
Submitted:
19 July 2024
Posted:
22 July 2024
You are already at the latest version
Abstract
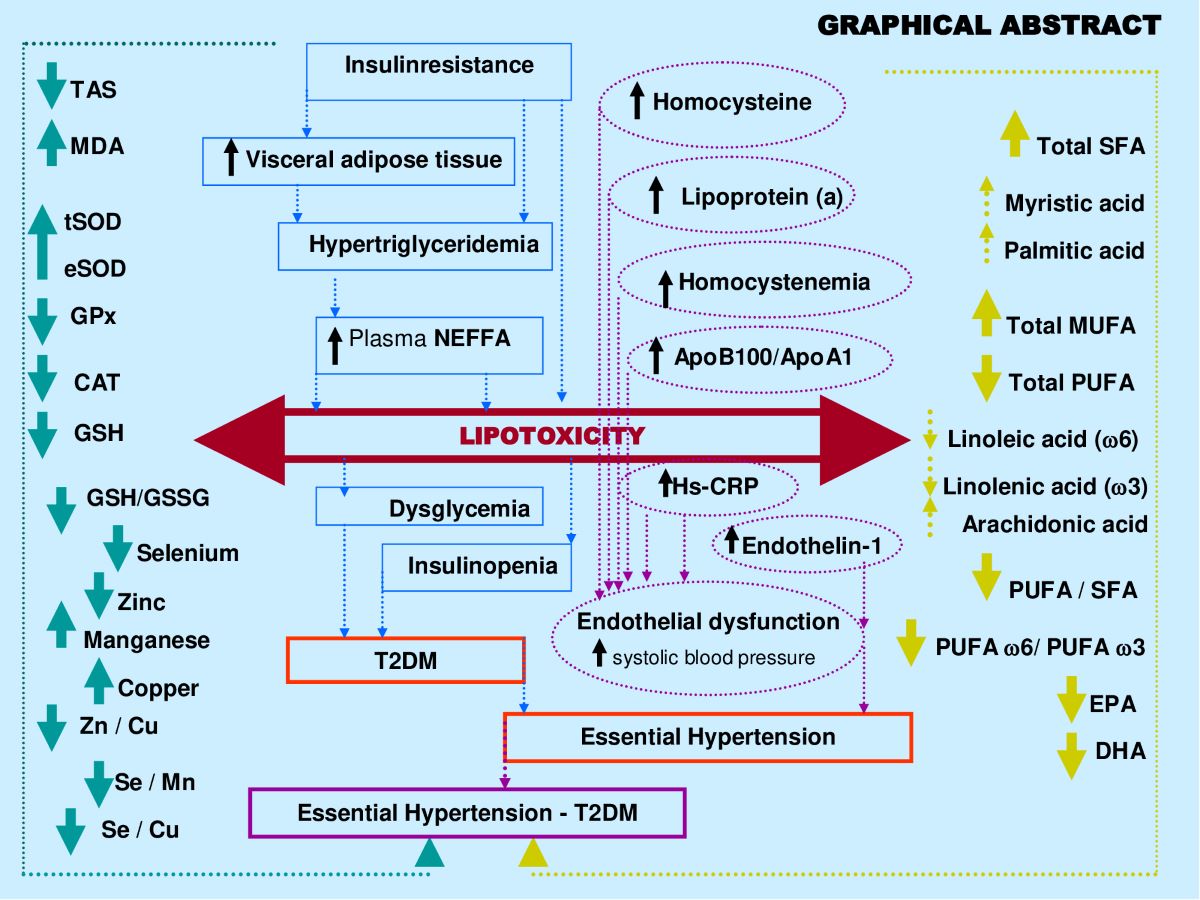
The coexistence of SAH with T2DM is a common comorbidity. In this present study, we investigated the link between altered plasma antioxidant trace elements (ATE: Manganese, Selenium, Zinc and Copper) and fatty acids ratio (FAR: polyunsaturated/saturated) imbalance as transition biomarkers between vascular pathology (SAH) to metabolic pathology (T2DM). Our data revealed strong correlation between plasma ATE and FAR profile which is modified during SAH-T2DM association compared to healthy group. This relationship is mediated by lipotoxicity (simultaneously prominent visceral adipose tissue lipolysis, significant flow of non-esterified free fatty acids release, TG-Chol-dyslipidemia, high association of Total SFA, Palmitic acid, Arachidonic acid and PUFA 6/ PUFA 3; drop in tandem of PUFA / SFA and EPA+DHA); oxidative stress (lipid peroxidation confirmed by TAS depletion and MDA raise, concurrently drop of Zn/Cu-SOD, GPx, GSH, Se, Zn, Se/Mn, Zn/Cu; concomitantly enhance Cu, Mn and Fe); endothelial dysfunction (endotheline-1 increase); athero-thrombogenesis risk (concomitantly raise of ApoB100/ApoA1, Ox-LDL, tHcy and Lp(a)) and inflammation (higher of Hs-CRP, Fibrinogen and Ferritin). Our study open to new therapeutic targets and to better dietary management, such as to establish dietary ATE and PUFA 6 / PUFA 3 or PUFA / SFA reference values for atherosclerotic risk prevention in hypertensive-diabetics patients.
Keywords:
Systemic arterial hypertension
; Type 2 diabetes mellitus
; Antioxidant trace elements
; Lipotoxicity
; Fatty acid Ratio
; Endothelial dysfunction
; Oxidative Stress
; Atherothrombogenic risk
; Insulinresistance
; inflammation
1. Introduction
In 2023, the World Health Organization established a report which highlights the incidence of Systemic Arterial Hypertension (SAH) would be responsible for 17 million deaths per year [1]. SAH is often a comorbidity associated with Type 2 Diabetes Mellitus (T2DM), having the Cardiometabolic syndrome (CMS) in common. The increase of chronic SAH and T2DM related to cardiovascular mortality is attributable to aggravating of CMS risk factors, but with similarities and differences [2]. SAH is primarily metabolic and vascular disorders, recognized as ischemic heart failure and stroke, mainly increased triglycerides and cholesterol, visceral adiposity, fasting glucose intolerance, and elevated blood pressure (BP). Insulin resistance remains the major pivotal factor of CMS in both SAH and T2DM [3].
Between 2000 and 2019, epidemiological statistics from the GBD (global burden of metabolic disease) study show an increase the mortality level for T2DM and SAH linked to dyslipidemia and hyperglycemia which generate glucolipotoxicity and a vascular haemodynamic disorder in diabetic β-cell damage [4] and hypertension [5], intimately interfere with endothelial function [6]. The interaction between SAH and T2DM is multifactorial complexity related to endothelial dysfunction and athero-thromboembolic process which represents the major contributor to ischemic heart disease and myocardial infarction [7]. Several studies have shown that interactions between dyslipidemia and lipotoxicity are closely influenced by fatty acid (FA) ratio, mainly polyunsaturated/saturated FA (PUFA/SFA) unbalanced [8].
Besides, Lipotoxicity-mediated endothelial dysfunction is caused by abnormally high levels of saturated FA, such as palmitic acid and cholesterol [9]. Recently, the American Health and Nutrition study was based on examination survey highlighted a strong correlation between dietary intake of SFA, and it has been shown that dietary ω-3 fatty acid deficiency contributes to the development of hypertension [10]. Randle demonstrated that plasma FA levels increase under fasting or T2DM and lead to insulin resistance by inhibition of carbohydrates oxidation [11]. Numerous studies have shown in T2DM, the increase SFA to PUFA ratio was positively correlated with low-grade inflammation, insulinipenia and atherosclerosis [12]. Some studies has revealed that alpha linolenic acid (C18: 3 n3) to linoleic acid (C18: 2 n6) ratio and docosahexaenoic (DHA) omega 3 (C22: 6 n3) to eicosapentaenoic (EPA) omega 3 (C20: 5 n3) ratio [13] have been extensively studied for their anti-atherosclerotic vascular benefits in T2DM [14]. In addition, PUFA-3 modulates blood pressure and regulates vascular hemodynamics by incorporating into the red blood cells membranes [15]. On the other hand, excess SFA in the bloodstream is recognized as predictive of coronary insufficiency [16] associated with venous thrombosis [17], mainly lauric (C12: 0), myristic (C14: 0) and palmitic (C16: 0).
Moreover, several studies have shown that atherothrombotic events cause damage to endothelial function. In addition, several studies have shown that atherothrombotic complications caused by lipids lead to endothelial dysfunction - oxidative stress interactions. These events maintain other disorders related to plasma levels of Oxidized low-density lipoprotein (Ox-LDL) and homocysteine. Indeed, hyperhomocysteinemia and Ox-LDL are associated with increased intima arteries damage to promote thrombosis by collagen activation pathway [18]. Besides, Endothelin-1 (ET-1) has been associated with SAH, heart failure and atherosclerosis. ET-1 is produced and released by the vascular endothelium. It is a potent endogenous vasoconstrictor peptide in SAH. [19]. Atherothrombogenic effect is also explained by the interaction between Ox-LDL and lipoprotein (a). It is interesting to note that omega 3 fatty acids correct this vascular deleterious effect [20]. Otherwise, atherothrombotic state are correlated with glucolipotoxicity - Redox state interactions witch leads to oxidative stress (OS) and generates overproduction of reactive oxygen species (ROS) in SAH-T2DM comorbidity [21,22].
The involvement of sodium, potassium and calcium in prevention and treatment of SAH-T2DM have been the major clinical studies investigations. However, little attention has been given to the role of trace elements in SAH-T2DM aetiology, mainly related to antioxidant trace elements (ATE) profile [23]. The metabolic balance between ATE is an important factor in homodynamic homeostasis by modulating the carbohydrates and lipid enzymes metabolism involved in the BP regulation [24,25] by influencing the cell membranes permeability via the vascular endothelial protection [26,27].
Plasma Trace Elements unsteadiness is strongly involved in SAH and T2DM, mainly Selenium (Se), Copper (Cu), Zinc (Zn) and Manganese (Mn). It is observed that the imbalance between ATE is associated with an increased cardiovascular complications attributed to the therapeutic management difficulty [28]. On the other hand, fatty acid metabolism is regulated by ATE, particularly by zinc. Zn reduces the activity of Δ6 desaturases metabolising linoleic acid to arachidonic acid [29]. Zn is involved as regulators of cardiovascular System, and the gastro-intestinal lipid transport, prostaglandin metabolism and cell membranes integrity [30]. Zn affects phosphodiesterase activity as an insulin-mimetic effect, and regulates the lipolysis (fatty acids release) from adipose tissue [31]. Conversely, an increase in the plasma NEFA level disrupts the Zn binding to albumin since plasma albumin binds and transports both free fatty acids and Zn [32]. Also, Zn and Zn/Cu molar ratio are associated with the renin angiotensin-aldosterone system and lead to an elevation in systemic BP [33,34].
As regard to Cu, the deficiency of this trace element alters the saturated fatty acids to unsaturated fatty acids ratio. Indeed, this effect is exerted via the control of the expression of the genes involved in the synthesis of fatty acids and the metabolism of cholesterol, such as the SREBP-1 and SREBP-2 (sterol regulatory element binding proteins 1 and 2) genes, or the CYP7A1 gene encoding cholesterol 7-alpha hydroxylase in the liver [35,36]. SREBP-1 is specifically involved in regulating fatty acid synthesis, while SREBP-2 plays an important role in modulating cholesterol biosynthesis [37]. The SREBP-1c isoform is the main transcription factor used by insulin to activate the gene expression of lipogenic enzymes [38]. Studies involving rats have shown increased cholesterol levels in the body as a result of Cu deficiency [39]. Besides, Cu is correlated the activity of tyrosinase, a key enzyme in the norepinephrine synthesis (catecholamines), a major vasoconstriction neuromediator [40].
Manganese (Mn) is a trace element with an important role in endothelial function, lipid metabolism and ROS destruction. Mn is involved in the NO (Nitric Oxide) metabolism by its incorporation into the active site of arginine synthetase (urea cycle) [41,42]. Mn is implicated in the hepatic cholesterol and fatty acid regulation as a cofactor for mevalonate kinase and acetyl CoA carboxylase, respectively [43,44]. Mn is related to the AGEs (advanced glycation end products) [45]. Several studies have described that trace elements (Cu, Zn and Mn) are strongly involved in the protection of hypertensive subjects via the active site of superoxide dismutase (SOD-Mn; SOD-Cu/Zn) [46,47].
Selenium (Se) is an essential trace element strongly involved in OS defence. Se has been integrated into the glutathione peroxidase active site in cellular antioxidant defence [48,49]. Se deficiency is described in Iran Se-endemic areas, known as cardiomyopathy Keshan disease. This pathology is characterized as necrosis and fibrosis of the myocardium and leads to shock and congestive heart failure. The main risk factors found were related to loss of GPx-1 activity [50]. The endothelium dysfunction linked to the ATE disorder and FA ratio imbalance is poorly described in the literature and the mechanisms involved remain unsuccessfully elucidated in arterial hypertension with or without T2DM [51,52].
In this context, our investigation was conducted in four subjects groups: healthy, diabetics without SAH, hypertensive without T2DM and SAH - T2DM comorbidity. We sought to highlight interactions between CMS clusters, endothelium dysfunction, oxidative stress biomarkers related to the ATE profile and fatty acid ratio imbalance, particularly PUFA/SFA-PUFA-n3/PUFA-n6 ratios in the management of essential hypertension in diabetic subjects. To our knowledge, very few previous studies have considered the relationship between fatty acid ratios and ATE profile including their ratio to prevent complications of hypertension in a diabetic subject.
2. Results
2.1. Clinical Characterization according Cardiometabolic Syndrome of Cohort Study
The anthropometric data are summarized in Table 1. A relationship was observed between body mass index (BMI) and body fat percentage (BF), but not with body weight. The waist circumference (WC) traduced an abdominal adipose tissue depot, reflecting visceral adiposity is significantly increased (p<0.001) in participants groups II (Diabetics), III (Hypertensive) and IV (Hypertensive-diabetic) compared to group I (Healthy). A Positive and significant correlation was observed between WC and BMI in groups II, III and IV versus group I (r = +0.88, p<0.001). The WC/Waist Hips ratio and the BF mass percentage highlight an adipose tissue accretion in abdominal-trunk topographic in male participants, which corroborate the android obesity. In contrast a fat abdominal-ileal accumulation in female participants, indicating the gynoid obesity profile. This correlation was confirmed in all participants of groups II and IV versus group I. The strongly positive connection was established between BF and the WC/Waist Hips ratio in groups II and IV versus group I, but not with group III (r = +0.69).
An insulin resistance state (IR) is revealed in groups II and III and becomes more prominent in the IV group participants versus group I (Table 2). The Homa-IR index is increased by 192% and 374% in hypertensives groups (II and IV) versus control group, respectively (p<0.0001). The correlation is positive between SBP and the Homa-IR index in hypertensives groups (r = +0.58; r = +0.47; respectively). Concomitantly with IR state, a hyperinsulinism is observed in all groups compared to healthy group (Table 2). Fasting plasma insulin levels are increased by 88% in the III group, and become bursting in the IV group (+159%) vs control group (p<0.0001). The hyperinsulinism state is positively correlated with the Homa-IR index, WC and % of BF (r = 0.63; r = 0.71 and r = 0.94, respectively).
The metabolic parameters data mentioned in Table 2 indicate that subjects in the group III remained normoglycemic, on average 5.33±0.81 mmol/L (0.96±0.15 g/L) of fasting plasma glucose despite their abdominal adiposity. In contrast, in the group IV, although they are treated with metformin (2 to 3g of Glucophage/day) they persist hyperglycemic, on average 9.63±0.66 mmol /L (1.73±0.12 g/l). This reveals that the association of hypertension and diabetes aggravates hyperglycemia. These glycemic variations have a negative impact on HbA1C levels, reflecting long-term glycemic balance (6-8 weeks). The values recorded in Table 2 show that the HbA1C of group IV subjects is > 7% vs control group (p<0.001), a sign of poor metabolic balance. However, we did not find a correlation between SBP and HbA1C in the group IV. Patients with higher levels of endothelin-1 (ET-1) had a significantly greater cardiovascular risk in group IV (Table 2). Pearson correlation showed a very strong positive association between elevated plasma ET-1 endothelin levels and plasma concentrations of lipoprotein a (Lpa), homocysteine (Hcy), the ApoB100/ApoA1 ratio and oxidized low-density lipoprotein (Ox-LDL) in group IV versus healthy Group (r=0.645, r=0.751; r=0.528; r=0.811; p<0.0001, respectively).
The lipid profile observed in group III and group IV patients is altered, it affects both triglyceride and cholesterol metabolism. If we examine the results recorded in Table 2, dyslipidemia is found both in triglyceride and cholesterol plasma levels in all patients groups compared to control group. Hypercholesterolemia is marked by a significant HDL-c drop, whether in male or female subjects in the group IV. The values of TG/HDL-C ratio increased to 50-56% in the hypertensive patients (group III and IV) vs control group (Table 3). TG/HDL-C concentration and visceral adiposity (WC/WH ratio) were highly correlated (P<0.001; r = 0.99) in all groups II, III and IV vs healthy group. Paradoxically, LDL-c concentrations remained normal (< 4.14 mmol/L or < 1.60 g/l) in all groups vs control group (Table 2). However, the HDL-c/LDL-c ratio drops drastically to 57% in hypertensive and diabetic’s patients groups versus healthy groups (P<0.001, Table 3). The blood pressure values mentioned in Table 3 were increased on average by 24% in the IV group compared to control group. According to the World Health Organization criteria, groups IV and III are classified as grade I with moderate hypertension (140/90 to 159/99 mmHg; systolic blood pressure /diastolic blood pressure, respectively). In contrast, group II remained normotensive state.
