
Submitted:
09 July 2024
Posted:
10 July 2024
You are already at the latest version
Abstract
Amorphous Indomethacin has enhanced bioavailability over its’ crystalline forms, yet amorphous forms can still possess a wide variety of structures. Here, Empirical Potential Structure Refinement (EPSR) has been used to provide accurate molecular models on the structure of five different amorphous Indomethacin samples, that are consistent with their high energy x-ray diffraction patterns. It is found that the majority of molecules in amorphous Indomethacin are non-bonded or bonded to one neighboring molecule via a single hydrogen bond, in contrast the doubly bonded dimers found in the crystalline state. The EPSR models further indicate a substantial variation in hydrogen bonding between different amorphous forms, leading to a diversity of chain structures not found in any known crystal structures. The majority of hydrogen bonds are associated with the carboxylic acid group, although a significant number of amide hydrogen bonding interactions are also found in the models. Evidence of some dipole-dipole interactions are also observed in the more structurally ordered models. The results are consistent a distribution of Z-isomer intra-molecular type conformations in the more disordered structures, that dis-tort when stronger inter-molecular hydrogen bonding occurs. The findings are supported by 1H and 2H NMR studies of the hydrogen bond dynamics in amorphous Indomethacin.
Keywords:
Amorphous
; Pair Distribution Function
; Indomethacin
; X-ray diffraction
; Monte Carlo simulation
1. Introduction
Active pharmaceutical ingredients can exist in a variety of solid forms with a range of different intermolecular interactions that affects both their bioavailability and structure [1,2,3]. Here we consider the hydrogen bonded structure and properties of amorphous Indomethacin, which has been studied extensively in the literature due to the increased solubility over its’ crystalline forms [4,5,6]. Several previous studies have compared the effects of different amorphization methods and storage conditions on the properties and stability of Indomethacin. Cowley and Zografi [7] cryogenically ground five different starting crystal phases to produce amorphous forms that exhibited significant differences in stability. Yoshika et al. [8] showed that the crystallization rates and mechanisms differ above and below the glass transition temperature. Andronis et al. [4,9] studied the effects of water which changed the surface properties, crystallization rates and polymorph formation. Greco et al. [10] investigated the effects of processing and annealing on the dissolution of amorphous Indomethacin. Karmwar et al. [11] prepared amorphous samples by melt quenching, spray drying, ball milling, and cryo-milling that yielded different shapes in their x-ray halos indicating a variation in packing between molecules.
The Indomethacin molecule (C19H16ClNO4) is comprised of a largely hydrophobic indole and chlorobenzyl groups, and several hydrophilic groups: namely an amide, methoxyl and a carboxylic acid [12]. γ-Indomethacin is the stable crystalline form which exists only with Z isomers [13], where hydrogen bonded dimers are connected through their carboxylic acid groups. α-Indomethacin is a denser metastable crystalline form comprising of three different isomers Z, E and α3 [14] and δ-Indomethacin has recently been found to consist of a dimer of the Z and E isomers [15]. Three new polymorphs obtained from aqueous suspensions have also yet to be characterized [15,16]. Our previous x-ray study on amorphous Indomethacin found a range of disordered structures denoted I through V [17]. In the amorphous forms the chlorobenzyl ring showed evidence of distinct isomer orientations in samples 1 and II, where the first sharp diffraction peak (FSDP) and medium range ordering was found to be lower. However, for those amorphous samples with no preferred torsion angles of the chlorobenzyl ring (samples IV and V), enhanced medium range order attributed to inter-molecular hydrogen bonding was observed, and this was reflected as a 20% increase in the intensity of the FSDP.
The isomers of Indomethacin can be primarily identified from the hindered rotation of the partial double bond between the N1 and C2 atoms (see Figure 1). This can lead to much more potent anti-inflammatory activity associated with the Z-isomer compared to the E isomer [12]. Therefore, in this study we have performed Empirical Potential Structure Refinement modeling of our previously reported high energy x-ray diffraction data on different amorphous Indomethacin samples, to investigate relation between molecular conformation and the range of inter-molecular hydrogen bonding interactions. Previous EPSR studies have demonstrated subtle but important hydrogen bonding differences between liquid and amorphous pharmaceuticals and excipients [18,19]. In addition to the solid state NMR (ssNMR) studies on samples I-V that have previously been reported [17], new experiments with partially deuterium exchanged amorphous indomethacin are reported to further investigate the possible molecular conformations and inter-molecular bonding configurations.
2. Materials and Methods
Crystalline γ-Indomethacin (>98% from Tokyo Chemical Industry) was used as received without further purification. The acetonitrile (99.9% HPLC grade, Concord Technologies) and the deuterium oxide (99.9% atom D, Cambridge Isotopes Laboratories) were used as received without further purification. To prepare indomethacin deuterated at the exchangeable acid position, 300 mg of indomethacin (0.8 mmol) was dissolved in 7 mL of a 70:30 mixture of acetonitrile and deuterium oxide (110 mmol) under mild heating and stirring. The solution was allowed to stir on a hot plate under mild heating for approximately 3 hours prior to solvent removal by dry nitrogen purge, followed by vacuum drying, resulting in the α crystal form. To obtain the γ form, the indomethacin was recrystallized by dissolving in the minimum amount of warm acetonitrile and allowing the solution to slowly cool to room temperature, followed by storage overnight at 5 ºC. The amorphous indomethacin samples were prepared by melt quenching with liquid nitrogen.
The ssNMR spectra were collected using a 400 MHz Varian VNMRS system equipped with a 1.6 mm triple resonance HXY probe configured for 1H-13C-2H operation with resonant frequencies of 399.739, 100.524, and 61.363 MHz respectively. The 1H ssNMR spectra were collected with a 1.75 μs 1H 90-degree pulse, a 30 s recycle delay, and a magic angle spinning (MAS) speed of 20 kHz. The 2H spectra were collected with 1.75 μs 2H 90-degree pulse, a recycle delay of 3 s, 20k scans, an MAS speed of 5 kHz, a sweep width of 500 kHz, and an acquisition time of 8.192 ms. The 1H →13C cross polarization (CP)-MAS spectra were collected using a 2.25 μs 1H 90-degree pulse, between 1k and 8k scans, a recycle delay of 10 seconds, an MAS speed of 20 kHz and a CP contact time of 2 ms. The CP was achieved using a 100 kHz 13C spin-lock pulse, and a ramped power (5%) 1H spin-lock pulse optimized to the -1 spinning side band of the Hartman Hahn condition (80 kHz). During 13C and 2H data collection high power (140 kHz) two pulse phase modulated (TPPM) 1H decoupling with a 3.3 μs pulse width and 8-degree phase offset was used, to improve spectral resolution. The chemical shifts for 1H and 13C were indirectly referenced to TMS in the solid state by setting the resonances for 1H and 13C to 1.8 ppm and 38.48 ppm, respectively. The 2H chemical shifts were referenced by setting the 2H resonance of liquid D2O to 4.8 ppm. The 1H and 13C NMR data was processed using VnmrJ 4.2, and the 2H NMR data was processed using Topspin 4.1. The 2H NMR line shapes were fit and analyzed using DMFit [20].
To investigate the variation in hydrogen bonding between the different amorphous forms of Indomethacin measured in our high energy x-ray diffraction experiments on beamline 6-ID-D at the Advanced Photon Source, Empirical Potential Structural Refinement (EPSR) modeling [21] was used. The five samples were all prepared by melt quenching and their preparation and characterization has previously been reported in detail by Benmore et al. [17]. It is important to re-iterate here that an accurate data reduction procedure is essential in obtaining the x-ray structure factor S(Q) and associated pair distribution function G(r). A review of the current software available for this purpose has recently been carried out by Gallington et al. [22]. EPSR is a Monte Carlo semi-rigid body type simulation, whereby all atoms on the molecules are defined using harmonic force constants, and angular and dihedral angles are used to describe the molecular geometry and allowed intra-molecular rotations [23,24]. The algorithm initially uses Lennard-Jones reference potentials with Coulombic terms to describe the intermolecular interactions. As the simulation progresses an empirical potential is employed to modify these inter-molecular interactions. This term in the potential is determined by taking the difference between the experimental diffraction data and that predicted by the Monte Carlo model. The continuously perturbed potential drives the model structure towards the experimental data by random changes in atomic (molecular) coordinates, including molecular rotations if flexibility of that part of the molecule is allowed, with each step resulting in new configurations. The change is accepted if the potential energy decreases, or with a Boltzmann probability if it is greater, to avoid becoming stuck in local minima. Once good fits are found between the model structures and the measured x-ray structure factor the simulation is collected over a large number of configurations. When applied to x-ray diffraction data from amorphous pharmaceuticals, the scattering is mainly dominated by the carbon and oxygen atoms that define the molecular geometry and inter-molecular pair correlations.
