
Submitted:
18 July 2024
Posted:
19 July 2024
You are already at the latest version
Abstract
Opioids have served as a cornerstone in pain management for decades. However, the emergence of increasingly potent synthetic analogs brings forth a range of side effects, including respiratory depression, tolerance, dependence, constipation, and, more importantly, the development of severe and debilitating opioid use disorder (OUD). Search for therapeutics to mitigate OUD has been challenging and this has called for novel approaches that include design of small molecules targeting neuronal circuits involved in addiction (opioid, dopamine, serotonin, norepinephrine, and glutamate receptors, etc.) and development of biologics that target circulating opioids/opiates. In this review, we retrieved and discussed over two dozen of relevant patents filed in the past twelve (12) years that focus on novel approaches to produce therapeutics for OUD. The current review excluded patents on biologics and concentrated on small molecules, which will be discussed separately in a subsequent sequel. The chemical entities disclosed were highlighted and specific examples were provided where necessary. Although the number of patents in the realm of drug discovery for OUD is currently limited, we foresee a continued expansion in the quest for therapeutics for OUD in the years to come.
Keywords:
opioids
; opioid use disorder
; addiction
; patents
; novel compounds
; small molecules
1. Introduction
Opioids are the most effective medication used to treat moderate to severe acute pain [1]. However, opioids produce side effects such as tolerance, constipation, respiratory depression, dependence, and addiction [2]. The ability of opioids to produce dependence and addiction leads to opioid use disorder (OUD). OUD is a chronic relapsing disorder that is initially driven by brain reward neurocircuits and then engages anti-reward neurocircuits that drive adverse emotional states and relapse [3]. OUD is associated with other conditions like HIV, hepatitis C infection and transmission, bacterial endocarditis, and neonatal opioid withdrawal syndrome (NOWS). The average duration of OUD ranges between 10 and 20 years and 26.8 million people world-wide were estimated to be living with OUD in 2017 [4,5]. The abuse liability of opioids and their respiratory depressive effects are the major contributors to the opioid overdose epidemic. The United States reported 107,543 deaths from drug overdose in 2023, with 81,083 of these deaths attributed to opioids alone[6]. Opioid overdose and OUD have a huge economic burden which were estimated to cost the United States $1.02 trillion in 2017 [7].
The analgesic effects and abuse liability of opioids are mediated by the mu opioid receptor (MOR) [8]. The pharmacotherapies for the treatment of OUD and opioid withdrawal include buprenorphine, methadone, naloxone, naltrexone, and lofexidine [9,10]. Buprenorphine, a partial MOR agonist, and methadone, a full MOR agonist, are used for opioid replacement or detoxification therapy in the treatment of OUD. Combinations of buprenorphine and the MOR antagonist naloxone are used in both the detoxification and maintenance phases of OUD treatment while naltrexone (MOR antagonist) is used to prevent relapse. Naloxone is also used to reverse opioid-induced overdose.[3,9] Lofexidine, an alpha 2 adrenergic agonist, is used to treat opioid withdrawal symptoms.[10] However, there is a low retention rate for medication assisted treatment of OUD with buprenorphine/naloxone treatment having a six-month retention rate of 46% [11]. As a result, there is a need for the development of new treatments and approaches to curb the opioid epidemic.
In this article, we review recent advancements that have been made in the development of novel pharmacotherapies to treat OUD. Particularly, we review recent patents that were submitted in the last 12 years (since 2012, immediately before the start of the third wave of the opioid overdose epidemic) that focus on small molecules used to treat OUD [12]. To this end, we searched google patents, Lens.org, and SciFinder for all patents filed in the last 12 years with the following keywords: OUD, Substance Use Disorder (SUD), Opioids. We applied filters to remove all patents that do not claim the synthesis or preparation of novel small molecules. Where general formulas are provided, we encourage readers to refer to the patent for the exhaustive list of possible substituents or chemical space.
2.1. Small Molecules Targeting Opioid Receptors
In 2020, Kruegel and colleagues claimed a series of deuterated mitragynine analogs with general structure 1 (Figure 1) that target the MOR with potential use in patients afflicted with pain, depression, anxiety or mood disorder[13]. Mitragynine (2), a well-explored alkaloid isolated from the Mitragyna speciosa (kratom) leaves, is pharmacologically described as a partial agonist at the MOR representing an alternative to current treatment options for pain (with less propensity to cause dependence), opioid withdrawal or OUD [14]. One setback to the applicability of mitragynine is the accumulation of its toxic metabolite 3-dehydromitragynine (3) (3-DM). The present invention embodies deuterated analogs of mitragynine with improved toxicity profile. The patent reported the key compound, 3-deuteromitragynine (4) (3-d-M), as having a reduced propensity to form toxic metabolites in human liver microsomes and male BALB/c mice brain homogenates without compromising the ability to form the active MOR metabolite 7-hydroxymitragynine (5). In vivo pharmacokinetic studies with male 129S1 and C57BL/6 mice
also showed that deuteration at the 3 position reduces formation of the toxic metabolite 3-DM. Mechanistically, the ability of the 3-deuterated compounds to attenuate the conversion of mitragynine to 3-DM can be partially explained by the greater strength of the deuterium-carbon bond compared to the protium-carbon bond, and the resulting kinetic isotopic effect attenuates conversion of such 3-deuterated compounds to 3-DM or their analogous 3-dehydro metabolites compared to the analogous non-deuterated compounds. In an interesting patent filed by Haile and colleagues, they report a combination therapy using mitragynine and naltrexone as a treatment for substance use disorders [15]. The combination of mitragynine (3 mg/kg) and naltrexone (3 mg/kg) reduced alcohol (10%) self-administration and amount consumed, and induced greater numbers of cFOs expressing neurons in brain regions that mediate alcohol reinforcement than either drug alone in female Sprague-Dawley rats. Although the authors do not report any data on the use of mitragynine and naltrexone to treat OUD, they suggest that the combination of mitragynine and naltrexone may also reduce opioid self-administration.
A 2020 patent filed by Zhang[16] claimed some novel non-peptide opioid receptor modulators with the general formula shown in Figure 2. The compounds disclosed in the patent were amide derivatives of naltrexamine 7 that target the MOR as antagonists with potential use in OUD. Among the entities declared, NAP (8), NFP (9), NYP (10) and NMP (11) (Table 1) were the prominent and well-characterized compounds of the invention [17,18,19]. These compounds displayed sub-nanomolar binding affinity at the MOR (see Table 1). Whereas NAP had low permeability through the blood brain barrier (BBB) and acted mainly in the periphery, NFP and NYP permeated the BBB and acted centrally to reverse the effects of opioids and produced minimal withdrawal symptoms as compared with naloxone, suggesting that NFP and NYP may be beneficial in treating OUD. In vivo studies in male Swiss Webster mice showed that NFP (10 mg/kg) and NYP (10 mg/kg) antagonized the antinociceptive effects of morphine (10 mg/kg). In mice that were implanted with a 75 mg morphine pellet, NFP (10 mg/kg) produced significantly less withdrawal signs (shakes, jumps, paw tremors) than naltrexone (1 mg/kg) 72 h after pellet implantation[18], suggesting that NFP may be a better candidate than naltrexone to treat OUD.
In another ensuing patent from the same lab, Zhang claimed 6-α and 6β-naltrexamine derivatives represented by general structure 12 [20]. The compounds in the embodiment bear 5-member heterocyclic ring systems and had MOR antagonism with potential use for OUD (Figure 3). Preliminary data presented in the declarations in the patent showed that most of the compounds had desirable CNS permeability and retained MOR affinity. Interestingly three (3) of the MOR antagonists identified (compounds 13, 14 and 15) displayed the most potent in vivo antagonism against morphine (0.42, 1.62, 1.51 mg/kg respectively) and precipitated fewer withdrawal symptoms warranting further exploration as chemical probes in OUD.
Novel analogs of gliotoxin 17 as opioid antagonists were claimed in a patent filed by German and Hossain [21] for the potential treatment of alcohol dependence, opioid abuse, neurological disorders, neuropathic pain, and fibromyalgia. These compounds are represented by the general formula 16 below (Figure 4). The analog with desirable characteristics that was investigated in the German and Hossain invention included Compound 18 (compound 1b in patent) that displayed submicromolar binding affinity at the MOR, delta opioid receptor (DOR), and kappa opioid receptor (KOR) (Figure 4). Compound 18 crossed the BBB efficiently (over 80%) compared to other compounds in the patent. No in vitro functional or in vivo data was reported in this current disclosure.
German and team at Texas Tech University Health Sciences Center filed another patent claiming some novel selective KOR antagonists as treatments for addiction, alcohol dependence, OUD, and neuropathic pain [22]. The disclosure included compounds with the general formula I (19) and II (20) with representative examples AH-3-193 (compound 21) and AH-3-199 (compound 22) shown in Figure 4. In vitro binding assays showed that the compounds prepared in this declaration were KOR ligands with binding affinity in the submicromolar range (Figure 5). These compounds did not bind to the DOR or MOR up to 10 µM [23]. Compound 22 antagonized U50488-induced responses in the mouse thermal place preference test, suggesting that 22 is a KOR antagonist. In addition, the compounds in the present invention were shown to inhibit pain responses by 20-40% when measuring various rat pain responses such as audible vocalizations, ultrasonic vocalizations, and electronic von Frey.
In an invention by Peltier et. al.,[24] oxycodone analogs in which the allylic alcoholic function is conjugated to different moieties via an ester were declared as potential abuse-resistant opioid compounds. These compounds have the general formula 23 (Figure 6) with representative ester conjugate compound 24. No behavioral animal studies were presented in the current invention. The claim focuses mostly on the preparation of the analogs. In vivo studies presented in the embodiment were for pharmacokinetic studies showing plasma concentrations of liberated oxymorphone (active metabolite) after oral administration in rats.
In a disclosure filed by Medina and colleagues, a series of amides represented by the general formula 25 (Figure 7) were claimed as potential treatments for SUDs including OUD [25]. The profile of compounds declared were MOR antagonists, DOR agonists (cAMP), and KOR β-arrestin antagonists. Of the disclosed entities, compound 26 was the most potent at the listed receptors/targets displaying IC50 less than 300 nM. In another patent filed by Rice and colleagues, they also report novel selective opioid receptor agonists and antagonists (represented by general structure 27, Figure 8) as potential treatments for pain and OUD [26]. Two compounds in this disclosure compounds 28 and 29 had partial agonist effects at the MOR in the cAMP assay (% Emax = 67.3 and 89.5, respectively) and GTP assay (% Emax = 10.54 and 17.97, respectively). These two compounds produced antinociceptive effects in squirrels and monkeys but with less respiration depression compared to morphine.
Brents and colleagues filed a patent reporting the development of deuterated buprenorphine, 30 (Figure 9) as a protective agent for fetuses against opioid exposure [27]. The chronic use of opioids by pregnant women can cause the fetus or neonate to develop opioid dependence and withdrawal, and potentially life-long neurobehavioral alterations that negatively affect quality of life [28]. There are currently no FDA approved medications to protect the fetus from maternal exposure to opioids. Deuterated buprenorphine reduces production of norbuprenorphine (a full opioid agonist), a contributor to fetal opioid dependence and neonatal opioid withdrawal syndrome (NOWS) [29]. Deuterated buprenorphine (BUP-D2, 30) like buprenorphine had sub-nanomolar affinity to the human MOR, KOR, and DOR. Compound 30 produced partial agonist effects at the human MOR and KOR. Plasma concentrations of norbuprenorphine were lower in rats treated with 30 (4 mg/kg i.v.) relative to those dosed with buprenorphine (4 mg/kg i.v.). Deuterated buprenorphine 30 mitigated fentanyl-induced catalepsy in pregnant Sprague-Dawley rats compared to buprenorphine and decreased NOWS when the mother was exposed to fentanyl. Deuterated buprenorphine safeguards the mother from opioid-induced toxicity upon relapse and protects the fetus from opioid withdrawal or opioid overdose or exposure to full opioid agonists via the mother.
2.1. Small Molecules Targeting Non-Opioid Receptors
Drug therapies targeting the MOR have been the main treatments for pain. However, these opioids have a high abuse potential and lead to the development of tolerance. Hence, the need to target non-opioid receptors for treatments of OUD. Ibogaine 31, the major psychoactive alkaloid found in the root bark of Tabernanthe iboga, has been shown to have anti-addictive properties. However, ibogaine and its active metabolite noribogaine 32 have been associated with sudden death in humans which has been attributed to their ability to cause QT interval prolongation and arrythmias [30]. Sames et al. report the development of benzofuran-containing ibogaine analogs represented by general structures 33 and 34 (Figure 10) with representative examples 25-27 that have therapeutic-like effects in SUD rat models [31]. In a pro-arrhythmia assay, noribogaine produced pro-arrhythmia while oxa-noribogaine 35, epi-oxa-noribogaine 36, and desethyl-oxa-noribogaine 37 did not produce pro-arrhythmic effects. Administration of 40 mg/kg oxa-noribogaine significantly reduced morphine intake for seven consecutive days in adult male Fisher F-344 rats trained to self-administer morphine. In addition, administration of 40 mg/kg of compound 35 significantly reduced fentanyl self-administration for two consecutive days in adult male Fisher F-344 rats trained to self-administer fentanyl. Sub-chronic dosing with 5 mg/kg of compound 35 reduced morphine intake for eighteen days following the last administration of 35, suggesting that its effects last far beyond the exposure to the drug. In addition, repeated doses of oxa-noribogaine 35 reduced fentanyl-induced hyperalgesia. Overall, these results showed that oxa-iboga alkaloids have desirable persistent neuroplasticity and neuro-restorative effects.
In another patent, Sames and colleagues investigated structure-activity-relationships of ibogaine and its metabolite noribogaine at the serotonin transporter (SERT) and vesicular monoamine transporter 2 (VMAT2) as a treatment option for SUD and other neuropsychiatric illnesses [32]. Modulation of the targets SERT and VMAT2 by ibogaine analogs have been shown to offset the adverse behavioral effects of substances of abuse in preclinical studies[33]. Moreover, an open label observational clinical study demonstrated that a single ibogaine treatment significantly reduced opioid withdrawal symptoms and improved patient quality of life [34,35]. Preclinical studies in rodent models of SUD showing attenuation of the self-administration of opioids, cocaine, nicotine, and alcohol upon administration of ibogaine are consistent with clinical studies with ibogaine [36,37]. In this patent, Sames and his group focused on analogs represented by general formula 38 (Figure 11). The findings showed that 39 was approximately 5 and 3.5 times more potent at SERT and VMAT2 than noribogaine with Ki values of 0.035 ± 0.0067 µM and 0.10 ± 0.02 µM at SERT and VMAT2, respectively. 10-Ethoxyibogaine, 40 was found to be a potent VMAT2 inhibitor and a much less potent inhibitor of SERT. The reported compounds in this patent have varying affinities at SERT and VMAT2 and have potential for treatment of psychiatric and neurological disorders. No in vivo data were reported in this claim.
In a patent filed by Dolmetsch et al., the authors report the use of mavoglurant, 41 (Figure 12), a mGlu5 receptor (mGluR5) antagonist, as a treatment for SUD [38]. In preclinical studies mavoglurant was reported to have effects consistent with reduction of opioid use and prevention of relapse, and alleviation of the symptoms of depression and anxiety associated with OUD. The target mGlu5R has been implicated in the rewarding effects of heroin and ketamine [39]. Mavoglurant was first synthesized and reported in patents by Kuesters et. al. [40] and Gasparini et. al.[41]. Compound 42 is a representative example from the patent by Dolmetsch.
Mavoglurant (1 mg/kg, po) 41 and 2-Methyl-6-(phenylethynyl)-pyridine (10 mg/kg, po) 42 were shown to significantly reduce morphine tolerance in mice in the hotplate test. Although mavoglurant did not significantly reduce remifentanil self-administration in Sprague Dawley rats, mavoglurant administered at a dose of 10 mg/kg reduced cue-induced relapse to the self-administration of remifentanil. Suggesting that it is plausible to use mavoglurant to attenuate relapse in a SUD patient. The authors also conducted a randomized, subject- and investigator-blinded, parallel group, placebo-controlled study in patients with SUD. Although the results of the clinical trial are not reported in the patent, the authors claim the use of mavoglurant as a treatment for reducing opioid use, relapse, abstinence, anxiety or depression in a SUD patient. Similar to mavoglurant, Imbert and colleagues filed a patent for novel mGlu5 negative allosteric modulators, represented by general formula 43, for treating substance abuse [42]. Among the compounds declared in this patent, two of them (compounds 44 and 45, Figure 12) were the most explored. For instance, an acute injection of 44 significantly attenuated oxycodone self-administration and re-instatement which suggests that 44 like mavoglurant has potential utility in treating OUD.
A patent by Newman et al. provides novel selective Dopamine D3 receptor (D3R) antagonist/partial agonists (represented by one of the general formula 46, Figure 13) that can be used in concert with opioids to treat pain, OUD, and opioid withdrawal [43]. The D3R is upregulated in the brains of cocaine addicts and restricted to limbic brain regions involved in behavioral and emotional responses; as a result, D3R has been implicated as a target to develop therapies for SUD [44]. The authors identified compound (±)-47 as a novel selective D3R (Ki = 6.84 ± 1.18 nM) agent with the potential to be developed as a treatment for pain and OUD. Pretreatment with compound (±)-47 dose-dependently inhibited oxycodone self-administration and reversed naloxone-precipitated conditioned place aversion in chronic oxycodone-treated male Long-Evans rats. In addition, compound (±)-47 potentiated the antinociceptive effects of oxycodone in the hot-plate test in rats and rhesus monkeys.
D3R antagonists and partial agonists have been reported to attenuate opioid self-administration without compromising the opioid antinociceptive effects nor potentiating the opioid cardiovascular effects [45,46,47]. In another patent, Newman et. al. report dual-target MOR and D3R compounds as potential non-addictive treatments for pain [48]. The combination of the well-established antinociceptive effects of MOR agonists with the abuse potential modulating effects of D3R antagonists/partial agonists could be an ideal treatment for pain. These compounds with general formula 48 (Figure 14) were designed to have affinities at both the MOR and D3R rather than simultaneous binding to MOR and D3R in close proximity (MOR- D3R heteromers). Compounds 49, 50, and 51 (Figure 14) were identified as lead compounds from in vitro binding and functional studies. These compounds were found to be partial agonists at MOR and partial agonists or antagonists at D3R. Compound 50 produced antinociceptive effects but did not increase locomotor activity like morphine.