In this study, the atherothrombogenic risk was assessed by Lpa, Hcy, the ApoB100/ApoA1 ratio and Ox-LDL. The data mentioned in Table 3 shows that the Lp (a) levels are > 0.30g/L only in hypertensive groups (III and IV) but not in diabetics group compared to healthy group. Besides, the plasma concentrations of Hcy are extremely higher (> 15 µmol/L) in the hypertensive-diabetics patients (p< 0.001), but moderately in the other groups. The ApoB/ApoAI ratio tended to be significantly higher in hypertensive patients (III and IV groups), but not in diabetics patients (II) vs controls group (p< 0.001). The systemic lipid peroxidation was assessed by ox-LDL. The data mentioned in Table 3 show that ox-LDL levels are excessively higher in hypertensive patients and diabetics vs. healthy controls. The values are increased by 53%, 67% and 72%, respectively in groups II, III and IV; p< 0.001). The Pearson correlation coefficient revealed a positive association between ox-LDL levels and saturated fatty acids (SFA), such as myristic and palmitic acid in group IV (r=0.465, r=0.571; p<0.001, respectively). At opposite, the correlation analysis showed a negative association between ox-LDL levels with omega-3 and omega-6 polyunsaturated fatty acids (PUFA), such as Eicosapentaenoic acid and Docosahexaenoic acid in group III (r=-0.694, r=-0.758; p<0.001, respectively). It is interesting to note that higher tHcy and Lp (a) levels was positively correlated with significantly higher ox-LDL levels in group IV (r=0.418, r=0.509; p<0.001, respectively). There was a significant positive correlation between hCys and Selenium (r = + 0.882, p < 0.0001).
Plasma levels of Hs-CRP and fibrinogen characterize the systemic inflammatory assessment and process in hypertensive and diabetic patients (Table 2). CRPus plasma levels were positively correlated with both plasma Lp(a) and Hcy concentrations in group IV (r = +0.971; r = +0.815, respectively; p < 0.0001). On the other hand, the correlation is positive between CRPus and % MGC (r=+0.617; p < 0.0001); and between CRPus and Homa-IR (r=+0.481; p < 0.001). No particular disorder in fibrinogen plasma levels in all groups.
2.2. Plasma Fatty Acids Profile
The saturated, mono and polyunsaturated fatty acids data summarizes in Table 4. We noted that NEFAs (Non-esterified free fatty acids) are moderately elevated in the hypertensive patients and not altered in diabetic patients (+14%) vs control group. Indeed, the NEFAs are extremely increased in the group IV compared to group I (+40%; p < 0.001). Concurrently, total saturated fatty acids (SFA) are significantly higher in the group IV and III vs control group (+53%, +27%, respectively; < 0.001) with small change in group II (+16%). The plasma NEFAs profile disorder affects both saturated and unsaturated fatty acids. This concerns laurate (+65%), myristate (+145%), palmitate (+127%) and stearate (+71%). It should be noted that palmitate is the most dominant SFA in diabetic-hypertensive subjects (group IV). The fraction of monounsaturated fatty acids (MUFA), represented in our study by oleic acid (C18:1) is significantly reduced in the group IV (- 48% vs control, p < 0.001); while it remains normal in the diabetics subjects (group II). Concerning polyunsaturated fatty acids (total PUFA), we did not observe any difference between the group II and hypertensive groups (III and IV) versus Control group. On the other hand, linoleic acid (PUFA-ω6) is moderately increased in group IV and group II vs control group (+29% and +24%, respectively); linolenic acid (3-PUFA) is half reduced in the group IV (-48% vs. control group, p < 0.001) and moderately in the group II (-17% vs. control). Likewise, arachidonic acid (PUFA-ω6) is depleted in all groups (-50% in group II vs control group, p < 0.01), but more markedly in the group IV (-62% vs control group, p < 0.001). The ω3-PUFAs production resulting from the linolenic acid elongation, such as, EPA (eicosapentaenoic acid) and DHA (docosahexaenoic acid), we recorded a drastic decrease in group IV vs control group (-60%, p < 0.001) and moderately in the group II and III vs control group (-30%). We also observed that group IV showed a significant increase in the linoleic acid (ω6 PUFA)/linolenic acid (ω3 PUFA) ratio associated with a marked decrease in the linolenic and arachidonic acids. Furthermore, we noticed that the PUFA/SFA ratio is significantly reduced in the group IV (-48% vs. control group, p < 0.001) and no change in the group II vs. control group. At the same time, we noted an increase in the PUFA-ω6/PUFA- ω 3 ratio and a reduction in the EPA (PUFA- ω 3) / linolenic acid (PUFA- ω 3) ratio in the two groups of patients (Table 4 ). In the DH group, we found a strong correlation between the fall in the PUFA/SFA ratio and the reduction in the HDL-c/LDL-c ratio (r=+66). This correlation was also associated with the increase in ApoB100/ApoA1, TG/HDL-c ratios (r=-71; r=-68, p < 0.001; respectively) and Lp(a) concentrations in the group IV (r=- 0.5; p < 0.001). It is important to note the decrease in the PUFA/SFA ratio is negatively correlated with the increase in Ox-LDL levels in the hypertensive-diabetics group (r=- 0.54; p < 0.020), but moderately in the hypertensive and diabetics group (r=- 0.54; p < 0.075; r=- 0.14; p < 0.006; respectively).
2.3. Oxidative stress Status
2.3.1. Total plasma antioxidant activity (TPAA) and plasma antioxidant enzymatic profile
The TPAA levels are significantly reduced only in hypertensive subjects groups III and IV (Figure 2B), but moderately in diabetics group (II) compared to the control group (p< 0.001). The TPAA drop is inversely proportional to lipid peroxidation estimated by plasma TBARS levels and evaluated by MDA concentrations (Figure 2A) which are extremely high in hypertensive-diabetic patients (group IV). We also observed a significant increase in MDA levels in groups III and II compared to the control group (p<0.0001). Paradoxically, total plasma SOD activity is strongly increased (Figure 2C) proportional to erythrocyte SOD activity (Figure 2D). As expected, plasma total GPx activity (Figure 3A) was significantly reduced in group IV, but moderate in groups III and II (p<0.0001). Concomitantly, the total plasma catalase activity (Figure 3D) is significantly depleted in group III, but more marked in group IV (p<0.0001). The hypertensive subjects (group IV and III) exhibit significantly (p<0.0001) depletion of plasma glutathione (GSH) levels compared to group II (Figure 3B). The plasma oxidized-reduced glutathione ratio (GSH/GSSG) is extremely reduced (Figure 3C) in hypertensive subjects (groups IV and III) compared to group II (p<0.0001). The Pearson r coefficient summarized in Table 5 shows that the decrease in the PUFA/SFA ratio is positively correlated with the TPAA levels drop and increased MDA levels. Concomitantly, the reduction in the PUFA/SFA ratio is positively correlated with the decrease plasma total GPx activity and inversely corralled with increase total plasma SOD activity.
2.3.2. Plasma Antioxidant Trace Elements (PATE) Profile
Figure 4 and 5 show that PATE profile is modified during the development of systemic arterial hypertension (groups III and IV) and Type 2 Diabetes Mellitus (group II) compared to the control group. Indeed, plasma levels of selenium (Figure 5 A) and zinc (Figure 4 A) gradually decrease, while plasma levels of copper (Figure 4 B), manganese (Figure 4 C) and iron (Figure 4 D) gradually increase (P<0.0001). The Se/Mn (Figure 5 B), Se/Cu (Figure 5 C) and Zn/Cu (Figure 5 D) ratios are significantly lower in hypertensive-diabetics (group IV) compared to groups II and III (P<0.0001). Pearson correlations data summarized in Table 5 showed that the decrease in the PUFA/SFA ratio is positively associated with the reduction of Se and Zn. Conversely, the decline in the PUFA/SFA ratio is inversely associated with the increase in Cu, Mn and Fe in hypertensive-diabetics (group IV) compared to the other groups.
3. Discussion
The data from this study shows that crucial relationship between plasma antioxidant altered trace elements (ATE) and plasma fatty acid ratio unbalance can be considered as a new approach to management of systemic arterial hypertension (Groups III and IV) in diabetic subjects (Group II). Our investigation can be supported by four interconnected disorders between the ATE - FA ratio and other parameters that affect vascular endothelial dysfunction: i) Cardiometabolic syndrome, particularly lipids disorder are linked to insulin resistance and visceral adipose tissue (VAT) accumulation ; ii) Oxidative stress damage (ATE related to SOD, GPx, CAT and GSH) and a chronic inflammatory state (Hs-CRP and ferritin); iii) Saturated/unsaturated fatty acid ratio imbalance and Athero-thromboembolic risk (Hcy, Ox-LDL, ET-1 and Lp (a)). All these disorders are generated by glucolipotoxicity predisposing to heart failure which can progress to atherosclerotic ischemia and myocardial infarction in hypertensive diabetic patients [72]. In this study, some points need to be clarified.
i) The first major point is linked to the ATE - Fatty acids – lipid disorders
In our investigation, the lipid abnormalities represented by the increase in plasma triglycerides, total cholesterol, LDL-cholesterol associated with the depletion of plasma HDL-cholesterol in groups III and IV versus group control. These lipids profiles are significantly related to increase the non-esterified free fatty acids (NEFFA) and disturbances in the fatty acid ratio. We found a simultaneous depletion of the PUFA/SFA ratio and the % of linolenic acids (PUFA-ω3). Concurrently, Selenium and Zinc are decreased, while Manganese, Copper and iron are increased in groups III and IV versus group control. The mutual influences between ATE - Fatty acids - and metabolic syndrome cardiovascular Risk factors are poorly described in hypertensive-diabetic patients. However, some mechanisms have been proposed to elucidate the interactions between LDL-dyslipidemia and SFAs, particularly palmitic (C 16:0) and myristic (C 14:0) acids [73]. Among ATE, zinc and selenium are the minerals most correlated with lipid metabolism and cardiovascular risk factors in diabetic hypertensive subjects [74]. Indeed, zinc is an important cofactor in the activity of Δ6 desaturase, such as the hepatic stearoyl-CoA desaturase [75]. This enzyme converts stearate into monounsaturated fatty acid (oleate biosynthesis from stearate). Several studies have shown that zinc deficiency causes decreased linoleic acid (C18:2n−6) metabolism to arachidonic acid (C20:4n−6) via the intermediates, 7-linolenic acid (C18:3n−6) and dihomo-7-linolenic acid (C20:3n−6) [76].
In our investigation, we observed a decrease in the PUFA/SFA ratio simultaneously with the reduction of linolenic acid (PUFA ω-3). The PUFA/SFA ratio exhaustion is associated by a drop in zinc concentrations in group IV and III versus control group (Table 5). This depletion is concomitant with a marked increase in linoleic acid (PUFA ω -6) which may explain the rapid decrease in long-chain derivatives of EPA and DHA. If we take into account the competition between PUFA ω -6 and PUFA ω-3, this can be explained by the desaturation and elongation of the ∆-6 and ∆-5 desaturase pathways [77]. In this context, the overload in PUFA ω-6 would direct the synthesis pathway towards arachidonic acid (C20: 4; ω -6). In our study, we found a strong correlation between the PUFA/SFA ratio both with zinc levels in group IV and III compared to groups II and controls (Table 5). In contrast, our study did not show that plasma levels of Cu, Mn and iron significantly change the fatty acid profile. However, the Zn/Cu molar ratio is negatively correlated with the PUFA/SFA ratio in hypertensive groups III and IV without affected diabetic group (group II) compared to control group (Table 5).
In addition, it is important to emphasize that Zn/Cu molar ratio positively associated with TG/HDL-c ratio and inversely coupled with HDL-c/LDL-c in hypertensive groups. It is likely that the imbalance between Zn and Cu may be due to an alteration of linoleic acid (LA): gamma-linolenic acid and acide dihomo-gamma-linoléique (DGLA) ratio in dyslipidemia. Indeed, active conversion of LA to its metabolites (DGLA) depends on the trace elements levels (Zn higher and Cu lower) for increased desaturase activity [78]. Some studies have shown that the decrease in the Zn/Cu molar ratio correlated with fatty acids dyslipidemia and high blood pressure could significantly predict the risk of cardiovascular stroke [79,80].
Conversely, plasma copper levels are increased by 42% in group IV and 35% in group II, but very little in group III (hypertensive); while the % of total AGS are increased in all 03 groups. This highlights that copper is an indicator of the diabetes evolution towards hypertension and not inversely. This observation suggests that low appropriate incorporation of copper into Cu/Zn-SOD may not be sufficient to prevent lipid peroxidation in hypertensive-diabetic subjects (group IV and II). Curiously, copper and zinc metabolism are opposed during the transition from hypertension to diabetes. In this investigation we did not account for the consumption of trace elements in food intake, as this was not in the objectives of this study. However, some studies have described that plasma ATE levels depend closely on food intake [81]. In addition, previous work has shown that low dietary Se intake increases blood pressure [82].
ii) The second point is linked between ATE - Fatty acids – Oxidative stress (OxS).
It should be noted the important expansion of visceral adipose tissue (VAT) leading to significant lipolysis and the release of an important non-esterified fatty acids (NEFA) serum levels witch inaugurates all the disorders observed in the hypertensive-diabetic group (IV), which explains the dyslipidemia with strongly increased lipotoxicity. In this regard, lipid peroxidation plays a crucial role in the OxS onset. OxS is due to the antioxidant system failure (SOD, GPx, Se, Zn, Cu, and Mn) and the burst inflammation (Hs-CRP, Fibrinogen, Ferritin) in the hypertensive (groups III, IV) and diabetic groups (II). These physiological disruption increase ROS production and circulating levels of malondialdehyde which decrease total antioxidant protection (TAS). Our data are in agreement with many recent studies conducted in hypertensive-diabetic patients [83,84,85].
In this discussion point, our attention was focused particularly on Manganese. Interestingly, we observed that plasma Mn levels are significantly increased in both groups hypertensive (III, IV) and diabetics (II) compared to control group. This data can be explained by a competitive effect on transferrin between Mn and iron, since they are transported by this same protein. In our investigation, the iron balance in groups III and IV shows hyperferritinemia compared to control group. This is explains the mobilization of iron which, by binding to transferrin, maintaining Mn in unbound state and to leads Mn accumulation in blood [86]. This will result in a displacement of iron from its binding sites (ferritin) on membrane phospholipids integrating PUFAs and would lead to lipid peroxidation. It is important to note that iron is constantly pro-oxidant, whereas Mn was an anti-oxidant. This explains that the interactions between Mn, Fe and ferritin are intimately linked and that they can lead to dysregulation of mitochondrial levels of Fe, Mn, copper and zinc [87].
It appears that the balance of oxidative and reducing forces is therefore subtle in the hypertensive-diabetic patient [88]. Based on our new data, we will recommend not supplementing Mn in hypertensive-diabetic subjects if they have hyperferritinemia associated with dyslipidemia. On the other hand, if iron toxicity is not proven (absence of hypersidermia), Mn is protective against the polyunsaturated fatty acids peroxidation, as has been shown in positive studies [89]. In addition, Mn bioavailability lack in mitochondria induces SOD-Mn inactive, which can aggravate the OxS effects. Indeed, it is described that mitochondria are particularly vulnerable to OxS because they consume more than 90% of cellular oxygen. The production of superoxide anion is linked to an escape during the process carried out in the respiratory chain, globally proportional to its functioning [90].
Nevertheless, in this study, it seems that the activity of cytosolic SOD-Cu/Zn is not slowed down, at least not by lack of Cu and/or Zn. Similarly, in experimental models of ischemia/reperfusion, some authors have noted a specific reduction in Mn-SOD activity, without affecting CuZn-SOD activity, although an increase in the concentration of lipid peroxides at the mitochondrial level has been found to be increased [91]. It should be noted that erythrocytes, which are devoid of mitochondria, have a low Mn content in diabetic subjects [92]. Our data suggest that antioxidant defense could be preserved in the cytosol, while it would be altered in the mitochondria [93]. It is interesting to recall that Mn is involved in endothelial dysfunction via NO production, and that it was found to be elevated in the hypertensive-diabetic groups versus the control group, which may explain that Mn represents a transition trace element between vascular pathology (hypertension) and metabolic pathology (diabetes). This same observation had been noted in the evolution of diabetes towards renal dysfunction from hypertension [94].
It is essential to add that Mn bioavailability lack can explain the hyperglycaemia in hypertensive subject (group III) to can be associated with T2DM (group IV). This observation can be explained by Mn function a cofactor for some metalloenzymes (glycolysis, gluconeogenesis, Krebs cycle) can play a critical role in the glycemia regulation (pyruvate carboxylase, GTP oxaloacetate carboxylase, isocitrate dehydrogenase, malate dehydrogenase, phosphoenolpyruvate carboxykinase) [95]. In this study, special attention is given to selenium (Se). Few studies have focused on Se in the hypertension associated to diabetes (group IV). In this investigation, selenium represents the link between homocysteine (hCys), oxidative stress (GPx activity), fatty acid unbalance and inflammation (Hs-CRP) during the SAH evolution.
In our investigation, it does not appear that groups II, III, and IV are depleted in Se, because the serum Se levels are not lower than 80 µg/L, as has been described in other study [96]. This current study showed that there is a strong relationship between Se and fatty acid profile. We found a positive correlation between decreased plasma Se and concurrently falling n-3 PUFA levels, GPx activity and GSH/GSSG ratio in group IV and III compared to groups II and controls. Indeed, n-3 PUFAs are vulnerable to lipid peroxidation, which leads to OxS and inflammation. In agreement with our findings, Se supplementation has been described as protective against PUFA peroxidation via GPx activity [97,98]. Several studies describe that Se incorporated into selenoprotein P protects the oxidation of n-3 PUFAs and inflammation in cardiovascular disease [99].
It is important to emphasize that we found a positive association between Se plasma levels reduction and Se/Cu and Se/Mn depletion ratios in groups II and IV. Concomitantly, PUFA ω6/ PUFA ω3 ratio and serum fibrinogen levels are increase, which is an aggregating factor. We suppose that Se influence on platelet function and the thromboxane / prostacyclin balance [100,101]. Furthermore, previous studies have shown that a GPx activity drop in erythrocytes leads to an accumulation of hydrogen peroxide linked to lipid peroxidation which can cause an inhibition of SOD activity [102]. We found a positive correlation between PUFA / SFA ratio and GPx or GSH levels in these groups. Conversely, the correlation is negative between PUFA / SFA ratio and tSOD or eSOD or catalase (Table 5). Our data are similar to several studies [103]. It seems that antioxidant protection by TE against OxS damage in groups III and IV is important to protect endothelial cell membrane PUFAs from lipid peroxidation, which protects the vascular endothelium from atherosclerosis and thrombogenesis [104]. It has been described that Se actions are exerted through the p38 MAP kinase and NF-κB signaling pathways [105]. Besides, Se is able to inhibit the expression of endothelial adhesion molecules generated by the atherosclerotic process, such as VCAM-1 (vascular cell adhesion molecule-1), ICAM-1 (intercellular adhesion molecule-1) and E-selectin [106].
iii) The third crucial point is related to the ATE - Fatty acids – Atherothroboembolic risk.