Here, EPSR simulations were performed on 64 molecules within a cubic box under periodic boundary conditions. The starting configuration of our model was constructed from a random array of Z isomer molecules since this is the most common isomer, particularly at low density. In addition, molecular dynamics simulations of amorphous Indomethacin predicts that the Z isomer is more favorable than the E isomer by a factor of about 5.7 [1]. To allow other conformations in the model and improve the fit to the x-ray data, rotations of five molecular groups were enabled, including the rotation of the chlorobenzyl ring. The atom labels for the different atom types used in the simulation and the allowed molecular rotations are illustrated in Figure 1. To prevent unrealistic hydrogen bonding an additional “soft” minimum distance constraint of 2.6Å was applied for the oxygen-oxygen (O-O) interactions between adjacent molecules. This constraint increases the repulsive part of the inter-atomic potential but may come to equilibrium at a lower atom-atom distance if necessary, in order to maintain an adequate fit to the data.
The parameters associated with the starting Lennard-Jones reference potentials are shown in Table 1. For simplicity, only the most influential partial charges were employed based on the molecular dynamics simulations of Xiang & Anderson [12]. Namely, the negative acceptor oxygen and positive carbon charges, with the charge balance placed on the OH donor hydrogen. Best fits to the more disordered high-energy x-ray diffraction signals (from samples I, II) were obtained using a ~5% lower density and the semi-rigid molecular models, whereby rotation about the 5 specified axes was allowed. The more ordered signals from samples IV and V were better fit using rigid Z-isomer molecules with rotations restricted to only a few degrees. Sample III was fit with an intermediate density and but all 5 rotations enabled.
Following initial Monte Carlo equilibration, the empirical potential term was refined to improve agreement with scattering data, Once the goodness-of-fit parameter was minimized between the model and the experimental S(Q), structural data were collected over ensembles of at least 10,000 configurations. While the EPSR fit to the data does not necessarily give a unique structural 3D configuration of molecules, it does provide an important insight into the types of interactions that are likely in the disordered sate. Since x-rays are scattered by electrons, the S(Q)’s and corresponding PDF’s are most sensitive to the heavier atoms and in particular the orientations of the carbon rings, oxygens and the chlorine atom interactions.
3. Results
3.1. EPSR Models
In order to best fit the periodicity in the high-Q region of the x-ray diffraction pattern, the average intramolecular bond lengths of the molecule needed to be lengthened by 1 to 3% compared to the Z-isomer molecule in the γ-form [14]. This average C-X (where C=C, N or O) bond length is defined in real-space by the first peak in the pair distribution function D(r). The results of the fits are shown in Figure 2 and listed in Table 2.
The x-ray patterns for the different amorphous samples are qualitatively similar, however important quantitative differences exist. A strong test of the validity of the different models is demonstrated by taking differences between the less ordered forms (I and II) and more ordered forms (IV and V), as shown in Figure 3. The main difference between the measured amorphous structure factors occur in the low-Q region, which can be associated with the packing (and hydrogen bonding interactions) between molecules [25].
The O2H2-O1 hydrogen bonded distance in the γ-form is 2.67Å and is associated with the formation of carboxylic acid dimers. In α-Indomethacin the structure comprises of three molecules in the asymmetric unit, with each molecule having a different conformation, forming a trimer. The trimer comprises of a hydrogen-bonded carboxylic acid dimer, with the third molecule forming a hydrogen bond between the carboxylic acid and an amide carbonyl in the dimer, spanning O2H2-O1 distances between 2.59-2.74 Å. In contrast, our EPSR models of the amorphous forms show a wide range of O2H2-O1 distances, from 2.50-2.85 Å. The partial pair distribution function gO2O1(r) in Figure 4a shows the distribution of O2H2-O1 hydrogen bonding interactions as a function of distance in the different amorphous Indomethacin samples.
The EPSR models for the more-ordered samples (IV & V) exhibit strong hydrogen bonds in the form of a single intense peak at ~2.5Å, a minima at 3.2Å, and second shell maximum at ~3.5Å. The less-ordered samples (I & II) span a broad (and continuous) range of O2H2-O1 distances from 2.5 to 3.5Å. This variation in the average number of hydrogen bonded molecules as a function of distance between different amorphous forms, is reflected in the running coordination number, nO2-O1(r) in Figure 4b. Nevertheless, a value of nO2-O1(r)~0.38 was found for all the amorphous samples at a distance of r=3.2Å. This compares to the value of 1.0 in the crystalline γ- and α-forms.
In our amorphous models hydrogen bonding inter-molecular interactions were also found between the carboxyl acid oxygen O2 and the O3 amide oxygen, which is not found in crystalline forms and likely arises from the highly disordered arrangement of molecules. Interestingly, the strongest O2-O3 interactions are found in sample III which exhibited the sharpest FSDP, see Figure 5. In other words, the sample with the most inter-molecular hydrogen bonding of the models with the most flexibility, by allowing all five intra-molecular rotations. Although we note that the most ordered models, namely III, IV and V, all exhibit a O2-O3 coordination number, nO2-O3(r)~0.2 at a distance of r=3.2Å.
In addition, since the amide carbonyl atoms C4 and O1 have large and opposite charges of +0.7 and −0.6 (see Table 1) they have the potential to induce dipole−dipole bonds between adjacent Indomethacin molecules. Indeed, in models IV and V evidence of short intermolecular C4···O1 distances of ~3.2 Å are found, similar to those observed in the γ-crystalline form, but these interactions are largely absent in models I, II and III. The inter-molecular carboxyl acid oxygen donor O2-O3 amide oxygen acceptor partial pair distribution functions from our EPSR models and (b) corresponding running coordination numbers for the amorphous Indomethacin samples. The inter-molecular carboxyl acid oxygen donor O2-O3 amide oxygen acceptor partial pair distribution functions from our EPSR models and (b) corresponding running coordination numbers for the amorphous Indomethacin samples.
3.2. ssNMR Experiments
The 1H magic angle spinning (MAS) spectra of the natural abundance amorphous and crystalline γ- and α-Indomethacin samples are shown in Figure 6A along with partially deuterium exchanged amorphous indomethacin (overlaid grey spectrum). The reduction in the 1H MAS spectral intensity in the 10-14 ppm range for the amorphous d1-indomethacin in comparison to the natural abundance amorphous indomethacin is confirmation that for selective acid exchanged deuteration of the sample. The 1H MAS spectra clearly indicates that the amorphous indomethacin has hydrogen bonding components that are similar to the dimer carboxylic acid configuration found in γ-Indomethacin (~13 ppm) as well as the carboxylic acid hydrogen bonding to the carbonyl as observed in α-Indomethacin (~11 ppm) [26]. The 1H acid region in amorphous indomethacin is broad compared with the crystalline samples due to the larger dispersion of hydrogen bonding and acid environments found in disordered molecular solids.
The 2H MAS NMR of d1-Indomethacin (~50% deuterated at the carboxylic acid based on 1H MAS NMR integration) allowed for elucidation of dynamic processes and further structural probing of the amorphous indomethacin hydrogen bonding environment. The 2H MAS NMR of amorphous d1-Indomethacin is shown in Figure 6B along with a 1st order quadrupolar simulation using DMFit to estimate the 2H quadrupolar coupling constant [20]. Both the isotropic chemical shift and the quadrupolar coupling constant (CQ) can be of interest in studies of hydrogen bonds and any associated exchange or molecular motion dynamics. The deuterium quadrupolar coupling constants for carboxylic acids are typically 140-200 kHz and the quadrupole coupling constant for heavy water and common hydrates is ~220 kHz [27,28]. The residual quadrupolar interaction is directly influenced by translational diffusion, molecular rotation and exchange dynamics. The observation of a quadrupole coupling constant of 178 kHz for d1-Indomethacin indicates that that amorphous indomethacin below Tg (at room temperature) does not have any appreciable exchange dynamics and exists as distributions of rigid intermolecular hydrogen bonded molecular units.
