
Submitted:
10 July 2024
Posted:
11 July 2024
You are already at the latest version
Abstract
Protein degradation is a biological phenomena essential for cellular homeostasis and survivor. Selective protein degradation is performed by the ubiquitination system which selectively target proteins that need to be eliminated and lead them to proteasome degradation. In this review, we focus on the ubiquitin-conjugating enzyme E2 O (UBE2O) and highlight the role of UBE2O in many biological and physiological process. We further discuss the UBE2O implications in various human diseases, particularly in leukemias and solid cancers and suggest how UBE2O targeting could be novel strategy in the treatment of different human neoplasia.
Keywords:
UBE2O
; UPS
; ubiquitin
; protein degradation
; erythropoiesis
; leukemia
; solid tumors
1. Introduction
All cell types during their lifetime must be able to respond to metabolic requirements and environmental changes and, at the same time, to maintain their homeostasis. Protein degradation is fundamental for cellular homeostasis and is an essential control of cell signaling [1]. Much of the control of cell signaling occurs via protein ubiquitination and/or protein phosphorylation. While protein phosphorylation is reversible and allows proteins to be in two or more states and to interchange between them, protein ubiquitination provides the irreversible degradation of the targeted proteins, even though it is now appreciated that, under particular conditions, protein ubiquitination could be reversible via the activity of the de-ubiquitylating enzymes (DUBs) [2,3,4].
Ubiquitination is one of the most significant cellular post-transcriptional mechanisms and is involved in a wide range of key biological processes and cellular functions, such as protein–protein interaction, protein localization and expression levels [5]. Furthermore, ubiquitination can mediate DNA repair, gene transcription, inflammatory response, protein trafficking and angiogenesis [3,6].
Ubiquitin was first discovered in 1975 by Gideon and collaborators [7], who isolated and purified from bovine thymus a polypeptide with a high degree of evolutionary conservation, initially named ubiquitous immunopoietic polypeptide (UBIP). Since then, extensive research has been conducted and the discovery of ubiquitin pathway revolutionized our concept of intracellular protein degradation. Ubiquitin is a polypeptide of 8,5 kDa, composed by 76-residues. In human, ubiquitin is encoded by four genes; UBA52 and UBA80/RPS27A encode for ribosomal subunits, UBB and UBC encode for poly-ubiquitin precursors, which are then converted into single ubiquitin by de-ubiquitinating enzymes [8,9]. In addition, other pseudogenes have been identified as potential genes encoding for ubiquitin [10]. During the 1980s, ubiquitin has been described as a cofactor of a proteolytic system and Hershko and coworkers produced the initial understanding of the ubiquitin mediated protein degradation system, today known as the Ubiquitin Proteasome System (UPS) [11,12]. These discoveries led Aaron Ciechanover, Avram Hershko and Irwin Rose to win the Nobel prize for chemistry in 2004 [https://www.nobelprize.org/prizes/chemistry/2004/summary/].
Proteolysis of cellular proteins is a highly complex process regulated and carried out by a complex cascade of enzymes [13]. The degradation of proteins ubiquitin-mediated is a 2 steps process: i) multiple ubiquitin molecules are covalently attached to the lysine-residue of the target protein, ii) the tagged protein is degraded by the 26S proteasome. In the first step, the ubiquitin is first activated in its C-terminal Gly by the ubiquitin-activating enzyme, E1. The activated ubiquitin is then transferred from E1 to a Cys residue of an ubiquitin-conjugating enzyme, E2, which finally transfers ubiquitin from E1 to an ubiquitin-ligase enzyme, E3, to which the substrate protein is specifically bound [13,14]. In human, there are two genes encoding for E1s, at least 38 genes encoding for E2s and more than 600 genes encoding forE3s, thus providing different types of ubiquitination and the ubiquitination of selected, specific substrates [6,15,16,17]. The system has a hierarchical structure, in which a single E1 can activate and transfer ubiquitin to several species of E2s, and each E2 can interact and cooperate with several E3s [13].
2. The Atypical Ubiquitin-Conjugating Enzyme E2 O: UBE2O
One of the biggest in size E2s known nowadays, is the atypical ubiquitin-conjugating enzyme E2 O (UBE2O), with a molecular weight of 141 kDa [18], while most members of the E2 family have a molecular weight from 20 to 25 kDa [19]. UBE2O was first extracted from rabbit reticulocytes and named E2-230K, subsequently human UBE2O was cloned from liver tissues [20]. In human, UBE2O is ubiquitously expressed in all organs and almost all types of tissue, but it is preferentially expressed in brain, heart, liver tissue and skeletal muscle [18]. Furthermore, UBE2O is highly conserved in animals and plants [6], suggesting that its function is fundamental in many biological processes. Indeed, since its discovery, several studies have shown a plethora of cellular functions involving UBE2O, from cell cycle and proliferation to bone morphogenesis and erythroid differentiation [6,21].
UBE2O possesses three conserved regions (CR1, CR2 and CR3), a coiled-coil (CC) domain and a UBC domain, which enables UBE2O to interact with multiple E3 ligases. Interestingly, although its two nuclear localization sequences (NLSs), UBE2O is mainly localized within the cytoplasm [22,23]. Human UBE2O has 25 different cysteine residues and two of them are the main responsible of its catalytic activity: Cys617 localized in the CR2 domain, is necessary for the ubiquitination of the target substrates [24]; Cys1040 in the UBC domain, acts as the E2 active site [22].
Despite being first discovered and described as an E2 enzyme, most of the reported UBE2O substrates are catalyzed by UBE2O in a E3-independent manner [20,21]. Berleth and Pickart described an intramolecular thiol relay mechanism of UBE2O, by which it catalyzes substrates ubiquitination [25]. The CR1, CR2 and CC domains found in the N-terminal segment of UBE2O can interact with the E2 active site contained in the C-terminal fragment, thus leading to the mono-ubiquitination of the target (Figure 1, right side). This, demonstrates that UBE2O works as an E2/E3 hybrid enzyme and displays an E3 ligase activity [6,25,26]. In example, UBE2O was shown to ubiquitinate the AMP-activated protein kinase (AMPK), facilitating its degradation via the proteasome and thus activating the mTOR (mechanistic target of rapamycin) signaling pathway. Furthermore, as a downstream target of the UBE2O/AMPK/mTOR pathway, MYC transcriptionally promotes UBE2O expression, thus constituting a positive feedback loop that promotes cell proliferation, epithelial-mesenchymal transformation (EMT) in many types of humane cancers [27]. Similarly, UBE2O mono-ubiquitinates SMAD6 at lysine 174 during bone morphogenetic thus reducing SMAD6 inhibitory effect on the BMP/SMAD downstream pathway, essential during bone morphogenetic, heart and central nervous development [28]. Furthermore, UBE2O was observed to interfere with circadian rhythm, as well. Mechanistically, the Cys residue in the CR2 UBE2O domain interacts with BMAL1, essential for circadian oscillation [29], thus promoting its ubiquitination and degradation. Hence, UBE2O is a critical regulator of the circadian clock, whose dysregulation are associated with many diseases, including cancer, diabetes, and obesity [24].
Furthermore, UBE2O also displays non-enzymatic functions. By interacting with the TRAF domain of TNF receptor-associated factor 6 (TRAF6), the N-terminal fragment of UBE2O can block its polyubiquitination on lysine 63 and suppress the activation of NF-κB in a UBE2O dose-dependent manner (Figure 1, left side). The removal of UBC domain in UBE2O does not impact the inhibition of TRAF6 ubiquitination thus demonstrating the non-enzymatic functions of UBE2O and indicating UBE2O as a potent TRAF6 regulator [30].
