
Submitted:
16 July 2024
Posted:
17 July 2024
You are already at the latest version
Abstract
Half-metallic semi-Heusler compounds are currently at the forefront of scientific research due to their potential applications in spintronic devices. Unlike other semi-Heuslers, the p0(d0)-d compounds do not appear to crystallize in the typical variant of the C1b structure. We investigate this phenomenon in the p0-d Heusler compounds LiYGa and LiYGe, where Y varies between Ca and Zn, using first-principles ab-initio electronic band structure calculations. We examine the electronic and magnetic properties of these compounds in relation to the three possible C1b structures. Notably, LiVGa, LiVGe, LiMnGa, and LiCrGe are half-metallic ferromagnets across all three variations of the C1b lattice structure. Our findings will serve as a foundation for future experimental studies on these compounds.
Keywords:
Heusler compounds
; Ab-initio calculations
; First-principles
; Electronic structure
; magnetic materials
; Slater-Pauling rule
1. Introduction
In the early 20th century, German metallurgist Heusler, while exploring ways to improve the electrical conductivity of steel, made a groundbreaking discovery [1,2]. He identified a novel compound, Cu2MnAl. As the century progressed, advancements in instrumentation revealed that Cu2MnAl possesses a face-centered cubic (f.c.c.) lattice structure, akin to well-known semiconductors such as Si and GaAs. This lattice structure is also present in a variety of intermetallic compounds with unique properties, which became known as "Heusler compounds" or "Heusler alloys" [3,4]. Many Heusler compounds notably exhibit ferromagnetic properties with high Curie temperatures. They can be classified into four distinct families based on the number and valence of their constituent atoms:
- Semi-Heusler compounds, such as NiMnSb, follow the chemical formula. Here, X and Y represent transition metal atoms or lanthanides, while Z is a metalloid. The lattice structure of semi-Heusler compounds is denoted as "".
- Full-Heusler compounds, such as Co2MnSi, have the chemical formula , with X, Y, and Z atoms similar to those in semi-Heuslers. These compounds crystallize in the "" lattice structure.
- Inverse Heuslers are similar to full-Heuslers, but the valence of X is smaller than that of Y. Their lattice structure is known as "" or "."
In all these Heusler compound families, the metalloid atom Z plays a significant role.
In the early 21st century, interest in Heusler compounds has surged, primarily due to the discovery of half-metallicity as a common feature among various ferromagnetic and ferrimagnetic Heusler compounds [6,7,8,9]. Half-metallic compounds exhibit typical metallic behavior for majority spin electrons while displaying semiconducting characteristics for minority spin electrons [10]. This unique property results in a high degree of spin polarization at the Fermi level, making them particularly attractive for spintronics and magnetoelectronics applications by introducing novel functionalities to electronic devices. While other materials have been explored for their half-metallic properties, Heusler compounds offer distinct advantages due to their high Curie temperatures. Consequently, extensive research has focused on investigating their fundamental properties and potential applications [11,12,13,14,15]. Recent studies have suggested that certain magnetic Heusler compounds may exhibit even more unconventional behaviors beyond half-metallicity, such as spin-gapless semiconducting and spin-filtering properties, introducing entirely new functionalities with promising implications for various applications [16].
The use of first-principles calculations, also known as ab-initio calculations, is a powerful method to understand the properties of materials and predict the development of new compounds with tailored properties. Recently, an increasing number of extensive databases based on first-principles calculations have emerged, encompassing hundreds of magnetic Heusler compounds [17,18,19,20,21,22]. These compounds show significant promise in the fields of spintronics and magnetoelectronics. These databases complement studies that focus primarily on understanding the fundamental origins of these compounds properties, which typically investigate a relatively limited number of Heusler compounds [6,7,8,9].
Modern growth techniques have enabled the realization of thin film compounds that were initially conceived through theoretical predictions. For example, (CrV)TiAl, a quaternary Heusler compound, was predicted in Reference [23] to be a fully-compensated ferrimagnetic semiconductor. It was later successfully synthesized, and its distinctive magnetic properties were verified, as evidenced by research in Reference [24]. This success highlights the strong rationale for exploring novel Heusler compounds that might exhibit unique properties. As previously mentioned, when it comes to magnetic Heusler compounds, X typically represents a transition metal or rare earth element. However, there are cases within the family of semi-Heusler compounds where X can be entirely replaced by an alkali atom or an alkali-earth atom. These particular compounds are referred to as "-d or -d Heusler compounds. The term refers to the Li, Be, Na and Mg elements and the term refers to the K, Rb, Cs, Ca, Sr and Ba elements; the term refers to the character of the first empty states in the free atom. Damewood et al [25], and Dehghan and Davatolhagh [26] have studied using ab-initio electronic structure calculations the LiMnPt, SrVSb and KMnP compounds. In Reference [27] Dehghan and Davatolhagh created a database containing 420 -d Heusler compounds whenre X was one of K, Rb or Ks, Y was a transition metal atom, and Z was a a group-IV, -V or -VI element. Among the 420 compounds, 98 were identified as half-metals following the =-8 Slater-Pauling rule (=-18 when Y was Cu or Zn) [28]. In 2022, the same group expanded their database to cover als the case of -d semi-Heusler compounds wher X was one of Li, Be, Na or Mg and Z was a group-V or group-VI element [29].
Although databases are extremely useful, because of the large number of compounds studied, they do not analyze in depth the properties of the compounds. Motivated by the above-mentioned results, in the present study, our focus is directed towards the LiYGa and LiYGe compounds. Y is a 3d transition metal atom ranging from Sc to Zn. For reasons of completeness we have also taken into account the case where Y is Ca an alkaline-earth element. The rationale behind this focus lies in the remarkable properties exhibited by these compounds, rendering them well suited for applications in the fields of spintronics and magnetoelectronics. Our research comprehensively examines various facets of these compounds, including their structural, electronic and magnetic properties.