In our study, the mutual relationship between ATE-Fatty Acid and atherothroboembolic biomarkers (Ox-LDL, ET-1, Hcy and Lp(a)) are very complex. To our knowledge, no study has been investigated this interactions. Taking our data together, we found a significant synergistic increase in oxidized LDL, homocysteine, endothelin and lipoprotein (a) in hypertensive groups (III and IV).
Regarding oxidative modified LDL (Ox-LDL) pathway, our data has highlighted that Ox-LDL has been widely accepted as a primary mediator of atherosclerosis in hypertension [107]. Ox-LDL is implicated in the atheromatous plaques formation because it is dense and small size which allows it to infiltrate the arterial wall [108]. In this study, the relationship between Ox-LDL, ATE Plasma levels, PUFA/SFA ratio and PUFA-ω3/ω6 ratio may be relevant in determining the resistance of LDL to oxidative modification. Some studies have shown that plasma LDL-cholesterol undergoes strong oxidation in the presence of increased copper levels (case of groups II and IV) and a high ratio of PUFA ω6/PUFA ω3 [109]. It is important to emphasize that saturated fatty acid plays a major role in the Ox-LDL formation [110]. Indeed, in our study, our attention is focused between Ox-LDL and PUFA/SFA ratio simultaneously associated with zinc depletion and copper enhance. Concomitantly, we found a significant increased arachidonic acid and we observed a marked amplify in % linoleic acid (PUFA-ω6). This explains the strong Ox-LDL raise and decline in long-chain derivatives of PUFA-ω3 (EPA+DHA) in group IV. It should be noted the competition between PUFA-ω3 and PUFA-ω6 via to the desaturation and elongation pathways activated by ∆-6 and ∆-5 desaturases-zinc dependant [111]. The dietary zinc reduces the ∆-6 desaturases activity metabolizing linoleic acid into arachidonic acid [112]. It is highly likely in the hypertension-diabetes association, the excess in PUFA-ω6 would direct the synthesis arachidonic acid (C20:4; ω6) pathway, significantly increased in group IV compared to group II. The increase in arachidonic acid and simultaneously depletion of PUFA-ω3 and zinc may be explained by an overproduction of Ox-LDL, because PUFA-ω3 inhibits the Ox-LDL [113].
Conversely, the arachidonate enhance can lead either to the prostaglandins production converted to thromboxane A2 (vasoconstrictor and platelet aggregator factor) via the cyclooxygenase action [114], or leukotrienes synthesis under the 5-lipoxygenase effect [115]. Furthermore, previous studies have shown in diabetic subjects with or without coronary insufficiency that arachidonic acid is incorporated more into the membrane phospholipids of blood platelets [116]. In this event, the risk of platelet aggregation increases and could lead to the development of thrombosis [117]. On other side, we found a positive association between Ox-LDL – hyperhomocysteinemia and the decrease in total PUFAs, particularly EPA and DHA in groups II, III and IV versus control group. Our results are in agreement with several studies which prove that homocysteine levels influences the Ox-LDL synthesis and disrupts plasma levels of eicosanoids derived from PUFAs [118]. Some studies have shown that hyperhomocysteinemia - Ox-LDL interactions stimulates the macrophage migration (Ox-LDL is a signaling pathway of immune system) in the sub endothelial space of the vascular endothelium and leads to the foam cells formation that play a crucial role in atherosclerotic lesions [119,120]; which has been found in ischemic cardiopathy coronary insufficiency [121,122].
Regarding total homocysteine (tHcy) interactions in this investigation, we noticed the link between hyperhomocysteinemia, a sudden drop in EPA+DHA levels, and significantly lower plasma Zn and Se levels in group IV. Homocysteine accumulation in plasma may be due to either excessive production or its low catabolism. This result can be explained by the betaine-homocysteine methyltransferase and methionine synthase activities have altered to convert hCys to methionine since they are dependent on zinc [123]. Zn and Se deficiency appears to be the most plausible response that demonstrates the hCys accretion in the blood. This result also explains the plasma glutathione depletion (GSH and GSH/GSSG ratio) observed in groups III and IV. Through the methylation process, hCys appears to be crucial for the metabolism of polyunsaturated fatty acids and their distribution in tissues. It seems that methyl function deficiency due to hyperhomocystenemia explains a drop in the PUFAs synthesis due to an elongation lack of the carbon chains of linolenic acid (PUFA ω3) observed in group IV [124]. It is unclear what mechanisms are underlying the correlation between hCys-fatty acid ratio-ATE in endothelial dysfunction deserves further study, although its involvement in atherosclerosis and thrombogenesis seems plausible as an independent factor in hypertension [125]. The limited data currently available in the literature do not allow definitive conclusions to be drawn on the relationship between Hcy and PUFA ω3.
Regarding lipoprotein (a) pathway in this study, we showed an high association between increased Lp(a) levels and elevated PUFA ω6/ PUFA ω3 ratio in diabetic (group II) and hypertensive (groups III and IV) participants. Recently, studies dietary interventions support our data by the fact that the increase in serum Lp(a) levels is associated with a decrease in unsaturated fatty acids and an increase in saturated fatty acids [126,127]. It is interesting to note that serum Lp(a) levels increase are positively correlated with EPA plasma levels decrease of but not DHA plasma levels. Our results are confirmed by other studies [128]. As previously argued for OxS statut, an association has been observed between increased serum Lp(a) levels and serum Ox-LDL levels. In our study, we found a positive association between elevated Lp(a) and high plasma copper levels in group IV. Our observation has been found in other studies [129]. This oxidative process leads to oxidized Lp(a) involved in atheromatous plaques development in hypertension [130]. On the other hand, it is possible that statin treatment increases serum Lp(a) levels since the hypertensive patients in this study are treated with statins, but this data remains controversial [131].
Regarding endothelin-1 (ET-1) pathway, our investigation, we did not find an association between PUFA ω6 / PUFA ω3 or PUFA / SFA ratios and serum ET-I levels; however, increased amounts of ET-1 are associated with the Total SFA, particularly with palmitic acid in group III and IV. It is likely that endothelin is strongly secreted in the presence of saturated but not unsaturated fatty acids. Our study data can be explained by the activation of Protein kinase C (PKC) family signaling pathway induced by palmitic acid which allows the ET-1induction [132]. Previous studies have shown that PKC stimulatory effect on ET-1 gene expression has been found in brain microvascular endothelial cells [133]. The transcription factor AP-1 (activator protein-1-dependent), which plays an important role in ET-1 gene transactivation [134], is a potential target of the PKC signaling pathway [135]. Our data are validated by other studies [136]. Furthermore, we showed a strong association between increased serum ET-I levels and hyperhomocysteinemia, but not with serum Lp(a) levels in group IV. Interestingly, an interaction was found between elevated serum ET-I levels and zinc deficiency in group IV. Several studies have shown that Zinc Modulates Endothelin-1 Signalling in Vascular Endothelial and Smooth Muscle Cells [137]. In our study, the decrease in zinc levels due to deficiency or sequestration by SOD-Zn/Cu appears to increase endothelin synthesis [138], since OxS is described as a factor aggravating endothelin formation in hypertension [139].
4. Patients and Methods
It is necessary to note that cohort study is the same as used our previous investigation in hypertensive versus diabetic subjects, which confirms the similarity of participants at enrolments [8].
4.1. Informed Consent Statement and Ethical Considerations
This clinical study protocol (Algiers essential arterial hypertension Study) was approved by the Ethics Committee of Algerian Ministry of Public Health (ECAMPH) and conformed to the principles outlined in the declaration of Helsinki (http://www.wma.net). Ethical approval code: The permits and ethical rules have been achieved according to the Executive Decree no. 10–90 (10 March 2010) completing the Executive Decree no. 04–82 (18 March 2004) of the Algerian Government, establishing the terms and approval modalities. An informed consent form was signed by each participant.
4.2. Participants and Clinical Protocol Design
This clinical investigation was a randomized, multicenter cross-sectional and observational design study-Case-control was carried between September 2020 and October 2023. All participants were admitted to diabetology unit, Mohamed Seghir Nekkache Hôpital, and diabetology-cardiology unit, Bab El Oued University Hospital Center (UHC), Mohamed Lamine Debaghine (MLD) of Algiers, Algeria. All the study parameters measurements were evaluated in the Biochemistry and Genetics Laboratory, UHC-MLD of Algiers. We included in the study 714 adult participants, aged between 36 and 54 years, including 397 men (M) and 317 women (F). The sample size was estimated using the Coshrans’ formula. The all participant’s cohort was classified according to age and sex, with a sex ratio of men/women = 0.94. This clinical investigation was undertaken on (Figure 1):
- 209 Type-2 diabetes mellitus (T2DM) participants without hypertension (Group II)
- 107 Hypertensive participants without T2DM (Group III)
- 298 T2DM participants with Hypertension (Group IV).
- 100 Healthy participants (Group I), without pathologies and non-smokers.
Figure 1.
The clinical protocol design in Diabetics, Hypertensive and Hypertensive-diabetic participants compared to Healthy participants (control group). This clinical investigation was a randomized, multicenter cross-sectional and observational design study-Case-control was carried between September 2020 and October 2023. The sample size was estimated using the Coshrans’ formula. The all participant’s cohort was classified according to age and sex, with a sex ratio of men/women = 0.94. Diabetic participants were treated with metformin 300 mg/24 h, associated with a sulfonylurea. The Group IV was treated with a variable combination therapy: beta-blocker, calcium channel blocker, inhibitor of the angiotensin converting enzyme, and diuretic.
Figure 1.
The clinical protocol design in Diabetics, Hypertensive and Hypertensive-diabetic participants compared to Healthy participants (control group). This clinical investigation was a randomized, multicenter cross-sectional and observational design study-Case-control was carried between September 2020 and October 2023. The sample size was estimated using the Coshrans’ formula. The all participant’s cohort was classified according to age and sex, with a sex ratio of men/women = 0.94. Diabetic participants were treated with metformin 300 mg/24 h, associated with a sulfonylurea. The Group IV was treated with a variable combination therapy: beta-blocker, calcium channel blocker, inhibitor of the angiotensin converting enzyme, and diuretic.
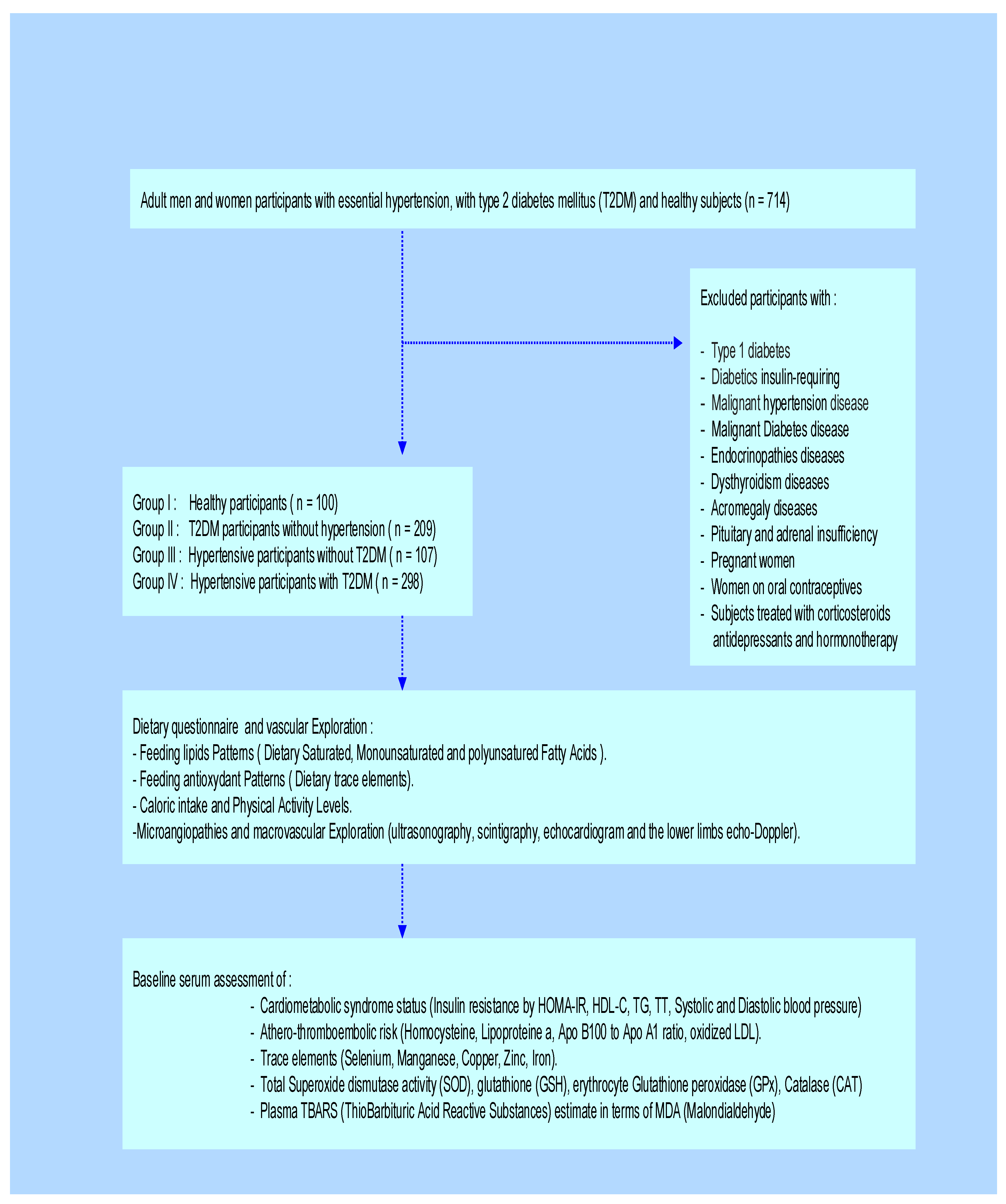
Diabetic participants were treated with metformin 300 mg/24 h, associated with a sulfonylurea. The Group IV was treated with a variable combination therapy: beta-blocker, calcium channel blocker, inhibitor of the angiotensin converting enzyme, and diuretic. No participants were insulin-requiring. The drug doses were stable throughout the study. The diabetes age and the presence of hypertension in Group IV were variable, between 5 and 10 years. In this study, we excluded all subjects with endocrinopathies, such as Cushing’s disease, dysthyroidism, acromegaly, pheochromocytoma, pituitary, and adrenal insufficiency. We also excluded pregnant women and those on oral contraceptives. Similarly, patients treated with corticosteroids, antidepressants, hormone therapy, and type 1 diabetics were excluded. In day hospital, we explored microangiopathies and macrovascular complications by ultrasonography, scintigraphy, echocardiogram, and the lower limbs echo-doppler. The participants benefited the supra-aortic trunks Doppler to calculate the intima-media diameter. All clinical explorations participants have been examined by the same physician.
4.3. Cardiometabolic Syndrome (CMS) Screening
CMS was confirmed according to the definition of the NCEP/ATPIII (National cholesterol education program third adult treatment panel /Adult Treatment Panel III) criteria [53]. The CMS was identified by the presence of three or more disorders of CMS clusters as follows: (1) visceral obesity; (2) high plasma triglyceride level; (3) low plasma HDL cholesterol level; (4) high fasting plasma glucose; (5) a blood pressure disturbance. Insulin resistance was calculated by the homeostasis model assessment insulin resistance (HOMA-IR) method: HOMA index = fasting glucose (mmol/L) x fasting insulin (mU/L)/22.5 [54]. The percentage of body fat (BF) was calculated using the formula: (1.2 x BMI) + (0.23 x age) - (10.8 x S) - 5.4 (S is the gender correction factor) [55]. The SBP (Systolic blood pressure) and DBP (Diastolic blood pressure) were measured in the prone position of the two arms, three times and two minutes after ten minutes of rest using a validated Omron 705 CP type BP monitor (Omron Healthcare Europe BV, Amsterdam, The Netherlands) [56]. Hypertension was measured via blood pressure defined based on the WHO (World Health Organization) standard definition as SBP of ≥ 140mmHg and/or DBP of ≥ 90mmHg and/or currently taking antihypertensive medications [57].
4.4. Plasma Samples and Biochemical Analysis
The participants were admitted to the hospital at 7 am after 12 h of fasting before they consumed their drugs (therapeutic treatment). Blood samples were centrifuged at 3000 rpm for 10 min, and plasma was obtained. Fasting plasma samples were immediately put on ice and kept frozen at - 80°C until analyses were performed. Fasting plasma glucose, triglycerides (TG), total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), Transaminases (ALT, AST), GGT, creatinine and uric acid were determined by enzymatic methods using an automatic biochemical analyzer (Cobas Integra 400® analyzer, Roche Diagnostics, Meylan, France). Plasma glycosylated haemoglobin (HbA1C) and Microalbuminuria were determined by turbidimetry (Roche Diagnostic Systems, Basel, Switzerland). The low-density lipoprotein cholesterol (LDL-C) was calculated using Friedewald’s formula [LDL-C (mg/dL) = TC - HDL-C - TG/5.0] applied to subjects with IRS [58]. The criterion for detecting low-grade inflammation has been determined by plasma high-sensitive C-reactive protein (Hs-CRP) level and ferritin assessed using immunoturbidimetric methods on chemical Synchron analyzer LX®20 PRO. The fibrinogen was evaluated by the chronometric Von Clauss methods using hemostasis analyzer ACL TOPTM (Biolabo, Maizy, France). Insulin concentrations were determined by RIA (RadioImmunoAssay) using commercially available kits (Human insulin specific RIA kit, EMD Millipore Corporation St. Louis, MO 63,103, USA). Apolipoprotein A1 (Apo A1), Apolipoprotein B100 (Apo B100) and Lp (a) lipoprotein were determined by Synchron LX®20 PRO analyzer. Homocysteinemia (Hcy) was assessed using FPIA (fluorescence polarization immuno assay) on Immulite 2000 analyzer Ref: L2KH02. The plasma Oxidized low-density lipoprotein (Ox-LDL) levels were assayed according to ELISA method previously described using OxiSelectTM LDLox Elisa Kits [59]. Endothelin-1 (ET-1) was quantified using commercially available ELISA kits (Morinaga and R&D System). Standards, reagents, and test samples were prepared and analyzed according to the manufacturer's instructions.
