
Submitted:
05 July 2024
Posted:
09 July 2024
Read the latest preprint version here
Abstract
1) Background: Impeded resolution of inflammation contributes substantially to the pathogenesis of Alzheimer's disease (AD); consequently, resolving inflammation is pivotal to the amelioration of AD pathology. This can potentially be achieved by the treatment with specialized pro-resolving lipid mediators (SPMs), which should resolve neuroinflammation in brains. 2) Methods: Here, we report the effects of long-term treatment with a SPM, maresin like 1 (MarL1), on AD pathogenesis in a transgenic 5xFAD mouse model. 3) Results: MarL1 treatment reduced Aβ overload, curbed the loss of neurons in brains especially cholinergic neurons associated with cleaved-caspase-3-associated apoptotic degeneration, reduced astrogliosis and microgliosis and the pro-inflammatory M1 polarization of microglia, curbed the AD-associated decline in an-ti-inflammatory Iba1+Arg-1+-M2 microglia, reduced astrogliosis-associated level of neuroin-flammatory TNF-α, inhibited phenotype switching of pro-inflammatory neurotoxic A1 astrocytes and N1 neutrophils, promoted polarization of inflammation-resolving neuroprotective A2 as-trocytes, promoted the blood–brain-barrier-associated tight-junction protein claudin-5 and de-creased neutrophil leakage in 5xFAD brains, and induced the switch of neutrophils toward the inflammation-resolving N2 phenotype. 4) Conclusion: Long-term administration of MarL1 mit-igates AD-related neuropathogenesis in brains by curbing neuroinflammation and neurodegen-eration. These findings provided the preclinical leads and mechanistic insights for the develop-ment of MarL1 into an effective modality to ameliorate AD pathogenesis.
Keywords:
Maresin-like
; Alzheimer's Disease
; neuroinflammation
; neuropathogenesis
; amyloid-β (Aβ)
; cholinergic neuron
; cleaved-caspase-3
; M1 or M2 microglia
; A1 or A2 astrocytes
; N1 or N2 neutrophil
1. Introduction
Alzheimer's disease (AD), the most prevalent form of dementia around the globe, develops over a period of several years, as a disease with both early and late onset and an increasing severity of cognitive decline [1]. AD affects more than 6.5 million people over the age of 65 years in the United States alone [2] and 50 million people worldwide [3]. The United Nations estimates are that the global AD population will increase to nearly 152 million by 2050 [3]. The main neuropathological characteristics of AD are the accumulation of amyloid-β (Aβ) peptide in brain parenchyma and perivascular regions as senile plaques, the presence of hyper-phosphorylated (phospho) tau protein as neurofibrillary tangles, and an accompanying synaptic and neuronal loss mainly in the hippocampal and cortical regions of the brain [4]. The early clinical signs of AD manifest as neurobehavioral symptoms such as difficulty remembering names, events, and recent conversations, followed by impaired communication skills, disorientation, confusion, and lack of judgment. In the later stages of the disease, progressive symptoms include difficulty in speaking, swallowing, and walking, together with dementia [5].
Understanding the molecular mechanisms of AD and targeting therapies for preclinical and clinical studies are indispensably aided by the use of transgenic animal models of AD that accurately recapitulate clinical pathology and cognitive decline with the symptomatic phase of AD [6,7]. The 5xFAD transgenic mouse model, developed in 2006, is still widely used because its amyloid plaque pathology is analogous to that found in human AD. 5xFAD mice overexpress humanized sequences of five AD-linked mutations: the Swedish (K670N/M671L), Florida (I716V), and London (V717I) mutations in APP and the M146L and L286V mutations in PSEN1 under the control of the neuron-specific thy1 promoter [6]. The APP transgene includes the 5' untranslated region and thus contains a putative interleukin-1beta translational enhancer element [6]. The most prominent feature of 5xFAD mice is that they present AD amyloid pathology and generate amyloid-β plaque formation with an accelerated rate of Aβ42 deposition in the cerebral cortex, subiculum, and deep cortical layers at an age as early as 2 months. This aggressive amyloid deposition of Aβ42 leads to memory deficits at 4 months and neuron loss, as indicated by a decrease in synaptic markers, such as synaptophysin, syntaxin, and postsynaptic density protein 95, at 9 months of age [6,8]. A significant loss of pyramidal neurons is evident in cortical Layer 5 and the subiculum from 9 months to 12 months of age [6], together with increases in amyloid Aβ42 plaque numbers at 6, 9, and 12 months of age in brain regions adjacent to Layer 5 and the subiculum, such as CA1/2, CA3, the dentate gyrus, Layers 4 and 6 of the cortex, and the midbrain [9]. 5xFAD mice show cardinal symptoms of AD, including cognitive deficits (learning and memory deficits) in Y-maze and Morris water maze test partially at 6 months and completely at 9 months of age [10,11] Some motor deficits, such as missing reflexes and rigidity of the fore and hind paws, also begin to appear at 9 months [12].
AD has a decade-long clinically silent prodromal phase associated with subtle changes in biochemical and cellular functions, including neuronal dystrophy [13], astrogliosis [14] , early onset of neurovascular dysfunction [15], and recruitment of microglia [16]. A need to shift focus to this silent prodromal phase has recently been acknowledged to prevent early loss of synapses in the brain and for exploration of potential targets for therapeutic AD interventions [17]. AD progression is significantly influenced by the neuroinflammatory state triggered by Aβ accumulation [18]. The innate immune response controls the Aβ levels in the brain [19]. Aβ can activate an inflammatory response involving escalations in microglial and astrocyte numbers for the uptake and clearance of Aβ from brains[19], even though Aβ primarily produced by neurons [20,21]
Microglial cells are highly dynamic and surveille the brain. They migrate toward dense-core Aβ plaques, and microgliosis occurs in the vicinity of Aβ [22]. Neuroinflammation is one of the major contributing factors to neurodegeneration in AD, and it starts as early as 2–3 months of age in 5xFAD mice [6]. Inflammatory M1-phenotype microglia contribute to and signify this neuroinflammation [23,24,25]. Microglia at the resting stage, as resident immune cells of the central nervous system, are constantly monitoring the microenvironment of the brain [26]. Upon injury, microglia can be activated to assume the M1 phenotype, becoming amoeboid and highly phagocytic in nature and expressing CD68 [27,28,29,30], MHC-II, and/or OX-6 as markers of inflammation [31,32]. By contrast, alternative M2 microglia help in immuno-resolution and repair processes in an injury and have neuroprotective effects [33,34]. The switching between M1 and M2 phenotypes depends on the severity and stage of the disease [35].
Astrocytes play a vital role in protecting and repairing neuronal damage in brains [36,37]. Astrocytes perform various essential functions including ion homeostasis, neurotransmitter buffering, secretion of neuroactive agents, synaptogenesis, and regulation of the blood brain barriers (BBBs). Astrocytes also use their end feet connections with the brain vasculature to vasodilate or constrict brain blood vessels to accommodate nutrient–waste exchange [38] through a waste clearance system known as the glymphatic system [38,39]. Recent research has also suggested that astrocytes may play a role in the development and progression of AD [40] by clearing the brain of amyloid-beta (Aβ), the peptide that forms the plaques that are a hallmark of AD. Astrocytes can normally phagocytose Aβ and are able to degrade it [38]; however, this ability is impaired in AD astrocytes, leading to Aβ accumulation in the brain [41]. Patients with AD have been reported to have reactive astrocytes in the hippocampus and entorhinal cortex, as indicated by a significantly higher mean number of glial fibrillary acidic protein positive (GFAP+) cells in the hippocampus [42,43]. These reactive astrocytes have been shown to produce high levels of inflammatory molecules, such as TNF-α and interleukin-1β, which may contribute to neuronal damage. AD is also associated with an additional GFAP-high state astrocyte type, termed disease-associated astrocytes (DAAs) [44], which exhibit an increased expression of pan-reactive and inflammatory A1 signatures in 5xFAD mice. DAAs are associated with diverse molecular pathways and are found positioned in proximity to Aβ plaques in AD-affected brains [6,45].
Reactive astrocytes display either detrimental or beneficial effects [46], depending on their level of reactivity. In mouse models of infection-induced by LPS and in middle cerebral artery occlusion, quiescent (A0) astrocytes undergo a transformation into reactive forms through the upregulation or downregulation of specific genes [47]. Gene expression profiles indicate the existence of reactive astrocytes in two distinct forms: A1 (inflammatory, neurotoxic) and A2 (inflammation-resolving, neuroprotective) [48,49]. Furthermore, the deposition of Aβ plaque triggers a response in A1 astrocytes, leading to an increased production of inflammatory mediators, such as chemokines, pro-inflammatory cytokines, and reactive oxygen species (ROS) that, in turn, damage neurons [50]. The presence of these pro-inflammatory signals creates a feedback loop that further activates astrocytes, thereby perpetuating the chronic inflammatory environment that is instrumental in the development and progression of AD. The A1 astrocytes, found surrounding amyloid plaques, release pro-inflammatory cytokines, such as TNF-α, IL-1, and IL-6, which contribute to the pathogenesis and progression of AD [51].
Recent studies have shed light on the molecular mechanisms underlying the formation and function of A2 astrocytes in AD. For instance, microglia-mediated signaling pathways have been implicated in the induction of A2 phenotype following brain injury [52]. Additionally, nuclear factor IA, a transcription factor, has been identified as a molecular switch that triggers the transition of astrocytes into the neuroprotective A2 state [53]. Furthermore, A2 astrocytes have been shown to upregulate various neuroprotective factors, including prokineticin-2, chitin-like 3, Frizzled class receptor 1, arginase 1, NF-E2-related factor 2 (Nrf2), pentraxin 3, sphingosine kinase 1, and transmembrane 4 L6 family member [54]. These factors contribute to synaptic maintenance, growth, and protection against neurodegenerative processes associated with AD. Conversely, other mediators undergo upregulation or downregulation in these distinct forms of reactive astrocytes. A2 astrocytes are characterized by their ability to dampen neuroinflammation, promote synaptic repair, and enhance neuronal survival. Studies have highlighted the induction of A2 astrocytes in various neurodegenerative conditions, including traumatic brain injury [55] and spinal cord injury [56]. However, their role in AD is still an emerging area of research. In the present study we characterized the A1 and A2 astrocytes and found that MarL1 treatment shifts the polarization of A1 (inflammatory, neurotoxic) to A2 (inflammation-resolving, neuroprotective) astrocytes demonstrating neuroprotective properties of MarL1 in 5xFAD mice model of AD.
Another cell type potentially involved in AD is the neutrophil, the most abundant circulating leukocyte, which plays a critical role as first responder to inflammation [57]. Recently, multiple reports have supported a role for the peripheral immune system, and especially neutrophils in AD [58,59]. Neutrophils can swarm to sites of inflammation but are not believed to cross the BBB [60,61,62]. A recent report has indicated an increase in the neutrophil population in AD brains and in a murine APP/PS1 model of AD [63]. Another study using 2-photon microscopy imaging reported that neutrophils infiltrate the brain parenchyma and migrate toward amyloid plaques in the 5xFAD mouse model, with a peak neutrophil infiltration occurring at 4 months of age in 5xFAD mice and at 6 months of age in the 3xTg-AD mouse model. This infiltration of neutrophils coincides with the onset of memory loss in cognitive tests [58]. A review article has summarized the role of a neutrophil granule protein, CAP37, as well as neutrophil elastase and cathepsin G, in neuroinflammation, with an emphasis on AD [64]. Analogous to the conventional categorization of macrophages or microglia into two major types, M1 and M2 phenotypes, neutrophils have been into categorized into pro-inflammatory N1 [65] and anti-inflammatory N2 subpopulations [66]. The N1 and N2 neutrophil populations reportedly have distinct transcriptomic profiles and functions [65]. Pro-inflammatory N1 neutrophils exhibit increased production of inflammatory cytokines/chemokines with elevated levels of ROS and NO, increased activity of protein and matrix-degrading enzymes, amplified chemotactic responses, and enhanced phosphorylated forms of ERK1/2 and p65 signaling molecules associated with an inflammatory phenotype. By contrast, N2 neutrophils demonstrate increased expression of CD206, Ym1 and Arg1, and have similar ROS and NO levels and equivalent chemotactic response to unstimulated controls but no changes in ERK1/2 and p65 signaling molecules [65]. We have determined the possible occurrence of N1 and N2 neutrophils in brains of 5xFAD mice with and without treatment as well as of wildtype control mice in this study.
The recognition of the importance of neuroinflammation in AD has led to increased focus on specialized pro-resolving mediators (SPMs) that effectively promote the resolution of inflammation [67]. One class of these compounds are the maresins, first discovered in 2009 by Serhan et al [68]. Maresins [68,69] and the related maresin -like (MarL) mediators [70] are derived from essential ω-3 docosahexaenoic acid (DHA) by the action of endogenous enzymatic systems [12- or 15-lipoxgenase (LO) for maresins; and 12/15-LO plus a cytochrome P450 CYP4F3 for MarL]. Maresin-1 (7R,14S-dihydroxy-4Z,8E,10E,12Z,16Z,19Z-DHA) reduces neuroinflammation, mitochondrial damage, and neuronal death, while enhancing neural functional recovery, macrophage phagocytosis of Aβ, and the switch in macrophage phenotype to the anti-inflammatory M2 type [71,72]. Maresin 1 also ameliorates the physiopathology of experimental autoimmune encephalomyelitis [73] and promotes neuroprotection and functional recovery after spinal cord injury by resolving inflammation [71]. Maresin 1 treatment has recently been reported to ameliorate Aβ42-induced cognitive decline and neuroinflammation in C57BL/6 mice [74]. Similarly, the intranasal instillation of maresin 1 in combination with resolvins and neuroprotectin/protectin D1 ameliorated memory deficits and gamma oscillation deficits, concurrently with a dramatic decrease in microglial activation in the AppNL-G-F/NL-G-F mouse model for AD [75]. Despite the protective effects of maresin 1 on the brain in 5xFAD transgenic mice, the underlying mechanisms remain abstruse.
By contrast, the maresin-like compound MarL1 (14S,22-dihydroxy-docosa-4Z,7Z,10Z,12E,16Z,19Z-hexaenoic acid) promotes the production of the regenerative angiogenic growth factor HGF and attenuates the production of the pro-inflammatory cytokine TNFα in vitro [70] in macrophages. MarL1 also enhances the macrophage-induced promotion of migration of epithelial cells and fibroblasts from scratch-wounded monolayer cultures, as well as stem cell transmigration [70]. Despite the growing evidence for pro-resolving and neuroprotective effects of maresin 1 and MarLs, the underlying mechanisms require further investigation for their therapeutic development. In the present study, we have hypothesized that long-term administration of MarL1 administration could mitigate AD-related brain neuropathogenesis by curbing neuroinflammation and neurodegeneration.
2. Materials and Methods
2.1. Animals
The Louisiana State University Health Science Center (LSUHSC) IACUC committee approved all the animal procedures, which are consistent with American Veterinary Medical Association guidelines. Mice were maintained at LSUHSC at a controlled temperature of 25 ± 2°C and 50%–65% humidity with a fixed 12:12 h light-dark cycle. We used a widely used model of AD—5x familial Alzheimer’s Disease (5xFAD) transgenic mice (Jackson Laboratory, Bar Harbor, ME, USA) with a C57BL/6J genetic background (MMRRC Strain #034840-JAX)—and compared them with wildtype (WT) control mice (C57BL/6J) [76].
2.2. Intranasal Treatment with Maresin-like 1
Male mice at the age of 1.5 months were randomly selected and equally divided into the following three groups: 1) WT (C57BL/6J mice), 2) 5xFAD mice treated with vehicle, and 3) 5xFAD mice treated with MarL1 plus vehicle. The MarL1 or vehicle was administered by the intranasal route 3 times per week to each mouse from 1.5 months to 9 months of age. Briefly, each mouse was temporarily immobilized for about 60 seconds by a slight anesthetization using isoflurane inhalation. During the immobilization, the mouse was held in the supine position and its nostrils were then instilled with 3 µL/nostril of MarL1 (100 ng per mouse for each administration) dissolved in the vehicle (0.05% dimethyl sulfoxide [DMSO] in sterile 0.9% saline) or with 3 µL the vehicle alone. The mouse was held at the same position for 20 more seconds to ensure liquid intranasal intake [75,77,78,79]. The mice were sacrificed at the age of 12.5 months.
2.3. Harvesting of Murine Brains for Immunohistology
The mice were anesthetized with 5% isoflurane and then transcardially perfused with 4% paraformaldehyde (PFA). The color change in the liver from a deep red to a paler shade was used as an indicator of a sufficient perfusion. The mice were then decapitated using a pair of sharp scissors and the brains were quickly removed from the cranium and immersed in 4% paraformaldehyde for fixing overnight at 4°C. Sections from the cortex and hippocampus regions were chosen for immunohistochemistry. After 24 h, the brains were transferred to 15% for 12 hours, then to 30% sucrose solution. After the brain tissue finally sank in the 30% sucrose solution, brain tissue blocks were made using optimal cutting temperature compound (OCT) liquid and cryo-molds for cryo-sectioning. Coronal section of brains were cut rostral-to-caudal serially at 20 μm thickness on a HM550 cryostat (Microm-HM 550, Thermo Fisher Scientific), mounted on Superfrost Plus glass slides (VWR, Radnor, PA, USA), and stored.
