
Submitted:
11 July 2024
Posted:
12 July 2024
You are already at the latest version
Abstract
Acute myocardial infarction (AMI) results from vulnerable plaque rupture, causing ischemic cardiomyocyte necrosis and intense inflammation. Paradoxically, this inflammation releases factors that aid heart repair. Recent findings suggest a role for extracellular vesicles (EVs) in intercellular communication during post-AMI cardiac repair. However, EVs’ tissue origin and chemokine profile in the blood of patients with AMI remains unclear. This study characterized EV tissue origin and chemokine receptor profile in the coronary and peripheral blood of patients with AMI.
The results reveal that vesicles isolated from coronary and peripheral blood plasma are enriched in tetraspanin (CD9) and express CD81+, CD90+, and CD144+. The vesicle size ranged between 145 and 162 nm, with the control group exhibiting smaller vesicles than the AMI group. Furthermore, all vesicles expressed CCR6+ and CXCR3+, whereas a small percentage expressed CCR4+. In addition, a decrease in CXCR3+ and CCR6+ positivity was observed in coronary and peripheral blood vesicles compared with the control. Non-difference was found in the coronary and peripheral blood for the CXCR3+ and CCR6+ markers.
In conclusion, our study demonstrates, for the first time, changes in the number of extracellular vesicles expressing CD144+, CXCR3+, and CCR6+ in the peripheral circulation of patients with AMI. Extracellular vesicles present in the peripheral circulation of patients with AMI hold excellent promise as a potential diagnostic tool.
Keywords:
Myocardial infarction
; extracellular vesicles
; CD144
; CCR6
; CXCR3
1. Introduction
Acute myocardial infarction (AMI) arises from complete or partial artery obstruction instigated by the rupture or erosion of a vulnerable plaque [1]. This event leads to cardiomyocyte death [2], which is a critical consequence considering the limited regenerative capacity of the adult mammalian heart. Therefore, AMI is characterized by the loss of significant myocardium, replaced by a collagen-based scar, ischemic cardiomyocytes, and a prominent inflammatory reaction, all of which are pivotal elements in heart repair and remodeling [3].
Extracellular vesicles (EVs) are classified into three subtypes based on size and biogenesis: exosomes, microvesicles, and apoptotic bodies. Among them, exosomes are the smallest (~100 nm in diameter), derived from the endosomal pathway, and they carry nucleic acids, proteins, lipids, and metabolites, which allow them to establish near and long-distance intercellular communication in health and disease [4,5,6]. Evidence indicates that EVs can attenuate cellular senescence, inflammation, and myocardial injury [7,8]. With the emergence of studies on exosome secretion by various cell types, including cardiomyocytes (CMs), endothelial cells (ECs), fibroblasts, and circulating progenitor cells (CPCs), protective effects on AMI patients receiving EVs have been demonstrated [9].
Recent findings also suggest the involvement of exosome-mediated intercellular communication in cardiac repair after AMI [10]. Thus, after AMI, it has been shown that EVs are secreted by diverse cardiac stem cells related to cardioprotective effects, including activation of regenerative signals (angiogenic and anti-inflammatory) and active participation in cardiac repair [11]. For instance, Chengwei et al. [12] revealed that EV release from cardiac mesenchymal stem cells promotes repair in AMI. Moreover, they found that the transfer of EV molecular cargo, including miRNA, accounts for cardioprotection in a mouse AMI model. EVs isolated from these cardiac mesenchymal stem cells were injected intramyocardially into the left ventricular wall (border zone) at three locations immediately after the left anterior descending ligation. Also, EVs isolated from mesenchymal cells promoted neovascularization and reduced infarct size after intramyocardial administration in a porcine model of chronic myocardial ischemic damage [13]. Additionally, EVs released from mesenchymal stem cells can directly reach the ischemic tissue, participating in cardiac regeneration and remodeling [14,15]
On the other hand, given their ease of retrieval from biological fluids like serum, plasma, and urine, EVs have also emerged as potential biomarkers for the evolution of AMI (reference). However, the tissue origin and chemokine profile of extracellular vesicles in patients with AMI’s coronary and peripheral blood remain unknown [16,17]. Knowing their origin is critical since several cardiovascular risk factors have been associated with differential cargo of circulating EVs [18], which differentially activate target cells [19] via transference of their cargo (proteins, mRNAs, miRNAs) to the recipient cells. Therefore, the chemokine content in circulating EVs is critical to understanding its potential beneficial effect in patients with AMI.
This study aimed to characterize serum-derived EVs’ tissue origin and chemokine profile in the coronary and peripheral blood of patients with AMI.
2. Materials and Methods
2.1. Subjects
Patients with AMI treated in “Centro Cardiovascular del Hospital Clinico Dr. Guillermo Grant Benavente” were included in this study. AMI was diagnosed according to the following criteria: i) Patient with a diagnosis of AMI with ST-segment elevation with less than 24 hours of evolution, with or not underwent primary percutaneous coronary or thrombolysis revascularization according to the criteria of the World Health Organization [20]. These criteria include: i) chest pain over 20 min. ii) electrocardiographic diagnosis of AMI with ST-segment elevation (12 leads). iii) Elevated troponin T and I level and creatine kinase-myocardial band (CKMB) isoenzyme. Other inclusion criteria for AMI patients included: i) Adults (up to 80), both sexes. ii) With or without treatment with acetylsalicylic acid, B-blockers, angiotensin-converting enzyme inhibitors, and statins. iii) AMI within 24 hours of the onset of symptoms. Exclusion criteria: i) Individuals with a history of previous myocardial infarction. (ii) Renal and hepatic impairment. (iii) Coagulation disorders. (iv) Moderate to severe heart failure from another cause, not ischemic. (v) Infectious or inflammatory processes active. (vi) Chronic inflammatory disease. (vii) Malignant disease. (viii) Recent trauma or surgical interventions. (ix) Blood transfusions in the last ten months.
As control subjects, we recruited age-matched individuals from the Universidad de Concepción who met the following inclusion criteria: i) Healthy subjects. ii) Adults (up to 80 years), both sexes. iii) Without a history of minor or significant cardiovascular events. Also, we used the following exclusion criteria: i) individuals with hypotension and infectious or inflammatory symptoms. ii) Consuming anticoagulants or any non-steroidal anti-inflammatory. iii) There was a blood donor during the week preceding the sampling. iv) To consume excess vitamin supplements. v) Pregnancy.
This study was approved by the Ethical Committee of the Servicio de Salud Concepción, Chile, and all included subjects signed the corresponding informed.
2.2. Echocardiography
The echocardiographic analysis was performed with a Vivid seven ultrasound machine (General Electric, Healthcare, Horten, Norway) with the patient in the left lateral position within 48 hours after AMI. The left ventricular volume and ejection fraction were calculated using images from an apical 4-chamber view. Two cameras were analyzed using the biplane Simpson’s method [21]. The left ventricle (LV) was evaluated along the division the American Society of Echocardiography recommends investigating the regional systolic function [22]
2.3. Samples and Analytical Procedures
Venous blood was drawn from the antecubital vein. Aliquots were collected in clean tubes. Samples were centrifuged at 1500 x g for 15 minutes, at 4º C, and serum was stored at 4º C and used within 24 hours for glucose and lipid profile, and serum aliquots were also stored at -70ºC.