4.5. Plasma Fatty Acids Extraction and Assay
The blood samples were taken on sodium oxalate. The total plasma lipids were separated by the Folch [60] and Dole [61] methods from 0.5 mL of plasma by adding 5 mL of a chloroform/methanol mixture (2:1 v/v) and 1 mL of 5% butyl-hydroxy-toluene in methanol.
The homogenate was purified on degreased filter paper (Durieux brand without ash N°114-110 m/m). After the extraction in the heptane phase, the dry residue containing the fatty acids was taken up in 50 µL of hexane, 1 µL of the solution obtained was injected into a stationary phase capillary column of polyethylene glycol (HP-Innowax type), 30 m length, 0.32 mm inside diameter, and 0.5 µm film thickness. The assessment of saturated fatty acids (SFA), Monounsaturated fatty acids (MUFA), polyunsaturated fatty acids (PUFA), Eicosapentaenoic acid (EPA), and Docosahexaenoic acid (DHA) were analyzed by gas chromatography on HP5890A (Hewlett-packard-normalk analyzer) series II equipped with a flame ionization detector. The carrier gas was nitrogen with a flow rate of 1 mL/min. The injector, detector, and column temperatures were 220°C, 275°C and 180°C, respectively. Adding internal standard or internal controls to samples allows the quantification of fatty acids within the sample by calculations using the area of known quantity of the internal standard peak relative to the area of the peak fatty acids. Internal standards dissolved in 1 mL/mg of dry chloroform: methanol
(2:1, v/v) containing butylated hydroxytoluene (BHT; 50 mg/L) as antioxidant. The loss in the total amount of fatty acids extraction by our method is estimated between 5 and 10%. The NEFFA were extracted by the Duncombe method [62] and determinates by microfluorimetry using a KONTRON analyzer, Power Supply SFM23, Augsburg, Germany.
4.6. Trace Elements Determination and Assessment Methods
Plasma trace elements (selenium, manganese, copper and zinc) were determined by the Flame Atomic Absorption Spectrometry (Flame-AAS) technique. This method was widely employed for elements determination [63]. Trace metal determinations were performed according to protocol method previously described [64] with a flameless atomic absorption spectrophotometer (PerkinElmerAnalyst 800®, Germany). Argon was used as the purging gas. One thousand µg ml−1 standard solutions of zinc, copper, manganese, and selenium were used to prepare the standard curves (Wako Pure Chemical Industries, Japan).
Zinc: A Shimadzu hollow-cathode zinc lamp was used as the source of a current of 10 mA. The spectrometer was operated at213.8 nm in the peak height mode with 1.1 nm slit width. Plasma was prepared by dilution with deionized distilled water, plasma in a dilution of 1/100. Zinc concentrations were calculated by linear regression lines.
Copper: A Shimadzu hollow-cathode copper lamp was used as the source of a current of 10 mA. The spectrometer was operated at 324.8 nm in the peak height mode with 2.2 nm slit width. Plasma was diluted 1/10 with 0.1 N nitric acid. The standard addition method was used.
Manganese: A Shimadzu hollow-cathode manganese lamp was used as the source of a current of 10 mA. The spectrometer was operated at 279.5 nm in the peak height mode with 1.1 nm slit width. The manganese concentration was calculated by the standard addition method.
Selenium: A Shimadzu hollow-cathode selenium lamp was used as the source of a current of 10 mA. The spectrometer was operated at 196.0 nm in the peak height mode with 2.2 nm slit width. Plasma samples were diluted (1/2) with 1.0% (w/v) nickel-0.l N nitric acid solution. The standard addition method was used. Duplicate measurements were made with each sample. All glassware was tested for contamination. The measurement of all trace elements were performed by the following heating program: drying by ramp mode from 20 to 200◦Cwith temperature increases of l°C/s, ashing at 60 A (500◦C) for 30 s, and atomization at 210 A (2000◦C) for 5 s. We also used Randox kits (Randox Laboratories, UK) to confirm the results obtained by biochemical methods [65].
Iron: the determination of iron in the serum was carried out by colorimetric method.
4.7. Plasma Oxidative Stress Biomarkers and Analytical Process
4.7.1. Total Blood Antioxidant status (TAS), Plasma Thiobarbituric Acid Reactive Substances (TBARS) and Plasma Malondialdehyde (MDA) Levels Quantification
The TAS was analysed by the method based on a test which measures the capacity of the biological fluids to inhibit the production of TBARS from sodium benzoate under the influence of the free oxygen radicals derived from Fenton’s reaction [66]. Among the end products formed during the peroxidation of polyunsaturated fatty acids mediated by free radicals, we assessed the plasma MDA and TBARS levels. These two biomarkers do not have the same oxidative stress specificity. However, it is the most frequently measured biomarkers lipid peroxidation [67] by MDA that is the prototype of the TBARS. The plasma TBARS were estimated according to the method described previously [68].
4.7.2. SOD, GPx, Catalase Activities, and Glutathione Levels Determination
The Superoxide dismutase activity (SOD) activity was measured both in plasma (total SOD) and in erythrocytes according to the method described previously [69]. The Glutathione peroxidase (GPx) activity was determined in erythrocytes according to the method described by previously [70]. The glutathione (GSH) levels were analysed by spectrophotometer methods [71]. The results of erythrocyte SOD and erythrocyte GPx were expressed in U/g Hb (unit/gr. of haemoglobin). The Hb was read at 541nm in a spectrophotometer (model 181 UV-vis, Hitachi, Ltd., Tokyo, Japan).
4.8. Atherothromboembolic Risk Assessment
The lipoprotein (a), homocysteine (hCys), Apo B100/Apo A1 ratio and ox-LDL have been used as atherothrombogenic biomarkers. The measurement methods have been described previously.
4.9. Statistical Analysis
Considering our investigation was randomised cohort, all data are measured normally distribution series. Results are presented as mean ± standard deviation (SD). All statistical analyses were performed with Epi-info version 5 and Statview version 5 (Abacus Concepts, Berkeley, USA). Student’s t-test and one-way ANOVA were used for the comparison both between the 03 groups (II, III and IV) and with the control participants (group I). P value less than 0.05 was considered statistically significant. Both methods are parametric and assume normality of the data and equality of variances across comparison groups. Pearson’s coefficient (r) correlation analysis was performed to quantify associations between the plasma PUFA/SFA-PUFA-n3/PUFA-n6 ratios, trace elements profile (Se, Zn, Cu, Mn, Iron), CMS clusters, oxidative stress biomarkers (TAS, SOD, GPx, CAT, GSH, GSSG) and the atherothrombogenic risk characterized by the levels of Ox- LDL, Homocysteine and Lp(a). The results were considered significant if p < 0.05 (*), very significant if p < 0.01 (**), or highly significant if p < 0.001 (***).
5. Conclusions
This study analyzes a clinical systemic arterial hypertension (SAH) with or without type 2 diabetes mellitus (T2DM). This study focuses on the multifactorial interactions between altered plasma trace element profile and plasma fatty acid ratio imbalance. The interconnection of these events is a very complex process that affects endothelial function, resulting from converging influences between metabolic, vascular damage, oxidative stress (OxS), atherothromboembolic risk and inflammatory factors. Unambiguously, our study reveals that plasma ATE (Se, Zn, Cu and Mn) dysregulation and the fatty acid ratio imbalance plays a crucial role in the management of systemic arterial hypertension with or without diabetes. Interactions between ATE and fatty acid ratio imbalance support the idea of a prognostic role as true predictive biomarkers that could be useful to screen for high risk factors for microangiopathies and macrovascular complications in hypertensive-diabetic patients. Nevertheless, we are convinced that our study has some limitations, particularly some medications, including oral antidiabetics (metformin); Angiotensin converting enzyme (ACE) inhibitors; antiplatelets; statins can affect plasma ATE and fatty acid levels.
Author Contributions
EA.K and I.G.: Methodology, Data curation, Investigation, Formal analysis, Validation, Statistical Analysis and Validation Software. AEM.H and S.A: recruitment of diabetic and hypertensive participants. EA.K.; M.M. and A.O.: Conceptualization, Investigation, Methodology. EA.K and I.G. Writing-original draft-review & editing. All authors have read and agreed to the published version of the manuscript.
Funding
The authors would like to acknowledge the financial support of the Algerian Agency for the Research & Development in Health (PNR No. 208/ANDRS and PNR No.41/ANDRS/2011) and the Algerian Ministry of Higher Education Program (No. D00L01UN160420200001).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the Corresponding author.
Acknowledgments
The authors thank all participants of this study for their cooperation and the Algerian Health Ministry.
Conflicts of Interest
The authors declare that they have no conflicts of interest related to this study.References.
References
- NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: a pooled analysis of 1201 population-representative studies with 104 million participants. Lancet 2021, 398, 957–980. [Google Scholar] [CrossRef] [PubMed]
- Gregg, E.W.; Buckley, J.; Ali, M.K.; Davies, J.; Flood, D.; Mehta, R.; Griffiths, B.; Lim, L.L.; Manne-Goehler, J.; Pearson-Stuttard, J.; Tandon, N.; Roglic, G.; Slama, S.; Shaw, J.E.; Global Health and Population Project on Access to Care for Cardiometabolic Diseases. Improving health outcomes of people with diabetes: target setting for the WHO Global Diabetes Compact. Lancet 2023, 401, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M. Relationships among insulin resistance, type 2 diabetes, essential hypertension, and cardiovascular disease: similarities and differences. J. Clin. Hypertens. (Greenwich) 2011, 13, 238–43. [Google Scholar] [CrossRef]
- Chew, N.W.S.; Ng, C.H.; Tan, D.J.H.; Kong, G.; Lin, C.; Chin, Y.H.; Lim, W.H.; Huang, D.Q.; Quek, J.; Fu, C.E.; et al. The global burden of metabolic disease: Data from 2000 to 2019. Cell Metab. 2023, 35, 414–428.e3. [Google Scholar] [CrossRef]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of β-Cell Lipo-and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies. Pharmacol. Rev. 2021, 73, 924–967. [Google Scholar] [CrossRef]
- Yu, Y.; Lyons, T.J. A lethal tetrad in diabetes: hyperglycemia, dyslipidemia, oxidative stress, and endothelial dysfunction. Am. J. Med. Sci. 2005, 330, 227–32. [Google Scholar] [CrossRef]
- Gouaref, I.; Bouazza, A.; Abderrhmane, S.A.; Koceir, E.A. Lipid Profile Modulates Cardiometabolic Risk Biomarkers Including Hypertension in People with Type-2 Diabetes: A Focus on Unbalanced Ratio of Plasma Polyunsaturated/Saturated Fatty Acids. Molecules 2020, 25, 4315. [Google Scholar] [CrossRef]
- Kim, J.A.; Montagnani, M.; Chandrasekran, S.; Quon, M.J. Role of lipotoxicity in endothelial dysfunction. Heart Fail. Clin. 2012, 8, 589–607. [Google Scholar] [CrossRef]
- Gou, R.; Gou, Y.; Qin, J.; Luo, T.; Gou, Q.; He, K.; Xiao, S.; Li, R.; Li, T.; Xiao, J.; et al. Association of dietary intake of saturated fatty acids with hypertension: 1999-2018 National Health and Nutrition Examination Survey. Front. Nutr. 2022, 9, 1006247. [Google Scholar] [CrossRef]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle: Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Poreba, M.; Rostoff, P.; Siniarski, A.; Mostowik, M.; Golebiowska-Wiatrak, R.; Nessler, J.; Undas, A.; Gajos, G. Relationship between polyunsaturated fatty acid composition in serum phospholipids, systemic low-grade inflammation, and glycemic control in patients with type 2 diabetes and atherosclerotic cardiovascular disease. Cardiovasc. Diabetol. 2018, 16, 17–29. [Google Scholar] [CrossRef]
- Wu, J.H.; Lemaitre, R.N.; King, I.B.; Song, X.; Psaty, B.M.; Siscovick, D.S.; Moza_arian, D. Circulating omega-6 polyunsaturated fatty acids and total and cause-specific mortality: The Cardiovascular Health Study. Circulation 2014, 130, 1245–1253. [Google Scholar] [CrossRef]
- Steffen, B.T.; Ste_en, L.M.; Zhou, X.; Ouyang, P.; Weir, N.L.; Tsai, M.Y. n-3 Fatty acids attenuate the risk of diabetes associated with elevated serum non esterified fatty acids: The multi-ethnic study of atherosclerosis. Diabetes Care 2015, 38, 575–580. [Google Scholar] [CrossRef]
- Colussi, G.; Catena, C.; Mos, L.; Sechi, L.A. The Metabolic Syndrome and the Membrane Content of Polyunsaturated Fatty Acids in Hypertensive Patients. Metab. Syndr. Relat. Disord. 2015, 13, 343–351. [Google Scholar] [CrossRef]
- Williams, C.M.; Salter, A. Saturated fatty acids and coronary heart disease risk: The debate goes on. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Zhai, Z.; Yang, Y.; Kuang, T.; Wang, C. Free fatty acids inhibit TM-EPCR expression through JNK pathway: an implication for the development of the prothrombotic state in metabolic syndrome. J. Thromb. Thrombolysis 2012, 34, 468–74. [Google Scholar] [CrossRef]
- Kassab, A.; Ajmi, T.; Issaoui, M.; Chaeib, L.; Miled, A.; Hammami, M. Homocysteine enhances LDL fatty acid peroxidation, promoting microalbuminuria in type 2 diabetes. Ann. Clin. Biochem. 2008, 45, 476–480. [Google Scholar] [CrossRef]
- Shreenivas, S.; Oparil, S. The role of endothelin-1 in human hypertension. Clin. Hemorheol. Microcirc. 2007, 37, 157–78. [Google Scholar]
- Rahmani, E.; Samimi, M.; Ebrahimi, F.A.; Foroozanfard, F.; Ahmadi, S.; Rahimi, M.; Jamilian, M.; Aghadavod, E.; Bahmani, F.; Taghizadeh, M.; et al. The effects of omega-3 fatty acids and vitamin E co-supplementation on gene expression of lipoprotein(a) and oxidized low-density lipoprotein, lipid profiles and biomarkers of oxidative stress in patients with polycystic ovary syndrome. Mol. Cell Endocrinol. 2017, 439, 247–255. [Google Scholar] [CrossRef]
- Amponsah-Offeh, M.; Diaba-Nuhoho, P.; Speier, S.; Morawietz, H. Oxidative Stress, Antioxidants and Hypertension. Antioxidants (Basel) 2023, 12, 281. [Google Scholar] [CrossRef] [PubMed]
- Petrie, J.R.; Guzik, T.J.; Touyz, R.M. Diabetes, Hypertension, and Cardiovascular Disease: Clinical Insights and Vascular Mechanisms. Can. J. Cardiol. 2018, 34, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Gouaref, I.; Bellahsene, Z.; Zekri, S.; Alamir, B.; Koceir, E.A. The link between trace elements and metabolic syndrome/oxidative stress in essential hypertension with or without type 2 diabetes. Ann. Biol. Clin. 2016, 74, 233–43. [Google Scholar] [CrossRef] [PubMed]
- Loyke, H.F. Effects of Elements in Human Blood Pressure Control. Biol. Trace Elem. Res. 2002, 85, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Youn, J.Y.; Cai, H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J. Hypertens. 2015, 33, 1128–36. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Banning, A.; Schnurr, K. Selenium-dependent enzymes in endothelial cell function. Antioxid. Redox Signal. 2003, 5, 205–15. [Google Scholar] [CrossRef]
- Sakhaei, F.; Keshvari, M.; Asgary, S.; Salehizadeh, L.; Rastqar, A.; Samsam-Shariat, SZ. Enzymatic antioxidant system and endothelial function in patients with metabolic syndrome. ARYA Atheroscler. 2020, 16, 94–101. [Google Scholar] [PubMed]
- Bastola, M.M.; Locatis, C.; Maisiak, R.; Fontelo, P. Selenium, copper, zinc and hypertension: an analysis of the National Health and Nutrition Examination Survey (2011- 2016). BMC Cardiovasc. Disord. 2020, 20, 45. [Google Scholar] [CrossRef]
- Stawarska, A.; Czerwonka, M. : Wyrębiak, R.; Wrzesień, R.; Bobrowska-Korczak, B. Zinc affects cholesterol oxidation products and fatty acids composition in rats’ serum. Nutrients 2021, 13, 1563. [Google Scholar] [CrossRef]
- Kitala, K.; Tanski, D.; Godlewski, J.; Krajewska-Włodarczyk, M.; Gromadziński, L.; Majewski, M. Copper and Zinc Particles as Regulators of Cardiovascular System Function-A Review. Nutrients 2023, 15, 3040. [Google Scholar] [CrossRef]
- Hernandez, M.C.; Rojas, P.; Carrasco, F.; Basfifer, K.; Codoce, J.; Inostroza, J.; Ruz, M. Zinc supplementation reduces free fatty acid concentration in patients with type 2 diabetes. Rev. Chil. Nutr. 2020, 47, 1000–1008. [Google Scholar]
- Coverdale, J.P.C.; Khazaipoul, S.; Arya, S.; Stewart, A.J.; Blindauer, C.A. Crosstalk between zinc and free fatty acids in plasma. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 532. [Google Scholar] [CrossRef]
- Kuruppu, D.; Hendrie, HC.; Yang, L.; Gao, S. Selenium levels and hypertension: a systematic review of the literature. Public Health Nutr. 2014, 17, 1342–52. [Google Scholar] [CrossRef]
- Tubek, S. Zinc balance normalization: an important mechanism of angiotensin-converting enzyme inhibitors and other drugs decreasing the activity of the rennin-angiotensin-aldosterone system. Biol. Trace Elem. Res. 2007, 115, 223–6. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Gasperkova, D.; Xu, J.; Baillie, R.; Lee, J.; Clarke, S. Copper deficiency induces hepatic fatty acid synthase gene transcription in rats by increasing the nuclear content of mature sterol regulatory element binding protein 1. J. Nutr. 2000, 130, 2915–2921. [Google Scholar] [CrossRef]
- Huster, D.; Purnat, T.D.; Burkhead, J.L.; Ralle, M.; Fiehn, O.; Stuckert, F.; Olson, N.E.; Teupser, D.; Lutsenko, S. High copper selectively alters lipid metabolism and cell cycle machinery in the mouse model of Wilson disease. J. Biol. Chem. 2007, 282, 8343–8355. [Google Scholar] [CrossRef] [PubMed]
- Morrell, A.; Tallino, S.; Yu, L.; Burkhead, J.L. The role of insufficient copper in lipid synthesis and fatty-liver disease. IUBMB Life 2017, 69, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Stelmańska, E. Regulation of extramitochondrial malic enzyme gene expression in lipogenic tissues. Postepy Hig. Med. Dosw. 2007, 61, 664–671. [Google Scholar]
- DiSilvestro, R.A.; Joseph, E.L.; Zhang, W.; Raimo, A.E.; Kim, Y.M. A randomized trial of copper supplementation effects on blood copper enzyme activities and parameters related to cardiovascular health. Metabolism 2012, 61, 1242–6. [Google Scholar] [CrossRef]
- Lutsenko, S.; Washington-Hughes, C.; Ralle, M.; Schmidt, K. Copper and the brain noradrenergic system. J. Biol. Inorg. Chem. 2019, 24, 1179–1188. [Google Scholar] [CrossRef]
- Singh, N.; Geethika, M.; Eswarappa, S.M.; Mugesh, G. Manganese-Based Nanozymes: Multienzyme Redox Activity and Effect on the Nitric Oxide Produced by Endothelial Nitric Oxide Synthase. Chemistry 2018, 24, 8393–8403. [Google Scholar] [CrossRef] [PubMed]
- Klimis-Zacas, D.; Kalea, A. Manganese: modulator of vascular function, structure, and metabolism. Cell Biol. Toxicol. 2008, 24, S–S130. [Google Scholar]
- Farkas, C.S. Manganese and hepatic cholesterol. N. Engl. J. Med. 1980, 302, 585. [Google Scholar] [PubMed]
- Lu, L.; Wang, M.; Liao, X.; Zhang, L.; Luo, X. Manganese influences the expression of fatty acid synthase and malic enzyme in cultured primary chicken hepatocytes. Br. J. Nutr. 2017, 118, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Pang, X.; Zhang, W.; Wu, W.; Zhao, J.; Yang, H.; Qu, W. Effects of zinc and manganese on advanced glycation end products (AGEs) formation and AGEs-mediated endothelial cell dysfunction. Life Sci. 2012, 90, 131–9. [Google Scholar] [CrossRef] [PubMed]
- Mondola, P.; Damiano, S.; Sasso, A.; Santillo, M. The Cu, Zn Superoxide Dismutase: Not Only a Dismutase Enzyme. Front. Physiol. 2016, 7, 594. [Google Scholar] [CrossRef] [PubMed]
- Cuzzocrea, S.; Mazzon, E.; Dugo, L.; Di Paola, R.; Caputi, AP.; Salvemini, D. Superoxide: a key player in hypertension. FASEB J. 2004, 18, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Loscalzo, J. The role of glutathione peroxidase-1 in health and disease. Free Radic Biol Med. 2022, 188, 146–161. [Google Scholar] [CrossRef] [PubMed]
- Chrissobolis, S.; Didion, S.P.; Kinzenbaw, D.A.; Schrader, L.I.; Dayal, S.; Lentz, S.R.; Faraci, F.M. Glutathione peroxidase-1 plays a major role in protecting against angiotensin II- induced vascular dysfunction. Hypertension 2008, 51, 872–7. [Google Scholar] [CrossRef]
- Lei, C.; Niu, X.; Ma, X.; Wei, J. Is selenium deficiency really the cause of Keshan disease? Environ. Geochem. Health. 2011, 33, 183–8. [Google Scholar] [CrossRef]
- Vivoli, G.; Bergomi, M.; Borella, P.; Rovesti, S. Trace elements in hypertension. In: Trace Elements in Man and Animals 10, edited by Roussel et al., Kluwer Academic/Plenum Publishers, New York, 2000, 581.