2.4. Immunofluorescence Staining
The mounted brain sections (containing cortex and hippocampus) were washed twice in phosphate-buffered saline (PBS), followed by two washes in PBS-T (PBS containing 0.05% Triton-X). The sections were then incubated in blocking buffer (1% BSA+0.5% Trition-X+0.02% Tween-20 in 1X PBS) at room temperature for 60 min. The sections were then treated with the following antibodies: NeuN (1:500, rabbit, cell signaling-12943S), amyloid-β (MOAB-2, 1:500, mouse host, monoclonal 6C3, Millipore-MABN254), Gr-1 (Ly-6G/Ly-6C monoclonal antibody (1:200, rat host, Invitrogen), ionized calcium-binding adapter molecule-1 (Iba-1, 1:500, rabbit, Fujifilm Wako-019-19741), iNOs (1:200, rat, Invitrogen eBioscience), Arg-1 (1:500, goat, Abcam-ab60176), GFAP (1:500, rabbit, Sigma-G9269), TNF-α (1:500, goat, Invitrogen-PA5-46945), anti-choline acetyltransferase (1:500, ChAT, goat, Millipore-sigma-AB144P), cleaved caspase-3 (cleaved-caspase-3, 1:500, rabbit, Cell Signaling-9661) antibody, CD68 (1:500, mouse, Santa Cruz Biotechnology-70761), C3 (1:500, rabbit, Invitrogen-PA5-21349), S100A10 (reconstituted in sterile PBS, 0.2 mg/mL, mouse, Biotechne-AF2377), and claudin-5 (1:500, mouse, Santa Cruz Biotechnology, sc-374221). The sections were then incubated with appropriate secondary antibodies (Alexa Fluor 488, 568, or 594; Invitrogen) compatible with the aforementioned primary antibodies, followed by the incubation with DAPI (1:10,000 dilution). A total of three to four sections of brain per slide from each of five to eight mice per group were used for the histological study. Co-stained (yellow-colored) cells by two protein/peptide biomarkers (green-colored and red-colored) were quantified by cell counting or Pearson’s coefficients obtained in ImageJ analysis (1.54J National Institutes of Health, Bethesda, MD).
2.5. Image Quantification
After immunofluorescence staining, fluorescent images were captured on ECHO Revolve fluorescent microscope using 4X, 10X, 20X, and 40X objectives, and mean intensity of fluorescence, number of immunoreactive cells, were quantified using the ImageJ software. The results were expressed in mean fluorescence intensity (MFI) as we conducted previously [80]. The counting of co-localized immune-positive cells for two markers was performed manually using the cell counter application of ImageJ. All quantifications were performed blinded to the experimental groups with the help of the ImageJ image analysis software.
2.6. Thioflavin S Staining and Analysis of Plaques
Thioflavin S (Sigma-Aldrich, T1892) is a fluorescent staining used for visualizing Aβ plaques in the brain [6]. The thioflavin S stained Aβ plaques appear bright green by fluorescence microscopy, making them easily identified. Briefly, brain sections (20 μm thickness) were incubated in 1% thioflavin S solution in deionized water for 10 min and washed with running water carefully for 5 min, then incubated in 1% acetic acid for 15 min and wash again in running water. The slides were dried by placing them on paper towels for few minutes and then dehydrated in an alcohol series of 70%, 80%, 95%, 100% ethanol, transferred to xylene, and then mounted with Permount mounting medium and dried overnight in the dark. Thioflavin-S-positive plaques were determined from the images taken by Discover-ECHO Revolve fluorescence microscope in a single plain at 4× magnification. The images were subjected to threshold processing (Otsu) using ImageJ, and the total number of plaques based on size (less than and greater than 100 μm) were analyzed in the cortical and hippocampal regions. For each animal, 6 fields from the cortex and from the hippocampus were imaged and analyzed.
2.7. Statistical Analysis
Results were expressed in Mean ± standard error of mean (SEM). Kruskal–Wallis one-way analysis of variance (ANOVA, non-parametric test) followed by Tukey’s multiple comparison post-hoc test was used to determine the statistical significance of differences between mouse groups of wildtype, 5xFAD, and 5xFAD + MarL1 (*p < 0.05, **p < 0.01, and ***p < 0.001). For all experiments at least three replicates were performed. The definition of the significance for various p values are described in the figure legends, together with the number of biological replicates (n) for each experiment. GraphPad Prism 9.0 (GraphPad, Boston, MA) was used for graphs and statistical analyses.
3. Results
3.1. Maresin-like1 Reduced Aβ Overload and Curbed Neuronal Population Loss in Brain Hippocampi of 5xFAD Mice
Amyloid beta deposition is most prominent in the hippocampus and cortex in the brain, especially in 5xFAD mice, as the Aβ deposition starts as early as 2 months of age [6,81]. Here, we examined the effect of long-term intranasal instillation of MarL1 on Aβ deposition and neuronal population in the hippocampus of 5xFAD mouse brains. We found a significantly more Aβ plaques in the hippocampus of the 5xAD mice than in the same brain regions in the wildtype mice (Figure 1, p < 0.001), and significant fewer Aβ1-42 plaques in the MarL1-treated 5xFAD mice than in the untreated 5xFAD mice (Figure 1, p < 0.001). We also evaluated the cerebral plaque in hippocampal and cortical sections by staining them with thioflavin-S (Thio-S) dye that detects the β-pleated sheet of the amyloid plaques [82]. The level of plaque deposition was quantified in the hippocampus and cortex by determining the number of plaques of size > 100 μm2 and size < 100 μm2 [83,84,85]. We found a markedly greater number of plaques in the cortex and hippocampus of 5xFAD mice than in the wildtype mice (Figure S1). The numbers of plaques of each size category were markedly lower in both the cortex and hippocampus of the MarL1-treated 5xFAD mice than in the same brain regions in the vehicle treated 5xFAD mice (Figure S1, p < 0.001). We found that GFAP+ astrocytes and Iba-1+ microglia engulfed the Aβ plaques and that those astrocytic and microglial cells formed clusters near Aβ plaques in 5xFAD mice (Figure S2).
We also examined Aβ-related neuronal losses by co-staining brain sections with MOAB-2 and with NeuN antibody. The NeuN+ neuronal population was significantly lower in the 5xFAD mice than in the wildtype mice (Figure 1B, p < 0.001). MarL1 treatment rescued the NeuN+ neuronal population in hippocampus (Figure 1B, p < 0.001), suggesting that MarL1 had a neuroprotective effect against AD pathogenesis in the brains of 5xFAD mice.
3.2. Maresin-like 1 Treatment of 5xFAD Mice Improved the Survival of Cholinergic Neurons and Decreased Cleaved-Caspase-3-Mediated Apoptotic Degeneration
Cholinergic neurons are vital for proper functioning of the brain, and they are integral for fine tuning of brain [86], maintenance of neuronal circuits balance [87]. They release the neurotransmitter acetylcholine (ACh), which is involved in regulating a wide range of brain functions including sensory processing, motor control, and cognitive processes such as motivation, behavioral flexibility, and associative learning [88]. These neurons are widely distributed throughout the brain, with a significant population found in the striatum, which contains the highest levels of ACh in the brain [89]. The loss of cholinergic neurons in the brain can have a detrimental effect on cognitive —a hallmark of AD [90,91].
Here, we investigated whether MarL1 treatment could curb the AD-associated decline in the cholinergic neuronal population in 5xFAD mice. We immunostained striatal brain sections with an antibody specific for choline acetyltransferase (ChAT), an enzyme that catalyzes the reversible synthesis of acetylcholine from acetyl CoA and choline at cholinergic synapses and therefore specifically stains cholinergic neurons [92]. We conducted the ChAT immunostaining together with cleaved-caspase-3 antibody to quantify the apoptotic ChAT+ neuronal population in the brain (Figure 2A). We found that the ChAT+ neuronal population was significantly smaller in the 5xFAD mice than in their wildtype littermates (p < 0.001), whereas MarL1 treatment significantly inhibited this decline in the ChAT+ population in 5xFAD mice compared to vehicle -treated 5xFAD mice (p < 0.001) (Figure 2B) in the striatum region. The number of apoptotic cleaved-caspase-3+ cells was also lower in the striatum region of 5xFAD mice treated with MarL1 than with vehicle (Figure 2B, p < 0.001), suggesting that MarL1 treatment may be effective in protecting cholinergic neurons from apoptosis and restoring their population in the brains of 5xFAD mice.
3.3. Maresin-like1 Attenuated the Pro-Inflammatory M1 Phenotypic Switching of Microglia by Inhibiting Iba-1+CD68+ Microglia in Brains of 5xFAD Mice
We investigated the effect of MarL1 on M1 phenotypic switching of microglia in the brains of 5xFAD mice by immunostaining assessment of the microglial phenotype using the microglial markers Iba-1 and CD68 for M1 microglia [27,28,29,30] in the brain (Figure 3A). Significantly greater immunoreactivities of Iba-1 were found in 5xFAD mice than in wildtype mice in hippocampus region (Figure 3B, p < 0.001), while the immunoreactivities of Iba-1 were significantly lower in MarL1-treated mice than in vehicle-treated 5xFAD mice (Figure 3B, p < 0.001).
We also quantified the levels of CD68 (scavenger receptor Class D), a phagocytic microglial marker on Iba-1+ microglial cells, in co-stained brain coronal sections [93]. The activation of CD68+ microglial cells was 3.25-fold higher in 5xFAD mice than in their wildtype littermates (Figure 3B, middle panel, p < 0.001), but this activation was significantly attenuated by 57% in the MarL1-treated 5xFAD mice (Figure 3B, middle panel, p < 0.001). The Iba-1+CD68+ cell count was significantly higher in 5xFAD brains than in wildtype controls (Figure 3B, right panel, p < 0.001), and was significantly lower in MarL1-treated 5xFAD mice than in vehicle-treated 5xFAD mice (Figure 3B, right panel, p < 0.05).
Microglia can be phenotypically categorized based on their circularity (i.e., the length of their processes) into different stages of activation ranging from ramified microglia (microglia at the resting stage, S0) to amoeboid microglia (microglia at the activated-stage, S1). Inter-stages also exist between S0 and S1 in the form of partially ramified microglia (having fewer processes) and partially amoeboid microglia (having 1-2 processes with a round shape) [94,95,96]. Notably, we observed an unusual phenomenon in the microglial phenotype in 5xFAD mice. We found that the microglial cells aggregated in the hippocampus and cortex to form a “cluster of cells” similar to those found near Aβ plaques and those microglia were partially or fully amoeboid in shape in the 5xFAD mice but did not show this morphology in the wildtype mice. We also observed some microglial aggregates in the cortex, but not in the hippocampus, of the MarL1-treated 5xFAD mice. The MarL1 treatment group showed more partially ramified microglial phenotypes than the vehicle-treated 5xFAD mice (Figure 3A).
We also quantified the numbers of microglial cells based on the morphology observed in Iba-1–immunostained sections. The number of ramified microglia was greater in the wildtype mice than in the 5xFAD and MarL1-treated mice (Figure 3C). The number of partially ramified microglia was significantly higher in the MarL1-treated 5xFAD mice than in the vehicle-treated 5xFAD or wildtype mice (Figure 3C, p < 0.001). The number of amoeboid microglia was significantly higher in the vehicle-treated 5xFAD mice than in the wildtype (Figure 3C, p < 0.001) and MarL1-treated 5xFAD mice (Figure 3C, p < 0.001). These results suggested that the transition of microglia from the ramified (S0) morphology to activated amoeboid (S1) morphology was greater in the 5xFAD mice than in the wildtype mice, while treatment with MarL1 attenuated that morphological transition.
3.4. Maresin-like 1 Curbed the AD Pathogenesis-Associated Decline in the M2 Microglial Population with an Anti-Inflammatory Alternatively Activated Phenotype in Brains of 5xFAD Mice
We determined the effect of MarL1 on the microglial phenotypic switch between M1 and M2 by investigating the M2 population in cortical brain sections. Immunostaining brain sections with Iba-1 and Arg-1, a biomarker for M2 microglia [97] (Figure 4A), revealed a significantly higher mean fluorescence intensity for Arg-1 in the brains of wildtype mice than of 5xFAD mice (Figure 4B, p < 0.001), but the population was significantly greater in MarL1-treated 5xFAD mice than in vehicle-treated 5xFAD mice; that is, the MarL1 treatment significantly restored the Arg1+Iba-1+ microglial population in 5xFAD mice (Figure 4B, p < 0.001). These results suggest that MarL1 treatment significantly shifted the polarization of the microglial population from M1 to M2, suggesting that MarL1 has a capacity for resolution of inflammation that could contribute to curbing AD pathogenesis.
3.5. Maresin-like1 Mitigated Astrogliosis-Associated Neuroinflammation in Brains of 5xFAD Mice
Cognitive dysfunction in AD is tightly linked to the interaction between neurodegeneration and neuroinflammation, where neuroinflammation contributes to and exacerbates neurodegeneration [98]. Microglia and astrocytes are the two main glial cells responsible for initiating and implementing innate immune responses in the brain, with astrocytes primarily involved in the immune and inflammatory responses [99]. The presence of AD-linked Aβ [100] triggers various cellular receptors and signaling pathways [101], particularly the advanced glycation end products receptor and the NF-κB pathway, thereby leading to the production of pro-inflammatory molecules, such as cytokines and chemokines, in astrocytes [102,103,104]. Since our results demonstrated that MarL1 inhibited microglial activation, we also investigated the effect of MarL1 on astrocyte population and activation in 5xFAD mice. We co-stained brain sections with the antibodies of active astrocyte marker GFAP and inflammation marker TNF-α (as shown in Figure 5A). The immunoreactivities of GFAP (Figure 5B upper panel, p < 0.001) and TNF-α (Figure 5B lower panel, p < 0.001) were significantly higher in 5xFAD mice than in wildtype mice, and they were significantly lower in the MarL1-treated 5xFAD mice than in the vehicle-treated 5xFAD mice (Figure 5B, p < 0.001 or p < 0.01. respectively). These results indicate that MarL1 treatment reduced AD-augmented neuroinflammation manifested by TNF-α expression, as well as suppressed the AD-increase of GFAP+ astrocytic population or astrogliosis in brains of 5xFAD mice.
3.6. Maresin-like1 Treatment Inhibited A1 Astrocytes and Promoted A2 Astrocytes in Brains of 5xFAD Mice
Astrocytes undergo significant changes in their structure, molecular composition, and function when exposed to various pathological triggers, and these alterations are highly influenced by specific contexts. Overall, the changes observed in astrocytes are characteristic of a reactive state, which is associated with their response to pathological condition [48,49]. Reactive astrocytes can play contrasting roles in the brain during disease progression. On one hand, reactive astrocytes can contribute to and exacerbate neuropathology and neurodegeneration, as they have the potential to exhibit pro-inflammatory and neurotoxic properties, which can further damage the surrounding neurons and tissues. This detrimental effect is particularly evident when reactive astrocytes adopt the A1 phenotype, triggered by signals from microglia or neurons. On the other hand, reactive astrocytes also possess the ability to provide neuroprotection and support tissue repair [37,105], as they can adopt an anti-inflammatory and neuroprotective A2 phenotype in response to specific signals from microglia or neurons. In this state, reactive astrocytes aid in reducing inflammation and promoting the survival and recovery of neurons. The duality observed in the behavior of reactive astrocytes arises from their ability to respond to different signaling cues in brains. These cues from microglia or neurons can influence astrocytes to adopt either the harmful A1 phenotype or the beneficial A2 phenotype [54,106,107,108]. In various neurodegenerative disorders [37,105] and in the aging brain [109], the number of A1 astrocytes is frequently elevated. Recent advancements in single-nucleus transcriptome analyses have unveiled additional phenotypes and subtypes of astrocytes, demonstrating the extensive diversity of reactive astrocytes and their association with specific diseases [54]. Here, we explored the astrocyte A1 and A2 subtypes populations in AD.
We conducted immunostaining of hippocampal sections using specific markers to analyze A1and A2 astrocyte subtypes. We co-stained GFAP and complement C3 in tissue sections to identify A1 astrocytes, as well as co-stained GFAP and S100A10 to identify A2 astrocytes [110]. The immunostaining results revealed significantly more GFAP+C3+ A1 astrocytes in 5xFAD mice than in the wildtype mice (Figure 6B, lower panel). However, treatment of 5xFAD mice with MarL1 noticeably reduced astroglial activation in the dentate gyrus compared to vehicle-treated 5xFAD mice (Figure 6, lower panel, p < 0.001), indicating that MarL1 treatment induced a shift in astrocyte polarization from A1 to A2. By contrast, examination of the hippocampal sections co-stained for GFAP and S100A10 (A2) revealed a lower expression in 5xFAD mice compared than in the wildtype mice. However, S100A10 expression was higher in MarL1-treated 5xFAD mice than in vehicle-treated 5xFAD mice (Figure 7), suggesting that MarL1 treatment promotes A2 astrocyte polarization in AD.
