
Submitted:
16 July 2024
Posted:
16 July 2024
You are already at the latest version
Abstract
In the field of quality analysis of food and flavoring products, gas chromatography – quadrupole mass spectrometry – ion mobility spectrometry (GC-QMS-IMS) is a powerful technique for the simultaneous detection of volatile organic compounds (VOC) by both QMS and IMS. GC is an established technique for the separation of complex VOC-rich foods such as beverages, dairy and fruit products as well as flavorings in general. While the subsequent detection by IMS features soft ionization of fragile compounds (e.g., terpenes) with characteristic drift times, MS provides the analytes’ m/z-values for database substance identification. A limitation of the prominently used static headspace-based GC-QMS-IMS systems is the substantially higher sensitivity of IMS in comparison to full scan QMS, which results in the detection of relevant VOC only in IMS, but not in the QMS analyzer. The present study describes a new prototypic trapped-headspace (THS)-GC-QMS-IMS setup, with optimized active flow regulation, able to gain valuable data of both detectors. This ultimately allows the combination of a soft ionization with m/z values obtained from database-searchable electron ionization (EI) spectra and as such, offers a substantial advantage in fields of quality control. The new setup features aligned retention times for IMS and MS and sufficient signal intensities for QMS and IMS. The analytical setup was evaluated with a set of mango puree samples from various provenances and the data was evaluated by suitable machine learning approaches. The results demonstrate that THS-GC-QMS-IMS allows for the classification of mango purees from different cultivars and can be a promising alternative method for authenticity control of food, flavors, and beverages.
Keywords:
HS-GC-MS-IMS
; ion mobility spectrometry
; volatile organic compounds
; trapped headspace sampling
; authentication
; mango puree
; chemometrics
1. Introduction
In the analysis of flavorings and volatile organic compounds (VOC) in general, gas chromatography (GC) is the method of choice for separation of a broad spectrum of VOC. There are numerous GC-based applications for the analysis of complex matrices such as essential oils, flavors, or fragrances, particularly in combination with mass spectrometry (MS). However, in the last decade, ion mobility spectrometry (IMS) has gained increasing attention for the analysis of food, beverages, and flavorings [1,2]. IMS systems in hyphenation with GC are mostly realized in the form of drift tube IMS systems (DTIMS), while only few systems base on differential mobility (DMS) or asymmetric field (AIMS/FAIMS) [3,4]. For the sake of simplicity, IMS is used in the context of DTIMS in the following. GC-IMS systems base on 3H, 63Ni, CD or UV ionization, whereas the ionization by 3H is among the most common, due to the low safety restrictions and the broad applicability [3,5,6]. Such systems offer a remarkably high sensitivity for polar and medium polar compounds, due to the soft ionization, and operate at ambient pressure. This simplifies their use substantially in contrast to vacuum-based MS detectors, with regard to robustness, ease of use and cost of systems. While IMS detectors commonly outperform standard quadrupole MS detectors in terms of sensitivity, one major drawback is the lack of comprehensive and commonly accepted databases. Whereas such libraries are widely used for substance identification in MS, this is not the case for IMS, where substances are typically identified relative to reference substances, comparing retention and drift time. For complex matrices, this is time consuming, tedious, and limited by the need for the availability of reference substances. In a previously published study, hyphenation of IMS and QMS showed a beneficial effect for substance identification, in combination with a soft ionization of the IMS and subsequently, valuable data for chemometric evaluation. Brendel et al. were able to show that a classification of selected citrus fruit juices is possible by both QMS and IMS data based on enrichment-free, static headspace GC-QMS-IMS, but match quality in MS databases was limited due to low QMS S/N ratios and the intense fragmentation in EI [2]. Additionally, Schanzmann et al. described the combination of thermal desorption with GC-MS-IMS for breath analysis. They were able to show a good accordance of MS and IMS retention times for a homologous series of ketones up to 2-decanone. Within the study, they were also able to identify five VOC, including ethanol, isoprene, acetone, 2-propanol, and 1-propanol, in breath samples with the NIST database [7]. This approach is typically not applicable for complex flavorings and food samples, as these show a broad range of VOC, ranging from ketones, esters to terpenes and sesquiterpenes, with boiling points up to 260 °C. In nearly all cases, QMS sensitivity was the limiting factor. To overcome this limitation, and to obtain m/z data also for complex matrices, a new trapped-headspace (THS)-GC-MS-IMS setup was designed. This involved the optimization of retention gap dimensions and APC pressure settings, in order to generate reasonable signal intensities on both detectors. The main challenge here was the balance between sufficient S/N in QMS and not overloading the IMS detector. In Figure 1, the THS-GC-MS-IMS setup and the flow directions are visualized. The hyphenation with the described trapped-headspace sampler is beneficial for sampling in static as well as dynamic headspace, in particular due to the constant control of the vial pressure and the ability of concentration steps.
1.1. Trapped Headspace Sampling
The basic principle for headspace sampling is the equilibration of analytes in sample solution and the vapor phase above [8]. The partition coefficient, describing the ratio of the analyte concentration between the phases, is shown in Equation 1 [9].Compounds with a low partition coefficient K, will be more abundant in the vapor phase, while compounds with a high partition coefficient will be more prevalent in the sample phase. Besides the analyte’s vaporization enthalpy and vapor pressure, the solubility in sample phase is particularly important for the partition coefficient. Thus, in method development, vial temperature, adjustment of pH-value, and the addition of salt are effective and important parameters [9].
where:
- K = partition coefficient of a defined compounds
- CS = concentration of the compound in sample phase
- CG = concentration of the compound in vapor phase
For headspace sampling, the vapor phase of a substance is sampled and analyzed. The sample is sealed in a vial, heated and brought to an equilibrium [8]. In static headspace sampling (SHS), a defined volume of the vapor phase is extracted using a syringe, resulting in analyte fractions in ppmv levels. This results in absolute amounts in the lower pg range on column. Paired with the system-immanent issue of high fragmentation behavior of sensitive compounds in EI mode and the limited sensitivity of QMS detectors in full scan, this poses a challenge for QMS based systems. Consequently, the majority of polar and medium polar analytes are detected mainly in IMS and only a limited number of these are detectable in the QMS detector. One elegant way around this limitation is trapped or dynamic headspace sampling (THS). It allows for a pre-concentration of the analytes up to sub-ppbv level [9]. While for matrices with higher analyte concentrations, classical SHS is an effective, feasible option, THS excels at samples with low abundant analytes in complex matrices [8]. This is of particular advantage in quality analytics of food and beverages, as the incubation temperature is limited due to the risk of transformation reactions and formation of artifacts.
In Figure 2, the principles of THS sampling are visualized. In a first step, the sample is pressurized and the volatiles are continuously evaporated to the vapor phase [10]. Subsequently, the vapor phase is trapped with a sorbent, extracting VOC from the gas phase [8,9]. The last step is the desorption of the trapped analytes and the transfer onto the GC column [10]. In multiple headspace extraction (MHE) mode, these steps are repeated several times, ensuring that the majority of the volatiles are extracted from the sample phase [9,10]. In MHE, the sample is equilibrated again after each sampling procedure. The trap material, such as TenaxTA or carbon-based sorbents, must fulfill several requirements. The material should be capable to extract a broad range of volatile samples and retain all analytes of interest, further allow for a fast injection on the GC column and should have a minimal carry-over effect of impurities to the sample [9]. For trapping, there are different approaches, such as cryogenic, electrically-cooled or with solid sorbents, as well as liquid films in solid support [11]. Applications for THS sampling are found in a broad spectrum of analytical tasks in foods, feeds, pharmaceuticals, and environmental samples [12,13,14,15,16].
1.2. Ion Mobility Spectrometry in VOC Analysis
Within the last decades, GC-hyphenated IMS gained increasing popularity for the analysis of VOC in all types of fields due to its high sensitivity in combination with a robust design. The applications range from process and quality control to explosives and drugs, foods, beverages, and flavor products [2,17,18,19]. The IMS cells operate at ambient pressure and 3H-based ionization of the analytes is a reaction of proton water clusters H+[H2O]x with the analytes, forming protonated monomers MH+[H2O]n-x as shown in Equation (2) and at higher analyte concentrations, dimers M2H+[H2O]n-x, as shown in Equation (3) [6]. The proton water clusters are formed within a reaction cascade, initiated by the tritium source (< 100 MBq).
The formation of proton water clusters, also called reactant ions, depends on residual moisture content of the gas atmosphere [3,6]. After ionization, monomers and dimers are accelerated into the drift tube by a weak electrical field against a defined flow of a drift or buffer gas, typically nitrogen. Due to collisions with drift gas molecules, ions are decelerated according to their collision cross section (CCS). Thus, drift time is dependent on mass, charge and structure, resulting in a characteristic drift time for each analyte. The soft, APcI-like ionization leads to excellent sensitivity ranges of DTIMS instruments, which makes them optimal tools for non-target strategies (NTS). These approaches are based on the complete (amenable) spectral fingerprint instead of using defined marker compounds [3].
In VOC profiling of foods and flavors, IMS is already used in a multitude of applications, ranging from quality analysis of olive oils, authenticity of honey, quality control of brewing hops, profiling of dairy products, such as kefir or for discrimination of different citrus juices [1,2,20,21,22,23].