Laezza et al.[49] in 2020 filed a patent to claim a series of novel tetrapeptide compounds with general structure 52 (Figure 15) that are inhibitors of fibroblast growth factor 13 (FGF13-1a and FGF13-1b). The patent included the preparation and the use thereof of compounds with potential applicability in the treatment of pain and combating SUD. The claimed compounds are purported to target a novel protein-protein interaction between Nav 1.6 and FGF13-1a/1b that selectively block peripheral pain transmission without affecting normal mechanical sensation. The prototype compound, PW0164 (53) (Figure 15), dose-dependently attenuates pain in capsaicin-induced pain models. PW0164 showed moderate DMPK profile; t1/2 = 1 hr PO (or 0.17 hr IV) and had no significant BBB penetration. These PW0164-like analogs claimed here are viewed as potential novel antinociceptives with the added advantage of causing less CNS side effects (less propensity for addiction or dependence).
In 2022, Booth filed a patent to claim enantiomerically pure 5-phenyl-2-aminotetralin (5-PAT) compounds with general formula 54 shown below (Figure 16) that target both 5-HT1A and 5-HT7 as partial agonists with potential utility as drugs that can treat or prevent OUD and other neuropsychiatric disorders.[50] The patent includes methods for preparation of the compounds and biological screening. Of the PAT compounds disclosed in the present embodiment, (2S)-5-FPT 55 (Figure 17) and (2S) 5-CPT 56 (Figure 17) were found to be high affinity dual partial agonists at the 5-HT1A (Ki = 4.0 ± 0.1 and 7.5 ± 1.5 nM, respectively) and 5-HT7 (Ki = 5.3 ± 2 and 1 ± 0.12 nM, respectively) receptors. (2S)-5-FPT tested in four heterogeneous models of repetitive and perseverative behavior showed that it eliminates both behaviors without modifying general locomotion suggestive of its potential to treat neuropsychiatric symptoms associated with drug withdrawal.
Rovati and his group at Rottapharm BNiotech claimed a first-in-class imidazole-2 receptor ligand, 2-phenyl-6-(1H-imidazol-1-yl) quinazoline (CR4056) 57 (Figure 18) when co-administered with opioids at a sub-analgesic dose, to be used as a prevention of abuse in an opioid non-addicted subject and to prevent opioid side effects like constipation, sedation, among others[51]. When co-administered with 1 mg/kg of morphine (subtherapeutic dose) in rats, CR4056 (po 10 – 400 mg/day) potentiated antinociceptive effects without triggering side effects such as abuse-related effects and constipation.
In some embodiments in the patent filed by Blahunka et al., [52] the compound designated as ASP8062 (58, Figure 19) was declared in a prior art by Shiraishi [53] as potential treatment for SUD including OUD. Unique to this compound is its ability to treat SUD without precipitating or worsening respiratory depression. ASP8062 is pharmacologically classified as an orally available GABAB positive allosteric modulator (PAM), an attractive target for OUD. In a phase 1 clinical study, the compound was found to be CNS permeable, safe, and well-tolerated. At a dose of 35 mg or 70 mg, ASP8062 displayed GABAergic pharmacodynamic effects that included increased slow-wave sleep and growth hormone release in humans. At 70mg dosing, the Cmax and AUC24 were found to be 165 ng/mL and 1570 ng/mL, respectively. In addition, ASP8062 suppressed morphine self-administration in a non-human primate self-administration model without potentiating morphine induced respiratory depression at high doses.
Kozikowski and Tueckmantel at Bright Minds Biosciences claimed a set of indole compounds that belong to the azepinoindole class of compounds and represented by the general formula 59(Figure 20) that interact with the 5-HT2A receptors, displaying less interaction at the cardiotoxic triggering 5-HT2B receptor and selective over 5-HT2C [54]. The patent sought to cover new chemical entities that mimic psilocybin 60 (or psilocin, 61) as treatment options for an array of neuropsychiatric disorders including depression, OUDs, etc. Of the chemical entities declared, compound 62 is the most promising and explored. In vitro preclinical studies reported in the patent shows the compounds despite being indoles, have low hERG risk and display limited off-target binding. Additionally, the compounds in the embodiment show favorable pharmacokinetic profile that included extrapolated human hepatic clearance of < 5.17 ml/min/kg for 62 (Figure 20). However, compound 62 is about 10-fold less potent in Head Twitch Response (HTR) compared to psilocin.
Flaherty et al. in a 2023 patent disclosed a series of pyrimidinone compounds (represented by general formula 63, Figure 21) that functionally inhibit neuronal adenylyl cyclase 1 (AC1), a novel drug target for unmet needs in autism, pain, opioid dependence, and alcohol use disorder. The compounds claimed were screened against adenylyl cyclase isoform 8 (AC8) as growing evidence suggest it is equally implicated in these disease pathways. The most potent compound was 67(Table 2). No in vivo or pharmacokinetic data were included.
In a patent filed by Wang et al. [56], a series of small molecules inhibiting receptor-type tyrosine-protein phosphatase delta (PTPRD) were claimed as potential treatment options for OUD. The molecules claimed were analogs of 7-butoxy illudalic acid (7-BIA, 68), a known PTPRD inhibitor. PTPRD, as a biological target, has been associated with several addiction-related effects including OUD. The compounds in the claim are represented by the general formula 69. Compound 70 was identified as the most potent inhibitor in the present disclosure with a sub-micromolar IC50 value (Figure 22).
Zhan and Zheng filed a patent demonstrating the use of butyrylcholinesterase (BChE) inhibitors as treatments for heroin use disorder [57]. Heroin is a prodrug whose effects are mediated by its metabolites, morphine and 6-monoacetylmorphine (6-MAM) [58]. BChE is responsible for hydrolyzing heroin into its active metabolites [59], as a result, selective inhibition of BChE will be an effective strategy to treat heroin abuse as well as rescue individuals from heroin overdose. The authors demonstrate that the BChE inhibitor ethopropazine (71, Figure 23) at 10 mg/kg and when combined with 0.3 mg/kg or 1 mg/kg naltrexone completely reversed heroin induced overdose. Pretreatment with the acetylcholinesterase inhibitor Galantamine (72, Figure 23) at 5 or 10 mg/kg did not reverse heroin induced overdose. Thus, selective inhibition of only BChE would be an ideal way to treat heroin use disorder and overdose. In this patent the authors do not report novel compounds, but demonstrate that selective BChE inhibitors like ethopropazine, cymserine, bisnorcymserine, phenethylnorcymserine, tacrine, pyridostigmine, physostigmine, neostigmine, rivastigmine, eptastigmine, iso-ompa, hetopropazine, bambuterol, and MF-8622 may be effective treatments for OUD and overdose.
Polymeropoulos and colleagues at Vanda Pharmaceuticals report the use of the neurokinin-1 receptor antagonist, tradipitant 73 (Figure 23), as treatment for individuals experiencing or likely to experience an undesired consequence of opioid use [60]. In a double-blind within-subject crossover design clinical trial, pre-treatment with tradipitant reduced desire for oxycodone in OUD patients. Tradipitant maintenance decreased opioid drug craving, pleasurable and drug seeking sensations.
Lindsley and colleagues reported the development of competitive and noncompetitive inhibitors of the muscarinic acetylcholine receptor M5 (mAChR M5) that have utility in the treatment of SUD [61,62]. The mAChR M5 is localized with dopaminergic neurons in the mesolimbic reward pathway, and deletion of the mAChR M5 has been shown to attenuate morphine reinforcement and withdrawal but not morphine analgesia [63,64]. VU6019650 (compound 74, Figure 23), an orthosteric antagonist at mAChR M5, was found to be over 100-fold selective for the human mAChR M5 over the human mAChR M1-4 with an IC50 of 36 nM. Pretreatment with 74, dose-dependently reduced oxycodone self-administration in rats [65]. Another compound VU6008667 (compound 75, Figure 24), a negative allosteric modulator (NAM) of mAChR M5, decreased oxycodone self-administration, attenuated cue-induced reinstatement of lever pressing following extinction from oxycodone self-administration and prevented acquisition of oxycodone self-administration behavior when administered to naïve rats [66]. VU6008667 75 did not attenuate naloxone-precipitated oxycodone withdrawal i.e., mAChR M5 antagonists or NAMs provide a novel non-opioid based treatment for OUD but not for opioid withdrawal.
In a patent filed by Lopez and colleagues in 2024, they report novel pannexin-1 modulators which can be used to treat OUD [67]. Pannexins are transmembrane proteins that form gap junctions in vertebrates allowing for cell-cell and cell-matrix interactions. The pannexin (PANX) family includes PANX1, PANX2, and PANX3. PANX1 is ubiquitously expressed in most cell types, including cells in the nervous system. Pannexins are involved in the regulation of ATP, as a result, regulation of pannexins can be beneficial to treat disorders associated with exacerbated activation of ATP like pain and opioid addiction [67]. The PANX-1 inhibitors PX004 (76) and PX011 (77) (Figure 24) produced antinociceptive effects in a rat model of neuropathic pain but produced significantly less opioid withdrawal effects compared to saline.
3. Conclusion and Expert Perspective
In this patent review, we provide an overview of over twenty-four (24) patents filed by industrial and academic institutions in the past decade (2012 and up) focusing on molecular probes that target various aspects of the pathophysiology of OUD. We delved into the small molecules category in which we reviewed patents filed for substances targeting established opioid receptors, as well as those targeting non-opioid systems within the brain.
The molecules covered in the patents are diverse in structure ranging from analogs of naturally occurring alkaloids (i.e., mitragynine, ibogaine), morphine analogs, endorphin analogs, and small peptides. The biological targets of the molecules claimed in the patents are in most cases established including opioid receptors (mu and kappa), dopamine receptors (D3), serotonin receptors (5-HTRs; 5-HT1A and 5-HT7) or transporters (SERT), VMAT, and glutamate receptors (mGluR5). A few of the molecules claimed did not directly treat SUD but provided alternatives to antinociception comparative to opioids with decreased tendency to cause addiction via activation of novel pathways such as Nav 1.6 and FGF-13-1a/1b.
Existing treatment for OUD integrates pharmacotherapies, such as long-acting formulations and combination therapies with digital health technologies reflecting a nuanced understanding of the diverse nature of OUD [68]. In the drug discovery arena, we witness a blend of innovative approaches that include the design of small molecules with unique mechanism(s) of action to combat the OUD problem. Overall, we expect that the interest in finding treatment options for OUD will continue given the projected effectiveness of non-opioid small molecules in preclinical studies.
Funding
This work was funded by National Institutes of Health National Institute on Drug Abuse [Grants DA25267, DA48353, and UG3/UH3 DA048353 01].
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Blondell, R.D.; Azadfard, M.; Wisniewski, A.M. Pharmacologic Therapy for Acute Pain. Am Fam Physician 2013, 87, 766–772.
- Benyamin, R.; Trescot, A.M.; Datta, S.; Buenaventura, R.; Adlaka, R.; Sehgal, N.; Glaser, S.E.; Vallejo, R. Opioid Complications and Side Effects. Pain Physician 2008, 11, S105-20.
- Strang, J.; Volkow, N.D.; Degenhardt, L.; Hickman, M.; Johnson, K.; Koob, G.F.; Marshall, B.D.L.; Tyndall, M.; Walsh, S.L. Opioid Use Disorder. Nat Rev Dis Primers 2020, 6, 3. [CrossRef]
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators Global, Regional, and National Incidence, Prevalence, and Years Lived with Disability for 354 Diseases and Injuries for 195 Countries and Territories, 1990-2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [CrossRef]
- Hser, Y.-I.; Huang, D.; Chou, C.-P.; Anglin, M.D. Trajectories of Heroin Addiction: Growth Mixture Modeling Results Based on a 33-Year Follow-up Study. Eval Rev 2007, 31, 548–563. [CrossRef]
- CDC-NCHS. U.S. Overdose Deaths Decrease in 2023, First Time Since 2018. Available online: https://www.cdc.gov/nchs/pressroom/nchs_press_releases/2024/20240515.htm (accessed on 21 May 2024).
- Florence, C.; Luo, F.; Rice, K. The Economic Burden of Opioid Use Disorder and Fatal Opioid Overdose in the United States, 2017. Drug Alcohol Depend 2021, 218, 108350. [CrossRef]
- Stein, C. Opioid Receptors. Annu Rev Med 2016, 67, 433–451. [CrossRef]
- Dugosh, K.; Abraham, A.; Seymour, B.; McLoyd, K.; Chalk, M.; Festinger, D. A Systematic Review on the Use of Psychosocial Interventions in Conjunction With Medications for the Treatment of Opioid Addiction. J Addict Med 2016, 10, 93–103. [CrossRef]
- Doughty, B.; Morgenson, D.; Brooks, T. Lofexidine: A Newly FDA-Approved, Nonopioid Treatment for Opioid Withdrawal. Annals of Pharmacotherapy 2019, 53, 746–753. [CrossRef]
- Timko, C.; Schultz, N.R.; Cucciare, M.A.; Vittorio, L.; Garrison-Diehn, C. Retention in Medication-Assisted Treatment for Opiate Dependence: A Systematic Review. J Addict Dis 2016, 35, 22–35. [CrossRef]
- Ciccarone, D. The Triple Wave Epidemic: Supply and Demand Drivers of the US Opioid Overdose Crisis. Int J Drug Policy 2019, 71, 183–188. [CrossRef]
- Kruegel, A.; Sames D.; Javiitch, J.; Majumdar, S.; inventors; The Trustees of Columbia University in the City of New York, The Research Foundation for Mental Hygiene, Inc., Sloan-Kettering Institute for Cancer Research, assignee. Deuterated Mitragynine Analogs As Safer Opioid Modulators In The Mitragynine Class. WIPO patent 2020160280A1. August 06,2020.
- Kruegel, A.C.; Gassaway, M.M.; Kapoor, A.; Váradi, A.; Majumdar, S.; Filizola, M.; Javitch, J.A.; Sames, D. Synthetic and Receptor Signaling Explorations of the Mitragyna Alkaloids: Mitragynine as an Atypical Molecular Framework for Opioid Receptor Modulators. J Am Chem Soc 2016, 138, 6754–6764. [CrossRef]
- Haile, C.N.; Kosten, T.A.; Das, J.; inventors; University of Houston Systems, assignee. Combination of Mitragynine and Naltrexone for Substance Use Disorders. WIPO patent WO2024/076660A1. April11, 2024..
- Zhang, Y.; inventors; Virginia Commonwealth University, assignee. Non-Peptide Opioid Receptor Modulators For Treatment Of Opioid Abuse And Addiction, Alcoholism And Neurological Disorders Associated With Opioid Receptors. WIPO patent 2020041159A1. February 27, 2020.
- Yuan, Y.; Elbegdorj, O.; Chen, J.; Akubathini, S.K.; Zhang, F.; Stevens, D.L.; Beletskaya, I.O.; Scoggins, K.L.; Zhang, Z.; Gerk, P.M.; et al. Design, Synthesis, and Biological Evaluation of 17-Cyclopropylmethyl-3,14β-Dihydroxy-4,5α-Epoxy-6β-[(4’-Pyridyl)Carboxamido]Morphinan Derivatives as Peripheral Selective μ Opioid Receptor Agents. J Med Chem 2012, 55, 10118–10129. [CrossRef]
- Zheng, Y.; Obeng, S.; Wang, H.; Jali, A.M.; Peddibhotla, B.; Williams, D.A.; Zou, C.; Stevens, D.L.; Dewey, W.L.; Akbarali, H.I.; et al. Design, Synthesis, and Biological Evaluation of the Third Generation 17-Cyclopropylmethyl-3,14β-Dihydroxy-4,5α-Epoxy-6β-[(4’-Pyridyl)Carboxamido]Morphinan (NAP) Derivatives as μ/κ Opioid Receptor Dual Selective Ligands. J Med Chem 2019, 62, 561–574. [CrossRef]
- Li, G.; Aschenbach, L.C.; Chen, J.; Cassidy, M.P.; Stevens, D.L.; Gabra, B.H.; Selley, D.E.; Dewey, W.L.; Westkaemper, R.B.; Zhang, Y. Design, Synthesis, and Biological Evaluation of 6alpha- and 6beta-N-Heterocyclic Substituted Naltrexamine Derivatives as Mu Opioid Receptor Selective Antagonists. J Med Chem 2009, 52, 1416–1427. [CrossRef]
- Zhang, Y.A.N.; inventor; Virginia Commonwealth University, assignee. Naltrexamine Derivatives Bearing 5-Member Heterocyclic Ring Systems as Opioid Receptor Modulators. WIPO Patent WO 2023/163969 A3. October 5, 2023.
- German, N.; Hossain, M.A.; inventors; Texas Tech University System, assignee. Novel Opioid Antagonists and Methods Related Thereto. US patent 20200131183A1. April 30, 2020.
- German, N.; Neugebauer, V.; Hossain, M.A.; Abbruscatto, T. inventors; Texas Tech University System, assignee. Novel Selective Kappa Opioid Receptor Antagonists And Methods Related Thereto For Treatment Of Addiction And Neuropathic Pain. WIPO Patent 2020092996A1. May 7, 2020.
- Shahbazi Nia, S.; Hossain, M.A.; Ji, G.; Jonnalagadda, S.K.; Obeng, S.; Rahman, M.A.; Sifat, A.E.; Nozohouri, S.; Blackwell, C.; Patel, D.; et al. Studies on Diketopiperazine and Dipeptide Analogs as Opioid Receptor Ligands. Eur J Med Chem 2023, 254, 115309. [CrossRef]
- Peltier, J.; Surman, M.; Golden, K.; Pasetto, P.;Jin, X.; Camara, F.; Jiang, X.; inventors; Waterville Valley Technologies, Inc., assignee. Preparation Of Oxycodone Analogs and Compositions for Preventing Opioid Abuse. US patent 20160326182A1. November 10, 2016.