2. Computational Details
Our research is dedicated to investigating the ground-state properties of Li-base semi-Heusler compounds. To accomplish this, we employ the full-potential nonorthogonal local- orbital minimum-basis band structure approach (FPLO) for our first-principles electronic band structure calculations, as outlined in References [30,31]. In these calculations, we apply the generalized gradient approximation (GGA) as the exchange-correlation functional within the Perdew-Burke-Ernzerhof (PBE) parameterization [32]. This choice is well-known for yielding precise outcomes, especially when dealing with half-metallic Heusler compounds, aligning closely with experimental observations [6,7]. To ensure the accuracy of our calculations, the total energy is converged to the 10th decimal point. Furthermore, a dense grid of k-points, specifically a grid, according to the Monkhorst-Pack scheme [33], is utilized for the integrals in reciprocal space.
3. Results and Discussion
3.1. Structural Properties
Semi-Heusler compounds as mentioned above crystallize in the lattice structure where one of the four inequivalnet sites is vacant. Depending on the sequence of the atoms there are three variants of the , which are widely known as the , and phases. All three phases are presented in Figure 1. Each A and C site (black spheres and black squares in the figure) are at the center of a cube surrounded by four B (pink spheres) and four D (green shperes) sites. Each A(C) site has as second-neighbors six C(A) sites. The same reasoning stands also for the nearest and next-nearest neighbors of the B and D sites. Thus in all cases the tetrahedral symmetry is present, and what alters is the local environment of the atoms and thus the interactions (hybridization) between orbitals located at nearest neighbors.
For all 22 compounds under study and for all three phases, we have determined the equilibrium lattice constant using total energy calculations; the former is the lattice constant for which the total energy is minimum. In Table 1 we gather our results. First, we have to note that the lattice constants range between 5.42 Å and 7.14 Å and thus are in the same range as most of the well-known Heusler compounds and binary semiconductors which is useful for practical applications where multilayers are used. The Ga compounds have slightly larger values of the equilibrium lattice constant than the corresponding Ge ones. This is expected since Ga has a slightly larger atomic radius as a free atom than Ge. If now, one keeps Ga or Ge fixed and varies Y between Ca and Zn the lattice constants follow the trend of the atomic radius of the free atoms. The equilibrium lattice constant decreases as we move from Ca to V and then increases as we move from V to Zn. If now one compares the three phases among them, the only safe conclusion is that the phase, where Li has four Y and four atoms as nearest neighbors corresponds also to the smallest equlibrium lattice constant. If we compare the and phases between them, there is no clear trend although in most cases the phase corresponds to larger lattice constants. In these two phases each Li atom has as nearest neighbors four vacant sites (Voids in Figure 1) and four Y or four atoms for the and phases, respectively.
Finally, we should comment on the relative stability of the three possible phases. In Table 1 we include the energy differences in eV units between the three phases expressed per formula unit. A minus sign means that the phase corresponding to the first total energy is the most stable among the two phases which are compared. The absolute energy differences in all cases are below 1 eV. Although this value may seem quite small, this is not really the case. At room temperature, the thermal energy much smaller than most of the values in Table 1. Thus at room temperature, the thermal energy provided at the compound is not enough to overcome the energy barrier and adopt another phase. One may envisage that this could be achieved by growing low-dimensional samples such as thin films and nanostructures.
To make clearer the behavior of the compounds in the last two columns of Table 1. we present the most and least stable structure for all compounds under study. We see that for both LiYGa and LiYGe compounds there is a clear trend. For most of them the phase is the most stable as is also the case for most of the -d and -d compounds studied in literature [25,26,27,29]. For the lighter Y elements the least stable is the phase. Ss we move to heavier elements, the phase first becomes energetically more favorable than the phase, and for the heaviest atoms its total energy becomes even lower than the phase and it is now the most stable phase.
3.2. Electronic Properties
At the equilibrium lattice constants, we performed electronic band structure calculations for all twenty two compounds within our study. Subsequently, we extracted the total density of states (DOS) per formula unit (f.u.), which is visually represented in Figure 2 for the LiYGa compounds and in Figure 3 for the LiYGe compounds. For each compound, we have performed calculations for all three , and phases presented in the three different columns in the two figures. In all cases there is also some DOS weight around -6 to -9 eV, which corresponds to one s-state per spin direction stemming from the Li atom and which is not shown in the figures.
Overall, the total DOS in the window which we show is governed by p-d hybrids created between the p-states of the Ga/Ge atoms and the d states of the transition-metal atom. In the case of the tetrahedral symmetry, the valence d states split into the double degenerate and the triple degenerate states. The latter transform following the same representation as the valence p states of the Ga/Ge atoms, and thus they are allowed to hybridize and create new states which spread across the transition metal and the Ga/Ge atoms. In the case of LiCaGa and LiCaGe compounds, the Ca atoms have no valence d-states and the DOS shown in Figure 2 and Figure 3 is due to the interaction p-p of the Ca and Ga/Ge valence p states. Finally in the case where Y is Cu all d states are occupied and thus are low in energy and the DOS at the Fermi level is very low. This is even more pronounced in the case of the LiZnGa and LiZnGe compounds where the Zn valence d states shift even deeper in energy and are below the energy window shown in the figures.