4. Discussion
Previous structural characterization methods of crystalline, liquid and amorphous forms of Indomethacin have included Raman and infra-red spectroscopy [29], nuclear magnetic resonance [28], x-ray crystallography [14] and molecular dynamics simulations [30]. The extraction of the pair distribution function from diffraction measurements provides yet another powerful tool capable of probing both intra- and intermolecular configurations of molecules, especially with regard to the most disordered forms [31,32,33]. The five amorphous Indomethacin samples modeled here have previously been characterized in detail using high energy x-ray diffraction, nuclear magnetic resonance, Raman scattering and differential scanning calorimetry [17]. From our EPSR models we can interrogate the variation in the model structures more thoroughly. A comparison of the ∠O3-N-Cl intra-molecular angle of the three different isomers found in α- and γ-Indomethacin are shown in Figure 7, along with the distribution of angles found in the EPSR models for all five amorphous samples. The most structured inter-molecular amorphous have the broadest range of intra-molecular conformations (around ~90o). This is a consequence of our EPSR constraints, since our models of samples IV and V used Z-isomer molecules with rotations limited to only a few degrees. In contrast, our models for our more disordered samples I, II and III, allowed five rotations within the Z-isomer molecules. This additional flexibility resulted in exhibit sharper peaks in the ∠O3-N-Cl angle distribution, and are attributed to preferred intra-molecular conformations. None of the models I, II or III found configurations with any significant population of E-isomers, suggesting that the pure amorphous forms likely act as better anti-inflammatory agents compared to the α-form. However, it has been pointed out that the distribution of Indomethacin conformations is sensitive to the physical environment, with the Z isomer conformation being preferred in solution and the E-isomer favored in inclusion complexes with β-cyclodextrin [34].
The broad range of molecular conformations observed in our models leads to a high degree of inter-molecular structural disorder associated with amorphous solids, and more complex hydrogen bonding patterns compared to the crystalline forms. Previous MD simulations on amorphous Indomethacin [12] have shown a much lower probability of carboxylic dimers than found in the crystals, and the most readily identified hydrogen bonded patterns to be short chains of Indomethacin molecules connected via carboxylic acid bonds. An analysis of the chain size distributions shows a high degree of consistency across all of our EPSR models (see Figure 8), with ~46% of Indomethacin molecules being non-bonded (isolated), and only ~39% bonded to one neighboring molecule via a single hydrogen bond, see Figure 9. The remaining 15% are associated with trimers, and bifurcated hydrogen bonds leading to a diversity of chain structures in the amorphous forms. This result compares to 21% non-bonded molecules, 31% singly hydrogen bonded molecules, and 48% with 2 or 3 hydrogen bonded neighbors in a previously reported molecular dynamics model of 10 mole.% water containing Indomethacin glass [12].
1H and 2H NMR supports the molecular model of amorphous indomethacin having a broad range of molecular conformations and inter-molecular structural disorder with complex hydrogen bonding patterns. Furthermore, deuteration and 2H NMR is shown as a promising direction for probing hydrogen bonding and molecular structure, motion and exchange dynamics in pharmaceutical compounds [35]. This is especially relevant with the increased interest in deuterated pharmaceutics [36]. Our future directions involve more closely integrating molecular computational of quadrupolar and chemical shift NMR quantities from ab initio computational model molecular configurations to better combine experimental and computational elucidation of complex distributions of conformations and inter-molecular distributions common to amorphous pharmaceutical compounds.
5. Conclusions
Semi-rigid molecular models fitted to five amorphous Indomethacin x-ray diffraction patterns have been interrogated to determine the main similarities and differences between the amorphous samples. All intra-molecular conformations are consistent with a wide distribution of configurations similar to the Z-isomer, but not the E-isomer. The majority of amorphous Indomethacin hydrogen bonds were found to include the donor carboxylic acid group and one of several hydrogen bond acceptor oxygen sites including the amide carbonyl oxygen, the methoxyl, and the carboxylic acid. Consequently, a wide range of hydrogen bonding strengths and interactions are found across the different models resulting in complex interaction patterns, although the primary hydrogen bond is found to be via carboxylic acid donor O2-O1 acceptor interactions. To a lesser extent O2-O3 amide hydrogen bonding interactions were also observed, and C4-O1 dipole-dipole interactions occurred in the more structurally ordered models. Overall, the majority of Indomethacin molecules were found to be either isolated (~46%) or form singly hydrogen bonded dimers (39%). Our amorphous models were consistent with our previous findings that there is competition between preferred intra-molecular conformations and stronger inter-molecular hydrogen bonding. From a wider perspective, this study shows that the method of EPSR modeling of high energy x-ray diffraction patterns from amorphous pharmaceuticals is a powerful tool role in exploring the range of possible molecular structures.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figures S1 and S2: 1H-13C CP-MAS ssNMR spectra. Figure S3: FT-IR spectra of Indomethacin polymorphs. Figures S1 and S5. FT-IR spectra of deuterated vs. natural abundance gamma.
Author Contributions
Conceptualization, C.J.B., J.L.Y. and S.R.B.; methodology, C.J.B., J.L.Y. and S.R.B.; formal analysis, C.J.B., J.L.Y., S.K.D., P.S. and C.D.S.; investigation, C.J.B., J.L.Y., S.K.D., P.S. and C.D.S.; writing—original draft preparation, C.J.B. and J.L.Y.; writing—review and editing, J.L.Y. and S.R.B.; All authors have read and agreed to the published version of the manuscript.
Funding
This research used resources of the Advanced Photon Source, a U.S. DOE Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The researchers and PI at ASU (JLY) would also like to acknowledge support from Arizona State University and the US National Science Foundation (NSF DMR BMAT 2105312 and NSF CHE 1856506).
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Byrn S.R., Z.G., Chen X.S., Solid State Properties of Pharmaceutical Material. 2017, Hoboken: John Wiley & Sons, Inc.
- Healy, A.M., et al., Pharmaceutical solvates, hydrates and amorphous forms: A special emphasis on cocrystals. Adv Drug Deliv Rev, 2017. 117: p. 25-46. [CrossRef]
- Hancock, B.C. and G. Zografi, Characteristics and significance of the amorphous state in pharmaceutical systems. J Pharm Sci, 1997. 86(1): p. 1-12. [CrossRef]
- Andronis, V., M. Yoshioka, and G. Zografi, Effects of sorbed water on the crystallization of indomethacin from the amorphous state. J Pharm Sci, 1997. 86(3): p. 346-51. [CrossRef]
- Bates, S., et al., Analysis of amorphous and nanocrystalline solids from their X-ray diffraction patterns. Pharm Res, 2006. 23(10): p. 2333-49. [CrossRef]
- Fukuoka, E., M. Makita, and S. Yamamura, Glassy state of pharmaceuticals. II. Bioinequivalence of glassy and crystalline indomethacin. Chem Pharm Bull (Tokyo), 1987. 35(7): p. 2943-8. [CrossRef]
- Crowley, K.J. and G. Zografi, Cryogenic grinding of indomethacin polymorphs and solvates: assessment of amorphous phase formation and amorphous phase physical stability. J Pharm Sci, 2002. 91(2): p. 492-507. [CrossRef]
- Yoshioka, M., B.C. Hancock, and G. Zografi, Crystallization of indomethacin from the amorphous state below and above its glass transition temperature. J Pharm Sci, 1994. 83(12): p. 1700-5. [CrossRef]
- Andronis, V. and G. Zografi, Crystal nucleation and growth of indomethacin polymorphs from the amorphous state. Journal of Non-crystalline Solids, 2000. 271: p. 236-248. [CrossRef]
- Greco, K. and R. Bogner, Crystallization of amorphous indomethacin during dissolution: effect of processing and annealing. Mol Pharm, 2010. 7(5): p. 1406-18. [CrossRef]
- Karmwar, P., et al., Investigation of properties and recrystallisation behaviour of amorphous indomethacin samples prepared by different methods. Int J Pharm, 2011. 417(1-2): p. 94-100.