Glucose, triglycerides, and total cholesterol were quantified by standardized methods (Roche Diagnostics, Mannheim, Germany) in Hitachi 917 autoanalyzer. Within-run precision (CV) for triglycerides and total cholesterol were 1.3 % and 1.1 %, respectively. Between-day precision (CV) was 2.4 % and 1.5 %, respectively. Laboratory bias was 1.1 % and -1.7 %, respectively. Low-density lipoprotein-cholesterol (LDL-C) level was determined as the difference between total cholesterol and the cholesterol contained in the supernatant obtained after selective precipitation of LDL with 10 g/l polyvinyl sulphate in polyethyleneglycol (M.W. 600; 2.5 % w/v; pH = 6.7). Within-run and between-day precision (CV) were 4.7 % and 5.0 %, respectively. HDL was isolated in the supernatant obtained following precipitation of apolipoprotein (apo) B-containing lipoproteins with 0.44 mmol/l phosphotungstic acid in the presence of magnesium ions. Within-run and between-day precision (CV) for HDL-C were 3.2 % and 3.8 %, respectively. Laboratory bias was -2.0 %. Quality control was performed by RIQA Program (Ireland). In the isolated LDL fractions, triglycerides and cholesterol were measured using the previously mentioned methods, phospholipids using the Bartlett method, and proteins using the Lowry method [23]. According to the manufacturer’s instructions, duplicate measurements of the LOX-1 serum levels were performed using an ELISA kit (Life Science Inc. USCN. Wuhan). The minimum detectable level of human LOX-1 is less than 0.04 ng/mL. C-reactive protein (CRP) was determined on an ILAB 600 autoanalyzer using a Quantex CRP Ultra-Sensitive commercial kit following the manufacturer’s instructions.
2.4. Isolation and Characterization of Human Plasma EVs
Venous blood samples were collected and aliquoted in 700 μl volumes and stored at -20°C until use. Frozen plasma was thawed and centrifuged at i) 2000 x g for 30 min at 4°C and ii) 12,000 x g for 45 min at 4°C, and then passed through a 0.22-μm CA syringe filter to remove cell debris and large extracellular vesicles, and used for small EVs isolation by size exclusion chromatography, as described in Contreras et al., 2023 [24]. Briefly describe these methods a bit more.
2.5. Size exclusion Chromatography (SEC)
For size exclusion chromatography (SEC), 500 μl of concentrated plasma was loaded onto the Sephadex column (G200/120 or G200/40 resin, Sigma® Chemical Company, MO, United States)). Once the sample reached the top of the column bed, elution with NaCl solution was initiated, and the eluate was collected by gravity. Each fraction was collected in volumes of 400 μl for the EV collection medium. Fractions were collected and stored at -20°C until use. Total protein concentration was determined for each fraction using the DC TM Protein Assay Kit (BioRad) following the manufacturer’s instructions.
After each separation, SEC columns were washed sequentially with 30 ml of 0.1 M NaOH solution (0.22 μm filtered and degassed by sonication), 60 ml of PBS, and 100 ml of filtered and degassed mobile phase for reuse. The fractions collected from the columns were subjected to gel electrophoresis with Coomassie Blue Staining. Samples were loaded onto 6% tris-glycine-SDS gels and electrophoresed at 180 V and 40 mA for 100 minutes. Subsequently, the gels were stained with Coomassie Stain at room temperature for 2 hours, followed by removal of excess stain through washes with a bleaching solution (20% methanol, 10% acetic acid, in distilled water).
2.6. Extracellular Vesicles Characterization
For Western blot analysis, 40 μl of non-standardized protein-homogenized from each fraction were separated by 10% SDS/PAGE gel and transferred to PVDF membranes. Membranes were blocked with nonfat milk (5% w/v, in TBS) for 1h at room temperature. Primary antibodies were diluted in blocking buffer and incubated overnight at 4°C with mouse anti-ALIX (1:500, SC-53540), polyclonal goat anti-TSG 101 (1:1000, SC- 6037), and mouse anti-CD9 (1:500, SC-13118) purchased from Santa Cruz Biotechnology. Then, the membranes were washed three times with TBS-T (0.05% w/v, TBS-Tween 20) for 10 min each and incubated with secondary antibody Goat Anti-Mouse IgG H&L (HRP) (1:1000, ab97023) or Goat Anti-Rabbit IgG (HRP) (1: 1500, ab97051), as previously described Contreras et al., 2023 [24].
Size distribution and concentration of isolated vesicles were measured using the NanoSight NS 300 instrument (Malvern Instruments Ltd., Malvern, UK), and data were analyzed with the NTA software (version 3.2 Dev Build 3.2.16). The detection threshold was set to 2, and blur and Max Jump Distance were set to auto. Background measurements were performed with an ultrafiltered NaCl solution, revealing the absence of particles. Samples processed by SEC were diluted in sterile and filtered NaCl solution to reduce the number of particles in the field of view below 140/frame. Readings were taken five times, captured over 60 s at 25 frames per second (fps), at a camera level set to 13, and with manual temperature monitoring.
2.7. Flow cytometry of EVs
To evaluate the enrichment of EVs isolated from the column, a flow cytometry analysis of the fractions was performed using the Exosome–Human CD63 Isolation/Detection Kit (Invitrogen, Carlsbad, CA, United States) according to manufacturer recommendations, and as previously described (references). Briefly, 25 μg EVs were incubated with 20 μl beads (Life Technologies, Carlsbad, CA, USA) overnight at 4°C. Next, glycine diluted in PBS (100 mM final concentration) was added, mixed gently, and kept for 45 min at room temperature. EVs/beads complex were washed twice with 1 ml of PBS/0.5% bovine serum albumin by centrifugation at 1500 g for 3 min at RT. The EVs/beads complexes were incubated with primary FITC-conjugated CD81 (Abcam, catalog no. 34162, clone MM2/57), CD90 (Abcam, catalog no. 95700, clone 5E10), CD144 (Abcam, catalog no. 272346, clone 55-7h1), CCR4 (Abcam, catalog no. 281322, clone EPR235002-85), CCR6 (Abcam, catalog no. 288307, clone EPR235002-85) or CXCR3 (Abcam, catalog no. 314293, clone EPR23845-44) for 1 hour at room temperature. A negative control antibody reaction was performed using latex beads, incubated with anti-CD63 or anti-CD81 for 1 hour at room temperature (RT). The labeled MVs/beads complex were pelleted and washed twice as above with 300 μl of PBS/0.5% bovine serum albumin. Finally, 100 μl pellets were resuspended in 200 μl of focusing fluid and subjected to flow cytometry using BD LSR Fortessa™ X-20.
2.8. Statistical Analysis
Data were analyzed using standard statistical software (SPSS version 25) and GraphPad Prism 9.0 (GraphPad Software Inc., CA). Values are expressed as mean, and S.E.M. The variables were analyzed using non-parametric ANOVA tests. Mann–Whitney tests were used for pair comparisons in cases with significant differences (p < 0.05).
3. Results
3.1. Studied Group
There were no differences in the average age, and most of the studied biochemical parameters were between controls and AMI patients. However, statistically significant differences were found in BMI, glycemia levels, and sLox-1 (p < 0.05 in all comparisons). Following the diagnosis criteria, patients with AMI exhibited elevated cardiac markers (Troponin, CK-Total, and CK-Mb) (Table 1). In contrast, lipid values, C-reactive protein (CRP), creatinine, and prothrombin time remained within normal ranges.
Extracellular vesicles were isolated from the plasma of healthy individuals (HC-P) by centrifugation and size exclusion chromatography. Eluted fractions were collected, and the total protein concentration was measured using a BSA standard curve (Figure 1A). Extrapolating the concentration values of the obtained fractions reveals that the total protein concentration peaks at 10 mg/mL between fractions 4, 5, and 6, with fraction 6 exhibiting the highest protein enrichment (Figure 1B).
Subsequently, the content was transferred to a membrane, and Western blotting was performed for Alix, Tsg101, and CD9. As shown in Figure 1C, fractions 3–6 display signals for Alix and CD9, whereas fraction 5 exhibits a signal for Tsg101. Fractions 4, 5, and 6 were enriched with EVs expressing all three proteins. At the same time, no expression of ALIX, Tsg101, or CD9 proteins was detected in the remaining fractions.
For the isolation of EVs using exclusion chromatography, we used the Exosome-Human CD63 Isolation/Detection kit [23] and labeled with anti-CD81, anti-CD90, and anti-CD144 (Figure 1D). Non-significant differences were noted in this signal across markers between the analyzed fractions (fractions 4, 5, and 6), with the signal being most abundant in fraction 5 in the flow cytometry analysis of these fractions (Figure 1D). Therefore, we used fractions 4 to 6 For the rest of the experiments.
