
Submitted:
16 July 2024
Posted:
17 July 2024
You are already at the latest version
Abstract
Idiopathic pulmonary fibrosis [IPF] is a chronic progressive disease characterised by the accumulation of scar tissue in the lung parenchyma. It primarily occurs in middle-aged and elderly adults and results in significant morbidity and mortality worldwide. The disease occurs due to repetitive lung epithelial injury, subsequent fibroblast activation and myofibroblast differentiation, resulting in excessive extracellular matrix deposition. This leads to scar formation and subsequent loss of lung function. Current treatment options for patients with IPF include the two anti-fibrotic drugs, pirfenidone and nintedanib, which can slow disease progression; however, there are currently no known cures for the disease. As such, novel methods and drug targets are warranted. In this review, we provide an up-to-date account of the importance of specific cytokines and the potential role of regulatory immune cells. We discuss their role in the pathogenesis of IPF and address some of the key gaps in knowledge.
Keywords:
Idiopathic pulmonary fibrosis
; myofibroblast
; extracellular matrix
; T cells
1. Introduction
Idiopathic pulmonary fibrosis (IPF) is the most common of the interstitial lung diseases [1]. Progressive scarring or fibrosis of the lungs is a characteristic of IPF, however, its etiology currently remains unknown [2]. In comparison to healthy lungs, IPF results in the expansion of bronchioles and alveoli, along with fibrosis in parenchyma lung tissue, resulting in defective gas exchange [3], as depicted in Figure 1.
Patients present with clinical symptoms including shortness of breath, an unrelenting dry cough, loss of appetite and fatigue. Due to symptomatic similarities to multiple other lung diseases, IPF is often challenging to diagnose and may be misdiagnosed [4]. Morbidity and mortality rates are extremely high, patients face a poor prognosis, with the average survival following diagnosis reported to be 2.5 - 5 years [5]. IPF incidence and prevalence have been steadily increasing since 2000 and are particularly high in Northern Ireland [6], occurrence increases with age, with those aged 65 or over being at the greatest risk, and the disease is more common in males than females [7]. The histopathological pattern defining IPF is defined as usual interstitial pneumonia (UIP) [8]. Several antifibrotic - drugs exist to slow disease progression, with pirfenidone and nintedanib currently approved for use in IPF [9]. Whilst these therapies slow progression, there is no cure for IPF. Currently, the only possible mechanism to increase life expectancy and alleviate the symptomatic burden is unilateral or bilateral lung transplantation. Very few patients are, however, eligible for lung transplantation, but the 5 years post-operative survival is reported at only 50% [10]. Hence, IPF poses a serious and increasing threat to human health and research aimed at uncovering novel treatments is crucial if we are to improve the devastating prognosis and decrease mortality among patients.
2. Pathogenesis
Although the exact cause of IPF is unknown, it is predicted that repeated micro-injuries to ageing alveoli lead to aberrant and unregulated repair mechanisms in the lungs [11]. These alveolar micro-injuries can be due to environmental factors such as cigarette smoke and repetitive dust inhalation, co-morbidities such as viral infections and gastroesophageal reflux disease, as well as genetic susceptibility [12]. The most prevalent known genetic component contributing to the development of IPF, is a gain-of-function mutation in the promoter region of the MUC5B gene, which is involved in the production of mucus in the lungs [13]. The two main risk factors for IPF are age and sex. Ageing alveoli tend to lose their shape, becoming dilated, and a decrease in gas exchange occurs; this in turn, can enhance fibrosis formation [14]. IPF prevails more in males than in females, with male patients also demonstrating higher mortality rates [15].
The body’s immune system mediates a tightly regulated lung tissue repair in healthy individuals. Clotting and inflammation are initiated in response to alveolar tissue damage by injury or infection. The infiltration of cells including erythrocytes and platelets, leads to clotting, followed by inflammation initiation by neutrophils, monocytes, dendritic cells and macrophages [16]. Inflammation is typically resolved by the recruitment of fibrocytes and the production of anti-inflammatory cytokines. Upon migration into the tissue, fibrocytes differentiate into fibroblasts, which then differentiate into myofibroblasts. Myofibroblasts are extracellular matrix (ECM)- producing cells, and subsequent ECM deposition leads to scar formation and tissue repair [16], as illustrated in Figure 2. The repair phase is concluded upon ECM degradation by fibroblasts and macrophages, completing the wound-healing process and restoring homeostasis in the lungs [17]. Regulatory T cells (Tregs) have demonstrated an essential role in healing, contributing to the repair phase by enhancing the proliferation of alveolar epithelial cells and impeding the pro-inflammatory activity of interferon-γ (IFN-γ) [18]. When this repair process is dysregulated, lung structure and function resolution does not occur, and fibrosis can form. Repeated micro-injuries can lead to the over-activation of fibroblasts and hence myofibroblasts. A consequent increase in ECM production and collagen deposition in the lungs leads to permanent scar tissue formation, known as fibrosis, as the accumulated ECM and collagen cannot be degraded quickly enough [19].
