
Submitted:
15 July 2024
Posted:
16 July 2024
You are already at the latest version
Abstract
Protein 53 (P53) shows different functions either to support or to contrast malignant growth. P53 history tells that discoveries of molecular features have contributed to the emergence of P53 functions. Conversely, the shift of paradigms demonstrates that P53 functions coexist even if are in conflict with one another.
Since 1979, five paradigms have been assembled then deconstructed by taking into account P53 functions of viral protein, oncogene, tumor suppressor gene, transcription factor, apoptotic factor.
By summarizing the past five disassembled P53 paradigms, in this investigation the current paradigm involving P53 in the modulation of a composite bio-morphological entity namely, tumor immune microenvironment (TIME) is analyzed.
The aims of this investigation were to provide an overview of solid and validated knowledge occurring in all P53 paradigms and to determine P53 abilities to modify TIME through manipulation of viruses.
As a result, definitive P53 evidence concerns its genetic localization, amino acids composition, its isoforms and subcellular expression in organelles. P53 “mutome” assembles all mutations affecting this complex protein. Lastly, both categories of tumor promoting and non-oncogenic viruses related to P53 were scrutinized.
In conclusion, paradigms teach that P53 is undruggable because its drugs inevitably target all P53 functions.
Keywords:
wtP53
; P53 paradigms
; P53 mutome
; tumor immune microenvironment (TIME) cytokines
; P53-related tumor promoting virus
; P53-related non-oncogenic viruses
1. Introduction
Protein 53 (P53) is the most popular protein favoring cancer development. In almost forty-five years, the number of P53 abilities is progressively implemented by bringing to light new aspects and proprieties.
On the occasion of the thirtieth anniversary of P53 discovery, two more than complete timelines were compiled by researchers who have contributed to the discovery of P53 features (see Timeline in ref. [1] and Figure 1 in ref. [2]) [1,2]. By examining the text of timelines, it emerges that contrasting functions have been reported for P53. At large, these conflicting abilities are the contingent issues that have led to the failure of numerous P53 dogmas regarding the functions and mechanisms of action by which this protein induces cancerous proliferation.
By facing these issues, Soussi T has already called into question the model of scientific paradigms discussed by the American physicist Thomas Kuhn in the book titled “The structure of scientific revolutions” [2,3]. For the propose of reconstructing the history of the different P53 functions, Soussi T used the shift of scientific paradigms and then, he associated P53 history with the history of theories proposed concerning the onset of cancer (see Figure 1 in ref. [2]) [2].
This question is still relevant today because the mixed activities of P53 reflect the fact that currently p53 has not passed the rigorous procedures to be validated as a diagnostic or therapeutic biomarker [4]. In actuality, P53 remains substantially undruggable and hence, in the USA or Europe there are no approved cancerous therapeutic drugs targeting P53 [5,6]. Lamivutide, statins or Zoledronic acid have been used in different clinical trials (CT) to target P53 in cancerous diseases. However, these medicinal products have been approved by U.S. Food and Drug Administration for treatments of non-cancerous diseases such as infection by human immunodeficiency virus (HIV) or for control of cholesterol and Calcium blood levels [7,8].
Therefore, by archiving P53 functions through timeline makes scientific conclusions unclear because they are providing heterogeneous features. In order to understand achievements and failures of therapeutic approaches targeting P53 functions, it could be useful to overcome the line of time by reporting the sequence of P53 paradigms revolutions. Indeed, the shift of P53 paradigms is independent of time because a paradigm has an indefinite period; conversely, it depends on scientific interpretation of discoveries and then, it is determined by validity of the investigation’s conclusions (Figure 1).
Mainly, the current review provides an overview of solid and validated knowledge occurring in all P53 paradigms.
By focusing on the current P53 paradigm, this study is aimed at assessing the abilities of P53 to modulate tumor immune microenvironment (TIME) through the usage of viral nanoparticles (np). Therefore, P53 related viruses are scrutinized whether these belong to tumor promoting or are part of the non-oncogenic virus category. Then, cytokines expressed in TIME are examined. Mostly, cytokines involved in P53 immune response during thyroid cancer development are investigated.
2. P53 History: Timeline Progression Vs Paradigm Revolutions
P53 Timeline
In 1979, the P53 story starts by the publication of seven investigations [9,10,11,12,13,14,15]. Aimed to find the principles governing cancerous growth, several teams have focused their main research interests on Simian Vacuolating 40 (SV40). This is a small thermophilic non-enveloped DNA virus belonging to the polyomaviridea family, highly stable even in extremal high environmental temperature [16,17]. Then, these investigators almost concurrently described an association between SV40 large T antigen with a polypeptide of molecular weight ranging between 48-55 kDa. Mostly, this protein has a facility to accumulate in the nuclear compartment of cancerous cells.
By the report of Lane & Crawford’s team from the United Kingdom (UK), the research community was informed about a protein that came from SV40 infection and was expressed in transformed cells [9]. Almost simultaneously, “Virology” journal published a study on a polypeptide with 48-55 kDa of molecular weight that immunoprecipitated either with serum against T antigen of SV40 or with antitumor serum [10].
In a period of two months after the American team of Linzer & Levine reported a 54 kDa protein showing similar features [11]. In the same month, through immunological approaches, a 53kDa protein associated with SV40 was found from the New York team of Lloyd Old [12]. Peculiarly, only four years after the publication of this report, this investigation was included among studies that have identified P53 [18].
In August of 1979, a 55 kDa protein with comparable properties was described from the French team of Kress [13].
Lastly, in October, Smith’s team from UK found similar traits; whereas, Linzer and colleagues connected SV40 genetic product to levels of a 54 kDA protein in cells infected or transformed by virus [14,15].
In 1983, during the first P53 Workshop in UK, this protein was officially called P53 because of its purported molecular mass. This is actually a misnomer since the human P53 has a molecular weight of 43.7 kDa [1].
P53 Paradigms
By cataloging P53 functions through the revolutions of scientific paradigms, five P53 paradigms have already become established over time and then, disassembled (Figure 1).
The first function reported for P53 falls within the scope of the viral paradigm. Indeed, human P53 was correlated to SV40 only and exclusively because this virus can activate cancerous proliferation through inhibition of P53 [19,20]. Since, this correlation aimed to find the cause of cancers, the P53 immuno-pathways activated during viral infections were considered of subordinate importance. In search of the cancer origin, the viral paradigm gave way to the oncogenic paradigm.
In fact, the second paradigm incorporated the investigations pointing to demonstrate the oncogenic function of P53 (Figure 1) [21,22,23]. The reasons for which P53 was retained an oncogene were based on transfection experiments demonstrating that co-operations of P53 with oncogenes of the RAS family induced cellular transformations.
Then, it was the turn of paradigms related to P53 functions of the tumor suppressor gene, transcription factor and apoptotic factor operating without transcriptional mechanisms (Figure 1).
The functions of the tumor suppressor gene came up because of P53 expressions in cancerous cells. Since P53 could be recognized in wild-type status (wtP53) but also and especially in mutated forms (mutP53), P53 gene was included among tumor suppressor genes [24,25,26,27].
The fourth P53 paradigm inferred to the ability of P53 to act as a transcription factor. In this condition, wtP53 can promote cell cycle arrest, apoptosis and DNA repair by arranging the distribution of target genes [6,28,29,30].
Lastly, antiproliferative effects of wtP53 without using transcription established the fifth P53 paradigm [6,31,32]. Specially, in mitochondria, wtP53 displays a direct pro-apoptotic role [31,33,34,35]. Indeed, wtP53 may modulate noncoding regulatory RNA molecules such as microRNA (miR), too. Notably, by removing a large intron of 30 kilobases (Kb), wtP53 can directly target the gene encoding miR34a (miR-34a). In cancerous cellular models, this wtP53 activity produces inactivation of miR-34a by prejudicing P53 apoptosis pathway [36,37].
To note, no drugs have been developed to target the P53 functions reported in the first two paradigms. The majority of therapies targeting P53 were developed under the auspices of third paradigm settled on wtP53 function of tumor suppressor. By using different strategies, P53 medicaments were based on a single principle: to work toward restoration of tumor suppressor function to prevent the onset and spread of cancerous diseases. Therefore, myriad compounds were initially identified as useful for this work as recently reported with great detail [6,8,38,39].
On this line, P53 tumor suppressor paradigm has brought to a continuous development of strategies encompassing from immuno to gene therapies by including also personalized drugs targeting selectively one mutP53 [6]. The main principle underpinning this type of therapeutic approach derived by the ability of wtP53 and its own mut forms to be immunogenic molecules [40]. Indeed, specific anti-P53 antibodies and P53 antigen-specific T cells were identified in patients affected by cancer [41]. More deeply, the working principle of these therapeutic approaches is the same that has led to the results reported from the team of Lloyd Old in 1979. Therefore, in 45 years, the principles have remained unchanged whereas the agents with which to target P53 immune-structures have been replaced.