- Xiang, T.-X. and B.D. Anderson, Molecular Dynamics Simulation of Amorphous Indomethacin. Molecular Pharmaceutics, 2013. 10(1): p. 102-114. [CrossRef]
- Kistenmacher, T.J. and R.E. Marsh, Crystal and molecular structure of an antiinflammatory agent, indomethacin, 1-(p-chlorobenzoyl)-5-methoxy-2-methylindole-3-acetic acid. Journal of the American Chemical Society, 1972. 94(4): p. 1340-1345. [CrossRef]
- Chen, X., et al., Reactivity differences of indomethacin solid forms with ammonia gas. J Am Chem Soc, 2002. 124(50): p. 15012-9. [CrossRef]
- Andrusenko, I., et al., Structure determination, thermal stability and dissolution rate of δ-indomethacin. International Journal of Pharmaceutics, 2021. 608: p. 121067. [CrossRef]
- Surwase, S.A., et al., Indomethacin: new polymorphs of an old drug. Molecular pharmaceutics, 2013. 10(12): p. 4472-4480. [CrossRef]
- Benmore, C.J., et al., A High Energy X-ray Diffraction Study of Amorphous Indomethacin. Journal of Pharmaceutical Sciences, 2022. 111(3): p. 818-824. [CrossRef]
- Benmore, C.J., et al. The Structure of Liquid and Glassy Carbamazepine. Quantum Beam Science, 2022. 6. [CrossRef]
- Benmore, C.J., et al., X-ray Diffraction of Water in Polyvinylpyrrolidone. Molecular Pharmaceutics, 2023. 20(7): p. 3645-3652. [CrossRef]
- Åman, K., P. Håkansson, and P.-O. Westlund, A general approach to the calculation of 2H2O NMR lineshapes in microheterogeneous systems: a distorted bicontinuous cubic phase. Physical Chemistry Chemical Physics, 2005. 7(7): p. 1394-1401. [CrossRef]
- Soper, A.K., Partial structure factors from disordered materials diffraction data: An approach using empirical potential structure refinement. Physical Review B, 2005. 72(10): p. 104204. [CrossRef]
- Gallington, L.C., et al. Review of Current Software for Analyzing Total X-ray Scattering Data from Liquids. Quantum Beam Science, 2023. 7. [CrossRef]
- Soper, A.K., Empirical potential Monte Carlo simulation of fluid structure. Chemical Physics, 1996. 202(2): p. 295-306. [CrossRef]
- Soper, A.K., Joint structure refinement of x-ray and neutron diffraction data on disordered materials: application to liquid water. Journal of Physics: Condensed Matter, 2007. 19(33): p. 335206.
- Benmore, C., L. Gallington, and E. Soignard, Intermediate range order in supercooled water. Molecular Physics, 2019. 117(18): p. 2470, 2476.
- Berglund, B. and R.W. Vaughan, Correlations between proton chemical shift tensors, deuterium quadrupole couplings, and bond distances for hydrogen bonds in solids. The Journal of Chemical Physics, 1980. 73(5): p. 2037-2043. [CrossRef]
- Di Martino, R.M.C., B.D. Maxwell, and T. Pirali, Deuterium in drug discovery: progress, opportunities and challenges. Nature Reviews Drug Discovery, 2023. 22(7): p. 562-584. [CrossRef]
- Lu, X., et al., Solid-state NMR analysis of crystalline and amorphous Indomethacin: An experimental protocol for full resonance assignments. J Pharm Biomed Anal, 2019. 165: p. 47-55. [CrossRef]
- Heinz, A., et al., Quantifying ternary mixtures of different solid-state forms of indomethacin by Raman and near-infrared spectroscopy. Eur J Pharm Sci, 2007. 32(3): p. 182-92. [CrossRef]
- Gerges, J. and F. Affouard, Insight From Molecular Dynamics Simulations on the Crystallization Tendency of Indomethacin Polymorphs in the Undercooled Liquid State. J Pharm Sci, 2020. 109(2): p. 1086-1095. [CrossRef]
- Terban, M.W. and S.J.L. Billinge, Structural Analysis of Molecular Materials Using the Pair Distribution Function. Chemical Reviews, 2022. 122(1): p. 1208-1272. [CrossRef]
- Benmore, C.J., 10.14 - X-ray and neutron diffraction from glasses and liquids, in Comprehensive Inorganic Chemistry III (Third Edition), J. Reedijk and K.R. Poeppelmeier, Editors. 2023, Elsevier: Oxford. p. 384-424.
- Chen, S., A.Y. Sheikh, and R. Ho, Evaluation of effects of pharmaceutical processing on structural disorders of active pharmaceutical ingredient crystals using nanoindentation and high-resolution total scattering pair distribution function analysis. J Pharm Sci, 2014. 103(12): p. 3879-3890. [CrossRef]
- Fronza, G., et al., 1H NMR and Molecular Modeling Study on the Inclusion Complex β-Cyclodextrin−Indomethacin. The Journal of Organic Chemistry, 1996. 61(3): p. 909-914. [CrossRef]
- Mantsch, H.H., H. Saitô, and I.C.P. Smith, Deuterium magnetic resonance, applications in chemistry, physics and biology. Progress in Nuclear Magnetic Resonance Spectroscopy, 1977. 11(4): p. 211-272. [CrossRef]
- Massiot, D., et al., Modelling one- and two-dimensional solid-state NMR spectra. Magnetic Resonance in Chemistry, 2002. 40(1): p. 70-76. [CrossRef]
Figure 1.
Left. Starting conformation of the Indomethacin molecule used in our EPSR models together with labels of the different atom types. Right. Overlay of Z, E and α3 isomers to illustrate the choice of the five specified allowed rotations denoted by curved arrows.
Figure 1.
Left. Starting conformation of the Indomethacin molecule used in our EPSR models together with labels of the different atom types. Right. Overlay of Z, E and α3 isomers to illustrate the choice of the five specified allowed rotations denoted by curved arrows.
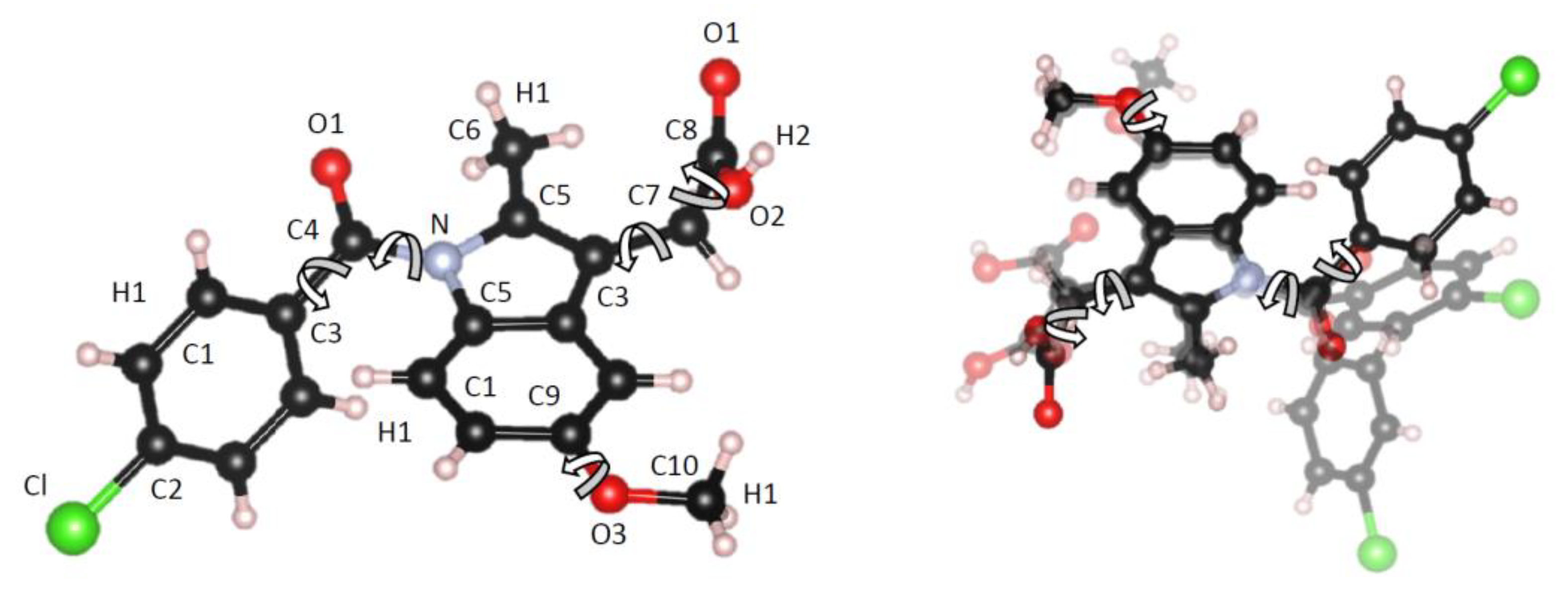
Figure 2.
