrogen.

Figure 7.
The pairwise RMSD from the dynamics of the FRAG2. The vertical and horizontal axes represent an ordered pair from the comparison between two frames. The color bar on the side indicates the RMSD values, where yellow represents the highest value and purple the lowests.
Figure 7.
The pairwise RMSD from the dynamics of the FRAG2. The vertical and horizontal axes represent an ordered pair from the comparison between two frames. The color bar on the side indicates the RMSD values, where yellow represents the highest value and purple the lowests.
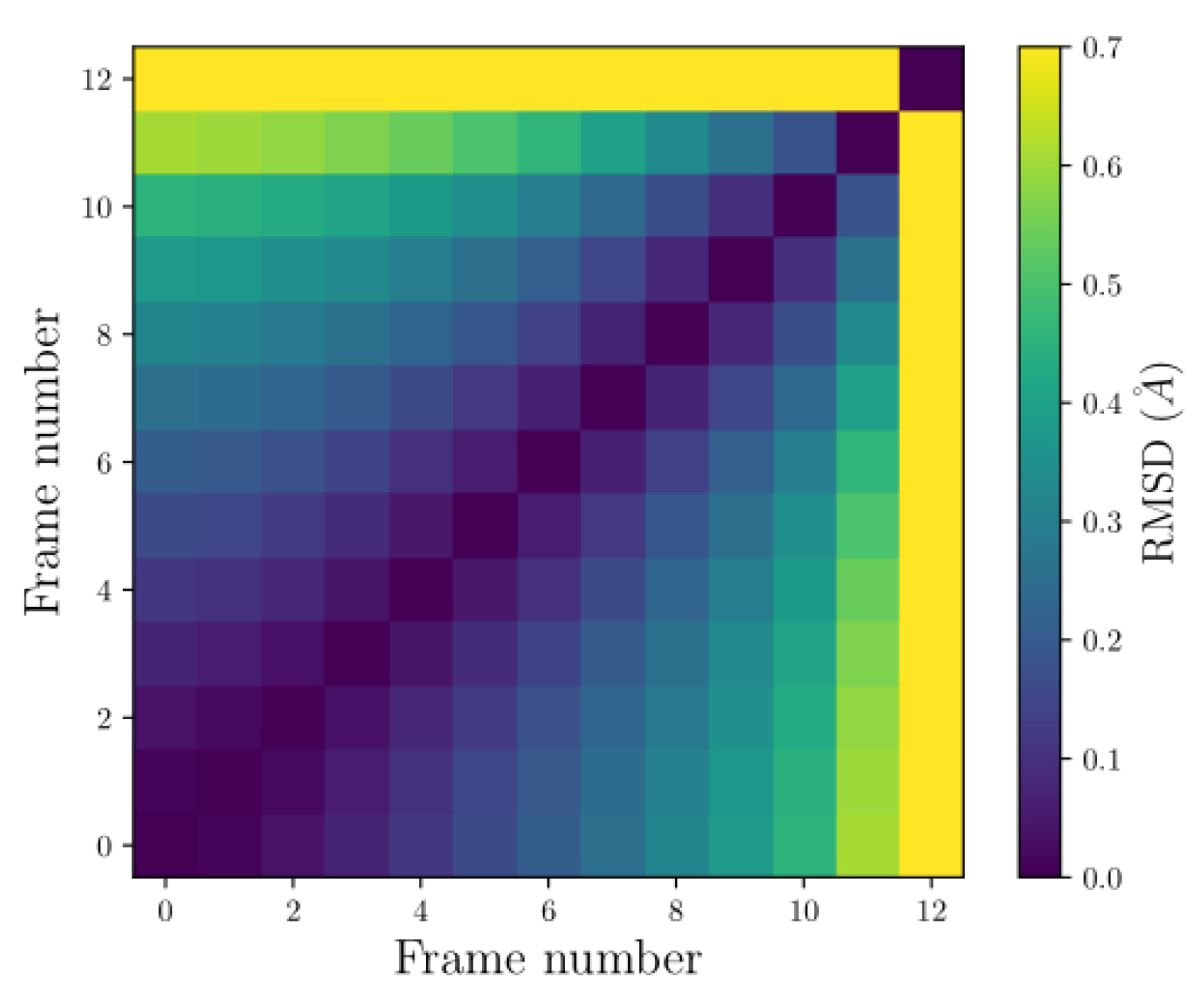
Figure 8.