3. Cytokines and IPF
One area that warrants consideration for developing novel IPF treatments is the influence of cytokines on fibrosis formation. TGFβ is a well-known pro-fibrotic cytokine, shown to be significantly increased in the plasma of IPF patients in comparison to that of healthy controls [20]. Tregs produce TGFβ as well as the cytokines IL-10 and IL-35 [21]. The production of TGFβ is believed to account for much of the pro-fibrotic properties of Tregs [22]. In a study by Bergeron et al., an increase in IL-10 in IPF lungs compared with controls was highlighted, which makes IL-10 another cytokine of interest and a potential target for IPF [23]. Multiple other interleukins have been described as profibrotic, including IL-1β, IL-4, IL-6, IL-13 and IL-17 [24]. Clinical trials targeting IL-4 and IL-13 did not yield promising results, and hence, we require further understanding of the complex interaction of the cytokine milieu which drives the fibrotic process [25]. The chemokine CCL2, known as monocyte chemoattractant protein-1 (MCP-1), has been found to be increased in IPF patient bronchoalveolar lavage (BAL) fluid samples compared to healthy controls and is thought to increase fibrosis by enhancing fibrocyte migration into the lung [26]. The pro-inflammatory chemokine CXCL1, has also been significantly increased in IPF patients with acute exacerbation [27]. CXCL1 is a chemokine known to attract neutrophils during inflammation and is also thought to play a part in enhancing fibrosis formation in the lungs [28]. On the contrary, the pro-inflammatory cytokine IFN-γ shows anti-fibrotic properties. Cytokines exhibiting anti-fibrotic properties have the potential to be enhanced as novel IPF treatments. IFN-γ has been tested in combination with pirfenidone, and shown to reduce fibrosis, opening a new avenue of potential cytokine treatments [29]. Therefore, further research into the effect of cytokines, both individually and in combination, on fibrotic progression could be beneficial in identifying promising therapeutic agents that either halt or reverse the pathogensis of IPF. Table 1 summarises some of the key cytokines of importance in IPF.
4. Regulatory Cells
Regulatory immune cells act to maintain homeostasis, controlling the immune response to injury and modulating the function and cytokine production of the immune cells involved. However, the exact function of regulatory immune cells in the context of IPF remains ambiguous, highlighting a current research gap in the literature [39]. Myeloid-derived suppressor cells (MDSCs) comprise a heterogeneous population of cells originating from the myeloid lineage; they significantly increase in number during inflammation and are involved in suppressing T-cell responses, preventing overactivation [40]. A protective, anti-fibrotic role for MDSCs has been suggested in IPF due to their anti-inflammatory functions [39]. Opposing roles have also, however, been suggested, with a reported MDSC increase in the peripheral blood of IPF patients, and they have been proposed to contribute to the decreased lung function during the course of IPF disease [41]. There are contradictory reports on the role of regulatory immune cell populations in IPF been reported in the literature, such as the role of macrophages and regulatory B cells (Bregs). Whilst macrophages are often considered as pro-fibrotic due to their production of transforming growth factor-β (TGF-β) [42], anti-inflammatory and anti-fibrotic properties have also been identified. They have been reported to suppress fibroblast differentiation into myofibroblasts [43] via the production of prostaglandin E2 [44,45]. Similarly, Breg’s produce TGF-β driving fibrosis formation [39]; but beyond this, it has been reported the number of Bregs was reduced in IPF patients compared with healthy controls, suggesting that they are not key drivers of IPF [46]. Therefore, it is evident that the role of regulatory immune cells in IPF remains undefined and further research is essential to decipher what is happening at a cellular level. The regulatory immune cell group of interest in IPF, are the regulatory T cells (Tregs). Tregs act to maintain homeostasis, suppressing undesirable immune responses and preventing autoimmunity by ensuring tolerance to self-antigens [47]. Tregs are characterised by the transcription factor FoxP3 and develop either in the thymus (known as natural or nTregs) or in the periphery (known as induced or iTregs) [48]. Tissue-resident Tregs have been described as both “Sentinels” and “Saboteurs” as they have made differing and contradictory contributions to various disease states [49]. The exact function of Tregs in IPF remains elusive, as multiple different behaviours have been reported in the literature. Artsen et al. showed that Treg concentration was inversely correlated with fibrosis formation. This alluded to the protective role of Tregs against fibrosis, as Tregs were able to suppress immune responses, preventing fibrosis development [50]. On the contrary, an increase in the number of activated Tregs was shown to drive fibrosis due to the secretion of the pro-fibrotic cytokines TGF- β and platelet-derived growth factor (PDGF)-B [51]. The reason for these contradictory functions is unknown. However, it has been suggested that factors including the specific subtype studied, the phase of fibrotic formation studied, the various antigens present on Tregs and the multi-functional nature of secreted factors, amongst other reasons, may have contributed to the variation between reported outcomes [52].