The x-ray structure factors of the five amorphous Indomethacin samples reported by Benmore et al. [17]. Left panel shows the S(Q) low-Q region encompassing the first sharp diffraction peak. The right panel shows the entire measured Q-range (circles) using the formalism Q[S(Q)-1] to emphasize high-Q, together with the EPSR model fits from this study (lines).
Figure 2.
The x-ray structure factors of the five amorphous Indomethacin samples reported by Benmore et al. [17]. Left panel shows the S(Q) low-Q region encompassing the first sharp diffraction peak. The right panel shows the entire measured Q-range (circles) using the formalism Q[S(Q)-1] to emphasize high-Q, together with the EPSR model fits from this study (lines).
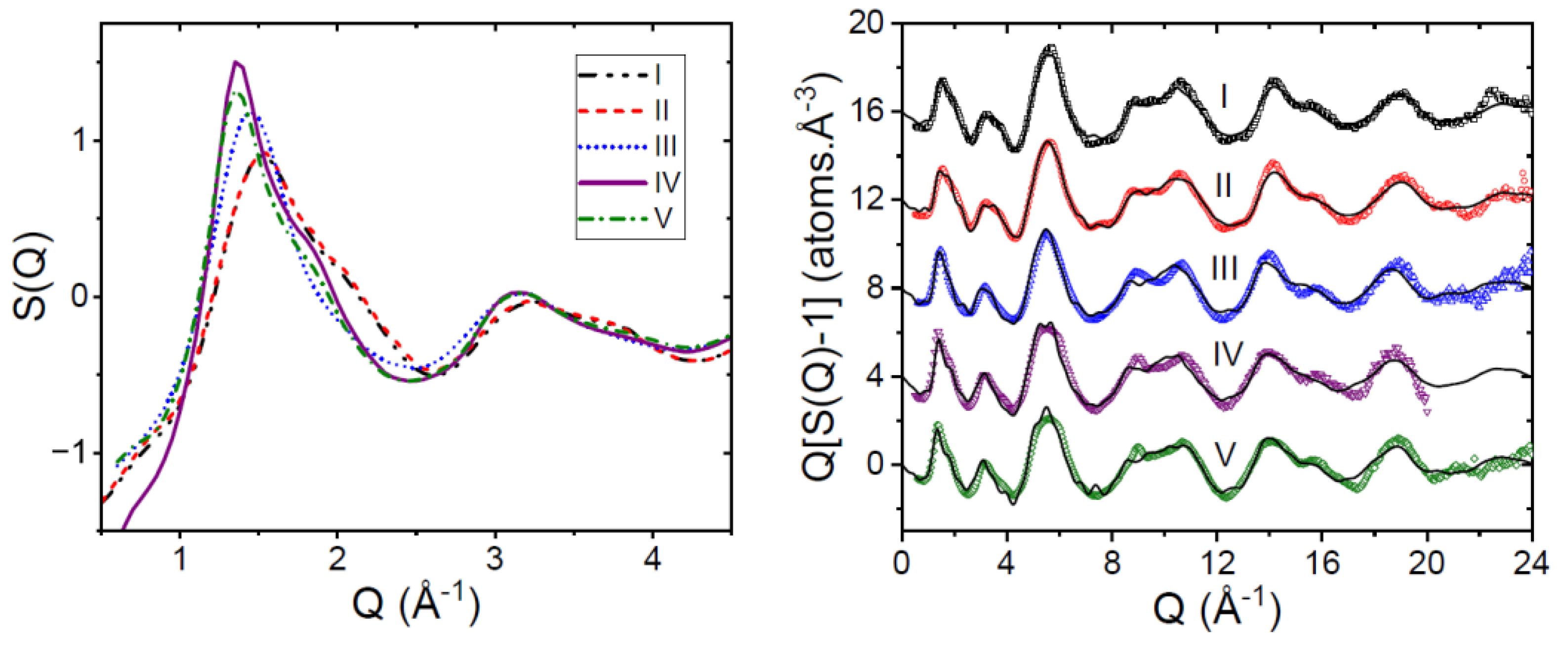
Figure 3.
Left. The difference between measured structure factors of amorphous Indomethacin samples (open circles) and compared to the same difference between EPSR models (red lines). Right. EPSR fits (blue lines) to the experimental real space x-ray pair distribution function D(r) (black circles) using the different sized Indomethacin molecules as described in Table 2.
Figure 3.
Left. The difference between measured structure factors of amorphous Indomethacin samples (open circles) and compared to the same difference between EPSR models (red lines). Right. EPSR fits (blue lines) to the experimental real space x-ray pair distribution function D(r) (black circles) using the different sized Indomethacin molecules as described in Table 2.
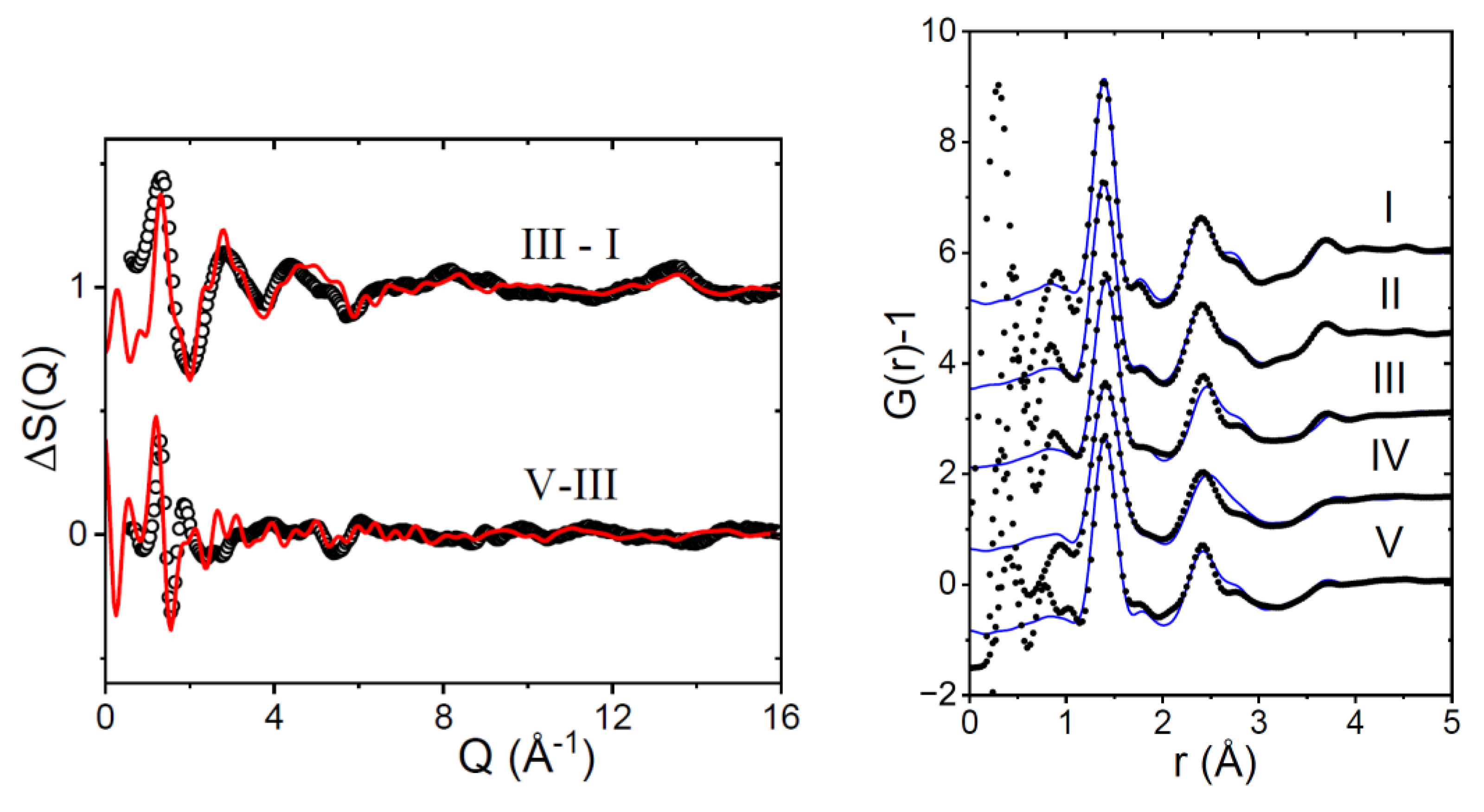
Figure 4.