- Medina, J.C.; Nerurkar, A.; Sadlowski, C.; Seidl, F.; Cheng, H.; Duquette, J.; Lee, J.; Holan, M.; Ding, P.; Wang, X.; et al., inventors; Epiodyne Inc., assignee. Opioid Receptor Modulators. WIPO Patent WO 2022/216750 A1. October 13, 2022.
- Rice, K.C.; Jacobson, A.; Sulima, A.; Chambers, D.; Roth, H.; inventors; US Health, assignee. Selective Opioid Receptor Agonists and Antagonists. WIPO patent 2024/026350A1. February 1, 2024.
- Brents, L.; Tobacyk, J.; Crooks, P.; Janganati, V.; inventors. Bio Ventures LLC, assignee. Deuterated Buprenorphine as a Protective Agent for Fetal Subjects Against Full-Agonist Opioid Exposure. WIPO patent 2024/036337A2. February 15, 2024.
- Griffin, B.A.; Caperton, C.O.; Russell, L.N.; Cabanlong, C. V; Wilson, C.D.; Urquhart, K.R.; Martins, B.S.; Zita, M.D.; Patton, A.L.; Alund, A.W.; et al. In Utero Exposure to Norbuprenorphine, a Major Metabolite of Buprenorphine, Induces Fetal Opioid Dependence and Leads to Neonatal Opioid Withdrawal Syndrome. J Pharmacol Exp Ther 2019, 370, 9–17. [CrossRef]
- Huang, P.; Kehner, G.B.; Cowan, A.; Liu-Chen, L.Y. Comparison of Pharmacological Activities of Buprenorphine and Norbuprenorphine: Norbuprenorphine Is a Potent Opioid Agonist. J Pharmacol Exp Ther 2001, 297, 688–695.
- Rubi, L.; Eckert, D.; Boehm, S.; Hilber, K.; Koenig, X. Anti-Addiction Drug Ibogaine Prolongs the Action Potential in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Cardiovasc Toxicol 2017, 17, 215–218. [CrossRef]
- Sames, D.; Hemby, S.; Havel, V.; inventors; The Trustees of Columbia University In The City Of New York, High Point University, assignee. Oxa-Ibogaine Analogues for Treatment of Substance Use Disorders. WIPO patent 2022178099A1. August 25, 2022.
- Sames, D.; Havel, V.; Hwu, C.; inventors; The Trustees Of Columbia University In The City Of New York, assignee. Ibogaine Analogs as Therapeutics for Neurological and Psychiatric Disorders. WIPO patent 2022020352A1. January 27, 2022.
- Kargbo, R.B. Ibogaine and Their Analogs as Therapeutics for Neurological and Psychiatric Disorders. ACS Med Chem Lett 2022, 13, 888–890. [CrossRef]
- Noller, G.E.; Frampton, C.M.; Yazar-Klosinski, B. Ibogaine Treatment Outcomes for Opioid Dependence from a Twelve-Month Follow-up Observational Study. Am J Drug Alcohol Abuse 2018, 44, 37–46. [CrossRef]
- Brown, T.K.; Alper, K. Treatment of Opioid Use Disorder with Ibogaine: Detoxification and Drug Use Outcomes. Am J Drug Alcohol Abuse 2018, 44, 24–36. [CrossRef]
- Belgers, M.; Leenaars, M.; Homberg, J.R.; Ritskes-Hoitinga, M.; Schellekens, A.F.A.; Hooijmans, C.R. Ibogaine and Addiction in the Animal Model, a Systematic Review and Meta-Analysis. Transl Psychiatry 2016, 6, e826. [CrossRef]
- Glick, S.D.; Maisonneuve, I.M.; Szumlinski, K.K. Mechanisms of Action of Ibogaine: Relevance to Putative Therapeutic Effects and Development of a Safer Iboga Alkaloid Congener. Alkaloids Chem Biol 2001, 56, 39–53. [CrossRef]
- Dolmetsch, R.C.E.; Gasparini, F.; Gomez-Mancilla, B.; inventors; Novartis A.G., assignee Use of mGluR5 Antagonists. WIPO patent 2022013809 A2. January 20, 2022.
- van der Kam, E.L.; de Vry, J.; Tzschentke, T.M. Effect of 2-Methyl-6-(Phenylethynyl) Pyridine on Intravenous Self-Administration of Ketamine and Heroin in the Rat. Behavioural pharmacology 2007, 18, 717–724. [CrossRef]
- Kuesters, E.; Acemoglu, M.; Lustenberger, P.; Sedelmeier, G.; Schmidt, B.; Penn, G.; inventors; Novartis A.G., assignee. Processes for the preparation of 4-oxo-octahydro-indole-1-carbocylic acid methyl ester and derivatives thereof. WIPO patent 2022013809A2. February 18, 2010.
- Gasparini, F.; Auberson, Y.; Ofner, S., inventors; Novartis A.G., Novartis Pharma Gmbh., Gasparini Fabrizio, Auberson Yves, Ofner Silvio, assignee. Acetylene Derivatives Having mGluR 5 Antagonistic Activity. CA patent 2732095A1. June 12, 2003.
- Imbert, B.; Sung, V.; Dolmetsch, R.E.; Clyde, H.W.; inventors; Tempero Bio Inc., assignee. Methods of Treating Substance Use Disorder with 4-(3-Cyanophenyl)-6-Pyridinylpyrimidine mGlu5 Negative Allosteric Modulators 2023. WIPO patent 2023/249789A1. December 28, 2023.
- Newman, A.H.; Kumar, V.; Shaik, A.B.; inventors; The United States of America, As Represented By The Secretary, Department Of Health And Human Services, assignee. Dopamine D3 Receptor Selective Antagonists/Partial Agonists and Uses Thereof. March 19, 2020.
- Segal, D.M.; Moraes, C.T.; Mash, D.C. Up-Regulation of D3 Dopamine Receptor MRNA in the Nucleus Accumbens of Human Cocaine Fatalities. Brain Res Mol Brain Res 1997, 45, 335–339. [CrossRef]
- You, Z.-B.; Bi, G.-H.; Galaj, E.; Kumar, V.; Cao, J.; Gadiano, A.; Rais, R.; Slusher, B.S.; Gardner, E.L.; Xi, Z.-X.; et al. Dopamine D3R Antagonist VK4-116 Attenuates Oxycodone Self-Administration and Reinstatement without Compromising Its Antinociceptive Effects. Neuropsychopharmacology 2019, 44, 1415–1424. [CrossRef]
- Jordan, C.J.; Humburg, B.A.; Thorndike, E.B.; Shaik, A.B.; Xi, Z.-X.; Baumann, M.H.; Newman, A.H.; Schindler, C.W. Newly Developed Dopamine D 3 Receptor Antagonists, R -VK4-40 and R -VK4-116, Do Not Potentiate Cardiovascular Effects of Cocaine or Oxycodone in Rats. Journal of Pharmacology and Experimental Therapeutics 2019, 371, 602–614. [CrossRef]
- Jordan, C.J.; Humburg, B.; Rice, M.; Bi, G.-H.; You, Z.-B.; Shaik, A.B.; Cao, J.; Bonifazi, A.; Gadiano, A.; Rais, R.; et al. The Highly Selective Dopamine D R Antagonist, R-VK4-40 Attenuates Oxycodone Reward and Augments Analgesia in Rodents. Neuropharmacology 2019, 158, 107597. [CrossRef]
- Newman, A.H.; Bonifazi, A.; Battiti, F.O.; inventors; The United States of America, As Represented By The Secretary, Department Of Health And Human Services, assignee. Dual-Target Mu Opioid and Dopamine D3 Receptors Ligands; Preparation and Use Thereof. WIPO patent 2022187206A1. September 09, 2024.
- Laezza, F.; Zhou, J.; Chung, J.M.; Wang, P.; La, J.; Folorunso, O.; Singh, A.; inventors; The Board of Regents of The University Of Texas System, assignee. Novel Non-Opioid Anti-Pain Medication. WIPO patent 2020154679 A1. July 30, 2020.
- Booth, R.; inventor; Northeastern University, assignee. Serotonin Receptor Modulators. WIPO patent 2022040395A2. February 24, 2022.
- Rovati, L.C.; inventor; Rottapharm Biotech S.R.L., assignee. Use of 2-phenyl-6-(1h-imidazol-1-yl) quinazoline for the prevention of abuse and of side effects of at least one opioid. WIPO patent WO2021104639 A1. June 3, 2021.
- Blahunka, P.; Gerard, M.; Mototsugu, I.; inventors; Astellas Pharma Global Dev Inc, assignee. Methods of Treating Substance Use Disorder. WIPO patent 2023028519 A2. March 2, 2023.
- Shiraishi, N.; Hoshii, H.; Hamaguchi, W.; Honjo, E.; Takuwa, T.; Kondo, Y.; Goto, T.; inventors; Astellas Pharma Inc., assignee. Sulfur-containing bicyclic compound. USPTO patent 9051339 B2. June 9, 2013.
- Kozikowski, A.; Tueckmantel, W.; inventors; Bright Minds Bioscienes, assignees. Azepinoindoles and Methods of Preparation Thereof 2023. WIPO patent 2023/212811 A1. November 9, 2023.
- Flaherty, D.P.; Watts, V.J.; Dwyer, T.S.; Annadka, S.; inventors; Purdue Research Foundation, assignee. Pyrazolyl Pyrimidinone Compounds and the Uses Thereof. US patent 20230250084 A1. August 10, 2023.
- Wang, W.; Prisinzano, T.; Uhl, G.; Henderson, I.; inventors; The Us Gov as Represented by the Department of Veterans Affairs , Wang Wei , Prisinzano Thomas; assignee. PTPRD Inhibitors and Uses Thereof. WIPO patent 2022232694A1. November 3, 2022.
- Zhan, C.; Zheng, F.; inventors; Univ Kentucky Res Found, assignee. Butyrylcholinesterase Inhibitors for Treatment of Opioid Use Disorder. US patent 20200368224A1. November 26, 2020.
- Inturrisi, C.E.; Schultz, M.; Shin, S.; Umans, J.G.; Angel, L.; Simon, E.J. Evidence from Opiate Binding Studies That Heroin Acts through Its Metabolites. Life Sci 1983, 33 Suppl 1, 773–776. [CrossRef]
- Lockridge, O.; Mottershaw-Jackson, N.; Eckerson, H.W.; La Du, B.N. Hydrolysis of Diacetylmorphine (Heroin) by Human Serum Cholinesterase. J Pharmacol Exp Ther 1980, 215, 1–8.
- Polymeropoulos MH, Birznieks GP, et al., inventors; Vanda Pharmaceuticals Inc., University Of Kentucky Research Foundation, assignee. Method Of Treatment With Tradipitant. WIPO patent 2019236852A1. December 12, 2019.
- Felts, A.; Han, C.; Capstick, R.; Orsi, D.; Whomble, D.; Lindsley C.; Conn, P.; inventors; Univ Vanderbilt, assignee. Arylsulfonyl Derivatives and Their Use as Muscarinic Acetylcholine Receptor M5 Inhibitors. WIPO patent 2021/257977A1. December 23, 2021.
- Lindsley, W.; Jones, K.C.; Conn, P.J.; Han, C.; Felts, A.S.; Orsi, D.L.; Engers, J.L.; Li, J.; Capstick, R.A.; Whomble, D.L.; Temple, K.J.; inventors; Univ Vanderbilt, assignee. Competitive and Noncompetitive Inhibitors of the Muscarinic Acetylcholine Receptor M5. WIPO patent 2021237038 A1. November 25, 2021.
- Basile, A.S.; Fedorova, I.; Zapata, A.; Liu, X.; Shippenberg, T.; Duttaroy, A.; Yamada, M.; Wess, J. Deletion of the M5 Muscarinic Acetylcholine Receptor Attenuates Morphine Reinforcement and Withdrawal but Not Morphine Analgesia. Proc Natl Acad Sci U S A 2002, 99, 11452–11457. [CrossRef]
- Bender, A.M.; Garrison, A.T.; Lindsley, C.W. The Muscarinic Acetylcholine Receptor M5: Therapeutic Implications and Allosteric Modulation. ACS Chem Neurosci 2019, 10, 1025–1034. [CrossRef]
- Garrison, A.T.; Orsi, D.L.; Capstick, R.A.; Whomble, D.; Li, J.; Carter, T.R.; Felts, A.S.; Vinson, P.N.; Rodriguez, A.L.; Han, A.; et al. Development of VU6019650: A Potent, Highly Selective, and Systemically Active Orthosteric Antagonist of the M5 Muscarinic Acetylcholine Receptor for the Treatment of Opioid Use Disorder. J Med Chem 2022, 65, 6273–6286. [CrossRef]
- Teal, L.B.; Bubser, M.; Duncan, E.; Gould, R.W.; Lindsley, C.W.; Jones, C.K. Selective M5 Muscarinic Acetylcholine Receptor Negative Allosteric Modulator VU6008667 Blocks Acquisition of Opioid Self-Administration. Neuropharmacology 2023, 227, 109424. [CrossRef]
- Lopez, D.A.; Gross, G.M.; inventors; Pannex Therapeutics Inc, assignee. Pannexin-1 Modulators and Methods of Treating Disorders in which Pannexin-1 is Implicated. WIPO patent 2024049929 A2. March 7, 2024.
- Buresh, M.; Stern, R.; Rastegar, D. Treatment of Opioid Use Disorder in Primary Care. BMJ 2021, n784. [CrossRef]
Figure 1.
General structure of mitragynine analogs 1 claimed in the patent by Kruegel and colleagues: Most analogs → X = N, R1 = 9-OMe, R2 = 7-OH, R3 = 3-D with examples of selected compounds (mitragynine 2, 3-dehydromitragynine (3-DM) 3, 3-Deuteromitragynine 4, 7-hydroxymitragynine, 5) highlighted in the patent filed by Kruegel et al.[13].
Figure 1.
General structure of mitragynine analogs 1 claimed in the patent by Kruegel and colleagues: Most analogs → X = N, R1 = 9-OMe, R2 = 7-OH, R3 = 3-D with examples of selected compounds (mitragynine 2, 3-dehydromitragynine (3-DM) 3, 3-Deuteromitragynine 4, 7-hydroxymitragynine, 5) highlighted in the patent filed by Kruegel et al.[13].
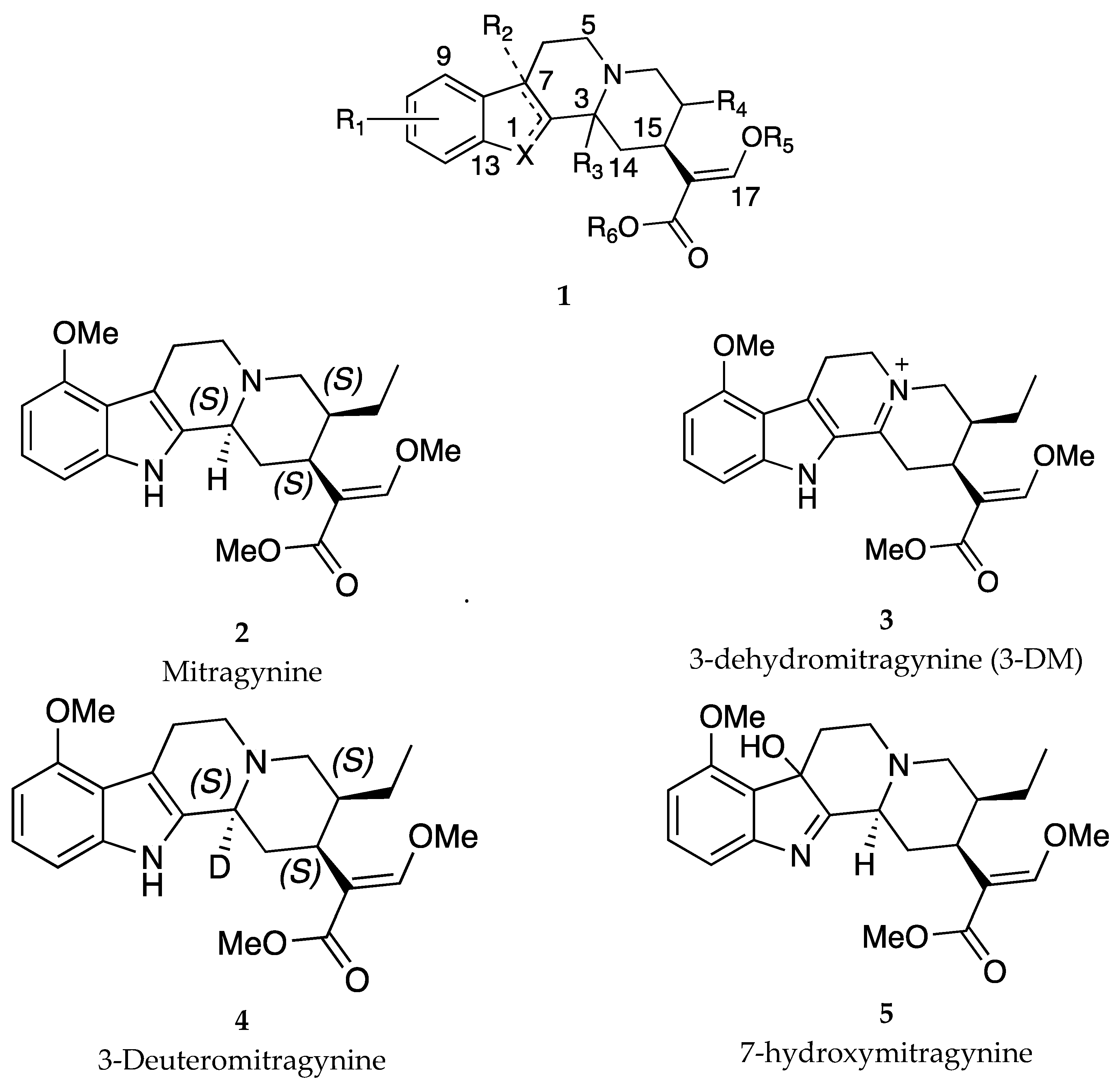
Figure 2.