3. What Remains of the Five P53 Disassembled Paradigms: Evidences, Functions and Hypotheses
Though paradigms shifted, some P53 evidence were anyhow dragged in subsequent paradigms and so, they are definitive data. However, by re-assembling in the context of a new paradigm, these ultimate evidences are able to generate new functions.
At this time, there are definitive evidences about P53 genetic localization, number of residues of amino acids (aa) composing wtP53, and its expression in subcellular organelles [34,35,42]. Conversely, wtP53 functional domains and their nano- structures are still to be defined [35,43,44,45].
By consisting of 20Kb, P53 gene is placed in correspondence to the short arm of chromosome 17 at 13.1 locus (Figure 2). By spanning from 7,687,490-7,668,421 loci, P53 gene is built by a sequence of 19,070 bp that includes 11 exons and 10 introns [46,47] (Figure 2). The fourth exon is highly conserved during the evolution; however, it manifests haplotype diversity either in Caucasians, Africans or Asians [47,48].
P53 gene encodes a sequence of 393 aa showing several functional domains [49]. By taking into account recent data coming up about P53 human transcript, wtP53 aa sequence can be subdivided in five, six or even in seven functional domains (Figure 2) [35,43,44,45].
Nevertheless, from N-terminal to C-terminal of P53 aa sequence, there can be comprehensively found two transactivation domains (TAD), namely, TAD-1 (1-39 aa) and TAD-2 (40-60 aa), a proline-rich domain (PRD, 61-93), a DNA-binding domain (DBD, 94-292), a hinge domain (HD, 293-322 aa), an oligomerization domain also named tetramerization domain (OD, TET, 323-355), and C-terminal regulatory domain (CTR, 356-393) (see Figure 1 in Ref. [35]) [35]. Differences in subdivision of wtP53 domains are caused by TAD segment (1-60 aa) because it is composed by TAD-1 and TAD-2 domains (Figure 2). Further, there are three segments for which the number of residues is not overlapping in recent data [35,43,44,45]. The first segment is corresponding to 94-102 aa sequence because it can even be included in DBD domain (Figure 2). The second segment is encompassing from 294 to 322 aa, it makes up HD domain and it is alternatively namely “linker” [44] (Figure 2). This sequence counts about 29 aa that can even be included in TD/OD/TET domain (Figure 2). Indeed, in this region, Bakker MJ et al. have reported a pre-tetramerization loop composed by residues, ranging between 292 and 325 aa and corresponding to an intrinsically disordered region (IDR) required by tetramerization mechanism (see Figure 1. In ref. [45]) [45]. Computational techniques and molecular dynamics have provided to show the flexibility of IDR. Transmission electron microscopy images have supplied IDR nanostructure by showing that tridimensional conformation varies [45,50]. Machine learning techniques have contributed to demonstrate that IDR conformational landscape is mainly altered by restricting the end terminals [8,45].
The third segment spans from 354 to 364 aa, it can be included in CTD/CTR/REG domain; it is a part of IDR since it allows the p53 monomers to link, in so far, it increases DNA binding affinity (Figure 2) [45,51].
In cellular compartments, P53 can be equally found in both the nucleus and cytoplasm. In fact, cytoplasmatic organelles such as mitochondria, lysosomes and endoplasmic reticulum may express P53, too [52,53,54,55]. To note that in the cytoplasm, wtP53 retains its exonuclease activity that matches with that endonuclease in in vitro biochemical analysis [34,56].
Furthermore, it is now known that P53 has two homologs namely, P63 and P73, respectively. The first, is located on the long arm of chromosome 3 at q 27-29 region; whereas the second, lies on the short arm of chromosome 1 at p36 loci [57,58]. P53 and its own homologs show a quite similar sequence for the genomic trait concerning the DNA binding domain. For this reason, their protein products are included among the P53 protein family. However, the high similarity of sequences does not guarantee overlapping functions [59].
On the other hand, twelve isoforms of P53 aminoacidic sequence have been identified namely FLP53, P53β, P53γ, Δ40α, Δ40β, Δ40γ, Δ133α, Δ133β, Δ133γ, Δ160α, Δ160β and Δ160γ, respectively [60]. These proteins are particularly abundant in cancerous tissues pertaining to several organs such as thyroid and head neck [61,62].
Since P53 isoforms play a role to modulate P53 expression, these are the best candidates as regulator molecules by tissue specific mode [63]. This is especially due to expressions of P53 isoforms that appear tissue and cancer specific. In fact, P53 isoforms show a distinct subcellular localization in connection to benign growth or malignant histotypes [61,62,64].
The high genomic instability of P53 has been unequivocally proven since more than 2000 different forms of mutP53 proteins have been screened [65]. Indicatively, P53 alterations are predominantly missense mutations that may determine either loss or gain of function (LOF and GOF, respectively) as well as inhibition of wtP53 allele that is indicated as mutation by dominant-negative inhibition (DNI). However, this categorization of P53 mutations results insufficient to include the wide spectrum of mutP53 and their own distinctive work. In fact, some mutants showing GOF activity may retain DNI ability [65].
Hence, the concept of “mutome” has been associated to mutP53 to explain the remarkable functional differences among P53 mutated forms [66]. P53 “mutome” is a new notion of complex protein that picks up old and novel oncogenic P53 functions by including tumor progression, metastatic growth and drug resistant effects [65,66,67].
At large, P53 mutations are considered as variable categories potentially associated each in order to identify P53 molecular diseases. By demonstrating that mutp53 forms contribute in varying degrees to regulate P53 functions, the P53 molecular diseases have been distinguished in P53 caused diseases and P53 linked diseases [65]. The concept of P53 “mutome” has the greatest achievement of enclosing all known as well as unknown P53 mutations in a unique categorization since the genetic alterations are inter-dependent with each other [65,66,67].
4. The Last P53 Paradigm
A new P53 paradigm is currently assembling on the basis of P53 abilities to modulate cytokines and growth factors during cancer development (Figure 1). In fact, P53 shows features of immunomodulator agent and it plays a role in the regulation of TIME [68,69].
TIME is a complex bio-morphological entity built up by distinct cellular types, molecules such as cytokines and np.
Morphologically, TIME is identified as extracellular “stroma” of cancerous proliferations and it includes cells such as fibroblasts, fibrocytes, immune-cells, extracellular matrix and blood vessels [68,70,71,72].
Thyroid cellular growth could represent an ideal model to individualize the role of TIME in the context of both benign and malignant proliferations [73,74].
In brief, the stromal capsule plays a crucial role to distinguish benign from malignant thyroid proliferations. Further, the absence of a stromal capsule in non-goitrogenic hyperplastic nodules allows to distinguish these lesions from benign adenomas. This distinction is highly significant because the absence of fibrosis ensures complete healing of hyperplastic lesions. Conversely, thyroid adenomas provided by capsule do not spontaneously heal.
During thyroid benign and malignant cellular growth, TIME cells share propensity to express cytokines such as interleukin 6 (IL-6), ligands for tumor necrosis factors such as CD30 ligand (CD30L) and even a mesenchyme-derived pleiotropic cytokine namely, hepatocyte growth factor (HGF) [75,76,77]. In ex vivo thyroid cancer tissues, cellular and subcellular localization of IL-6, CD30L and HGF have been identified [76,78]. Indeed, for some time now these molecules have been detected in stroma/TIME associated with thyroid cancerous proliferations belonging to follicular cells [76,78]. HGF, IL-6 and CD30L work as ligands for receptors such as c-met protooncogene, IL-6 receptor (IL-6R) and CD30, or antigen K1 [76,78]. By taking into consideration the expression of cognate receptors, the distribution of these ligands was evaluated in the context of stroma/TIME and in carcinomatous cells. As a result, immune-expressions of ligands were arranged according to those of the receptors: ligands were specially detected in stroma/TIME; whereas, receptors were found in follicular epithelial cells [76,78]. More deeply, the passage from the benign cellular growth to malignant proliferations was marked by a change in distribution of ligands [76,78]. Peculiarly, IL-6, CD30L and HGF settlements transited from stroma/TIME to epithelial cells. In fact, these ligands were found more frequently in malignant follicular cells (see Figure 1 in ref. [76] and [78]) [76,78].