1.3. VOC Analysis of Mangos
Mango fruits (Mangifera Indica L.) are considered as the “King of Fruits” and particularly mango purees are of substantial value for food and beverage industry [24]. There are more than 100 cultivars available, which differ in taste and flavor. In general, the flavor is described as fruity, sweet and floral, but some cultivars display different, citrus and terpene notes. Due to that flavor profile, the Indian cultivar ‘Alphonso’ is one of the most popular and shows an increasing demand on the world market [25]. Although there are numerous studies on mangos and their volatiles, most of these focus on regional cultivars or on farm-grown fruits and not on partially processed commercial products, such as purees or pulps [26,27,28]. Mango fruits display a complex volatile profile with more than 50 compounds, including alcohols, ketones, aldehydes, esters, monoterpenes and sesquiterpenes [27,29,30,31]. With regard to VOC composition, the cultivars can be distinguished into three groups. The first group features green, herbal flavors and 3-carene as the most abundant monoterpene, the second with rosin flavors and α-terpinolene as the major monoterpene compound and the last with (Z)-β-ocimene as the predominant monoterpene, covering sweet, terpene and citrus notes, such as the cultivar ‘Alphonso’ [32].
The majority of published studies on the VOC profiles of mango fruits and products hereof are target-based approaches and are dominated by SHS-GC-MS applications, mostly with solid phase microextraction (SPME) enrichment, Arrow, or other enrichment steps [27,33,34,35].
In previously published non-targeted studies, Tandel et al. applied PCA and HCA to HS-SPME-GC-MS data and different extraction methods of Indian mango cultivars, while Farag et al. employed PCA to analyze HS-SPME-GC-MS data of Egyptian mango cultivars [27,33]. Further, Shimizu et al. reported on a volatile profiling of 17 different mango cultivars using HS-SPME-GC-MS in combination with PCA [26].
So far, the literature on the use of GC-IMS in the context of mango fruits, purees, or other low-processed mango products is scarce. A number of authors reported on GC-IMS applications for post-harvest effects and focused on the terpene profiles in relation to fruit quality [36,37]. However, these studies focused specifically on Chinese cultivars and on the ripening process from green fruits to ripe fruits and used separate instruments for IMS and MS.
Consequently, the aim of the present study was to demonstrate the potential of THS sampling in combination with simultaneous GC-MS-IMS detection. While the study focused on the non-target approach to differentiate Mangifera indica L. cultivars, relevant metabolites were confirmed via MS in parallel to generate a more detailed insight. The prototypic system described here allows both approaches from one single injection. An additional source of information for the non-target approach is resulting from the use of a chiral GC column, which generates substantially more characteristic signals.
2. Materials and Methods
2.1. Reagents and Mango Samples
In total, 30 different commercial mango pulp and concentrate samples were measured for this study. The samples were taken by independent auditors as part of routine audits of SGF International's Voluntary Control System in the years 2019 to 2023 and were kindly provided by SGF International e.V. (Saulheim, Germany). The sample set included 10 samples of the cultivar ‘Alphonso’ (from India), one of the cultivar ‘Kent’ (from Peru), two of the cultivar ‘Criollo’ (from Peru), seven of the cultivar ‘Tommy’ (from Mexico), including six concentrates and ten samples of the cultivar ‘Totapuri’ (from India), with three concentrates. Samples were shipped and stored at –18 °C and defrosted slowly at room temperature. Subsequently, 2 g of each sample were placed in a 20 mL-headspace vial and closed tightly with a screw-cap with butyl/PTFE septa. All samples were prepared and measured in duplicate. Reference substances were ethyl butyrate, α-pinene, D-limonene, α-terpineol (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany), and β-caryophyllene (Carl Roth GmbH + Co. KG, Karlsruhe, Germany). Reference chemicals were dissolved in fresh canola oil as neutral matrix.
2.2. Instrumentation (HS-GC-MS-IMS)
Oven temperature of the HS 20 headspace sampler (Shimadzu Corporation, Kyoto, Japan) was set to 50 °C, transfer and sample line were operated at 150 °C. Trap cooling temperature was -10 °C and desorption temperature was set to 250 °C. Equilibrium temperature was set to 25 °C. Multi injection was set to 5. Chromatographic separation was performed with a Nexis™ GC-2030 (Shimadzu Corporation, Kyoto, Japan) and a chiral BGB 174 capillary column (30 m × 0.25 mm × 0.25 μm, BGB Analytik Vertrieb GmbH, Rheinfelden), using helium as carrier gas in constant pressure mode with 180 kPa and splitter advanced pressure controller (APC) pressure of 38 kPa. Oven program started at 40 °C initial temperature, followed by a temperature ramp of 1 °C/min to 80 °C, 4 °C/min to 120 °C and 6 °C/min to 160 °C, holding for 5 min, resulting in 45 min per run. At the end of the analytical column, the column gas flow was split by a SilFlow GC 4 port splitter plate (Trajan Scientific and Medical, Ringwood, Australia) into two retention gaps (0.7 m length and 0.15 mm inner diameter to IMS and 1.6 m length and 0.15 mm inner diameter to MS, respectively). Transfer lines were operated both at 220 °C to MS and to IMS (Hillesheim GmbH, Waghäusel) respectively. Ion source temperature of the QP2020 NX MSD (Shimadzu Corporation, Kyoto, Japan) was set to 200 °C, electron ionization energy was 70 eV, emission current 150 µA and scan range m/z 35 to m/z 400 and a duty cycle of 300 ms.
The OEM-Focus-IMS cell with an 3H ionization source (100 MBq ß-emission) was operated at 100 °C. The drift tube diameter was 15.2 mm with a length of 98 mm, consisting of stainless steel and PEEK. IMS was operated in positive ion mode at a constant voltage of 2.5 kV. Injection voltage was set to 2,500 V, blocking voltage to 70 V. Drift gas was nitrogen with a purity of 99.9999 % and flow was controlled using a mass flow controller at 150 mL/min (Voegtlin Instruments AG, Aesch, Switzerland). Each spectrum was averaged over six scans, using a repetition rate of 21 ms. The injection pulse was set to a width of 100 μs, sampling frequency was 228 kHz.
2.3. Data Processing and Evaluation
MS data was analyzed using GCMSsolutions 4.53 (Shimadzu Corporation, Kyoto, Japan) and for substance identification, NIST/EPA/NIH Mass Spectral Library 23 from the NIST of the U.S. Department of Commerce was used. For retention time comparison, the TIC chromatograms were integrated and the retention times at peak maximum were evaluated. In IMS, the spectra were processed as described in the following paragraph.
Python version 3.8.8 and the package gc-ims-tools version 1.7. were used for data preprocessing, multivariate analysis and visualization of the IMS spectra [38]. Preprocessing is a crucial step in multivariate data analysis and important to obtain most of the biological information of a sample. To reduce the data size, each spectrum was treated by a “db3” wavelet compression with a level of 3. All spectra were aligned in drift time dimensions and normalized to the reactant ion peak (RIP). The alignment is particularly important, as the drift time is dependent on pressure in the IMS cell. Consequently, variations in the atmospheric pressure may lead to shifts in the drift time of spectra over different days. Further, spectra were cropped to the areas with sample information, in regions of 100 s to 2,700 s in the retention time axis and between 1.03 and 2.5 (ca. 6.5 – 16,25 ms) in the RIP relative drift time axis. Afterwards a baseline correction was applied to the dataset, using asymmetric least squares, with weighting of 0.001 and smoothing set at 107. Subsequently, automated 2D peak detection by persistent homology (PH) was used for an objective and reproducible determination of the retention time [39]. The last preprocessing steps were Pareto scaling to reduce the influence of large peaks in comparison to smaller peaks and finally, mean centering.
3. Results & Discussion
3.1. Performance of the THS-GC-MS-IMS System
Our group recently reported on the application of simultaneous SHS-GC-QMS-IMS in brewing hops and Citrus juices with limitations from inferior signal intensities of the QMS data in comparison to the IMS data [2,22]. To overcome these limitations, THS sampling was applied to mango samples and the simultaneous GC-QMS (Fig. 3a) and GC-IMS (Figure 3b) spectra of a cv. ‘Totapuri’ mango sample are visualized in Figure 3. For a majority of substances detected by IMS, now also signals in the QMS detector are sufficiently abundant. More than 80 compounds were detected in the samples and show sufficient S/N ratios, both in MS and IMS spectra, while still the information of both detectors is complementary to a certain extent. This number is in line with previously published profiling studies of mango fruits, using static HS-SPME-GC-MS [26,35]. While the IMS system is able to detect the intact ion species at low trace levels, the EI-QMS features high-fragmenting m/z information, optimally suited for database referral. As to date, no substance databases for GC-IMS systems are commercially available, only the simultaneously generated full scan MS data was used for substance confirmation. To evaluate the synchronicity of both IMS and MS retention time, a number of relevant substances was selected. Figure 3 shows the exemplary compounds: ethyl acetate (1), α-pinene (2), ethyl butyrate (3), β-myrcene (4), D-limonene (5), trans-β-ocimene (6), acetic acid (7), nonanal (8), α-terpineol (9), and β-caryophyllene (10).