General formula 6 for non-peptide opioid compounds claimed by Zhang [16]. R1, R2, R3, and R4 = variable substituents including hydrogens, halogens, and alkyl groups and the structure of the naltrexamine 7.
Figure 2.
General formula 6 for non-peptide opioid compounds claimed by Zhang [16]. R1, R2, R3, and R4 = variable substituents including hydrogens, halogens, and alkyl groups and the structure of the naltrexamine 7.
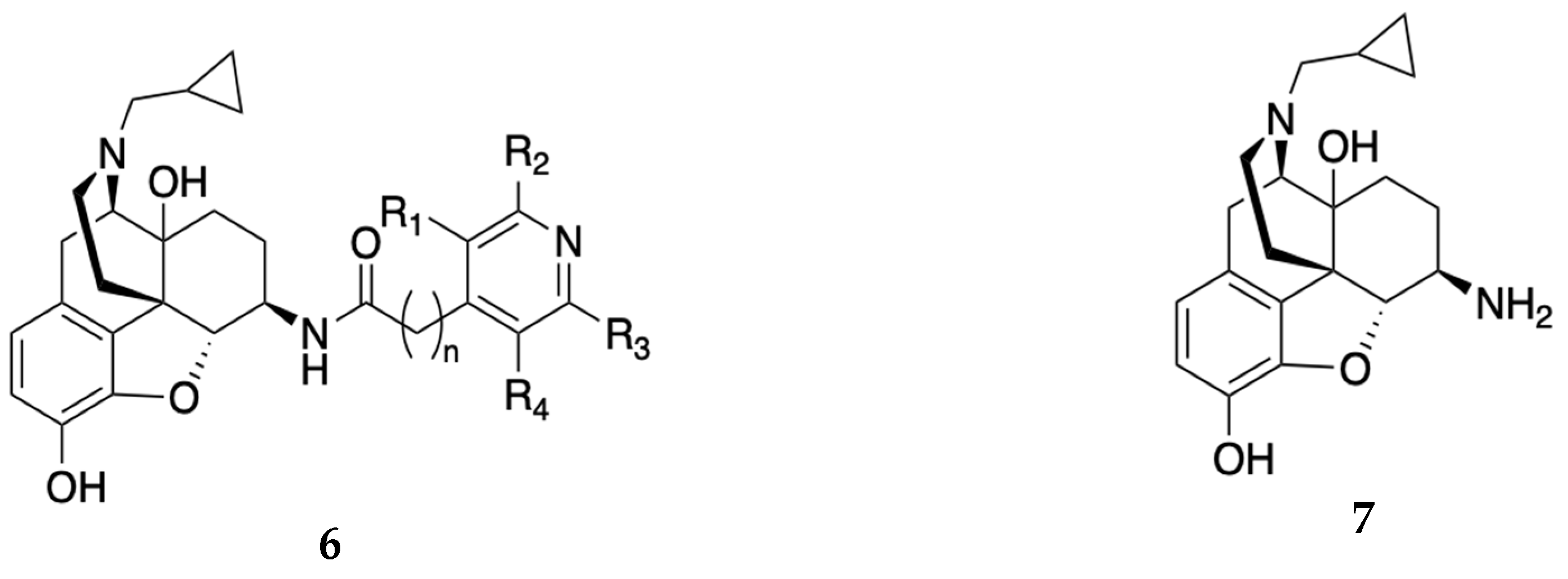
Figure 3.
A series of non-peptide opioid receptor modulators having the general formula 12 and represented by compounds 13, 14, and 15 and their respective binding affinities. M = variable alkyl chain of length 0-10 while X1-X5 found in R are independently C, N, O or S with M attached to any of X1-X5.
Figure 3.
A series of non-peptide opioid receptor modulators having the general formula 12 and represented by compounds 13, 14, and 15 and their respective binding affinities. M = variable alkyl chain of length 0-10 while X1-X5 found in R are independently C, N, O or S with M attached to any of X1-X5.
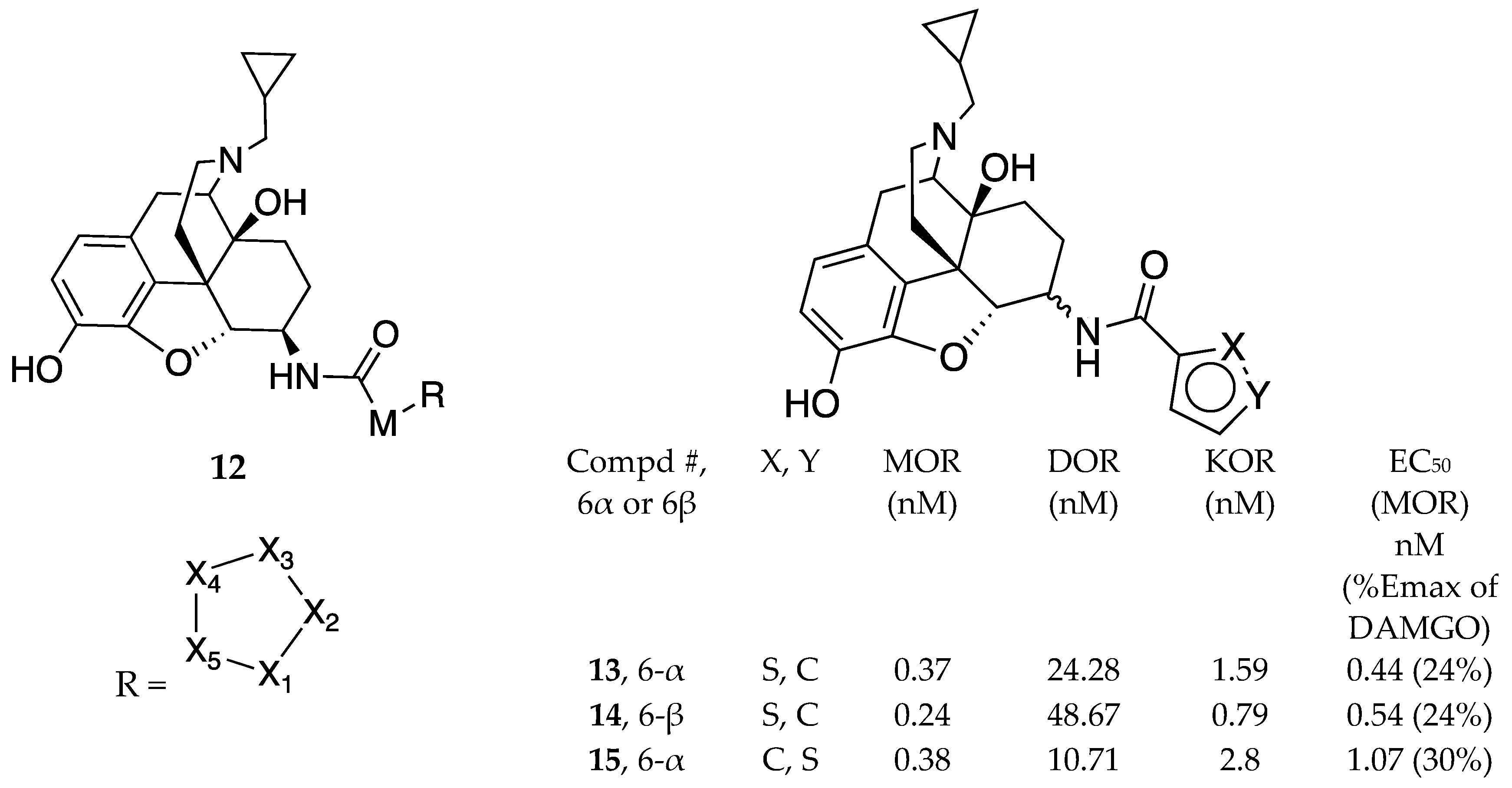
Figure 4.
General structure of gliotoxin analogs 16, Gliotoxin 17 and compound 18 (with their binding affinity at opioid receptors). R1 = OMe and R2 = CH3.
Figure 4.
General structure of gliotoxin analogs 16, Gliotoxin 17 and compound 18 (with their binding affinity at opioid receptors). R1 = OMe and R2 = CH3.
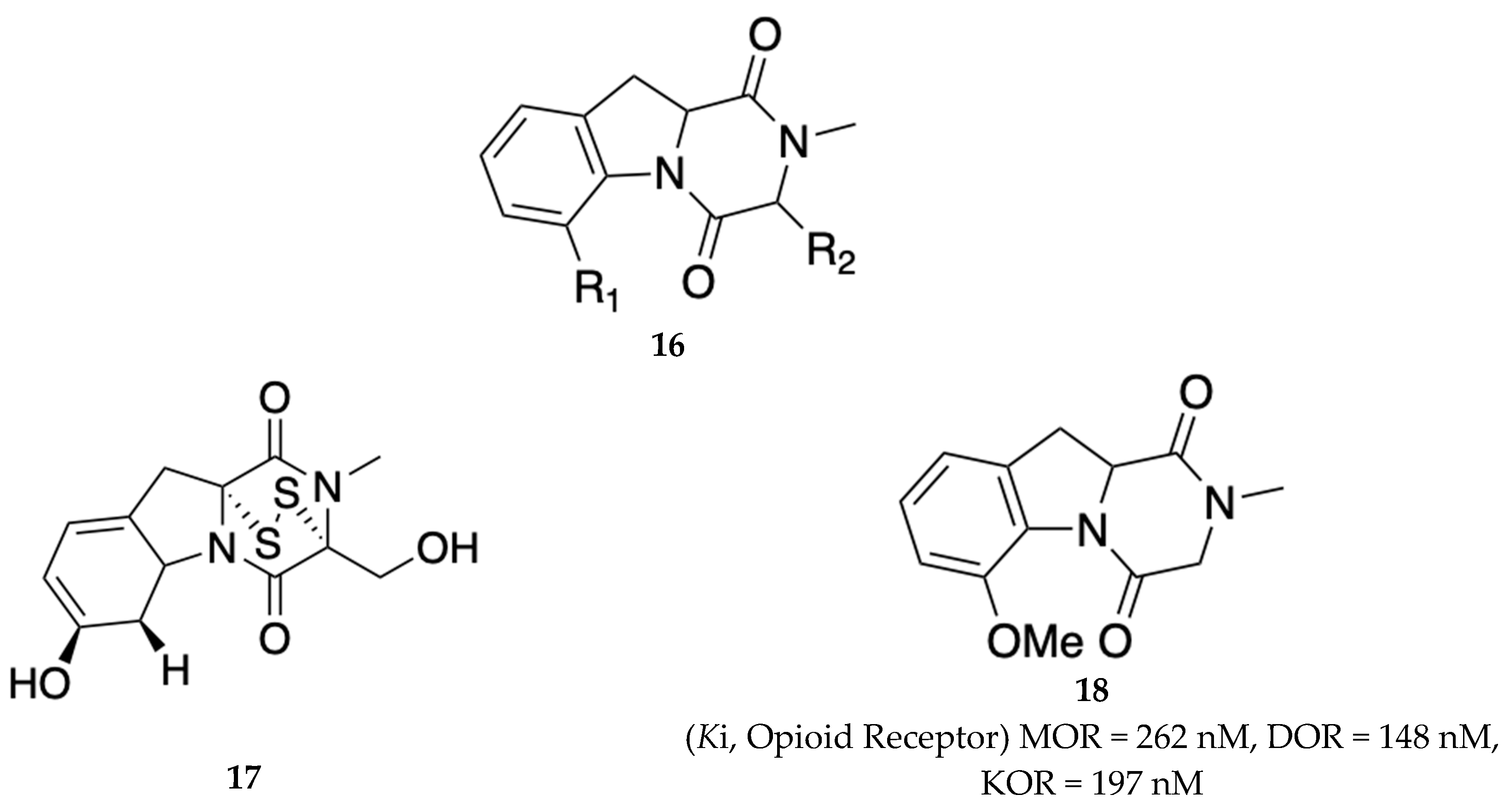
Figure 5.
General formula (I) 19 & formula (II) 20 of the selective KOR antagonists claimed by German and colleagues [22]. Respective examples are included herein for formula I and II as AH-3-193 (21) and AH-3-199 (22). Formula I & II: R1 = variable H, halogen, NO2, alkyl, cycloalkyl, trifluoroalkyl, substituted phenyl, carboxylic acid, alkyl ester of carboxylic acid, and acetyl. R2: variable H, OH, alkyloxy, alkyl ester, amine, alkylamine, dialkylamine, thio, thioalkyl, and alkyl ethers. R3: H, benzyl, substituted benzyl, methyl, alkyl carbamate, alkyl, prenyl, and cycloalkyl. R4: H, alkyl, acetyl, cycloalkyl, benzyl, and substituted benzyl. R5: alkyl ester of carboxylic acid, alkyl ether, alkylamide, amine, monoalkylamine, dialkyl amine, and cycloalkyl.
Figure 5.
General formula (I) 19 & formula (II) 20 of the selective KOR antagonists claimed by German and colleagues [22]. Respective examples are included herein for formula I and II as AH-3-193 (21) and AH-3-199 (22). Formula I & II: R1 = variable H, halogen, NO2, alkyl, cycloalkyl, trifluoroalkyl, substituted phenyl, carboxylic acid, alkyl ester of carboxylic acid, and acetyl. R2: variable H, OH, alkyloxy, alkyl ester, amine, alkylamine, dialkylamine, thio, thioalkyl, and alkyl ethers. R3: H, benzyl, substituted benzyl, methyl, alkyl carbamate, alkyl, prenyl, and cycloalkyl. R4: H, alkyl, acetyl, cycloalkyl, benzyl, and substituted benzyl. R5: alkyl ester of carboxylic acid, alkyl ether, alkylamide, amine, monoalkylamine, dialkyl amine, and cycloalkyl.
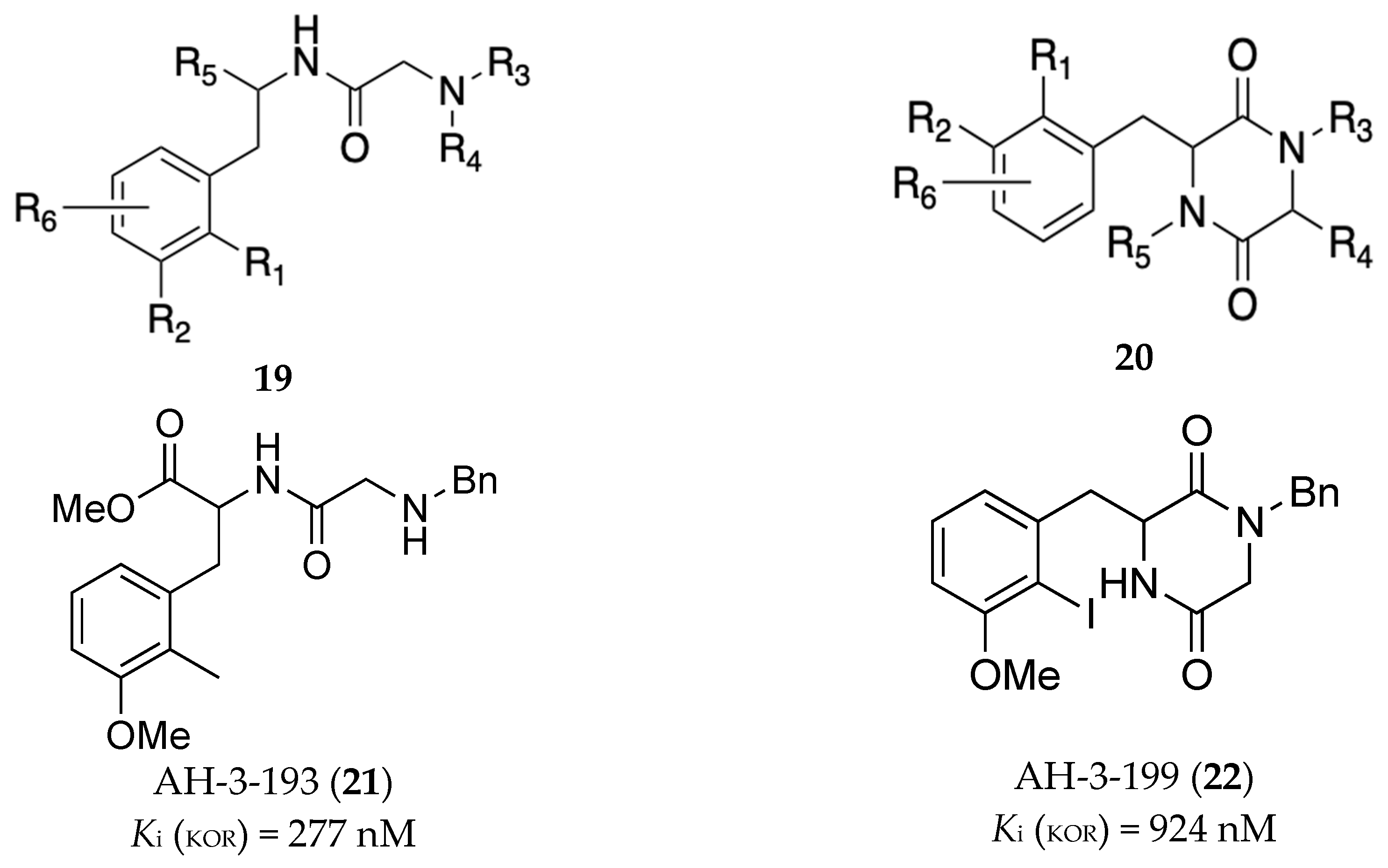
Figure 6.
The general formula 23 for oxycodone analogs claimed by Peltier et al.[24]. Compound 24 is a representative example of the compounds claimed. R1 =OH, alkoxy, and —OC(O)-alkoxy; R2 =OH and —OC(O)-variable R3 = variable substituent as described in patent.
Figure 6.
The general formula 23 for oxycodone analogs claimed by Peltier et al.[24]. Compound 24 is a representative example of the compounds claimed. R1 =OH, alkoxy, and —OC(O)-alkoxy; R2 =OH and —OC(O)-variable R3 = variable substituent as described in patent.