5. Conclusion
IPF is a devastating lung disease with high morbidity, causing a large economic and healthcare burden worldwide. Both genetic and epigenetic factors contribute to IPF pathogenesis; however, while our understanding of the disease has improved in recent years, the precise cause remains unknown. Unfortunately, limited treatment options exist, and there is currently no known cure for the disease. The antifibrotic drugs pirfenidone and nintedanib can slow functional decline in IPF, but adverse reactions limit their usefulness. Regulatory cells control and mediate immune responses. Therefore, the modulation of their activity to drive repair and limit fibrosis is an enticing prospect. However, we need a more thorough understanding of the pathogenesis of IPF, including the interplay between inflammation, immune mechanisms and dysregulated repair mechanisms, and how regulatory cells mediate this. This will facilitate the development of novel, more effective treatment options for patients suffering from IPF.
Author Contributions
FJ and AMR wrote the review article, RJI, AMR, RB, DS, BS and NC edited the review article. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Conflicts of Interest
The authors declare no conflict of interest
References
- Antoniou KM, Margaritopoulos GA, Tomassetti S, Bonella F, Costabel U, Poletti V. Interstitial lung disease. Eur Respir Rev. 2014 Mar 1;23(131):40–54.
- Barratt, S.L.; Creamer, A.; Hayton, C.; Chaudhuri, N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. J. Clin. Med. 2018, 7, 201. [Google Scholar] [CrossRef] [PubMed]
- Glassberg, M.K. Overview of idiopathic pulmonary fibrosis, evidence-based guidelines, and recent developments in the treatment landscape. Am J Manag Care. 2019, 25, S195–S203. [Google Scholar] [PubMed]
- Lancaster, L.; Bonella, F.; Inoue, Y.; Cottin, V.; Siddall, J.; Small, M.; Langley, J. Idiopathic pulmonary fibrosis: Physician and patient perspectives on the pathway to care from symptom recognition to diagnosis and disease burden. Respirology 2021, 27, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, H.; Kobayashi, T.; Azuma, A. Idiopathic Pulmonary Fibrosis: Treatment and Prognosis. Clin. Med. Insights: Circ. Respir. Pulm. Med. 2015, 9s1, 179–185. [Google Scholar] [CrossRef]
- Strongman, H.; Kausar, I.; Maher, T.M. Incidence, Prevalence, and Survival of Patients with Idiopathic Pulmonary Fibrosis in the UK. Adv. Ther. 2018, 35, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Nalysnyk, L.; Cid-Ruzafa, J.; Rotella, P.; Esser, D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur. Respir. Rev. 2012, 21, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018 Sep 1;198(5):e44–68.