- Tubek, S. Role of Trace Elements in Primary Arterial Hypertension: Is Mineral Water Style or Prophylaxis? Biol. Trace Elem. Res. 2006, 114, 1–4. [Google Scholar] [CrossRef]
- Expert panel on detection evaluation treatment of high blood cholesterol in adults. Executive summary of the third report of the National cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III). JAMA 2001, 285, 2486–97.
- Bonora, E.; Targher, G.; Alberiche, M.; Bonadonna, R.C.; Saggiani, F.; Zenere, M.B.; Monauni, T.; Muggeo, M. Homeostasis model assessment closely mirrors the glucose clamp technique in the assessment of insulin sensitivity: Studies in subjects with various degrees of glucose tolerance and insulin sensitivity. Diabetes Care 2000, 23, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Deurenberg, P.; Westrate, J.A.; Seidell, J.C. Body mass index as a measure of body fatness: Age- and sex-specific prediction formulas. Br. J. Nutr. 1991, 65, 105–111. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.; Mee, F.; Atkins, N.; Thomas, M. Evaluation of three devices for self measurement of blood pressure according to the revised British Hypertension Society Protocol: The Omron HEM-705CP, Philips HP5332, and Nissei DS-175. Blood Press. Monit. 1996, 1, 55–61. [Google Scholar]
- Bangalore, S.; Gong, Y.; Cooper-DeHoff, R.M.; Pepine, C.J.; Messerli, F.H. 2014 Eighth Joint National Committee panel recommendation for blood pressure targets revisited: results from the INVEST study. J. Am. Coll. Cardiol. 2014, 64, 784–93. [Google Scholar] [CrossRef]
- Knopfholz, J.; Disserol, C.C.; Pierin, A.J.; Schirr, F.L.; Streisky, L.; Takito, L.L.; Massucheto Ledesma, P.; Faria-Neto, J.R.; Olandoski, M.; da Cunha, C.L.; et al. Validation of the Friedewald formula in patients with metabolic syndrome. Cholesterol 2014, 2014, 261878. [Google Scholar] [CrossRef] [PubMed]
- Itabe, H.; Ueda, M. Measurement of plasma oxidized low-density lipoprotein and its clinical implications. J. Atheroscler. Thromb. 2007, 14, 1–11. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Dole, V.P.; Meinertz, H. Micro-determination of long chain fatty acids in plasma and tissues. J. Biol. Chem. 1960, 235, 2595–2599. [Google Scholar] [CrossRef]
- Duncombe, W.G.; Rising, T.J. Quantitative extraction and determination of non esterified fatty acids in plasma. J. Lipid Res. 1973, 14, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, J.; Bellanger, J.; Bienvenu, F.; Chappuis, P.; Favier, A. Recommended method for assaying serum zinc with flame atomic absorption. Ann. Biol. Clin. 1986, 44, 77–87. [Google Scholar]
- Aihara, K.; Nishi, Y.; Hatano, S.; Kihara, M.; Yoshimitsu, K.; Takeichi, N.; Ito, T.; Ezaki, H.; Usui, T. Zinc, copper, manganese, and selenium metabolism in thyroid disease. Am. J. Clin. Nutr. 1984, 40, 26–35. [Google Scholar] [CrossRef]
- Beckett, J.M.; Hartley, T.F.; Ball, M.J. Evaluation of the Randox colorimetric serum copper and zinc assays against atomic absorption spectroscopy. Ann. Clin. Biochem. 2009, 46, 322–6. [Google Scholar] [CrossRef]
- Koracevic, D.; Koracevic, G.; Djordjevic, V.; Andrejevic, S.; Cosic, V. Method for the measurement of antioxidant activity in human fluids. J. Clin. Pathol. 2001, 54, 356–61. [Google Scholar] [CrossRef]
- Tsikas, D. Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: Analytical and biological challenges. Anal. Biochem. 2017, 524, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Ghani, M.A.; Barril, C.; Bedgood, D.R., Jr; Prenzler, P.D. Measurement of antioxidant activity with the thiobarbituric acid reactive substances assay. Food Chem. 2017, 230, 195–207. [Google Scholar] [CrossRef]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–55. [Google Scholar] [CrossRef]
- Paglia, D.E.; Valentine, W.N. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J. Lab. Clin. Med. 1967, 70, 158–69. [Google Scholar]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2006, 1, 3159–65. [Google Scholar] [CrossRef]
- Cerf, M.E. Cardiac Glucolipotoxicity and Cardiovascular Outcomes. Medicina (Kaunas). 2018, 54, 70. [Google Scholar] [CrossRef]
- Karupaiah, T.; Tan, C.H.; Chinna, K.; Sundram, K. The chain length of dietary saturated fatty acids affects human postprandial lipemia. J. Am. Coll. Nutr. 2011, 30, 511–21. [Google Scholar] [CrossRef]
- Khajeh, M.; Hassanizadeh, S.; Pourteymour Fard Tabrizi, F.; Hassanizadeh, R.; Vajdi, M.; Askari, G. Effect of Zinc Supplementation on Lipid Profile and Body Composition in Patients with Type 2 Diabetes Mellitus: A GRADE-Assessed Systematic Review and Dose-Response Meta-analysis. Biol. Trace Elem. Res. 2024. [Google Scholar] [CrossRef]
- Kröger, J.; Schulze, MB. Recent insights into the relation of Δ5 desaturase and Δ6 desaturase activity to the development of type 2 diabetes. Curr. Opin. Lipidol. 2012, 23, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Knez, M.; Stangoulis, J.C.R.; Glibetic, M.; Tako, E. The Linoleic Acid: Dihomo-γ-Linolenic Acid Ratio (LA: DGLA)-An Emerging Biomarker of Zn Status. Nutrients 2017, 9, 825. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Kothapalli, K.S.; Brenna, J.T. Desaturase and elongase-limiting endogenous long-chain polyunsaturated fatty acid biosynthesis. Curr. Opin. Clin. Nutr. Metab. Care. 2016, 19, 103–10. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.C.; Rojas, P.; Carrasco, F.; Basfifer, K.; Codoce, J.; Inostroza, J.; Ruz, M. Zinc supplementation reduces free fatty acid concentration in patients with type 2 diabetes. Rev. Chil. Nutr. 2020, 47, 1000–1008. [Google Scholar]
- Mansoori, A.; Ghiasi Hafezi, S.; Ansari, A.; Arab Yousefabadi, S.; Kolahi Ahari, R.; Darroudi, S.; Eshaghnezhad, M.; Ferns, G.; Ghayour-Mobarhan, M.; Esmaily, H.; Effati, S. Serum Zinc and Copper Concentrations and Dyslipidemia as Risk Factors of Cardiovascular Disease in Adults: Data Mining Techniques. Biol. Trace Elem. Res. 2024. [Google Scholar] [CrossRef]
- Mirończuk, A.; Kapica-Topczewska, K.; Socha, K.; Soroczyńska, J.; Jamiołkowski, J.; Kułakowska, A.; Kochanowicz, J. Selenium, Copper, Zinc Concentrations and Cu/Zn, Cu/Se Molar Ratios in the Serum of Patients with Acute Ischemic Stroke in North eastern Poland - A New Insight into Stroke Pathophysiology. Nutrients 2021, 13, 2139. [Google Scholar] [CrossRef]
- Hu, X.F.; Sharin, T.; Chan, H.M. Dietary and blood selenium are inversely associated with the prevalence of stroke among Inuit in Canada. J. Trace Elem. Med. Biol. Organ. Soc. Miner. Trace Elem. (GMS) 2017, 44, 322–330. [Google Scholar] [CrossRef]
- Xie, C.; Xian, J.; Zeng, M.; Cai, Z.; Li, S.; Zhao, Y.; Shi, Z. Regional Difference in the Association between the Trajectory of Selenium Intake and Hypertension: A 20-Year Cohort Study. Nutrients 2021, 13, 1501. [Google Scholar] [CrossRef] [PubMed]
- Franco, C.; Sciatti, E.; Favero, G.; Bonomini, F.; Vizzardi, E.; Rezzani, R. Essential Hypertension and Oxidative Stress: Novel Future Perspectives. Int. J. Mol. Sci. 2022, 23, 14489. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Camargo, L.L.; Rios, F.J.; Alves-Lopes, R.; Montezano, A.C.; Touyz, R.M. Oxidative Stress and Hypertension. Circ. Res. 2021, 128, 993–1020. [Google Scholar] [CrossRef] [PubMed]
- Sinha, N.; Dabla, P.K. Oxidative stress and antioxidants in hypertension-a current review. Curr. Hypertens. Rev 2015, 11, 132–42. [Google Scholar] [CrossRef] [PubMed]
- Fitsanakis, V.A.; Zhang, N.; Garcia, S.; Aschner, M. Manganese (Mn) and Iron (Fe): Interdependency of Transport and Regulation. Neurotox. Res. 2010, 18, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Jouihan, H.A.; Cobine, P.A.; Cooksey, R.C.; Hoagland, E.A.; Boudina, S.; Abel, E.D.; Winge, D.R.; McClain, D.A. Iron-mediated inhibition of mitochondrial manganese uptake mediates mitochondrial dysfunction in a mouse model of hemochromatosis. Mol. Med. 2008, 14, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Cui, Z.; Lu, W.; Wang, P.; Wang, J.; Zhou, Z.; Zhang, N.; Wang, Z.; Lin, T.; Song, Y.; et al. Association between serum manganese levels and diabetes in Chinese adults with hypertension. J. Clin. Hypertens (Greenwich). 2022, 24, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Rauhala, P.; Chiueh, C.C. Effects of atypical antioxidative agents, S-nitroso glutathione and manganese, on brain lipid peroxidation induced by iron leaking from tissue disruption. Ann. N Y. Acad. Sci. 2000, 899, 238–54. [Google Scholar] [CrossRef] [PubMed]
- Friederich, M.; Hansell, P.; Palm, F. Diabetes, oxidative stress, nitric oxide and mitochondria function. Curr. Diabetes Rev. 2009, 5, 120–144. [Google Scholar] [CrossRef]
- Malecki, E.A.; Greger, J.L. Manganese protects against heart mitochondrial lipid peroxidation in rats fed high levels of polyunsaturated fatty acids. J. Nutrition. 1996, 126, 27–33. [Google Scholar] [CrossRef]
- Ekmekcioglu, C.; Prohaska, C.; Pomazal, K.; Steffan, I.; Schernthaner, G.; Marktl, X. Concentrations of seven trace elements in different hematological matrices in patients with type 2 diabetes as compared to healthy controls. Biol. Trace Elem. Res. 2001, 79, 205–19. [Google Scholar] [CrossRef] [PubMed]
- Leverve, X.M.; Guigas, B.; Detaille, D.; Batandier, C.; Koceir, E.A.; Chauvin, C.; Fontaine, E.; Wiernsperger, N.F. Mitochondrial metabolism and type-2 diabetes: a specific target of metformin. Diabetes Metab. 2003, 29, 6S88–6S94. [Google Scholar] [CrossRef]
- Koh, E.S.; Kim, S.J.; Yoon, H.E.; Chung, J.H.; Chung, S.; Park, C.W.; Chang, Y.S.; Shin, S.J. Association of blood manganese level with diabetes and renal dysfunction: a cross-sectional study of the Korean general population. BMC Endocr. Disord. 2014, 14, 24. [Google Scholar] [CrossRef]
- Wiernsperger, N.; Rapin, J.R. Trace elements in glucometabolic disorders: an update. Diabetol. Metab. Syndr. 2010, 2, 70. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–68. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, H.M.; Folmer, M.; Wang, S.; Lie, O.; Maage, A.; Mundal, H.H.; Ydersbond, T.A. Supplementary selenium influences the response to fatty acid-induced oxidative stress in humans. Biol. Trace Elem. Res. 1997, 60, 51–68. [Google Scholar] [CrossRef]
- Wen, Y.; Zhang, L.; Li, S.; Wang, T.; Jiang, K.; Zhao, L.; Zhu, Y.; Zhao, W.; Lei, X.; Sharma, M.; et al. Effect of dietary selenium intake on CVD: a retrospective cohort study based on China Health and Nutrition Survey (CHNS) data. Public Health Nutr. 2024, 27, e122. [Google Scholar] [CrossRef]
- Mattmiller, S.A.; Carlson, B.A.; Sordillo, L.M. Regulation of inflammation by selenium and selenoproteins: impact on eicosanoid biosynthesis. J. Nutr. Sci 2013, 2, e28. [Google Scholar] [CrossRef]
- Hampel, G.; Watanabe, K.; Weksler, B.B.; Jaffe, E.A. Selenium deficiency inhibits prostacyclin release and enhances production of platelet activating factor by human endothelial cells. Biochem. Biophys. Acta. 1989, 1006, 151–158. [Google Scholar] [CrossRef]
- Haberland, A.; Neubert, K.; Kruse, I.; Behne, D.; Schimk, I. Consequences of long-term selenium-deficient diet on the prostacyclin and thromboxane release from rat aorta. Biol. Trace Elem. Res. 2001, 81, 71–8. [Google Scholar] [CrossRef]
- Montazerifar, F.; Hashemi, M.; Karajibani, M.; Sanadgol, H.; Dikshit, M. Evaluation of lipid peroxidation and erythrocyte glutathione peroxidase and superoxide dismutase in hemodialysis patients. Saudi J. Kidney Dis. Transpl. 2012, 23, 274–9. [Google Scholar] [PubMed]