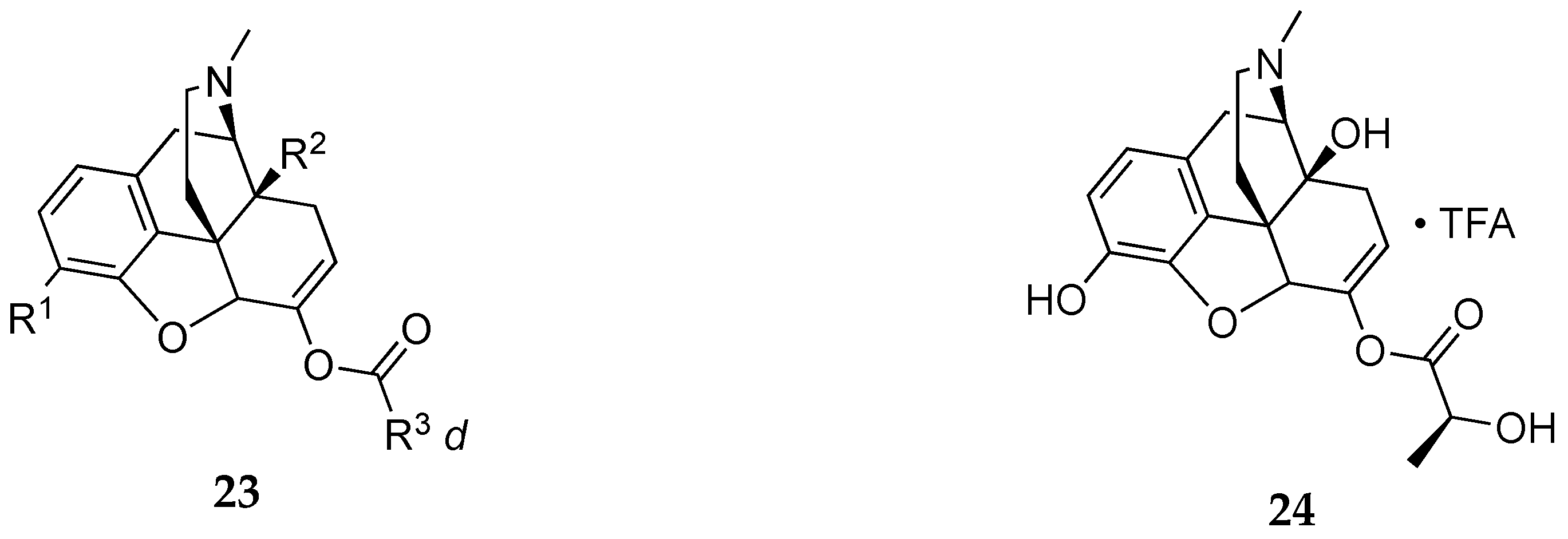
Figure 7.
General formula, 25, of compounds claimed by Medina and colleagues [25]. Structure of representative compound, 26. R1, R2 = C6 - C10 aryl, heteroaryl R3 = -X-R3a where X = a bond or alkylene, R3a = cycloalkyl, R4 = H, alkyl or haloalkyl, R5 and R6 are each independently selected from H, C1-6 alkyl, and C1-6 haloalkyl groups.
Figure 7.
General formula, 25, of compounds claimed by Medina and colleagues [25]. Structure of representative compound, 26. R1, R2 = C6 - C10 aryl, heteroaryl R3 = -X-R3a where X = a bond or alkylene, R3a = cycloalkyl, R4 = H, alkyl or haloalkyl, R5 and R6 are each independently selected from H, C1-6 alkyl, and C1-6 haloalkyl groups.
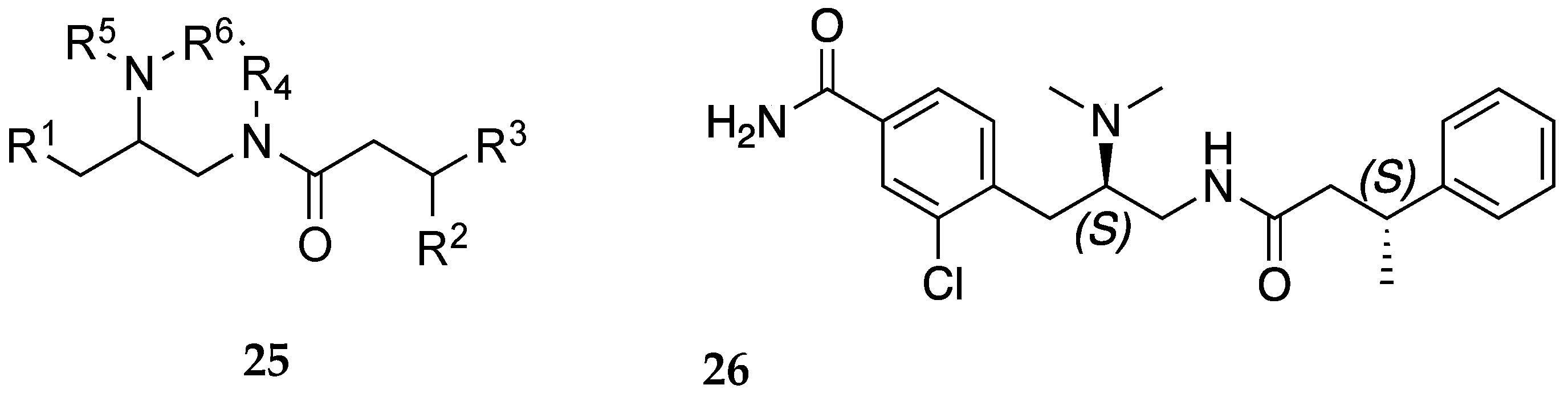
Figure 8.
General formula, 27, of compounds claimed by Rice and colleagues [26]. X is -OR1, -NR1R2, -CO2R1, -CONR1R2, -(CR1R2)m1OH, an amide, a nitrogen-containing heteroaryl, or Ar-NHCHO; wherein each R1 is H, OH, an ether, an ester, a carboxyl, a substituted or unsubstituted C1-C30 alkyl, a substituted or unsubstituted C3-C30 cycloalkyl, or a substituted or unsubstituted C5-C30 aryl; each R2 is H, OH, an ether, an ester, a substituted or unsubstituted C1-C30 alkyl, a substituted or unsubstituted C3-C30 cycloalkyl, a substituted or unsubstituted C2-C30 alkanoyl, a substituted or unsubstituted C4-C30 cycloalkanoyl, or a substituted or unsubstituted C5-C30 aryl; Ar is a substituted or unsubstituted C5-C30 aryl and n is an integer of 1 to 10. J1 and J2 are each independently, H or a halogen. Structures of representative compounds 28 and 29 are also shown.
Figure 8.
General formula, 27, of compounds claimed by Rice and colleagues [26]. X is -OR1, -NR1R2, -CO2R1, -CONR1R2, -(CR1R2)m1OH, an amide, a nitrogen-containing heteroaryl, or Ar-NHCHO; wherein each R1 is H, OH, an ether, an ester, a carboxyl, a substituted or unsubstituted C1-C30 alkyl, a substituted or unsubstituted C3-C30 cycloalkyl, or a substituted or unsubstituted C5-C30 aryl; each R2 is H, OH, an ether, an ester, a substituted or unsubstituted C1-C30 alkyl, a substituted or unsubstituted C3-C30 cycloalkyl, a substituted or unsubstituted C2-C30 alkanoyl, a substituted or unsubstituted C4-C30 cycloalkanoyl, or a substituted or unsubstituted C5-C30 aryl; Ar is a substituted or unsubstituted C5-C30 aryl and n is an integer of 1 to 10. J1 and J2 are each independently, H or a halogen. Structures of representative compounds 28 and 29 are also shown.
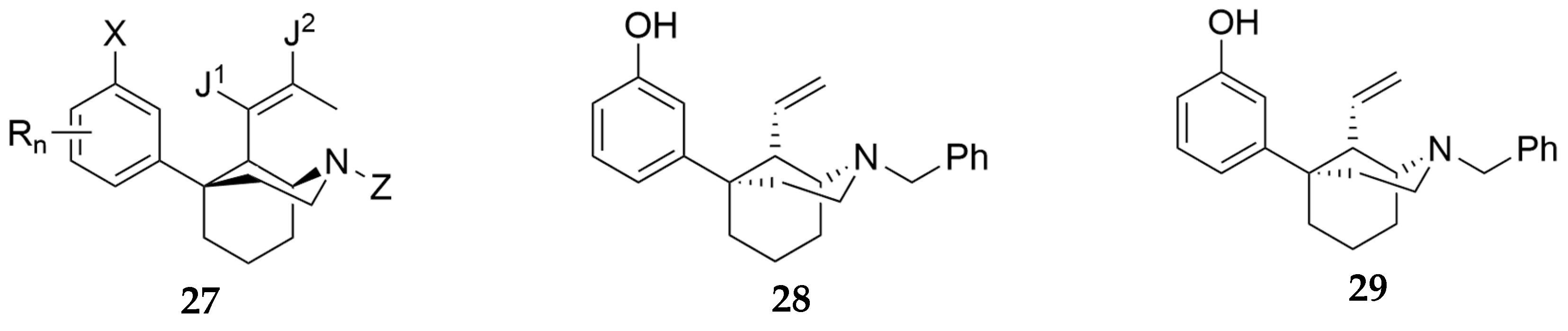
Figure 9.
Structure of compound 30 (BUP-D2) reported in the patent by Brents and colleagues [27].
Figure 9.
Structure of compound 30 (BUP-D2) reported in the patent by Brents and colleagues [27].
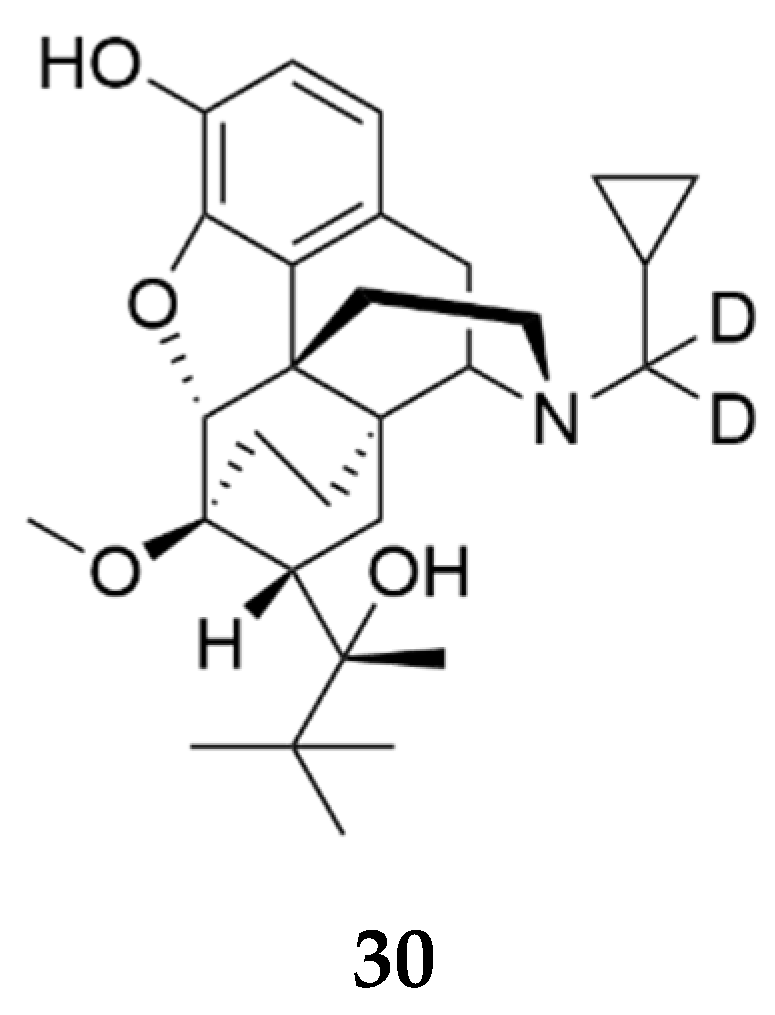
Figure 10.
Structures of Ibogaine 31, noribogaine 32, general structure of the ibogaine analogs (33,34), oxa-noribogaine 35, epi-noribogaine 36, and desethyl-oxa-noribogaine 37 claimed in the patent by Sames et al.[31]. In general formula 1a, 33, A = ring structure with or without substitution, X1 and X2 can be N, C or S, Y1, Y2 = alkyl groups. In general formula 1b, 34, Y1 = alkyl group, R1 = mostly ethyl.
Figure 10.
Structures of Ibogaine 31, noribogaine 32, general structure of the ibogaine analogs (33,34), oxa-noribogaine 35, epi-noribogaine 36, and desethyl-oxa-noribogaine 37 claimed in the patent by Sames et al.[31]. In general formula 1a, 33, A = ring structure with or without substitution, X1 and X2 can be N, C or S, Y1, Y2 = alkyl groups. In general formula 1b, 34, Y1 = alkyl group, R1 = mostly ethyl.
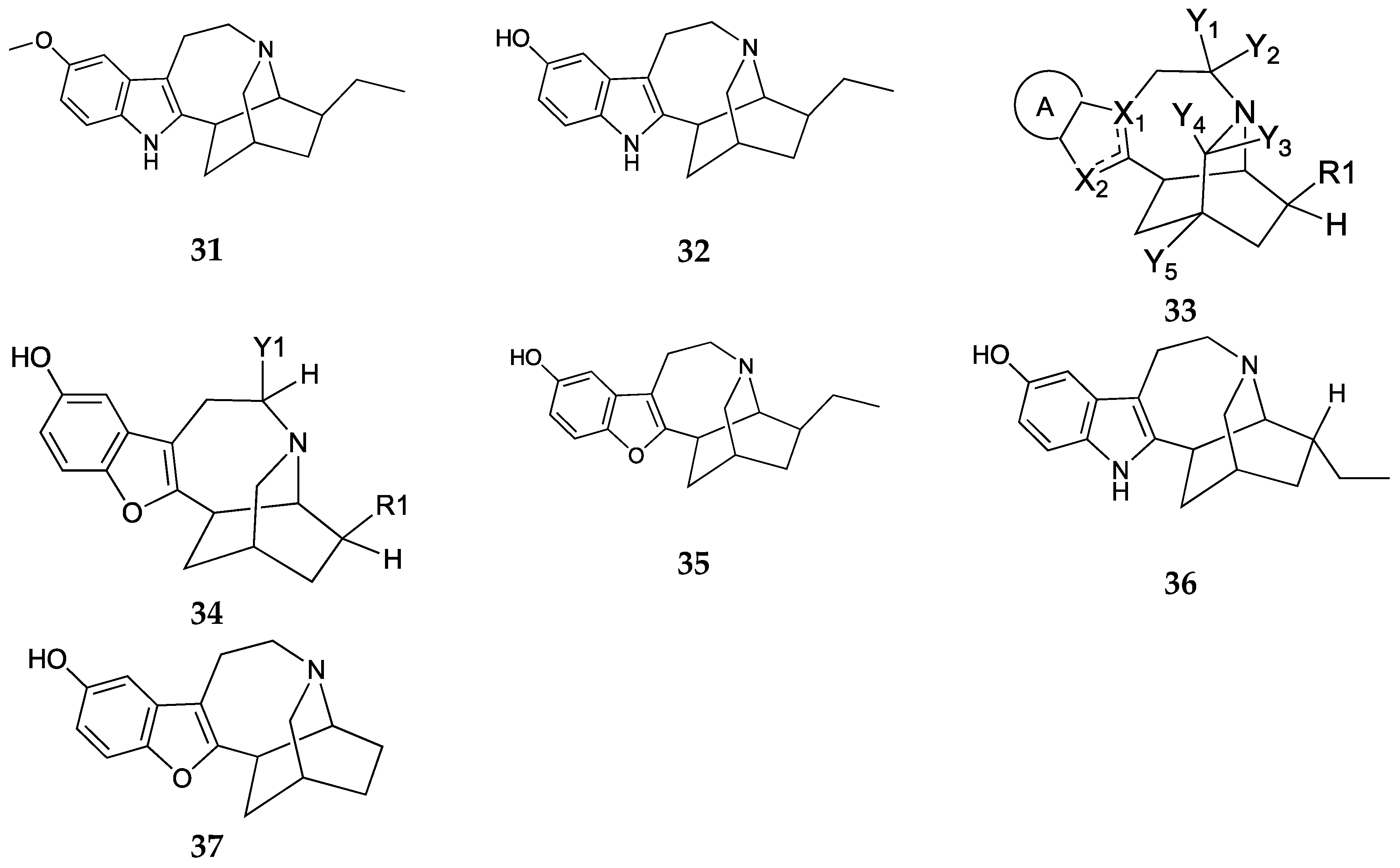
Figure 11.
General formula 38 and representative examples (39 and 40) of ibogaine analogs claimed by Sames et al.[32]. In general formula, R1, R2, R3, and R4 = variable substituents (halogens, alkyl groups), Y1, Y2 = variable alkyl/aryl groups, X1 = alkyl group, Z1 and Z2 = variable alkyl halide.
Figure 11.
General formula 38 and representative examples (39 and 40) of ibogaine analogs claimed by Sames et al.[32]. In general formula, R1, R2, R3, and R4 = variable substituents (halogens, alkyl groups), Y1, Y2 = variable alkyl/aryl groups, X1 = alkyl group, Z1 and Z2 = variable alkyl halide.
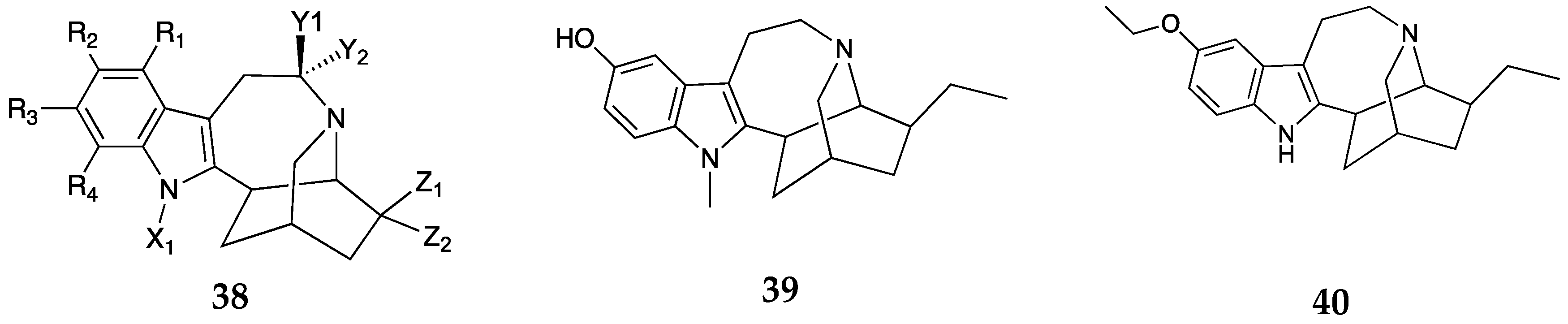
Figure 12.