3.2. Characteristics of Peripheric and Coronary EVs from AMI (MI-P and MI-C) and Controls (HC-P)
We then counted the number of the isolated EVs and measured their size using NTA in the fractions pool (F4, F5, F6). EVs from the control group (HC-P) have 3.2 x 1011 particles/mL with a modal size of 106.3 ± 3 nm (n = 5). In contrast, in the coronary blood samples (MI-C) from patients with AMI, the concentration of EVs was significantly higher, 9 x 1012 particles/mL, without changes in the modal size of 112.8 ± 4 nm (n = 8). In addition, EVs from the peripheral blood group of patients with AMI yielded 9.9 x 1012 particles/mL with a modal size of 121.4 ± 5 nm (Figure 2B) (n = 5). We then separate the size of the particles in the 10th, 50th, and 90th percentiles. Interestingly, for the 10th percentile, there were significant differences in the size of the EVs when comparing those isolated from blood coronary of AMI (MI-C) versus those in the control group (HC-P) (Figure 2C). In addition, at the 50th and 90th percentiles, EVs isolated from the control group were significantly smaller (124 ± 3 nm) than those isolated from coronary blood (134 ± 2 nm), without significant changes compared with those isolated from peripheral blood (MI-P) (141 ± 3 nm) (Figure 2C). No significant differences were found in the 90th percentile of EV size between control and AMI samples (coronary or periphery blood samples) (Figure 2C).
3.3. Immunophenotype Characterization of Extracellular Vesicles by Flow Cytometry
EVs were isolated by size exclusion chromatography, incubated with the exosome-human CD63 isolation/detection kit, and labeled with anti-CD90, anti-CD81, and anti-CD144 antibodies. EVs isolated from the plasma control and AMI patients (coronary and peripheral blood) were positive for CD81, CD90, and CD144 labeling (Figure 3).
Individual analysis of these markers showed that CD81 was differentially expressed in the EV from controls (17%) versus AMI patients, both in the coronary (24%) and peripheral (21%) source of the EVs (Figure 3B), reaching statistical significance between controls and peripheral source of EVs. At the same time, no significant differences were found in CD81+ EVs from the coronary and peripheral sources in AMI patients.
Similar findings were found in the analysis of CD144+ (Figure 3B). Thus, EVs from control have less proportion of CD144+ EVs (6%) compared with AMI patients, again reaching significant differences with the peripheral EVs of AMI patients (8%, p<0.05).
Furthermore, most EVs isolated from controls and AMI patients (coronary or peripheral source) were CD90+ (Figure 3B), showing non-significant differences in the ANOVA comparative analysis.
3.3. Characterization of the Chemokine Receptor Profile of Extracellular Vesicles by Flow Cytometry
EVs were labeled with anti-CCR4, anti-CCR6, and anti-CXCR3 antibodies, as shown in representative dot plots (Figure 4). CCR6+ EVs represented 5 to 10% of those isolated from control and AMI patients. At the same time, only 0.3% to 0.5% were CXCR4+ EVs in the studied groups. The proportion of CXCR3+ and CCR6+ EVs in the control group was significantly lower than their counterpart in the peripheral EVs from AMI patients (Figure 4B). However, no differences were found in the proportion of these chemokine receptors in EVs isolated from coronary samples in AMI patients compared with peripheral samples belonging to AMI or control subjects.
4. Discussion
Several studies have revealed that extracellular vesicles released during AMI exhibit specific molecular signatures reflective of their tissue origin and pathophysiological state. Our study demonstrates that AMI transiently increases the circulating levels of CD144+, CCR6+, and CXCR3+ EVs. Indeed, those EVs have differential characteristics in terms of size, number, and proportion of CD81+ EVs, CD144+, or CXCR3+ when comparing MI-P versus control. At the same time, MI-C parameters were not significantly different compared with MI-P or controls. These results align with previous studies suggesting that hypoxia, inflammation, or injury leads to increased secretion of EVs by cardiomyocytes and other cardiac cells (reference).
Therefore, our results suggest a differential population of EVs in the periphery and cardiac origin after AMI. We speculate that those in the periphery may be derived from the endothelium (express CD144). The sample was taken within 24 hours of the onset of the infarction, and the observed differences in microvesicle levels are associated with more significant destruction of vascular tissue during the infarction. This suggests that EVs identified after AMI can be used as an early marker of cardiac response.
Plasma-isolated vesicles were positive for CD90 and CD144. Differences were observed only in the CD144+ samples isolated from the control group and the MI-P. These findings are consistent with the literature, indicating that under conditions of damage such as AMI, there is an increased release of CD144, signifying a defined marker for endothelial dysfunction (CD144+). CD144, also known as VE-cadherin, is a constitutive marker of endothelial cells [17,18]. Levels of CD144+ and CD31+CD41 endothelial microparticles (EMPs) serve as an index of endothelial injury in various diseases [25,26,27]. Thus, elevated levels of CD144+ EMPs may reflect endothelial dysfunction and structural vascular damage in ischemic cardiovascular diseases (ICVD). Also, CD144+ EMPs are significantly associated with indices of neurological damage [25,26,27], supporting their utility as independent biomarkers of ICVD severity to assess the extent of ischemia.
EVs from the endothelium are notable for their capacity to mirror endothelial dysfunction [26,27], correlate with cardiovascular outcomes, and predict disease severity [28] and plaque rupture in acute coronary syndrome patients [28]. Associations between circulating EVs and angiographic lesions have also been reported and correlated with a higher calculated 10-year Framingham Coronary Artery Disease risk [28,29,30,31]. Therefore, CD144+ EVs could be a marker of future ventricular arrhythmia and AMI or a biomarker of ischemic severity in the clinic.
On the other hand, our work shows that in AMI, circulating EVs express chemokine receptors (CCR6 and CXCR3) and can be detected in the peripheral circulation of patients, being weakly positive for CCR4+. However, significant differences were observed when comparing the percentages of CCR6+ and CXCR3+ EVs from coronary and peripheral blood between the infarction and control groups. Currently, there are no reports detailing the expression of any of these receptors on the surface of vesicles from patients with AMI. Studies by Yang et al. (2019) show that CD4+CXCR5+CXCR3- exosomes increase during acute rejection following renal transplantation. These follicular helper T cell-derived exosomes promote B cell proliferation and differentiation in antibody-mediated rejection [33]. Similarly, Ciullo et al. demonstrated using an in vivo rat AMI model that a cell sheet of mesenchymal stem cells (MSCs) primed with exosomes overexpressing CXCR4 (ExoCXCR4) was able to preserve heart function when applied soon after coronary artery ligation, whereas the MSC sheet exposed to control exosomes was ineffective. These results indicate that MSC-derived ExoCXCR4 enhances MSC-mediated cardioprotection.
The literature details that the CCR6 receptor controls the release of inflammatory monocytes from the bone marrow into the circulating blood and their subsequent recruitment to the atherosclerotic plaque by interacting with its chemokine ligand CCL20 [34]. During atherogenesis, endothelial dysfunction can result in a local increase in CCL20, which may direct the migration of circulating CCR6+ cells to the subendothelial space. Monocyte-derived macrophages are sources of inflammatory cytokines such as TNFα, which can induce endothelial cells to express CCL20. In addition, macrophages themselves are producers of CCL20. CCL20 levels increase locally in the vessel wall, attracting more circulating monocytes to the plaque and promoting atherosclerosis through a positive feedback loop [35,36]. These results suggest mechanisms of recruitment of vesicles to the ischemic myocardium, as the ligand-receptor interaction exists.
Interestingly, it could also be shown that the receptor for CCL20, the chemokine receptor CCR6, plays an essential role in the recruitment of T cells to ischemic brain tissues [37]. CCR6 is highly expressed in lymphocytes, a cell type that has also been shown to contribute to ischemia-reperfusion injury after AMI [38]. As the role of CCR6 in AMI and cardiac injury, while plausible, is elusive, we set out to investigate this further in mice with or without CCR6 deficiency in a transient left anterior descending coronary artery ligation model and found that CCR6 deficiency increases the infarct size, followed by decreased ejection fraction and stroke volume. Furthermore, CCR6 deficiency increases apoptosis and the infiltration of inflammatory cells in the injured heart [38].