Most of the compounds, as can be easily deduced by the DOS in the two figures, are semiconductors (identical DOS for both spin directions), and only a few are magnetic. For magnetism to be favorable, usually the Stoner criterion must be valid, and the DOS at the Fermi level in the magnetic case should result to lower total energy than in the non-magnetic case. We will discuss in detail the magnetic properties in the next subsection. Among the magnetic compounds a few are half-metals or almost half-metals possesing a gap in the spin-down band structure. A detalined analysis of the DOS around the Fermi level reveals that perfect half metallicity is found in the case of LiCaGe in both and phases, LiScGa only in the phase (in the phase it is close to half-metallicity), and LiVGa/LiVGe/LiCrGe in all three phases. The LiMnGa and LiMnGe compounds are also close to half-metallicity, except LiMnGa in phase which is a perfect half-metal, since the Fermi level crosses slightly the band just below or just above the spin-down energy gap.
Finally, we should shortly comment on the behavior of the DOS as a function of the three phases. Overall, the DOS between the and phases is similar. The common feature of these two phases is that the Y and Ga/Ge atoms are nearest neighbors. This leads to similar bonding between the valence states of these atoms and thus to similar DOS. The Li atoms contribute only with the s states very deep in energy. Thus, its different local environment in the two phases plays no crucial role in the shape of the DOS. The shape of the DOS for the phase where the Y and Ga/Ge atoms are next-nearest neighbors has more pronounced differences. Thus one can conclude that the reason the most stable structure for all twenty two compounds, as presented in Table 1, is either the or the phase is the stronger bonding between the Y and Ga/Ge atoms which are now nearest neighbors.
3.3. Magnetic Properties
As discussed above, most of the compounds are semiconductors. In Table 2, we provide the computed atomic and total spin magnetic moments for all magnetic compounds employing their equilibrium lattice constants for each phase. In all cases the Ga/Ge atoms carry a large portion of the total spin magnetic moment as expected since the Ga/Ge-p – Y- hybrids have considerable weight also at the Ga/Ge sites. Only in the case of heavier transition metal atoms like V, Cr, Mn and Fe, the spin magnetic moment is mainly concentrated at the Y atom. As it is also the case for most of the Heusler compounds, the Y and Ga/Ge spin magnetic moments are antiparallel when Y is a transition metal atom with the exception of Sc. Li atoms also carry small spin magnetic moments which can be safely neglected with the exception of the LiMnGa and LiFeGa compounds. Here we should also mention that for LiCaGa, LiCaGe and LiScGa the spin magnetic moments in Table 2 have negative sign. Although this has no sence since all are parallel for these three compounds, we adopted this convention in order to be consisten with the Slater-Pauling rule discussed in the next paragraph since these compounds have less than eight valence electrons per formula unit.
Following the discussion of the total spin magnetic moments per formula unit in References [6,28], we focus now on the behavior of the total spin magnetic moment per formula unit. To make our discussion clear we provide in Table 2 the total number of valence electrons per unit cell (which coincides with the per formula unit value), and the total spin magnetic moment expected by the Slater-Pauling rules. In Figure 4 we have plotted the total spin magnetic for all compounds under study as function of the Y element and compare it with the values expected from the Slater-Pauling rule. For the magnetic compounds (total spin magnetic moment does not vanish) when the calculated and the Slater-Pauling rule derived total spin magnetic moments coincide, one could expect that perfect half-metallicity is present. This is true if we compare the total spin magnetic moments with the DOS presented in the previous subsection. Interestingly, as can be deduced from Figure 4 the are four compounds LiVGa, LiMnGa, LiVGe and LiCrGe which have either integer values of their total spin magnetic moments or almost integer in all three phases and thus are either perfect half-metals or almost perfect half-metals, respectively. These compounds are of particular interest since they could find applications in spintronic and magnetoelectronic devices, since half-metallicity is independent of the grown phase.
To elucidate the origin of the spin-down gap in the half-metallic compounds and thus the origin of the Slater-Pauling rule, we should perform an analysis similar to the one for other half-metallic Heusler compounds [28]. When Y is Ca to Ni, in the spin-down electronic band structure, there are exactly four fully occupied states. The first one stems from the s valence state of Li and the following three stem from the p- hybridization between the Ga/Ge and the Y atoms (p-p in the case where Y is Ca). Thus the number of non compensated spins which coincides with the total spin magnetic moment per unit cell is . When Y is Cu and Zn, for perfect half-metallicity to occur, all d valence staes should be occupied and the Slater-Pauling rule becomes now . Of course as discussed above when Y is Co, Ni, Cr or Zn the DOS at the Fermi level for the non-magnetic case is quite low and thus no tendency to magnetism is present.
4. Summary and Conclusions
Half-metallic semi-Heusler compounds are at the forefront of current scientific research due to their potential use in spintronic devices. Unlike other semi-Heuslers, -d compounds can crystallize in three different variations of the lattice structure known as , and phases where the sequence of the atoms in the unit cell changes. Using state-of-the-art ab initio electronic band structure calculations, we focus on the LiYGa and LiYGe -d Heusler compounds, where Y ranges from Ca to Zn. We examined the structural, electronic and magnetic properties of these compounds in relation to the three possible variants of the structure. Our results suggest that all compounds prefer to crystallize in the and phases. For both and phases each compound has similar properties didacted by the fact that the Y and Ga/Ge atoms are nearest neighbors. Among the studied compounds the ones being half-metallic magnets follow a Slater-Pauling behavior with respect to their total spin magnetic moment which can be explained by a band-structure analysis. Notably, LiVGa, LiVGe, LiMnGa, and LiCrGe are (almost) half-metallic ferromagnets across all three phases and thus are of particular interest for applications.
We expect our results to pave the way for further experimental and theoretical studies of these compounds which are susceptible of finding several applications in spintronics and magnetoelectronics.