The plethora of specialized features and multifunctional domains within the UBE2O protein demonstrates the important role of UBE2O in many aspects of cells life. Consequently, alteration in UBE2O expression levels might represent an important factor to be evaluated at onset and progression of human diseases, due to UBE2O ability to interfere with a broad spectrum of molecular targets and functions.
3. Role for UBE2O in the Hematological Field
Constant removal of surplus and damaged peptides, as well as maintenance of the protein homeostasis, is essential for proper cellular function. Therefore, alterations in the regulation of protein turnover have a role in the pathogenesis of a number of different types of solid cancers and diseases. Dysregulation of this proteolysis-regulating machinery can result in uncontrolled cell proliferation, accumulation of harmful proteins and genetic instability, leading ultimately to malignancy [31]. Dysregulations of several members of UPS have been identified in hematological malignancies.
Several therapies that have been developed for the treatment of the hematological diseases are based on the manipulation of the UPS members. The most relevant example is bortezomib, a selective inhibitor of the 26S proteasome which revolutionized the therapy of Multiple Myeloma (MM) and Mantle Cell Lymphoma (MCL) becoming and agent used in first-line [32]. The deubiquitinating enzyme USP1 was reported to be involved in Fanconi anemia (FA), a rare hematological disorder caused by the genetic loss of key factors of DNA repair [33]. Moreover, it has been shown that the inhibition of the deubiquitinating enzyme USP10 inhibits proliferation in chronic myeloid leukemia (CML) cells, both in imatinib-sensitive and imatinib-resistance cells [34,35]. On the other hand, many therapeutic strategies directly regulate protein degradation, such as thalidomide analogs. The E3 ubiquitin ligase cereblon (CRBN) was identified as the main binding partner of thalidomide. Fang et al. [36], showed that Lenalidomide, the main clinically used thalidomide-derived compound, acts through CRBN and has cytotoxic effects in cells of both myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). In addition, the latest years, a CRBN-based PROTACs (PROteolysis Targeting Chimeras) have gained attention showing promising results in inducing degradation of disease-associated proteins [32]
Here we report the therapeutic opportunities and strategies based on the activation or inactivation of UBE2O enzyme, describing its potential roles in the hematological filed.
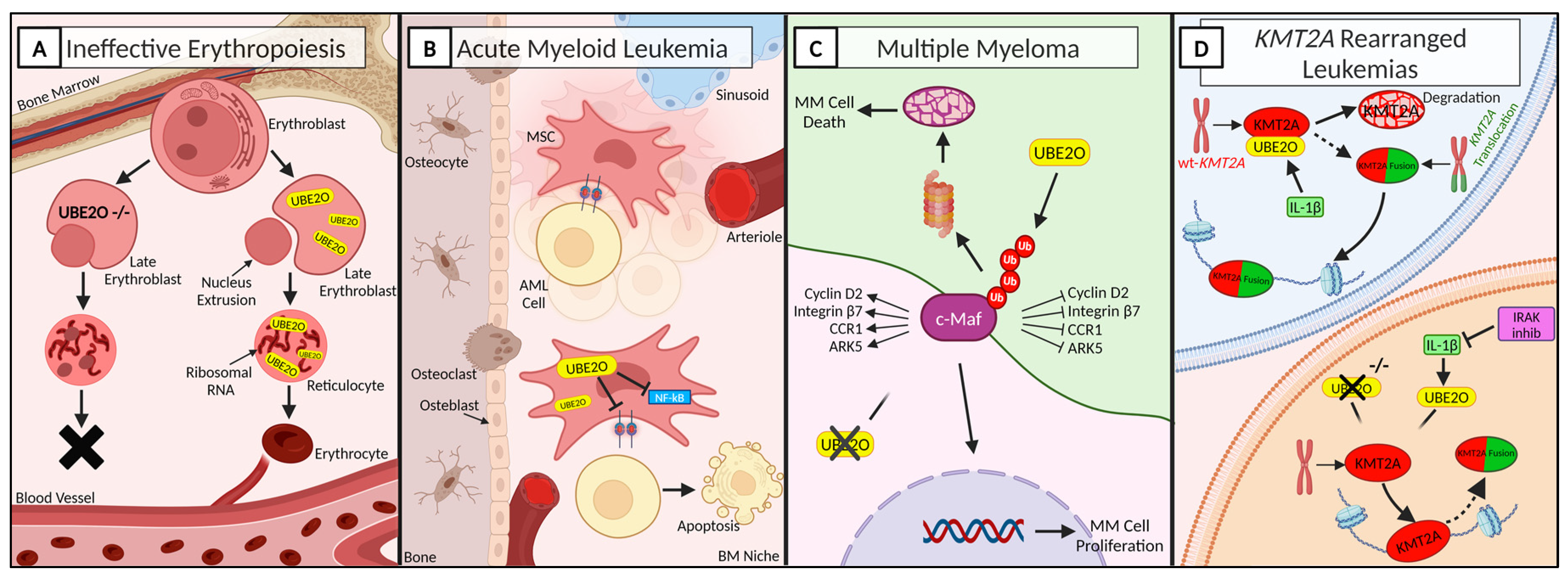
Figure 1.
UBE2O in hematology under physiological and pathological conditions. A fine regulation of protein turnover and degradation is essential for keeping cells’ homeostasis. Alteration at this level, The importance of UBE2O in many aspects of hematology. A) UBE2O is essential for a proper erythroid differentiation. Hence, UBE2O silencing leads to incapability of generating red blood cells. UBE2O: ubiquitin conjugating enzyme E2 O. B) Overexpression of UBE2O in AML cells inhibits leukemic cells proliferation and induces apoptosis. AML: Acute Myeloid Leukemia; BM: Bone Marrow; MSC: Mesenchymal Stromal Cell. C) UBE2O activity promotes c-Maf polyubiquitination and subsequent apoptosis of MM cells. ARK: AMPK-related protein kinase 5; CCR1: C-C Motif Chemokine Receptor 1; MM: Multiple Myeloma. D) UBE2O depletion promotes the stability of wild-type KMT2A in hematological neoplasia characterized by KMT2A rearrangements. IL-1β: Interleukin-1β; IRAK: Interleukin-1 Receptor-Associated Kinases; KMT2A: Lysine Methyltransferase 2A; WT: Wild-Type. Created with www.BioRender.com.
Figure 1.
UBE2O in hematology under physiological and pathological conditions. A fine regulation of protein turnover and degradation is essential for keeping cells’ homeostasis. Alteration at this level, The importance of UBE2O in many aspects of hematology. A) UBE2O is essential for a proper erythroid differentiation. Hence, UBE2O silencing leads to incapability of generating red blood cells. UBE2O: ubiquitin conjugating enzyme E2 O. B) Overexpression of UBE2O in AML cells inhibits leukemic cells proliferation and induces apoptosis. AML: Acute Myeloid Leukemia; BM: Bone Marrow; MSC: Mesenchymal Stromal Cell. C) UBE2O activity promotes c-Maf polyubiquitination and subsequent apoptosis of MM cells. ARK: AMPK-related protein kinase 5; CCR1: C-C Motif Chemokine Receptor 1; MM: Multiple Myeloma. D) UBE2O depletion promotes the stability of wild-type KMT2A in hematological neoplasia characterized by KMT2A rearrangements. IL-1β: Interleukin-1β; IRAK: Interleukin-1 Receptor-Associated Kinases; KMT2A: Lysine Methyltransferase 2A; WT: Wild-Type. Created with www.BioRender.com.