- Cameli, P.; Refini, R.M.; Bergantini, L.; D’alessandro, M.; Alonzi, V.; Magnoni, C.; Rottoli, P.; Sestini, P.; Bargagli, E. Long-Term Follow-Up of Patients With Idiopathic Pulmonary Fibrosis Treated With Pirfenidone or Nintedanib: A Real-Life Comparison Study. Front. Mol. Biosci. 2020, 7. [Google Scholar] [CrossRef]
- Glass, D.S.; Grossfeld, D.; Renna, H.A.; Agarwala, P.; Spiegler, P.; DeLeon, J.; Reiss, A.B. Idiopathic pulmonary fibrosis: Current and future treatment. Clin. Respir. J. 2022, 16, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: pathogenesis and management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef]
- Guo, H.; Sun, J.; Zhang, S.; Nie, Y.; Zhou, S.; Zeng, Y. Progress in understanding and treating idiopathic pulmonary fibrosis: recent insights and emerging therapies. Front. Pharmacol. 2023, 14, 1205948. [Google Scholar] [CrossRef]
- Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017 May 13;389(10082):1941–52.
- López-Ramírez, C.; Valdivia, L.S.; Portal, J.A.R. Causes of Pulmonary Fibrosis in the Elderly. Med Sci. 2018, 6, 58. [Google Scholar] [CrossRef] [PubMed]
- Caminati, A.; Madotto, F.; Conti, S.; Cesana, G.; Mantovani, L.; Harari, S. The natural history of idiopathic pulmonary fibrosis in a large European population: the role of age, sex and comorbidities. Intern. Emerg. Med. 2021, 16, 1793–1802. [Google Scholar] [CrossRef] [PubMed]
- Florez-Sampedro, L.; Song, S.; Melgert, B.N. The diversity of myeloid immune cells shaping wound repair and fibrosis in the lung. Regeneration 2018, 5, 3–25. [Google Scholar] [CrossRef]
- Adhyatmika, A.; Putri, K.S.S.; Beljaars, L.; Melgert, B.N. The Elusive Antifibrotic Macrophage. Front. Med. 2015, 2, 81. [Google Scholar] [CrossRef]
- Cipolla, E.M.; Alcorn, J.F. Repair of the Lung by Regulatory T Cells. Am. J. Respir. Cell Mol. Biol. 2020, 63, 405–407. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Evrard, S.M.; D′Audigier, C.; Mauge, L.; Israël-Biet, D.; Guerin, C.L.; Bieche, I.; Kovacic, J.C.; Fischer, A.; Gaussem, P.; Smadja, D.M. The profibrotic cytokine transforming growth factor-β1 increases endothelial progenitor cell angiogenic properties. J. Thromb. Haemost. 2012, 10, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Arce-Sillas, A.; Álvarez-Luquín, D.D.; Tamaya-Domínguez, B.; Gomez-Fuentes, S.; Trejo-García, A.; Melo-Salas, M.; Cárdenas, G.; Rodríguez-Ramírez, J.; Adalid-Peralta, L. Regulatory T Cells: Molecular Actions on Effector Cells in Immune Regulation. J. Immunol. Res. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular Mechanisms of Treg-Mediated T Cell Suppression. Front. Immunol. 2012, 3, 51. [Google Scholar] [CrossRef]
- Bergeron A, Soler P, Kambouchner M, Loiseau P, Milleron B, Valeyre D, et al. Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-beta and IL-10. Eur Respir J. 2003 Jul;22(1):69–76.
- She, Y.X.; Yu, Q.Y.; Tang, X.X. Role of interleukins in the pathogenesis of pulmonary fibrosis. Cell Death Discov. 2021, 7, 1–10. [Google Scholar] [CrossRef]
- Ma, H.; Liu, S.; Li, S.; Xia, Y. Targeting Growth Factor and Cytokine Pathways to Treat Idiopathic Pulmonary Fibrosis. Front. Pharmacol. 2022, 13, 918771. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.H.G.; Paliogiannis, P.; Nasrallah, G.K.; Giordo, R.; Eid, A.H.; Fois, A.G.; Zinellu, A.; Mangoni, A.A.; Pintus, G. Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cell. Mol. Life Sci. 2020, 78, 2031–2057. [Google Scholar] [CrossRef] [PubMed]
- Schupp, J.C.; Binder, H.; Jäger, B.; Cillis, G.; Zissel, G.; Müller-Quernheim, J.; Prasse, A. Macrophage Activation in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. PLOS ONE 2015, 10, e0116775–e0116775. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-L.; Yin, R.; Wang, S.-N.; Ying, R. A Review of CXCL1 in Cardiac Fibrosis. Front. Cardiovasc. Med. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.N.; Chen, X.; Foda, H.D.; Smaldone, G.C.; Hasaneen, N.A. Interferon-γ enhances the antifibrotic effects of pirfenidone by attenuating IPF lung fibroblast activation and differentiation. Respir. Res. 2019, 20, 1–14. [Google Scholar] [CrossRef]
- Gabay, C. Interleukin-6 and chronic inflammation. Arthritis Res. Ther. 2006, 8, S3–S3. [Google Scholar] [CrossRef] [PubMed]
- Fielding, C.A.; Jones, G.W.; McLoughlin, R.M.; McLeod, L.; Hammond, V.J.; Uceda, J.; Williams, A.S.; Lambie, M.; Foster, T.L.; Liao, C.-T.; et al. Interleukin-6 Signaling Drives Fibrosis in Unresolved Inflammation. Immunity 2014, 40, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Singh S, Anshita D, Ravichandiran V. MCP-1: Function, regulation, and involvement in disease. Int Immunopharmacol. 2021 Dec;101(Pt B):107598.