3.7. Maresin-like1 Treatment Promoted the Expression of BBB-Associated Tight-Junction Protein Claudin-5, Decreased Infiltration of Neutrophils in 5xFAD Brains, and Induced the Switch of Neutrophils toward the Inflammation-Resolving N2 Phenotype
Conventionally, AD has primarily been considered as a neurodegenerative disorder, but new reports suggest the role of dysfunction of the blood-brain barriers and linked neuroinflammation in close association with peripheral inflammatory signals [60,61,62,111,112] . Patients with early cognitive decline or mild cognitive impairment (MCI) have been reported to show BBB breakdown in the hippocampus (CA1, CA3, and dentate gyrus and parahippocampal gyrus), an area crucial for memory and learning [113]. As a result, the BBB can no longer effectively clear Aβ, and that peptide then accumulates in the brain and blood vessels [114], leading to increased expression of adhesion molecules on brain blood vessels and the release of inflammatory substances, such as complement system peptides, chemokines, and cytokines, that potentially promote leukocyte recruitment [114].
The resident immune cells in brains, such as microglia and astrocytes, when activated, may release tissue danger signals, cytokines, and chemokines that recruit leukocytes from the peripheral circulation to inflamed areas in brains. Both experimental and clinical evidence indicates that neutrophils and T-cells migrate into the brain in AD [115,116,117,118]. Recent studies using two-photon laser scanning microscopy have revealed that vascular deposition of Aβ in brains promotes intraluminal adherence and movement of neutrophils in brain blood vessels and that neutrophils preferentially extravasate in the parenchyma with the areas of Aβ deposits [119]. We explored the possibility of neutrophil infiltration in the brain by immunostaining cortical sections with Gr-1 (a marker for myeloid differentiation, present primarily in neutrophils and transiently in monocytes/macrophages) and claudin-5 (a tight-junction protein present in endothelial cells of vessels in brain for vasculature), as shown in Figure 8A. We found a significantly greater influx of neutrophils in 5xFAD mice than in wildtype mice (Figure 8B, p < 0.001). The MarL1 treatment significantly reduced this influx compared to vehicle-treated 5xFAD mice (Figure 8B, p < 0.001). We also observed a phenomenon in 5xFAD mice called “neutrophil swarming” which is the accumulation of large numbers of neutrophils to neutralize large microbes and clusters of microbes in a targeted area [120,121]. Here, we noticed a massive number of neutrophil aggregations in the cortex of 5xFAD mice brain. One possibility is that neutrophil swarming may occur due to presence of Aβ plaques in the cortex of 5xFAD mice. Here, we report the first evidence for “neutrophil swarming” in the cortex of the brains of 5xFAD mice. We did not observe neutrophil swarming in MarL1-treated 5xFAD mice, although the neutrophil clusters that are not swarming were evident in the cortical sections (Figure 8A).
The BBB integrity was further assessed using the tight-junction protein marker claudin-5, which is vital for maintaining the integrity of the endothelial cells lining brain blood vessels, and staining endothelial cells in the blood vessels in the brain [122]. The mean claudin-5 intensities were significantly lower in 5xFAD mice than in wildtype mice. The MarL1 treatment restored the BBB integrity by increasing the claudin-5 levels in brain blood vessels in 5xFAD mice (Figure 8B). These results suggest that MarL1 treatment may provide a protective effect on the BBB integrity and decrease the infiltration of immune cells into the brain.
We also categorized the infiltrating neutrophil population into N1-neutrophils, a pro-inflammatory phenotype [65], and N2-neutrophils, anti-inflammatory phenotype of pro-resolution of inflammation [66]. Immunostaining of coronal brain sections using the co-localization of Gr-1 and iNOS (Gr-1+iNOS+) revealed more Gr-1+iNOS+ N1 neutrophils in 5xFAD mice than in wildtype mice (Figure 9A), while MarL1 treatment reversed this trend in the hippocampal region. The Pearson’s coefficients between the mean fluorescence intensities, MFIs of Gr-1+ cells and iNOS+ cells are consistent with this trend about the co-localization Gr-1+ and iNOS+ (Figure 9B right panel, p < 0.01 or 0.05). By contrast, the microimages of Gr-1 and Arg1 co-localized brain sections showed less N2 neutrophils in 5xFAD mice than in wildtype mice (Figure 10A) and MarL1 treatment replenished the N2 population above that in vehicle-treated 5xFAD mice. This direct microscopic observation is consistent with the analysis of Pearson’s coefficients between MFIs of Gr-1+ cells and Arg1+ cells in the microimages (Figure 10B right panel, p < 0.001 or 0.01). These results suggest that MarL1 treatment polarizes the shift of N1 to N2 neutrophil population in neurodegenerative events.
4. Discussion
AD is characterized as a progressive neurodegenerative disorder and the accumulation of amyloid beta plaques and tau tangles in the brain are considered to be key pathological features of AD [123]; however, recent studies have shown that AD has a multifactorial etiology and that neuroinflammation plays a central role in its etiopathogenesis, owing to its capacity to exacerbate Aβ and Tau pathologies [123,124,125]. Evidence of increased microglia activation (inflammation) in the brains of AD patients with elevated levels of pro-inflammatory cytokines in serum and in post-mortem brains supports the link between neuroinflammation and AD [126,127]. In the present study, we examined the effect of MarL1 on the extent of inflammation in brains of transgenic 5xFAD mice.
We evaluated the effect of intranasal instillation of the MarL1 mediator from the age of 1.5 to 9 months in 5xFAD mice. Intranasal instillation of drugs is well recognized to partly bypass the BBB to deliver drugs to brains more efficiently than ip or iv methods, thereby increasing drug bioavailability in the brain, while also delivering drugs noninvasively and to the blood circulation as well [77,128,129,130]. At the age of 12.5 months, 5xFAD mice show a significant reduction in levels of oligomeric Aβ42 and Aβ plaques in cortex and hippocampal regions of the brain and in the loss of NeuN+ neurons in CA3 and dentate gyrus regions of hippocampus (Figure 1). Quantification of the Aβ plaque numbers based on sizes greater than and less than 100 μm2 area [83,84,85] using thioflavin-S staining revealed a marked reduction in the number of Aβ plaques following MarL1 treatment of 5xFAD mice (Figure S1). The neuroprotective effect of MarL1 was mediated by restoring the cholinergic neurons in striatum and decreasing the apoptotic cleaved caspase-3+ neurons in brain. Other studies have shown increased activation of cleaved caspase-3 in the hippocampus of AD patients and significant increases in synaptic pre-caspase-3 and active caspase-3 (i.e., cleaved caspase-3) expression levels in the postsynaptic density fractions [131]. These findings suggest that MarL1 has a neuroprotective effect in the brain.
Neuroinflammation is thought to play an essential role in neurodegeneration by contributing to neuronal damage and synaptic loss, with microglia and astrocytes as the key players [132,133,134]. Microglia, as the resident phagocytes in the brain, constantly use their highly motile processes to surveille brain regions for the presence of pathogens and cellular debris and simultaneously provide factors that support tissue maintenance [134]. The M2 phenotype microglia are also involved in the protection and remodeling of synapses for proper maintenance of the plasticity of neuronal circuits and release of trophic factors, including brain-derived neurotrophic factors, which contribute to memory formation [135,136]. Upon activation due to pathological triggers, such as neuronal death or protein aggregates, microglia change phenotypically to an amoeboid state and begin to migrate to the lesion, where they initiate an innate immune response [35]. Activated microglia (M1) are “pro-inflammatory” in nature and increase the expressions of markers involved in microglial activation, such as CD68 [27,28,29,30], CD86, CD45, CX3CR1, CD11b, and MHC-II [137].
Histological examinations of AD brains show that microglial cells are found in close association with Aβ deposits and are associated with dense-core plaques [138,139,140]; however, fewer microglial cells are found in the vicinity of the diffuse Aβ deposits in the cerebral cortex of AD patients [141,142]. The number of microglia and their size directly increases in proportion to plaque dimension, and Aβ deposition has been reported to recruit a microglial population to allow for the accumulation of these cells at the periphery of amyloid deposits [143,144]. Microglia have the ability to attach themselves to soluble oligomers and fibrils of Aβ by means of various receptors, such as CD36, α6β1 integrin, CD47, CD14, and class A scavenger receptor A1, as well as toll-like receptors (TLR2, TLR4, TLR6, and TLR9) [143,144]. Our results demonstrate enhanced microglial phagocytosis of Aβ plaques (Figure S2), and we found microglia in clusters in 5xFAD mice. These aggregates showed that microglial accumulation and proliferation in the brain could cause neuroinflammation, while MarL1 treatment reduced the Aβ accumulation thereby decreasing the microglial activation.
Enhanced microglial activation leads to increases in the expression of pro-inflammatory markers, such as interleukin-1β, TNF-α, and iNOs, thereby exacerbating neuroinflammation [126,145]. Our data demonstrate that MarL1 treatment attenuated microglial activation by decreasing the population of Iba-1 positive microglia and CD68 expression in the hippocampus (CA1, dentate gyrus) of the 5xFAD mouse brain. We phenotypically characterized microglia and calculated the number of microglia in different states as: 1) ramified, 2) partially- ramified, 3) partially-amoeboid, 4) and fully-amoeboid microglia [146,147]. We found that MarL1 reduced the level of amoeboid microglia, suggesting its inflammation-resolving and anti-inflammatory properties are a consequence of suppression of microglial activation (Figure 3).
Specialized pro-resolving lipid mediators are reported to modulate immunity and inflammation by resolving inflammation by triggering a biochemical paradigm shift commonly referred to as the “lipid mediator class switch” and skewing the M1/M2 macrophage balance toward the anti-inflammatory M2 phenotype, with replacement of injured cells and restoration of the normal functions of tissues [67]. Consistent with these reports, MarL1 treatment caused a shift in the M1/M2 population in brain as indicated by a surging Arg-1+ microglial population in the brain (Figure 4). These finding suggest that MarL1 is a potent immunoresolvent that is effective in brains
We found hypertrophy of astrocytes interacting with Aβ plaques in hippocampus in 5xFAD mice (Figure S2), as well as increased expression of TNFα in astrocytes in 5xFAD mice (Figure 5). These responses suggest that the mutations present in 5xFAD mice disrupt the mechanisms that would normally protect against and repair neuronal damage in the brain—functions that are typically carried out by astrocytes [36,37]. The MarL1 treatment attenuated the GFAP-high state astrocytes in brains of 5xFAD mice and reduced Aβ accumulation. Our characterization of A1 and A2 astrocytes in the 5xFAD mouse model of AD revealed that MarL1 treatment shifts the polarization of A1 (neurotoxic) astrocytes to the A2 (neuroprotective) form, thereby demonstrating a potential reason for the neuroprotective action of MarL1 (Figure 6 and Figure 7). These results demonstrate that MarL1 mitigates the astrocyte hypertrophy and inflammatory cytokine production and increases Aβ clearance in brains.
In this study we explored the concept of neutrophil migration in brain parenchyma. Here, we observed the phenomenon of “neutrophil swarming” [148] in the cortex, as shown in Figure 8, as evidence of neutrophil aggregation in the 5xFAD mouse brain. This is the first immunohistochemical report of neutrophil swarming in the brain cortex in the literature. Co-saining of Gr-1 (a marker for neutrophils) and claudin-5 (a marker for vasculature) showed that Gr-1+ neutrophils migrated into the brain and formed swarms, as neutrophils are nonresident cells in brains. We could not find neutrophil swarms in the brains of MarL1-treated 5xFAD mice, although neutrophils aggregate were evident than wildtype mice. No swarms were detected in the brains of the wildtype littermates. These findings suggests that neutrophil infiltration into brains could contribute to AD pathogenesis since neutrophils produce reactive oxygen species and degradation enzymes that can cause neuroinflammation and neurodegeneration[57,58,59]. Furthermore, our findings revealed that MarL1 treatment inhibited the AD-linked neutrophil infiltration and swarm in the brains of 5xFAD mice.
We also characterized neutrophils into pro-inflammatory N1 [65] and anti-inflammatory N2 subpopulations [66]. The N1 and N2 neutrophil populations reportedly have distinct transcriptomic profiles and functions [65]. Pro-inflammatory N1 neutrophils exhibit increased production of inflammatory cytokines/chemokines with elevated levels of ROS and NO, increased activity of protein and matrix-degrading enzymes, amplified chemotactic responses, and enhanced phosphorylated forms of ERK1/2 and p65 signaling molecules associated with an inflammatory phenotype. By contrast, N2 neutrophils demonstrate increased expression of CD206, Ym1 and Arg1, and have similar ROS and NO levels and equivalent chemotactic response to unstimulated controls but no changes in ERK1/2 and p65 signaling molecules [65]. In our study, we found that Gr-1+iNOS+ N1 neutrophils increased in 5xFAD mice. We also found that MarL1 treatment dampens the elevation of N1 neutrophils in brain (Figure 9). Furthermore, Gr-1+Arg1+ N2 neutrophils were amplified with MarL1 treatment and were lower in 5xFAD mice than in wildtype controls (Figure 10). These results demonstrate that chronic MarL1 treatment polarizes a shift from N1 to N2 in 5xFAD mice, suggesting resolution of inflammation in the brain.
Taken together, our results provide the first evidence that MarL1 was effective in inhibiting Aβ pathology in brain. However, the 5xFAD mouse model is an aggressive early onset transgenic AD model [8,149,150,151,152], therefore, the protection effects of MarL1 need to be explored using less aggressive AD models, including late-onset AD models [152,153]. Furthermore, the different doses of MarL1 and their safety should be evaluated for future clinical studies. Further research is needed to fully understand the mechanisms involved in the neuroprotection imparted by MarL1 in AD. MarL1-specific receptor targeted research could be a promising strategy for developing new treatments for this debilitating disease.
5. Conclusions
The long-term administration of MarL1 in 5xFAD mice had a positive impact on reducing the neuropathology associated with Alzheimer's disease in the brains of 5xFAD mice—an animal model deemed appropriate for the study of early onset AD and Aβ pathology. This study highlights the potential use of MarL1 as a therapeutic lead for the treatment of AD. Further research is needed to fully understand the underlying mechanisms of action and to further optimize the treatment efficacy. MarL1 treatment efficiency and its involved mechanisms should also be studied using the animal models available for AD Tau pathology.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: MarL1 treatment prevented or reduced the elevation of thioflavin-S positive cerebral plaques in brains of 5xFAD mice; Figure S2: The interaction between astrocytes (a-c), microglia (d-f) and amyloid beta plaques in cortex and hippocampus of 5xFAD mice.
Author Contributions
Conceptualization, S.H.; Methodology, S.H., Y.L., P.S., Y.K., Y.Z., W.L; Validation, P.S., Y.L., S.S., Y.K., Y.Z., N.L., A.-R.M. and S.H.; Formal analysis P.S., Y.L., N.L. and S.H.; Investigation, P.S., Y.L., Y.K., Y.Z., N.L. and S.H.; Resources, Y.K., Y.Z., W.L., and S.H.; Data curation, P.S., Y.L., Y.K., Y.Z., N.L. and S.H.; Writing - original draft, P.S., Y.L., S.S. and S.H.; Writing - review & editing, P.S., Y.L., S.S., Y.K., Y.Z., N.L., A.-R.M. and S.H.; Visualization, P.S., Y.L. and S.H.; Supervision: Y.K. and S.H.; Project administration, Y.K. and S.H.; Funding acquisition, Y.K. and S.H.
Funding
This research was funded by LSU Health-New Orleans research enhancement fund (to S.H.), USA National Institute of Health grants 1R21AG060430, 1R21AG066119, and 1R21AG068756 (to S.H.) and the Japan Society for the Promotion of Science JSPS KAKENHI, Grant Number JP15H05904 (to Y. K.).
Institutional Review Board Statement
The animal use protocols [#3616 (2018-2021), #3876 (2021 to present)] have been authorized and approved by the Institutional Animal Care and Use Committee of Louisiana State University, Health, New Orleans, USA.
Data Availability Statement
Data is contained within the article.
Acknowledgments
We are very grateful to Professor Nicholas G Bazan, the director of Neuroscience Center of Excellence (NCE), School of Medicine, LSU, Health-New Orleans, USA for the strong support to make this research possible.
Conflicts of Interest
The authors do not have any conflict of interest.