- Pedro-Botet, J.; Covas, M.I.; Martin, S.; Rubiés-Prat, J. Decreased endogenous antioxidant enzymatic status in essential hypertension. J. Hum. Hypertens. 2000, 14, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Hennig, B.; Chow, K.C. Lipid peroxidation and endothelial cell injury: implications in atherosclerosis. Free Radic Biol Med. 1988, 4, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.T.; Zhou, L.N.; Huang, C.J.; Hua, X.; Jian, R.; Su, B.H. Selenium inhibits high glucose - and high insulin-induced adhesion molecule expression in vascular endothelial cells. Arch. Med. Res. 2008, 39, 373–9. [Google Scholar] [CrossRef] [PubMed]
- Xun, P.; Liu, K.; Morris, J.S.; Daviglus, M.L.; He, K. Longitudinal association between toenail selenium levels and measures of subclinical atherosclerosis : the CARDIA trace element study. Atherosclerosis 2010, 210, 662–7. [Google Scholar] [CrossRef] [PubMed]
- Trpkovic, A.; Resanovic, I.; Stanimirovic, J.; Radak, D.; Mousa, S.A.; Cenic-Milosevic, D.; Jevremovic, D.; Isenovic, E.R. Oxidized low-density lipoprotein as a biomarker of cardiovascular diseases. Crit. Rev. Clin. Lab. Sci. 2015, 52, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhou, Y.; Nabavi, S.M.; Sahebkar, A.; Little, P.J.; Xu, S.; Weng, J.; Ge, J. Mechanisms of Oxidized LDL-Mediated Endothelial Dysfunction and Its Consequences for the Development of Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 925923. [Google Scholar] [CrossRef]
- Seppanen, C.M.; Cho, H.; Csallany, A.S. Comparison between High-PUFA and Low-PUFA Fats on Lipid Peroxidation and LDL Oxidation. Food and Nutrition Sciences 2013, 04, 572–579. [Google Scholar] [CrossRef]
- Staprans, I.; Pan, X.M.; Rapp, J.H.; Feingold, K.R. The role of dietary oxidized cholesterol and oxidized fatty acids in the development of atherosclerosis. Mol. Nutr. Food Res. 2005, 49, 1075–82. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Kothapalli, K.S.; Brenna, J.T. Desaturase and elongase limiting endogenous long-chain polyunsaturated fatty acid biosynthesis. Curr. Opin. Clin. Nutr. Metab. Care. 2016, 19, 103–10. [Google Scholar] [CrossRef]
- Chimhashu, T.; Malan, L.; Baumgartner, J.; van Jaarsveld, P.J.; Galetti, V.; Moretti, D.; Smuts, C.M.; Zimmermann, M.B. Sensitivity of fatty acid desaturation and elongation to plasma zinc concentration: A randomised controlled trial in beninese children. Br. J. Nutr. 2018, 119, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Sherratt, S.C.R.; Juliano, R.A.; Mason, R.P. Eicosapentaenoic acid (EPA) has optimal chain length and degree of unsaturation to inhibit oxidation of small dense LDL and membrane cholesterol domains as compared to related fatty acids in vitro. Biochim. Biophys. Acta. Biomembr. 2020, 1862, 183254. [Google Scholar] [CrossRef]
- Erkan, L.G.; Guvenc, G.; Altinbas, B.; Niaz, N.; Yalcin, M. The effects of centrally injected arachidonic acid on respiratory system: Involvement of cyclooxygenase to thromboxane signaling pathway. Respir. Physiol. Neurobiol. 2016, 225, 1–7. [Google Scholar] [CrossRef]
- Meng, H.; McClendon, C.L.; Dai, Z.; Li, K.; Zhang, X.; He, S.; Shang, E.; Liu, Y.; Lai, L. Discovery of Novel 15-Lipoxygenase Activators To Shift the Human Arachidonic Acid Metabolic Network toward Inflammation Resolution. J. Med. Chem. 2016, 59, 4202–9. [Google Scholar] [CrossRef]
- Tretjakovs, P.; Kalnins, U.; Dabina, I.; Erglis, A.; Dinne, I.; Jurka, A.; Latkovskis, G.; Zvaigzne, A.; Pirags, V. Nitric oxide production and arachidonic acid metabolism in platelet mem-branes of coronary heart disease patients with and without diabetes. Med. Princ. Pract. 2003, 12, 10–6. [Google Scholar] [CrossRef] [PubMed]
- Nabli, N.; Slimene, M.; Bouslama, A.; Omezzine, A.; Laradi, S.; Garcia, I.; Drai, J.; Barnier, E.; Boughzala, E.; Hammami, M.; et al. Arachidonate to saturated fatty acid ratio of circulating sterides and phospholipids: can it be risk marker of coronary stenosis ? A Tunisian study. Ann. Biol. Clin. 2001, 59, 743–9. [Google Scholar]
- Al-Shabrawey, M.; Elmarakby, A.; Samra, Y.; Moustafa, M.; Looney, S.W.; Maddipati, K.R.; Tawfik, A. Hyperhomocysteinemia dysregulates plasma levels of polyunsaturated fatty acids-derived eicosanoids. Life Res. (Auckl). 2022, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- de Souza, A.W.; Silva, N.P.; de Carvalho, J.F.; D'Almeida, V.; Noguti, M.A.; Sato, E.I. Impact of hypertension and hyperhomocysteinemia on arterial thrombosis in primary anti phospholipid syndrome. Lupus 2007, 16, 782–7. [Google Scholar] [CrossRef] [PubMed]
- Koubaa, N.; Nakbi, A.; Smaoui, M.; Abid, N.; Chaaba, R.; Abid, M.; Hammami, M. Hyperhomocysteinemia and elevated ox-LDL in Tunisian type 2 diabetic patients: role of genetic and dietary factors. Clin. Biochem. 2007, 40, 1007–14. [Google Scholar] [CrossRef]
- Toshima, S.; Hasegawa, A.; Kurabayashi, M.; Itabe, H.; Takano, T.; Sugano, J.; Shimamura, K.; Kimura, J.; Michishita, I.; Suzuki, T.; et al. Circulating oxidized low density lipoprotein levels. A biochemical risk marker for coronary heart disease. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2243–7. [Google Scholar] [CrossRef]
- Holvoet, P.; Mertens, A.; Verhamme, P.; Bogaerts, K.; Beyens, G.; Verhaeghe, R.; Collen, D.; Muls, E.; Van de Werf, F. Circulating oxidized LDL is a useful marker for identifying patients with coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 844–8. [Google Scholar] [CrossRef] [PubMed]
- Millian, N.S.; Garrow, TA. Human betaine-homocysteine methyltransferase is a zinc metalloenzyme. Arch. Biochem. Biophys. 1998, 356, 93–8. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Kirsch, H.; Domergue, F.; Abbadi, A.; Sperling, P.; Bauer, J.; Cirpus, P.; Zank, T.K.; Moreau, H.; Roscoe, T.J; et al. Novel fatty acid elongases and their use for the reconstitution of docosahexaenoic acid biosynthesis. J. Lipid. Res. 2004, 45, 1899–909. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, G.; Laganà, A.S. The Link between Homocysteine and Omega-3 Polyunsaturated Fatty Acid: Critical Appraisal and Future Directions. Biomolecules. 2020, 10, 219. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.M.; Sapp, P.A.; Kris-Etherton, P.; Petersen, K. Effects of saturated fatty acid consumption on lipoprotein(a): a systematic review and meta-analysis of randomized controlled trials. Am. J. Clin. Nutr 2024, S0002-9165(24)00591-4. [Google Scholar] [CrossRef] [PubMed]
- Law, H.G.; Meyers, F.J.; Berglund, L.; Enkhmaa, B. Lipoprotein(a) and diet-a challenge for a role of saturated fat in cardiovascular disease risk reduction? Am. J. Clin. Nutr. 2023 118, 23–26. [CrossRef]
- Ward, N.C.; Ying, Q.; Chan, D.C.; Pang, J.; Mori, T.A.; Schultz, C.J.; Dwivedi, G.; Francis, R.J.; Watts, G.F. Improved arterial inflammation with high dose omega-3 fatty acids in patients with elevated lipoprotein(a): Selective effect of eicosapentaenoic acid? J. Clin. Lipidol. 2023, 17, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Yamashita, T.; Kusuhara, M.; Yonemura, A.; Ito, T.; Higashi, K.; Ayaori, M.; Ohmori, R.; Nakamura, H.; Ohsuzu, F. The susceptibility of lipoprotein (a) to copper oxidation is correlated with the susceptibility of autologous low density lipoprotein to oxidation. Clin. Biochem. 2003, 36, 113–20. [Google Scholar] [CrossRef]
- Antonicelli, R.; Testa, R.; Bonfigli, A.R.; Sirolla, C.; Pieri, C.; Marra, M.; Marcovina, S.M. Relationship between lipoprotein (a) levels, oxidative stress, and blood pressure levels in patients with essential hypertension. Clin. Exp. Med. 2001, 1, 145–150. [Google Scholar] [CrossRef]
- Tsimikas, S.; Gordts, P.L.S.M.; Nora, C.; Yeang, C.; Witztum, J.L. Statin therapy increases lipoprotein(a) levels. Eur. Heart J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, W.S.; Wang, X.; Xu, L.; Yang, X.C. Palmitic Acid Increases Endothelin-1 Expression in Vascular Endothelial Cells through the Induction of Endoplasmic Reticulum Stress and Protein Kinase C Signaling. Cardiology 2018, 140, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Yakubu, M.A.; Leffler, C.W. Regulation of ET-1 biosynthesis in cerebral microvascular endothelial cells by vasoactive agents and PKC. Am. J. Physiol. 1999, 276, C300–C305. [Google Scholar] [CrossRef] [PubMed]
- Delerive, P.; Martin-Nizard, F.; Chinetti, G.; Trottein, F.; Fruchart, J.C.; Najib, J.; Duriez, P.; Staels, B. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ. Res. 1999, 85, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Baier-Bitterlich, G.; Uberall, F.; Bauer, B.; Fresser, F.; Wachter, H.; Grunicke, H.; Utermann, G.; Altman, A.; Baier, G. Protein kinase C-θ isoenzyme selective stimulation of the transcription factor complex AP-1 in T lymphocytes. Mol. Cell. Biol 1996, 16, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.S.; Therkelsen, K.; Møller, J.M.; Dyerberg, J.; Schmidt, E.B. n-3 fatty acids do not decrease plasma endothelin levels in healthy individuals. Scand. J. Clin. Lab. Invest. 1997, 57, 495–9. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, H.; Moridaira, K.; Wada, O. Zinc deficiency further increases the enhanced expression of endothelin-1 in glomeruli of the obstructed kidney. Kidney Int. 2000, 58, 575–86. [Google Scholar] [CrossRef]
- López-Ongil, S.; Senchak, V.; Saura, M.; Zaragoza, C.; Ames, M.; Ballermann, B.; Rodríguez-Puyol, M.; Rodríguez-Puyol, D.; Lowenstein, C.J. Superoxide regulation of endothelin-converting enzyme. J. Biol. Chem. 2000, 275, 26423–7. [Google Scholar] [CrossRef]
- Lankhorst, S.; Kappers, M.H.; van Esch, J.H.; Danser, A.H.; van den Meiracker, A.H. Hypertension during vascular endothelial growth factor inhibition: focus on nitric oxide, endothelin-1, and oxidative stress. Antioxid. Redox Signal. 2014, 20, 135–45. [Google Scholar] [CrossRef]
Figure 2.
Total antioxidant status and SOD antioxidant activities in plasma of different trial groups. A. MDA (malondialdehyde) levels; B. TAS (total antioxidant status); C. Total SOD (Superoxide dismutase); D. Erythrocytes SOD1. Group I: Healthy controls participants; Group II: Diabetics participants; Group III: Hypertensive participants; Group IV: Hypertensive-diabetic participants. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 2.
Total antioxidant status and SOD antioxidant activities in plasma of different trial groups. A. MDA (malondialdehyde) levels; B. TAS (total antioxidant status); C. Total SOD (Superoxide dismutase); D. Erythrocytes SOD1. Group I: Healthy controls participants; Group II: Diabetics participants; Group III: Hypertensive participants; Group IV: Hypertensive-diabetic participants. *p < 0.05; **p < 0.01; ***p < 0.001.
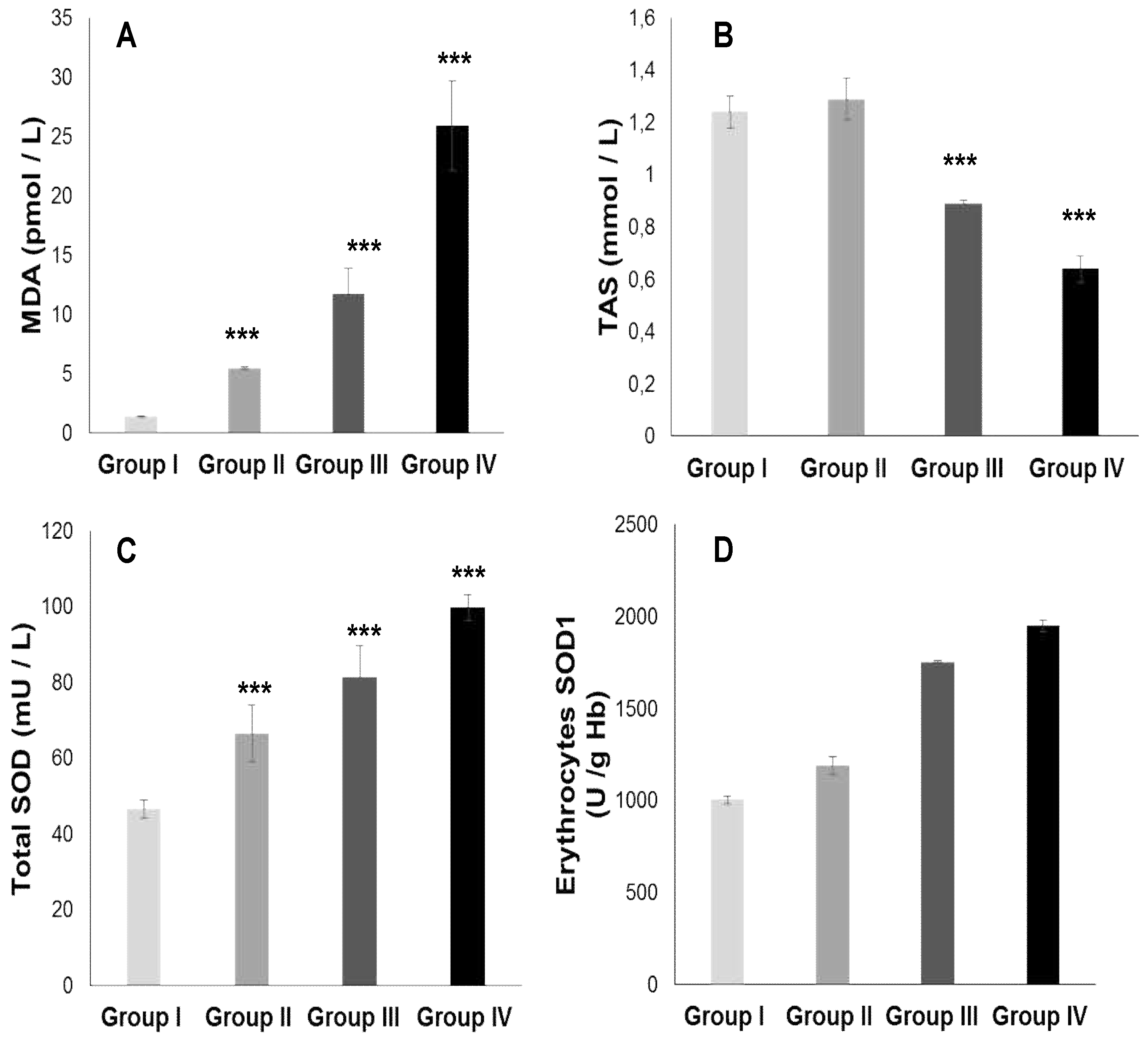
Figure 3.
GPx (Glutathione peroxidase) and CAT (Catalase) antioxidant activities, and GSH (Glutathione) status in plasma of different trial groups. A. GPx; B. GSH; C. GSH/GSSG (reduced glutathione / oxidized glutathione); D. CAT. Group I: Healthy controls participants; Group II: Diabetics participants; Group III: Hypertensive participants; Group IV: Hypertensive-diabetic participants. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 3.
GPx (Glutathione peroxidase) and CAT (Catalase) antioxidant activities, and GSH (Glutathione) status in plasma of different trial groups. A. GPx; B. GSH; C. GSH/GSSG (reduced glutathione / oxidized glutathione); D. CAT. Group I: Healthy controls participants; Group II: Diabetics participants; Group III: Hypertensive participants; Group IV: Hypertensive-diabetic participants. *p < 0.05; **p < 0.01; ***p < 0.001.
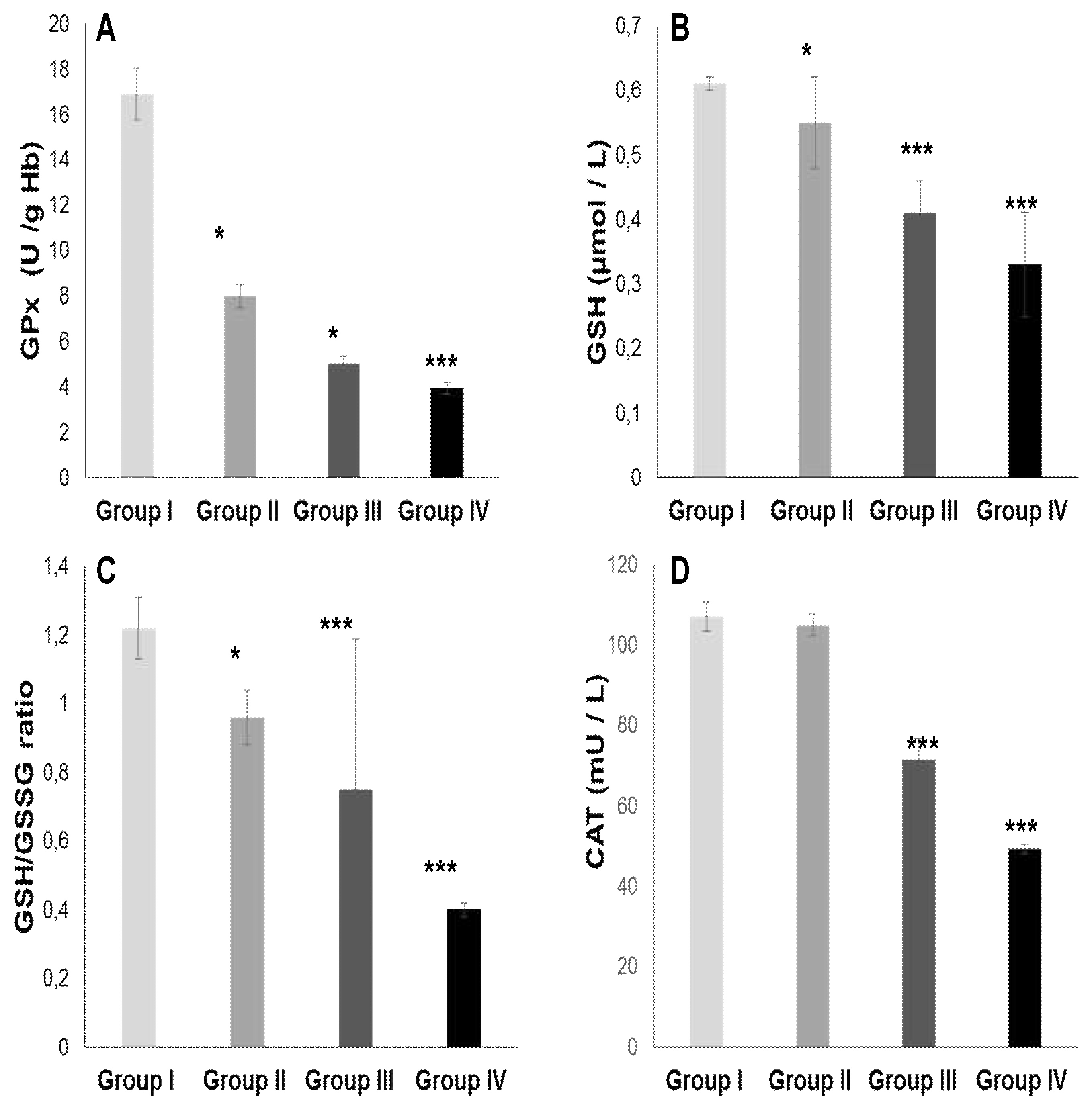
Figure 4.