- Scott, M.K.D.; Quinn, K.; Li, Q.; Carroll, R.; Warsinske, H.; Vallania, F.; Chen, S.; A Carns, M.; Aren, K.; Sun, J.; et al. Increased monocyte count as a cellular biomarker for poor outcomes in fibrotic diseases: a retrospective, multicentre cohort study. Lancet Respir. Med. 2019, 7, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Segel MJ, Izbicki G, Cohen PY, Or R, Christensen TG, Wallach-Dayan SB, et al. Role of interferon-gamma in the evolution of murine bleomycin lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2003 Dec;285(6):L1255-62.
- Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004 Feb;75(2):163–89.
- Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, et al. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med. 2010 Mar 15;207(3):535–52.
- Ge, Y.; Huang, M.; Yao, Y.-M. Biology of Interleukin-17 and Its Pathophysiological Significance in Sepsis. Front. Immunol. 2020, 11, 1558. [Google Scholar] [CrossRef]
- Ke, Y.; Liu, K.; Huang, G.-Q.; Cui, Y.; Kaplan, H.J.; Shao, H.; Sun, D. Anti-Inflammatory Role of IL-17 in Experimental Autoimmune Uveitis. 2009; 182, 3183–3190. [Google Scholar] [CrossRef]
- van Geffen C, Deißler A, Quante M, Renz H, Hartl D, Kolahian S. Regulatory Immune Cells in Idiopathic Pulmonary Fibrosis: Friends or Foes? Front Immunol. 2021;12:663203.
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Greiffo, F.R.; Frankenberger, M.; Bandres, J.; Heinzelmann, K.; Neurohr, C.; Hatz, R.; Hartl, D.; Behr, J.; Eickelberg, O. Peripheral blood myeloid-derived suppressor cells reflect disease status in idiopathic pulmonary fibrosis. Eur. Respir. J. 2016, 48, 1171–1183. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef]
- Ishikawa, G.; Liu, A.; Herzog, E.L. Evolving Perspectives on Innate Immune Mechanisms of IPF. Front. Mol. Biosci. 2021, 8. [Google Scholar] [CrossRef]
- Garrison, G.; Huang, S.K.; Okunishi, K.; Scott, J.P.; Penke, L.R.K.; Scruggs, A.M.; Peters-Golden, M. Reversal of Myofibroblast Differentiation by Prostaglandin E2. Am. J. Respir. Cell Mol. Biol. 2013, 48, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, S.; Matsumoto, M.; Takeda, K.; Akira, S. Lipopolysaccharide-Dependent Prostaglandin E2 Production Is Regulated by the Glutathione-Dependent Prostaglandin E2 Synthase Gene Induced by the Toll-Like Receptor 4/MyD88/NF-IL6 Pathway. J. Immunol. 2002, 168, 5811–5816. [Google Scholar] [CrossRef] [PubMed]
- Asai, Y.; Chiba, H.; Nishikiori, H.; Kamekura, R.; Yabe, H.; Kondo, S.; Miyajima, S.; Shigehara, K.; Ichimiya, S.; Takahashi, H. Aberrant populations of circulating T follicular helper cells and regulatory B cells underlying idiopathic pulmonary fibrosis. Respir. Res. 2019, 20, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shevyrev D, Tereshchenko V. Treg Heterogeneity, Function, and Homeostasis. Front Immunol. 2019;10:3100.