Left panel. The inter-molecular carboxyl acid oxygen donor O2-O1 oxygen acceptor partial pair distribution functions from our EPSR models. The right panel shows corresponding running coordination numbers for the amorphous Indomethacin samples.
Figure 4.
Left panel. The inter-molecular carboxyl acid oxygen donor O2-O1 oxygen acceptor partial pair distribution functions from our EPSR models. The right panel shows corresponding running coordination numbers for the amorphous Indomethacin samples.
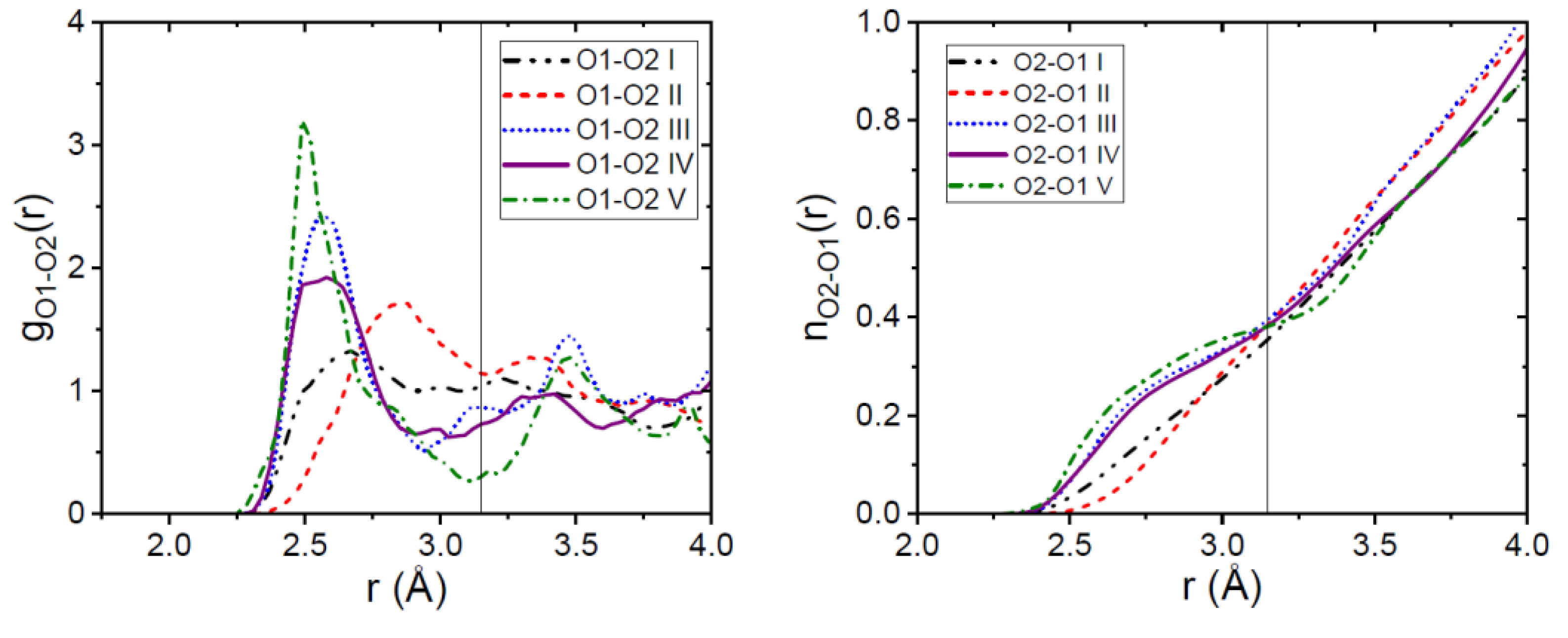
Figure 5.
Left panel. The inter-molecular carboxyl acid oxygen donor O2-O3 amide oxygen acceptor partial pair distribution functions from our EPSR models. The right panel shows the corresponding running coordination numbers for the amorphous Indomethacin samples.
Figure 5.
Left panel. The inter-molecular carboxyl acid oxygen donor O2-O3 amide oxygen acceptor partial pair distribution functions from our EPSR models. The right panel shows the corresponding running coordination numbers for the amorphous Indomethacin samples.
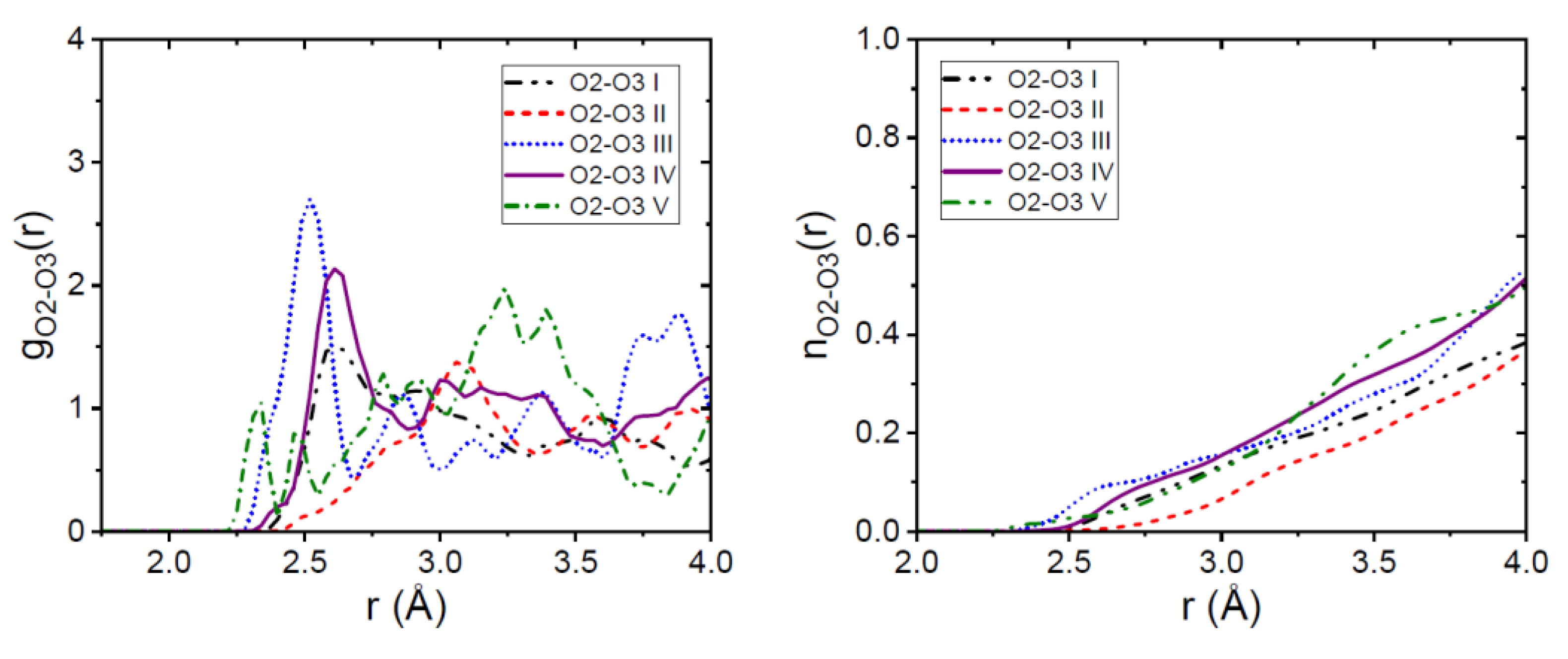
Figure 6.