References
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer's disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef] [PubMed]
- 2022 Alzheimer's disease facts and figures. Alzheimers Dement 2022, 18, 700–789. [CrossRef] [PubMed]
- Giridharan, V.V.; Barichello De Quevedo, C.E.; Petronilho, F. Microbiota-gut-brain axis in the Alzheimer's disease pathology - an overview. Neurosci Res 2022, 181, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Boyle, P.A.; Yu, L.; Wilson, R.S.; Leurgans, S.E.; Schneider, J.A.; Bennett, D.A. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol 2018, 83, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Sanchez-Varo, R.; Mejias-Ortega, M.; Fernandez-Valenzuela, J.J.; Nunez-Diaz, C.; Caceres-Palomo, L.; Vegas-Gomez, L.; Sanchez-Mejias, E.; Trujillo-Estrada, L.; Garcia-Leon, J.A.; Moreno-Gonzalez, I.; et al. Transgenic Mouse Models of Alzheimer's Disease: An Integrative Analysis. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Oblak, A.L.; Lin, P.B.; Kotredes, K.P.; Pandey, R.S.; Garceau, D.; Williams, H.M.; Uyar, A.; O'Rourke, R.; O'Rourke, S.; Ingraham, C.; et al. Comprehensive Evaluation of the 5XFAD Mouse Model for Preclinical Testing Applications: A MODEL-AD Study. Front Aging Neurosci 2021, 13, 713726. [Google Scholar] [CrossRef]
- Eimer, W.A.; Vassar, R. Neuron loss in the 5XFAD mouse model of Alzheimer's disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol Neurodegener 2013, 8, 2. [Google Scholar] [CrossRef]
- Devi, L.; Ohno, M. Genetic reductions of beta-site amyloid precursor protein-cleaving enzyme 1 and amyloid-beta ameliorate impairment of conditioned taste aversion memory in 5XFAD Alzheimer's disease model mice. Eur J Neurosci 2010, 31, 110–118. [Google Scholar] [CrossRef]
- Neuner, S.M.; Heuer, S.E.; Huentelman, M.J.; O'Connell, K.M.S.; Kaczorowski, C.C. Harnessing Genetic Complexity to Enhance Translatability of Alzheimer's Disease Mouse Models: A Path toward Precision Medicine. Neuron 2019, 101, 399–411.e395. [Google Scholar] [CrossRef] [PubMed]
- O'Leary, T.P.; Brown, R.E. Visuo-spatial learning and memory impairments in the 5xFAD mouse model of Alzheimer's disease: Effects of age, sex, albinism, and motor impairments. Genes Brain Behav 2022, 21, e12794. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef] [PubMed]
- Luo, J. TGF-beta as a Key Modulator of Astrocyte Reactivity: Disease Relevance and Therapeutic Implications. Biomedicines 2022, 10. [Google Scholar] [CrossRef]
- Lecordier, S.; Pons, V.; Rivest, S.; ElAli, A. Multifocal Cerebral Microinfarcts Modulate Early Alzheimer's Disease Pathology in a Sex-Dependent Manner. Front Immunol 2021, 12, 813536. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer's disease model. J Exp Med 2020, 217. [Google Scholar] [CrossRef]
- Epelbaum, S.; Genthon, R.; Cavedo, E.; Habert, M.O.; Lamari, F.; Gagliardi, G.; Lista, S.; Teichmann, M.; Bakardjian, H.; Hampel, H.; et al. Preclinical Alzheimer's disease: A systematic review of the cohorts underlying the concept. Alzheimers Dement 2017, 13, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Garbuz, D.G.; Zatsepina, O.G.; Evgen'ev, M.B. Beta Amyloid, Tau Protein, and Neuroinflammation: An Attempt to Integrate Different Hypotheses of Alzheimer's Disease Pathogenesis. Mol Biol (Mosk) 2021, 55, 734–747. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. A Bridge Between the Innate Immunity System and Amyloid-beta Production in Alzheimer's Disease. Neurosci Bull 2021, 37, 898–901. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease--a double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef]
- Calhoun, M.E.; Burgermeister, P.; Phinney, A.L.; Stalder, M.; Tolnay, M.; Wiederhold, K.H.; Abramowski, D.; Sturchler-Pierrat, C.; Sommer, B.; Staufenbiel, M.; et al. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci U S A 1999, 96, 14088–14093. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; de Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature 2008, 451, 720–724. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front Aging Neurosci 2022, 14, 815347. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, P.; Arango-Lievano, M.; Neves, I.D.; Rousset, M.C.; Baranger, K.; Rivera, S.; Jeanneteau, F.; Claeysen, S.; Marchi, N. Cerebrovascular pathology during the progression of experimental Alzheimer's disease. Neurobiol Dis 2016, 88, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Landel, V.; Baranger, K.; Virard, I.; Loriod, B.; Khrestchatisky, M.; Rivera, S.; Benech, P.; Feron, F. Temporal gene profiling of the 5XFAD transgenic mouse model highlights the importance of microglial activation in Alzheimer's disease. Mol Neurodegener 2014, 9, 33. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Kobayashi, K.; Imagama, S.; Ohgomori, T.; Hirano, K.; Uchimura, K.; Sakamoto, K.; Hirakawa, A.; Takeuchi, H.; Suzumura, A.; Ishiguro, N.; et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis 2013, 4, e525. [Google Scholar] [CrossRef] [PubMed]
- Morganti, J.M.; Riparip, L.K.; Rosi, S. Call Off the Dog(ma): M1/M2 Polarization Is Concurrent following Traumatic Brain Injury. PLoS One 2016, 11, e0148001. [Google Scholar] [CrossRef]
- Koizumi, T.; Taguchi, K.; Mizuta, I.; Toba, H.; Ohigashi, M.; Onishi, O.; Ikoma, K.; Miyata, S.; Nakata, T.; Tanaka, M.; et al. Transiently proliferating perivascular microglia harbor M1 type and precede cerebrovascular changes in a chronic hypertension model. J Neuroinflammation 2019, 16, 79. [Google Scholar] [CrossRef]
- Hashimoto, A.; Karim, M.R.; Kuramochi, M.; Izawa, T.; Kuwamura, M.; Yamate, J. Characterization of Macrophages and Myofibroblasts Appearing in Dibutyltin Dichloride-Induced Rat Pancreatic Fibrosis. Toxicol Pathol 2020, 48, 509–523. [Google Scholar] [CrossRef]
- Bottcher, C.; Schlickeiser, S.; Sneeboer, M.A.M.; Kunkel, D.; Knop, A.; Paza, E.; Fidzinski, P.; Kraus, L.; Snijders, G.J.L.; Kahn, R.S.; et al. Human microglia regional heterogeneity and phenotypes determined by multiplexed single-cell mass cytometry. Nat Neurosci 2019, 22, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Leal, W. Why microglia kill neurons after neural disorders? The friendly fire hypothesis. Neural Regen Res 2019, 14, 1499–1502. [Google Scholar] [CrossRef] [PubMed]
- Varin, A.; Gordon, S. Alternative activation of macrophages: immune function and cellular biology. Immunobiology 2009, 214, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 2009, 27, 451–483. [Google Scholar] [CrossRef] [PubMed]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front Cell Neurosci 2020, 14, 198. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.; Choi, Y.K. The Role of Astrocytes in the Central Nervous System Focused on BK Channel and Heme Oxygenase Metabolites: A Review. Antioxidants (Basel) 2019, 8. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Deneen, B. The Emerging Nature of Astrocyte Diversity. Annu Rev Neurosci 2019, 42, 187–207. [Google Scholar] [CrossRef]
- Jessen, N.A.; Munk, A.S.; Lundgaard, I.; Nedergaard, M. The Glymphatic System: A Beginner's Guide. Neurochem Res 2015, 40, 2583–2599. [Google Scholar] [CrossRef]
- Monterey, M.D.; Wei, H.; Wu, X.; Wu, J.Q. The Many Faces of Astrocytes in Alzheimer's Disease. Front Neurol 2021, 12, 619626. [Google Scholar] [CrossRef]
- Westergard, T.; Rothstein, J.D. Astrocyte Diversity: Current Insights and Future Directions. Neurochem Res 2020, 45, 1298–1305. [Google Scholar] [CrossRef] [PubMed]
- Vanzani, M.C.; Iacono, R.F.; Caccuri, R.L.; Berria, M.I. Immunochemical and morphometric features of astrocyte reactivity vs. plaque location in Alzheimer's disease. Medicina (B Aires) 2005, 65, 213–218. [Google Scholar]
- Muramori, F.; Kobayashi, K.; Nakamura, I. A quantitative study of neurofibrillary tangles, senile plaques and astrocytes in the hippocampal subdivisions and entorhinal cortex in Alzheimer's disease, normal controls and non-Alzheimer neuropsychiatric diseases. Psychiatry Clin Neurosci 1998, 52, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-associated astrocytes in Alzheimer's disease and aging. Nat Neurosci 2020, 23, 701–706. [Google Scholar] [CrossRef] [PubMed]
- von Bohlen und Halbach, O. Immunohistological markers for proliferative events, gliogenesis, and neurogenesis within the adult hippocampus. Cell Tissue Res 2011, 345, 1–19. [Google Scholar] [CrossRef]
- Ding, Z.B.; Song, L.J.; Wang, Q.; Kumar, G.; Yan, Y.Q.; Ma, C.G. Astrocytes: a double-edged sword in neurodegenerative diseases. Neural Regen Res 2021, 16, 1702–1710. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J Neurosci 2012, 32, 6391–6410. [Google Scholar] [CrossRef]
- Ben Haim, L.; Carrillo-de Sauvage, M.A.; Ceyzeriat, K.; Escartin, C. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front Cell Neurosci 2015, 9, 278. [Google Scholar] [CrossRef]
- Escartin, C.; Guillemaud, O.; Carrillo-de Sauvage, M.A. Questions and (some) answers on reactive astrocytes. Glia 2019, 67, 2221–2247. [Google Scholar] [CrossRef]
- Cai, Z.; Wan, C.Q.; Liu, Z. Astrocyte and Alzheimer's disease. J Neurol 2017, 264, 2068–2074. [Google Scholar] [CrossRef]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of microglia and astrocytes: a roadway to neuroinflammation and Alzheimer's disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef]
- Shinozaki, Y.; Shibata, K.; Yoshida, K.; Shigetomi, E.; Gachet, C.; Ikenaka, K.; Tanaka, K.F.; Koizumi, S. Transformation of Astrocytes to a Neuroprotective Phenotype by Microglia via P2Y(1) Receptor Downregulation. Cell Rep 2017, 19, 1151–1164. [Google Scholar] [CrossRef]
- Tchieu, J.; Calder, E.L.; Guttikonda, S.R.; Gutzwiller, E.M.; Aromolaran, K.A.; Steinbeck, J.A.; Goldstein, P.A.; Studer, L. NFIA is a gliogenic switch enabling rapid derivation of functional human astrocytes from pluripotent stem cells. Nat Biotechnol 2019, 37, 267–275. [Google Scholar] [CrossRef]
- Neal, M.; Luo, J.; Harischandra, D.S.; Gordon, R.; Sarkar, S.; Jin, H.; Anantharam, V.; Desaubry, L.; Kanthasamy, A.; Kanthasamy, A. Prokineticin-2 promotes chemotaxis and alternative A2 reactivity of astrocytes. Glia 2018, 66, 2137–2157. [Google Scholar] [CrossRef]
- Healy, L.M.; Antel, J.P. Sphingosine-1-Phosphate Receptors in the Central Nervous and Immune Systems. Curr Drug Targets 2016, 17, 1841–1850. [Google Scholar] [CrossRef]
- Hodgetts, S.I.; Harvey, A.R. Neurotrophic Factors Used to Treat Spinal Cord Injury. Vitam Horm 2017, 104, 405–457. [Google Scholar] [CrossRef]
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front Physiol 2018, 9, 113. [Google Scholar] [CrossRef]
- Baik, S.H.; Cha, M.Y.; Hyun, Y.M.; Cho, H.; Hamza, B.; Kim, D.K.; Han, S.H.; Choi, H.; Kim, K.H.; Moon, M.; et al. Migration of neutrophils targeting amyloid plaques in Alzheimer's disease mouse model. Neurobiol Aging 2014, 35, 1286–1292. [Google Scholar] [CrossRef]
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer's disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med 2015, 21, 880–886. [Google Scholar] [CrossRef]
- Santos-Lima, B.; Pietronigro, E.C.; Terrabuio, E.; Zenaro, E.; Constantin, G. The role of neutrophils in the dysfunction of central nervous system barriers. Front Aging Neurosci 2022, 14, 965169. [Google Scholar] [CrossRef]
- Rossi, B.; Constantin, G.; Zenaro, E. The emerging role of neutrophils in neurodegeneration. Immunobiology 2020, 225, 151865. [Google Scholar] [CrossRef]
- Rossi, B.; Santos-Lima, B.; Terrabuio, E.; Zenaro, E.; Constantin, G. Common Peripheral Immunity Mechanisms in Multiple Sclerosis and Alzheimer's Disease. Front Immunol 2021, 12, 639369. [Google Scholar] [CrossRef]
- Smyth, L.C.D.; Murray, H.C.; Hill, M.; van Leeuwen, E.; Highet, B.; Magon, N.J.; Osanlouy, M.; Mathiesen, S.N.; Mockett, B.; Singh-Bains, M.K.; et al. Neutrophil-vascular interactions drive myeloperoxidase accumulation in the brain in Alzheimer's disease. Acta Neuropathol Commun 2022, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Stock, A.J.; Kasus-Jacobi, A.; Pereira, H.A. The role of neutrophil granule proteins in neuroinflammation and Alzheimer's disease. J Neuroinflammation 2018, 15, 240. [Google Scholar] [CrossRef]
- Mihaila, A.C.; Ciortan, L.; Macarie, R.D.; Vadana, M.; Cecoltan, S.; Preda, M.B.; Hudita, A.; Gan, A.M.; Jakobsson, G.; Tucureanu, M.M.; et al. Transcriptional Profiling and Functional Analysis of N1/N2 Neutrophils Reveal an Immunomodulatory Effect of S100A9-Blockade on the Pro-Inflammatory N1 Subpopulation. Front Immunol 2021, 12, 708770. [Google Scholar] [CrossRef] [PubMed]
- Cuartero, M.I.; Ballesteros, I.; Moraga, A.; Nombela, F.; Vivancos, J.; Hamilton, J.A.; Corbi, A.L.; Lizasoain, I.; Moro, M.A. N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARgamma agonist rosiglitazone. Stroke 2013, 44, 3498–3508. [Google Scholar] [CrossRef]
- Chiurchiu, V.; Maccarrone, M. Bioactive lipids as modulators of immunity, inflammation and emotions. Curr Opin Pharmacol 2016, 29, 54–62. [Google Scholar] [CrossRef]
- Serhan, C.N.; Yang, R.; Martinod, K.; Kasuga, K.; Pillai, P.S.; Porter, T.F.; Oh, S.F.; Spite, M. Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J Exp Med 2009, 206, 15–23. [Google Scholar] [CrossRef]
- Serhan, C.N.; Dalli, J.; Karamnov, S.; Choi, A.; Park, C.K.; Xu, Z.Z.; Ji, R.R.; Zhu, M.; Petasis, N.A. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. Faseb J 2012, 26, 1755–1765. [Google Scholar] [CrossRef]
- Hong, S.; Lu, Y.; Tian, H.; Alapure, B.V.; Wang, Q.; Bunnell, B.A.; Laborde, J.M. Maresin-like lipid mediators are produced by leukocytes and platelets and rescue reparative function of diabetes-impaired macrophages. Chem Biol 2014, 21, 1318–1329. [Google Scholar] [CrossRef]
- Francos-Quijorna, I.; Santos-Nogueira, E.; Gronert, K.; Sullivan, A.B.; Kopp, M.A.; Brommer, B.; David, S.; Schwab, J.M.; Lopez-Vales, R. Maresin 1 Promotes Inflammatory Resolution, Neuroprotection, and Functional Neurological Recovery After Spinal Cord Injury. J Neurosci 2017, 37, 11731–11743. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, X.; Hjorth, E.; Colas, R.A.; Schroeder, L.; Granholm, A.C.; Serhan, C.N.; Schultzberg, M. Pro-Resolving Lipid Mediators Improve Neuronal Survival and Increase Abeta42 Phagocytosis. Mol Neurobiol 2016, 53, 2733–2749. [Google Scholar] [CrossRef]
- Sanchez-Fernandez, A.; Zandee, S.; Mastrogiovanni, M.; Charabati, M.; Rubbo, H.; Prat, A.; Lopez-Vales, R. Administration of Maresin-1 ameliorates the physiopathology of experimental autoimmune encephalomyelitis. J Neuroinflammation 2022, 19, 27. [Google Scholar] [CrossRef]
- Yin, P.; Wang, X.; Wang, S.; Wei, Y.; Feng, J.; Zhu, M. Maresin 1 Improves Cognitive Decline and Ameliorates Inflammation in a Mouse Model of Alzheimer's Disease. Front Cell Neurosci 2019, 13, 466. [Google Scholar] [CrossRef]
- Emre, C.; Arroyo-Garcia, L.E.; Do, K.V.; Jun, B.; Ohshima, M.; Alcalde, S.G.; Cothern, M.L.; Maioli, S.; Nilsson, P.; Hjorth, E.; et al. Intranasal delivery of pro-resolving lipid mediators rescues memory and gamma oscillation impairment in App(NL-G-F/NL-G-F) mice. Commun Biol 2022, 5, 245. [Google Scholar] [CrossRef]
- Van Dam, D.; De Deyn, P.P. Animal models in the drug discovery pipeline for Alzheimer's disease. Br J Pharmacol 2011, 164, 1285–1300. [Google Scholar] [CrossRef]