3.1. UBE2O Regulates Proteome Remodeling during Terminal Erythroid Differentiation
Erythropoiesis consists in the maturation and differentiation of multipotent hematopoietic stem cells to unipotent erythroid progenitors [37]. During terminal differentiation, erythrocytes precursors undergo an extensive remodeling of their organelles, including nucleus, mitochondria and ribosomes, in order to make space to globin, which is fundamental for oxygen transportation and constitutes ~98% of the cytosol [38]. This reorganization deeply involves the ubiquitination and de-ubiquitination system [39]. One of the main actors is UBE2O, which is specifically induced and active in terminally differentiating reticulocytes, resulting in massive protein degradation [40,41]. Interestingly, despite UBE2O being the second most abundant mRNA in mouse reticulocytes, it is abundant in reticulocytes while it is low or absent in undifferentiated cells, suggesting that in erythroid cells UBE2O is reticulocyte-specific [40]. Nguyen et al. [41] gained mechanistic insight to the function of UBE2O during red blood cells (RBC) maturation (Figure 2A). Taking advantage of UBE2O-null mouse model, they demonstrated that UBE2O deficiency associates with a defective mechanism of protein ubiquitination, leading to extensive intracellular accumulation of ribosomal proteins and development of microcytic hypochromic anemia in mice. Moreover, UBE2O has been shown to selectively ubiquitinate unassembled α-globin molecules that fail to assemble with β-globin in reticulocytes [42]. Thus, UBE2O has a prominent role in the regulation of RBC maturation, also acting as a quality control factor in reticulocytes, suggesting a putative role for UBE2O in the treatment of hematological malignancies characterized by ineffective erythropoiesis, such as β-thalassemia and myelodysplastic syndromes.
3.2. UBE2O Overexpression Inhibits Acute Myeloid Leukemia Progression
Acute myeloid leukemia (AML) is an aggressive hematological malignancy characterized by the accumulation of immature myeloid progenitors [43]. Although very few studies investigated the involvement of UBE2O in acute leukemias, many studies addressed the role of bone marrow (BM) microenvironment in supporting leukemia, demonstrating that BM microenvironment can be remodeled to support leukemogenesis and impede normal hematopoiesis [44,45]. Tian et al., identified around 1,000 genes with altered expression in AML-mesenchymal stromal cells (MSCs) and select UBE2O as the target gene of their study [46,47]. Lentiviral overexpression of UBE2O in AML-MSCs cells demonstrated that high levels of UBE2O inhibit the proliferation of MSCs via deactivation of NF-kB pathway. Furthermore, they showed that UBE2O-overexpressing cells, due to a lower adhesion capability, led to a decreased growth of AML cells and prolonged survival of mice (Figure 2B). These results provide the theoretical basis for a BM microenvironment-based therapeutic strategy to control disease progression. The demonstration that UBE2O overexpression in MSCs inhibits proliferation of leukemic cells, suggests that the induction of UBE2O overexpression could be investigated as a strategy to control disease progression.
3.3. UBE2O Knock-Down Reduces Cell Proliferation in KMT2A Rearranged Leukemias
Contrary to the hematological malignancies evaluated so far, for which the upregulation of UBE2O could represent a novel therapeutic strategy, other types of leukemia would benefit from the inhibition of UBE2O.
KMT2A (also known as MLL, mixed lineage leukemia or myeloid-lymphoid leukemia) belongs to the group of the KMT genes and encodes a DNA-binding protein methylating histone H3 lys4 (H3K4), the lysine methyltransferase 2A [48,49]. Rearrangements involving KMT2A have been shown to occur in precursor in B-acute lymphoblastic leukemia (B-ALL), T-acute lymphoblastic leukemia (T-ALL), acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), mixed lineage (biphenotypic) leukemia (MPAL) and secondary leukemia [50]. The KMT2A rearrangements are typically associated with poor prognosis and a disease progression often accompanied by a drift or switch in lineage, such as from lymphoid to myeloid [51]. The rearrangements of KMT2A, located on chromosome 11q23s, usually occurs as a single mutation [52,53], however it can also be present along with a cooperative mutation, especially PI3K-RAS, KRAS, NRAS and TP53 [50,52,54,55].
Taking advantage of multidimensional protein identification technology (MudPIT) assays and coimmunoprecipitation assays, Liang et al. [23] showed that UBE2O is the most abundant protein specifically interacting with wild-type KMT2A. The wilt-type KMT2A is less stable than the chimeric KMT2A-r, thus suggesting that stabilizing the KMT2A protein in leukemic cells could have a therapeutic potential [23,56]. Specifically, they demonstrated that interleukin-1 β (IL-1β) increases the KMT2A-UBE2O interaction and induces polyubiquitination of wild-type KMT2A, leading to its degradation. Furthermore, the interleukin-1 receptor-associated kinases (IRAK) could phosphorylate UBE2O, thereby enhancing its interaction with wild-type KMT2A (Figure 2D). Hence, downregulation of UBE2O levels could potentially improve the stability of wild-type KMT2A. RNA-seq analysis of UBE2O knocked-down cells revealed the downregulation of a plethora of genes, also associated with cell cycle activation or cellular response to growth factor stimulus [6]. In conclusion, in KMT2A-related diseases, UBE2O depletion increase the stability of wilt-type KMT2A and consequently downregulate a common subset of KMT2A chimera target genes and reduce leukemic cells proliferation
3.4. UBE2O Induces Apoptosis in Multiple Myeloma Cells
Multiple myeloma (MM) is one of the most commonly diagnosed blood cancers [57]. One of the genes commonly involved in pathophysiology of MM is the transcription factor c-Maf that belongs to the Maf family. c-Maf is overepressed in more than 50% of MM patients and in MM cell lines [58,59]. Zhang et al. [60] observed that c-Maf significantly enhances myeloma cell proliferation in vitro and tumor formation in vivo. UBE2O interacts with c-Maf and mediates the polyubiquitination of c-Maf at K48 inducing its degradation through the ubiquitin proteasome pathways [60,61]. Moreover, UBE2O also downregulates the transcriptional activity of c-Maf and the expression of c-Maf downstream genes, including cyclin D2, integrin β7, CCR1 (C-C Motif Chemokine Receptor 1) and ARK5 (AMPK-related protein kinase 5), which are responsible for cell cycle progress, MM cell proliferation, and myeloma cell invasion [61,62]. c-Maf destabilization and degradation impairs the growth of MM. Furthermore, UBE2O has been found to be downregulated in MM cells and restoration of UBE2O levels and activity induces MM cell apoptosis and suppressed cell proliferation both in vitro and in vivo, suggesting that UBE2O acts as a tumor suppressor against MM and might therefore be a potential therapeutic strategy for the treatment of multiple myeloma (Figure 2C) [61].
4. UBE2O Acts as a Pro-Oncogenic Factor in Many Human Cancers
The deregulation of UBE2O and its subsequent abnormal expression occur in many types of tumor and are associated with several human diseases [21,27,63,64]. UBE2O is localized in the 17q25 chromosome region, which has been found to be amplified in different cancers, including breast, prostate, gastric, kidney and ovarian cancers [21,65,66,67]. All this evidence supports the strong connection existing between UBE2O deregulation and tumor initiation and progression.