Zn (Zinc), Cu (Cooper), Mn (Manganese) and Iron plasma levels in different trial groups. A. Zn; B. Cu; C. Mn; D. Iron. Group I: Healthy controls participants; Group II: Diabetics participants; Group III: Hypertensive participants; Group IV: Hypertensive-diabetic participants. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 4.
Zn (Zinc), Cu (Cooper), Mn (Manganese) and Iron plasma levels in different trial groups. A. Zn; B. Cu; C. Mn; D. Iron. Group I: Healthy controls participants; Group II: Diabetics participants; Group III: Hypertensive participants; Group IV: Hypertensive-diabetic participants. *p < 0.05; **p < 0.01; ***p < 0.001.
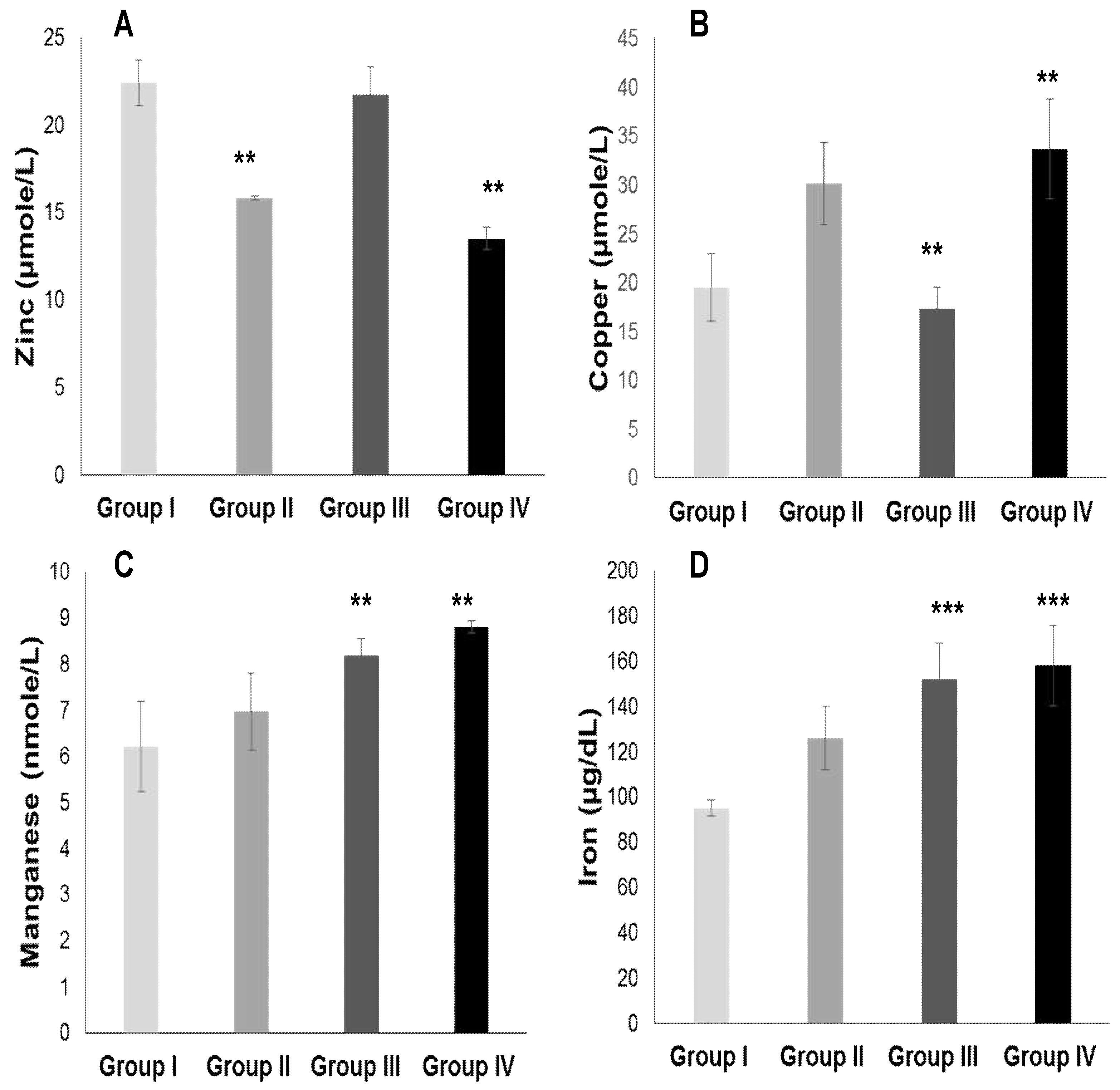
Figure 5.
Se (Selenium) plasma levels in different trial groups. A. Se; B. Se/Mn ratio; C. Se/Cu ratio; D. Zn/Cu ratio. Group I: Healthy controls participants; Group II: Diabetics participants; Group III: Hypertensive participants; Group IV: Hypertensive-diabetic participants. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 5.
Se (Selenium) plasma levels in different trial groups. A. Se; B. Se/Mn ratio; C. Se/Cu ratio; D. Zn/Cu ratio. Group I: Healthy controls participants; Group II: Diabetics participants; Group III: Hypertensive participants; Group IV: Hypertensive-diabetic participants. *p < 0.05; **p < 0.01; ***p < 0.001.
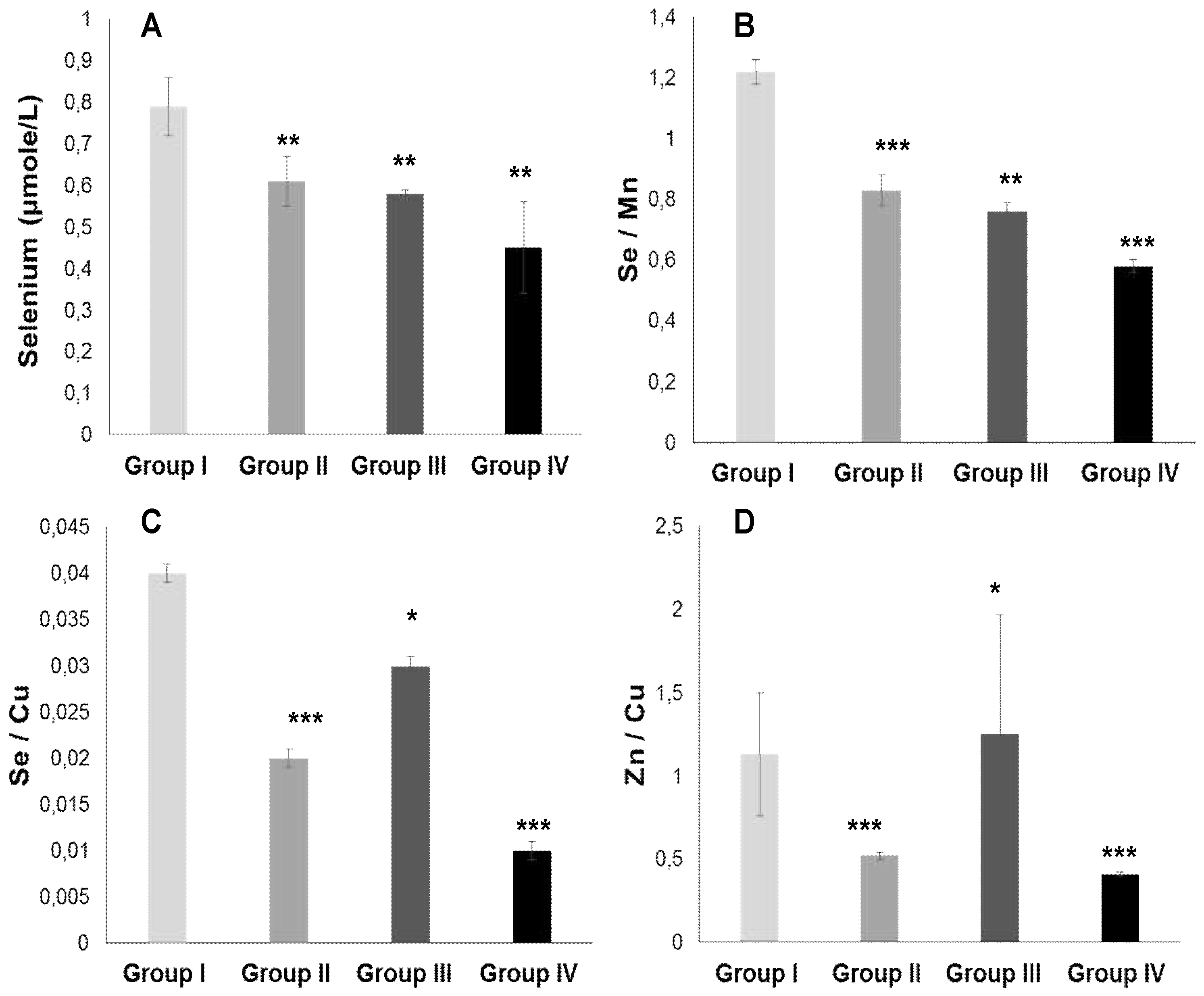

Table 1.
Participant’s cohort characterization according anthropometric status in various clinical trial groups.
Table 1.
Participant’s cohort characterization according anthropometric status in various clinical trial groups.
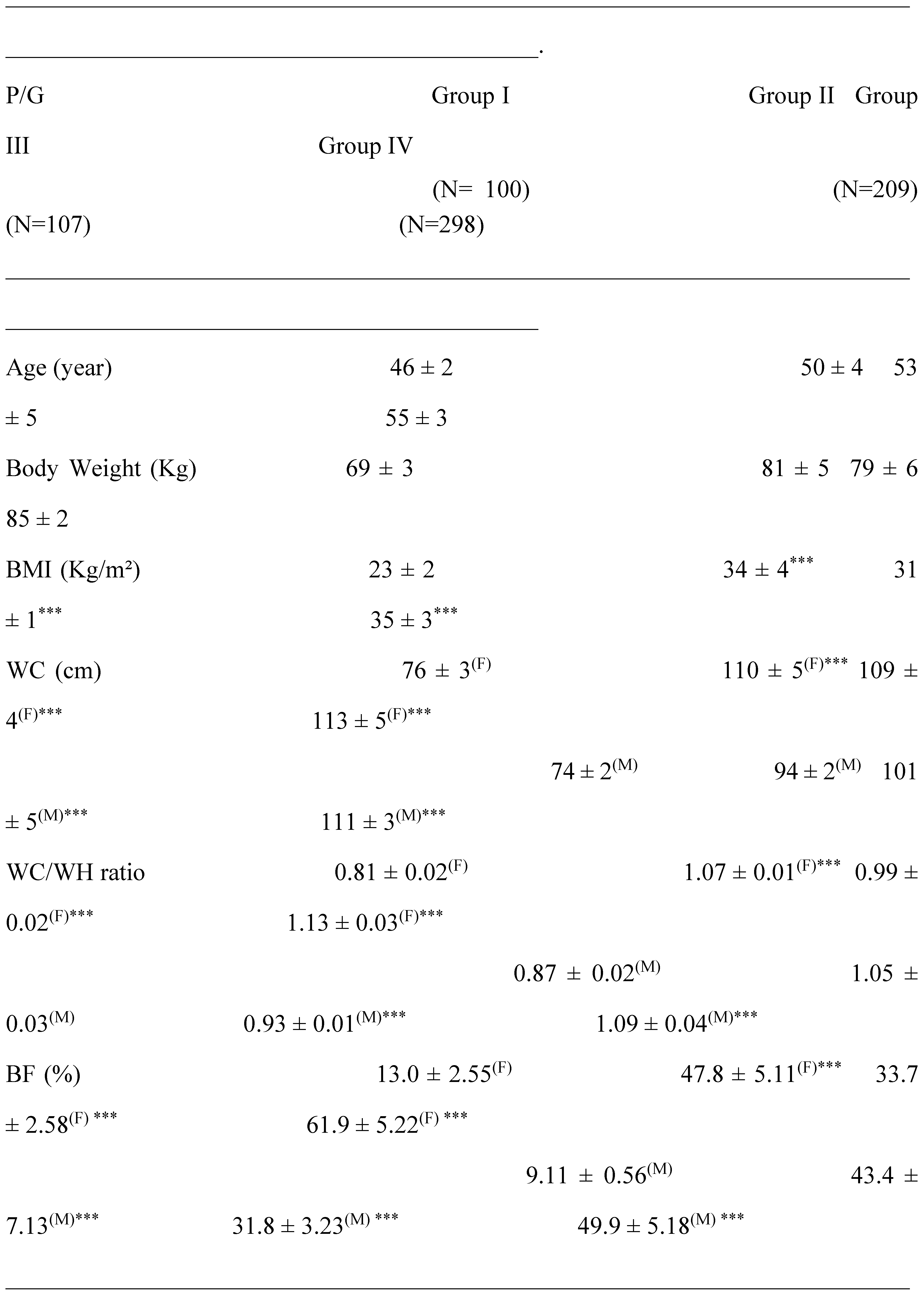
P: parameters; G: group; Group I: Healthy controls participants; Group II: Diabetics participants; Group III: Hypertensive participants; Group IV: Hypertensive-diabetic participants; M: male; F: female; N: total number of participants; BMI: Body mass index; WC: Waist Circumference; WH: Waist Hips; BF: body fat percentage. The mean values are assigned from the standard error to the mean (X ± SD). The degree of significance is calculated for a risk of error α = 5%. The comparison of means is established both between the groups II, III and IV versus control group. *: p<0.05; **: p<0.01; ***: p<0.001.
Table 2.
Participant’s cohort classification according Cardiometabolic Syndrome in various clinical trial groups.
Table 2.
Participant’s cohort classification according Cardiometabolic Syndrome in various clinical trial groups.



P: parameters; G: group; Group I: Healthy controls participants; Group II: Diabetics participants without hypertension; Group III: Hypertensive participants without DT2; Group IV: Hypertensive-diabetic participants; DT2: Type 2 Diabetes; HOMA: Homeostasis Model Assessment; C: cholesterol; HDL: high density lipoprotein; LDL: low density lipoprotein; Hs-CRP: High sensitive C reactive Protein; AST: Aspartate aminotransferase; ALT: Alanine aminotransferase; GGT: Gamma-glutamyl transferase. The comparison of means is established both between the groups II, III and IV versus control group. *: p<0.05; **: p<0.01; ***: p<0.001.
Table 3.
Participant’s cohort screening according cardiovascular and athero-thrombogenic profile in various clinical trial groups.
Table 3.
Participant’s cohort screening according cardiovascular and athero-thrombogenic profile in various clinical trial groups.
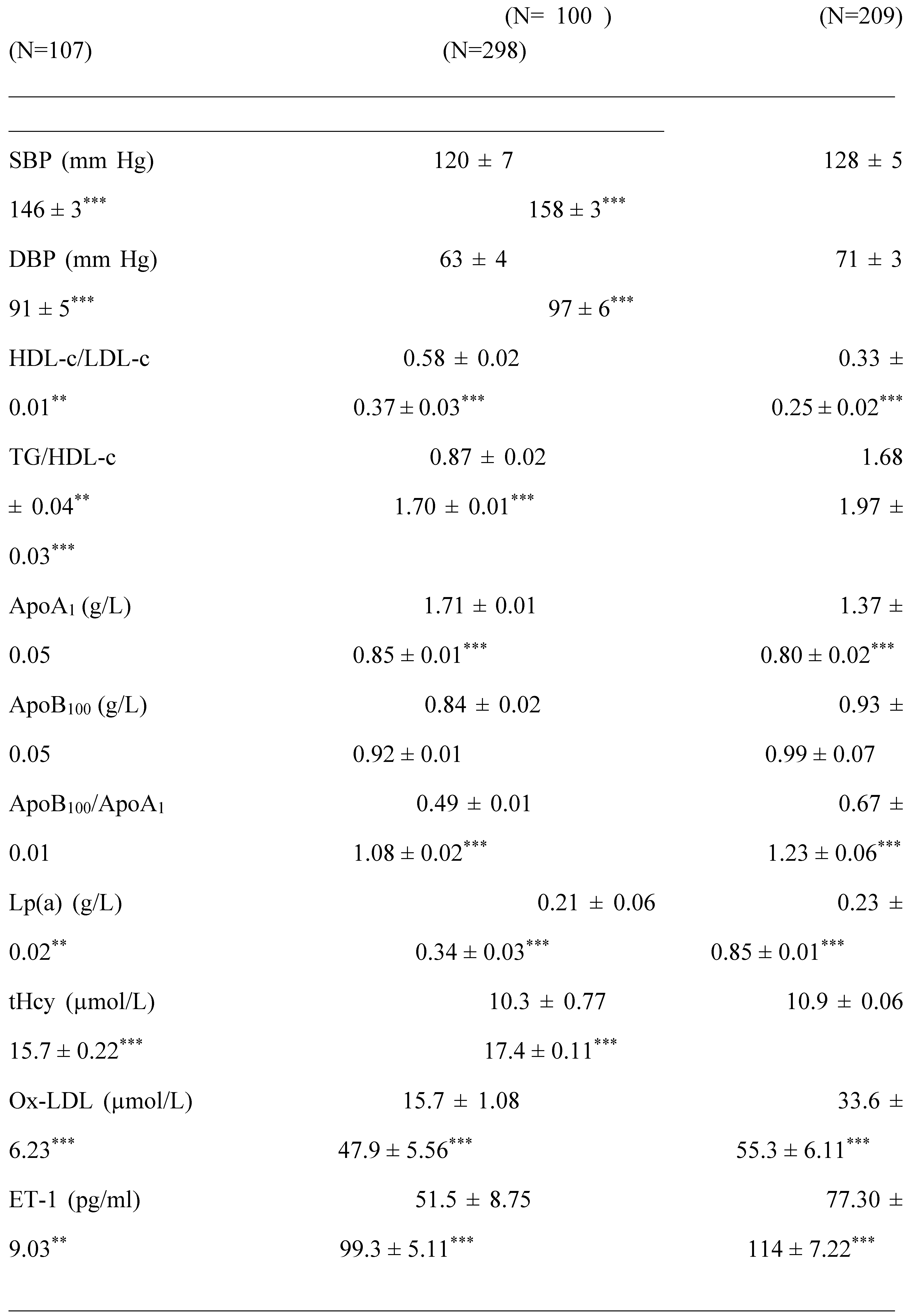
P: parameters; G: group; Group I: Healthy controls participants; Group II: Diabetics participants; Group III: Hypertensive participants; Group IV: Hypertensive-diabetic participants; SBP: systolic blood pressure; DBP: diastolic blood pressure. TG: Triglycerides; ApoB100: Apoprotein B; ApoA1: Apoprotein A; Lp (a): Lipoprotein (a); tHcy: Total homocysteine. Ox-LDL: oxidized low-density lipoprotein. ET-1: Endothelin-1.The comparison of means is established both between the groups II, III and IV versus control group. *: p<0.05; **: p<0.01; ***: p<0.001.