- Southam, D.S.; Dolovich, M.; O'Byrne, P.M.; Inman, M.D. Distribution of intranasal instillations in mice: effects of volume, time, body position, and anesthesia. Am J Physiol Lung Cell Mol Physiol 2002, 282, L833–L839. [Google Scholar] [CrossRef]
- Torika, N.; Asraf, K.; Cohen, H.; Fleisher-Berkovich, S. Intranasal telmisartan ameliorates brain pathology in five familial Alzheimer's disease mice. Brain Behav Immun 2017, 64, 80–90. [Google Scholar] [CrossRef]
- Xu, J.; Tao, J.; Wang, J. Design and Application in Delivery System of Intranasal Antidepressants. Front Bioeng Biotechnol 2020, 8, 626882. [Google Scholar] [CrossRef]
- Hong, S.; Nagayach, A.; Lu, Y.; Peng, H.; Duong, Q.A.; Pham, N.B.; Vuong, C.A.; Bazan, N.G. A high fat, sugar, and salt Western diet induces motor-muscular and sensory dysfunctions and neurodegeneration in mice during aging: Ameliorative action of metformin. CNS Neurosci Ther 2021, 27, 1458–1471. [Google Scholar] [CrossRef]
- Crouzin, N.; Baranger, K.; Cavalier, M.; Marchalant, Y.; Cohen-Solal, C.; Roman, F.S.; Khrestchatisky, M.; Rivera, S.; Feron, F.; Vignes, M. Area-specific alterations of synaptic plasticity in the 5XFAD mouse model of Alzheimer's disease: dissociation between somatosensory cortex and hippocampus. PLoS One 2013, 8, e74667. [Google Scholar] [CrossRef] [PubMed]
- Bussiere, T.; Bard, F.; Barbour, R.; Grajeda, H.; Guido, T.; Khan, K.; Schenk, D.; Games, D.; Seubert, P.; Buttini, M. Morphological characterization of Thioflavin-S-positive amyloid plaques in transgenic Alzheimer mice and effect of passive Abeta immunotherapy on their clearance. Am J Pathol 2004, 165, 987–995. [Google Scholar] [CrossRef]
- de Sousa, D.M.B.; Benedetti, A.; Altendorfer, B.; Mrowetz, H.; Unger, M.S.; Schallmoser, K.; Aigner, L.; Kniewallner, K.M. Immune-mediated platelet depletion augments Alzheimer's disease neuropathological hallmarks in APP-PS1 mice. Aging (Albany NY) 2023, 15, 630–649. [Google Scholar] [CrossRef]
- Liu, P.; Reichl, J.H.; Rao, E.R.; McNellis, B.M.; Huang, E.S.; Hemmy, L.S.; Forster, C.L.; Kuskowski, M.A.; Borchelt, D.R.; Vassar, R.; et al. Quantitative Comparison of Dense-Core Amyloid Plaque Accumulation in Amyloid-beta Protein Precursor Transgenic Mice. J Alzheimers Dis 2017, 56, 743–761. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.I.; Wong, E.; Joghee, R.; Balbaa, A.; Vitanova, K.S.; Stringer, K.M.; Vanshoiack, A.; Phelan, S.J.; Launchbury, F.; Desai, S.; et al. Plaque contact and unimpaired Trem2 is required for the microglial response to amyloid pathology. Cell Rep 2022, 41, 111686. [Google Scholar] [CrossRef]
- Bonsi, P.; Cuomo, D.; Martella, G.; Madeo, G.; Schirinzi, T.; Puglisi, F.; Ponterio, G.; Pisani, A. Centrality of striatal cholinergic transmission in Basal Ganglia function. Front Neuroanat 2011, 5, 6. [Google Scholar] [CrossRef]
- Zhou, K.; Cherra, S.J., 3rd; Goncharov, A.; Jin, Y. Asynchronous Cholinergic Drive Correlates with Excitation-Inhibition Imbalance via a Neuronal Ca(2+) Sensor Protein. Cell Rep 2017, 19, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Hashimoto, K.; Kobayashi, K. Cholinergic regulation of object recognition memory. Front Behav Neurosci 2022, 16, 996089. [Google Scholar] [CrossRef]
- Lim, S.A.; Kang, U.J.; McGehee, D.S. Striatal cholinergic interneuron regulation and circuit effects. Front Synaptic Neurosci 2014, 6, 22. [Google Scholar] [CrossRef]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Mesulam, M. The cholinergic lesion of Alzheimer's disease: pivotal factor or side show? Learn Mem 2004, 11, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Pang, P.; Chen, W.; Zhu, H.; Henok, K.A.; Li, H.; Wu, Z.; Ke, X.; Wu, J.; Zhang, T.; et al. The Lesion Analysis of Cholinergic Neurons in 5XFAD Mouse Model in the Three-Dimensional Level of Whole Brain. Mol Neurobiol 2018, 55, 4115–4125. [Google Scholar] [CrossRef] [PubMed]
- Hopperton, K.E.; Mohammad, D.; Trepanier, M.O.; Giuliano, V.; Bazinet, R.P. Markers of microglia in post-mortem brain samples from patients with Alzheimer's disease: a systematic review. Mol Psychiatry 2018, 23, 177–198. [Google Scholar] [CrossRef] [PubMed]
- Sheets, K.G.; Jun, B.; Zhou, Y.; Zhu, M.; Petasis, N.A.; Gordon, W.C.; Bazan, N.G. Microglial ramification and redistribution concomitant with the attenuation of choroidal neovascularization by neuroprotectin D1. Mol Vis 2013, 19, 1747–1759. [Google Scholar]
- Crews, F.T.; Lawrimore, C.J.; Walter, T.J.; Coleman, L.G., Jr. The role of neuroimmune signaling in alcoholism. Neuropharmacology 2017, 122, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Choi, S.; Bikkannavar, P.; Cordeiro, M.F. Microglia: Key Players in Retinal Ageing and Neurodegeneration. Front Cell Neurosci 2022, 16, 804782. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D.; Olschowka, J.A.; O'Banion, M.K. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener 2020, 9, 42. [Google Scholar] [CrossRef]
- Gotoh, M.; Miyamoto, Y.; Ikeshima-Kataoka, H. Astrocytic Neuroimmunological Roles Interacting with Microglial Cells in Neurodegenerative Diseases. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Huijbers, W.; Mormino, E.C.; Schultz, A.P.; Wigman, S.; Ward, A.M.; Larvie, M.; Amariglio, R.E.; Marshall, G.A.; Rentz, D.M.; Johnson, K.A.; et al. Amyloid-beta deposition in mild cognitive impairment is associated with increased hippocampal activity, atrophy and clinical progression. Brain 2015, 138, 1023–1035. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Cellular Receptors of Amyloid beta Oligomers (AbetaOs) in Alzheimer's Disease. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med 2003, 9, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.J.; Xiao, M.; Chen, R.; Cai, Z. Metabolic Dysfunction of Astrocyte: An Initiating Factor in Beta-amyloid Pathology? Aging Neurodegener 2013, 1, 7–14. [Google Scholar]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Hinkle, J.T.; Dawson, V.L.; Dawson, T.M. The A1 astrocyte paradigm: New avenues for pharmacological intervention in neurodegeneration. Mov Disord 2019, 34, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., 2nd; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat Neurosci 2019, 22, 1635–1648. [Google Scholar] [CrossRef]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson's disease. Nat Med 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Munch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U S A 2018, 115, E1896–E1905. [Google Scholar] [CrossRef]
- Liu, Y.X.; Sun, H.; Guo, W.Y. Astrocyte polarization in glaucoma: a new opportunity. Neural Regen Res 2022, 17, 2582–2588. [Google Scholar] [CrossRef]
- Morris, J.K.; Honea, R.A.; Vidoni, E.D.; Swerdlow, R.H.; Burns, J.M. Is Alzheimer's disease a systemic disease? Biochim Biophys Acta 2014, 1842, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.J. A systemic view of Alzheimer disease - insights from amyloid-beta metabolism beyond the brain. Nat Rev Neurol 2017, 13, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D'Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer's disease. Neurobiol Dis 2017, 107, 41–56. [Google Scholar] [CrossRef]
- Abbott, A. Dementia: a problem for our age. Nature 2011, 475, S2–S4. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Pietronigro, E.; Zenaro, E.; Constantin, G. Imaging of Leukocyte Trafficking in Alzheimer's Disease. Front Immunol 2016, 7, 33. [Google Scholar] [CrossRef]
- Song, Z.; Bhattacharya, S.; Clemens, R.A.; Dinauer, M.C. Molecular regulation of neutrophil swarming in health and disease: Lessons from the phagocyte oxidase. iScience 2023, 26, 108034. [Google Scholar] [CrossRef] [PubMed]
- Lammermann, T. In the eye of the neutrophil swarm-navigation signals that bring neutrophils together in inflamed and infected tissues. J Leukoc Biol 2016, 100, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol 2003, 161, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Shadfar, S.; Hwang, C.J.; Lim, M.S.; Choi, D.Y.; Hong, J.T. Involvement of inflammation in Alzheimer's disease pathogenesis and therapeutic potential of anti-inflammatory agents. Arch Pharm Res 2015, 38, 2106–2119. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Grundke-Iqbal, I. Alzheimer's disease, a multifactorial disorder seeking multitherapies. Alzheimers Dement 2010, 6, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Carreiras, M.C.; Mendes, E.; Perry, M.J.; Francisco, A.P.; Marco-Contelles, J. The multifactorial nature of Alzheimer's disease for developing potential therapeutics. Curr Top Med Chem 2013, 13, 1745–1770. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar-Colucci, S.; Landreth, G.E. Microglia and inflammation in Alzheimer's disease. CNS Neurol Disord Drug Targets 2010, 9, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer's disease. Ann Transl Med 2015, 3, 136. [Google Scholar] [CrossRef] [PubMed]
- Erdo, F.; Bors, L.A.; Farkas, D.; Bajza, A.; Gizurarson, S. Evaluation of intranasal delivery route of drug administration for brain targeting. Brain Res Bull 2018, 143, 155–170. [Google Scholar] [CrossRef]
- Lochhead, J.J.; Thorne, R.G. Intranasal delivery of biologics to the central nervous system. Adv Drug Deliv Rev 2012, 64, 614–628. [Google Scholar] [CrossRef]
- Chauhan, M.B.; Chauhan, N.B. Brain Uptake of Neurotherapeutics after Intranasal versus Intraperitoneal Delivery in Mice. J Neurol Neurosurg 2015, 2. [Google Scholar] [CrossRef]
- Louneva, N.; Cohen, J.W.; Han, L.Y.; Talbot, K.; Wilson, R.S.; Bennett, D.A.; Trojanowski, J.Q.; Arnold, S.E. Caspase-3 is enriched in postsynaptic densities and increased in Alzheimer's disease. Am J Pathol 2008, 173, 1488–1495. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.; Wilcockson, D.C.; Campion, S.; Lunnon, K.; Perry, V.H. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci 2005, 25, 9275–9284. [Google Scholar] [CrossRef] [PubMed]
- Sil, S.; Ghosh, T. Role of cox-2 mediated neuroinflammation on the neurodegeneration and cognitive impairments in colchicine induced rat model of Alzheimer's Disease. J Neuroimmunol 2016, 291, 115–124. [Google Scholar] [CrossRef]
- Singhal, G.; Jaehne, E.J.; Corrigan, F.; Toben, C.; Baune, B.T. Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front Neurosci 2014, 8, 315. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Prados, J.C.; Traves, P.G.; Cuenca, J.; Rico, D.; Aragones, J.; Martin-Sanz, P.; Cascante, M.; Bosca, L. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol 2010, 185, 605–614. [Google Scholar] [CrossRef]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: mechanism and potential therapeutic targets. Signal Transduct Target Ther 2023, 8, 359. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol 2009, 27, 119–145. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Hao, C.; Munoz, D.G. Role of microglia in senile plaque formation. Neurobiol Aging 1995, 16, 797–804. [Google Scholar] [CrossRef]
- Stalder, M.; Phinney, A.; Probst, A.; Sommer, B.; Staufenbiel, M.; Jucker, M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol 1999, 154, 1673–1684. [Google Scholar] [CrossRef]
- Yan, P.; Bero, A.W.; Cirrito, J.R.; Xiao, Q.; Hu, X.; Wang, Y.; Gonzales, E.; Holtzman, D.M.; Lee, J.M. Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J Neurosci 2009, 29, 10706–10714. [Google Scholar] [CrossRef]
- Perlmutter, L.S.; Barron, E.; Chui, H.C. Morphologic association between microglia and senile plaque amyloid in Alzheimer's disease. Neurosci Lett 1990, 119, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Cras, P.; Kawai, M.; Siedlak, S.; Mulvihill, P.; Gambetti, P.; Lowery, D.; Gonzalez-DeWhitt, P.; Greenberg, B.; Perry, G. Neuronal and microglial involvement in beta-amyloid protein deposition in Alzheimer's disease. Am J Pathol 1990, 137, 241–246. [Google Scholar] [PubMed]
- Wegiel, J.; Imaki, H.; Wang, K.C.; Wegiel, J.; Wronska, A.; Osuchowski, M.; Rubenstein, R. Origin and turnover of microglial cells in fibrillar plaques of APPsw transgenic mice. Acta Neuropathol 2003, 105, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Bornemann, K.D.; Wiederhold, K.H.; Pauli, C.; Ermini, F.; Stalder, M.; Schnell, L.; Sommer, B.; Jucker, M.; Staufenbiel, M. Abeta-induced inflammatory processes in microglia cells of APP23 transgenic mice. Am J Pathol 2001, 158, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer's disease. Int J Neurosci 2014, 124, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, P.; Vaibhav, K.; Tabassum, R.; Khan, A.; Ishrat, T.; Khan, M.M.; Ahmad, A.; Islam, F.; Safhi, M.M.; Islam, F. Anti-apoptotic and anti-inflammatory effect of Piperine on 6-OHDA induced Parkinson's rat model. J Nutr Biochem 2013, 24, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Franco-Bocanegra, D.K.; Gourari, Y.; McAuley, C.; Chatelet, D.S.; Johnston, D.A.; Nicoll, J.A.R.; Boche, D. Microglial morphology in Alzheimer's disease and after Abeta immunotherapy. Sci Rep 2021, 11, 15955. [Google Scholar] [CrossRef] [PubMed]
- Poplimont, H.; Georgantzoglou, A.; Boulch, M.; Walker, H.A.; Coombs, C.; Papaleonidopoulou, F.; Sarris, M. Neutrophil Swarming in Damaged Tissue Is Orchestrated by Connexins and Cooperative Calcium Alarm Signals. Curr Biol 2020, 30, 2761–2776.e2767. [Google Scholar] [CrossRef]
- Kotredes, K.P.; Pandey, R.S.; Persohn, S.; Elderidge, K.; Burton, C.P.; Miner, E.W.; Haynes, K.A.; Santos, D.F.S.; Williams, S.P.; Heaton, N.; et al. Characterizing molecular and synaptic signatures in mouse models of late-onset Alzheimer's disease independent of amyloid and tau pathology. Alzheimers Dement 2024, 20, 4126–4146. [Google Scholar] [CrossRef]
- Oblak, A.L.; Forner, S.; Territo, P.R.; Sasner, M.; Carter, G.W.; Howell, G.R.; Sukoff-Rizzo, S.J.; Logsdon, B.A.; Mangravite, L.M.; Mortazavi, A.; et al. Model organism development and evaluation for late-onset Alzheimer's disease: MODEL-AD. Alzheimers Dement (N Y) 2020, 6, e12110. [Google Scholar] [CrossRef]
- Sukoff Rizzo, S.J.; Masters, A.; Onos, K.D.; Quinney, S.; Sasner, M.; Oblak, A.; Lamb, B.T.; Territo, P.R.; consortium, M.-A. Improving preclinical to clinical translation in Alzheimer's disease research. Alzheimers Dement (N Y) 2020, 6, e12038. [Google Scholar] [CrossRef] [PubMed]
- Forner, S.; Kawauchi, S.; Balderrama-Gutierrez, G.; Kramar, E.A.; Matheos, D.P.; Phan, J.; Javonillo, D.I.; Tran, K.M.; Hingco, E.; da Cunha, C.; et al. Systematic phenotyping and characterization of the 5xFAD mouse model of Alzheimer's disease. Sci Data 2021, 8, 270. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wei, J.; Ma, X.; Xia, B.; Shakir, N.; Zhang, J.K.; Zhang, L.; Cui, Y.; Ferguson, D.; Qiu, S.; et al. Disrupted Maturation of Prefrontal Layer 5 Neuronal Circuits in an Alzheimer's Mouse Model of Amyloid Deposition. Neurosci Bull 2023, 39, 881–892. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
MarL1 treatment ameliorated AD neuropathology in brains of 5xFAD mice. (A) Immunostaining of NeuN (green) and Amyloid-β1-42 (red) in CA3 and dentate gyrus (DG) of hippocampus. White arrows mark some Aβ1-42 deposition in hippocampal regions. Scale bar: 180 μm. (B) Quantification of NeuN+ and Amyloid-β1-42+ staining intensities of hippocampus (Mean fluorescence intensity, MFI). Data are Means ± SEM. ***p < 0.001.