In Table 1, a comparison of the retention time of both IMS and QMS, and the MS-match quality of the identified VOCs are displayed. When comparing the QMS and IMS retention times of the selected compounds, the majority are matching with only low deviations. As observed in previous studies, analytes with a lower boiling point featured slightly better correspondence of the retention times [2,7] than those with higher boiling points. By optimized retention gaps, the retention time difference was improved substantially. Schanzmann et al. describe a hyphenation of thermal desorption with GC-MS-IMS for breath analysis. There, a homologous series of ketones was evaluated, with regard to retention times in MS and IMS. Retention times up to 2-octanone were in reasonable accordance, but the difference of MS to IMS was reported to increase with growing chain lengths up to 0.24 min at 2-decanone [7].
Brendel et al. reported a comparable observation for terpenes and terpenoids. In that study, IMS retention values of α-pinene and β-myrcene were in matching with the QMS signals. However, the higher boiling β-caryophyllene and α-terpineol showed a substantial difference of 0.18 min and 0.19 min, respectively [2]. In the present study, we were able to optimize this particular pair of substances to retention time differences of 0.05 min and 0.15 min respectively. One crucial factor in this context is the IMS cell temperature, particularly for higher boiling volatile compounds. Commercially available IMS instruments are limited to cell temperatures below 100 °C, which may facilitate condensation effects of high-boiling compounds in the cell. This adversely affects the peak shape of high boiling volatile compounds, such as sesquiterpenes significantly.
In Table 2, VOC detected are listed. A broad profile of compounds was detected including esters, aldehydes, terpenes and sesquiterpenes. While IMS has its strengths for polar and medium polar compounds, such as aldehydes and esters, the MS detection was advantageous in the detection of substances of lower polarity and higher boiling points of above 250 °C, such as sesquiterpenes. The lower IMS cell temperatures in relation to the higher EI source temperature are presumably one important driver here. For IMS, these substances feature limited separation due to strong tailing effects. One general option to overcome this limitation is to increase IMS cell temperatures, but such IMS systems are not yet commercially available. Studies on a prototypic high-temperature IMS (HT-IMS) demonstrated significant improvements in peak shape for high boiling VOC [40].
As previously published, SHS-SPME-GC-MS studies reported similar VOC in mango fruits, however were limited to achiral gas chromatography [26,27,29,30]. Chiral GC separation however features an excellent performance for enantiomers and particularly, for those of sesquiterpenes. Chiral GC applications have a high potential and are considered an emerging topic in the field of ”greener” quality analytics of food and flavorings, even though approaches have limited applicability yet [41]. In the present study, we employed a ß-cyclodextrin-based BGB-174 chiral GC column with regard to the generation of an enantiomeric fingerprint for the non-targeted data analysis. The underlying hypothesis was that an increase in relevant features based on chiral information of the respective samples should increase the differentiation power of exploratory data analysis approaches or machine learning models, as described in the following section.
3.2. Exploratory Data Evaluation of Non-Targeted IMS Data
The principal rationale for using IMS data for the exploratory data analysis is the significantly higher overall sensitivity of the IMS over the QMS detector, resulting in higher number of signals detected, in particular for the polar and medium polar species. Among other, this is due to the softer ionization process in the IMS vs. the hard EI ionization in QMS. EI spectra are dominated by high numbers of often similar fragment ions for different molecular species and as such, add variance to the data that is not associated with discriminative information, but rather increases inter-class variance. IMS generates nearly no fragment ions and the second-order data from the drift time domain increases the number of “useful” signals. These additional signals are important features for the subsequent data analysis, typically leading to more meaningful and more robust principal component analysis (PCA) models. This was already demonstrated by Brendel et al., where typically, the IMS data generated more robust models [2,22].
For the subsequent evaluation of the non-targeted data resulting from the GC-IMS domain, PCA was employed as a first step. Our toolbox gc-ims-tools allows not only to perform PCA on the preprocessed data, but furthermore adds a substantial step in understanding the character of the underlying data by backwards projection of the respective loadings to the original data space [38,42].
Figure 4 (a) shows a scores plot of the second and third principal components. The visualization reveals the position of samples in the variable space.
PC2, covering 19.0 % of variance, and PC3, covering 14.1 % of variance, were selected due to the highest explained variance from the different mango cultivars. While PC2 shows a separation of cv. ‘Alphonso’ and the other classes, PC3 separates cvs. ‘Totapuri’, ‘Tommy’, and ‘Criollo’. The backwards projected PCA loadings to the original data space with the dimensions of retention time × normalized drift time are visualized in Figure 4 b. This allows for a direct connection between the original data and the PCA scores plot and show in a figurative way the contribution or relevance of individual substance signals for the separation. High positive (red) or negative values (blue) indicate a significant correlation, while values around zero are less relevant. For PC2, this is visualized with the arrow in Figure 4 (a), with red for high positive loadings and blue for high negative loadings. For the separation of Alphonso mango vs other cultivars, several compounds display a substantial impact. Based on the QMS spectra of the dataset, 17 relevant components with a high influence on PC2 were confirmed via the NIST database. These are visualized in Figure 4 (b). The loadings plot shows the importance of PC2 for the separation of the cultivars. Cv. ‘Totapuri’ and the cv. ‘Criollo’ feature a higher amount and number of sesquiterpenes, including α-gurjunene, β-caryophyllene, and δ-cadinene. Further, the two cultivars show a higher abundance of α-pinene and β-pinene, β-myrcene, as well as α-phellandrene, and β-phellandrene. With a high negative correlation in the PCA loadings, 2,5-dimethyl-4-methoxy-3(2H)-furanone was important for the separation of the cv. ‘Alphonso’. This compound was already described as a particularly important flavor constituent for this cultivar in previous studies [43,44].
Another observation was the differentiability of minimal processed pulps and concentrate pulps via the VOC profile: the scores plot of PC1 and PC2 is shown in Figure 5 (a) and shows a separation between the samples of cv. ‘Tommy’, and two samples of cv. ‘Totapuri’ relative to all of the other mango samples. These particular samples were concentrated pulps, while the remaining samples were minimally processed pulps. To identify the compounds which are responsible for the observed separation on PC1, the loadings plot is again backwards-projected onto the original data space (Figure 5 (b)). Acetic acid, ethanol, and 3-carene displayed a significant positive correlation, while ethyl acetate, 2,5-dimethyl-4-methoxy-3(2H)-furanone, and trans-β-ocimene featured low abundances. These VOC were identified for all analyzed samples of the cv. ‘Tommy’, but also for two samples of the cv. ‘Totapuri’, which also feature high positive scores on PC1. In comparison to the other minimally processed cv. ‘Totapuri’ samples, both concentrate samples featured lower abundances of a number of esters (e.g., ethyl propanoate, propyl acetate, methyl butyrate) and alcohols (e.g., isobutanol, isopentyl alcohol). However, these compounds did not show significant impact on PC1, which indicates a lower variance of these compounds relative to the substances with higher abundances. This underlines that the relevant information for the separation described by the respective PC is not only the mere presences of the signals, but rather is found within the ratios of the peaks. For instance, acetic acid indicated a higher abundance in the concentrates relative to the minimally processed samples. This underlines the importance of good feature selection for potentially following supervised methods and is in line with a recently published study of our group on the selection of relevant features [42].
To explain the observation of the two clusters for the cv. ‘Totapuri’, hierarchical cluster analysis (HCA), based on Euclidean distances was applied to the dataset. This approach evaluates the distances between the individual samples. The higher the distance, the higher the sample difference and vice versa. The corresponding dendrogram is visualized in Figure 6 and shows two main clusters. The first contains all minimally processed pulp samples and the second one includes all concentrated pulp samples.
The separation can be explained by the processing employed in the concentration step and the utilization of flavorings for concentrated products. The concentrated samples particularly displayed higher amount of acetic acid, which is in little dose reported as an useful product for fruit flavoring and on the other hand, contained less fruity esters, such as propyl acetate and methyl butyrate [32,45,46,47].
Additionally, HCA separated the cluster of the minimally processsed pulp samples into two subgroups. The first subgroup includes the samples of the cv. ‘Alphonso’, and the second subgroup contains the other cultivars. As already mentioned, the cv. ‘Alphonso’ showed a quite characteristic profile with a number of lactones, such as (e.g., γ-butyrolactone, γ-hexalactone) and 2,5-dimethyl-4-methoxy-3(2H)-furanone. The subgroup of the other cultivars displayed a further clustering of the cv. ‘Totapuri’ and the other cultivars. Cvs. ‘Kent’, ‘Tommy’, and ‘Criollo’ featured the monoterpene 3-carene, which was not detected in the Totapuri cultivar, where trans- β-ocimene was the predominant terpene.
4. Conclusions
This study evaluated the potential of chiral THS-GC-QMS-IMS for nontarget analysis of complex food and beverage samples, in combination with chemometric data evaluation. The instrumentation offers the combination of soft ionization in IMS and valuable m/z value information of the QMS detector. The simultaneous data acquisition shows highly comparable retention times, and more than 80 compounds were identified within the study. The chiral column was used with a focus on the generation of additional features for non-target screening, rather than with a view on the differentiation of individual compounds. Using PCA, the combination of PCA scores plots and backwards-projected loadings plots, revealed characteristic VOC for the different mango cultivars and allowed further for the differentiation of minimally processed and concentrated samples. HCA offers first indications for similarities in the VOC profiles of the mango samples and allows a clear separation of concentrated vs minimally processed samples and furthermore, differentiates between the different cultivars. The dual detection in combination with trapped headspace sampling generates a complex fingerprint of the sample, reducing the need for two separate systems, but at the same time, generates complementary information from one single injection. While these preliminary results are promising, a larger set of samples is required, with regard to cultivars, provenance, and processing methods.