In the latest years much attention has been paid to evaluating the role of UBE2O overexpression in the pathogenesis and progression of breast and prostate cancers. In 2017 Vila et al. [65], demonstrated the high levels of UBE2O promote tumor initiation in mouse models of breast and prostate cancers in a AMPKα2 dependent manner, in mice models. Mechanistically, UBE2O ubiquitinates AMPKα2, facilitating its degradation which in turn determines the activation of mTORC1 signaling pathway. The axis UBE2O/ AMPKα2/mTORC1 favorites then the transcriptional activity of the oncoprotein MYC, generating a positive feedback loop that promotes cancer cell proliferation and epithelial–mesenchymal transformation (EMT) [27] and endows BC cells with cancer stemness properties. Furthermore, pharmacological inhibition of UBE2O reduced its protumor activity, through the restoration of AMPKα2 [27,65].
Furthermore, in-silico analysis of TCGA database, demonstrated that UBE2O amplification is frequently found in many types of gastric and lung cancers [6]. In lung cancer, which is the deadliest malignancy worldwide, UBE2O has been showed to play a putative essential role in radioresistance. Huang et al. [68] showed that UBE2O ubiquitinates MAX interactor 1 (Mxi1), whose degradation results in increased sensitivity to squamous cell carcinoma and malignant lymphoma in mouse models [69]. Consistently, UBE2O overexpression promotes lung cancer proliferation and radioresistance and predicts poor prognosis patients through negative regulation of Mxi1 [68] . Hence, genetical or pharmacological inhibition of UBE2O could be a potential strategy for treating lung cancer.
Recent studies also highlighted the potential of targeting UBE2O/HADHA axis in the treatment of hepatocellular carcinoma (HCC), the most common primary malignant liver tumor and the sixth highest cause of cancer-related death [70]. HADHA is a mitochondrial β-oxidation enzyme and is downregulated in HCC. UBE2O expression negatively correlates with HADHA levels, as UBE2O can mediate the ubiquitination and degradation of HADHA [71]. Through the regulation of HADHA, UBE2O modulates lipid metabolic reprogramming and promotes HCC with poor survival. Liver-specific deletion of UBE2O inhibits HCC growth and metastasis and is sufficient to prevent metabolic reprogramming in mice models [71]. In addition, novel integrating data from proteomic, mass spectrometry, and survival analysis, identified the interferon induced protein (IFIT3), a mediator of interferon (IFN) signaling and inhibitor of cell proliferation and migration [72], as a ubiquitination substrate of UBE2O that recognizes the lysin 236 of IFIT3. In HCC, high expression of IFIT3 increases the effectiveness of IFN therapy by upregulating the IFN-α signaling pathway and response [73]. Li et al. [74] demonstrated that the knockdown of UBE2O enhances the efficacy of IFN-α signaling, suggesting that targeting UBE2O could be a promising strategy to increase therapeutic effect of interferon-α.
These observations highlighted a pro-oncogenic role of UBE2O and support the concept of UBE2O targeting as a potential therapeutic strategy in diseases where metabolic reprogramming and proteome remodeling play a role.
5. UBE2O Role in Non-Oncologic Diseases
In addition to hematological diseases and cancers, UBE2O has been found involved in many other malignancies. In 2019 Vila et al. [75] demonstrated that UBE2O expression is universally upregulated in animal models exhibiting insulin resistance and metabolic disorders and significantly elevated in obese subjects with type 2 diabetes. AMPK is a key sensor and a crucial regulator of metabolism and energy balance [76]. As already reported, UBE2O ubiquitinates AMPKα2 for its degradation, therefore UBE2O inhibition could improve metabolic balance through AMPK pathway activity. Through UBE2O knockout models, they managed to improve insulin sensitivity in diet-induced type 2 diabetes mice and demonstrated that UBE2O ablation protects mice against diet-induced obesity and metabolic syndrome. These evidences suggest that the development of drugs interfering with UBE2O-AMPK interaction or the pharmacological inhibition of UBE2O activity could represent a powerful strategy for contributing to muscle insulin sensitivity, ameliorating diabetes and metabolic health.
Also, dysfunctions in the UPS are associated with the accumulation of misfolded or damaged proteins in the brain, which is the hallmark of age-related neurodegenerative diseases, such as Alzheimer Diseases (AD), Parkinson’s and Huntington’s diseases. The peak expression of UBE2O happens in the second week postanal, then its expression is gradually reduced with age, consistent with decline of UPS with aging Cheng et al. [77] recently demonstrated that UBE2O is reduced in the cortex and hippocampus of AD mice and reduced levels of UBE2O are associated with increased neuronal death. Mechanistically, UBE2O should contribute to reduce amyloid-β (Aβ) deposit, but accumulation of Aβ protein precursor (AβPP) with pathogenic mutation for AD, reduces he expression of UBE2P. These data suggest that UBE2O is dysregulated in AD neurons and age-associated reduction of UBE2O may facilitate neuronal death in AD, and indicate that increasing UBE2O expression in neurons may have therapeutic potential for AD.
Figure 3.
UBE2O in non-blood-diseases. Overview of UBE2O mechanism of action in non-hematological diseases: lower left: hepatocellular carcinoma; upper left: lung cancer; center: Alzheimer disease; upper right: breast cancer; lower right: type 2 diabetes. Aβ: amyloid-β; AD: Alzheimer diseases; AMPKα2: AMP-activated protein kinase alpha 2; HADHA: hydroxyacyl-CoA dehydrogenase; IFIT3: the interferon induced protein; IFN-α: interferon-alpha; mTORC1: mammalian target of rapamycin complex 1; Mxi1: MAX interactor 1; MYC: also known as cMyc. Created with www.BioRender.com.
Figure 3.
UBE2O in non-blood-diseases. Overview of UBE2O mechanism of action in non-hematological diseases: lower left: hepatocellular carcinoma; upper left: lung cancer; center: Alzheimer disease; upper right: breast cancer; lower right: type 2 diabetes. Aβ: amyloid-β; AD: Alzheimer diseases; AMPKα2: AMP-activated protein kinase alpha 2; HADHA: hydroxyacyl-CoA dehydrogenase; IFIT3: the interferon induced protein; IFN-α: interferon-alpha; mTORC1: mammalian target of rapamycin complex 1; Mxi1: MAX interactor 1; MYC: also known as cMyc. Created with www.BioRender.com.

6. Conclusions
The primary role of protein degradation in cell homeostasis and disease onset, became much more evident in the recent years attracting the interest of many studies. The biological and clinical relevance of E2s to the pathogenesis and progression of diseases and cancer suggest that E2s potentially hold great therapeutic promises as druggable targets [21,78]. Despite recent progress in the development of additional small-molecule E2 inhibitors [79,80], no such E2-targeting therapy has yet made its way to clinical trials [21]. Arsenic, which can crosslink adjacent cysteines within the catalytic approach of UBE2O could serve as the basis of an alternative approach to inhibiting E2 activity and is currently being tested against various form of cancer in clinical trials (hhtps://clinicaltrials.gov) [21,65]. It is also expected that E2-targeting therapeutics will be more efficacious against diseases and cancer when used in combination with current chemotherapy regimens.
While on the one hand the development of drugs design to block UBE2O activity could provide a relevant mechanism for the inhibition of progression of many tumors and for the improvement of insulin sensitivity and systemic physiology; on the other hand, UBE2O is required for erythropoiesis and have showed a favorable role in some hematological diseases. Although further studies are needed, UBE2O regulation could be a promising strategy in the treatment of different human neoplasia.
Author Contributions
Conceptualization, B.M. and D. C.; writing-original draft preparation, B.M., and A.M.; writing and editing, B.M. and D.C.; visualization, B.M.; supervision, D.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interests.