Figure 1.
MarL1 treatment ameliorated AD neuropathology in brains of 5xFAD mice. (A) Immunostaining of NeuN (green) and Amyloid-β1-42 (red) in CA3 and dentate gyrus (DG) of hippocampus. White arrows mark some Aβ1-42 deposition in hippocampal regions. Scale bar: 180 μm. (B) Quantification of NeuN+ and Amyloid-β1-42+ staining intensities of hippocampus (Mean fluorescence intensity, MFI). Data are Means ± SEM. ***p < 0.001.
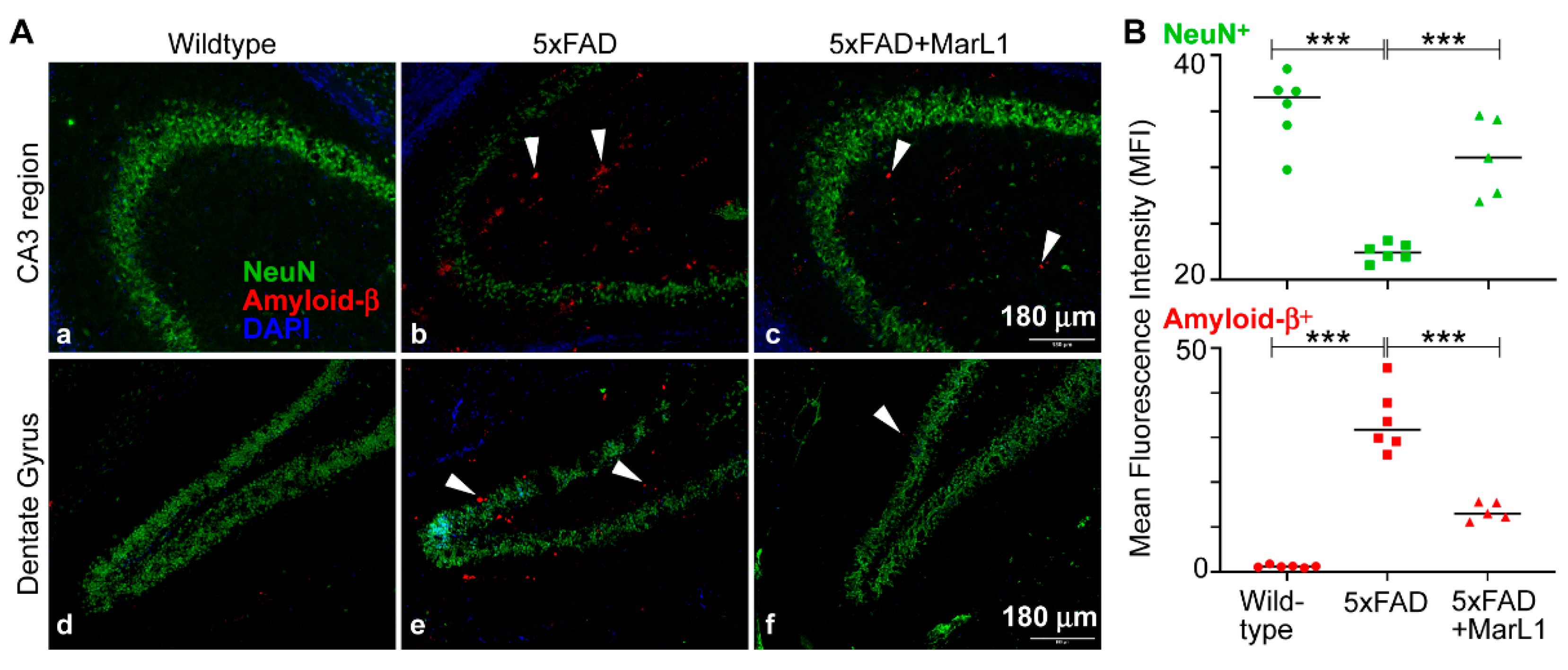
Figure 2.
MarL1 protected cholinergic neurons (ChAT+) and inhibited apoptotic cleaved-caspase-3 activity in brains of 5xFAD mice. (A) Immunostaining of ChAT (green) and cleaved-caspase-3 (red) in striatum (Panels a-c: 10X magnification; scale bar: 180 µm. White arrows mark cleaved-caspase-3+ cholinergic neurons in zoomed-in images (Panels d-f). Scale bar: 35 µm. (B) Quantification of ChAT and caspase-3 in striatum. Left: Mean fluorescence intensity MFI for ChAT+; middle: MFI for cleaved-caspase-3+; right: count of cells stained positive for both ChAT and cleaved-caspase-3. Data are Means ± SEM. *** p < 0.001, and *p < 0.05.
Figure 2.
MarL1 protected cholinergic neurons (ChAT+) and inhibited apoptotic cleaved-caspase-3 activity in brains of 5xFAD mice. (A) Immunostaining of ChAT (green) and cleaved-caspase-3 (red) in striatum (Panels a-c: 10X magnification; scale bar: 180 µm. White arrows mark cleaved-caspase-3+ cholinergic neurons in zoomed-in images (Panels d-f). Scale bar: 35 µm. (B) Quantification of ChAT and caspase-3 in striatum. Left: Mean fluorescence intensity MFI for ChAT+; middle: MFI for cleaved-caspase-3+; right: count of cells stained positive for both ChAT and cleaved-caspase-3. Data are Means ± SEM. *** p < 0.001, and *p < 0.05.
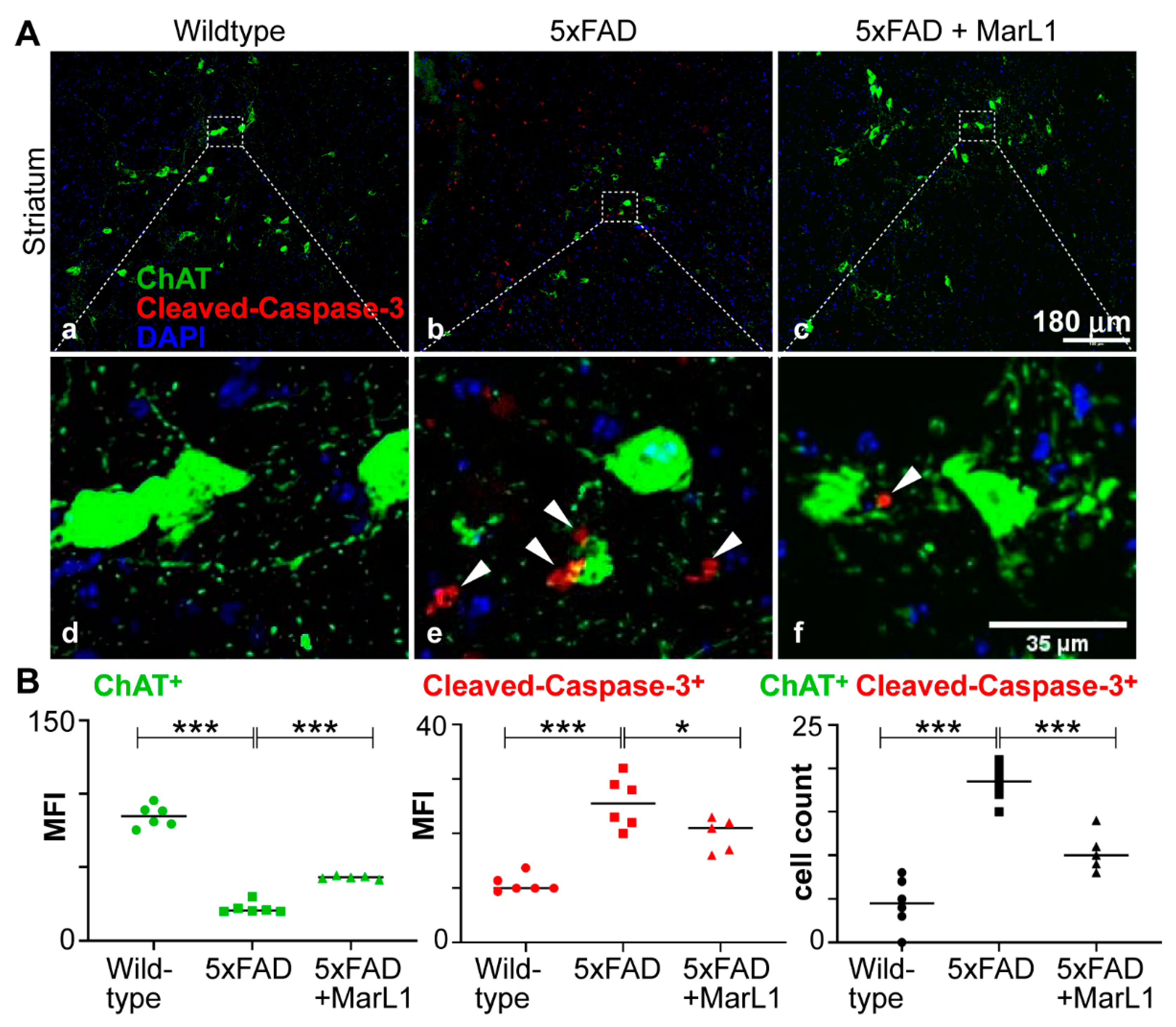
Figure 3.
MarL1 suppressed pro-inflammatory M1 phenotype polarization of microglia in brains of 5xFAD mice. (A) Immunostaining of microglia with Iba-1 (green) and CD68 (red) in CA1 region of hippocampus from 5xFAD transgenic mice (Panels a-c: 10X magnification; scale bar: 180 µm. Panels d-f: zoomed-in images; scale bar: 30 µm). White arrows mark Iba-1+CD68+ microglia. (B) Quantification of Iba-1+ and CD68+ in hippocampus. Left: mean fluorescence intensity MFI of Iba-1+; middle: MFI of CD68+; right: count of microglia stained positive for both Iba-1+ and CD68+; (C) Quantification of microglia based on phenotype characterization (ramified, partially ramified, partially amoeboid, amoeboid) in hippocampus. Data are Means ± SEM. *** p < 0.001, ** p <0.01, and * p < 0.05.
Figure 3.
MarL1 suppressed pro-inflammatory M1 phenotype polarization of microglia in brains of 5xFAD mice. (A) Immunostaining of microglia with Iba-1 (green) and CD68 (red) in CA1 region of hippocampus from 5xFAD transgenic mice (Panels a-c: 10X magnification; scale bar: 180 µm. Panels d-f: zoomed-in images; scale bar: 30 µm). White arrows mark Iba-1+CD68+ microglia. (B) Quantification of Iba-1+ and CD68+ in hippocampus. Left: mean fluorescence intensity MFI of Iba-1+; middle: MFI of CD68+; right: count of microglia stained positive for both Iba-1+ and CD68+; (C) Quantification of microglia based on phenotype characterization (ramified, partially ramified, partially amoeboid, amoeboid) in hippocampus. Data are Means ± SEM. *** p < 0.001, ** p <0.01, and * p < 0.05.
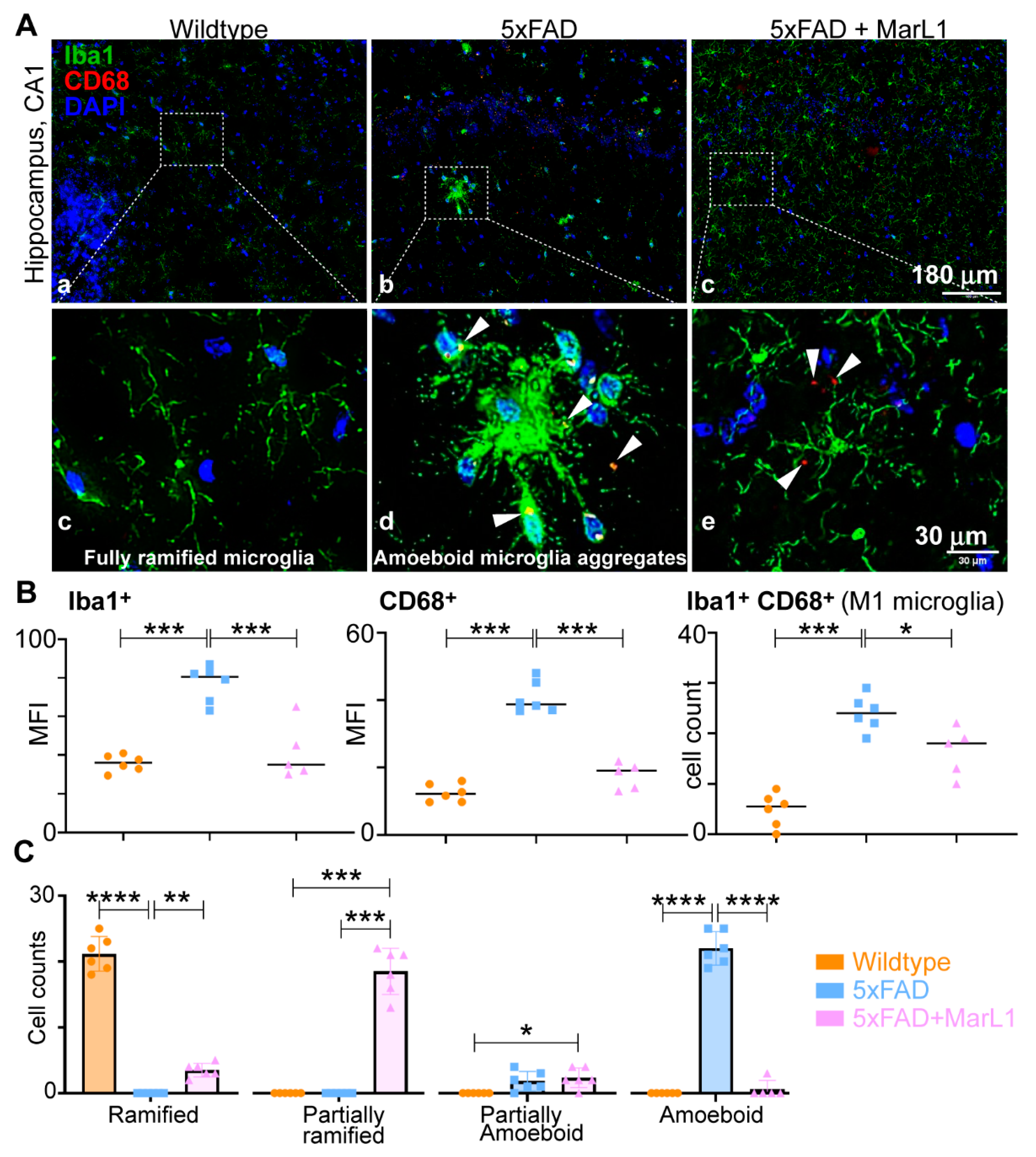
Figure 4.
MarL1 promoted anti-inflammatory M2 phenotype polarization of microglia in brains of 5xFAD mice. (A) Immunostaining of microglia with Iba-1 (green) and Arg1 (red) in cortex (Panels a-c: 20X magnification; scale bar: 90 µm. Panels d-f: zoomed-in images; scale bar: 30 µm). White arrows mark Iba1+Arg1+ microglia. Red arrows mark microglial aggregation in cortex of 5xFAD mice. (B) Quantification of Iba-1 and Arg1 in cortex. Left: mean fluorescence intensity MFI of Iba1+; middle: MFI of Arg1+; right: count of microglia stained positive for both Iba1 and Arg1 in cortex. Data are Means ± SEM. *** p < 0.001, and ** p < 0.01.
Figure 4.
MarL1 promoted anti-inflammatory M2 phenotype polarization of microglia in brains of 5xFAD mice. (A) Immunostaining of microglia with Iba-1 (green) and Arg1 (red) in cortex (Panels a-c: 20X magnification; scale bar: 90 µm. Panels d-f: zoomed-in images; scale bar: 30 µm). White arrows mark Iba1+Arg1+ microglia. Red arrows mark microglial aggregation in cortex of 5xFAD mice. (B) Quantification of Iba-1 and Arg1 in cortex. Left: mean fluorescence intensity MFI of Iba1+; middle: MFI of Arg1+; right: count of microglia stained positive for both Iba1 and Arg1 in cortex. Data are Means ± SEM. *** p < 0.001, and ** p < 0.01.