Structure of mavoglurant 41 [40,41], and a representative example 42, (patent from Dolmetsch et al.) and the general structure 43 of the mGlu5 negative allosteric modulators claimed by Imbert et al. where R is H, Me, F, Cl or cyano, [42].
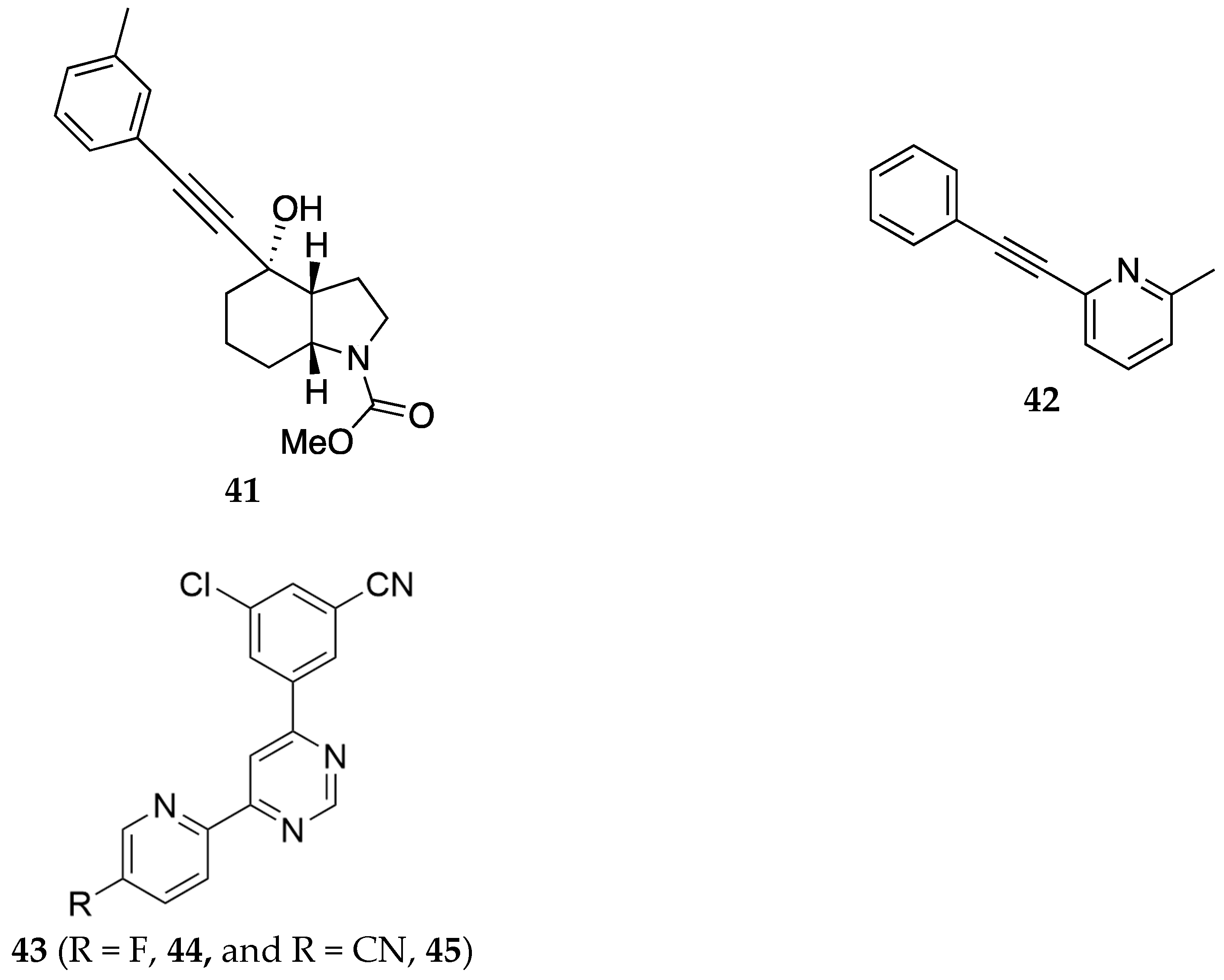
Figure 13.
General formula 46, of compounds claimed by Newman et al. and structure of key compound 47 [43]. Where in general formula Ar = heteroaryl with variable substituents, n, m = 0-4 (1 -2 specifically), R1 = H or halogen, R2 = R3 = H, C1-C3 alkoxy, or halogen, R4, R5 = H, OH or halogen.
Figure 13.
General formula 46, of compounds claimed by Newman et al. and structure of key compound 47 [43]. Where in general formula Ar = heteroaryl with variable substituents, n, m = 0-4 (1 -2 specifically), R1 = H or halogen, R2 = R3 = H, C1-C3 alkoxy, or halogen, R4, R5 = H, OH or halogen.
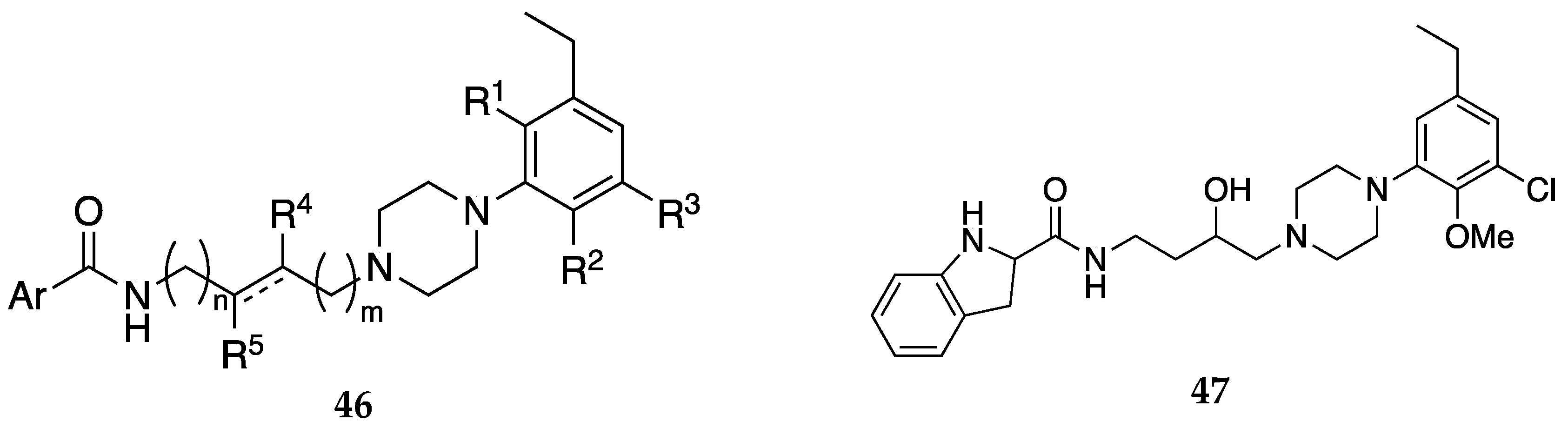
Figure 14.
General formula 48 and examples of compounds with dual activity at MOR and D3R claimed by Newman et al. [48]. Compounds 50 and 51 are representative examples. Rx = CN, -(C=O)-N(Rz)2 wherein Rz = H, C1-C6 alkyl, haloalkyl, C2-C6 alkanoyl; Ra = C1-C6 alkyl, C1-C6 haloalkyl, halogen, OH, NH2, NO2, CN, -COOH, CHO, CONH2, variable alkoxy, haloalkoxy; n = 0-3; Y1 is -NH- or a piperazinyl group attached to the core structure and L1 through the nitrogen atoms; L1 is a covalent bond or a linking group; Y2 is a covalent bond or a 5-6-membered heterocyclic group comprising 1 or 2 nitrogen atoms; L2 is a covalent bond or a linking group; and Ar1 is an aryl or a heteroaryl, wherein when Ar1 is phenyl, the phenyl is substituted by 2, 3, or 4 substituents (see patent for exhaustive list).
Figure 14.
General formula 48 and examples of compounds with dual activity at MOR and D3R claimed by Newman et al. [48]. Compounds 50 and 51 are representative examples. Rx = CN, -(C=O)-N(Rz)2 wherein Rz = H, C1-C6 alkyl, haloalkyl, C2-C6 alkanoyl; Ra = C1-C6 alkyl, C1-C6 haloalkyl, halogen, OH, NH2, NO2, CN, -COOH, CHO, CONH2, variable alkoxy, haloalkoxy; n = 0-3; Y1 is -NH- or a piperazinyl group attached to the core structure and L1 through the nitrogen atoms; L1 is a covalent bond or a linking group; Y2 is a covalent bond or a 5-6-membered heterocyclic group comprising 1 or 2 nitrogen atoms; L2 is a covalent bond or a linking group; and Ar1 is an aryl or a heteroaryl, wherein when Ar1 is phenyl, the phenyl is substituted by 2, 3, or 4 substituents (see patent for exhaustive list).
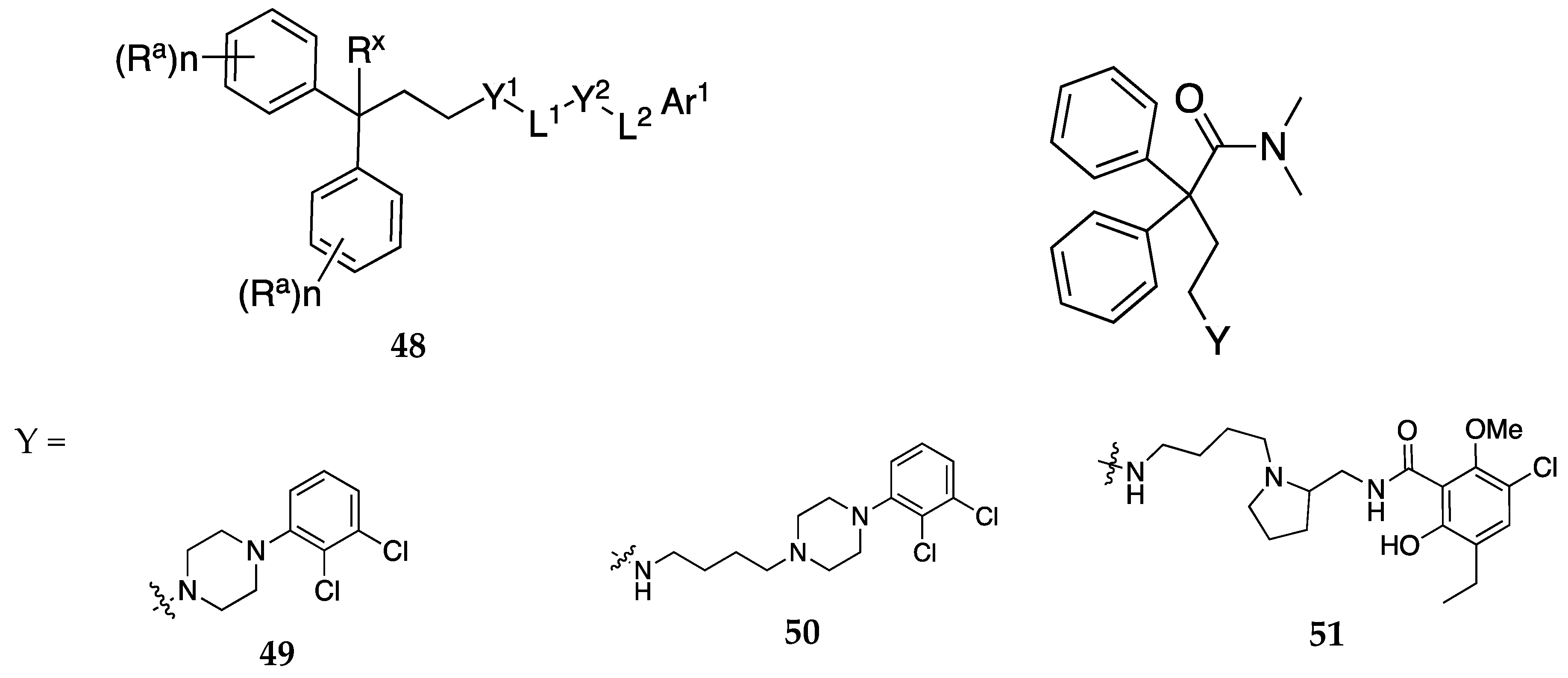
Figure 15.
General structure of tetrapeptides 52 and structure of prototype compound 53 (PW0164) claimed by Laezza and group [49]. In general formula 52, R1 = H, alkyl, cycloalkyl, aryl, heteroaryl, R4CO-, R5NHCO-, R6OCO-, R7SC>2-, or Fmoc; where R4, R5, R6 and R7 = alkyl, cycloalkyl, alkenyl, aryl, heteroaryl, adamantyl, and benzyl, which are further substituted with -OH, -CN, -NH2, and halogen; R2 is H, alkyl, aryl, heteroaryl, cycloalkyl, or Boc; R3 is OH, alkoxy, allyloxy, -NR8R9; wherein R8 and R9 = H, alkyl, aryl, and heteroaryl; or R8 and R9 are optionally joined to form a N-containing heterocycle with 1-3 heteroatoms; and wherein R1 and R2 are not both hydrogen.
Figure 15.
General structure of tetrapeptides 52 and structure of prototype compound 53 (PW0164) claimed by Laezza and group [49]. In general formula 52, R1 = H, alkyl, cycloalkyl, aryl, heteroaryl, R4CO-, R5NHCO-, R6OCO-, R7SC>2-, or Fmoc; where R4, R5, R6 and R7 = alkyl, cycloalkyl, alkenyl, aryl, heteroaryl, adamantyl, and benzyl, which are further substituted with -OH, -CN, -NH2, and halogen; R2 is H, alkyl, aryl, heteroaryl, cycloalkyl, or Boc; R3 is OH, alkoxy, allyloxy, -NR8R9; wherein R8 and R9 = H, alkyl, aryl, and heteroaryl; or R8 and R9 are optionally joined to form a N-containing heterocycle with 1-3 heteroatoms; and wherein R1 and R2 are not both hydrogen.
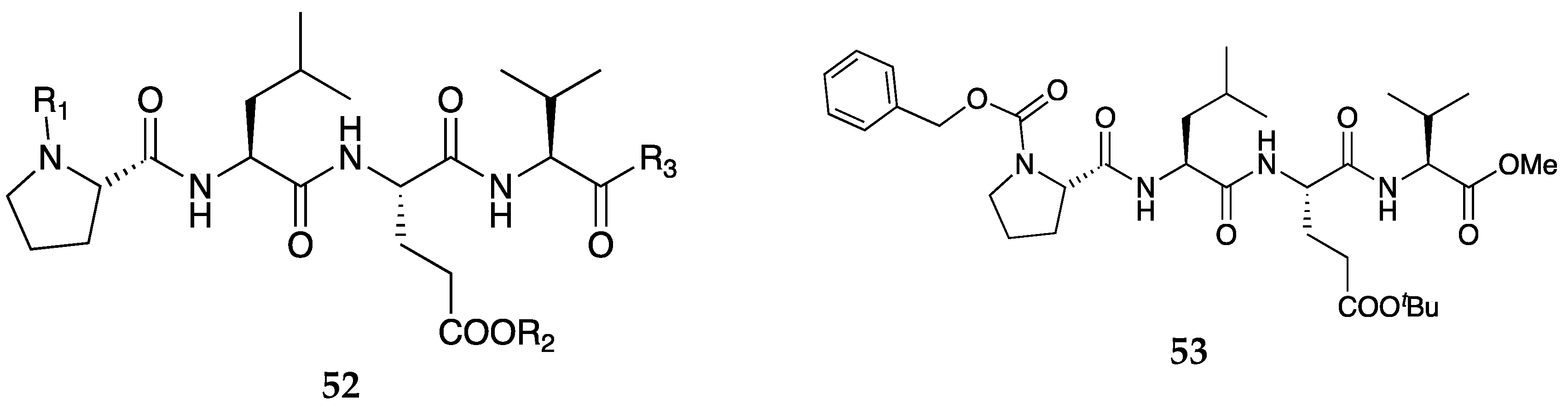
Figure 16.
General formula 54 and structure of analogs of 5-phenyl-2-aminotetralin (5-PAT) claimed by Booth [50]. R1 and R2 = H or alkyl substituent, cyclized R1 and R2 into a ring. R3-R7 = variable substituents on aromatic ring including H, halogen, and alkyl groups, R8, R9 and R10 = variable substituents including halogens, alkyl groups, and heterocyclics.
Figure 16.
General formula 54 and structure of analogs of 5-phenyl-2-aminotetralin (5-PAT) claimed by Booth [50]. R1 and R2 = H or alkyl substituent, cyclized R1 and R2 into a ring. R3-R7 = variable substituents on aromatic ring including H, halogen, and alkyl groups, R8, R9 and R10 = variable substituents including halogens, alkyl groups, and heterocyclics.