Several studies have reported increased levels of EVs in the plasma of patients with AMI compared with healthy individuals or patients with stable coronary artery disease (references). Furthermore, the specific molecular composition of these EVs, including cardiac-specific markers and inflammatory molecules, could provide valuable information for risk stratification and disease progression monitoring. In addition to their diagnostic potential, EVs may play a role in the pathophysiology of AMI and cardiac repair processes. In contrast, EVs derived from cardiac or regenerative cells may carry factors that stimulate angiogenesis, tissue repair, and cardioprotection [36]. All these findings indicate that EVs are an emerging area of research with potential clinical implications in the short term.
Our study has several limitations concerning understanding the molecular mechanisms behind EVs’ origin and release/synthesis. Additionally, we need to look further into the analysis of EVs cargo. Furthermore, what is the biological effect of EVs during a heart attack, and how they can contribute to the cardiac repair process.
In conclusion, our study demonstrates, for the first time, changes in the number of vesicles expressing CD144+, CXCR3+, and CCR6+ in the peripheral circulation of patients with AMI, suggesting that the presence of EVs in the peripheral circulation of patients with AMI holds excellent promise as a potential diagnostic tool.
Author Contributions
CA and EGG conceptualized the study and lead the research team. AM, ENL, and PL performed most of the experiments in this manuscript. CR, PAZ, ENL, and LR perform selected experiments. AM, CA, and PL were responsible for patient recruitment. CA, EGG, CR, and CE edited the manuscript. All co-authors approved the final version of this manuscript.
Acknowledgments
This study was funded by CONICYT PCI PII20150053 and VRIDEnlace Universidad de Concepción, 2018.072.039-1.0. CE is supported by FONDECYT 1240295. EN-L is funded by FONDECYT 1211480, VO, FONDECYT de Iniciación 11190522. EG-G is supported by FONDECYTde Iniciación 11170710.
Conflicts of Interest
None to declare.
References
- Saleh M, Ambrose JA. Understanding myocardial infarction. F1000Res. 2018;7.
- Reinecke H, Minami E, Zhu WZ, Laflamme MA. Cardiogenic differentiation and transdifferentiation of progenitor cells. Circ Res. 2008;103(10):1058-71.
- Moreno Ruiz N. Modificación de los criterios de Sgarbossa para el diagnóstico de infarto agudo de miocardio en presencia de bloqueo de rama izquierda. Revista de la Facultad de Medicina 2015;63(1):151-4.
- Martinez-Greene, J.A.; Hernandez-Ortega, K.; Quiroz-Baez, R.; Resendis-Antonio, O.; Pichardo-Casas, I.; Sinclair, D.A.; Budnik, B.; Hidalgo-Miranda, A.; Uribe-Querol, E.; Ramos-Godinez, M.D.P.; et al. Quantitative proteomic analysis of extracellular vesicle subgroups isolated by an optimized method combining polymer-based precipitation and size exclusion chromatography. J Extracell Vesicles 2021, 10, e12087. [CrossRef]
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.M.; Molaei, F.; Alahari, S.K. Exosomes: composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol Cancer 2019, 18, 75. [CrossRef]
- Zhu, J.; Lu, K.; Zhang, N.; Zhao, Y.; Ma, Q.; Shen, J.; Lin, Y.; Xiang, P.; Tang, Y.; Hu, X.; et al. Myocardial reparative functions of exosomes from mesenchymal stem cells are enhanced by hypoxia treatment of the cells via transferring microRNA-210 in an nSMase2-dependent way. Artif Cells Nanomed Biotechnol 2018, 46, 1659-1670. [CrossRef]
- Giricz, Z.; Varga, Z.V.; Baranyai, T.; Sipos, P.; Paloczi, K.; Kittel, A.; Buzas, E.I.; Ferdinandy, P. Cardioprotection by remote ischemic preconditioning of the rat heart is mediated by extracellular vesicles. J Mol Cell Cardiol 2014, 68, 75-78. [CrossRef]
- Mackie, A.R.; Klyachko, E.; Thorne, T.; Schultz, K.M.; Millay, M.; Ito, A.; Kamide, C.E.; Liu, T.; Gupta, R.; Sahoo, S.; et al. Sonic hedgehog-modified human CD34+ cells preserve cardiac function after acute myocardial infarction. Circ Res 2012, 111, 312-321. [CrossRef]
- de Couto, G.; Gallet, R.; Cambier, L.; Jaghatspanyan, E.; Makkar, N.; Dawkins, J.F.; Berman, B.P.; Marban, E. Exosomal MicroRNA Transfer Into Macrophages Mediates Cellular Postconditioning. Circulation 2017, 136, 200-214. [CrossRef]
- Sahoo, S.; Losordo, D.W. Exosomes and cardiac repair after myocardial infarction. Circ Res 2014, 114, 333-344. [CrossRef]
- Lazar, E.; Benedek, T.; Korodi, S.; Rat, N.; Lo, J.; Benedek, I. Stem cell-derived exosomes - an emerging tool for myocardial regeneration. World J Stem Cells 2018, 10, 106-115. [CrossRef]
- Ju, C.; Shen, Y.; Ma, G.; Liu, Y.; Cai, J.; Kim, I.M.; Weintraub, N.L.; Liu, N.; Tang, Y. Transplantation of Cardiac Mesenchymal Stem Cell-Derived Exosomes Promotes Repair in Ischemic Myocardium. J Cardiovasc Transl Res 2018, 11, 420-428. [CrossRef]
- Yao, J.; Huang, K.; Zhu, D.; Chen, T.; Jiang, Y.; Zhang, J.; Mi, L.; Xuan, H.; Hu, S.; Li, J.; et al. A Minimally Invasive Exosome Spray Repairs Heart after Myocardial Infarction. ACS Nano 2021, 15, 11099-11111. [CrossRef]
- Huang, L.; Yang, L.; Ding, Y.; Jiang, X.; Xia, Z.; You, Z. Human umbilical cord mesenchymal stem cells-derived exosomes transfers microRNA-19a to protect cardiomyocytes from acute myocardial infarction by targeting SOX6. Cell Cycle 2020, 19, 339-353. [CrossRef]
- Vicencio, J.M.; Yellon, D.M.; Sivaraman, V.; Das, D.; Boi-Doku, C.; Arjun, S.; Zheng, Y.; Riquelme, J.A.; Kearney, J.; Sharma, V.; et al. Plasma exosomes protect the myocardium from ischemia-reperfusion injury. J Am Coll Cardiol 2015, 65, 1525-1536. [CrossRef]
- Jansen, F.; Yang, X.; Proebsting, S.; Hoelscher, M.; Przybilla, D.; Baumann, K.; Schmitz, T.; Dolf, A.; Endl, E.; Franklin, B.S.; et al. MicroRNA expression in circulating microvesicles predicts cardiovascular events in patients with coronary artery disease. J Am Heart Assoc 2014, 3, e001249. [CrossRef]
- Sinning, J.M.; Losch, J.; Walenta, K.; Bohm, M.; Nickenig, G.; Werner, N. Circulating CD31+/Annexin V+ microparticles correlate with cardiovascular outcomes. Eur Heart J 2011, 32, 2034-2041. [CrossRef]
- Nozaki, T.; Sugiyama, S.; Koga, H.; Sugamura, K.; Ohba, K.; Matsuzawa, Y.; Sumida, H.; Matsui, K.; Jinnouchi, H.; Ogawa, H. Significance of a multiple biomarkers strategy including endothelial dysfunction to improve risk stratification for cardiovascular events in patients at high risk for coronary heart disease. J Am Coll Cardiol 2009, 54, 601-608. [CrossRef]
- Jimenez, J.J.; Jy, W.; Mauro, L.M.; Soderland, C.; Horstman, L.L.; Ahn, Y.S. Endothelial cells release phenotypically and quantitatively distinct microparticles in activation and apoptosis. Thromb Res 2003, 109, 175-180. [CrossRef]
- World Health Organization. Cardiovascular Diseases. Available online: https://www.who.int/health-topics/cardiovasculardiseases#tab=tab_1 (accessed on 15 March 2020).