This approach may be of substantial importance for a plethora of other applications in quality control of food, beverage, and flavoring, where characteristic VOC profiles are found.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: MS (a) and IMS spectrum (b) of cv. “Alphonso”; Figure S2: MS (a) and IMS spectrum (b) of cv. “Kent”; Figure S3: MS (a) and IMS spectrum (b) of cv. “Criollo”; Figure S4: MS (a) and IMS spectrum (b) of cv. “Tommy”; Figure S5: PC3 loadings plot of PCA.
Author Contributions
Conceptualization, P.W. and L.B.; sample provision, M.J.; methodology, L.B.; software, L.B.; data curation, L.B.; writing—original draft preparation, L.B.; writing—review and editing, P.W., S.S, M.J. and S.R.; supervision, P.W.; funding acquisition, P.W. and S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by by the Federal Ministry of Education and Research (BMBF), Berlin, Germany., grant number 13FH138KX0 (“Deep Authent”).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Gerhardt, N.; Birkenmeier, M.; Schwolow, S.; Rohn, S.; Weller, P. Volatile-Compound Fingerprinting by Headspace-Gas-Chromatography Ion-Mobility Spectrometry (HS-GC-IMS) as a Benchtop Alternative to 1H NMR Profiling for Assessment of the Authenticity of Honey. Anal. Chem. 2018, 90, 1777–1785. [CrossRef]
- Brendel, R.; Schwolow, S.; Rohn, S.; Weller, P. Volatilomic Profiling of Citrus Juices by Dual-Detection HS-GC-MS-IMS and Machine Learning-An Alternative Authentication Approach. J. Agric. Food Chem. 2021, 69, 1727–1738. [CrossRef]
- Capitain, C.; Weller, P. Non-Targeted Screening Approaches for Profiling of Volatile Organic Compounds Based on Gas Chromatography-Ion Mobility Spectroscopy (GC-IMS) and Machine Learning. Molecules 2021, 26. [CrossRef]
- Karpas, Z. Applications of ion mobility spectrometry (IMS) in the field of foodomics. Food Research International 2013, 54, 1146–1151. [CrossRef]
- Parastar, H.; Weller, P. Towards greener volatilomics: Is GC-IMS the new Swiss army knife of gas phase analysis? TrAC Trends in Analytical Chemistry 2024, 170, 117438. [CrossRef]
- Borsdorf, H.; Eiceman, G.A. Ion Mobility Spectrometry: Principles and Applications. Applied Spectroscopy Reviews 2006, 41, 323–375. [CrossRef]
- Schanzmann, H.; Ruzsanyi, V.; Ahmad-Nejad, P.; Telgheder, U.; Sielemann, S. A novel coupling technique based on thermal desorption gas chromatography with mass spectrometry and ion mobility spectrometry for breath analysis. J. Breath Res. 2023, 18. [CrossRef]
- Kremser, A.; Jochmann, M.A.; Schmidt, T.C. Systematic comparison of static and dynamic headspace sampling techniques for gas chromatography. Anal. Bioanal. Chem. 2016, 408, 6567–6579. [CrossRef]
- Gas chromatography; Poole, C.F., Ed., 2nd ed.; Elsevier: Cambridge, 2021, ISBN 9780128206751.
- Soria, A.C.; García-Sarrió, M.J.; Sanz, M.L. Volatile sampling by headspace techniques. TrAC Trends in Analytical Chemistry 2015, 71, 85–99. [CrossRef]
- Costa Freitas, A.M.; Gomes da Silva, M.D.R.; Cabrita, M.J. Sampling Techniques for the Determination of Volatile Components in Grape Juice, Wine and Alcoholic Beverages. Comprehensive sampling and sample preparation: Analytical techniques for scientists; Academic Press: Amsterdam, Netherlands, 2012; pp 27–41, ISBN 9780123813749.
- Ikem, A. Measurement of volatile organic compounds in bottled and tap waters by purge and trap GC–MS: Are drinking water types different? Journal of Food Composition and Analysis 2010, 23, 70–77. [CrossRef]
- Schulz, K.; Dressler, J.; Sohnius, E.-M.; Lachenmeier, D.W. Determination of volatile constituents in spirits using headspace trap technology. Journal of Chromatography A 2007, 1145, 204–209. [CrossRef]
- Manzini, S.; Durante, C.; Baschieri, C.; Cocchi, M.; Sighinolfi, S.; Totaro, S.; Marchetti, A. Optimization of a Dynamic Headspace-Thermal Desorption-Gas Chromatography/Mass Spectrometry procedure for the determination of furfurals in vinegars. Talanta 2011, 85, 863–869. [CrossRef]
- Jeleń, H.; Gracka, A.; Myśków, B. Static Headspace Extraction with Compounds Trapping for the Analysis of Volatile Lipid Oxidation Products. Food Anal. Methods 2017, 10, 2729–2734. [CrossRef]
- Soria, A.C.; García-Sarrió, M.J.; Ruiz-Matute, A.I.; Sanz, M.L. Headspace Techniques for Volatile Sampling. Green Extraction Techniques - Principles, Advances and Applications; Elsevier, 2017; pp 255–278, ISBN 9780128110829.
- Christmann, J.; Weber, M.; Rohn, S.; Weller, P. Nontargeted Volatile Metabolite Screening and Microbial Contamination Detection in Fermentation Processes by Headspace GC-IMS. Anal. Chem. 2024. [CrossRef]
- Sammarco, G.; Bardin, D.; Quaini, F.; Dall'Asta, C.; Christmann, J.; Weller, P.; Suman, M. A geographical origin assessment of Italian hazelnuts: Gas chromatography-ion mobility spectrometry coupled with multivariate statistical analysis and data fusion approach. Food Res. Int. 2023, 171, 113085. [CrossRef]
- Babis, J.S.; Sperline, R.P.; Knight, A.K.; Jones, D.A.; Gresham, C.A.; Denton, M.B. Performance evaluation of a miniature ion mobility spectrometer drift cell for application in hand-held explosives detection ion mobility spectrometers. Anal. Bioanal. Chem. 2009, 395, 411–419. [CrossRef]
- Capitain, C.C.; Nejati, F.; Zischka, M.; Berzak, M.; Junne, S.; Neubauer, P.; Weller, P. Volatilomics-Based Microbiome Evaluation of Fermented Dairy by Prototypic Headspace-Gas Chromatography-High-Temperature Ion Mobility Spectrometry (HS-GC-HTIMS) and Non-Negative Matrix Factorization (NNMF). Metabolites 2022, 12. [CrossRef]
- Gerhardt, N.; Schwolow, S.; Rohn, S.; Pérez-Cacho, P.R.; Galán-Soldevilla, H.; Arce, L.; Weller, P. Quality assessment of olive oils based on temperature-ramped HS-GC-IMS and sensory evaluation: Comparison of different processing approaches by LDA, kNN, and SVM. Food Chem. 2019, 278, 720–728. [CrossRef]
- Brendel, R.; Schwolow, S.; Rohn, S.; Weller, P. Gas-phase volatilomic approaches for quality control of brewing hops based on simultaneous GC-MS-IMS and machine learning. Anal. Bioanal. Chem. 2020, 412, 7085–7097. [CrossRef]
- Schanzmann, H.; Augustini, A.L.R.M.; Sanders, D.; Dahlheimer, M.; Wigger, M.; Zech, P.-M.; Sielemann, S. Differentiation of Monofloral Honey Using Volatile Organic Compounds by HS-GCxIMS. Molecules 2022, 27. [CrossRef]
- Lauricella, M.; Emanuele, S.; Calvaruso, G.; Giuliano, M.; D'Anneo, A. Multifaceted Health Benefits of Mangifera indica L. (Mango): The Inestimable Value of Orchards Recently Planted in Sicilian Rural Areas. Nutrients 2017, 9. [CrossRef]
- Tharanathan, R.N.; Yashoda, H.M.; Prabha, T.N. Mango (Mangifera indica L.), “The King of Fruits”—An Overview. Food Reviews International 2006, 22, 95–123. [CrossRef]
- Shimizu, K.; Matsukawa, T.; Kanematsu, R.; Itoh, K.; Kanzaki, S.; Shigeoka, S.; Kajiyama, S.'i. Volatile profiling of fruits of 17 mango cultivars by HS-SPME-GC/MS combined with principal component analysis. Biosci. Biotechnol. Biochem. 2021, 85, 1789–1797. [CrossRef]
- Tandel, J.; Tandel, Y.; Kapadia, C.; Singh, S.; Gandhi, K.; Datta, R.; Singh, S.; Yirgu, A. Nontargeted Metabolite Profiling of the Most Prominent Indian Mango (Mangifera indica L.) Cultivars Using Different Extraction Methods. ACS Omega 2023, 8, 40184–40205. [CrossRef]
- Musharraf, S.G.; Uddin, J.; Siddiqui, A.J.; Akram, M.I. Quantification of aroma constituents of mango sap from different Pakistan mango cultivars using gas chromatography triple quadrupole mass spectrometry. Food Chem. 2016, 196, 1355–1360. [CrossRef]
- Pino, J.A.; Mesa, J.; Muñoz, Y.; Martí, M.P.; Marbot, R. Volatile components from mango (Mangifera indica L.) cultivars. J. Agric. Food Chem. 2005, 53, 2213–2223. [CrossRef]
- Mahattanatawee, K.; Goodner, K.; Baldwin, E.A. Volatile constituents and character impact compounds of selected Florida's tropical fruit. Proc Fla State Hort Soc 2005, 118.