References
- Pickart, C.M. Back to the Future with Ubiquitin. Cell 2004, 116, 181–190. [CrossRef]
- Wilkinson, K.D. Essay. Cell 2004, 119, 741–745. [CrossRef]
- Hershko, A.; Ciechanover, A. THE UBIQUITIN SYSTEM. Annu. Rev. Biochem. 1998, 67, 425–479. [CrossRef]
- Nalepa, G.; Rolfe, M.; Harper, J.W. Drug Discovery in the Ubiquitin–Proteasome System. Nat Rev Drug Discov 2006, 5, 596–613. [CrossRef]
- Mukhopadhyay, D.; Riezman, H. Proteasome-Independent Functions of Ubiquitin in Endocytosis and Signaling. Science 2007, 315, 201–205. [CrossRef]
- Ullah, K.; Zubia, E.; Narayan, M.; Yang, J.; Xu, G. Diverse Roles of the E2/E3 Hybrid Enzyme UBE 2O in the Regulation of Protein Ubiquitination, Cellular Functions, and Disease Onset. The FEBS Journal 2019, 286, 2018–2034. [CrossRef]
- Goldstein, G.; Scheid, M.; Hammerling, U.; Schlesinger, D.H.; Niall, H.D.; Boyse, E.A. Isolation of a Polypeptide That Has Lymphocyte-Differentiating Properties and Is Probably Represented Universally in Living Cells. Proc. Natl. Acad. Sci. U.S.A. 1975, 72, 11–15. [CrossRef]
- Dubois, M.-L.; Meller, A.; Samandi, S.; Brunelle, M.; Frion, J.; Brunet, M.A.; Toupin, A.; Beaudoin, M.C.; Jacques, J.-F.; Lévesque, D.; et al. UBB Pseudogene 4 Encodes Functional Ubiquitin Variants. Nat Commun 2020, 11, 1306. [CrossRef]
- Clague, M.J.; Heride, C.; Urbé, S. The Demographics of the Ubiquitin System. Trends in Cell Biology 2015, 25, 417–426. [CrossRef]
- Nijman, S.M.B.; Luna-Vargas, M.P.A.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.G.; Sixma, T.K.; Bernards, R. A Genomic and Functional Inventory of Deubiquitinating Enzymes. Cell 2005, 123, 773–786. [CrossRef]
- Ciechanover, A.; Elias, S.; Heller, H.; Ferber, S.; Hershko, A. Characterization of the Heat-Stable Polypeptide of the ATP-Dependent Proteolytic System from Reticulocytes. Journal of Biological Chemistry 1980, 255, 7525–7528. [CrossRef]
- Kresge, N.; Simoni, R.D.; Hill, R.L. The Discovery of Ubiquitin-Mediated Proteolysis by Aaron Ciechanover, Avram Hershko, and Irwin Rose. Journal of Biological Chemistry 2006, 281, e32–e36. [CrossRef]
- Ciechanover, A. The Ubiquitin-Proteasome Pathway: On Protein Death and Cell Life. The EMBO Journal 1998, 17, 7151–7160. [CrossRef]
- Breitschopf, K. A Novel Site for Ubiquitination: The N-Terminal Residue, and Not Internal Lysines of MyoD, Is Essential for Conjugation and Degradation of the Protein. The EMBO Journal 1998, 17, 5964–5973. [CrossRef]
- Schulman, B.A.; Wade Harper, J. Ubiquitin-like Protein Activation by E1 Enzymes: The Apex for Downstream Signalling Pathways. Nat Rev Mol Cell Biol 2009, 10, 319–331. [CrossRef]
- Sun, Y. E3 Ubiquitin Ligases as Cancer Targets and Biomarkers. Neoplasia 2006, 8, 645–654. [CrossRef]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 Enzymes: More than Just Middle Men. Cell Res 2016, 26, 423–440. [CrossRef]
- Yokota, T.; Nagai, H.; Harada, H.; Mine, N.; Terada, Y.; Fujiwara, H.; Yabe, A.; Miyazaki, K.; Emi, M. Identification, Tissue Expression, and Chromosomal Position of a Novel Gene Encoding Human Ubiquitin-Conjugating Enzyme E2-230k. Gene 2001, 267, 95–100. [CrossRef]
- Wijk, S.J.L.; Timmers, H.T.M. The Family of Ubiquitin-conjugating Enzymes (E2s): Deciding between Life and Death of Proteins. FASEB j. 2010, 24, 981–993. [CrossRef]
- Klemperer, N.S.; Berleth, E.S.; Pickart, C.M. A Novel, Arsenite-Sensitive E2 of the Ubiquitin Pathway: Purification and Properties. Biochemistry 1989, 28, 6035–6041. [CrossRef]
- Hormaechea-Agulla, D.; Kim, Y.; Song, M.S.; Song, S.J. New Insights into the Role of E2s in the Pathogenesis of Diseases: Lessons Learned from UBE2O.
- Mashtalir, N.; Daou, S.; Barbour, H.; Sen, N.N.; Gagnon, J.; Hammond-Martel, I.; Dar, H.H.; Therrien, M.; Affar, E.B. Autodeubiquitination Protects the Tumor Suppressor BAP1 from Cytoplasmic Sequestration Mediated by the Atypical Ubiquitin Ligase UBE2O. Molecular Cell 2014, 54, 392–406. [CrossRef]
- Liang, K.; Volk, A.G.; Haug, J.S.; Marshall, S.A.; Woodfin, A.R.; Bartom, E.T.; Gilmore, J.M.; Florens, L.; Washburn, M.P.; Sullivan, K.D.; et al. Therapeutic Targeting of MLL Degradation Pathways in MLL-Rearranged Leukemia. Cell 2017, 168, 59-72.e13. [CrossRef]
- Chen, S.; Yang, J.; Zhang, Y.; Duan, C.; Liu, Q.; Huang, Z.; Xu, Y.; Zhou, L.; Xu, G. Ubiquitin-Conjugating Enzyme UBE2O Regulates Cellular Clock Function by Promoting the Degradation of the Transcription Factor BMAL1. Journal of Biological Chemistry 2018, 293, 11296–11309. [CrossRef]
- Berleth, E.S.; Pickart, C.M. Mechanism of Ubiquitin Conjugating Enzyme E2-230K: Catalysis Involving a Thiol Relay? Biochemistry 1996, 35, 1664–1671. [CrossRef]
- Zhang, X.; Zhang, J.; Bauer, A.; Zhang, L.; Selinger, D.W.; Lu, C.X.; Ten Dijke, P. Fine-Tuning BMP7 Signalling in Adipogenesis by UBE2O/E2-230K-Mediated Monoubiquitination of SMAD6. EMBO J 2013, 32, 996–1007. [CrossRef]