- Dhamne C, Chung Y, Alousi AM, Cooper LJN, Tran DQ. Peripheral and thymic foxp3(+) regulatory T cells in search of origin, distinction, and function. Front Immunol. 2013;4:253.
- Lee, J.; Kim, D.; Min, B. Tissue Resident Foxp3+ Regulatory T Cells: Sentinels and Saboteurs in Health and Disease. Front. Immunol. 2022, 13, 865593. [Google Scholar] [CrossRef] [PubMed]
- Artsen, A.M.; Rytel, M.; Liang, R.; King, G.E.; Meyn, L.; Abramowitch, S.D.; Moalli, P.A. Mesh induced fibrosis: The protective role of T regulatory cells. Acta Biomater. 2019, 96, 203–210. [Google Scholar] [CrossRef]
- Hou, Z.; Ye, Q.; Qiu, M.; Hao, Y.; Han, J.; Zeng, H. Increased activated regulatory T cells proportion correlate with the severity of idiopathic pulmonary fibrosis. Respir. Res. 2017, 18, 170. [Google Scholar] [CrossRef]
- Wang, F.; Xia, H.; Yao, S. Regulatory T cells are a double-edged sword in pulmonary fibrosis. Int. Immunopharmacol. 2020, 84, 106443. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author[s] and contributor[s] and not of MDPI and/or the editor[s]. MDPI and/or the editor[s] disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
Figure 1.
Alveolar Differences between healthy and IPF Lungs. A) Healthy lungs with functioning alveoli, bronchioles and capillary network allowing for normal gas exchange. B) Damaged lungs from a patient with IPF, showing damaged and expanded bronchioles and alveoli, fibrosis formation between parenchyma lung tissue and a consequent decrease in gas exchange. Adapted from Glassberg (2019) [3], using Biorender [www.biorender.com].
Figure 1.
Alveolar Differences between healthy and IPF Lungs. A) Healthy lungs with functioning alveoli, bronchioles and capillary network allowing for normal gas exchange. B) Damaged lungs from a patient with IPF, showing damaged and expanded bronchioles and alveoli, fibrosis formation between parenchyma lung tissue and a consequent decrease in gas exchange. Adapted from Glassberg (2019) [3], using Biorender [www.biorender.com].
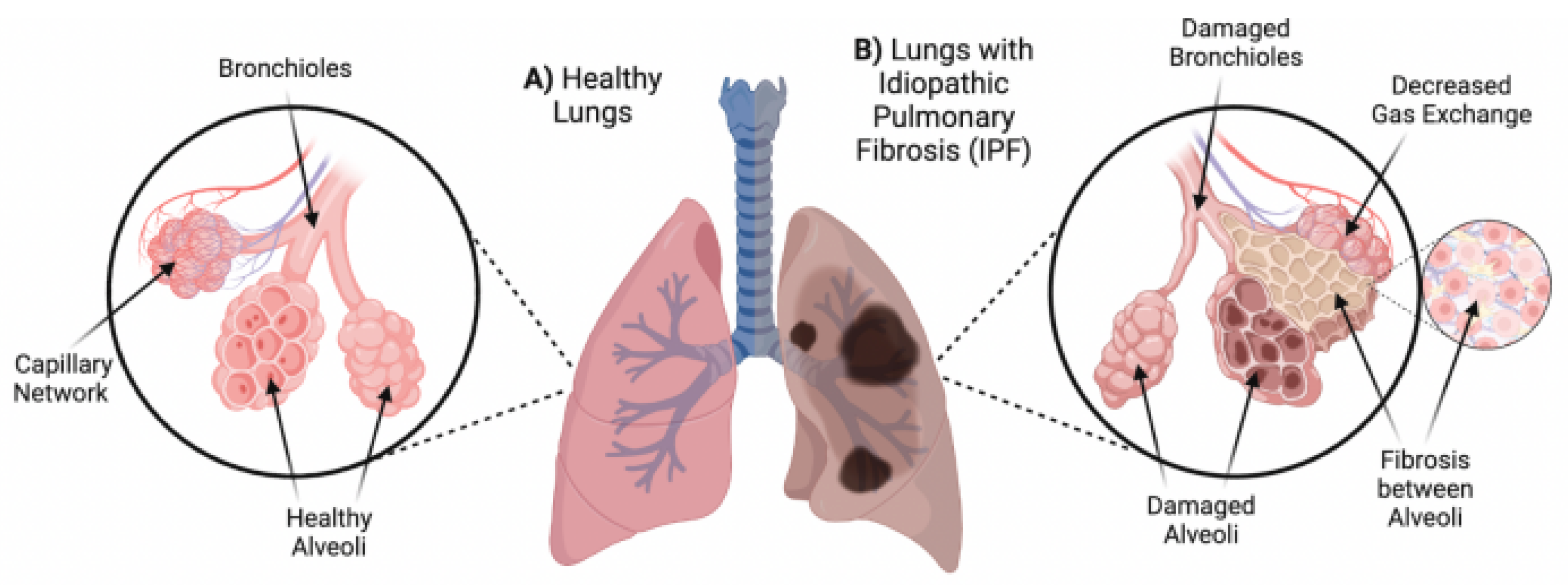
Figure 2.