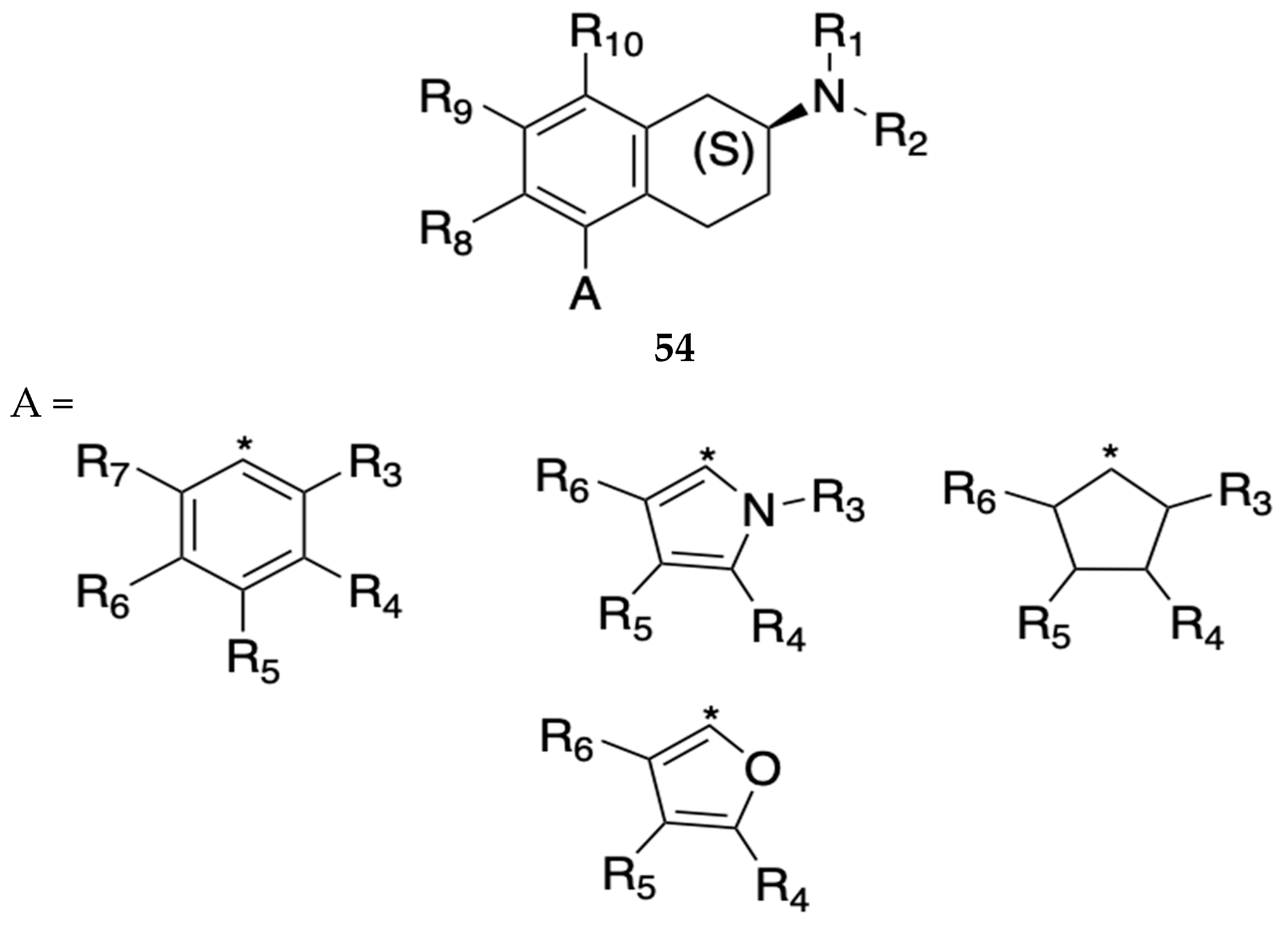
Figure 17.
Structure of PAT compounds (2S) 5-FPT 55 and (2S) 5-CPT 56 claimed by Booth showing respective binding affinity to 5-HT1A and 5-HT7 [50].
Figure 17.
Structure of PAT compounds (2S) 5-FPT 55 and (2S) 5-CPT 56 claimed by Booth showing respective binding affinity to 5-HT1A and 5-HT7 [50].

Figure 18.
Structure of imidazole-2 receptor ligand, 2-phenyl-6-(1H-imidazol-1-yl) quinazoline (CR4056, 57) claimed by Rovati [51].
Figure 18.
Structure of imidazole-2 receptor ligand, 2-phenyl-6-(1H-imidazol-1-yl) quinazoline (CR4056, 57) claimed by Rovati [51].
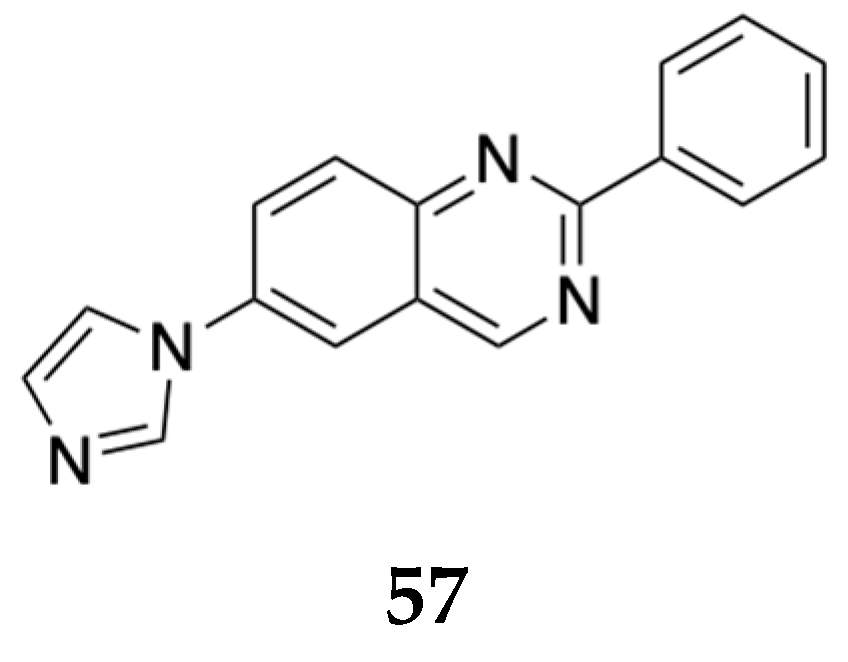
Figure 19.
The chemical structure of ASP8062 (58) claimed by Blahunka and colleagues [52].
Figure 19.
The chemical structure of ASP8062 (58) claimed by Blahunka and colleagues [52].
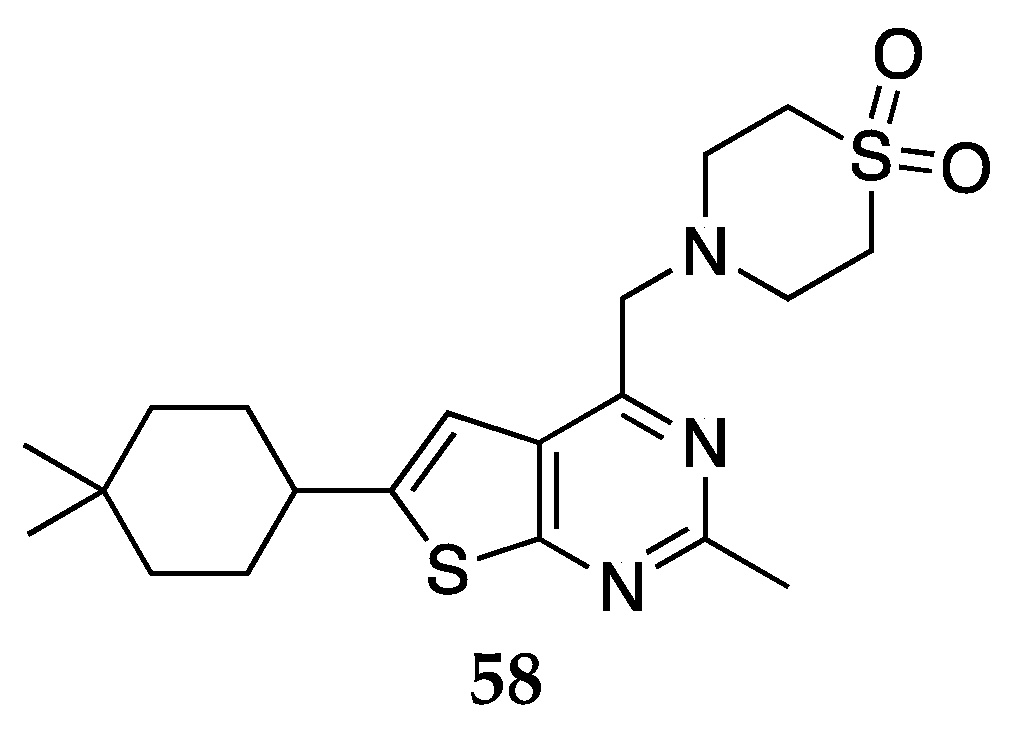
Figure 20.
General structure 59 of compounds claimed by Kozikowski and Tueckmantel, [54]. Structure of psilocybin 60, psilocin 61 and representative example of compounds claimed 62. Summary of associated EC50 values for 61 and 62 at 5-HT2A,2B and 2C are included. For 59, R1 = H, alkyl, alkenyl, alynyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, e1 or e2 form a chain of 2 to 4 carbon atoms to which are attached independent substituents similar to R1 ; R2 = alkyl, alkenyl, alynyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, etc.; a, b = H, halogen, lower alkyl, CHF2, CF3, OCH3, OCHF2, OCF3, SCHF3, amine, CN etc. ; R3 = R1 plus it can be attached to N in azetidine or pyrrolidine ring; each c, d, e, and f = H or lower alkyl group; c1 and c2 together form a part of spirofused cyclopropane/cyclobutene; Z = H, alkyl, variable substituents (see patent for exhaustive list).
Figure 20.
General structure 59 of compounds claimed by Kozikowski and Tueckmantel, [54]. Structure of psilocybin 60, psilocin 61 and representative example of compounds claimed 62. Summary of associated EC50 values for 61 and 62 at 5-HT2A,2B and 2C are included. For 59, R1 = H, alkyl, alkenyl, alynyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, e1 or e2 form a chain of 2 to 4 carbon atoms to which are attached independent substituents similar to R1 ; R2 = alkyl, alkenyl, alynyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, etc.; a, b = H, halogen, lower alkyl, CHF2, CF3, OCH3, OCHF2, OCF3, SCHF3, amine, CN etc. ; R3 = R1 plus it can be attached to N in azetidine or pyrrolidine ring; each c, d, e, and f = H or lower alkyl group; c1 and c2 together form a part of spirofused cyclopropane/cyclobutene; Z = H, alkyl, variable substituents (see patent for exhaustive list).
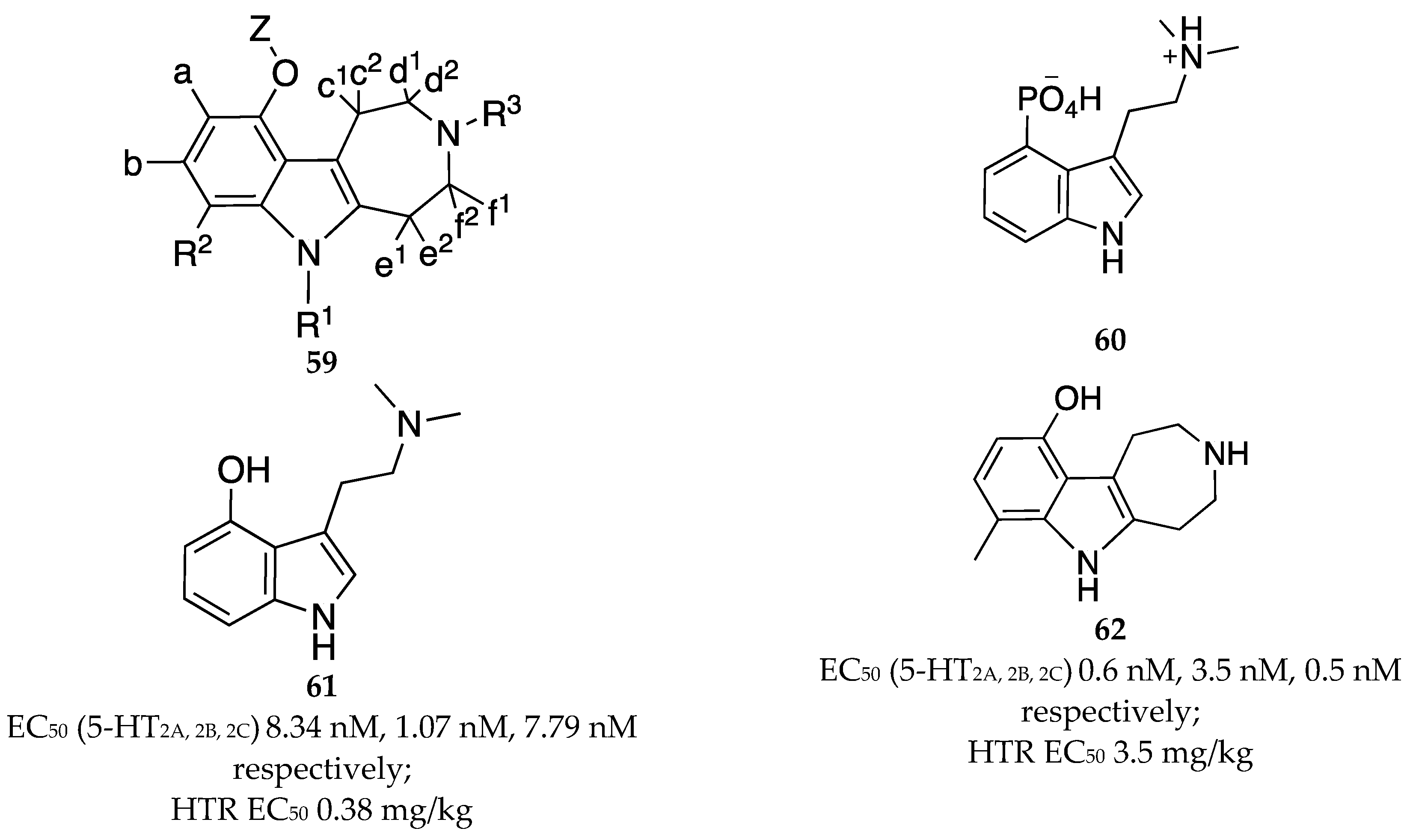
Figure 21.
The general structure 63 of a series of non-opioid pyrimidinone compounds disclosed by Flaherty and his group [55] [ R1 = C1-C12 alkyl, R2 = C1-C12 alkyl, and R3 = variable including alkyl, cyclic ring, heterocyclyl, acyl etc].
Figure 21.
The general structure 63 of a series of non-opioid pyrimidinone compounds disclosed by Flaherty and his group [55] [ R1 = C1-C12 alkyl, R2 = C1-C12 alkyl, and R3 = variable including alkyl, cyclic ring, heterocyclyl, acyl etc].
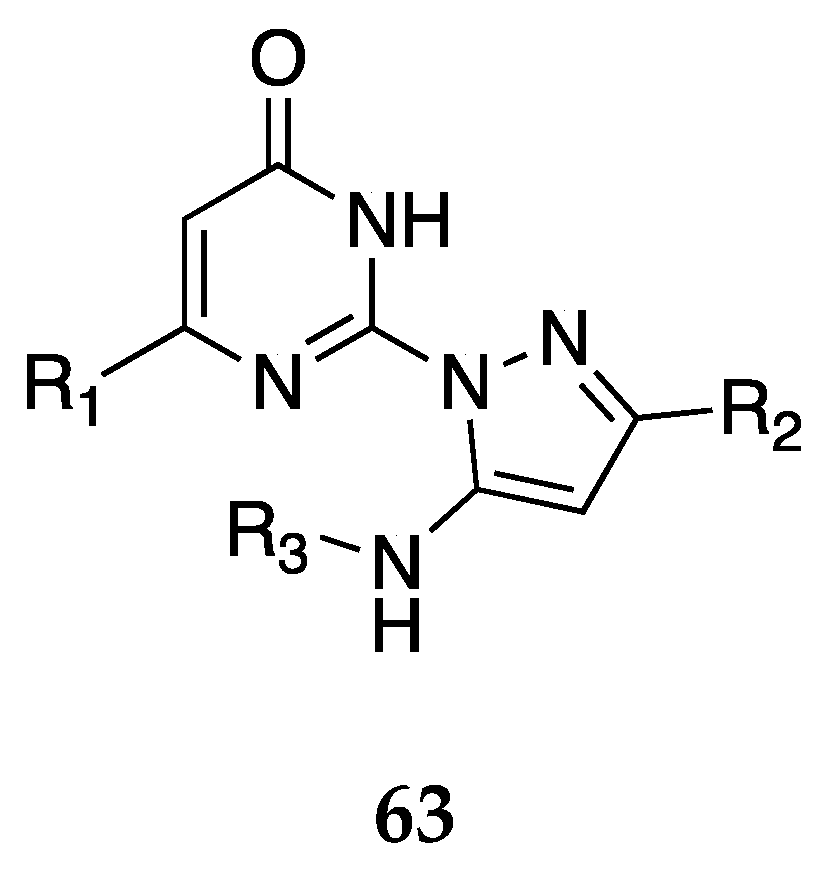
Figure 22.
Structure of 7-BIA 68. General structure of PTPRD analogs claimed in the present patent 69 [56]. Representative example of the most potent PTPRD inhibitor claimed, 70 with associated IC50 at the hPTPRD target. R1, R2, = H, C1-C4 alkyl, C2-C4 alkenyl, C1-C4 haloalkyl, cyanoalkyl, C1-C4 hydroxyalkyl, C2-C4 alkoxy, C2-C4 haloalkoxy etc R3 = variable C1-C9 alkyl, etc.
Figure 22.
Structure of 7-BIA 68. General structure of PTPRD analogs claimed in the present patent 69 [56]. Representative example of the most potent PTPRD inhibitor claimed, 70 with associated IC50 at the hPTPRD target. R1, R2, = H, C1-C4 alkyl, C2-C4 alkenyl, C1-C4 haloalkyl, cyanoalkyl, C1-C4 hydroxyalkyl, C2-C4 alkoxy, C2-C4 haloalkoxy etc R3 = variable C1-C9 alkyl, etc.