- Liu, X.; Ma, F.; Liu, C.; Zhu, K.; Li, W.; Xu, Y.; Li, G.; Niu, Z.; Liu, J.; Chen, D.; et al. UBE2O Promotes the Proliferation, EMT and Stemness Properties of Breast Cancer Cells through the UBE2O/AMPKα2/mTORC1-MYC Positive Feedback Loop. Cell Death Dis 2020, 11, 10. [CrossRef]
- Wang, R.N.; Green, J.; Wang, Z.; Deng, Y.; Qiao, M.; Peabody, M.; Zhang, Q.; Ye, J.; Yan, Z.; Denduluri, S.; et al. Bone Morphogenetic Protein (BMP) Signaling in Development and Human Diseases. Genes & Diseases 2014, 1, 87–105. [CrossRef]
- Abe, Y.O.; Yoshitane, H.; Kim, D.W.; Kawakami, S.; Koebis, M.; Nakao, K.; Aiba, A.; Kim, J.K.; Fukada, Y. Rhythmic Transcription of Bmal1 Stabilizes the Circadian Timekeeping System in Mammals. Nat Commun 2022, 13, 4652. [CrossRef]
- Zhang, X.; Zhang, J.; Zhang, L.; Van Dam, H.; Ten Dijke, P. UBE2O Negatively Regulates TRAF6-Mediated NF-κB Activation by Inhibiting TRAF6 Polyubiquitination. Cell Res 2013, 23, 366–377. [CrossRef]
- Nakayama, K.I.; Nakayama, K. Ubiquitin Ligases: Cell-Cycle Control and Cancer. Nat Rev Cancer 2006, 6, 369–381. [CrossRef]
- Drula, R.; Iluta, S.; Gulei, D.; Iuga, C.; Dima, D.; Ghiaur, G.; Buzoianu, A.D.; Ciechanover, A.; Tomuleasa, C. Exploiting the Ubiquitin System in Myeloid Malignancies. From Basic Research to Drug Discovery in MDS and AML. Blood Reviews 2022, 56, 100971. [CrossRef]
- Nijman, S.M.B.; Huang, T.T.; Dirac, A.M.G.; Brummelkamp, T.R.; Kerkhoven, R.M.; D’Andrea, A.D.; Bernards, R. The Deubiquitinating Enzyme USP1 Regulates the Fanconi Anemia Pathway. Molecular Cell 2005, 17, 331–339. [CrossRef]
- Liao, Y.; Liu, N.; Xia, X.; Guo, Z.; Li, Y.; Jiang, L.; Zhou, R.; Tang, D.; Huang, H.; Liu, J. USP10 Modulates the SKP2/Bcr-Abl Axis via Stabilizing SKP2 in Chronic Myeloid Leukemia. Cell Discov 2019, 5, 24. [CrossRef]
- Shibata, N.; Ohoka, N.; Tsuji, G.; Demizu, Y.; Miyawaza, K.; Ui-Tei, K.; Akiyama, T.; Naito, M. Deubiquitylase USP25 Prevents Degradation of BCR-ABL Protein and Ensures Proliferation of Ph-Positive Leukemia Cells. Oncogene 2020, 39, 3867–3878. [CrossRef]
- Fang, J.; Liu, X.; Bolanos, L.; Barker, B.; Rigolino, C.; Cortelezzi, A.; Oliva, E.N.; Cuzzola, M.; Grimes, H.L.; Fontanillo, C.; et al. A Calcium- and Calpain-Dependent Pathway Determines the Response to Lenalidomide in Myelodysplastic Syndromes. Nat Med 2016, 22, 727–734. [CrossRef]
- Sarodaya, N.; Karapurkar, J.; Kim, K.-S.; Hong, S.-H.; Ramakrishna, S. The Role of Deubiquitinating Enzymes in Hematopoiesis and Hematological Malignancies. Cancers 2020, 12, 1103. [CrossRef]
- Vinchi, F. Erythroid Differentiation: A Matter of Proteome Remodeling. HemaSphere 2018, 2, e26. [CrossRef]
- Wefes, I.; Mastrandrea, L.D.; Haldeman, M.; Koury, S.T.; Tamburlin, J.; Pickart, C.M.; Finley, D. Induction of Ubiquitin-Conjugating Enzymes during Terminal Erythroid Differentiation. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 4982–4986. [CrossRef]
- Haldeman, M.T.; Finley, D.; Pickart, C.M. Dynamics of Ubiquitin Conjugation during Erythroid Differentiation in Vitro. Journal of Biological Chemistry 1995, 270, 9507–9516. [CrossRef]
- Nguyen, A.T.; Prado, M.A.; Schmidt, P.J.; Sendamarai, A.K.; Wilson-Grady, J.T.; Min, M.; Campagna, D.R.; Tian, G.; Shi, Y.; Dederer, V.; et al. UBE2O Remodels the Proteome during Terminal Erythroid Differentiation. Science 2017, 357, eaan0218. [CrossRef]
- Yanagitani, K.; Juszkiewicz, S.; Hegde, R.S. UBE2O Is a Quality Control Factor for Orphans of Multiprotein Complexes. Science 2017, 357, 472–475. [CrossRef]
- Estey, E.; Döhner, H. Acute Myeloid Leukaemia. The Lancet 2006, 368, 1894–1907. [CrossRef]
- Wang, J.; Wang, P.; Zhang, T.; Gao, Z.; Wang, J.; Feng, M.; Yin, R.; Zhang, H. Molecular Mechanisms for Stemness Maintenance of Acute Myeloid Leukemia Stem Cells. Blood Science 2019, 1, 77–83. [CrossRef]
- Colmone, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic Cells Create Bone Marrow Niches That Disrupt the Behavior of Normal Hematopoietic Progenitor Cells. Science 2008, 322, 1861–1865. [CrossRef]
- Tian, C.; Chen, Z.; Wang, L.; Si, J.; Kang, J.; Li, Y.; Zheng, Y.; Gao, Y.; Nuermaimaiti, R.; You, M.J.; et al. Over Expression of Ubiquitin-conjugating Enzyme E2O in Bone Marrow Mesenchymal Stromal Cells Partially Attenuates Acute Myeloid Leukaemia Progression. Br J Haematol 2023, 200, 476–488. [CrossRef]
- Tian, C. Low-Expression of UBE2O in Bone Marrow Mesenchymal Stromal Cells Promotes Acute Myeloid Leukemia Progression. Blood 2022, 140, 11423–11423. [CrossRef]
- Castiglioni, S.; Di Fede, E.; Bernardelli, C.; Lettieri, A.; Parodi, C.; Grazioli, P.; Colombo, E.A.; Ancona, S.; Milani, D.; Ottaviano, E.; et al. KMT2A: Umbrella Gene for Multiple Diseases. Genes 2022, 13, 514. [CrossRef]
- Tkachuk, D.C.; Kohler, S.; Cleary, M.L. Involvement of a Homolog of Drosophila Trithorax by 11q23 Chromosomal Translocations in Acute Leukemias. Cell 1992, 71, 691–700. [CrossRef]