Alveolar immune mechanisms leading to lung fibrosis. (1) Lung injury or infection leads to damaged alveolar epithelial cells (AECs), in turn resulting in initiation of clotting by erythrocytes and platelets. (2) Following injury, inflammatory responses are triggered by pro-inflammatory cytokines and other stimuli. Neutrophils and monocytes are recruited from the bloodstream to the site of inflammation, and activation of macrophages and dendritic cells occurs to fight infection (3). Fibrocytes migrate from the bloodstream and differentiate into fibroblasts in response to inflammation. Fibroblasts become activated and differentiate into the extracellular matrix (ECM)-producing cells, known as myofibroblasts (8). Myofibroblasts deposit ECM and collagen, forming a scar. In healthy lung tissue, ECM is degraded following repair, and homoeostasis is restored. Repeated micro-injuries and subsequent dysregulation, however, of these immune responses has the potential to lead to accumulation of ECM and collagen deposition, resulting in alveolar damage and fibrosis. Image adapted from Florez-Sampedro et al., (2017) [16], using Biorender [www.biorender.com].
Figure 2.
Alveolar immune mechanisms leading to lung fibrosis. (1) Lung injury or infection leads to damaged alveolar epithelial cells (AECs), in turn resulting in initiation of clotting by erythrocytes and platelets. (2) Following injury, inflammatory responses are triggered by pro-inflammatory cytokines and other stimuli. Neutrophils and monocytes are recruited from the bloodstream to the site of inflammation, and activation of macrophages and dendritic cells occurs to fight infection (3). Fibrocytes migrate from the bloodstream and differentiate into fibroblasts in response to inflammation. Fibroblasts become activated and differentiate into the extracellular matrix (ECM)-producing cells, known as myofibroblasts (8). Myofibroblasts deposit ECM and collagen, forming a scar. In healthy lung tissue, ECM is degraded following repair, and homoeostasis is restored. Repeated micro-injuries and subsequent dysregulation, however, of these immune responses has the potential to lead to accumulation of ECM and collagen deposition, resulting in alveolar damage and fibrosis. Image adapted from Florez-Sampedro et al., (2017) [16], using Biorender [www.biorender.com].