(A) 1H solid-state MAS (nr = 20 kHz) NMR spectra of alpha, gamma and amorphous indomethacin. A selectively acid deutermium enriched amorphous indomethacin sample was made through 1H to 2H exchange prior to melt quenching. The 1H solid-state MAS (nr = 20 kHz) NMR spectrum of the (grey) partially acid deuterated amorphous indomethacin sample (d1-indomethacin) is overlaid with the (black) standard amorphous indomethacin and shows a reduction in 1H signal due to the partial acid deuteration. Dotted vertical lines are shown at 12.7 and 11.5 ppm and labeled (a) and (c), respectively. These are the approximate 1H chemical shifts for the acid (C2OH) protons in a (a) hydrogen bonded dicarboxylic acid environment (gamma-indomethacin) and (c) in a weaker hydrogen bonded carbonyl - carboxylic acid enviroment. (B) 2H solid-state MAS (nr = 5 kHz) NMR spectra (black) and DMFit simulation (Blue) of amorphous d1-indomethacin (~50% 2H exchanged at the acid site). The best fit 2H quadrupolar tensor gave a coupling constant (CQ) of 178 kHz with no asymmetry (h = 0) with a 2H chemical shift of 8 ppm and a linewidth of 10 ppm. Above the full 2H spectrum and simulation is a zoomed in plot to more clearly show the centerband and first few satellite transitions (black) and associated simulation fit (blue) of amorphous d1-indomethacin.
Figure 6.
(A) 1H solid-state MAS (nr = 20 kHz) NMR spectra of alpha, gamma and amorphous indomethacin. A selectively acid deutermium enriched amorphous indomethacin sample was made through 1H to 2H exchange prior to melt quenching. The 1H solid-state MAS (nr = 20 kHz) NMR spectrum of the (grey) partially acid deuterated amorphous indomethacin sample (d1-indomethacin) is overlaid with the (black) standard amorphous indomethacin and shows a reduction in 1H signal due to the partial acid deuteration. Dotted vertical lines are shown at 12.7 and 11.5 ppm and labeled (a) and (c), respectively. These are the approximate 1H chemical shifts for the acid (C2OH) protons in a (a) hydrogen bonded dicarboxylic acid environment (gamma-indomethacin) and (c) in a weaker hydrogen bonded carbonyl - carboxylic acid enviroment. (B) 2H solid-state MAS (nr = 5 kHz) NMR spectra (black) and DMFit simulation (Blue) of amorphous d1-indomethacin (~50% 2H exchanged at the acid site). The best fit 2H quadrupolar tensor gave a coupling constant (CQ) of 178 kHz with no asymmetry (h = 0) with a 2H chemical shift of 8 ppm and a linewidth of 10 ppm. Above the full 2H spectrum and simulation is a zoomed in plot to more clearly show the centerband and first few satellite transitions (black) and associated simulation fit (blue) of amorphous d1-indomethacin.
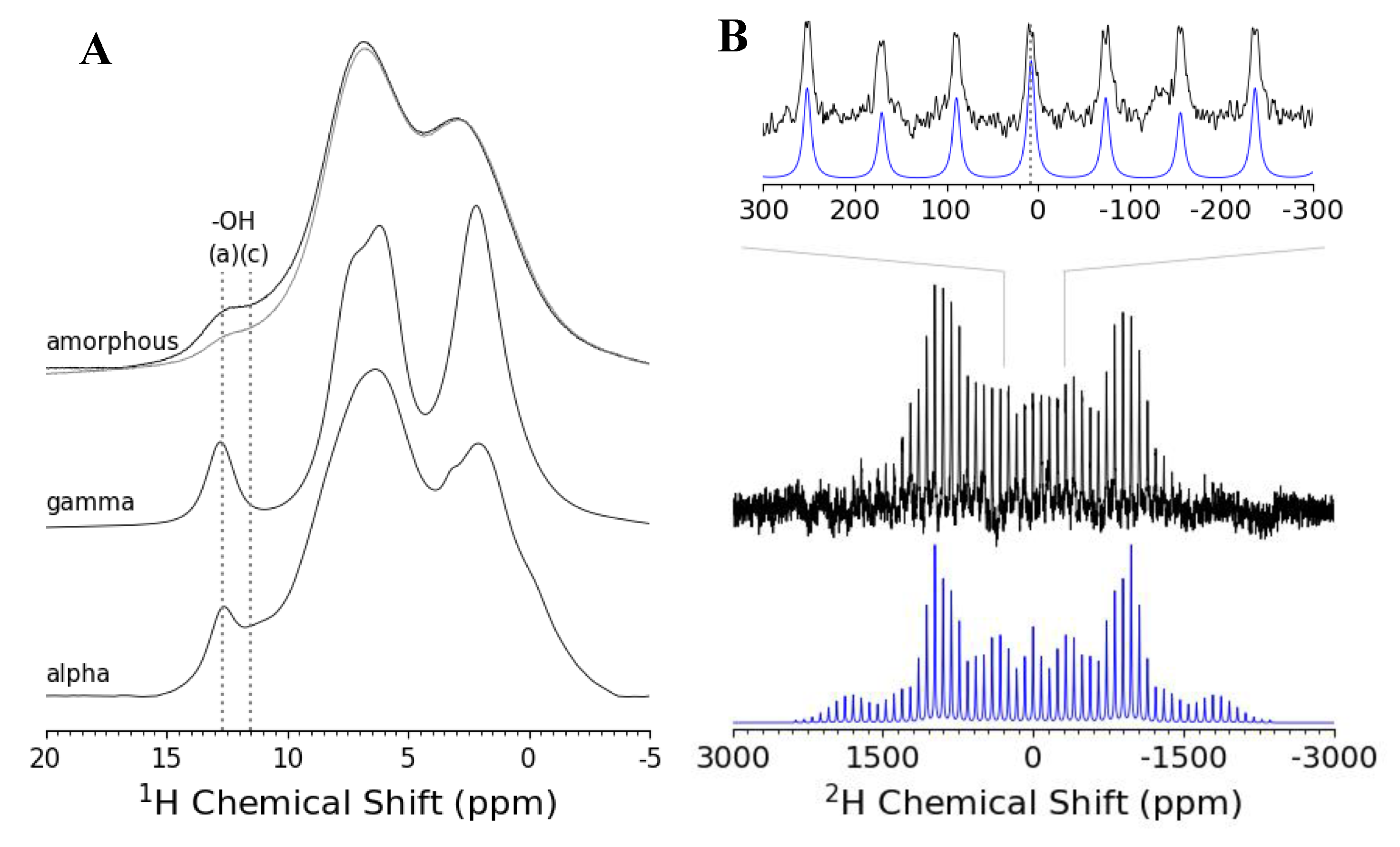
Figure 7.
The intra-molecular ∠O3-N-Cl angle determined from the EPSR models of the five samples. The angles associated with the three different isomers found in the crystal are denoted on the top axis.
Figure 7.
The intra-molecular ∠O3-N-Cl angle determined from the EPSR models of the five samples. The angles associated with the three different isomers found in the crystal are denoted on the top axis.
Figure 8.
The percent of number of hydrogen bonds per molecule as a function of chain size for our amorphous Indomethacin samples.
Figure 8.
The percent of number of hydrogen bonds per molecule as a function of chain size for our amorphous Indomethacin samples.