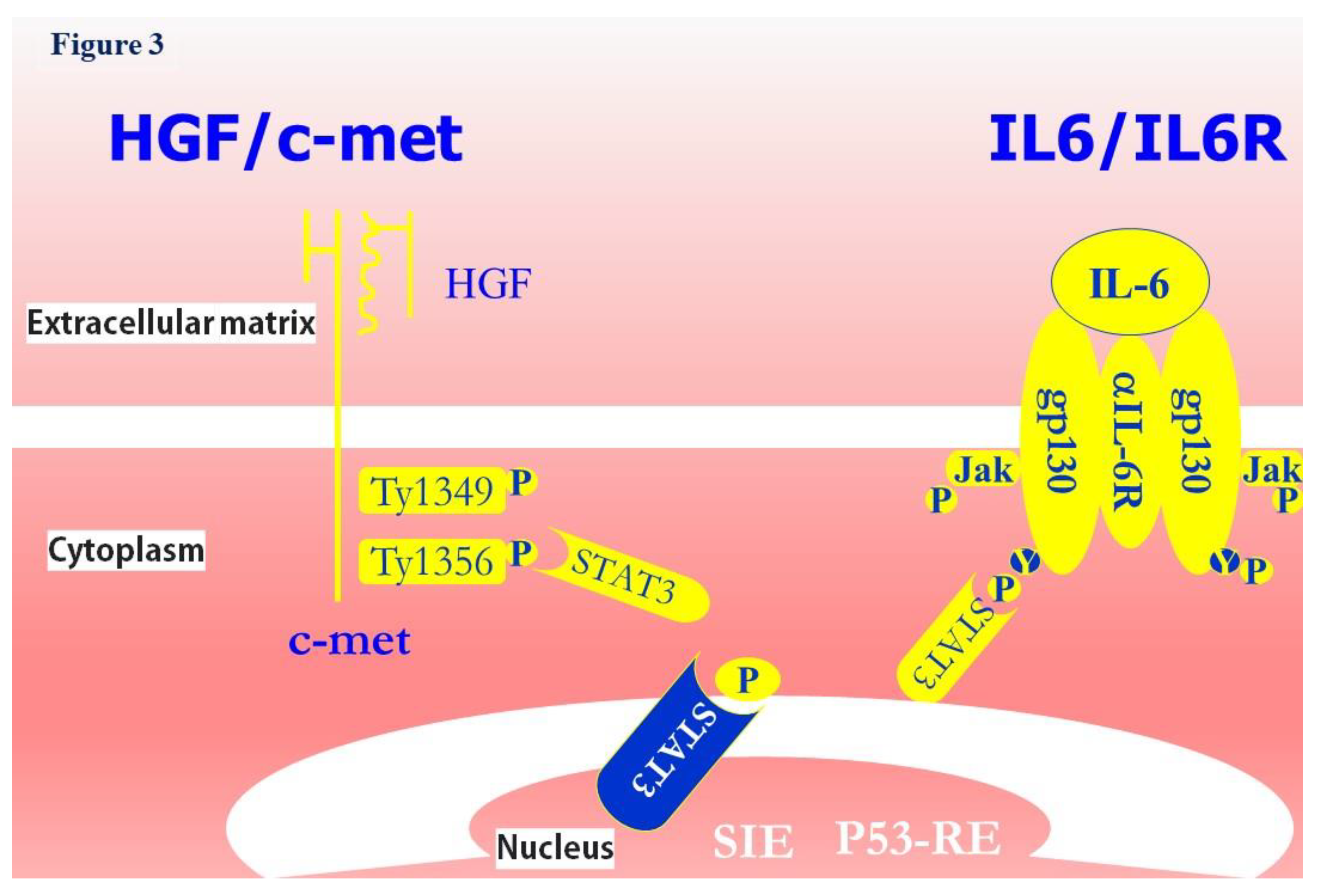
The role of wtP53 in TIME associated with c-met, CD30 and IL-6 has been broadly reported in cancerous cells [72,79,80,81,82]. Therefore, in view of new therapeutic targets, the translational approaches of wP53 functions in TIME milieu have to consider the expressions of these cytokines. In support of this argument there are HGF and IL-6 effects mediated by signal transducer and activator of transcription (STAT), specially STAT3 [83,84,85,86].
Both HGF/c-met and IL-6/IL-6R axes can recruit STAT3 effector (Figure 3). HGF/c-met axis is initiated by the interaction of receptor and ligand which is also present in extracellular matrix [78,83,84]. The heterodimeric tyrosine kinase c-met is auto-phosphorylated for which it provides binding sites for molecules showing SH2 groups. By acting as intracellular transducers, these groups recruit and phosphorylate cytoplasmatic STAT3 that migrates into the nucleus (Figure 3) [83,84]. Even IL-6/IL-6R axis is initiated by the interaction of receptor and ligand which is present in extracellular matrix [76,85]. After binding IL-6R, IL-6 induces homodimerization and autophosphorylation of gp-130, therefore, Janus protein tyrosine kinases are activated and STAT 3 is phosphorylated. In such form, STAT3 transits from the cytosol to the nucleus [85] (Figure 3). Once inside the nucleus, STAT3 can bind sis-inducible elements but also a P53 responsive element (P53-RE) [83,84,85,86] (Figure 3).
By phosphorylation STAT3 migrates to nucleus and subsequently it binds SIE to induce cellular morphogenetic response. Further, STAT3 can bind a P53 promoter Abbreviations. HGF: hepatocyte growth factor; c-met: mesenchymal epithelial transition factor; IL6: interleukin 6; IL6R: interleukin 6 receptor; gp130: glycoprotein 130; JAK: Janus chinasi; STAT3: signal transducer and activator of transcription 3; SIE: sis-inducible element; P53-RE: P53 responsive element.
STAT3 orchestration by HGF/c-met and IL-6/IL-6R axes have been also proved through simultaneous cellular expression of these molecules in malignant and benign proliferation pertaining to thyroid gland and pituitary adenomas [87,88]. Further, HGF/c-met/STAT3 pathway is expressed in colorectal tumors [89].
5. Time Viral Np: Tumor Promoting And Non-Oncogenic Viruses
Among np isolated in TIME, viral strings of acid nucleic are of great importance. This is because these np are able to drive cells toward growth or apoptosis through P53 manipulation [72,90,91,92].
By finding viral np in TIME, the first P53 paradigm is re-evaluating and thereby, P53 pathways activated via viral infections are re-assessing, too by the scientific outlook of P53 TIME paradigm [72,93,94,95].
Actually, both categories of tumor promoting and non-oncogenic viruses are associated with wtP53 [94,95].
Among tumor promoting viruses, in particular DNA viruses, such as SV40 and human papilloma viruses, have been related to P53 [91,96]. Conversely, DNA, RNA and even retro-viruses are listed in the category of non-oncogenic viruses associated with wtP53 [94,95]. Human herpes simplex virus-1(HSV-1) and three members belonging to the poxvirus family namely, smallpox, vaccinia and Tanapox viruses, are recorded in the category of non-oncogenic DNA viruses [94,97,98,99]. Among non-oncogenic RNA viruses, wtP53 is relevant to the Influenza A virus (IAV) and members of the Flavivirus family such as Zika virus (ZIKV) and the west Nile viruses [94,100,101,102]. Lastly, lentivirus HIV-1 of the retrovirus family can interact with wtP53, too [103].
By rejoining strategies that viruses put in play for viral replication, survival and spreading, it is irrefutable that viruses manipulate wtP53 for their own support. However, now it is also appearing quite clearly that wtP53 can stop viral spreading by molecular remodeling of TIME molecules [72].
To induce cancerous proliferation, viruses manipulate P53 through three pathways. Firstly, DNA tumor promoting viruses use transcription mechanisms that host cell employs to replicate DNA [94]. In this way, an aberrant entrance in S phase is aided that will be promptly annulled by wtP53 that promotes apoptosis [104]. However, by binding wtP53, these viruses are going right to inactivate wtP53, the reason why the host cells grow abnormally. Secondly, oncogenic viruses can use “DNA mimicry”. By this option, SV40 inhibits P53 transcription since P53-RE are included in T antigen [1,105,106]. Therefore, P53 transcriptional regulation is repressed and then, cells move toward an uncontrolled proliferation [105]. On this road, wtP53 may have a “helper” function for the benefit of SV40 due to providing transcriptional promoting proteins [107], Lastly, SV40 can activate “p53 host defense mechanisms”, too [94]. This pathway is based on a competitive binding between P53 and a host protein, namely Sp1, which is crucial for SV40 assembly [94]. By this direction it might explain why in the first phase of infection, wtP53 is detected in cells in which SV40 is unexpressed.
Different pathways are activated by non-oncogenic viruses associated with P53. In regard to HSV-1, wtP53 plays two conflicting roles. In fact, wtP53 can support efficient HSV-1 replication but can even reduce its expression [97]. Conversely, to gain advantage to proliferate, non-oncogenic DNA poxviruses downregulate wtP53 [94]. In fact, by exploiting either phosphorylation or acetylation pathways these produce reduction of wtP53 expression [94].
In the course of infection by IAV and West Nile virus, wtP53 can act both as an antiviral and as a promoter of viral infection [94]. The relationship between wtP53 and ZIKV remains constantly monitored, especially because this virus has been associated with increasing incidence of congenital microcephaly (CM). Indeed, wtP53 has already been related to abnormal development of the nervous system (NS) in animal models and human fetuses [101,108].