View of FRAG 3 composed of FRAG1, in which the amide group of one chitin unit was modified to NH3+, and a Cl- was connected through strong hydrogen bonds to the NH3+ group and a near OH group. Red=oxygen, blue=nitrogen, grey=carbon, white=hydrogen.
Figure 8.
View of FRAG 3 composed of FRAG1, in which the amide group of one chitin unit was modified to NH3+, and a Cl- was connected through strong hydrogen bonds to the NH3+ group and a near OH group. Red=oxygen, blue=nitrogen, grey=carbon, white=hydrogen.
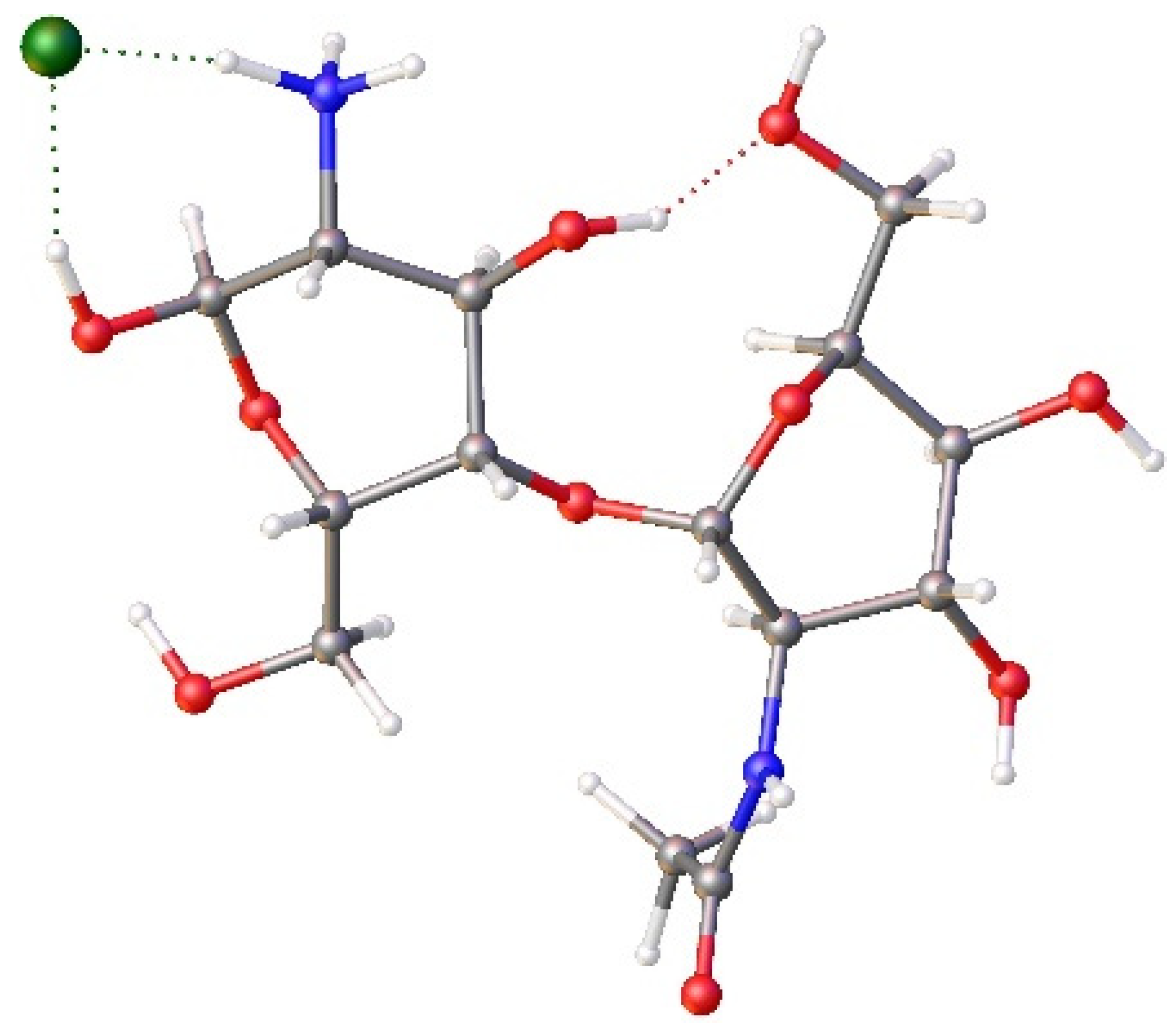
Figure 9.