- Pandit, S.S.; Chidley, H.G.; Kulkarni, R.S.; Pujari, K.H.; Giri, A.P.; Gupta, V.S. Cultivar relationships in mango based on fruit volatile profiles. Food Chem. 2009, 114, 363–372. [CrossRef]
- Ziegler, H. Flavourings: Production, composition, applications, regulations, 2nd, completely rev ed.; Wiley: Chichester, Weinheim, 2007, ISBN 3527611452.
- Farag, M.A.; Dokalahy, E.U.; Eissa, T.F.; Kamal, I.M.; Zayed, A. Chemometrics-Based Aroma Discrimination of 14 Egyptian Mango Fruits of Different Cultivars and Origins, and Their Response to Probiotics Analyzed via SPME Coupled to GC-MS. ACS Omega 2022, 7, 2377–2390. [CrossRef]
- Indrati, N.; Sumpavapol, P.; Samakradhamrongthai, R.S.; Phonsatta, N.; Poungsombat, P.; Khoomrung, S.; Panya, A. Volatile and non-volatile compound profiles of commercial sweet pickled mango and its correlation with consumer preference. Int J of Food Sci Tech 2022, 57, 3760–3770. [CrossRef]
- Bonneau, A.; Boulanger, R.; Lebrun, M.; Maraval, I.; Gunata, Z. Aroma compounds in fresh and dried mango fruit (M angifera indica L . cv. K ent): impact of drying on volatile composition. Int J of Food Sci Tech 2016, 51, 789–800. [CrossRef]
- Li, L.; Yi, P.; Sun, J.; Tang, J.; Liu, G.; Bi, J.; Teng, J.; Hu, M.; Yuan, F.; He, X.; et al. Genome-wide transcriptome analysis uncovers gene networks regulating fruit quality and volatile compounds in mango cultivar 'Tainong' during postharvest. Food Res. Int. 2023, 165, 112531. [CrossRef]
- Xie, H.; Meng, L.; Guo, Y.; Xiao, H.; Jiang, L.; Zhang, Z.; Song, H.; Shi, X. Effects of Volatile Flavour Compound Variations on the Varying Aroma of Mangoes 'Tainong' and 'Hongyu' during Storage. Molecules 2023, 28. [CrossRef]
- Christmann, J.; Rohn, S.; Weller, P. gc-ims-tools – A new Python package for chemometric analysis of GC–IMS data. Food Chem. 2022, 224, 133476. [CrossRef]
- Parastar, H.; Christmann, J.; Weller, P. Automated 2D peak detection in gas chromatography-ion mobility spectrometry through persistent homology. Analytica Chimica Acta 2024, 1289, 342204. [CrossRef]
- Capitain, C.C.; Zischka, M.; Sirkeci, C.; Weller, P. Evaluation of IMS drift tube temperature on the peak shape of high boiling fragrance compounds towards allergen detection in complex cosmetic products and essential oils. Talanta 2023, 257, 124397.
- D'Orazio, G.; Fanali, C.; Asensio-Ramos, M.; Fanali, S. Chiral separations in food analysis. TrAC Trends in Analytical Chemistry 2017, 96, 151–171. [CrossRef]
- Christmann, J.; Rohn, S.; Weller, P. Finding features - variable extraction strategies for dimensionality reduction and marker compounds identification in GC-IMS data. Food Research International 2022, 161, 111779. [CrossRef]
- Kulkarni, R.; Chidley, H.; Deshpande, A.; Schmidt, A.; Pujari, K.; Giri, A.; Gershenzon, J.; Gupta, V. An oxidoreductase from 'Alphonso' mango catalyzing biosynthesis of furaneol and reduction of reactive carbonyls. Springerplus 2013, 2, 494. [CrossRef]
- Kallio, H.P. Historical Review on the Identification of Mesifurane, 2,5-Dimethyl-4-methoxy-3(2 H)-furanone, and Its Occurrence in Berries and Fruits. J. Agric. Food Chem. 2018, 66, 2553–2560. [CrossRef]
- Zhang, W.; Zhu, G.; Zhu, G. The imitation and creation of a mango flavor. Food Sci. Technol 2022, 42. [CrossRef]
- Surburg, H.; Panten, J. Common fragrance and flavor materials: Preparation, properties and uses, 6 completely revised and updated edition; Wiley-VCH: Weinheim, 2016, ISBN 978-3-527-69317-7.
- Burdock, G.A.; Fenaroli, G. Fenaroli's handbook of flavor ingredients, 6th ed.; CRC Press/Taylor & Francis Group: Boca Raton Fla., 2010, ISBN 9780429150838.
Figure 1.
THS-GC-MS-IMS setup and flow directions.

Figure 2.
Schematic visualization of multiple injection trapped headspace sampling.
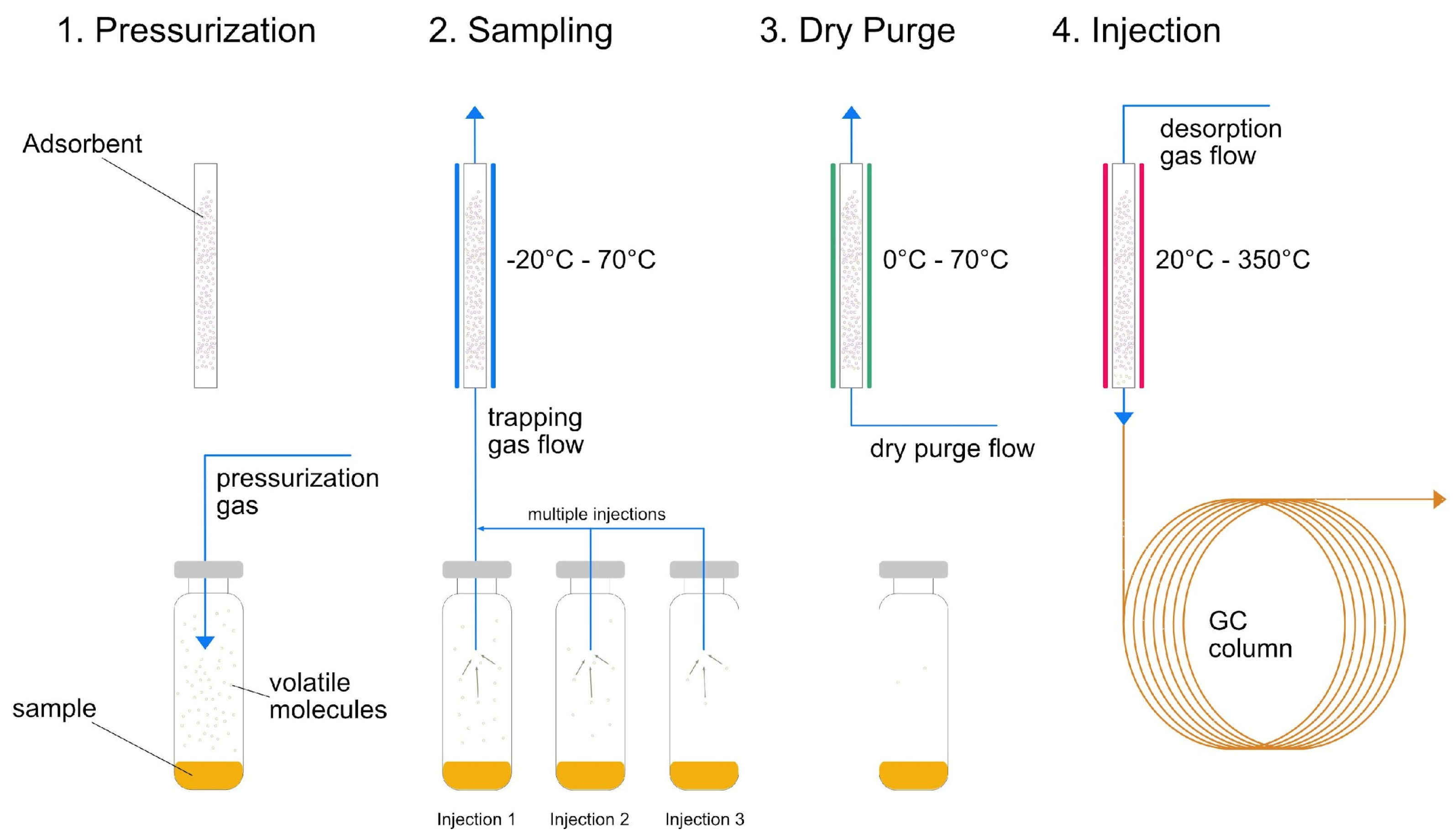
Figure 3.