- Górecki, M.; Kozioł, I.; Kopystecka, A.; Budzyńska, J.; Zawitkowska, J.; Lejman, M. Updates in KMT2A Gene Rearrangement in Pediatric Acute Lymphoblastic Leukemia. Biomedicines 2023, 11, 821. [CrossRef]
- Rossi, J.G.; Bernasconi, A.R.; Alonso, C.N.; Rubio, P.L.; Gallego, M.S.; Carrara, C.A.; Guitter, M.R.; Eberle, S.E.; Cocce, M.; Zubizarreta, P.A.; et al. Lineage Switch in Childhood Acute Leukemia: An Unusual Event with Poor Outcome. American J Hematol 2012, 87, 890–897. [CrossRef]
- for The St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project; Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; et al. The Landscape of Somatic Mutations in Infant MLL-Rearranged Acute Lymphoblastic Leukemias. Nat Genet 2015, 47, 330–337. [CrossRef]
- Agraz-Doblas, A.; Bueno, C.; Bashford-Rogers, R.; Roy, A.; Schneider, P.; Bardini, M.; Ballerini, P.; Cazzaniga, G.; Moreno, T.; Revilla, C.; et al. Unraveling the Cellular Origin and Clinical Prognostic Markers of Infant B-Cell Acute Lymphoblastic Leukemia Using Genome-Wide Analysis. Haematologica 2019, 104, 1176–1188. [CrossRef]
- Prelle, C.; Bursen, A.; Dingermann, T.; Marschalek, R. Secondary Mutations in t(4;11) Leukemia Patients. Leukemia 2013, 27, 1425–1427. [CrossRef]
- Lanza, C.; Gaidano, G.; Cimino, G.; Pastore, C.; Nomdedeu, J.; Volpe, G.; Vivenza, C.; Parvis, G.; Mazza, U.; Basso, G.; et al. Distribution ofTP53 Mutations among Acute Leukemias withMLL Rearrangements. Genes Chromosom. Cancer 1996, 15, 48–53. [CrossRef]
- Poreba, E.; Lesniewicz, K.; Durzynska, J. Aberrant Activity of Histone–Lysine N-Methyltransferase 2 (KMT2) Complexes in Oncogenesis. IJMS 2020, 21, 9340. [CrossRef]
- Charliński, G.; Jurczyszyn, A. Non-Secretory Multiple Myeloma: Diagnosis and Management. Adv Clin Exp Med 2021, 31, 95–100. [CrossRef]
- Lv, Y.; Xing, F. Regulatory Roles of an Atypical Ubiquitin Ligase UBE2O in Orphans of Multi-Protein Complexes for Degradation. Turkish Journal of Biology 2022, 46, 186–194. [CrossRef]
- Hideshima, T.; Bergsagel, P.L.; Kuehl, W.M.; Anderson, K.C. Advances in Biology of Multiple Myeloma: Clinical Applications. Blood 2004, 104, 607–618. [CrossRef]
- Zhang, Z.; Tong, J.; Tang, X.; Juan, J.; Cao, B.; Hurren, R.; Chen, G.; Taylor, P.; Xu, X.; Shi, C.; et al. The Ubiquitin Ligase HERC4 Mediates C-Maf Ubiquitination and Delays the Growth of Multiple Myeloma Xenografts in Nude Mice. Blood 2016, 127, 1676–1686. [CrossRef]
- Xu, Y.; Zhang, Z.; Li, J.; Tong, J.; Cao, B.; Taylor, P.; Tang, X.; Wu, D.; Moran, M.F.; Zeng, Y.; et al. The Ubiquitin-Conjugating Enzyme UBE2O Modulates c-Maf Stability and Induces Myeloma Cell Apoptosis. J Hematol Oncol 2017, 10, 132. [CrossRef]
- Hurt, E.M.; Wiestner, A.; Rosenwald, A.; Shaffer, A.L.; Campo, E.; Grogan, T.; Bergsagel, P.L.; Kuehl, W.M.; Staudt, L.M. Overexpression of C-Maf Is a Frequent Oncogenic Event in Multiple Myeloma That Promotes Proliferation and Pathological Interactions with Bone Marrow Stroma. Cancer Cell 2004, 5, 191–199. [CrossRef]
- Rice, K.L.; Lin, X.; Wolniak, K.; Ebert, B.L.; Berkofsky-Fessler, W.; Buzzai, M.; Sun, Y.; Xi, C.; Elkin, P.; Levine, R.; et al. Analysis of Genomic Aberrations and Gene Expression Profiling Identifies Novel Lesions and Pathways in Myeloproliferative Neoplasms. Blood Cancer Journal 2011, 1, e40–e40. [CrossRef]
- Toffoli, S.; Bar, I.; Abdel-Sater, F.; Delrée, P.; Hilbert, P.; Cavallin, F.; Moreau, F.; Van Criekinge, W.; Lacroix-Triki, M.; Campone, M.; et al. Identification by Array Comparative Genomic Hybridization of a New Amplicon on Chromosome 17q Highly Recurrent in BRCA1 Mutated Triple Negative Breast Cancer. Breast Cancer Res 2014, 16, 466. [CrossRef]
- Vila, I.K.; Yao, Y.; Kim, G.; Xia, W.; Kim, H.; Kim, S.-J.; Park, M.-K.; Hwang, J.P.; González-Billalabeitia, E.; Hung, M.-C.; et al. A UBE2O-AMPKα2 Axis That Promotes Tumor Initiation and Progression Offers Opportunities for Therapy. Cancer Cell 2017, 31, 208–224. [CrossRef]
- Wang, X.; Li, X.; Cheng, Y.; Sun, X.; Sun, X.; Self, S.; Kooperberg, C.; Dai, J.Y. Copy Number Alterations Detected by Whole-Exome and Whole-Genome Sequencing of Esophageal Adenocarcinoma. Hum Genomics 2015, 9, 22. [CrossRef]
- Briffa, R.; Um, I.; Faratian, D.; Zhou, Y.; Turnbull, A.K.; Langdon, S.P.; Harrison, D.J. Multi-Scale Genomic, Transcriptomic and Proteomic Analysis of Colorectal Cancer Cell Lines to Identify Novel Biomarkers. PLoS ONE 2015, 10, e0144708. [CrossRef]
- Huang, Y.; Yang, X.; Lu, Y.; Zhao, Y.; Meng, R.; Zhang, S.; Dong, X.; Xu, S.; Wu, G. UBE2O Targets Mxi1 for Ubiquitination and Degradation to Promote Lung Cancer Progression and Radioresistance. Cell Death Differ 2021, 28, 671–684. [CrossRef]
- Schreiber-Agus, N.; Meng, Y.; Hoang, T.; Hou, H.; Chen, K.; Greenberg, R.; Cordon-Cardo, C.; Lee, H.-W.; DePinho, R.A. Role of Mxi1 in Ageing Organ Systems and the Regulation of Normal and Neoplastic Growth. Nature 1998, 393, 483–487. [CrossRef]