The pairwise RMSD from the dynamics of the FRAG3. The vertical and horizontal axes represent an ordered pair by comparing two frames. The color bar on the side indicates the RMSD values, where yellow represents the highest value and purple is the lowest.
Figure 9.
The pairwise RMSD from the dynamics of the FRAG3. The vertical and horizontal axes represent an ordered pair by comparing two frames. The color bar on the side indicates the RMSD values, where yellow represents the highest value and purple is the lowest.

Figure 10.
View of FRAG4 composed of FRAG3 with Sr2+, two Cl- and a fructose molecule. Red=oxygen, blue=nitrogen, grey=carbon, white=hydrogen.
Figure 10.
View of FRAG4 composed of FRAG3 with Sr2+, two Cl- and a fructose molecule. Red=oxygen, blue=nitrogen, grey=carbon, white=hydrogen.
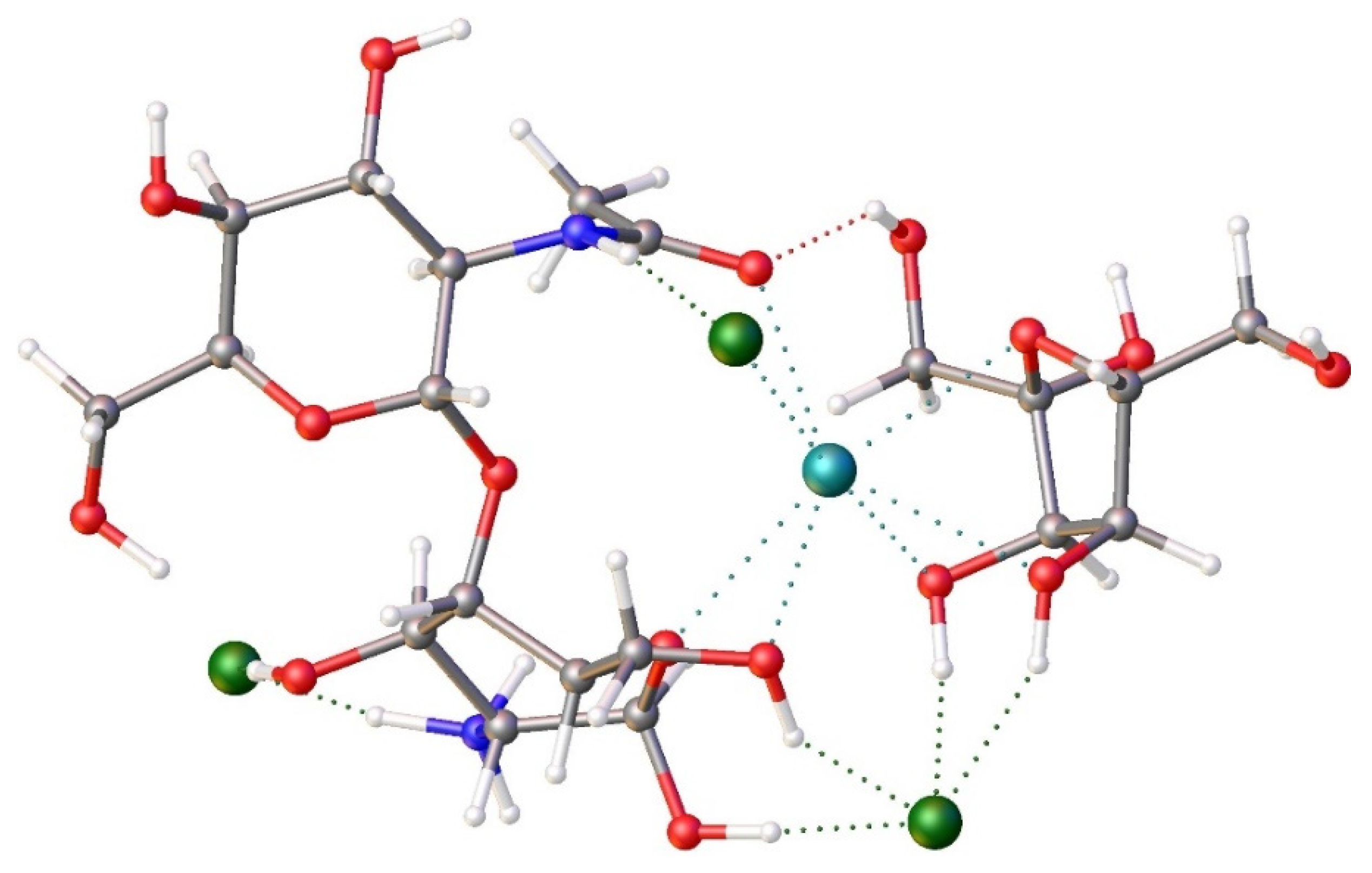
Figure 11.
The pairwise RMSD from the dynamics of the FRAG4. The vertical and horizontal axes represent an ordered pair by comparing two frames. The color bar on the side indicates the RMSD values, where yellow represents the highest value and purple is the lowest.
Figure 11.
The pairwise RMSD from the dynamics of the FRAG4. The vertical and horizontal axes represent an ordered pair by comparing two frames. The color bar on the side indicates the RMSD values, where yellow represents the highest value and purple is the lowest.
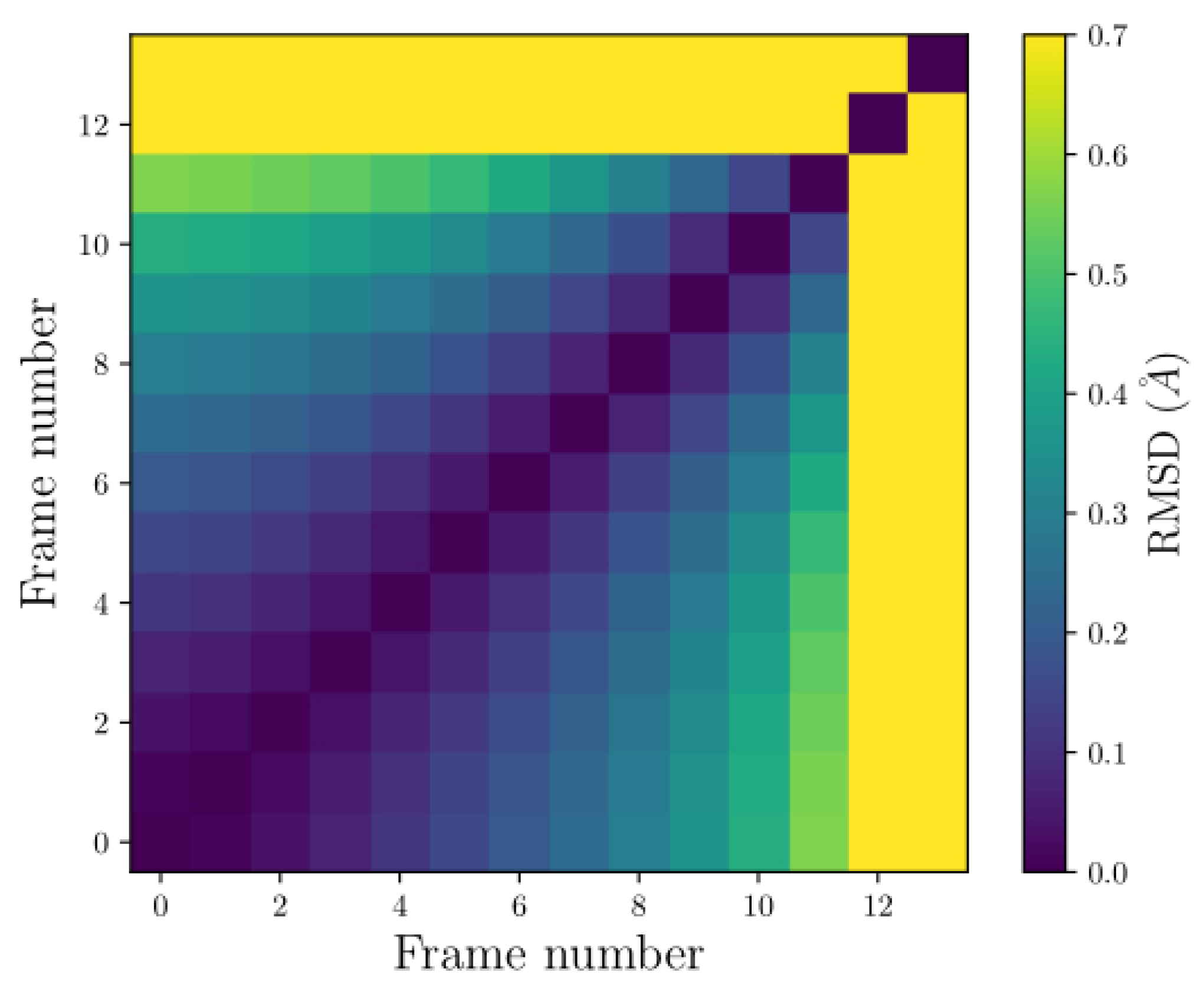