Table 4.
Participant’s cohort screening according plasma fatty acids profile in various clinical trial groups.
Table 4.
Participant’s cohort screening according plasma fatty acids profile in various clinical trial groups.
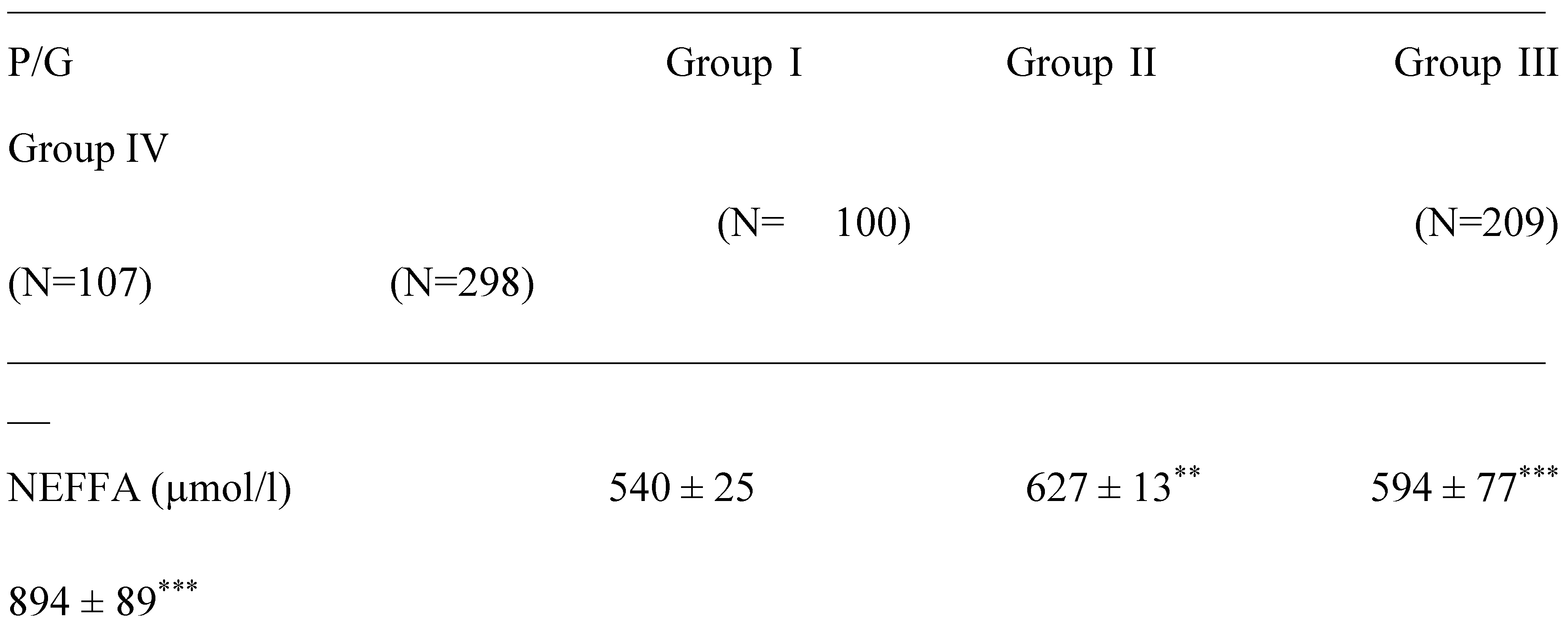
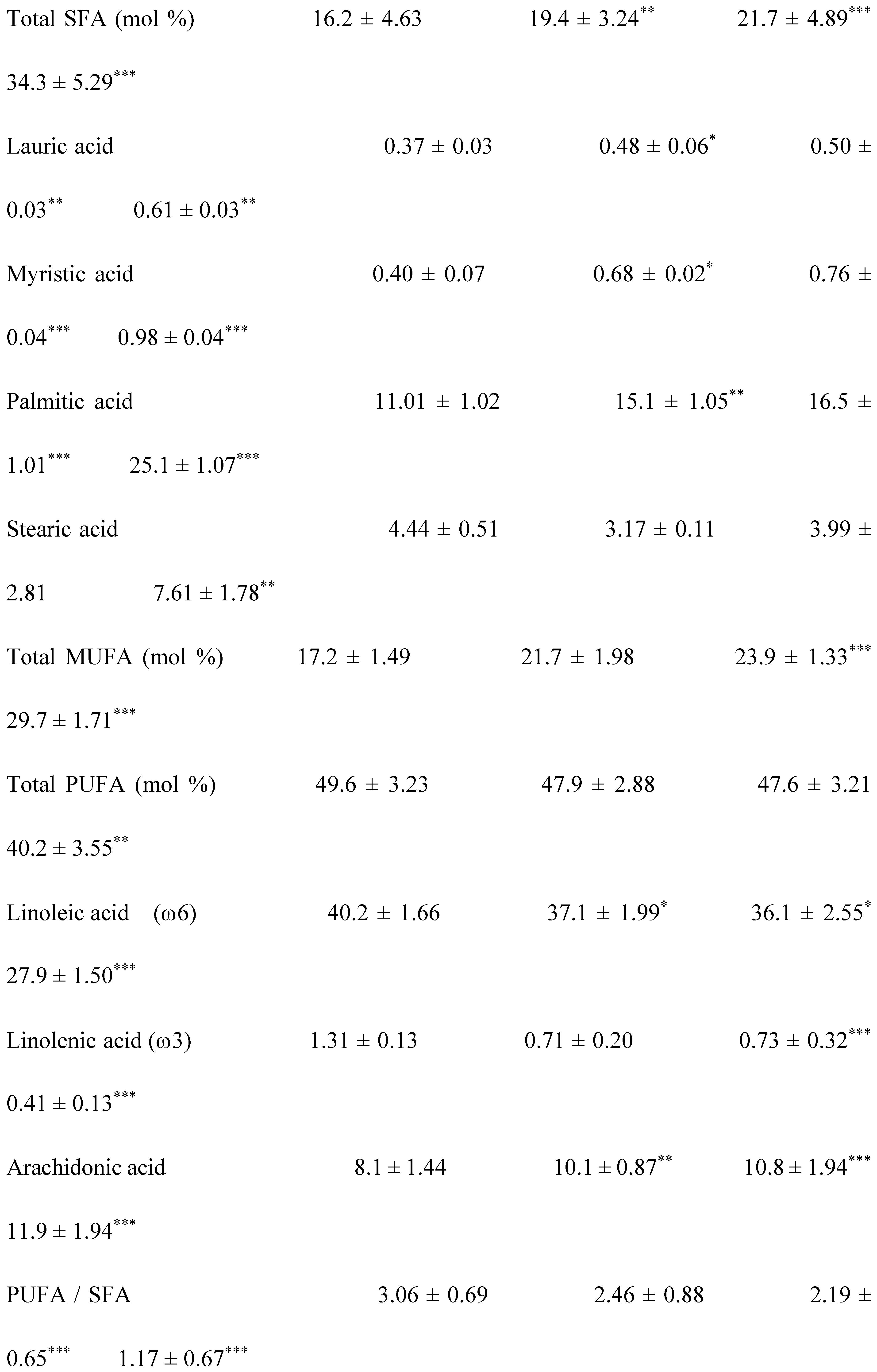
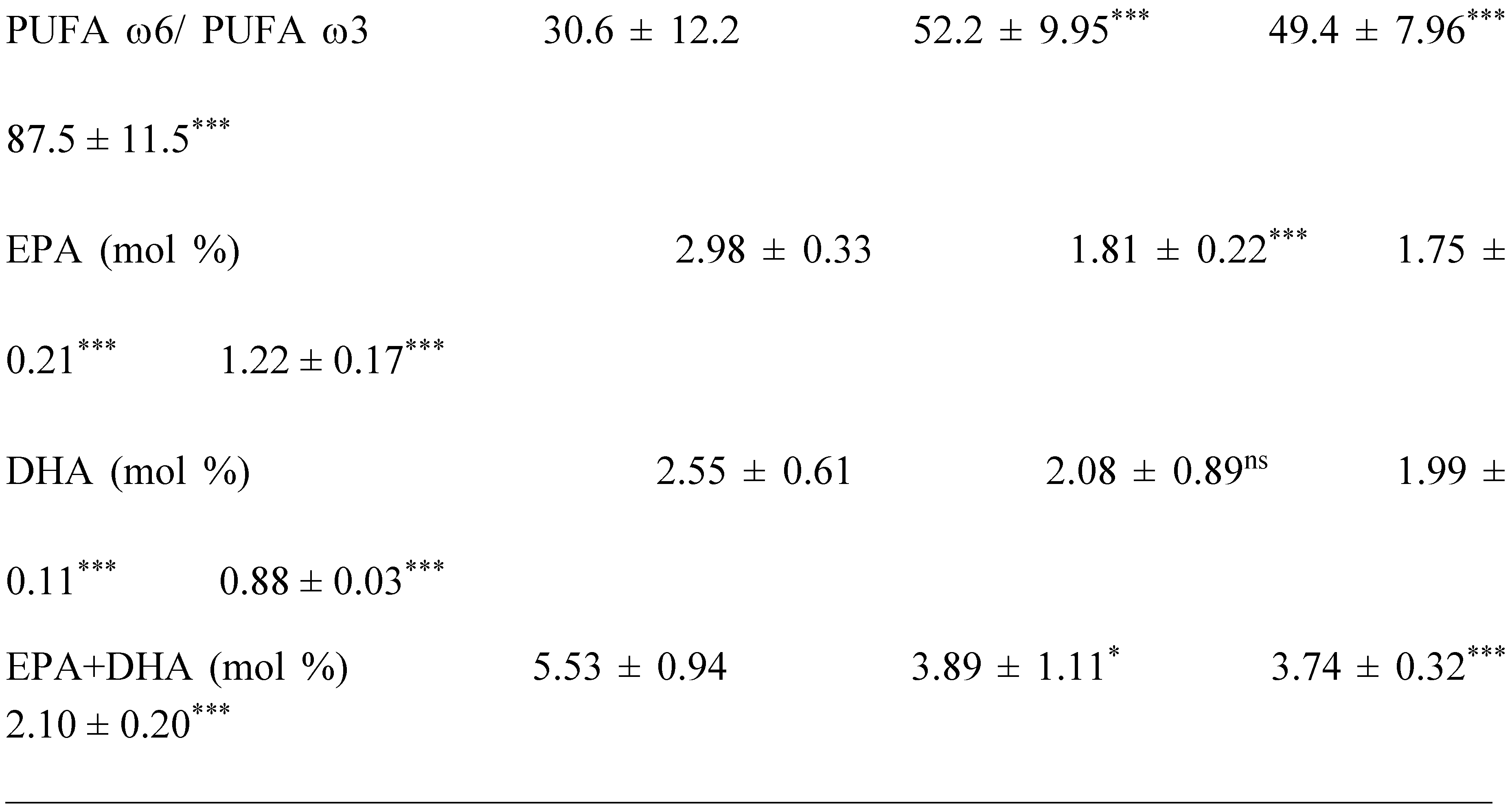
P: parameters; G: group; Group I: Healthy controls participants; Group II: Diabetics participants; Group III: Hypertensive participants; Group IV: Hypertensive-diabetic participants; NEFFA : Non-esterified free fatty acids; SFA : saturated fatty acids ; MUFA: Monounsaturated fatty acids ; PUFA : polyunsaturated fatty acids; EPA : Eicosapentaenoic acid; DHA : Docosahexaenoic acid. The percentage (%) of fatty acids (saturated and unsaturated) was calculated according from the total FA lipid class respectively. The comparison of means is established between the groups II, III and IV versus control group. *: p<0.05; **: p<0.01; ***: p<0.001. .
Table 5.
Pearson correlation between the decrease in the PUFA/SFA ratio and Oxidative stress biomarkers in various clinical trial groups.
Table 5.
Pearson correlation between the decrease in the PUFA/SFA ratio and Oxidative stress biomarkers in various clinical trial groups.

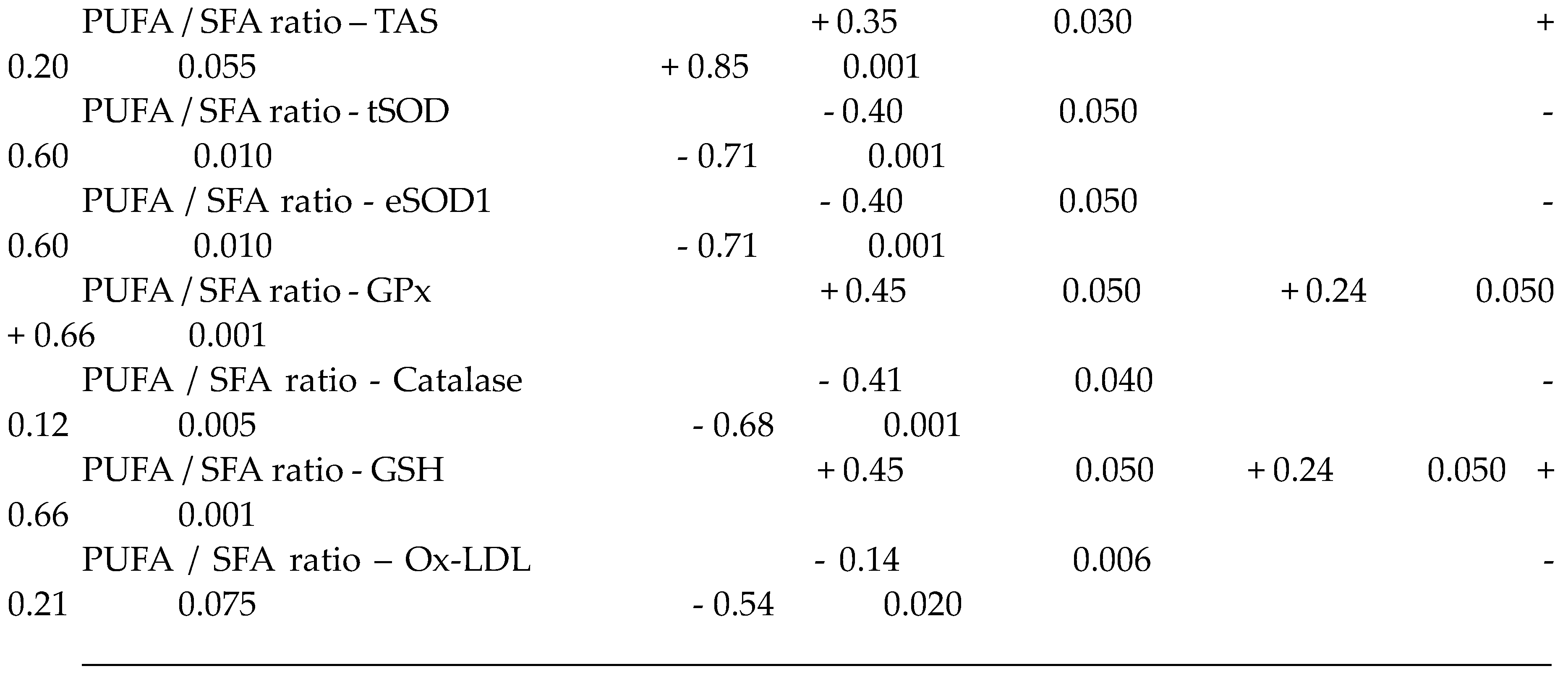
P: Parameters; G: Group; Group I: Healthy controls participants; Group II: Diabetics participants; Group III: Hypertensive participants; Group IV: Hypertensive-diabetic participants; PUFA: polyunsaturated fatty acids; SFA: saturated fatty acids. Se: Selenium; Cu: Copper; Zn: Zinc; Mn: Manganese. TAS: Total antioxidant status; tSOD: Total Superoxide dismutase; eSOD1: Erythrocyte Superoxide dismutase-1; GPx: Glutathione Peroxidase; GSH: Reduced glutathione; Ox-LDL: oxidized low-density lipoprotein. The analysis correlation between two parameters was determined by Pearson's coefficient (r). The results were considered significant (*p <0.05), very significant (**p <0.01) or highly significant (***p <0.001.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