Funding
This research received no external funding
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The author declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| DOS | Density of States |
| f.u. | formula unit |
| FPLO | Full-potential nonorthogonal local-orbital minimum- basis band structure approach |
| GGA | Generalized gradient approximation |
| PBE | Perdew Burke Ernzerhof |
References
- Heusler, F. Über magnetische manganlegierungen. Verh. Dtsch. Phys. Ges. 1903, 12, 219. [Google Scholar]
- Heusler, F.; Take, E. The nature of the Heusler alloys. Phys. Z. 1912, 13, 897. [Google Scholar] [CrossRef]
- Webster, P.J.; Ziebeck, K.R.A. Alloys and Compounds of d-Elements with Main Group Elements. Part 2. In Landolt-Börnstein, New Series, Group III, vol 19c; Editor Wijn, H.R.J.; Springer: Berlin. 1988; pp. 75–184.
- Ziebeck, K.R.A.; Neumann, K.-U. Magnetic Properties of Metals. In Landolt-Börnstein, New Series, Group III, vol 32/c. Editor Wijn, H.R.J.; Springer: Berlin. 2001; pp. 64–414.
- Graf, T.; Felser, C.; Parkin, S.S.P. Simple rules for the understanding of Heusler compounds. Progress in Solid State Chemistry 2011, 39, 1. [Google Scholar] [CrossRef]
- Galanakis, I.; Dederichs, P.H.; Papanikolaou, N. Origin and Properties of the Gap in the Half-Ferromagnetic Heusler Alloys. Phys. Rev. B 2002, 66, 134428. [Google Scholar] [CrossRef]
- Galanakis, I.; Dederichs, P.H.; Papanikolaou, N. Slater-Pauling Behavior and Origin of the Half-Metallicity of the Full-Heusler Alloys. Phys. Rev. B 2002, 66, 174429. [Google Scholar] [CrossRef]
- Skaftouros, S.; Özdoğan, K.; Şaşıoğlu, E.; Galanakis, I. Generalized Slater-Pauling rule for the inverse Heusler compounds. Phys. Rev. B 2013, 87, 024420. [Google Scholar] [CrossRef]
- Özdoğan, K.; Şaşıoğlu, E.; . Galanakis, I. Slater-Pauling behavior in LiMgPdSn-type multifunctional quaternary Heusler materials: Half-metallicity, spin-gapless and magnetic semiconductors. J. Appl. Phys. 2013, 113, 193903. [Google Scholar] [CrossRef]
- Katsnelson, M.I.; Irkhin, V. Yu; Chioncel, L.; Lichtenstein, A.I.; de Groot, R.A. Half-metallic ferromagnets: From band structure to many-body effects. Rev. Mod. Phys. 2008, 80, 315. [Google Scholar] [CrossRef]
- Hirohata, A.; Takanashi, K. Perspectives of Heusler compounds. J. Phys. D: Appl. Phys. 2014, 47, 193001. [Google Scholar] [CrossRef]
- Half-metallic Alloys. Fundamentals and Applications. In Lectures Notes in Physics, vol. 676. Editors Galanakis, I.; Dederichs, P.H. Springer: Berlin Heidelberg. 2005.
- Spintronics. From Materials to Devices. Editors Felser, C.; Fecher, G.H. Spintronics. From Materials to Devices. Editors Felser, C.; Fecher, G.H., Springer. 2013.
- Half-metallic Materials and Their Properties. In Series on Materials for Engineering, vol. 2. Editors Fong, C.Y.; Pask, J.E.; Yang, L.H. Imperial College Press. 2013.
- Heusler Alloys. Properties, Growth, Applications. In Springer Series in Materials Science, vol. 222. Editors Felser, C.; Hirohata, A. Springer International Publishing. 2018.
- Galanakis, I.; Özdoğan, K.; Şaşıoğlu, E. Spin-filter and spin-gapless semiconductors: The case of Heusler compounds. AIP Adv. 2016, 6, 055606. [Google Scholar] [CrossRef]
- Gillessen, M.; Dronskowski, R. A combinatorial study of full Heusler alloys by first-principles computational methods. J. Comput. Chem. 2009, 30, 1290. [Google Scholar] [CrossRef] [PubMed]
- Gillessen, M.; Dronskowski, R. A combinatorial study of inverse Heusler alloys by first-principles computational methods. J. Comput. Chem. 2010, 31, 612. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Hegde, V.I.; Munira, K.; Xie, Y.; Keshavarz, S.; Mildebrath, D.T.; Wolverton, C.; Ghosh, A.W.; Butler, W.H. Computational investigation of half-Heusler compounds for spintronics applications. Phys. Rev. B 2017, 95, 024411. [Google Scholar] [CrossRef]