Exemplary visualization of a ‘Totapuri’ mango sample and the simultaneous sample data of MS (a) and IMS (b) with selected peaks. The peaks are ethyl acetate (1), α-pinene (2), ethyl butyrate (3), β-myrcene (4), D-limonene (5), trans-β-ocimene (6), acetic acid (7), nonanal (8), α-terpineol (9), β-caryophyllene (10). Dimers are marked with #.
Figure 3.
Exemplary visualization of a ‘Totapuri’ mango sample and the simultaneous sample data of MS (a) and IMS (b) with selected peaks. The peaks are ethyl acetate (1), α-pinene (2), ethyl butyrate (3), β-myrcene (4), D-limonene (5), trans-β-ocimene (6), acetic acid (7), nonanal (8), α-terpineol (9), β-caryophyllene (10). Dimers are marked with #.
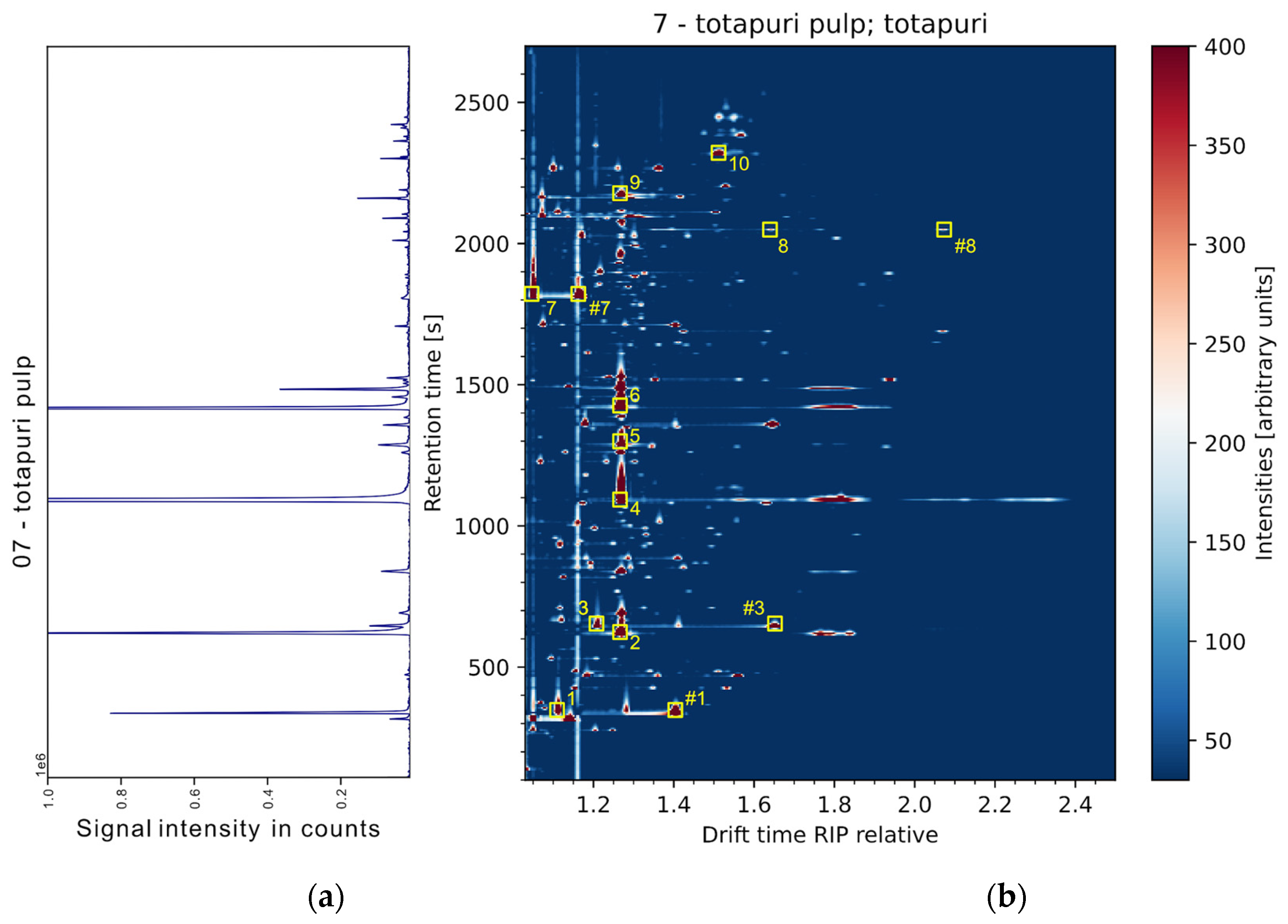
Figure 4.
(a) PCA scores plot of PC2 and PC3. The arrow indicates the direction of the loadings influence of PC2. (b) PCA loadings of PC2 with selected, (MS-) confirmed compounds. The compounds are α-pinene (1), camphene (2), β-pinene (3), β-myrcene (4), α-phellandrene (5), trans-β-ocimene (6), 2-butenoic acid, ethyl ester, (E) (7), β-phellandrene (8), 2-pronanone,1-methoxy (9) , 3-penten-2-one (10), 2,5-dimethyl-4-methoxy-3(2H)-furanone (11), α-terpineol (12), α-copaene (13), α-gurjunene (14), β-caryophyllene (15), 1,4,7,-cycloundecatriene, 1,5,9,9-tetramethyl-, Z,Z,Z- (16), and β-selinene (17). Dimers are marked #.
Figure 4.
(a) PCA scores plot of PC2 and PC3. The arrow indicates the direction of the loadings influence of PC2. (b) PCA loadings of PC2 with selected, (MS-) confirmed compounds. The compounds are α-pinene (1), camphene (2), β-pinene (3), β-myrcene (4), α-phellandrene (5), trans-β-ocimene (6), 2-butenoic acid, ethyl ester, (E) (7), β-phellandrene (8), 2-pronanone,1-methoxy (9) , 3-penten-2-one (10), 2,5-dimethyl-4-methoxy-3(2H)-furanone (11), α-terpineol (12), α-copaene (13), α-gurjunene (14), β-caryophyllene (15), 1,4,7,-cycloundecatriene, 1,5,9,9-tetramethyl-, Z,Z,Z- (16), and β-selinene (17). Dimers are marked #.
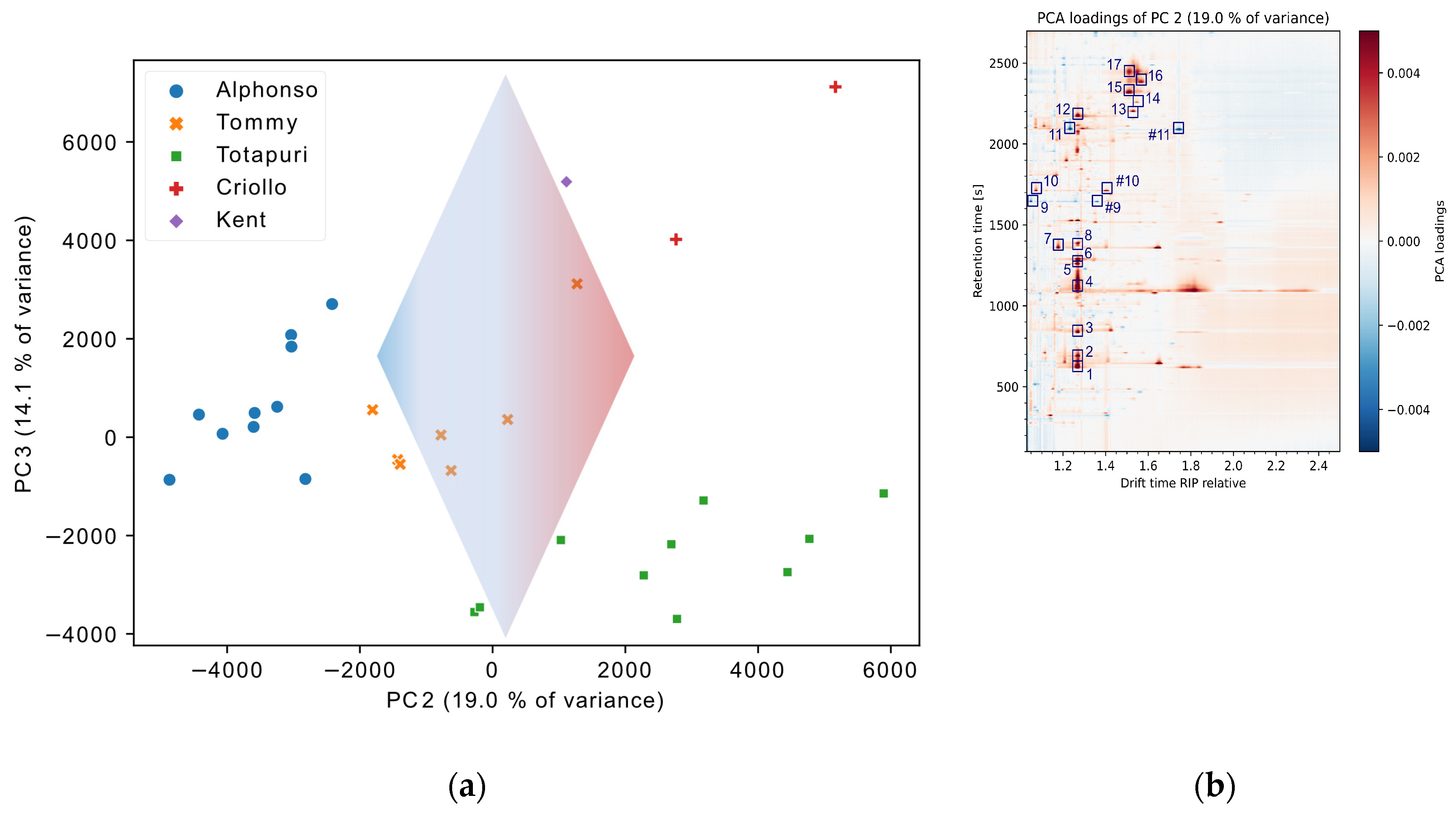
Figure 5.