- Otero-Ortega, L.; Alonso-Lopez, E.; Perez-Mato, M.; Laso-Garcia, F.; Gomez-de Frutos, M.C.; Diekhorst, L.; Garcia-Bermejo, M.L.; Conde-Moreno, E.; Fuentes, B.; Alonso de Lecinana, M.; et al. Similarities and Differences in Extracellular Vesicle Profiles between Ischaemic Stroke and Myocardial Infarction. Biomedicines 2020, 9. [CrossRef]
- Barile, L.; Lionetti, V.; Cervio, E.; Matteucci, M.; Gherghiceanu, M.; Popescu, L.M.; Torre, T.; Siclari, F.; Moccetti, T.; Vassalli, G. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc Res 2014, 103, 530-541. [CrossRef]
- Oporto, K.; Radojkovic, C.; Mellisho, E.A.; Zuniga, F.; Ormazabal, V.; Guzman-Gutierrez, E.; Nova-Lamperti, E.; Rodriguez-Alvarez, L.; Aranda, M.; Escudero, C.; et al. Adenosine promoted angiogenesis mediated by the release of small extracellular vesicles from human endothelial progenitor cells. Microvasc Res 2023, 148, 104498. [CrossRef]
- Contreras, H.; Alarcón-Zapata, A.; Nova-Lamperti, E.; Ormazabal, V.; Varas-Godoy, M.; Salomon, C.; Zuniga, F. Comparative study of size exclusion chromatography for isolation of small extracellular vesicle from cell-conditioned media, plasma, urine, and saliva. Front. Nanotechnol 2023, 5, 1146772. [CrossRef]
- Vion, A.C.; Ramkhelawon, B.; Loyer, X.; Chironi, G.; Devue, C.; Loirand, G.; Tedgui, A.; Lehoux, S.; Boulanger, C.M. Shear stress regulates endothelial microparticle release. Circ Res 2013, 112, 1323-1333. [CrossRef]
- Al Faraj, A.; Gazeau, F.; Wilhelm, C.; Devue, C.; Guerin, C.L.; Pechoux, C.; Paradis, V.; Clement, O.; Boulanger, C.M.; Rautou, P.E. Endothelial cell-derived microparticles loaded with iron oxide nanoparticles: feasibility of MR imaging monitoring in mice. Radiology 2012, 263, 169-178. [CrossRef]
- Werner, N.; Wassmann, S.; Ahlers, P.; Kosiol, S.; Nickenig, G. Circulating CD31+/annexin V+ apoptotic microparticles correlate with coronary endothelial function in patients with coronary artery disease. Arterioscler Thromb Vasc Biol 2006, 26, 112-116. [CrossRef]
- van Ierssel, S.H.; Hoymans, V.Y.; Van Craenenbroeck, E.M.; Van Tendeloo, V.F.; Vrints, C.J.; Jorens, P.G.; Conraads, V.M. Endothelial microparticles (EMP) for the assessment of endothelial function: an in vitro and in vivo study on possible interference of plasma lipids. PLoS One 2012, 7, e31496. [CrossRef]
- Amabile, N.; Heiss, C.; Real, W.M.; Minasi, P.; McGlothlin, D.; Rame, E.J.; Grossman, W.; De Marco, T.; Yeghiazarians, Y. Circulating endothelial microparticle levels predict hemodynamic severity of pulmonary hypertension. Am J Respir Crit Care Med 2008, 177, 1268-1275. [CrossRef]
- Zacharia, E.; Antonopoulos, A.S.; Oikonomou, E.; Papageorgiou, N.; Pallantza, Z.; Miliou, A.; Mystakidi, V.C.; Simantiris, S.; Kriebardis, A.; Orologas, N.; et al. Plasma signature of apoptotic microvesicles is associated with endothelial dysfunction and plaque rupture in acute coronary syndromes. J Mol Cell Cardiol 2020, 138, 110-114. [CrossRef]
- Bernal-Mizrachi, L.; Jy, W.; Fierro, C.; Macdonough, R.; Velazques, H.A.; Purow, J.; Jimenez, J.J.; Horstman, L.L.; Ferreira, A.; de Marchena, E.; et al. Endothelial microparticles correlate with high-risk angiographic lesions in acute coronary syndromes. Int J Cardiol 2004, 97, 439-446. [CrossRef]
- Yang, J.; Bi, L.; He, X.; Wang, Z.; Qian, Y.; Xiao, L.; Shi, B. Follicular Helper T Cell Derived Exosomes Promote B Cell Proliferation and Differentiation in Antibody-Mediated Rejection after Renal Transplantation. Biomed Res Int 2019, 2019, 6387924. [CrossRef]
- Ciullo, A.; Biemmi, V.; Milano, G.; Bolis, S.; Cervio, E.; Fertig, E.T.; Gherghiceanu, M.; Moccetti, T.; Camici, G.G.; Vassalli, G.; et al. Exosomal Expression of CXCR4 Targets Cardioprotective Vesicles to Myocardial Infarction and Improves Outcome after Systemic Administration. Int J Mol Sci 2019, 20. [CrossRef]
- Wan, W.; Murphy, P.M. Regulation of atherogenesis by chemokine receptor CCR6. Trends Cardiovasc Med 2011, 21, 140-144. [CrossRef]
- Arunachalam, P.; Ludewig, P.; Melich, P.; Arumugam, T.V.; Gerloff, C.; Prinz, I.; Magnus, T.; Gelderblom, M. CCR6 (CC Chemokine Receptor 6) Is Essential for the Migration of Detrimental Natural Interleukin-17-Producing gammadelta T Cells in Stroke. Stroke 2017, 48, 1957-1965. [CrossRef]
- Hofmann, U.; Frantz, S. Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ Res 2015, 116, 354-367. [CrossRef]
- Schumacher, D.; Liehn, E.A.; Singh, A.; Curaj, A.; Wijnands, E.; Lira, S.A.; Tacke, F.; Jankowski, J.; Biessen, E.A.L.; van der Vorst, E.P.C. CCR6 Deficiency Increases Infarct Size after Murine Acute Myocardial Infarction. Biomedicines 2021, 9. [CrossRef]
- Feng, G.; Bajpai, G.; Ma, P.; Koenig, A.; Bredemeyer, A.; Lokshina, I.; Lai, L.; Forster, I.; Leuschner, F.; Kreisel, D.; et al. CCL17 Aggravates Myocardial Injury by Suppressing Recruitment of Regulatory T Cells. Circulation 2022, 145, 765-782. [CrossRef]
Figure 1.
Protein profile fractions, Western blot analysis Flow cytometry analysis of EVs isolated from plasma fractions. Consecutive fractions were collected to complete 400 μL elution volume per fraction. Each combined fraction was concentrated in a centrifugal rotary evaporator at 4°C overnight and resuspended in 200 μL. (A and B) Protein elution profile for the EV-collection medium. (C) Western blot analysis for the Alix, Tgs 101 and CD9 expression of the collected fractions from column. (D). Small CD63 positive EVs were captured from each fraction 4, 5, and 6 employing Dynabeads® coated with anti-CD63 and stained with anti CD90 or anti CD81 or anti CD144 (Lef panel). Represents small EV-beads-CD63 positive staining.
Figure 1.
Protein profile fractions, Western blot analysis Flow cytometry analysis of EVs isolated from plasma fractions. Consecutive fractions were collected to complete 400 μL elution volume per fraction. Each combined fraction was concentrated in a centrifugal rotary evaporator at 4°C overnight and resuspended in 200 μL. (A and B) Protein elution profile for the EV-collection medium. (C) Western blot analysis for the Alix, Tgs 101 and CD9 expression of the collected fractions from column. (D). Small CD63 positive EVs were captured from each fraction 4, 5, and 6 employing Dynabeads® coated with anti-CD63 and stained with anti CD90 or anti CD81 or anti CD144 (Lef panel). Represents small EV-beads-CD63 positive staining.