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73, 4–13. [CrossRef]
- Ma, M.; Zhang, C.; Cao, R.; Tang, D.; Sang, X.; Zou, S.; Wang, X.; Xu, H.; Liu, G.; Dai, L.; et al. UBE2O Promotes Lipid Metabolic Reprogramming and Liver Cancer Progression by Mediating HADHA Ubiquitination. Oncogene 2022, 41, 5199–5213. [CrossRef]
- Chikhalya, A.; Dittmann, M.; Zheng, Y.; Sohn, S.-Y.; Rice, C.M.; Hearing, P. Human IFIT3 Protein Induces Interferon Signaling and Inhibits Adenovirus Immediate Early Gene Expression. mBio 2021, 12, e02829-21. [CrossRef]
- Yang, Y.; Zhou, Y.; Hou, J.; Bai, C.; Li, Z.; Fan, J.; Ng, I.O.L.; Zhou, W.; Sun, H.; Dong, Q.; et al. Hepatic IFIT3 Predicts Interferon-α Therapeutic Response in Patients of Hepatocellular Carcinoma. Hepatology 2017, 66, 152–166. [CrossRef]
- Li, H.; Liu, Y.; Cheng, C.; Wu, Y.; Liang, S.-H.; Wu, L.; Wang, H.; Tu, C.; Yao, H.-H.; Meng, F.-Z.; et al. UBE2O Reduces the Effectiveness of Interferon-α via Degradation of IFIT3 in Hepatocellular Carcinoma. Cell Death Dis 2023, 14, 854. [CrossRef]
- Vila, I.K.; Park, M.K.; Setijono, S.R.; Yao, Y.; Kim, H.; Badin, P.-M.; Choi, S.; Narkar, V.; Choi, S.-W.; Chung, J.; et al. A Muscle-Specific UBE2O/AMPKα2 Axis Promotes Insulin Resistance and Metabolic Syndrome in Obesity. JCI Insight 2019, 4, e128269. [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat Rev Mol Cell Biol 2012, 13, 251–262. [CrossRef]
- Cheng, J.; Zheng, H.; Liu, C.; Jin, J.; Xing, Z.; Wu, Y. Age-Associated UBE2O Reduction Promotes Neuronal Death in Alzheimer’s Disease. JAD 2023, 93, 1083–1093. [CrossRef]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in Disease Pathogenesis and Treatment. Nat Med 2014, 20, 1242–1253. [CrossRef]
- Chen, H.; Wu, G.; Gao, S.; Guo, R.; Zhao, Z.; Yuan, H.; Liu, S.; Wu, J.; Lu, X.; Yuan, X.; et al. Discovery of Potent Small-Molecule Inhibitors of Ubiquitin-Conjugating Enzyme UbcH5c from α-Santonin Derivatives. J. Med. Chem. 2017, 60, 6828–6852. [CrossRef]
- Morreale, F.E.; Testa, A.; Chaugule, V.K.; Bortoluzzi, A.; Ciulli, A.; Walden, H. Mind the Metal: A Fragment Library-Derived Zinc Impurity Binds the E2 Ubiquitin-Conjugating Enzyme Ube2T and Induces Structural Rearrangements. J. Med. Chem. 2017, 60, 8183–8191. [CrossRef]
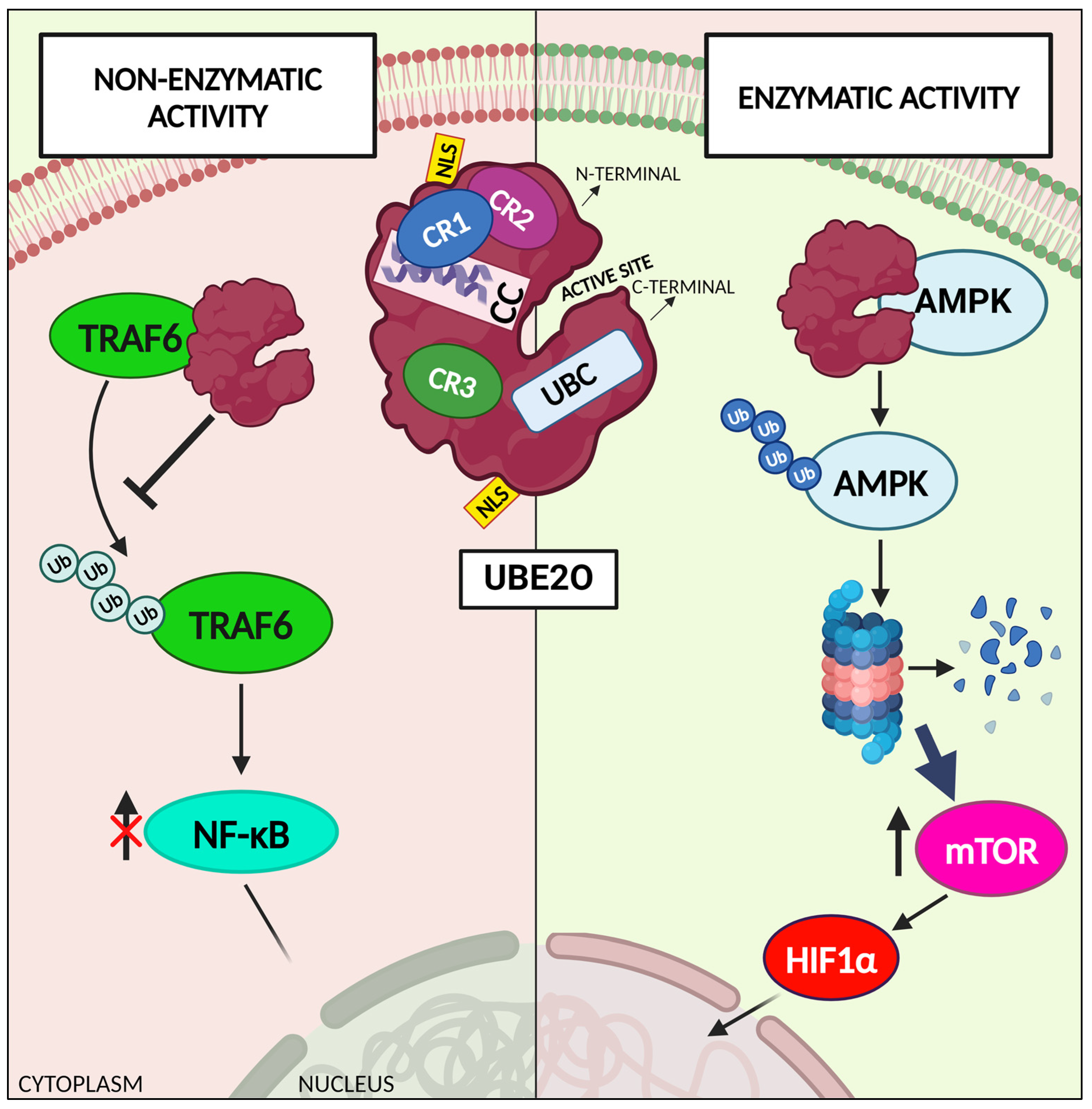
Figure 1.
UBE2O exerts both enzymatic functions and non-enzymatic functions. The most studied and reported function of UBE2O, is that as a hybrid E2/E3 enzyme it catalyzes substrates ubiquitination. Nonetheless, UBE2O has been showed to exert also non-enzymatic functions. Figure 1 shows two examples of UBE2O different functions. The enzymatic function is explained through the depiction of the AMPK-mTOR pathway, in which UBE2O catalyzes the ubiquitination of AMPK, thus inducing its degradation and the activation of mTOR and subsequently of HIF1α. The non-enzymatic function of UBE2O is represented through the schematization of the interaction of UBE2O N-terminal fraction with TRAF6, thus impeding its ubiquitination and negatively regulating NF-κB pathway. AMPK: 5′ adenosine monophosphate-activated protein kinase; CC: coiled-coil region; CR1/2/3: conserved region 1/2/3; HIF1α: hypoxia-inducible factor 1-alpha; mTOR: mammalian target of rapamycin; NLS: nuclear localization sequence; NF-kB: nuclear factor kappa-light enhancer of activated B cells; TRAF6: tumor necrosis factor receptor associated factor 6; UB: ubiquitin; UBE2O: ubiquitin conjugating enzyme E2 O; UBC: ubiquitin-conjugating core domain. Created with www.BioRender.com.
Figure 1.
UBE2O exerts both enzymatic functions and non-enzymatic functions. The most studied and reported function of UBE2O, is that as a hybrid E2/E3 enzyme it catalyzes substrates ubiquitination. Nonetheless, UBE2O has been showed to exert also non-enzymatic functions. Figure 1 shows two examples of UBE2O different functions. The enzymatic function is explained through the depiction of the AMPK-mTOR pathway, in which UBE2O catalyzes the ubiquitination of AMPK, thus inducing its degradation and the activation of mTOR and subsequently of HIF1α. The non-enzymatic function of UBE2O is represented through the schematization of the interaction of UBE2O N-terminal fraction with TRAF6, thus impeding its ubiquitination and negatively regulating NF-κB pathway. AMPK: 5′ adenosine monophosphate-activated protein kinase; CC: coiled-coil region; CR1/2/3: conserved region 1/2/3; HIF1α: hypoxia-inducible factor 1-alpha; mTOR: mammalian target of rapamycin; NLS: nuclear localization sequence; NF-kB: nuclear factor kappa-light enhancer of activated B cells; TRAF6: tumor necrosis factor receptor associated factor 6; UB: ubiquitin; UBE2O: ubiquitin conjugating enzyme E2 O; UBC: ubiquitin-conjugating core domain. Created with www.BioRender.com.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