In animal models, Trp53 knockout mice developed abnormalities in the neural tube [109]. Human fetuses affected by NS malformations showed reduced amounts of wtP53 in both NS including dysplastic neuronal cells and placenta; whereas, high wtP53 amounts were detected in non-malformed organs [108]. As a consequence, this organ-specific distribution of wtP53 is indicative of wtP53 role in normal fetal development of SN [108].
Depending on the infection stage, HIV-1 turns wtP53 abilities to its advantage. HIV-1 can contribute to wtP53 suppression; however, it can also facilitate wtP53 activation [103]. In the early phase of infection, HIV-1promotes wtP53 suppression by interaction of nanostructures belonging to both HIV-1 and wtP53, therefore, the viral transcription is improved [94]. In the later stage, HIV-1 induces wtP53 expression without interacting through wtP53 [94]. In fact, to support its own viral replication, HIV-1 prevents binds among wtP53 and its inhibitors [110].
This has several relapses in cancerous theories because P53 may arrest the cancerous proliferation by TIME activation. Further it has pharmacological implications because of viral therapeutic agents should be useful in both infective and cancerous diseases. Indeed, Lamivutide was in a clinical trial (CT) to cure cancerous lesions. Essentially, the CT namely, NCT03144804 was designed by Lamivutide usage for metastatic colorectal cancer. To date, this phase 2 study was completed showing as a result that Lamivutide should help to prevent the growth and spread of cancerous cells.
6. Conclusion And PerspectiveS
P53 is a multifunctional aminoacidic sequence that shows different functions in malignant proliferation and in viral inflammatory diseases.
The lesson of paradigms teaches P53 functions can’t be catalogued in a table as independent discoveries. In fact, the shift of paradigms displays that P53 functions are synchronous, even if they appear discordant. P53 paradigms are independent by time: information, systematically examined and quantified in each paradigm, can be re-used in the future in a new context for new usages. Further, the interpretation of the tremendous data reporting P53 functions through paradigms explains why P53 is actually undruggable. This is because drugs inevitably target all P53 functions to a greater or lesser degree. The future prospectives concern P53 translational researches to find medicaments able to modulate multiple P53 functions to achieve the desired effects.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: title; Table S1: title; Video S1: title.
Author Contributions
Each author has made substantial contributions to the conception of the work; interpretation of data and has approved the submitted version and agrees to be personally accountable for the author’s own contributions and for ensuring that questions related to the accuracy or integrity of any part of the work.
Conflicts of Interest
The authors have read and confirmed their agreement with the MDPI authorship and conflict of interest criteria. The authors also confirm this article is unique and not under consideration or published in any other publication. The authors have nothing to disclose.
Appendix A
The appendix is an optional section that can contain details and data supplemental to the main text—for example, explanations of experimental details that would disrupt the flow of the main text but nonetheless remain crucial to understanding and reproducing the research shown; figures of replicates for experiments of which representative data is shown in the main text can be added here if brief, or as Supplementary data. Mathematical proofs of results not central to the paper can be added as an appendix.
Appendix B
All appendix sections must be cited in the main text. In the appendices, Figures, Tables, etc. should be labeled starting with “A”—e.g., Figure A1, Figure A2, etc.
References
- Levine, A.J.; Oren, M. The First 30 Years of P53: Growing Ever More Complex. Nat Rev Cancer 2009, 9, 749–758. [CrossRef]
- Soussi, T. The History of P53: A Perfect Example of the Drawbacks of Scientific Paradigms. EMBO Reports 2010, 11, 822–826. [CrossRef]
- Kuhn, T.S.; Hacking, I. The Structure of Scientific Revolutions; Fourth edition.; The University of Chicago Press: Chicago ; London, 2012; ISBN 9780226458113.
- Trovato, M. Update on International Medical Taxonomies of Biomarkers and Their Applications in Management of Thyroid Cancers. Diagnostics 2022, 12, 662. [CrossRef]
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-Function Mutant P53 in Cancer Progression and Therapy. Journal of Molecular Cell Biology 2020, 12, 674–687. [CrossRef]
- Hassin, O.; Oren, M. Drugging P53 in Cancer: One Protein, Many Targets. Nat Rev Drug Discov 2023, 22, 127–144. [CrossRef]
- Nishikawa, S.; Iwakuma, T. Drugs Targeting P53 Mutations with FDA Approval and in Clinical Trials. Cancers 2023, 15, 429. [CrossRef]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting P53 Pathways: Mechanisms, Structures, and Advances in Therapy. Sig Transduct Target Ther 2023, 8, 92. [CrossRef]
- Lane, D.P.; Crawford, L.V. T Antigen Is Bound to a Host Protein in SY40-Transformed Cells. Nature 1979, 278, 261–263. [CrossRef]
- Melero, JoséA.; Stitt, D.T.; Mangel, W.F.; Carroll, R.B. Identification of New Polypeptide Species (48–55K) Immunoprecipitable by Antiserum to Purified Large T Antigen and Present in SV40-Infected and -Transformed Cells. Virology 1979, 93, 466–480. [CrossRef]
- Linzer, D.I.H.; Levine, A.J. Characterization of a 54K Dalton Cellular SV40 Tumor Antigen Present in SV40-Transformed Cells and Uninfected Embryonal Carcinoma Cells. Cell 1979, 17, 43–52. [CrossRef]
- DeLeo, A.B.; Jay, G.; Appella, E.; Dubois, G.C.; Law, L.W.; Old, L.J. Detection of a Transformation-Related Antigen in Chemically Induced Sarcomas and Other Transformed Cells of the Mouse. Proc. Natl. Acad. Sci. U.S.A. 1979, 76, 2420–2424. [CrossRef]
- Kress, M.; May, E.; Cassingena, R.; May, P. Simian Virus 40-Transformed Cells Express New Species of Proteins Precipitable by Anti-Simian Virus 40 Tumor Serum. J Virol 1979, 31, 472–483. [CrossRef]
- Smith, A.E.; Smith, R.; Paucha, E. Characterization of Different Tumor Antigens Present in Cells Transformed by Simian Virus 40. Cell 1979, 18, 335–346. [CrossRef]
- Linzer, D.I.H.; Maltzman, W.; Levine, A.J. The SV40 a Gene Product Is Required for the Production of a 54,000 MW Cellular Tumor Antigen. Virology 1979, 98, 308–318. [CrossRef]
- Buck, C.B.; Van Doorslaer, K.; Peretti, A.; Geoghegan, E.M.; Tisza, M.J.; An, P.; Katz, J.P.; Pipas, J.M.; McBride, A.A.; Camus, A.C.; et al. The Ancient Evolutionary History of Polyomaviruses. PLoS Pathog 2016, 12, e1005574. [CrossRef]
- Ehlers, B.; Anoh, A.E.; Ben Salem, N.; Broll, S.; Couacy-Hymann, E.; Fischer, D.; Gedvilaite, A.; Ingenhütt, N.; Liebmann, S.; Martin, M.; et al. Novel Polyomaviruses in Mammals from Multiple Orders and Reassessment of Polyomavirus Evolution and Taxonomy. Viruses 2019, 11, 930. [CrossRef]
- Crawford, L. The 53,000-Dalton Cellular Protein and Its Role in Transformation. Int Rev Exp Pathol 1983, 25, 1–50.
- Wolf, D.; Rotter, V. Inactivation of P53 Gene Expression by an Insertion of Moloney Murine Leukemia Virus-Like DNA Sequences. Molecular and Cellular Biology 1984, 4, 1402–1410. [CrossRef]
- Mietz, J.A.; Unger, T.; Huibregtse, J.M.; Howley, P.M. The Transcriptional Transactivation Function of Wild-Type P53 Is Inhibited by SV40 Large T-Antigen and by HPV-16 E6 Oncoprotein. The EMBO Journal 1992, 11, 5013–5020. [CrossRef]
- Eliyahu, D.; Raz, A.; Gruss, P.; Givol, D.; Oren, M. Participation of P53 Cellular Tumour Antigen in Transformation of Normal Embryonic Cells. Nature 1984, 312, 646–649. [CrossRef]
- Jenkins, J.R.; Rudge, K.; Currie, G.A. Cellular Immortalization by a cDNA Clone Encoding the Transformation-Associated Phosphoprotein P53. Nature 1984, 312, 651–654. [CrossRef]
- Parada, L.F.; Land, H.; Weinberg, R.A.; Wolf, D.; Rotter, V. Cooperation between Gene Encoding P53 Tumour Antigen and Ras in Cellular Transformation. Nature 1984, 312, 649–651. [CrossRef]
- Wolf, D.; Rotter, V. Major Deletions in the Gene Encoding the P53 Tumor Antigen Cause Lack of P53 Expression in HL-60 Cells. Proc. Natl. Acad. Sci. U.S.A. 1985, 82, 790–794. [CrossRef]
- Eliyahu, D.; Goldfinger, N.; Pinhasi-Kimhi, O.; Shaulsky, G.; Skurnik, Y.; Arai, N.; Rotter, V.; Oren, M. Meth A Fibrosarcoma Cells Express Two Transforming Mutant P53 Species. Oncogene 1988, 3, 313–321.