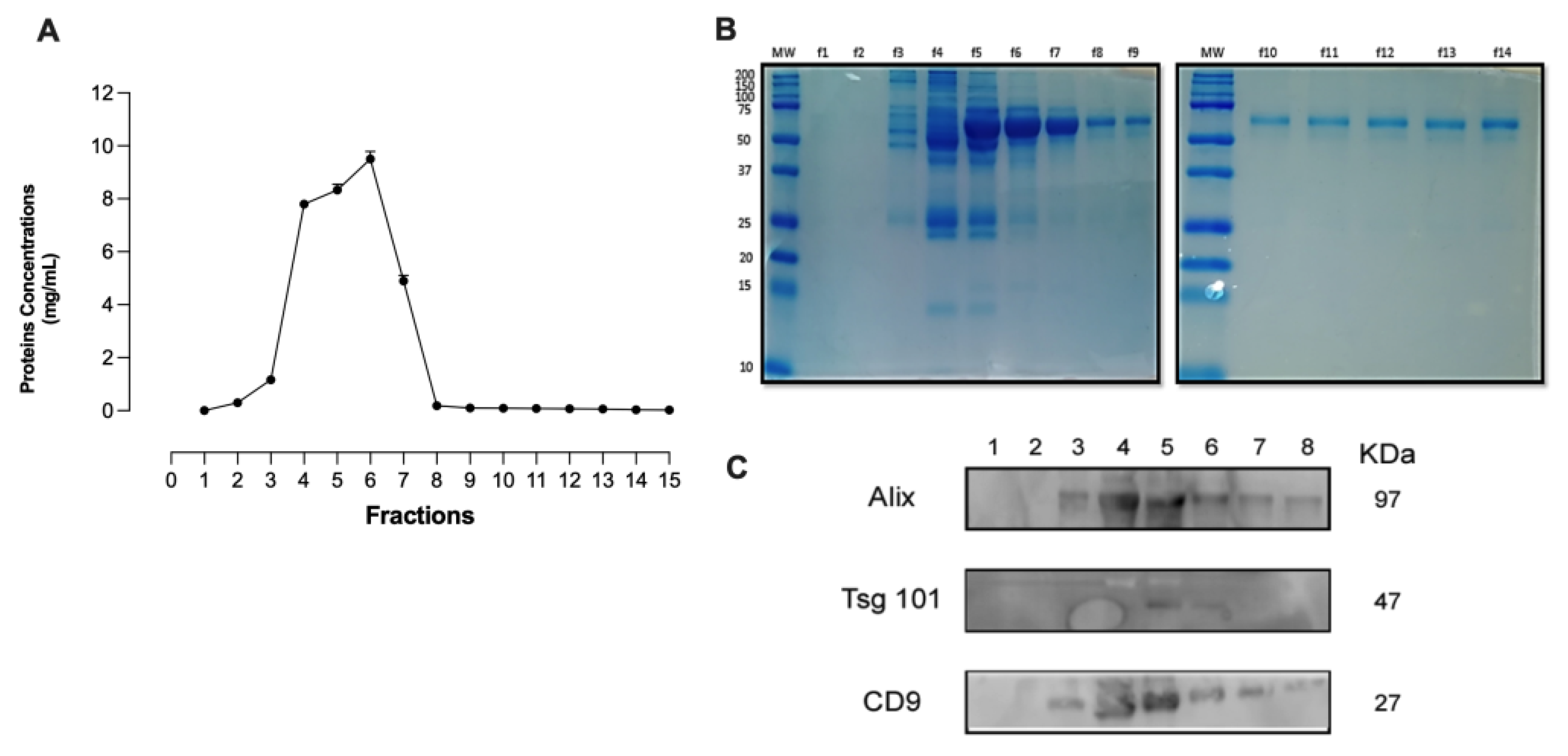
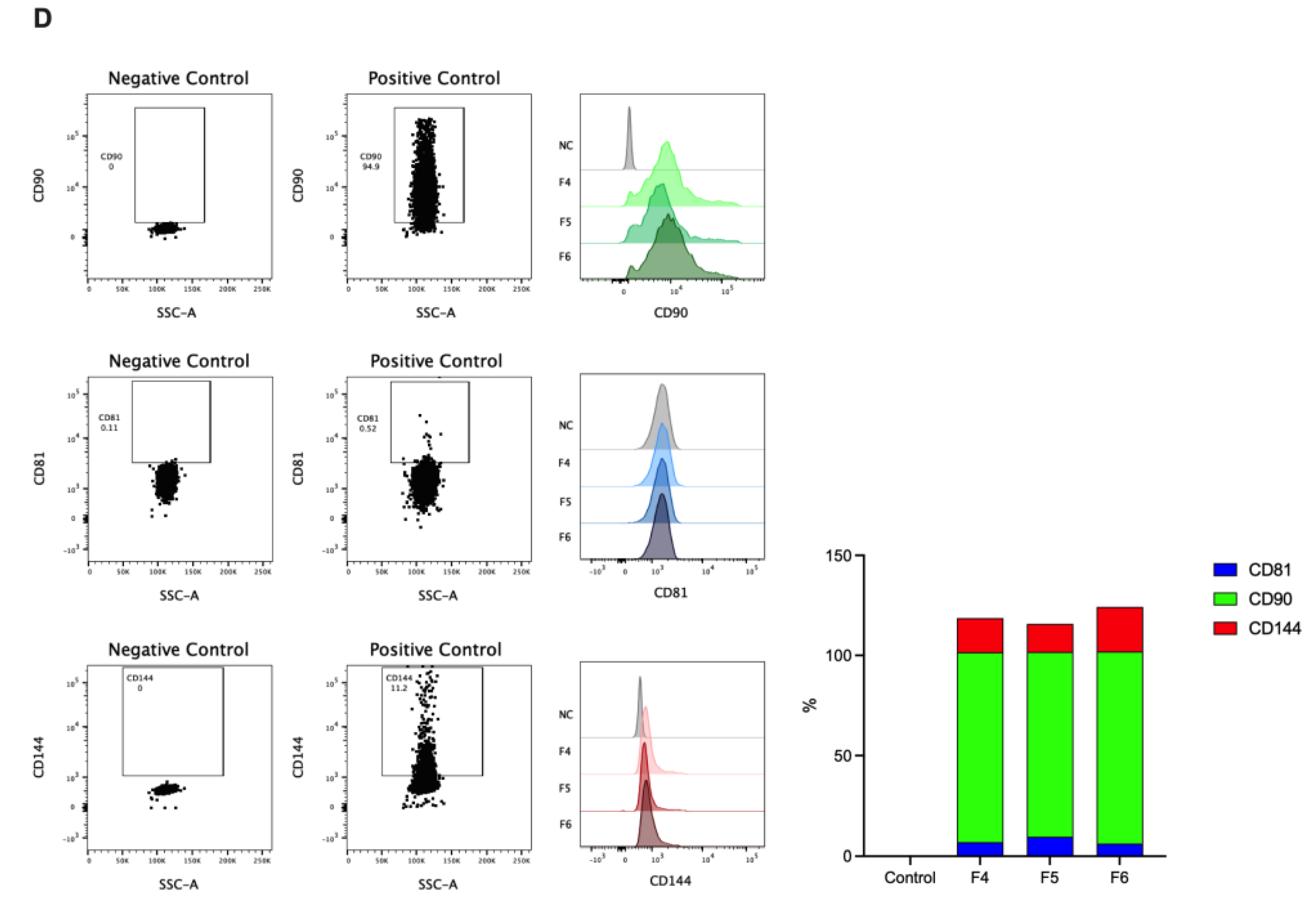
Figure 2.
Analysis of MV concentration and size distribution of EVs isolated from plasma. NTA data shows size distribution of EVs isolated from the control group (HC-P), coronary blood (MI-C), and peripheral blood (MI-P) of patients with myocardial infarction (A). Bar graphs compare the concentration, mean size, and mode size of MVs in plasma (B). In C, the size distribution of MV from plasma and serum is shown as D10, D50, and D90 values. Values are mean ± standard deviation (n = 5-8 in each group).
Figure 2.