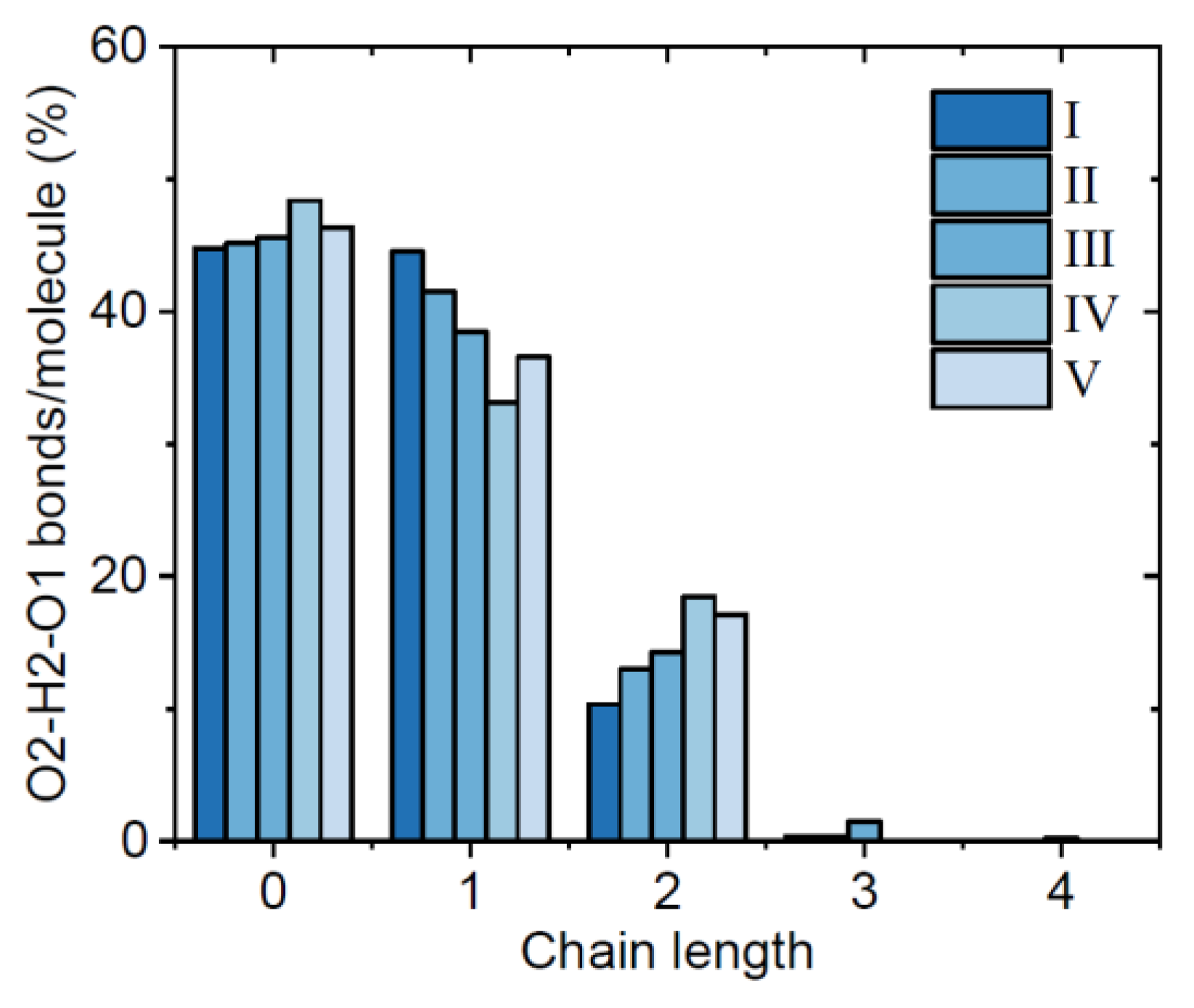
Figure 9.
Snapshots of (a) A carboxylic acid dimer found in γ-Indomethacin, (b) a singly hydrogen bonded molecular pair, and a (c) hydrogen bond between the carboxylic acid and an amide carbonyl in found in our EPSR models.
Figure 9.
Snapshots of (a) A carboxylic acid dimer found in γ-Indomethacin, (b) a singly hydrogen bonded molecular pair, and a (c) hydrogen bond between the carboxylic acid and an amide carbonyl in found in our EPSR models.
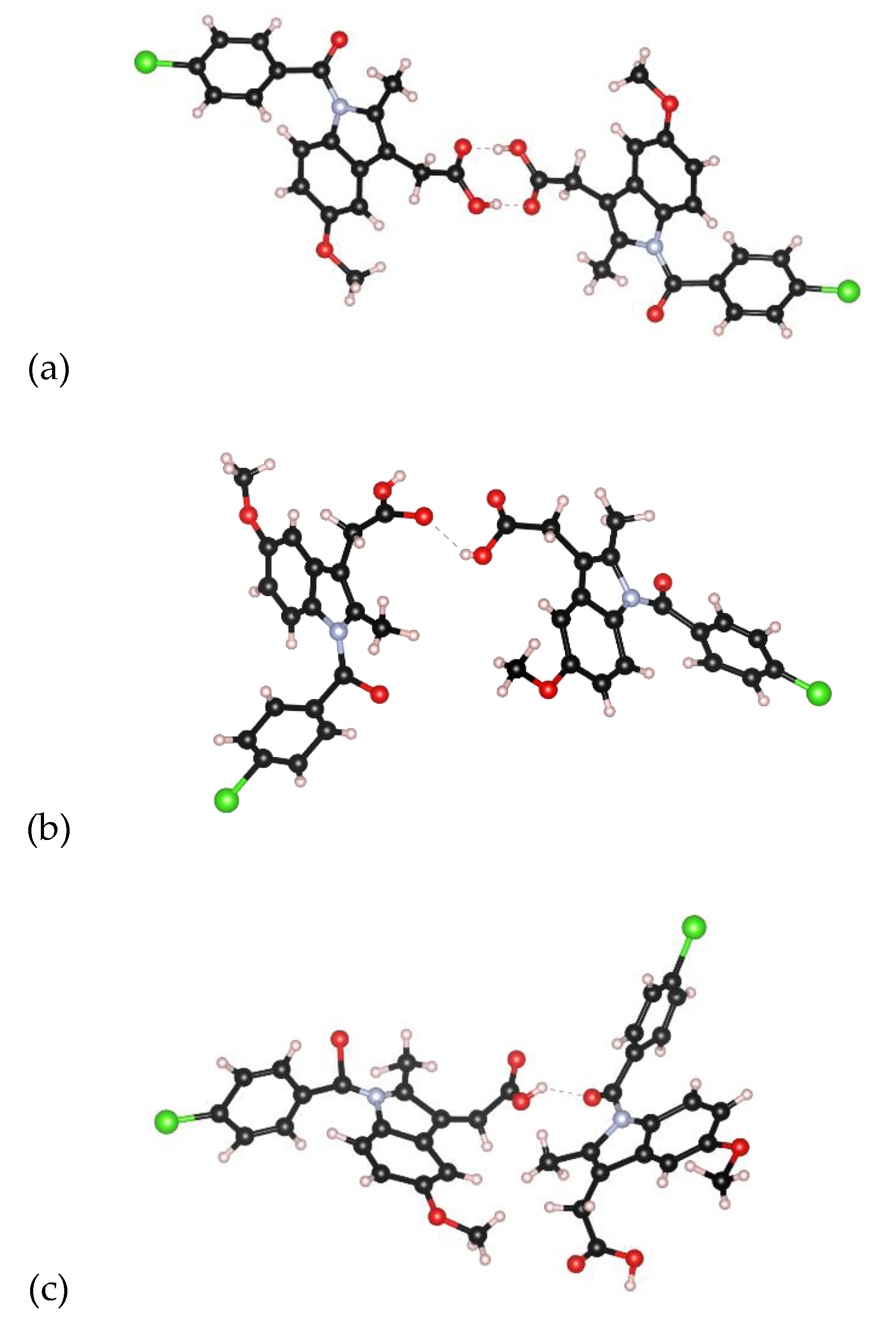

Table 1.
Starting Lennard-Jones parameters and partial charges.
| Atom | ε (KJ/mol) | σ (Å) | Partial charge, Q |
|---|---|---|---|
| O1 (acceptor), O2 (donor) | 0.65 | 3.1 | -0.6 |
| O3 (acceptor) | 0.65 | 3.1 | -0.4 |
| C4, C8 | 0.8 | 3.7 | +0.7 |
| H2 | 0 | 0 | +0.8 |
| H1 | 0 | 0 | 0.0 |
| C1,C2,C3,C5,C6,C7,C9,C10 | 0.8 | 3.7 | 0.0 |
| Cl | 0.8 | 3.2 | 0.0 |
Table 2.
EPSR simulation box parameters, densities and average C-C, C-O & C-N intra-molecular bond lengths compared to the crystal used in this study.
Table 2.
EPSR simulation box parameters, densities and average C-C, C-O & C-N intra-molecular bond lengths compared to the crystal used in this study.
| Sample | Atomic number density (atomsÅ-3) | Number of rotations within molecule | Intra-molecular C-X bond length or expansion (Å) |
| γ-form (crystal) | 0.0952 | None (Z isomer fixed geometry) | 1.380 (initial bond) |
| I | 0.0900 | 5 | +0.015 |
| II | 0.0900 | 5 | +0.015 |
| III | 0.0925 | 5 | +0.036 |
| IV | 0.0950 | None (slight variation) | +0.041 |
| V | 0.0950 | None (slight variation) | +0.036 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