Figure 12.
Graphs of energy versus time for the three simulations studied: a) FRAG2, b) FRAG3, and c) FRAG4. Each point in the graph represents an energy difference between the initial energy and that at the marked time instant. The initial energy was arbitrarily set to zero. Time is measured in picoseconds and energy in Hartrees.
Figure 12.
Graphs of energy versus time for the three simulations studied: a) FRAG2, b) FRAG3, and c) FRAG4. Each point in the graph represents an energy difference between the initial energy and that at the marked time instant. The initial energy was arbitrarily set to zero. Time is measured in picoseconds and energy in Hartrees.
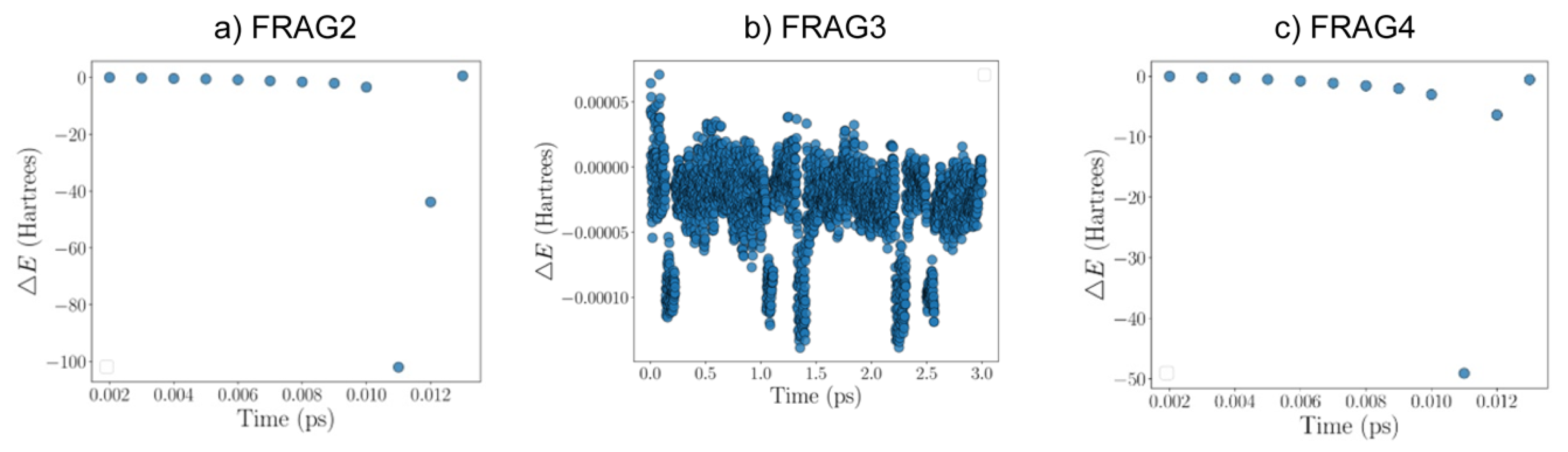
Figure 13.