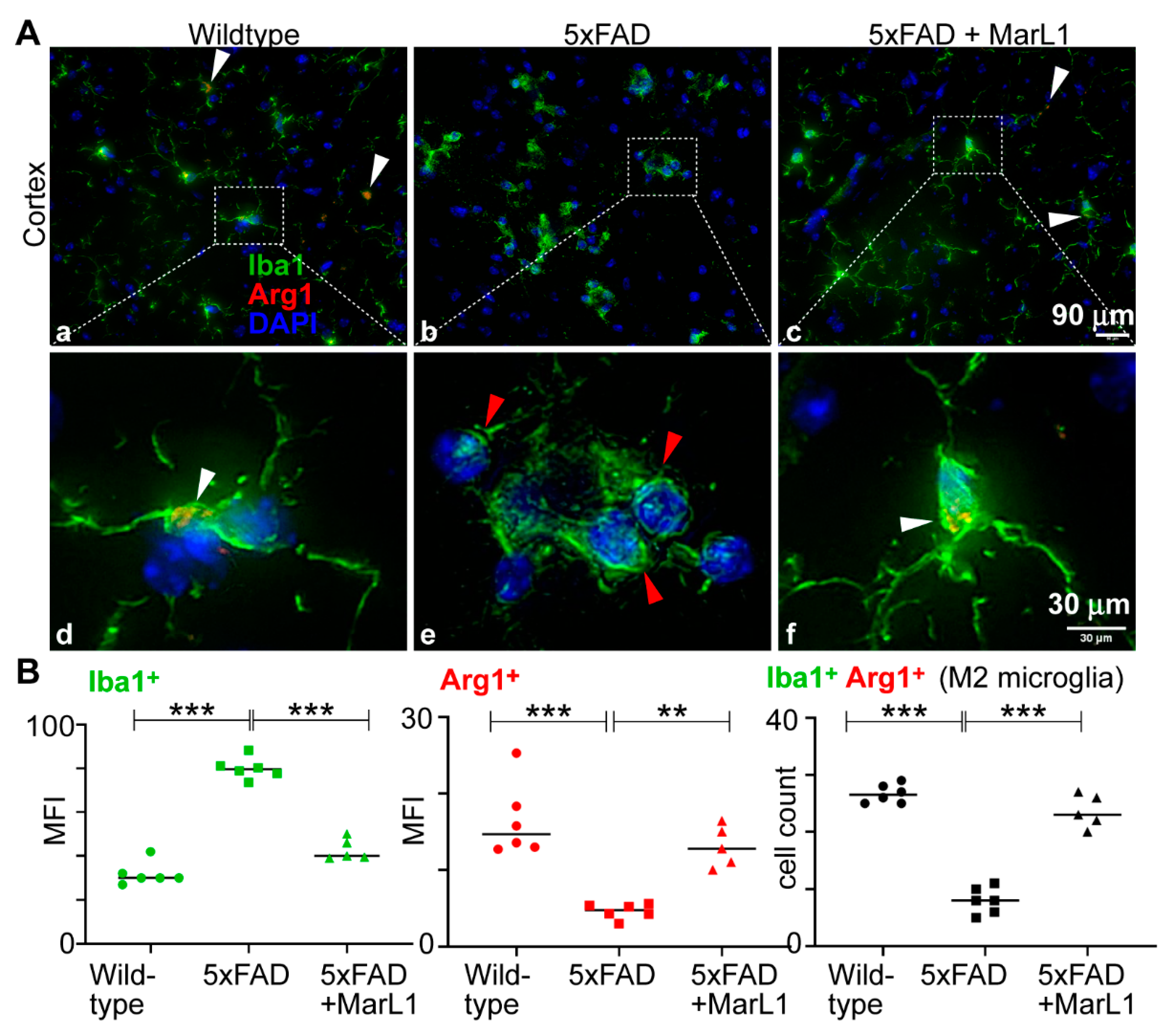
Figure 5.
MarL1 treatment mitigated AD-augmented TNF-α expression and astrogliosis in brains of 5xFAD mice. (A) Immunostaining of GFAP (green) and TNF-α (red) in cortex. White arrows mark GFAP+TNF-α+ astrocytes in cortex (Panels a-c: 10X magnification; scale bar: 180 µm. Panels d-f: zoomed-in images; scale bar: 30 µm). (B) Quantification of GFAP+ and TNF-α+ in mean fluorescence intensity MFI in cortex. Data are Means ± SEM. *** p < 0.001, and ** p < 0.01.
Figure 5.
MarL1 treatment mitigated AD-augmented TNF-α expression and astrogliosis in brains of 5xFAD mice. (A) Immunostaining of GFAP (green) and TNF-α (red) in cortex. White arrows mark GFAP+TNF-α+ astrocytes in cortex (Panels a-c: 10X magnification; scale bar: 180 µm. Panels d-f: zoomed-in images; scale bar: 30 µm). (B) Quantification of GFAP+ and TNF-α+ in mean fluorescence intensity MFI in cortex. Data are Means ± SEM. *** p < 0.001, and ** p < 0.01.
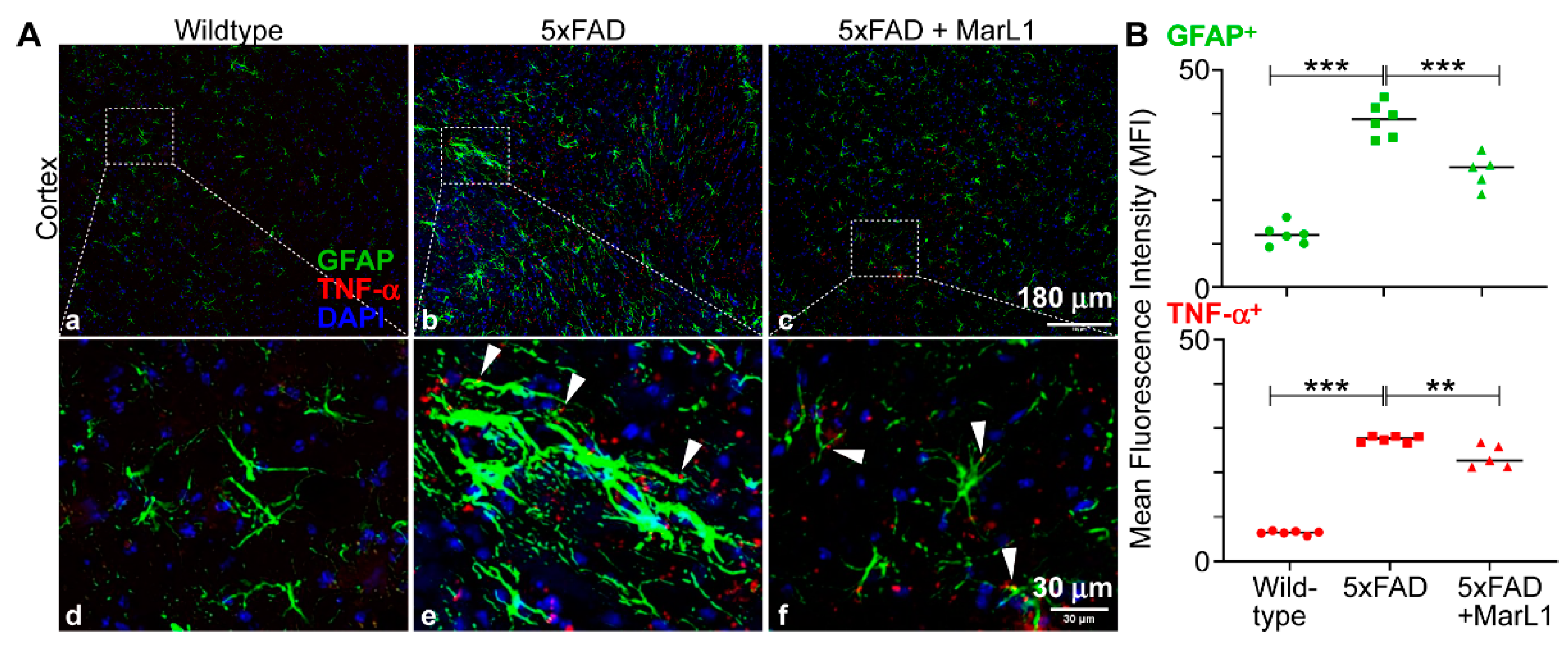
Figure 6.
MarL1 treatment suppressed AD-associated pro-inflammatory A1 phenotypic polarization of astrocytes in brains of 5xFAD mice. (A) Immunostaining of GFAP (green) and Complement C3 (red) in dentate gyrus of hippocampus. Panels a-c: 20X magnification; scale bar: 90 µm. White arrows mark GFAP and complement C3 astrocytes co-expression in zoomed-in images (panels d-f); scale bar: 30 µm. (B) Quantification of GFAP+ and C3+ in mean fluorescence intensity MFI in dentate gyri. Left: MFI of GFAP+; middle: MFI of C3+; right: Pearson’s coefficient for quantification of co-localization of GFAP and C3. Data are Means ± SEM. *** p < 0.001, and ** p < 0.01.
Figure 6.
MarL1 treatment suppressed AD-associated pro-inflammatory A1 phenotypic polarization of astrocytes in brains of 5xFAD mice. (A) Immunostaining of GFAP (green) and Complement C3 (red) in dentate gyrus of hippocampus. Panels a-c: 20X magnification; scale bar: 90 µm. White arrows mark GFAP and complement C3 astrocytes co-expression in zoomed-in images (panels d-f); scale bar: 30 µm. (B) Quantification of GFAP+ and C3+ in mean fluorescence intensity MFI in dentate gyri. Left: MFI of GFAP+; middle: MFI of C3+; right: Pearson’s coefficient for quantification of co-localization of GFAP and C3. Data are Means ± SEM. *** p < 0.001, and ** p < 0.01.
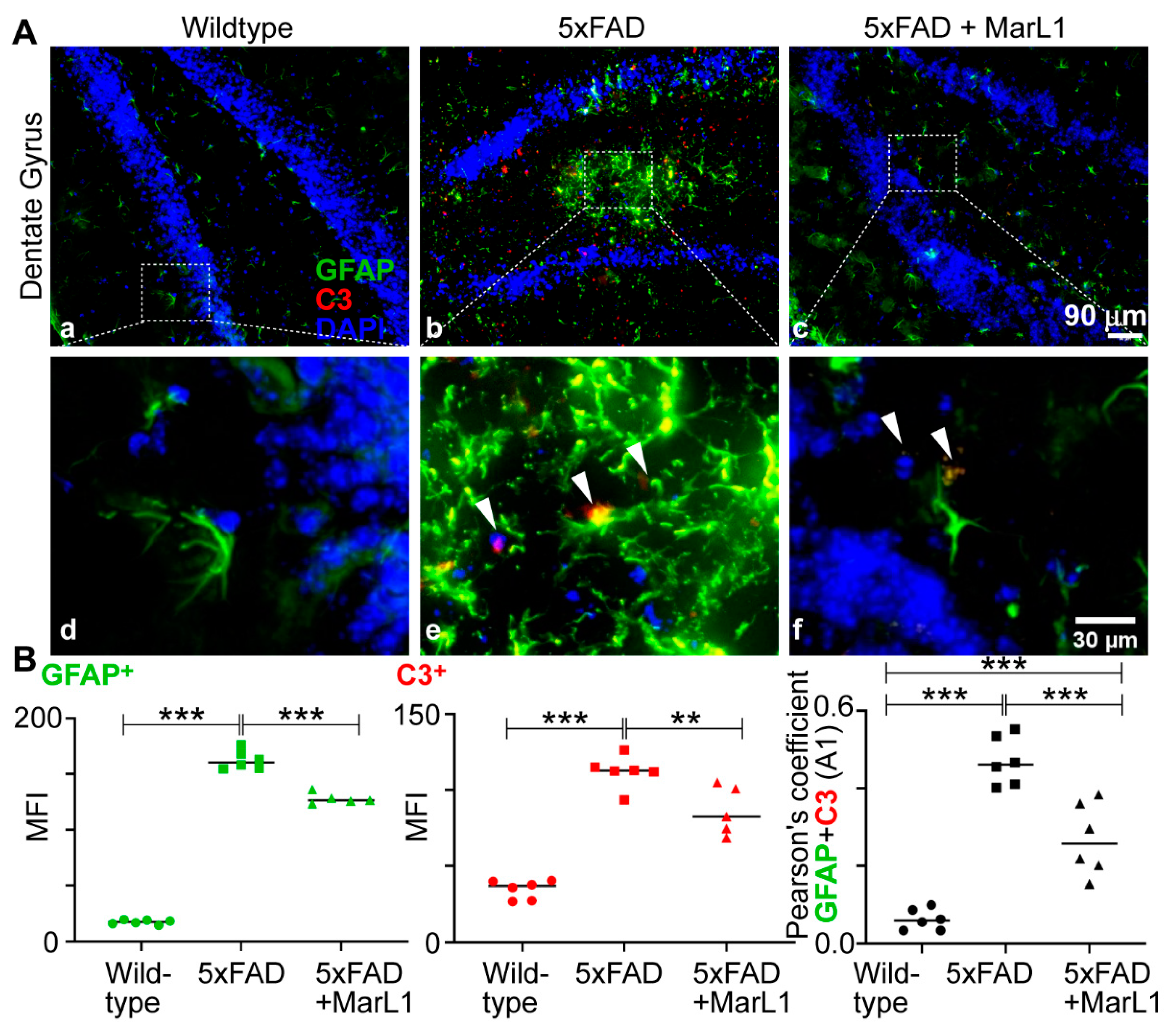
Figure 7.
MarL1 treatment reversed AD-associated diminish of anti-inflammatory A2 phenotypic polarization of astrocytes in brains of 5xFAD mice. (A) Immunostaining of GFAP (green) and A2 marker S100A10 (red) in dentate gyrus of hippocampus. White arrows mark GFAP and S100A10 positive astrocytes in dentate gyri (Panels a-c: 20X magnification; scale bar, 90 µm. Panels d-e: zoomed-in images; scale bar: 30 µm). (B) Quantification of GFAP+ and S100A10+ in mean fluorescence intensity MFI in dentate gyri. Left: MFI of GFAP+; middle: MFI of s100A10+; right: Pearson’s coefficient for quantification of co-localization of GFAP and s100A10. Data are Means ± SEM. *** p < 0.001, and *p < 0.05.
Figure 7.
MarL1 treatment reversed AD-associated diminish of anti-inflammatory A2 phenotypic polarization of astrocytes in brains of 5xFAD mice. (A) Immunostaining of GFAP (green) and A2 marker S100A10 (red) in dentate gyrus of hippocampus. White arrows mark GFAP and S100A10 positive astrocytes in dentate gyri (Panels a-c: 20X magnification; scale bar, 90 µm. Panels d-e: zoomed-in images; scale bar: 30 µm). (B) Quantification of GFAP+ and S100A10+ in mean fluorescence intensity MFI in dentate gyri. Left: MFI of GFAP+; middle: MFI of s100A10+; right: Pearson’s coefficient for quantification of co-localization of GFAP and s100A10. Data are Means ± SEM. *** p < 0.001, and *p < 0.05.
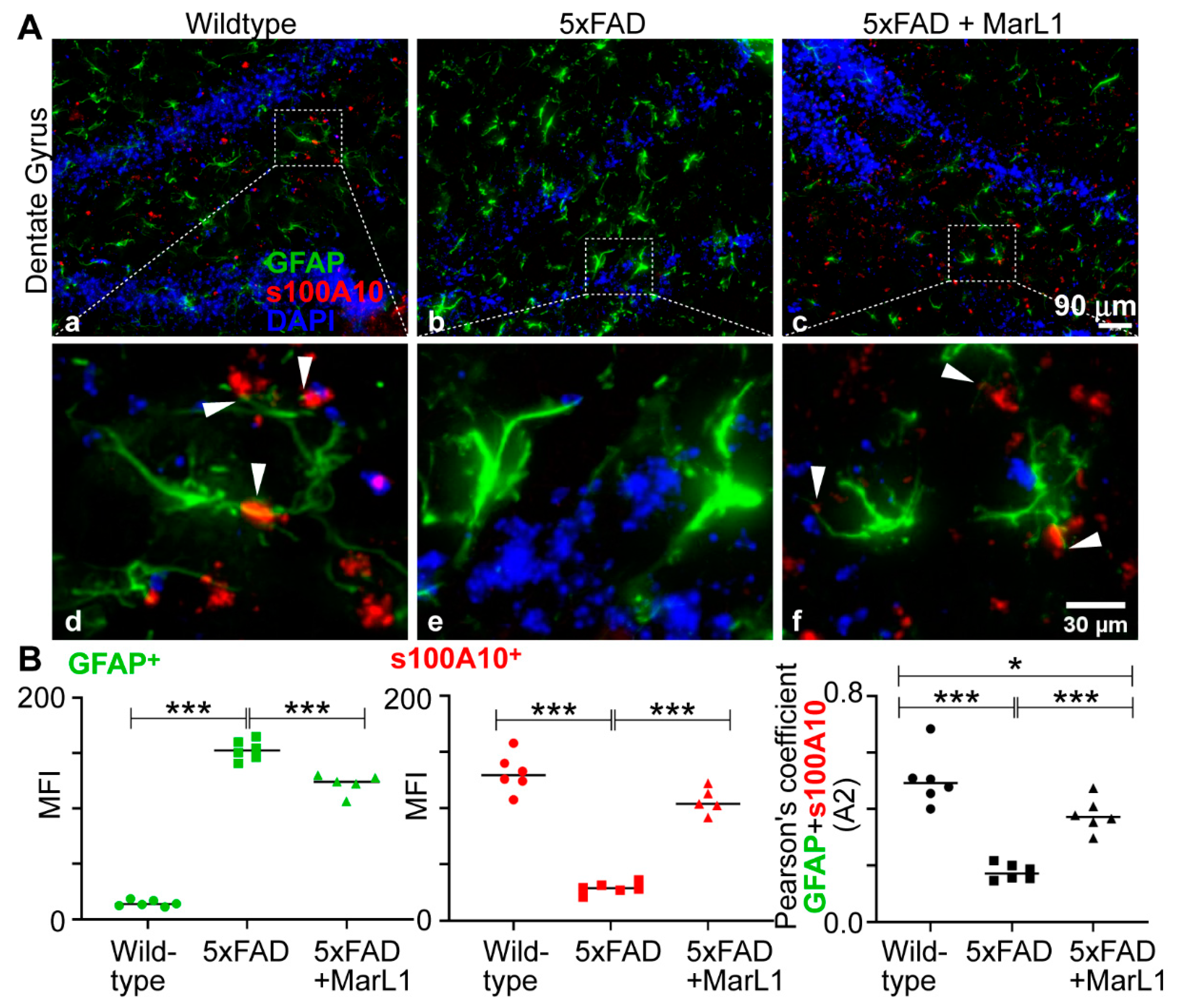
Figure 8.