- Sanvito, S.; Oses, C.; Xue, J.; Tiwari, A.; Zic, M.; Archer, T.; Tozman, P.; Venkatesan, M.; Coey, M.; Curtarolo, S. Accelerated discovery of new magnets in the Heusler alloy family. Sci. Adv. 2017, 3, e1602241. [Google Scholar] [CrossRef] [PubMed]
- Faleev, S.V.; Ferrante, Y.; Jeong, J.; Samant, M.G.; Jones, B.; Parkin, S.S.P. Unified explanation of chemical ordering, the Slater-Pauling rule, and half-metallicity in full Heusler compounds. Phys. Rev. B 2017, 95, 045140. [Google Scholar] [CrossRef]
- Faleev, S.V.; Ferrante, Y.; Jeong, J.; Samant, M.G.; Jones, B.; Parkin, S.S.P. Heusler compounds with perpendicular magnetic anisotropy and large tunneling magnetoresistance. Phys. Rev. Mater. 2017, 1, 024402. [Google Scholar] [CrossRef]
- Galanakis, I.; Özdoğan, K.; Şaşıoğlu, E. High-TC fully compensated ferrimagnetic semiconductors as spin-filter materials: the case of CrVXAl (X = Ti, Zr, Hf) Heusler compounds. J. Phys.: Condens. Matter 2014, 26, 086003. [Google Scholar] [PubMed]
- Venkateswara, Y.; Gupta, S.; Samatham, S.S.; Varma, M.R.; Enamullah, *!!! REPLACE !!!*; Suresh, K.G.; Alam, A. Competing magnetic and spin-gapless semiconducting behavior in fully compensated ferrimagnetic CrVTiAl: Theory and experiment. Phys. Rev. B 2018, 97, 054407. [Google Scholar] [CrossRef]
- Damewood, L.; Busemeyer, B.; Shaughnessy, M.; Fong, C.Y.; Yang, L.H.; Felser, C. Stabilizing and increasing the magnetic moment of half-metals: The role of Li in half-Heusler LiMnZ (Z=N,P,Si). Phys. Rev. B 2015, 91, 064409. [Google Scholar] [CrossRef]
- Dehghan, A.; Davatolhagh, S. d0-d half-Heusler alloys: A potential class of advanced spintronic materials. J. All. Comp. 2019, 773, 132. [Google Scholar] [CrossRef]
- Dehghan, A.; Davatolhagh, S. First principles study of d0-d half-Heusler alloys containing group-IV, -V, and -VI sp atoms as prospective half-metals for real spintronic applications. Mat. Chem. Phys. 2021, 273, 125064. [Google Scholar] [CrossRef]
- Galanakis, I. Slater–Pauling Behavior in Half-Metallic Heusler Compounds. Nanomaterials 2023, 13, 2010. [Google Scholar] [CrossRef]
- Javari, A.R.; Davatolhagh, S.; Dehghan, A. Half-metallic p0-d half-Heusler alloys with stable structure in ferromagnetic state. J. Phys. Chem. Solids 2022, 166, 110702. [Google Scholar] [CrossRef]
- Koepernik, K.; Eschrig, H. Full-potential nonorthogonal local-orbital minimum-basis band-structure scheme. Phys. Rev. B 1999, 59, 1743. [Google Scholar] [CrossRef]
- Kopernik, K. Full Potential Local Orbital Minimum Basis Bandstructure Scheme User’s Manual (http://www.fplo.de/download/doc.pdf).
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of the three possible phases of the structure adopted by the semi-Heusler compounds. The black spheres, pink spheres, black squares, and green spheres are widely called A, B, C, and D sites respectively. The large cube in the figure contains exactly four primitive unit cells.
Figure 1.
Schematic representation of the three possible phases of the structure adopted by the semi-Heusler compounds. The black spheres, pink spheres, black squares, and green spheres are widely called A, B, C, and D sites respectively. The large cube in the figure contains exactly four primitive unit cells.
Figure 2.
Total density of states (DOS) for the LiYGa compounds for all three phases. The zero energy has been assigned to the Fermi level. Positive(Negative) DOS values correspond to the spin-up(spin-down) electrons.
Figure 2.
Total density of states (DOS) for the LiYGa compounds for all three phases. The zero energy has been assigned to the Fermi level. Positive(Negative) DOS values correspond to the spin-up(spin-down) electrons.
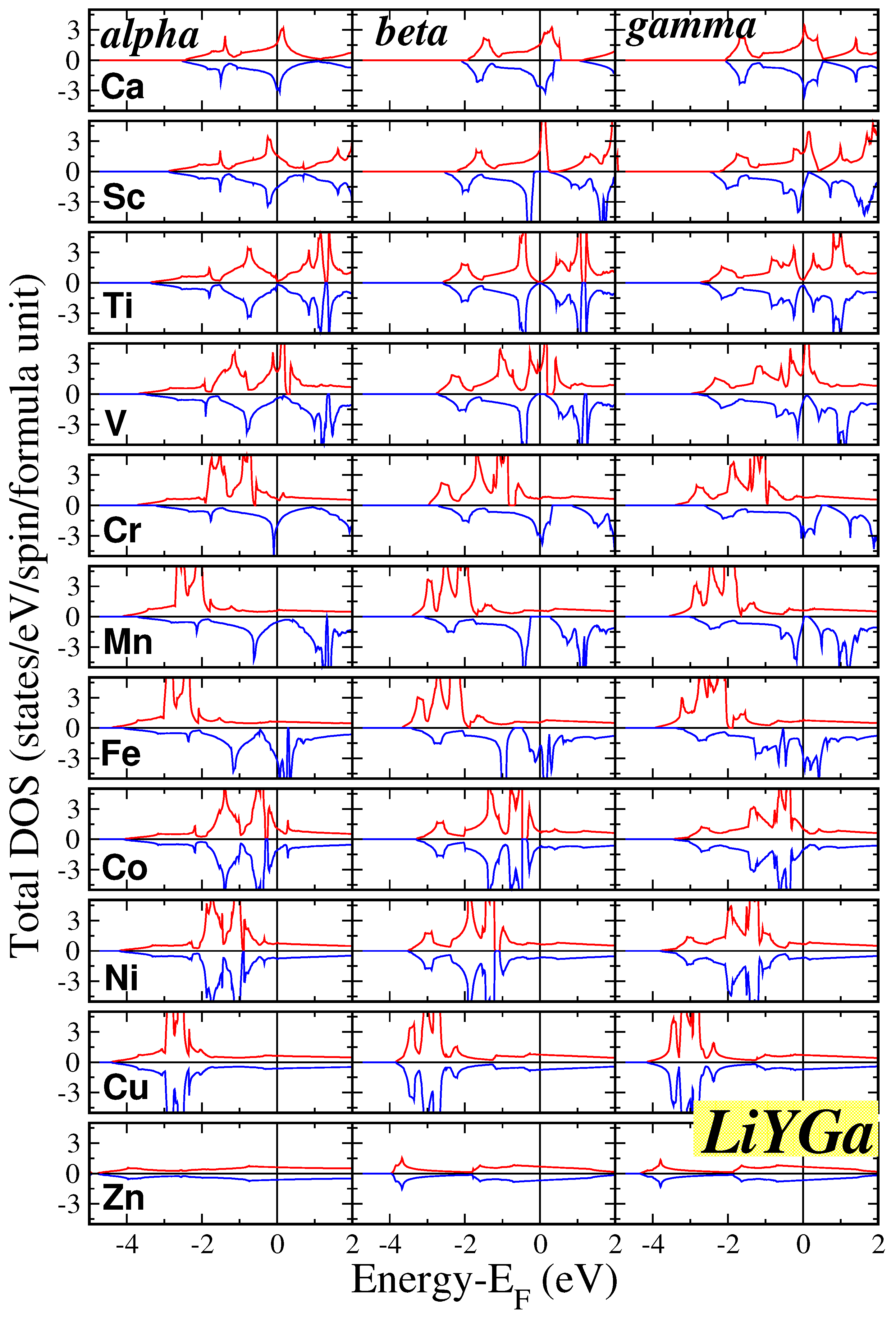
Figure 3.