SHG images measured in different points of the chitosan-based NPs, a)-c) before the Sr loading, d)-f) after the Sr loading (Sr-NPs).
Figure 13.
SHG images measured in different points of the chitosan-based NPs, a)-c) before the Sr loading, d)-f) after the Sr loading (Sr-NPs).


Table 1.
Energy Dispersive Spectroscopy (EDS) analysis for the Sr-NPs, the chitosan-based NPs loaded with SrCl2 alone, and chitosan-based NPs loaded with SrCl2 and fructose. The relative abundances are averaged with respect to different points of the sample.
Table 1.
Energy Dispersive Spectroscopy (EDS) analysis for the Sr-NPs, the chitosan-based NPs loaded with SrCl2 alone, and chitosan-based NPs loaded with SrCl2 and fructose. The relative abundances are averaged with respect to different points of the sample.
| Sr-NPs | ||
| Element | Weight% | Standard deviation |
| Cl | 2.9 | 0.1 |
| Sr | 0.6 | 0.1 |
| Chitosan-based NPs + SrCl2 | ||
| Cl | 1.9 | 0.1 |
| Sr | Not present | Not present |
| Chitosan-based NPs + SrCl2 + Fructose | ||
| Cl | 1.5 | 0.1 |
| Sr | Not present | Not present |
Table 2.
Bond lengths (Å) of the chitin unit from the X-ray data (XRD) and the fragment of chitosan simulated at B3LYP/6–31G(d) level of calculations (FRAG1).
Table 2.
Bond lengths (Å) of the chitin unit from the X-ray data (XRD) and the fragment of chitosan simulated at B3LYP/6–31G(d) level of calculations (FRAG1).
| XRD | FRAG1 | XRD | FRAG1 | ||
|---|---|---|---|---|---|
| C1-C2 | 1.46(4) | 1.55 | C4-O11 | 1.45(4) | 1.43 |
| C1-O1 | 1.47(3) | 1.41 | C5-C6 | 1.48(5) | 1.52 |
| C1-O5 | 1.32(4) | 1.40 | C5-O5 | 1.47(3) | 1.42 |
| C2-C3 | 1.51(2) | 1.54 | C6-O6 | 1.42(5) | 1.41 |
| C2-N1 | 1.43(4) | 1.45 | C7-C8 | 1.49(2) | 1.52 |
| C3-C4 | 1.50(2) | 1.53 | C7-N1 | 1.38(4) | 1.38 |
| C3-O3 | 1.40(3) | 1.42 | C7-O7 | 1.31(4) | 1.22 |
| C4-C5 | 1.51(4) | 1.54 | O1-C42 | 1.45(4) | 1.41 |
1-X,-Y,-1/2+Z; 2-X,-Y,1/2+Z.
Table 3.
Bond lengths (Å) of the chitin unit from the X-ray data (XRD) and the fragment of chitosan interacting with SrCl2 and Fructose (FRAG2) simulated at B3LYP/6–31G(d) level of calculations.
Table 3.