MarL1 attenuated the AD-associated compromise of blood–brain-barrier tight-junctions as well as neutrophil infiltration into brains of 5xFAD mice. (A) Immunostaining of Gr-1 (green) for neutrophils and Claudin-5 (red) for tight-junctions of the vasculatures in cortex. Panels a-c show images from cortex (4X magnification; scale bar: 460 µm). Panels d-f show zoomed-in images; scale bar: 65 µm. White arrows mark some Gr-1+ cells outside the vasculature in parenchyma in zoomed-in images. Yellow arrows mark some claudin-5+ vasculatures. Neutrophil swarming is evident in panels b and e. (B) Quantification of Gr-1+ and Claudin-5+ in MFI in cortex. Data are Means ± SEM. *** p < 0.001, and *p < 0.05.
Figure 8.
MarL1 attenuated the AD-associated compromise of blood–brain-barrier tight-junctions as well as neutrophil infiltration into brains of 5xFAD mice. (A) Immunostaining of Gr-1 (green) for neutrophils and Claudin-5 (red) for tight-junctions of the vasculatures in cortex. Panels a-c show images from cortex (4X magnification; scale bar: 460 µm). Panels d-f show zoomed-in images; scale bar: 65 µm. White arrows mark some Gr-1+ cells outside the vasculature in parenchyma in zoomed-in images. Yellow arrows mark some claudin-5+ vasculatures. Neutrophil swarming is evident in panels b and e. (B) Quantification of Gr-1+ and Claudin-5+ in MFI in cortex. Data are Means ± SEM. *** p < 0.001, and *p < 0.05.
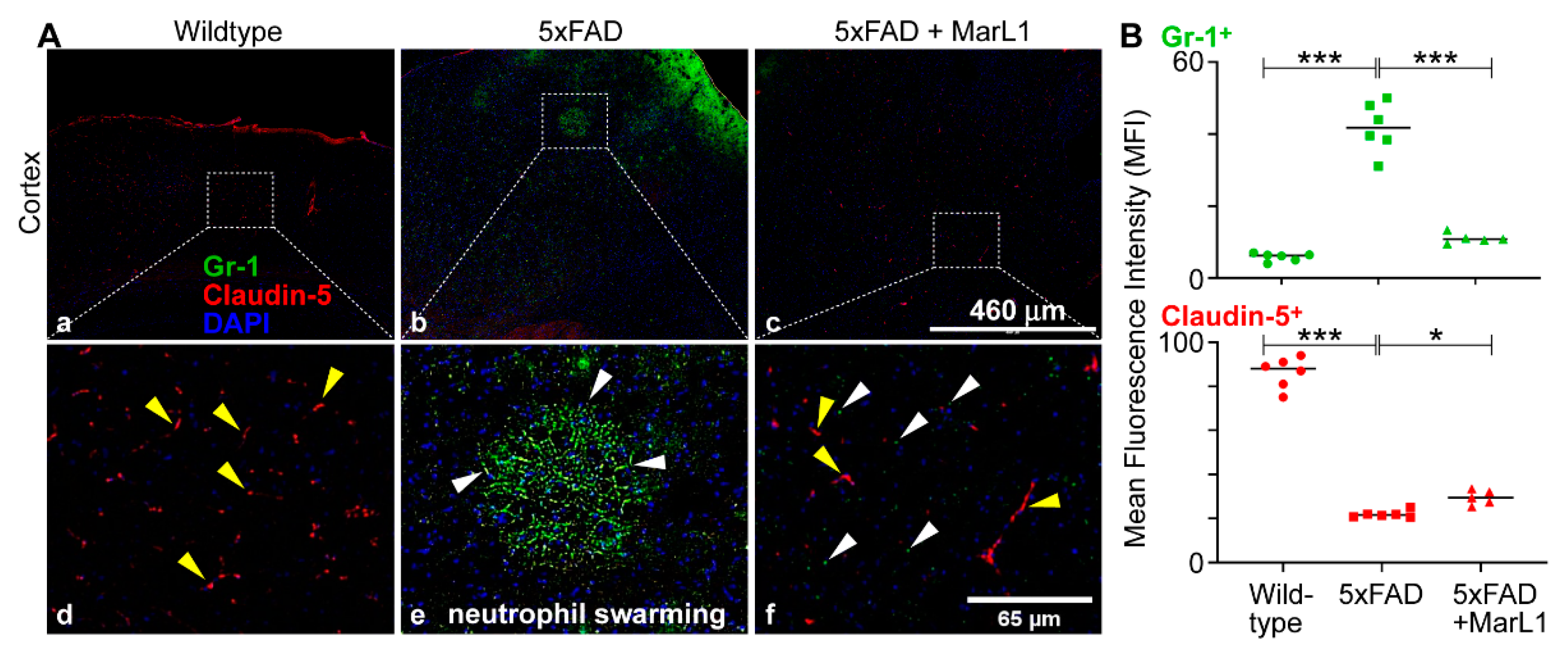
Figure 9.
MarL1 treatment suppressed pro-inflammatory N1 polarization of neutrophils infiltrated into AD-pathogenic brains in 5xFAD mice. (A) Immunostaining of Gr-1 (green) for neutrophils and iNOs (red), an inflammatory marker. Panels a-c show hippocampus (4X magnification, scale bar: 460 µm). Panels d-f show zoomed-in images; scale bar: 40 µm. White arrows mark some Gr-1+iNOs+ cells and yellow arrows mark only Gr-1 positive cells in zoomed-in panels. (B) Quantification of Gr-1+ and iNOs+ in hippocampus. Left: MFI of Gr-1+; middle: MFI of iNOs+; right: Pearson’s coefficient for quantification of co-localization of Gr-1 and iNOs. *** p < 0.001, ** p <0.01, and *p < 0.05.
Figure 9.
MarL1 treatment suppressed pro-inflammatory N1 polarization of neutrophils infiltrated into AD-pathogenic brains in 5xFAD mice. (A) Immunostaining of Gr-1 (green) for neutrophils and iNOs (red), an inflammatory marker. Panels a-c show hippocampus (4X magnification, scale bar: 460 µm). Panels d-f show zoomed-in images; scale bar: 40 µm. White arrows mark some Gr-1+iNOs+ cells and yellow arrows mark only Gr-1 positive cells in zoomed-in panels. (B) Quantification of Gr-1+ and iNOs+ in hippocampus. Left: MFI of Gr-1+; middle: MFI of iNOs+; right: Pearson’s coefficient for quantification of co-localization of Gr-1 and iNOs. *** p < 0.001, ** p <0.01, and *p < 0.05.
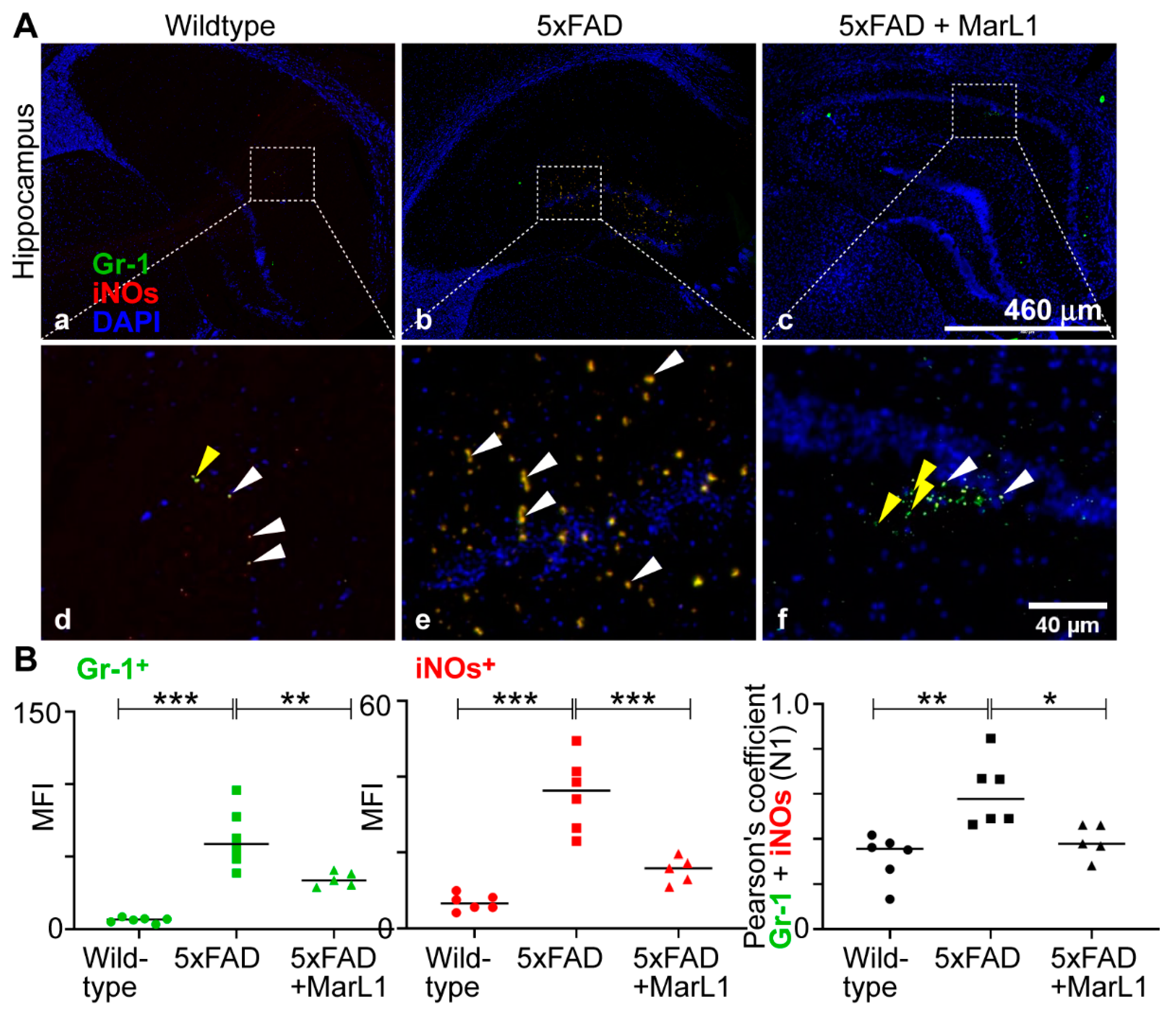
Figure 10.
MarL1 treatment induced anti-inflammatory N2 phenotypic polarization of neutrophils infiltrated into AD-pathogenic brains in 5xFAD mice. (A) Immunostaining of Gr-1 (green) for neutrophils and Arg1(red), an anti-inflammatory marker. Panels a-c show hippocampus (4X magnification, scale bar: 460 µm). Panels d-f show zoomed-in images; scale bar: 40 µm. White arrows mark Gr-1+ cells and yellow arrows mark Gr-1+Arg1+ cells in zoomed-in panels. (B) Quantification of Gr-1+ and Arg1+ in hippocampus. Left: MFI of Gr-1+; middle: MFI of Arg1+; right: Pearson’s coefficient for quantification of co-localization of Gr-1 and Arg1. Data are Means ± SEM. *** p < 0.001, ** p < 0.01, and *p < 0.05.
Figure 10.
MarL1 treatment induced anti-inflammatory N2 phenotypic polarization of neutrophils infiltrated into AD-pathogenic brains in 5xFAD mice. (A) Immunostaining of Gr-1 (green) for neutrophils and Arg1(red), an anti-inflammatory marker. Panels a-c show hippocampus (4X magnification, scale bar: 460 µm). Panels d-f show zoomed-in images; scale bar: 40 µm. White arrows mark Gr-1+ cells and yellow arrows mark Gr-1+Arg1+ cells in zoomed-in panels. (B) Quantification of Gr-1+ and Arg1+ in hippocampus. Left: MFI of Gr-1+; middle: MFI of Arg1+; right: Pearson’s coefficient for quantification of co-localization of Gr-1 and Arg1. Data are Means ± SEM. *** p < 0.001, ** p < 0.01, and *p < 0.05.
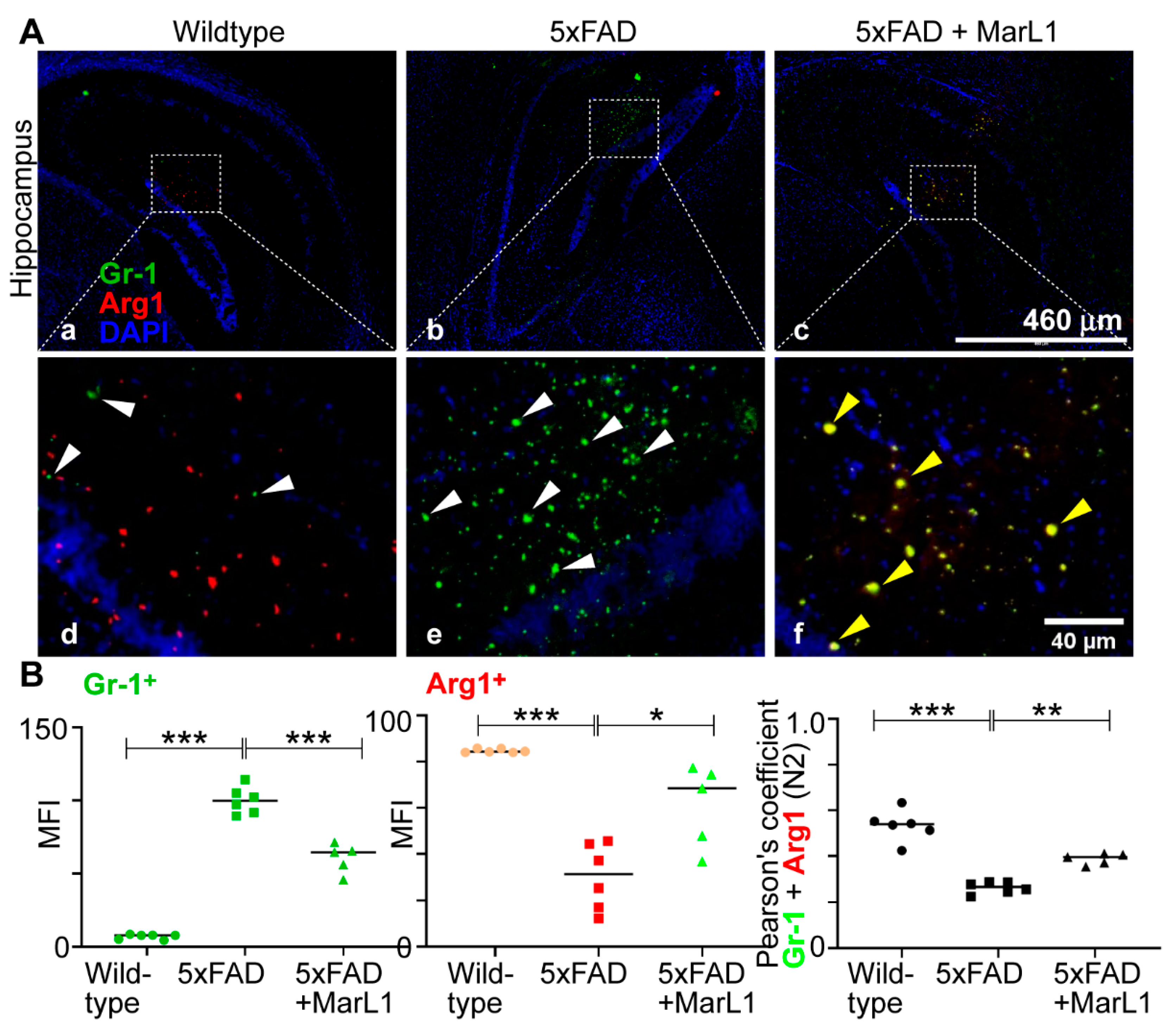
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