Similar to Figure 2 for the LiYGe compounds.
Figure 3.
Similar to Figure 2 for the LiYGe compounds.
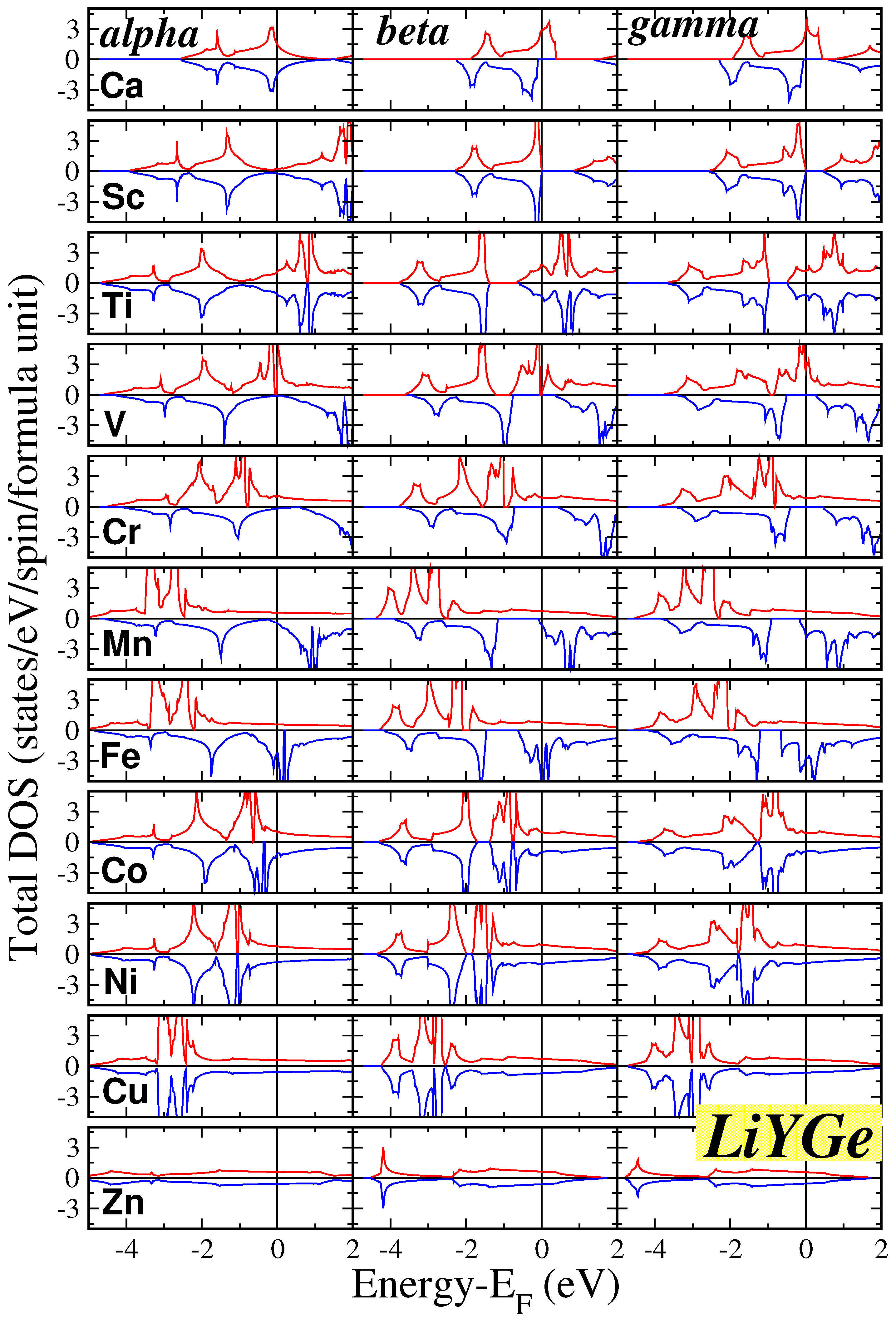
Figure 4.