(a) PCA scatter plot of PC1 and PC2. The arrow indicates the direction of the loadings influence of PC1 (b) PCA loadings of PC1 with selected, (MS-) confirmed compounds. Marked by rectangle: concentrate. The compounds are ethanol (1), ethyl acetate (2), β-pinene (3), β-myrcene, (4), α-phellandrene (5), trans-β-ocimene (6), 2,3-butanedion (7), β-phellandrene (8), acetic acid (9), 3-penten-2-one (10), and 2,5-dimethyl-4-methoxy-3(2H)-furanone (11).
Figure 5.
(a) PCA scatter plot of PC1 and PC2. The arrow indicates the direction of the loadings influence of PC1 (b) PCA loadings of PC1 with selected, (MS-) confirmed compounds. Marked by rectangle: concentrate. The compounds are ethanol (1), ethyl acetate (2), β-pinene (3), β-myrcene, (4), α-phellandrene (5), trans-β-ocimene (6), 2,3-butanedion (7), β-phellandrene (8), acetic acid (9), 3-penten-2-one (10), and 2,5-dimethyl-4-methoxy-3(2H)-furanone (11).
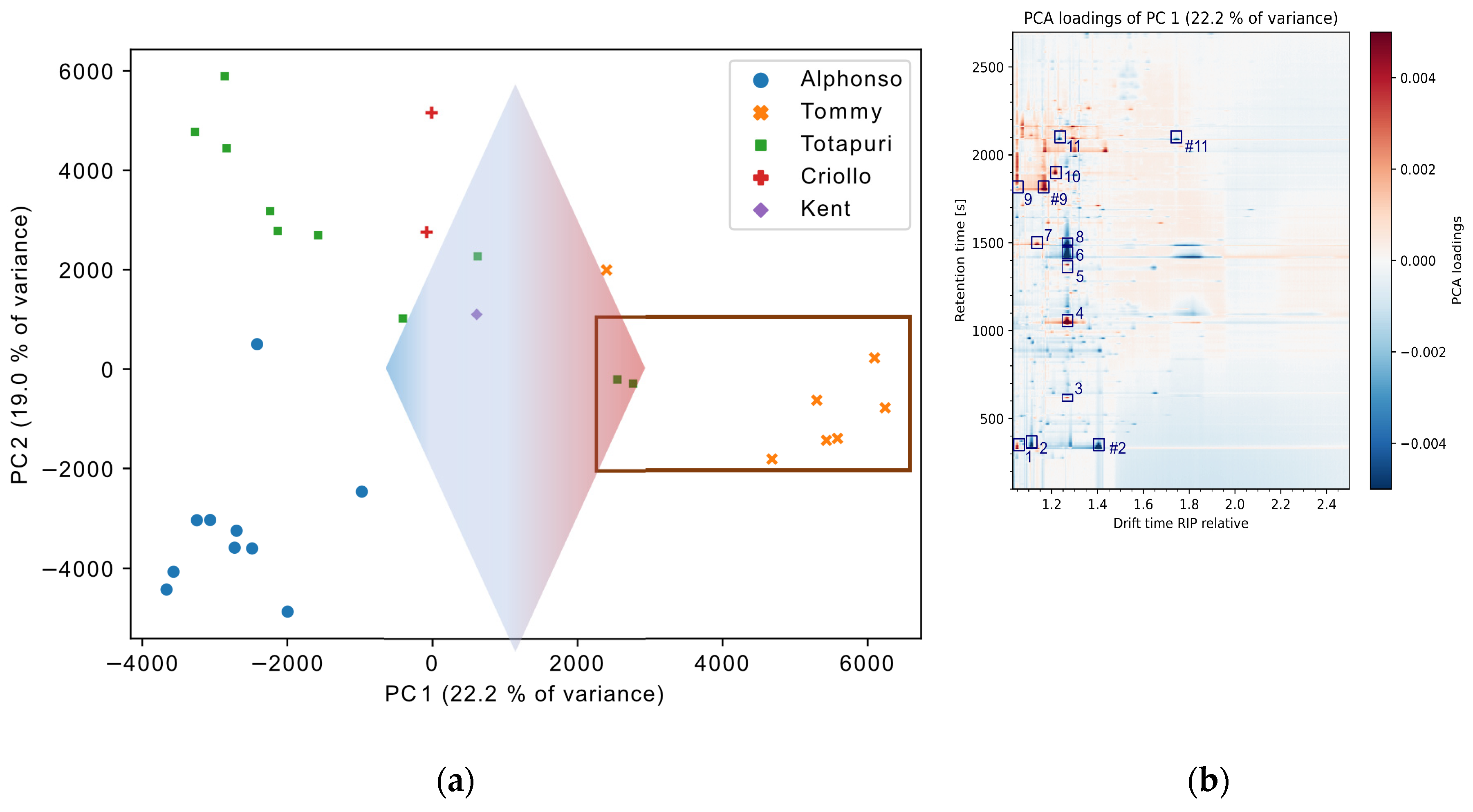
Figure 6.
HCA dendrogram, displaying the distances of the different mango samples.
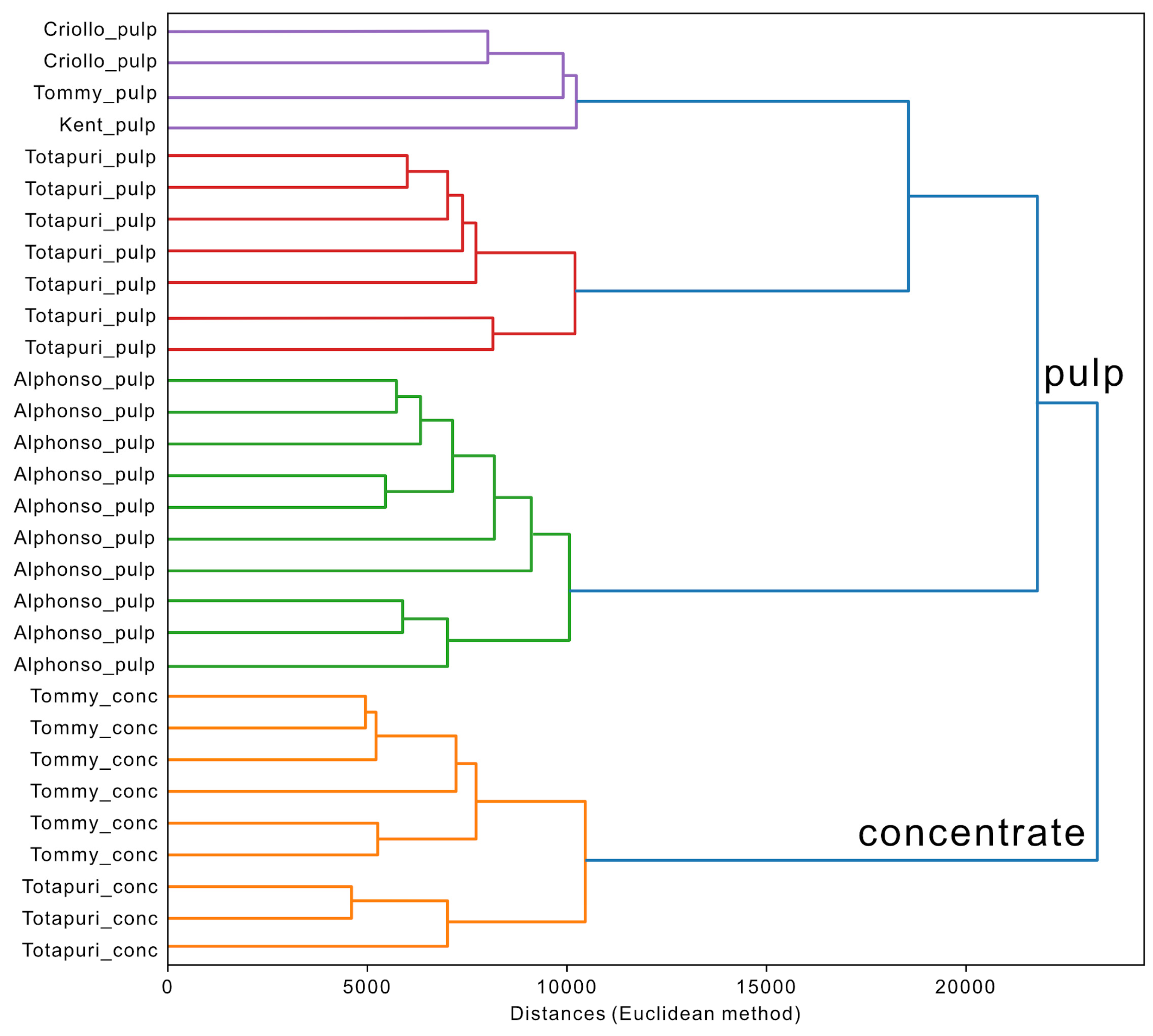

Table 1.