- Baker, S.J.; Fearon, E.R.; Nigro, J.M.; Hamilton, S.R.; Preisinger, A.C.; Jessup, J.M.; vanTuinen, P.; Ledbetter, D.H.; Barker, D.F.; Nakamura, Y.; et al. Chromosome 17 Deletions and P53 Gene Mutations in Colorectal Carcinomas. Science 1989, 244, 217–221. [CrossRef]
- Finlay, C.A.; Hinds, P.W.; Levine, A.J. The P53 Proto-Oncogene Can Act as a Suppressor of Transformation. Cell 1989, 57, 1083–1093. [CrossRef]
- Kastan, M.B.; Canman, C.E.; Leonard, C.J. P53, Cell Cycle Control and Apoptosis: Implications for Cancer. Cancer Metast Rev 1995, 14, 3–15. [CrossRef]
- Fridman, J.S.; Lowe, S.W. Control of Apoptosis by P53. Oncogene 2003, 22, 9030–9040. [CrossRef]
- Williams, A.B.; Schumacher, B. P53 in the DNA-Damage-Repair Process. Cold Spring Harb Perspect Med 2016, 6, a026070. [CrossRef]
- Speidel, D. Transcription-Independent P53 Apoptosis: An Alternative Route to Death. Trends in Cell Biology 2010, 20, 14–24. [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting P53 in Context. Cell 2017, 170, 1062–1078. [CrossRef]
- Moll, U.M.; Marchenko, N.; Zhang, X. P53 and Nur77/TR3 – Transcription Factors That Directly Target Mitochondria for Cell Death Induction. Oncogene 2006, 25, 4725–4743. [CrossRef]
- Ho, T.; Tan, B.X.; Lane, D. How the Other Half Lives: What P53 Does When It Is Not Being a Transcription Factor. IJMS 2019, 21, 13. [CrossRef]
- Shen, J.; Wang, Q.; Mao, Y.; Gao, W.; Duan, S. Targeting the P53 Signaling Pathway in Cancers: Molecular Mechanisms and Clinical Studies. MedComm 2023, 4, e288. [CrossRef]
- Raver-Shapira, N.; Marciano, E.; Meiri, E.; Spector, Y.; Rosenfeld, N.; Moskovits, N.; Bentwich, Z.; Oren, M. Transcriptional Activation of miR-34a Contributes to P53-Mediated Apoptosis. Molecular Cell 2007, 26, 731–743. [CrossRef]
- Chang, T.-C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of miR-34a by P53 Broadly Influences Gene Expression and Promotes Apoptosis. Mol Cell 2007, 26, 745–752. [CrossRef]
- Tuval, A.; Strandgren, C.; Heldin, A.; Palomar-Siles, M.; Wiman, K.G. Pharmacological Reactivation of P53 in the Era of Precision Anticancer Medicine. Nat Rev Clin Oncol 2024, 21, 106–120. [CrossRef]
- Liu, Y.; Su, Z.; Tavana, O.; Gu, W. Understanding the Complexity of P53 in a New Era of Tumor Suppression. Cancer Cell 2024, 42, 946–967. [CrossRef]
- Levine, A.J. P53 and The Immune Response: 40 Years of Exploration—A Plan for the Future. IJMS 2020, 21, 541. [CrossRef]
- Pedersen, A.E.; Stryhn, A.; Justesen, S.; Harndahl, M.; Rasmussen, S.; Donskov, F.; Claesson, M.H.; Pedersen, J.W.; Wandall, H.H.; Svane, I.M.; et al. Wildtype P53-Specific Antibody and T-Cell Responses in Cancer Patients. Journal of Immunotherapy 2011, 34, 629–640. [CrossRef]
- The TP53 database. Available online: https://tp53.isb-cgc.org/ (accessed on 01 July 2024).
- Nishimura, M.; Takizawa, Y.; Nozawa, K.; Kurumizaka, H. Structural Basis for P53 Binding to Its Nucleosomal Target DNA Sequence. PNAS Nexus 2022, 1, pgac177. [CrossRef]
- Gregory, E.; Daughdrill, G.W. Sequence Properties of an Intramolecular Interaction That Inhibits P53 DNA Binding. Biomolecules 2022, 12, 1558. [CrossRef]
- Bakker, M.J.; Sørensen, H.V.; Skepö, M. Exploring the Role of Globular Domain Locations on an Intrinsically Disordered Region of P53: A Molecular Dynamics Investigation. J. Chem. Theory Comput. 2024, 20, 1423–1433. [CrossRef]
- https://www.ncbi.nlm.nih.gov/nuccore/NC_000017.11?report=fasta&from=7668421&to=7687490&strand=true (accessed on 01 July 2024).
- Belyi, V.A.; Ak, P.; Markert, E.; Wang, H.; Hu, W.; Puzio-Kuter, A.; Levine, A.J. The Origins and Evolution of the P53 Family of Genes. Cold Spring Harbor Perspectives in Biology 2010, 2, a001198–a001198. [CrossRef]
- Ruggeri, R.M.; Vicchio, T.M.; Giovinazzo, S.; Certo, R.; Alibrandi, A.; Trimarchi, F.; Benvenga, S.; Trovato, M. TP53 Polymorphism May Contribute to Genetic Susceptibility to Develop Hashimoto’s Thyroiditis. J Endocrinol Invest 2015, 38, 1175–1182. [CrossRef]
- https://www.ncbi.nlm.nih.gov/gene/7157/ (accessed on 01 July 2024).
- Solares, M.J.; Jonaid, G.M.; Luqiu, W.Y.; Berry, S.; Khadela, J.; Liang, Y.; Evans, M.C.; Pridham, K.J.; Dearnaley, W.J.; Sheng, Z.; et al. High-Resolution Imaging of Human Cancer Proteins Using Microprocessor Materials. ChemBioChem 2022, 23, e202200310. [CrossRef]
- Gencel-Augusto, J.; Lozano, G. P53 Tetramerization: At the Center of the Dominant-Negative Effect of Mutant P53. Genes Dev. 2020, 34, 1128–1146. [CrossRef]
- Vaseva, A.V.; Moll, U.M. The Mitochondrial P53 Pathway. Biochimica et Biophysica Acta (BBA) - Bioenergetics 2009, 1787, 414–420. [CrossRef]
- Yuan, X.-M.; Li, W.; Dalen, H.; Lotem, J.; Kama, R.; Sachs, L.; Brunk, U.T. Lysosomal Destabilization in P53-Induced Apoptosis. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 6286–6291. [CrossRef]
- Yamashita, G.; Takano, N.; Kazama, H.; Tsukahara, K.; Miyazawa, K. P53 Regulates Lysosomal Membrane Permeabilization as Well as Cytoprotective Autophagy in Response to DNA-Damaging Drugs. Cell Death Discov. 2022, 8, 1–11. [CrossRef]
- Giorgi, C.; Bonora, M.; Sorrentino, G.; Missiroli, S.; Poletti, F.; Suski, J.M.; Galindo Ramirez, F.; Rizzuto, R.; Di Virgilio, F.; Zito, E.; et al. P53 at the Endoplasmic Reticulum Regulates Apoptosis in a Ca 2+ -Dependent Manner. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 1779–1784. [CrossRef]
- Gila, L.; Elena, N.; Yechezkel, S.; Mary, B. P53-Associated 3′→5′ Exonuclease Activity in Nuclear and Cytoplasmic Compartments of Cells. Oncogene 2003, 22, 233–245. [CrossRef]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dötsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. P63, a P53 Homolog at 3q27–29, Encodes Multiple Products with Transactivating, Death-Inducing, and Dominant-Negative Activities. Molecular Cell 1998, 2, 305–316. [CrossRef]
- Kaghad, M.; Bonnet, H.; Yang, A.; Creancier, L.; Biscan, J.-C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.-M.; Dumont, X.; et al. Monoallelically Expressed Gene Related to P53 at 1p36, a Region Frequently Deleted in Neuroblastoma and Other Human Cancers. Cell 1997, 90, 809–819. [CrossRef]
- Bourdon, J.-C. P53 and Its Isoforms in Cancer. Br J Cancer 2007, 97, 277–282. [CrossRef]
- Khoury, M.P.; Bourdon, J.-C. The Isoforms of the P53 Protein. Cold Spring Harbor Perspectives in Biology 2010, 2, a000927–a000927. [CrossRef]
- Trovato, M.C.; Ruggeri, R.M.; Scardigno, M.; Sturniolo, G.; Vita, R.; Vitarelli, E.; Arena, G.; Gambadoro, O.; Sturniolo, G.; Trimarchi, F.; et al. Immunoreactions for P53 Isoforms Are Associated with Ultrastructural Proliferative Profiles in Benign Thyroid Nodules. Histology and Histopathology 2016, 1079–1087. [CrossRef]
- Trovato, M.; Ruggeri, R.; Guzzo, E.; Certo, R.; Alibrandi, A.; Scifo, S.; Scardigno, M.; Vitarelli, E.; Arena, G.; Gambadoro, O.; et al. Expression of P53 and Isoforms in Beningn and Malignant Lesions of the Head and Neck. Histology and Histopathology 2017, 371–377. [CrossRef]
- Bourdon, J.-C.; Surget, S.; Khoury, M.P. Uncovering the Role of P53 Splice Variants in Human Malignancy: A Clinical Perspective. OTT 2013, 57. [CrossRef]
- Vieler, M.; Sanyal, S. P53 Isoforms and Their Implications in Cancer. Cancers 2018, 10, 288. [CrossRef]
- Stiewe, T.; Haran, T.E. How Mutations Shape P53 Interactions with the Genome to Promote Tumorigenesis and Drug Resistance. Drug Resistance Updates 2018, 38, 27–43. [CrossRef]
- Kotler, E.; Segal, E.; Oren, M. Functional Characterization of the P53 “Mutome.” Molecular & Cellular Oncology 2018, 5, e1511207. [CrossRef]
- Bauer, M.R.; Krämer, A.; Settanni, G.; Jones, R.N.; Ni, X.; Khan Tareque, R.; Fersht, A.R.; Spencer, J.; Joerger, A.C. Targeting Cavity-Creating P53 Cancer Mutations with Small-Molecule Stabilizers: The Y220X Paradigm. ACS Chem. Biol. 2020, 15, 657–668. [CrossRef]
- Blagih, J.; Buck, M.D.; Vousden, K.H. P53, Cancer and the Immune Response. Journal of Cell Science 2020, 133, jcs237453. [CrossRef]
- Truffi, M.; Sorrentino, L.; Corsi, F. Fibroblasts in the Tumor Microenvironment. In Tumor Microenvironment; Birbrair, A., Ed.; Springer International Publishing: Cham, 2020; Vol. 1234, pp. 15–29 ISBN 9783030371838.