Total spin magnetic per formula unit in units as a function of the Y chemical element. The red lines represent the ideal Slater Pauling rules for half-metallicity: =-8 for Y = Ca to Ni and =-18 for Y = Cu or Zn.
Figure 4.
Total spin magnetic per formula unit in units as a function of the Y chemical element. The red lines represent the ideal Slater Pauling rules for half-metallicity: =-8 for Y = Ca to Ni and =-18 for Y = Cu or Zn.
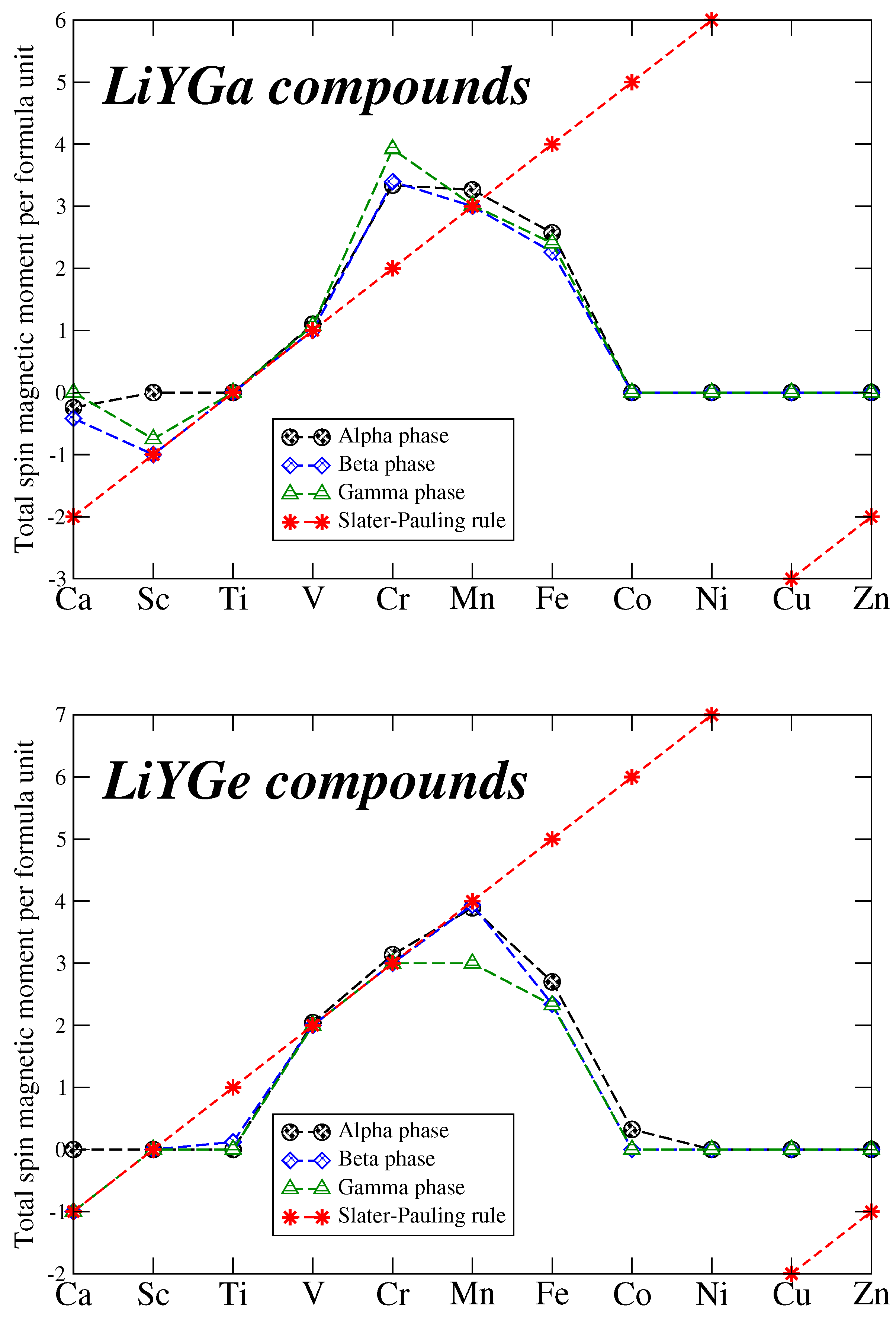

Table 1.
We provide the equilibrium lattice constants for all compounds under study in all three , and phases. The next three columns present the total energy difference between the two phases using for each one the equilibrium lattice constant.
Table 1.
We provide the equilibrium lattice constants for all compounds under study in all three , and phases. The next three columns present the total energy difference between the two phases using for each one the equilibrium lattice constant.