Analysis of MV concentration and size distribution of EVs isolated from plasma. NTA data shows size distribution of EVs isolated from the control group (HC-P), coronary blood (MI-C), and peripheral blood (MI-P) of patients with myocardial infarction (A). Bar graphs compare the concentration, mean size, and mode size of MVs in plasma (B). In C, the size distribution of MV from plasma and serum is shown as D10, D50, and D90 values. Values are mean ± standard deviation (n = 5-8 in each group).
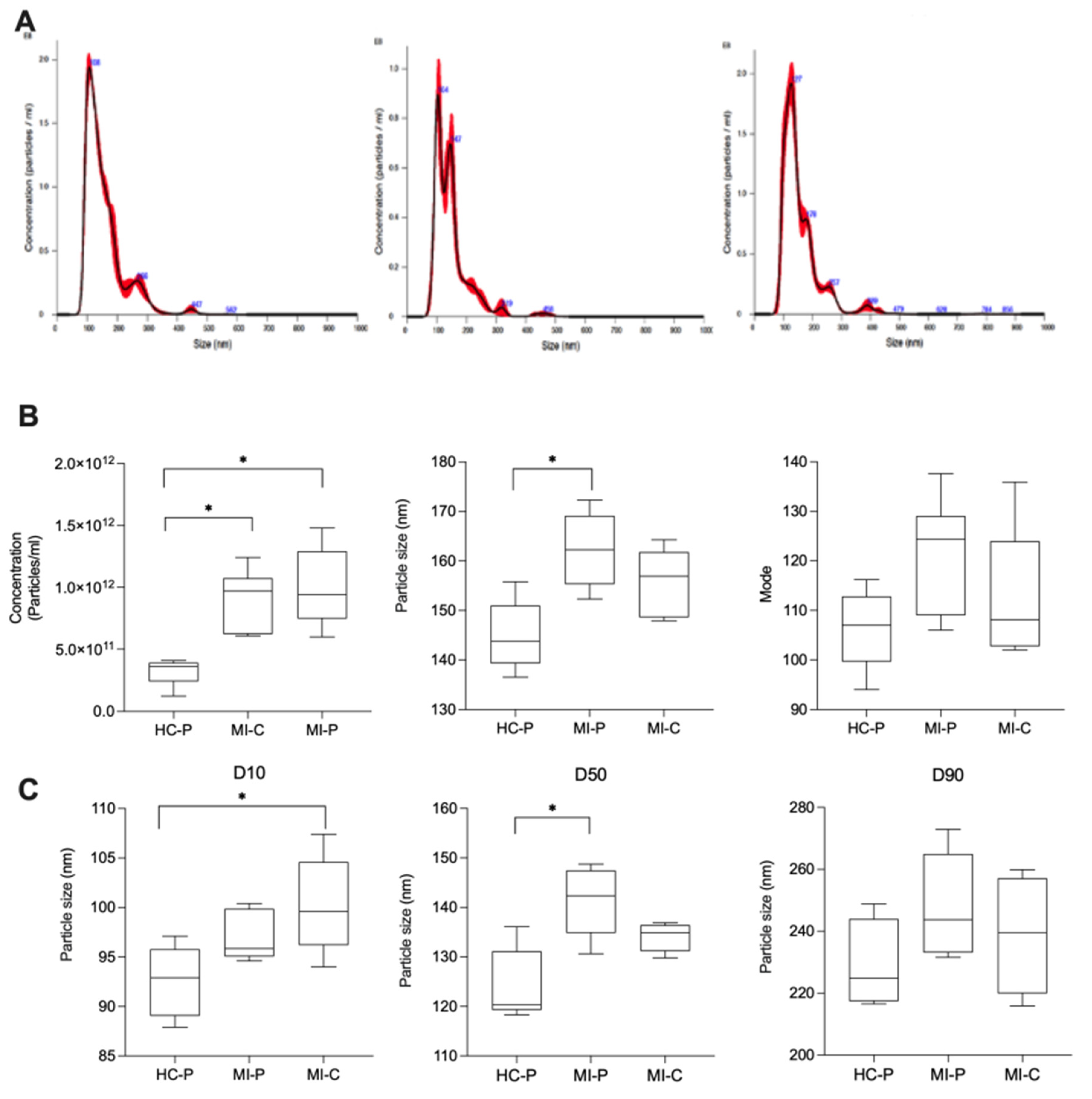
Figure 3.
Flow cytometry characterizes derived EVs from fractions of plasma. In the up panel, flow cytometry was used to identify CCR4, CCR6, or CXCR3 antigens in EVs isolated from plasma from the control group (HC-P), coronary blood (MI-C), and peripheral blood (MI-P) of a patient with myocardial infarct (A). The lower panel bar graphs compare the % of CD90, CD81, and CD144. % are mean ± standard deviation (n = 5-8 in each group).
Figure 3.
Flow cytometry characterizes derived EVs from fractions of plasma. In the up panel, flow cytometry was used to identify CCR4, CCR6, or CXCR3 antigens in EVs isolated from plasma from the control group (HC-P), coronary blood (MI-C), and peripheral blood (MI-P) of a patient with myocardial infarct (A). The lower panel bar graphs compare the % of CD90, CD81, and CD144. % are mean ± standard deviation (n = 5-8 in each group).
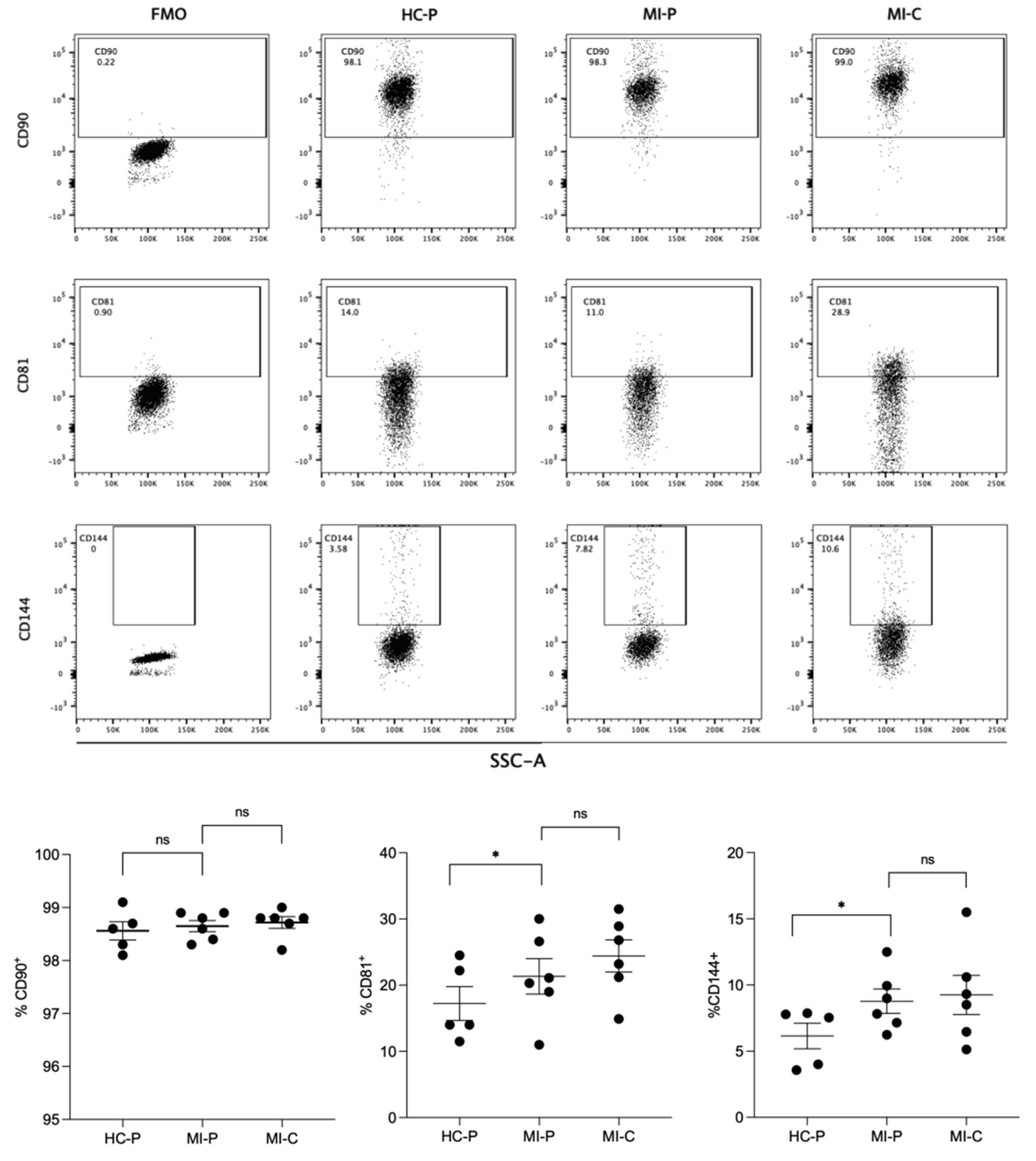
Figure 4.