Bond lengths (Å) of the chitin unit from the X-ray data (XRD) and the fragment of chitosan interacting with SrCl2 and Fructose (FRAG2) simulated at B3LYP/6–31G(d) level of calculations.
| XRD | FRAG2 | XRD | FRAG2 | ||
|---|---|---|---|---|---|
| C1-C2 | 1.46(4) | 1.54 | C4-O11 | 1.45(4) | 1.42 |
| C1-O1 | 1.47(3) | 1.43 | C5-C6 | 1.48(5) | 1.53 |
| C1-O5 | 1.32(4) | 1.40 | C5-O5 | 1.47(3) | 1.44 |
| C2-C3 | 1.51(2) | 1.53 | C6-O6 | 1.42(5) | 1.41 |
| C2-N1 | 1.43(4) | 1.46 | C7-C8 | 1.49(2) | 1.52 |
| C3-C4 | 1.50(2) | 1.55 | C7-N1 | 1.38(4) | 1.35 |
| C3-O3 | 1.40(3) | 1.42 | C7-O7 | 1.31(4) | 1.25 |
| C4-C5 | 1.51(4) | 1.54 | O1-C42 | 1.45(4) | 1.42 |
1-X,-Y,-1/2+Z; 2-X,-Y,1/2+Z.
Table 4.
Relevant distances (Å) of the chitin unit from the X-ray data (XRD) and the fragment of chitosan with an NH3+ group and a Cl- anion, simulated at B3LYP/6–31G(d) level of calculations (FRAG3).
Table 4.
Relevant distances (Å) of the chitin unit from the X-ray data (XRD) and the fragment of chitosan with an NH3+ group and a Cl- anion, simulated at B3LYP/6–31G(d) level of calculations (FRAG3).
| XRD | FRAG4 | XRD | FRAG4 | ||
|---|---|---|---|---|---|
| C1-C2 | 1.46(4) | 1.55 | C4-O11 | 1.45(4) | 1.42 |
| C1-O1 | 1.47(3) | 1.41 | C5-C6 | 1.48(5) | 1.53 |
| C1-O5 | 1.32(4) | 1.40 | C5-O5 | 1.47(3) | 1.44 |
| C2-C3 | 1.51(2) | 1.54 | C6-O6 | 1.42(5) | 1.42 |
| C2-N1 | 1.43(4) | 1.45 | C7-C8 | 1.49(2) | 1.52 |
| C3-C4 | 1.50(2) | 1.53 | C7-N1 | 1.38(4) | 1.38 |
| C3-O3 | 1.40(3) | 1.42 | C7-O7 | 1.31(4) | 1.22 |
| C4-C5 | 1.51(4) | 1.54 | O1-C42 | 1.45(4) | 1.43 |
1-X,-Y,-1/2+Z; 2-X,-Y,1/2+Z.
Table 5.
Relevant distances (Å) of the chitin unit from the X-ray data (XRD) and the fragment of chitosan loaded with a Cl anion, simulated at B3LYP/6–31G(d) level of calculations (FRAG4).
Table 5.
Relevant distances (Å) of the chitin unit from the X-ray data (XRD) and the fragment of chitosan loaded with a Cl anion, simulated at B3LYP/6–31G(d) level of calculations (FRAG4).
| XRD | FRAG4 | XRD | FRAG4 | ||
|---|---|---|---|---|---|
| C1-C2 | 1.46(4) | 1.55 | C4-O11 | 1.45(4) | 1.42 |
| C1-O1 | 1.47(3) | 1.41 | C5-C6 | 1.48(5) | 1.53 |
| C1-O5 | 1.32(4) | 1.40 | C5-O5 | 1.47(3) | 1.44 |
| C2-C3 | 1.51(2) | 1.54 | C6-O6 | 1.42(5) | 1.42 |
| C2-N1 | 1.43(4) | 1.45 | C7-C8 | 1.49(2) | 1.52 |
| C3-C4 | 1.50(2) | 1.53 | C7-N1 | 1.38(4) | 1.38 |
| C3-O3 | 1.40(3) | 1.42 | C7-O7 | 1.31(4) | 1.22 |
| C4-C5 | 1.51(4) | 1.54 | O1-C42 | 1.45(4) | 1.43 |
1-X,-Y,-1/2+Z; 2-X,-Y,1/2+Z.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