- Boesch, M.; Baty, F.; Rumpold, H.; Sopper, S.; Wolf, D.; Brutsche, M.H. Fibroblasts in Cancer: Defining Target Structures for Therapeutic Intervention. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 2019, 1872, 111–121. [CrossRef]
- Fu, T.; Dai, L.-J.; Wu, S.-Y.; Xiao, Y.; Ma, D.; Jiang, Y.-Z.; Shao, Z.-M. Spatial Architecture of the Immune Microenvironment Orchestrates Tumor Immunity and Therapeutic Response. J Hematol Oncol 2021, 14, 98. [CrossRef]
- Kumari, S.; Sharma, S.; Advani, D.; Khosla, A.; Kumar, P.; Ambasta, R.K. Unboxing the Molecular Modalities of Mutagens in Cancer. Environ Sci Pollut Res 2022, 29, 62111–62159. [CrossRef]
- MacDonald, L.; Jenkins, J.; Purvis, G.; Lee, J.; Franco, A.T. The Thyroid Tumor Microenvironment: Potential Targets for Therapeutic Intervention and Prognostication. HORM CANC 2020, 11, 205–217. [CrossRef]
- Fozzatti, L.; Cheng, S. Tumor Cells and Cancer-Associated Fibroblasts: A Synergistic Crosstalk to Promote Thyroid Cancer. Endocrinol Metab 2020, 35, 673–680. [CrossRef]
- Trovato, M.; Sciacchitano, S.; Facciolà, A.; Valenti, A.; Visalli, G.; Di Pietro, A. Interleukin 6 Signalling as a Valuable Cornerstone for Molecular Medicine (Review). Int J Mol Med 2021, 47, 107. [CrossRef]
- Ruggeri, R.M.; Villari, D.; Simone, A.; Scarfì, R.; Attard, M.; Orlandi, F.; Barresi, G.; Trimarchi, F.; Trovato, M.; Benvenga, S. Co-Expression of Interleukin-6 (IL-6) and Interleukin-6 Receptor (IL-6R) in Thyroid Nodules Is Associated with Co-Expression of CD30 Ligand/CD30 Receptor. J Endocrinol Invest 2002, 25, 959–966. [CrossRef]
- Trovato, M.; Campennì, A.; Giovinazzo, S.; Siracusa, M.; Ruggeri, R.M. Hepatocyte Growth Factor/C-Met Axis in Thyroid Cancer: From Diagnostic Biomarker to Therapeutic Target. Biomark�Insights 2017, 12, 117727191770112. [CrossRef]
- Trovato, M.; Villari, D.; Bartolone, L.; Spinella, S.; Simone, A.; Violi, M.A.; Trimarchi, F.; Batolo, D.; Benvenga, S. Expression of the Hepatocyte Growth Factor and c-Met in Normal Thyroid, Non-Neoplastic, and Neoplastic Nodules. Thyroid 1998, 8, 125–131. [CrossRef]
- Hwang, C.-I.; Matoso, A.; Corney, D.C.; Flesken-Nikitin, A.; Körner, S.; Wang, W.; Boccaccio, C.; Thorgeirsson, S.S.; Comoglio, P.M.; Hermeking, H.; et al. Wild-Type P53 Controls Cell Motility and Invasion by Dual Regulation of MET Expression. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 14240–14245. [CrossRef]
- Rassidakis, G.Z. The Emerging Role of CD30 and P53 as Novel Targets for Therapy in Anaplastic Large Cell Lymphoma. Front Biosci 2016, 8, 61–71. [CrossRef]
- Sistigu, A.; Di Modugno, F.; Manic, G.; Nisticò, P. Deciphering the Loop of Epithelial-Mesenchymal Transition, Inflammatory Cytokines and Cancer Immunoediting. Cytokine & Growth Factor Reviews 2017, 36, 67–77. [CrossRef]
- Phan, T.T.T.; Truong, N.V.; Wu, W.-G.; Su, Y.-C.; Hsu, T.-S.; Lin, L.-Y. Tumor Suppressor P53 Mediates Interleukin-6 Expression to Enable Cancer Cell Evasion of Genotoxic Stress. Cell Death Discov. 2023, 9, 340. [CrossRef]
- Boccaccio, C.; Andò, M.; Tamagnone, L.; Bardelli, A.; Michieli, P.; Battistini, C.; Comoglio, P.M. Induction of Epithelial Tubules by Growth Factor HGF Depends on the STAT Pathway. Nature 1998, 391, 285–288. [CrossRef]
- Maffè, A.; Comoglio, P.M. HGF Controls Branched Morphogenesis in Tubular Glands. Eur J Morphol 1998, 36 Suppl, 74–81.
- Hirano, T. Interleukin 6 and Its Receptor: Ten Years Later. International Reviews of Immunology 1998, 16, 249–284. [CrossRef]
- Niu, G.; Wright, K.L.; Ma, Y.; Wright, G.M.; Huang, M.; Irby, R.; Briggs, J.; Karras, J.; Cress, W.D.; Pardoll, D.; et al. Role of Stat3 in Regulating P53 Expression and Function. Molecular and Cellular Biology 2005, 25, 7432–7440. [CrossRef]
- Trovato, M.; Grosso, M.; Vitarelli, E.; Ruggeri, R.; Alesci, S.; Trimarchi, F.; Barresi, G.; Benvenga, S. Distinctive Expression of STAT3 in Papillary Thyroid Carcinomas and a Subset of Follicular Adenomas. Histology and Histopathology 2003, 393–399. [CrossRef]
- Trovato, M.; Torre, M.L.; Ragonese, M.; Simone, A.; Scarfì, R.; Barresi, V.; Giuffrè, G.; Benvenga, S.; Angileri, F.F.; Tuccari, G.; et al. HGF/c-Met System Targeting PI3K/AKT and STAT3/Phosphorylated-STAT3 Pathways in Pituitary Adenomas: An Immunohistochemical Characterization in View of Targeted Therapies. Endocrine 2013, 44, 735–743. [CrossRef]
- Trovato, M.; Vitarelli, E.; Grosso, M.; Alesci, S.; Benvenga, S.; Trimarchi, F.; Barresi, G. Immunohistochemical Expression of HGF, c-MET and Transcription Factor STAT3 in Colorectal Tumors. Eur J Histochem 2004, 48, 291–297.