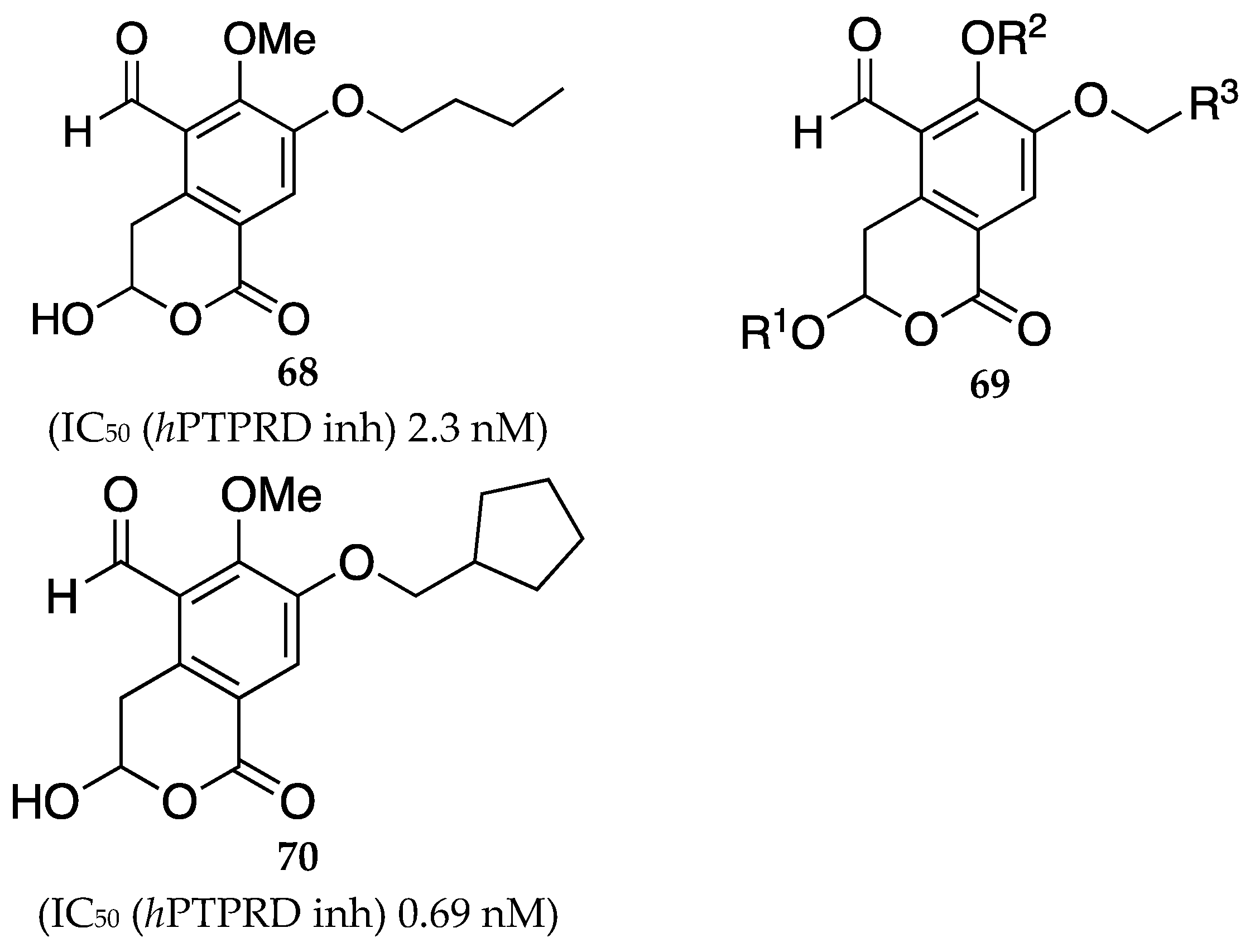
Figure 23.
Structures of some BChE inhibitors highlighted by Zhang and his colleagues (Ethnopropazine 71 and Galantamine 72) [57] and the structure of tradipitant 73 reported by Polymeropoulos and colleagues [60].
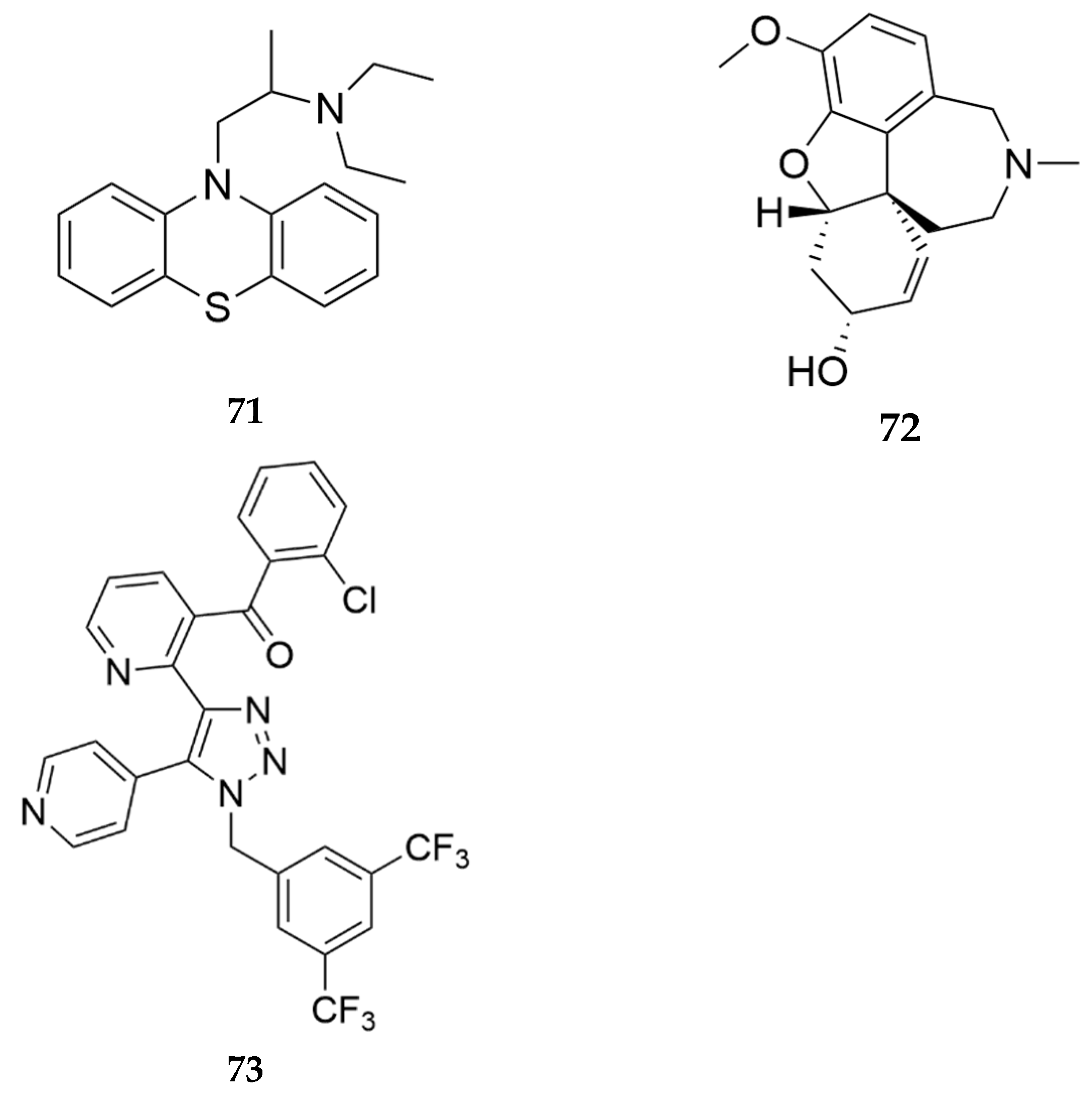
Figure 24.
Structures of mAChR M5 inhibitors (74 & 75) reported by Lindsley et al. [61,62] and pannexin-1 modulators (76 & 77) reported by Lopez et al. [67]. [Compounds 74, 75, 76, and 77 were reported in the patent and other publications as VU6019650, VU6008667, PX004, and PX011, respectively].
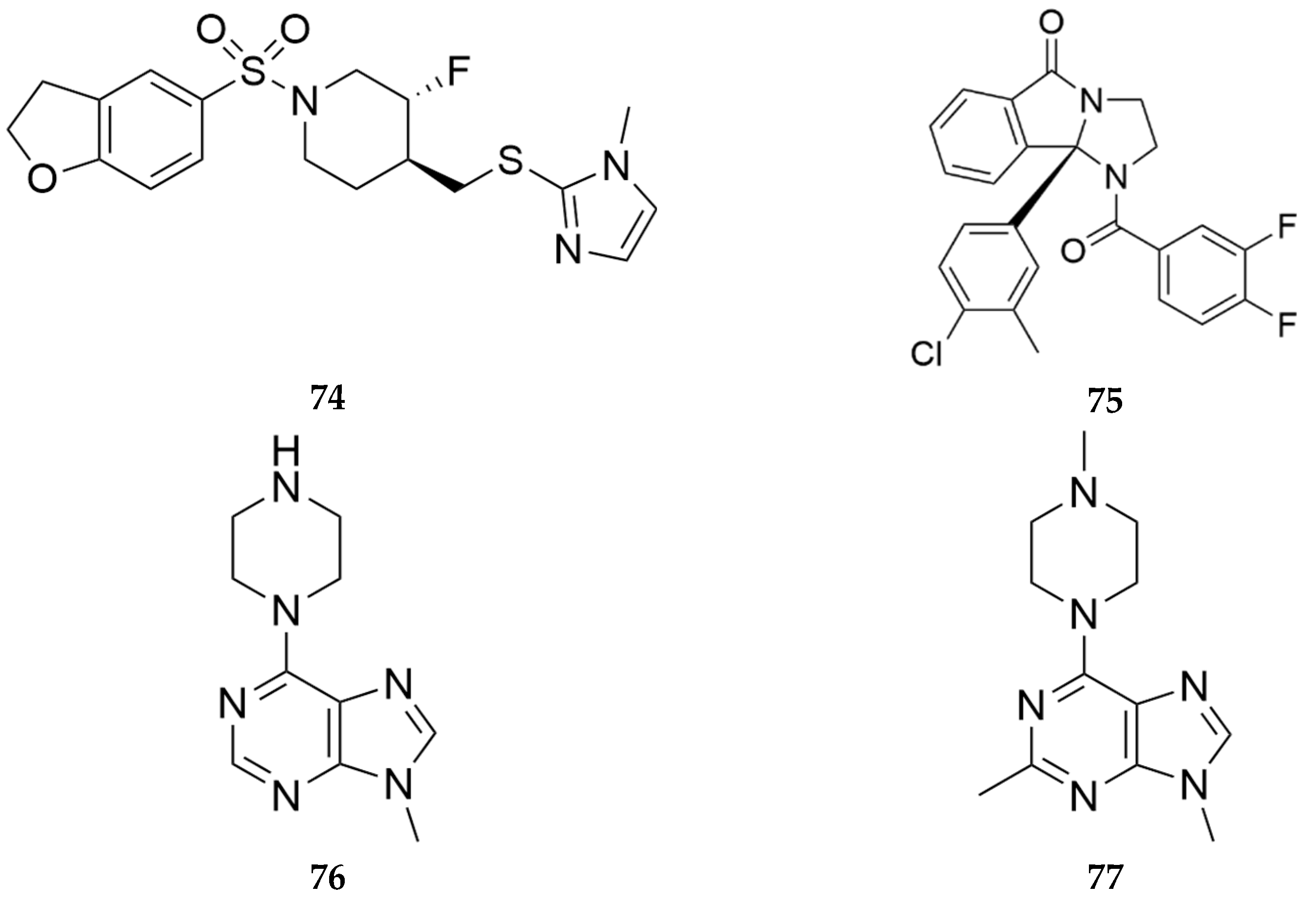

Table 1.
The binding affinity and efficacy of key naltrexamine analogs claimed in the patent filed by Zhang [16].
Table 1.
The binding affinity and efficacy of key naltrexamine analogs claimed in the patent filed by Zhang [16].
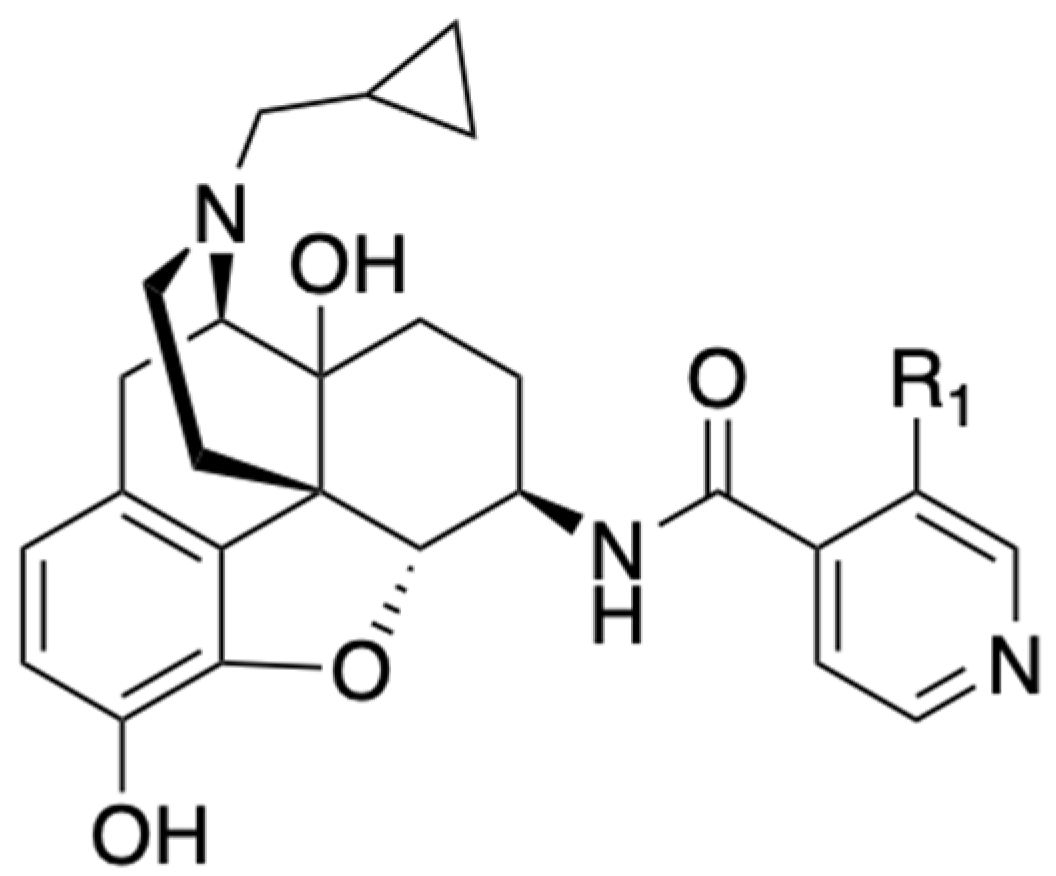
| Compound | Ki (nM) MOR | EC50 (nM) [MOR 35S-GPT(γS) ] (% of DAMGO) |
| NAP (8); R1 = H, | 0.37 | 1.14 (22.72%) |
| NFP (9); R1 = F, | 0.36 | 1.20 (34.97%) |
| NYP (10); R1 = CN, | 0.87 | 1.28 (26.05%) |
| NMP (11); R1 = CH3 | 0.58 | 1.52 (30.63%) |
Table 2.
A table of representative examples of the various scaffolds claimed by Flaherty in this embodiment (64-65: Scaffold A, 66: Scaffold B and 67: Scaffold C) with their respective IC50 values at AC1 and AC8 plus max % inhibition at AC8 [55].
Table 2.
A table of representative examples of the various scaffolds claimed by Flaherty in this embodiment (64-65: Scaffold A, 66: Scaffold B and 67: Scaffold C) with their respective IC50 values at AC1 and AC8 plus max % inhibition at AC8 [55].
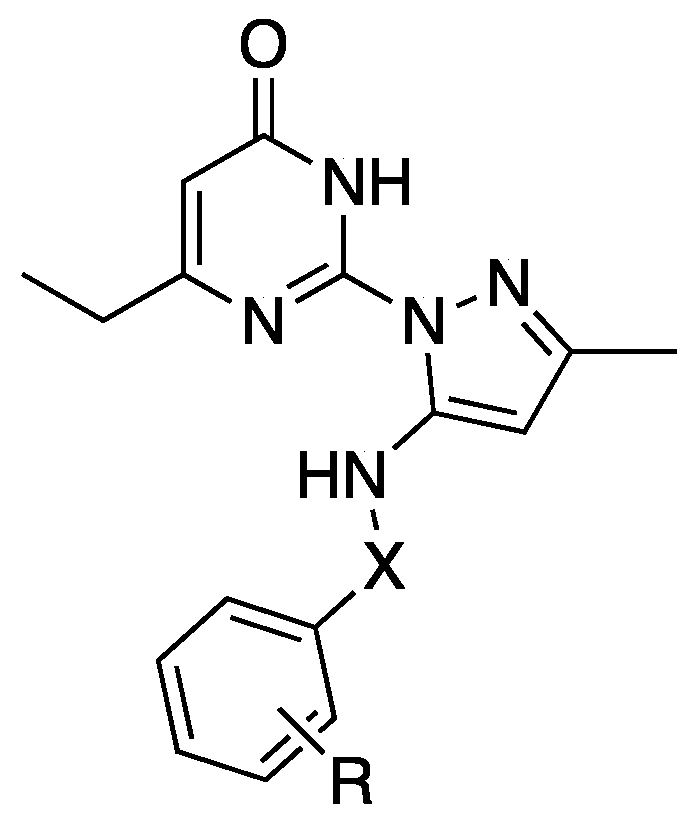
| # | X | R | IC50, AC1 | IC50, AC8 | Max AC8 Inh (%) |
| 64 | C(O) | 4-Cl | 0.3 | 9.5 | 32 |
| 65 | C(O) | 2-F | 0.4 | 5.5 | 60 |
| 66 | -CH2C(O)- | 3-CH3 | 10 | 50 | 59 |
| 67 | -CH2 | 3-CH3 | 0.2 | 3.3 | 31 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