Flow cytometry of chemokine receptors express EVs isolated from fraction plasma. In the up panel, flow cytometry was used to identify CCR4, CCR6, or CXCR3 antigens in fractions of plasma from the control group (HC-P), coronary blood (MI-C) and peripheral blood (MI-P) of patients with myocardial infarct. The lower panel bar graphs compare the % CCR4, CCR6, or CXCR3. % are mean ± standard deviation (n = 5-8 in each group).
Figure 4.
Flow cytometry of chemokine receptors express EVs isolated from fraction plasma. In the up panel, flow cytometry was used to identify CCR4, CCR6, or CXCR3 antigens in fractions of plasma from the control group (HC-P), coronary blood (MI-C) and peripheral blood (MI-P) of patients with myocardial infarct. The lower panel bar graphs compare the % CCR4, CCR6, or CXCR3. % are mean ± standard deviation (n = 5-8 in each group).
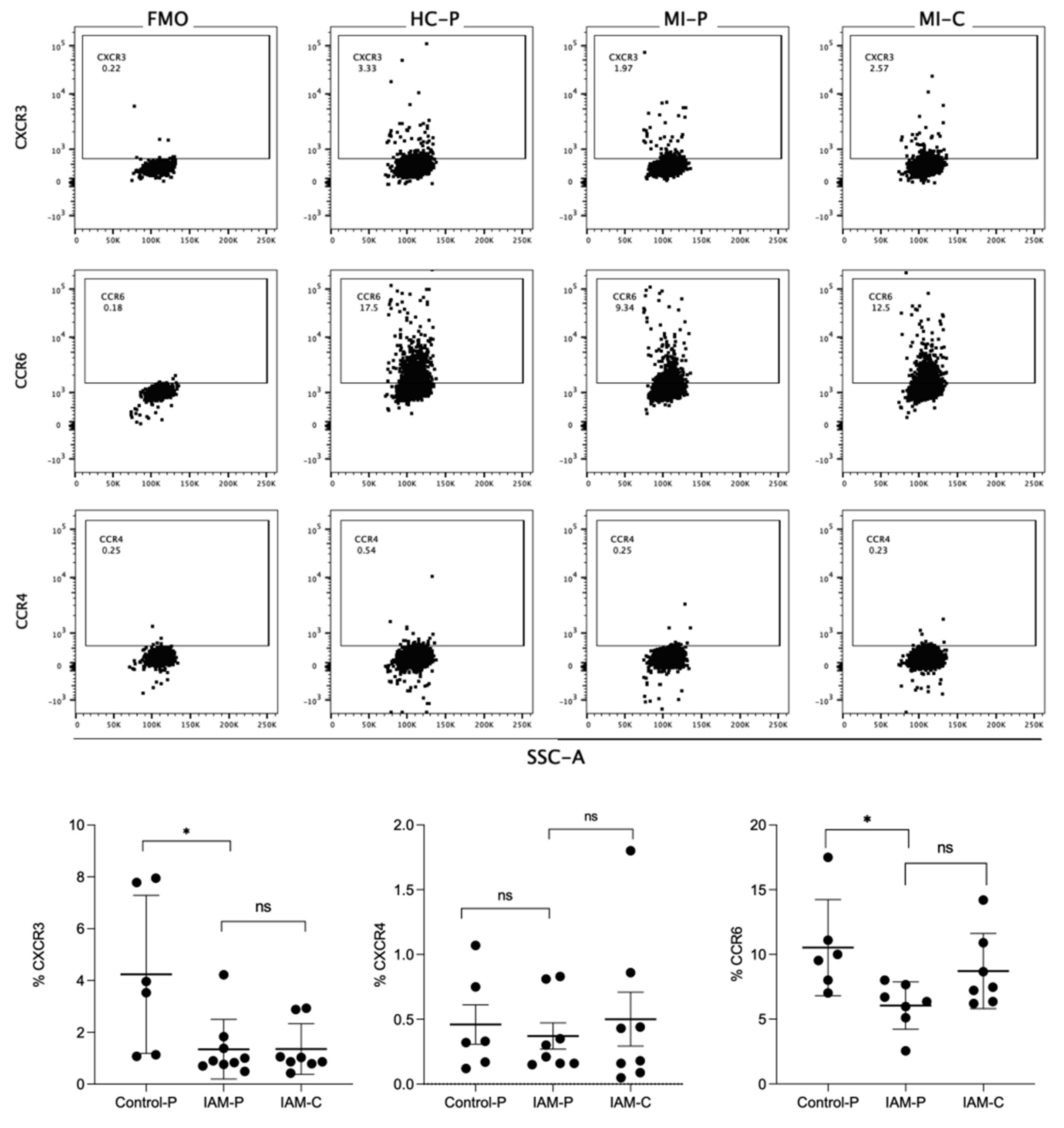

Table 1.
Clinical and biochemical characteristics from control subjects and Acute myocardial infarction (AMI) patients.
Table 1.
Clinical and biochemical characteristics from control subjects and Acute myocardial infarction (AMI) patients.
| CONTROL | IAM | |
| n | 8 | 10 |
| Gender (F/M) | 5/3 (62%/38%) | 6/4 (60%/40%) |
| Age | 60 ± 2,4 | 64,8 ± 5,1 |
| Weight (kg) | 74,9 ± 5,7 | 70,1 ± 4,2 |
| Height (m) | 1,6 ± 0,1 | 1,6 ± 1,4 |
| BMI | 29,6 ±1,9 | 26,6 ±1,2* |
| Waist Circumference (cm) | 95,5 ± 4,6 | 102,6 ± 3,3 |
| Glycemia (mg/dl) | 76,4 ± 5,6 | 176,0 ± 45,0* |
| Total Cholesterol (mg/dl) | 171,7± 14,5 | 181,5± 12,7 |
| Triglycerides | 130,4 ± 7,6 | 149,6 ± 27,9 |
| LDL Cholesterol (mg/dl) | 119,3 ± 20,8 | 107,8 ± 12,4 |
| HDL Cholesterol (mg/dl) | 38,1 ± 5,1 | 43,0 ± 3,9 |
| sLox-1 | 156,7 ± 55 | 294±70,3* |
| Troponin (ng/ml) | - | 238.262 ± 72494 |
| CK-Total (U/L) | - | 2.205 ± 677 |
| CK-Mb (ng/ml) | - | 219,8 ± 64,7 |
| CRP (mg/dl) | - | 1,6 ± 0,5 |
| Previous Myocardial Infarction | ||
| Yes | - | 0 (0%) |
| No | - | 10 (100%) |
| Diagnosis | ||
| AMI with ST-segment elevation in inferior wall | - | 5 (50%) |
| AMI with ST-segment elevation in anterior wall | - | 4 (40%) |
| AMI with ST-segment elevation in anteroseptal area | - | 1 (10%) |
| TIMI Score | ||
| I | - | 0 |
| II | - | 0 |
| III | - | 10 (100%) |
| KILLIP score | ||
| I | - | 5 (50%) |
| II | - | 5 (50%) |
| III | - | 0 (0%) |
| IV | - | 0 (0%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