- Li, S.; Kong, L.; Yu, X.; Zheng, Y. Host–Virus Interactions: From the Perspectives of Epigenetics. Reviews in Medical Virology 2014, 24, 223–241. [CrossRef]
- Balakrishnan, L.; Milavetz, B. Epigenetic Regulation of Viral Biological Processes. Viruses 2017, 9, 346. [CrossRef]
- Łasut-Szyszka, B.; Rusin, M. The Wheel of P53 Helps to Drive the Immune System. IJMS 2023, 24, 7645. [CrossRef]
- Lazo, P.A.; Santos, C.R. Interference with P53 Functions in Human Viral Infections, a Target for Novel Antiviral Strategies? Reviews in Medical Virology 2011, 21, 285–300. [CrossRef]
- Aloni-Grinstein, R.; Charni-Natan, M.; Solomon, H.; Rotter, V. P53 and the Viral Connection: Back into the Future ‡. Cancers 2018, 10, 178. [CrossRef]
- Harford, J.B. A Second Career for P53 as A Broad-Spectrum Antiviral? Viruses 2023, 15, 2377. [CrossRef]
- Durzynska, J.; Lesniewicz, K.; Poreba, E. Human Papillomaviruses in Epigenetic Regulations. Mutation Research/Reviews in Mutation Research 2017, 772, 36–50. [CrossRef]
- Maruzuru, Y.; Fujii, H.; Oyama, M.; Kozuka-Hata, H.; Kato, A.; Kawaguchi, Y. Roles of P53 in Herpes Simplex Virus 1 Replication. J Virol 2013, 87, 9323–9332. [CrossRef]
- Brown, B.; Fricke, I.; Imarogbe, C.; Padrón González, A.A.; Batista, O.A.; Mensah, P.; Chacon-Cruz, E. Immunopathogenesis of Orthopoxviridae: Insights into Immunology from Smallpox to Monkeypox (Mpox). Explor Immunol 2023, 525–553. [CrossRef]
- Martin, C.K.; Samolej, J.; Olson, A.T.; Bertoli, C.; Wiebe, M.S.; De Bruin, R.A.M.; Mercer, J. Vaccinia Virus Arrests and Shifts the Cell Cycle. Viruses 2022, 14, 431. [CrossRef]
- Turpin, E.; Luke, K.; Jones, J.; Tumpey, T.; Konan, K.; Schultz-Cherry, S. Influenza Virus Infection Increases P53 Activity: Role of P53 in Cell Death and Viral Replication. J Virol 2005, 79, 8802–8811. [CrossRef]
- Li, P.; Jiang, H.; Peng, H.; Zeng, W.; Zhong, Y.; He, M.; Xie, L.; Chen, J.; Guo, D.; Wu, J.; et al. Non-Structural Protein 5 of Zika Virus Interacts with P53 in Human Neural Progenitor Cells and Induces P53-Mediated Apoptosis. Virol. Sin. 2021, 36, 1411–1420. [CrossRef]
- Yang, M.-R.; Lee, S.R.; Oh, W.; Lee, E.-W.; Yeh, J.-Y.; Nah, J.-J.; Joo, Y.-S.; Shin, J.; Lee, H.-W.; Pyo, S.; et al. West Nile Virus Capsid Protein Induces P53-Mediated Apoptosis via the Sequestration of HDM2 to the Nucleolus. Cell Microbiol 2007, 0, 070816152918002-???. [CrossRef]
- Yaseen, M.M.; Abuharfeil, N.M.; Darmani, H. The Role of P53 in HIV Infection. Curr HIV/AIDS Rep 2023, 20, 419–427. [CrossRef]
- Levine, A.J. The Common Mechanisms of Transformation by the Small DNA Tumor Viruses: The Inactivation of Tumor Suppressor Gene Products: P53. Virology 2009, 384, 285–293. [CrossRef]
- Liu, X.; Marmorstein, R. When Viral Oncoprotein Meets Tumor Suppressor: A Structural View. Genes Dev. 2006, 20, 2332–2337. [CrossRef]
- Zhu, J.Y.; Abate, M.; Rice, P.W.; Cole, C.N. The Ability of Simian Virus 40 Large T Antigen to Immortalize Primary Mouse Embryo Fibroblasts Cosegregates with Its Ability to Bind to P53. J Virol 1991, 65, 6872–6880. [CrossRef]
- Hermannstädter, A.; Ziegler, C.; Kühl, M.; Deppert, W.; Tolstonog, G.V. Wild-Type P53 Enhances Efficiency of Simian Virus 40 Large-T-Antigen-Induced Cellular Transformation. J Virol 2009, 83, 10106–10118. [CrossRef]
- Trovato, M.; D’Armiento, M.; Lavra, L.; Ulivieri, A.; Dominici, R.; Vitarelli, E.; Grosso, M.; Vecchione, R.; Barresi, G.; Sciacchitano, S. Expression of P53/Hgf/c-Met/STAT3 Signal in Fetuses with Neural Tube Defects. Virchows Arch 2007, 450, 203–210. [CrossRef]
- Danilova, N.; Sakamoto, K.M.; Lin, S. P53 Family in Development. Mechanisms of Development 2008, 125, 919–931. [CrossRef]
- Izumi, T.; Io, K.; Matsui, M.; Shirakawa, K.; Shinohara, M.; Nagai, Y.; Kawahara, M.; Kobayashi, M.; Kondoh, H.; Misawa, N.; et al. HIV-1 Viral Infectivity Factor Interacts with TP53 to Induce G2 Cell Cycle Arrest and Positively Regulate Viral Replication. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 20798–20803. [CrossRef]
Figure 1.
Legend: Data are taken from Ref. *[9,10,11,12,13,14,15,19,20]; **[21,22,23]; ***[24,25,26,27]; ^[6,28,29,30]; ^^[6,31,32,33,34,35]; ^^^[68,69].
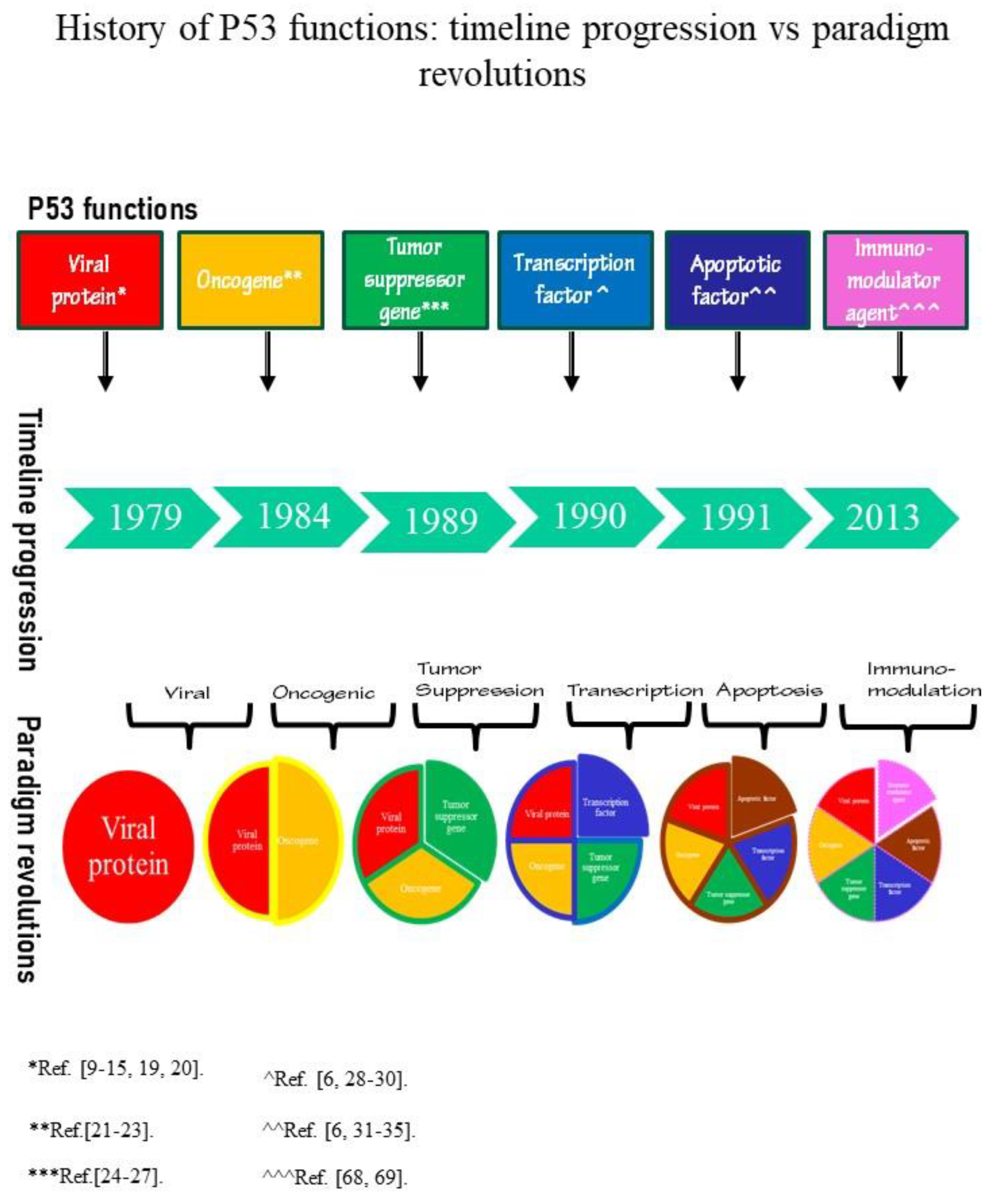
Figure 2.