| Compound | Lattice constant a in Å | Energy difference in eV | Most stable |
Least stable |
||||
| Li | -phase | -phase | -phase | phase | phase | |||
| LiCaGa | 6.72 | 7.00 | 7.14 | -0.13 | -0.86 | -0.73 | ||
| LiScGa | 6.13 | 6.42 | 6.52 | -0.17 | -1.04 | -0.87 | ||
| LiTiGa | 5.80 | 6.06 | 6.17 | -0.28 | -1.00 | -0.72 | ||
| LiVGa | 5.69 | 5.93 | 6.03 | -0.30 | -0.67 | -0.37 | ||
| LiCrGa | 5.84 | 6.08 | 6.17 | -0.35 | -0.36 | -0.01 | ||
| LiMnGa | 5.78 | 5.95 | 5.99 | -0.31 | -0.28 | 0.03 | ||
| LiFeGa | 5.66 | 5.81 | 5.89 | -0.42 | -0.06 | 0.36 | ||
| LiCoGa | 5.44 | 5.63 | 5.66 | -0.66 | -0.04 | 0.63 | ||
| LiNiGa | 5.55 | 5.69 | 5.71 | -0.55 | 0.19 | 0.74 | ||
| LiCuGa | 5.75 | 5.87 | 5.87 | -0.32 | 0.29 | 0.61 | ||
| LiZnGa | 5.97 | 6.06 | 6.05 | -0.32 | 0.09 | 0.41 | ||
| LiCaGe | 6.49 | 6.86 | 6.94 | -0.08 | -1.10 | -1.03 | ||
| LiScGe | 5.98 | 6.31 | 6.38 | -0.41 | -1.24 | -0.83 | ||
| LiTiGe | 5.72 | 6.01 | 6.10 | -0.48 | -0.94 | -0.46 | ||
| LiVGe | 5.69 | 5.96 | 5.98 | -0.61 | -0.76 | -0.15 | ||
| LiCrGe | 5.72 | 5.94 | 5.95 | -0.59 | -0.51 | 0.08 | ||
| LiMnGe | 5.76 | 5.99 | 5.96 | -0.49 | -0.30 | 0.19 | ||
| LiFeGe | 5.59 | 5.75 | 5.76 | -0.62 | -0.04 | 0.58 | ||
| LiCoGe | 5.42 | 5.59 | 5.58 | -0.93 | 0.06 | 0.98 | ||
| LiNiGe | 5.49 | 5.65 | 5.62 | -0.68 | 0.25 | 0.93 | ||
| LiCuGe | 5.67 | 5.81 | 5.77 | -0.47 | 0.31 | 0.78 | ||
| LiZnGe | 5.88 | 6.00 | 5.96 | -0.58 | 0.06 | 0.64 | ||
Table 2.
For the compounds being magnetic we provide the atom-resolved spin magnetic moments in units as well as the total spin magnetic moment per formula unit which coincides with the per unit cell value. The last two columns are the total number of valence electrons in the unit cell, , as well as the ideal total spin magnetic moment if the Slater-Pauling rules were valid, . The cases not-presented in the table have both zero atomic and total spin magnetic moments.
Table 2.
For the compounds being magnetic we provide the atom-resolved spin magnetic moments in units as well as the total spin magnetic moment per formula unit which coincides with the per unit cell value. The last two columns are the total number of valence electrons in the unit cell, , as well as the ideal total spin magnetic moment if the Slater-Pauling rules were valid, . The cases not-presented in the table have both zero atomic and total spin magnetic moments.
| Compound | phase | ||||||
| LiCaGa | -0.062 | -0.046 | -0.134 | -0.242 | 6 | -2 | |
| -0.068 | -0.066 | -0.282 | -0.417 | 6 | -2 | ||
| LiCaGe | -0.006 | -0.091 | -0.903 | -1.000 | 7 | -1 | |
| 0.008 | -0.176 | -0.832 | -1.000 | 7 | -1 | ||
| LiScGa | -0.076 | -0.458 | -0.466 | -1.000 | 7 | -1 | |
| -0.089 | -0.388 | -0.271 | -0.748 | 7 | -1 | ||
| LiTiGe | 0.008 | 0.135 | -0.022 | 0.121 | 9 | 1 | |
| LiVGa | 0.006 | 1.332 | -0.237 | 1.101 | 9 | 1 | |
| -0.066 | 1.288 | -0.222 | 1.000 | 9 | 1 | ||
| -0.001 | 1.311 | -0.232 | 1.078 | 9 | 1 | ||
| LiVGe | 0.066 | 2.404 | -0.426 | 2.044 | 10 | 2 | |
| 0.023 | 2.438 | -0.461 | 2.000 | 10 | 2 | ||
| 0.104 | 2.362 | -0.466 | 2.000 | 10 | 2 | ||
| LiCrGa | -0.073 | 3.835 | - 0.422 | 3.340 | 10 | 2 | |
| -0.190 | 3.933 | -0.348 | 3.396 | 10 | 2 | ||
| -0.002 | 4.203 | -0.280 | 3.921 | 10 | 2 | ||
| LiCrGe | -0.013 | 3.747 | - 0.596 | 3.138 | 11 | 3 | |
| -0.030 | 3.697 | -0.667 | 3.000 | 11 | 3 | ||
| 0.076 | 3.636 | -0.712 | 3.000 | 11 | 3 | ||
| LiMnGa | -0.204 | 3.979 | -0.511 | 3.264 | 11 | 3 | |
| -0.227 | 3.830 | -0.603 | 3.000 | 11 | 3 | ||
| -0.125 | 3.768 | -0.625 | 3.018 | 11 | 3 | ||
| LiMnGe | -0.028 | 4.264 | -0.343 | 3.892 | 12 | 4 | |
| -0.034 | 4.290 | -0.317 | 3.940 | 12 | 4 | ||
| 0.089 | 4.054 | -0.427 | 3.717 | 12 | 4 | ||
| LiFeGa | -0.160 | 2.986 | -0.254 | 2.572 | 12 | 4 | |
| -0.154 | 2.748 | -0.333 | 2.261 | 12 | 4 | ||
| -0.093 | 2.785 | -0.287 | 2.406 | 12 | 4 | ||
| LiFeGe | -0.088 | 2.978 | -0.189 | 2.701 | 13 | 5 | |
| -0.055 | 2.640 | -0.244 | 2.342 | 13 | 5 | ||
| 0.024 | 2.604 | -0.254 | 2.326 | 13 | 5 | ||
| LiCoGe | -0.018 | 0.395 | -0.053 | 0.324 | 14 | 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