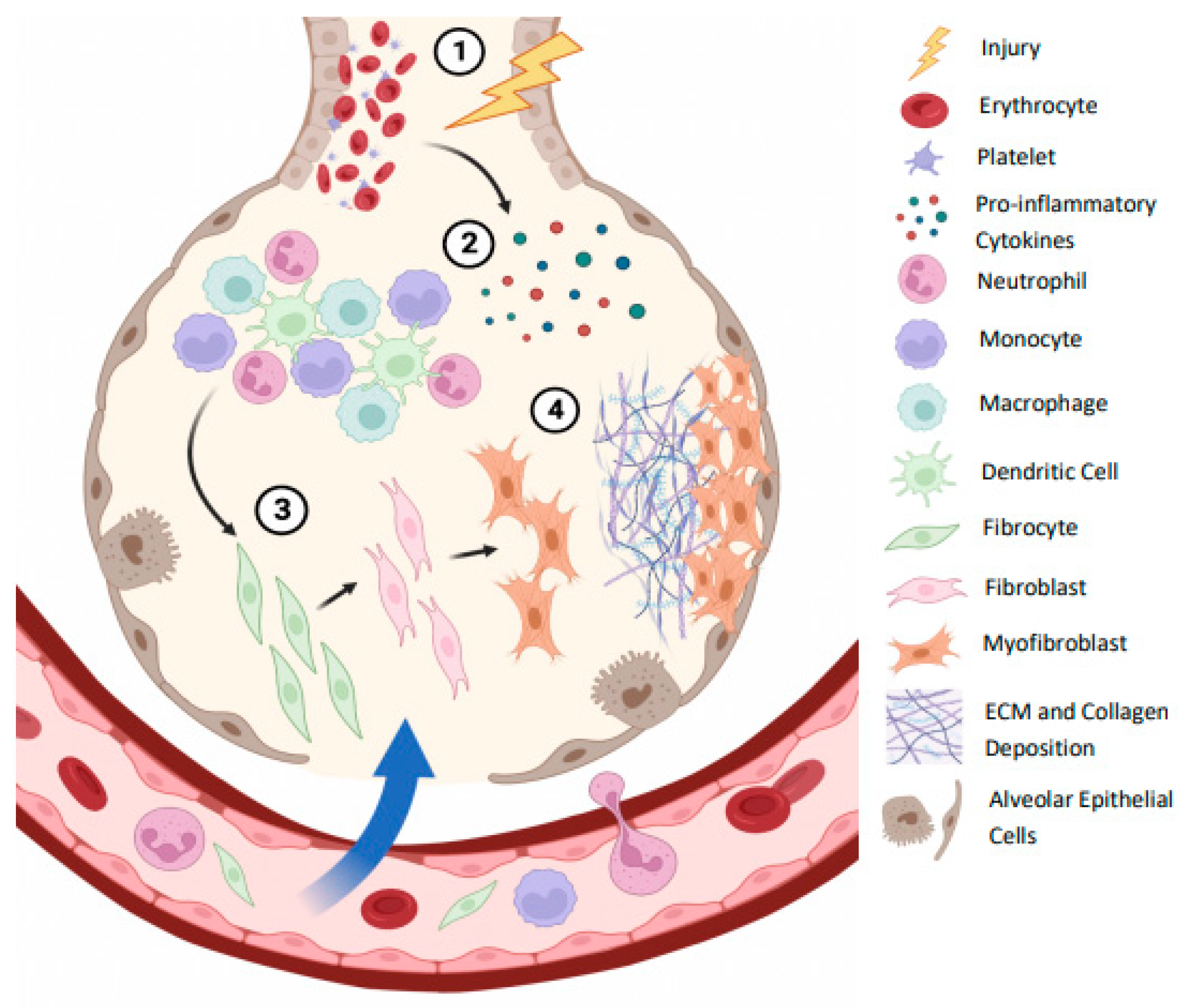

Table 1.
Key cytokines and their involvement in IPF.
| Cytokine | Main Functions | Relevance to Study | Supporting Literature |
|---|---|---|---|
| IL-6 | Pro-inflammatory cytokine Involved in the transition from acute to chronic inflammatory responses |
Possesses pro-fibrotic properties Previously significantly increased in IPF patients Suggested to drive fibrosis in unresolved inflammation |
[24,30,31] |
| CXCL1 | Pro-inflammatory chemokine Involved in chemoattraction of neutrophils to the site of inflammation |
Previously significantly increased in IPF patients with acute exacerbation | [27,28] |
| MCP-1 | Pro-inflammatory chemokine Role in recruitment and activation of monocytes |
Previously significantly increased in IPF patients Monocytes suggested as a possible marker of IPF severity |
[26,32,33] |
| TGF-b | Both pro- and anti-inflammatory properties Essential in wound healing and tissue repair |
Pro-fibrotic cytokine and key mediator in fibrotic progression Significantly increased in IPF Produced and secreted by Tregs |
[20,21,22] |
| IFN- | Pro-inflammatory cytokine Involved in the activation and regulation of numerous immune cells |
Typically anti-fibrotic Potential to be combined with anti-fibrotic drug pirfenidone Suggested pro-fibrotic nature in certain contexts |
[29,34,35] |
| IL-10 | Anti-inflammatory cytokine Maintains homeostasis by suppressing immune activity |
Both pro- and anti-fibrotic effects Previously significantly increased in IPF patients Produced and secreted by Tregs |
[21,23,26] |
|
IL-17 (IL-17A) |
Both pro- and anti-inflammatory properties Has implications in autoimmunity and chronic inflammation |
Has demonstrated pro-fibrotic properties Previously significantly increased in IPF patients |
[24,36,37,38] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