Legend A: Data are taken from Ref. [46,47]. *Data and abbreviations are taken from Ref. [43]. TAD: transcription activation domain (amino acid 1-60); TAD1: transcription activation domain 1 (amino acid 1-40); TAD2: transcription activation domain 2 (amino acid 41-60); PRR: proline-rich region (amino acid 61-93); DBD: DNA-binding domain (amino acid 102-293); TD: tetramerization domain (amino acid 323-353) and CTD: C-terminal domain (amino acid 364-393). ^Data and abbreviations are taken from Ref. [35]. TAD-1: transcription activation domain-1 (amino acid 1–39); TAD-2: transcription activation domain-2 (amino acid 40–60); PRD: proline-rich domain (amino acid 61–93), DBD: DNA-binding domain (amino acid 94–292); HD: hinge domain (amino acid 293–322); OD: oligomerization domain (amino acid 323–355) and CTR: C-terminal regulatory domain (amino acid 356–393). §Data and abbreviations are taken from Ref. [44]. TAD1: transcription activation domain-1 (amino acid 1–39); TAD2: transcription activation domain-2 (amino acid 40–60); PRR: proline rich region (amino acid 61–93), DBD/core: DNA-binding or Core domain (amino acid 94–292); linker (amino acid 293–322); TET: tetramerization domain (amino acid 323–355) and REG: regulatory domain (amino acid 356–393). #Data and abbreviations are taken from Ref. [45]. TAD1: transcription activation domain 1 (amino acid 1-40); TAD2: transcription activation domain 2 (amino acid 41-60); PRD: proline-rich domain (amino acid 64–92); DBD: DNA-binding domain (amino acid 94–292); TET: tetramerization domain (amino acid 325–356) and REG: regulatory domain (amino acid 356–393).
Figure 2.
Legend A: Data are taken from Ref. [46,47]. *Data and abbreviations are taken from Ref. [43]. TAD: transcription activation domain (amino acid 1-60); TAD1: transcription activation domain 1 (amino acid 1-40); TAD2: transcription activation domain 2 (amino acid 41-60); PRR: proline-rich region (amino acid 61-93); DBD: DNA-binding domain (amino acid 102-293); TD: tetramerization domain (amino acid 323-353) and CTD: C-terminal domain (amino acid 364-393). ^Data and abbreviations are taken from Ref. [35]. TAD-1: transcription activation domain-1 (amino acid 1–39); TAD-2: transcription activation domain-2 (amino acid 40–60); PRD: proline-rich domain (amino acid 61–93), DBD: DNA-binding domain (amino acid 94–292); HD: hinge domain (amino acid 293–322); OD: oligomerization domain (amino acid 323–355) and CTR: C-terminal regulatory domain (amino acid 356–393). §Data and abbreviations are taken from Ref. [44]. TAD1: transcription activation domain-1 (amino acid 1–39); TAD2: transcription activation domain-2 (amino acid 40–60); PRR: proline rich region (amino acid 61–93), DBD/core: DNA-binding or Core domain (amino acid 94–292); linker (amino acid 293–322); TET: tetramerization domain (amino acid 323–355) and REG: regulatory domain (amino acid 356–393). #Data and abbreviations are taken from Ref. [45]. TAD1: transcription activation domain 1 (amino acid 1-40); TAD2: transcription activation domain 2 (amino acid 41-60); PRD: proline-rich domain (amino acid 64–92); DBD: DNA-binding domain (amino acid 94–292); TET: tetramerization domain (amino acid 325–356) and REG: regulatory domain (amino acid 356–393).
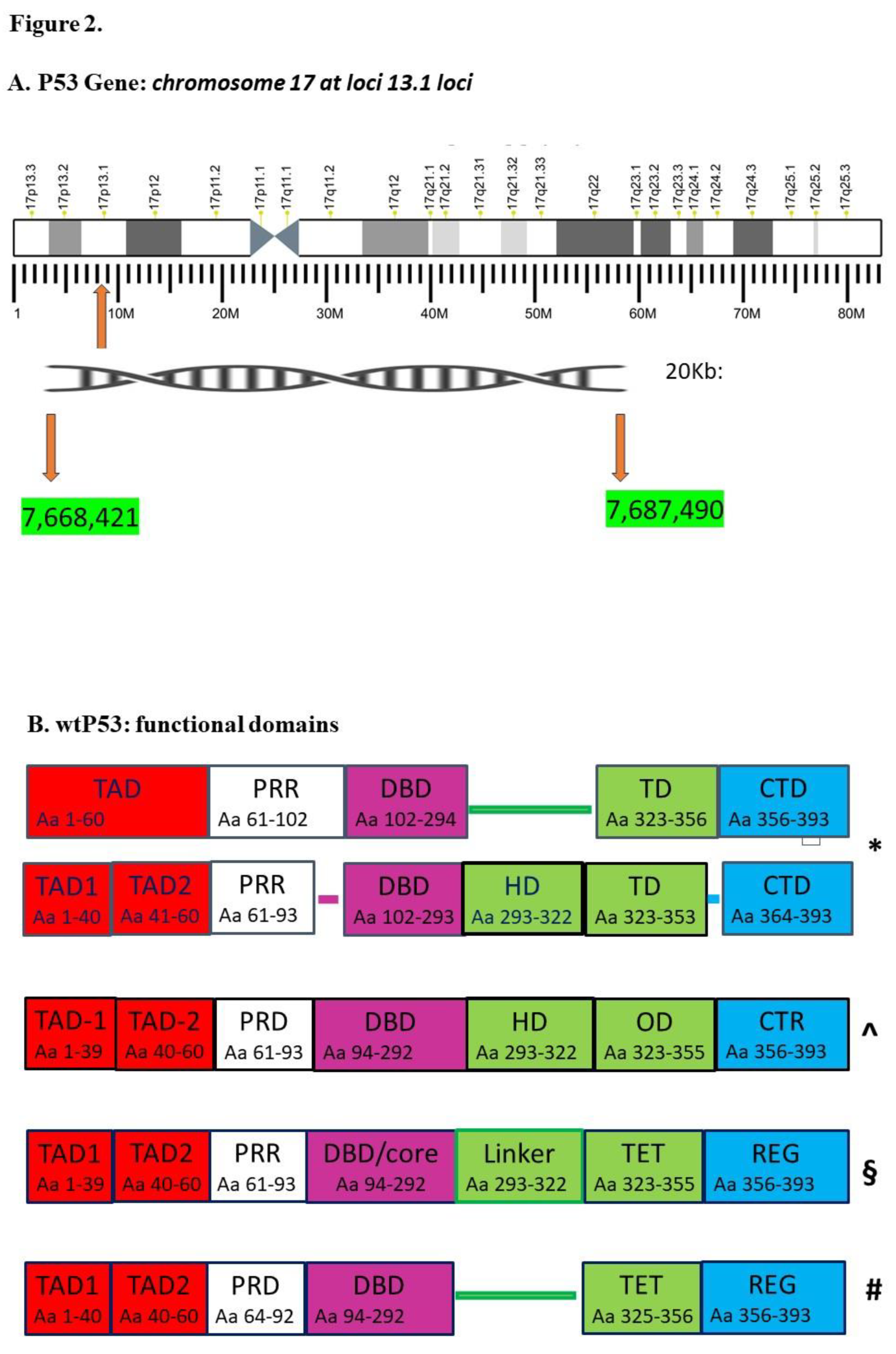
Figure 3.
Title: From HGF and IL-6 to P53 pathways. Legend: HGF/c-met interaction leads to phosphorylation of two tyrosine residues (Tyr 1349 and Tyr1356) recruiting substates by SH2 domain. IL-6/IL-6R binding induce autophosphorylation of gp-130 and activation of Janus protein tyrosine kinases.
Figure 3.
Title: From HGF and IL-6 to P53 pathways. Legend: HGF/c-met interaction leads to phosphorylation of two tyrosine residues (Tyr 1349 and Tyr1356) recruiting substates by SH2 domain. IL-6/IL-6R binding induce autophosphorylation of gp-130 and activation of Janus protein tyrosine kinases.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