Retention time of IMS, MS, their retention time difference and the MS match quality of the compounds.
Table 1.
Retention time of IMS, MS, their retention time difference and the MS match quality of the compounds.
| Volatile compound | Retention time IMS [min] |
Retention time MS [min] | RT Difference MS-IMS [min] |
MS match quality [%] |
|---|---|---|---|---|
| ethyl acetate | 5.53 ± 0.02 | 5.50 ± 0.04 | 0.03 | 97 |
| α-pinene | 10.28 ± 0.01 | 10.23 ± 0.01 | 0.05 | 96 |
| ethyl butyrate | 10.70 ± 0.02 | 10.66 ± 0.02 | 0.04 | 95 |
| β-myrcene | 18.15 ± 0.02 | 18.12 ± 0.03 | 0.03 | 95 |
| D-limonene | 21.42 ± 0.01 | 21.38 ± 0.01 | 0.04 | 95 |
| trans-β-ocimene | 23.60 ± 0.03 | 23.56 ± 0.01 | 0.04 | 95 |
| acetic acid | 30.10 ± 0.04 | 30.05 ± 0.05 | 0.05 | 95 |
| nonanal | 34.11 ± 0.02 | 34.02 ± 0.01 | 0.07 | 95 |
| α-terpineol | 36.06 ± 0.03 | 36.01 ± 0.01 | 0.05 | 95 |
| β-caryophyllene | 38.51 ± 0.01 | 38.36 ± 0.01 | 0.15 | 96 |
Table 2.
Compounds detected in the different cultivars; x: detected both in IMS and QMS; IMS: detected in IMS only; QMS: detected in MS only; n.d.: not detected.
Table 2.
Compounds detected in the different cultivars; x: detected both in IMS and QMS; IMS: detected in IMS only; QMS: detected in MS only; n.d.: not detected.
|
Mango cultivar |
||||||
| Nr. | Volatile compound | Alphonso | Totapuri | Criollo | Tommy | Kent |
| 1 | ethanol | x | x | x | x | x |
| 2 | ethyl acetate | x | x | x | x | x |
| 3 | unidentified | IMS only | IMS only | IMS only | IMS only | IMS only |
| 4 | ethylcyclohexane | x | x | x | x. | x |
| 5 | ethyl propanoate | x | x (pulp only) |
x | x (pulp only) |
x |
| 6 | propyl acetate | x | x (pulp only) |
IMS only | n.d. | IMS only |
| 7 | methyl butyrate | IMS only | x (pulp only) |
x | x (pulp only) |
x |
| 8 | unidentified | IMS only | IMS only | IMS only | IMS only | IMS only |
| 9 | unidentified | IMS only | IMS only | IMS only | IMS only | IMS only |
| 10 | α-pinene | x | x | x | x | x |
| 11 | ethyl butyrate | x | x (pulp only) |
x | x | x |
| 12 | unidentified | IMS only | IMS only | IMS only | IMS only | IMS only |
| 13 | camphene | x | x | x | x | x |
| 14 | 2-butenoic acid, methyl ester, (z)- | IMS only | x (pulp only) |
x | x (pulp only) |
x |
| 15 | butyl acetate | IMS only | x | x | x | x |
| 16 | unidentified | IMS only | IMS only | IMS only | IMS only | IMS only |
| 17 | β-pinene | x | x | x | x | n.d. |
| 18 | 1-butanol | x | x (pulp only) |
x | x | x |
| 19 | isovaleraldehyd | x | x | x | x | x |
| 20 | isobutanol | x | x (pulp only) |
x | x | IMS only |
| 21 | isobutyraldehyde | x | x | x | x | x |
| unidentified | IMS only | n.d. | n.d. | n.d. | IMS only | |
| 22 | pentanal | x | x | x | x | x |
| 23 | isopentyl alcohol | x | x (pulp only) |
x | x | x |
| 24 | 3-carene | n.d. | n.d. | x | x | x |
| 25 | ethyl cyclopropancarboxylate | n.d. | x (pulp only) |
x | x | x |
| 26 | β-myrcene | x | x | x | x | x |
| 27 | α-phellandrene | IMS only | x | x | x | x |
| 28 | 2-butanone | x | x | x | x | x |
| 29 | α-terpinene | IMS only | x | x | x | x |
| 30 | isobutyl butyrate | IMS only | x (pulp only) |
n.d. | n.d. | n.d. |
| 31 | d-limonene | x | x | x | x | x |
| 32 | unidentified | IMS only | IMS only | IMS only | IMS only | IMS only |
| 33 | 2-butenoic acid, ethyl ester, (e) | n.d. | x (pulp only) |
x | x | x |
| 34 | β-phellandrene | n.d. | x | x | x | x |
| 35 | trans-β-ocimene | x | x | x | n.d. | n.d. |
| 36 | 4-carene | n.d. | n.d. | x | x | x |
| 37 | 2,3-butanedion | x | x (conc. only) |
x | x | x |
| 38 | cis- β-ocimene | x | x | x | n.d. | n.d. |
| 39 | butyl butyrate | IMS only | x (pulp only) |
x | x | x |
| 40 | α-terpinolene | n.d. | x | x | x | x |
| 41 | ethyl hexanoate | n.d. | n.d. | x | x | n.d. |
| 42 | 2-pronanone,1-methoxy | x | n.d. | x | n.d. | IMS only. |
| 43 | 2-methylbutyl butyrate | n.d. | x | x | n.d. | n.d. |
| 44 | isoamyl butyrate | x | x (pulp only) |
x | x | x |
| 45 | 3-penten-2-one | x | x | x | x | x |
| 46 | unidentified | IMS only | IMS only | IMS only | IMS only | IMS only |
| 47 | unidentified | IMS only | IMS only | IMS only | IMS only | IMS only |
| 48 | acetic acid | x | x | x | n.d. | x |
| 49 | alloocimene | x | x | x | n.d. | n.d. |
| 50 | ethyl-3-hydroxbutyrate | x | x | x | x | x |
| 51 | p-1,3,8-menthatriene | x | x | n.d. | n.d. | n.d. |
| 52 | neo-alloocimene | x | x | x | n.d. | n.d. |
| 53 | nonanal | x | x | x | x | x |
| 54 | furfural | x | x | x | x | x |
| 55 | ethyl octanoate | n.d. | x (pulp only) |
x | x | x |
| 56 | trans-sabinene hydrate | n.d. | x | n.d. | n.d. | n.d. |
| 57 | 2,5-dimethyl-4-methoxy-3(2h)-furanone | x | n.d. | n.d. | n.d. | n.d. |
| 58 | acetoin | n.d. | x | x | x | x |
| 59 | β-terpineol | n.d. | x | n.d | x | n.d. |
| 60 | unidentified | IMS only | IMS only | IMS only | IMS only | IMS only |
| 61 | α-terpineol | n.d. | x | n.d. | x | n.d. |
| 62 | α-copaene | n.d. | x | n.d. | x | n.d. |
| 63 | α-gurjujene | n.d. | x | x | x | x |
| 64 | unidentified | IMS only | IMS only | IMS only | IMS only | IMS only |
| 65 | β-caryophyllene | x | x | x | x | x |
| 66 | α-guaiene | n.d. | x | n.d. | n.d. | n.d. |
| 67 | unidentified | n.d. | IMS only | IMS only | IMS only | IMS only |
| 68 | ethyl decanoate | n.d. | n.d. | x | n.d. | n.d. |
| 69 | 1,4,7,-cycloundecatriene, 1,5,9,9- tetramethyl-, z,z,z |
x | x | x | x | x |
| 70 | 4,5-di-epi-aristolochene | n.d. | x | x | x | n.d |
| 71 | γ-gurjujene | n.d. | x | x | n.d. | n.d |
| eremophilene | n.d. | n.d. | QMS only | n.d. | n.d | |
| 72 | β-selinene | n.d. | x | x | x (pulp only) |
n.d. |
| 73 | α-selinene | n.d. | x | x | x (pulp only) |
n.d. |
| 74 | α-bulnesene | n.d. | x | x | n.d. | n.d. |
| 75 | δ-cadinene | n.d. | x | x | n.d. | n.d. |
| 76 | β-cadinene | n.d. | QMS only | n.d. | n.d. | n.d. |
| 77 | γ-butyrolactone | x | n.d. | x | n.d. | n.d. |
| 78 | γ-hexalactone | x | n.d. | x | n.d. | IMS only |
| 79 | cis-calamenene | n.d. | n.d. | QMS only | n.d. | n.d. |
| 80 | γ-octalactone | x | n.d. | x | n.d. | n.d. |
| 81 | ethyl dodecanoate | x | n.d. | x | n.d. | n.d. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



