
Submitted:
20 July 2024
Posted:
22 July 2024
You are already at the latest version
Abstract
Chimeric antigen receptor T cells (CAR-Ts) have shown a remarkable efficacy in hematological malignancies, but limited responses in solid tumors. Among solid tumors, CAR-T cell therapy was particularly explored in brain tumors. CAR-T cells have shown a limited clinical efficacy in various types of brain tumors due to several factors that have hampered their activity, including tumor antigen heterogeneity (not all tumor cells express the target antigen), the limited access of CAR-T cells to brain tumor cells, limited CAR-T cell trafficking and in vivo persistence and the presence of a highly immunosuppressive tumor microenvironment. Despite these considerations, some recent studies have shown promising anti-tumor activity of GD2-CAR-T cells on diffuse midline gliomas and neuroblastomas and of CARv3-TEAM-E cells in glioblastomas. However, strategies are required to improve the effect of CAR-T cells in brain tumors, including advanced CAR-T cell design with multiple antigenic targeting and incorporation of combination therapies.
Keywords:
Immunotherapy
; Chimeric Antigen Receptor T-cells
; Nervous System Tumors
; Brain Tumors
; Glioblastoma
; Diffuse Midlime Glioma
; Neuroblastoma
1. Introduction
In the last years cancer treatment explored new strategies targeting molecules specifically expressed on tumor cells. One of these approaches was based on immunotherapy that, through different strategies, promotes the host’s immune system to specifically kill cancer cells, sparing normal cells. Among the numerous different types of immunotherapies developed in the last two decades, one of growing interest is represented by the generation of T lymphocytes engineered to express chimeric antigen receptors (CARs). CAR-T cells represent a very powerful tool to promote anti-cancer response based on two different mechanisms: promotion of the killing of tumor cells expressing a given antigen; induction of an immune response in the tumor microenvironment.
A fundamental property of CAR-T cells that supports their application to cancer therapy is represented by their capacity to promote recognition and killing of targets cells in a major histocompatibility complex (MHC)-independent manner, thus bypassing a physiological mechanism required by normal T cells; the bypass of MHC restriction by CAR-T cells is an important property since MHC downregulation represents one of the key mechanisms of immune escape adopted by cancer cells [1].
The basic structure of a CAR-T in its simplest form is represented by an antibody or ligand-derived ectodomain (conferring the antigen binding capacity) fused with a hinge, transmembrane domain, and an intracellular T-cell signaling domain. This simple structure corresponds to the first-generation CARs characterized by the presence of a single intracellular domain; however, the CAR-T cells engineered with first-generation CARs, although exhibiting an efficient anti-tumor potential, display in vivo a limited proliferation, survival and trafficking, thus limiting their antitumor efficacy [2]. (Figure 1) Given these limitations, two strategies have been adopted to improve CAR-T cell expansion and persistence consisting in the introduction of one or two costimulatory domains in the CAR structure, allowing the development of second-generation and third-generation CAR-T cells, respectively [2]. (Figure 1) Co-stimulatory domains commonly used for CAR-T cell generation are represented by CD28, 1-1BB, CD27 and ICOS [2]. Finally, more recently, CAR generation was further optimized through introduction of antitumor cytokines, such as interleukin-2, in the CAR structure, thus allowing the development of fourth-generation CAR-T cells [2]. (Figure 1)
The process of autologous CAR-T cell manufacturing involves various steps the usually take 2 weeks and include initial cell isolation, T cell activation, transduction of CAR transgene, cell expansion, final formulation and product release testing [3,4]. Most of CAR-T cell products are frozen, cryopreserved in liquid nitrogen and thawed at bedside prior to infusion into patients. Recently, abbreviated manufacturing procedures have been introduced, able to reduce the whole ex vivo process of manufacturing to 2-3 days [3].
CAR-T cells have been introduced in the clinical treatment of hematological malignancies, achieving impressive clinical results in relapsed/refractory B-cell malignancies, including lymphomas, leukemias and myeloma [5,6]. Following remarkable clinical outcomes in hematological malignancies, the FDA approved six CAR-T cell products for indications such as lymphoma, leukemia and myeloma [5,6].
However, CAR-T cell applications in the treatment of solid tumors remain limited, due to several constraints: the limited or suboptimal access of CAR-T cells to solid tumor cells in organs; the presence of a highly immunosuppressive tumor microenvironment; limited CAR-T cell trafficking and in vivo persistence; T cell exhaustion; tumor antigen heterogeneity (not all tumor cells express the target antigen); lack of suitable target antigens [7,8].
In spite these limitations, a growing number of phase I/II clinical trials are exploring CAR-T cell therapy in solid tumors. Brain tumors represent the most common solid tumor types undergoing clinical trial evaluation for CAR-T cell safety and efficacy [7,8].
The treatment of brain tumors with CAR-T cells show additional difficulties due to the semipermeable properties of blood brain barrier leading to a difficult access to the tumor mass [9]. CAR-T cell infusion to the brain include three different ways of administration: delivery via the blood, delivery via cerebro-spinal fluid and delivery in the tumor cavity (intracavitary) or in the brain ventricles (intraventricular). Two additional techniques of CAR-T administration directly in the CNS are represented by spinal intratechal infusion (SII) and disruption of the blood brain barrier by focal ultrasound; SII is a technique well-known in pediatric oncology used for intermittent infusion of chemotherapy [9]. Numerous trials exploring anti-CD19 CAR-T cells in hematologic malignancies have shown the capacity of engineered T cells to cross the blood-brain barrier [9]. However, studies in preclinical models have supported a superiority of loco-regional (intracavitary or intraventricular) CAR-T cell infusion compared to systemic intravenous infusion, in terms of both enhanced antitumor efficacy and reduced toxicity [10].
In this review we focus on recent studies based on CAR-T cell therapy in pediatric and adult brain tumors. The results of these studies, as well as the challenges of CAR-T cell therapy for brain cancers, are discussed and analyzed.
2. Experimental and Clinical Studies of CAR-T Cell Therapy in Brain Tumors
A consistent number of experimental and clinical studies have explored the targeting of brain tumors using CAR-T cells engineered to interact with surface antigens expressed on brain tumor cells. (Table 1)
2.1. HER-2
The EGFR family member HER-2, also known as ERBB2, is overexpressed in brain tumor cells and represents a potential tumor target of CAR-T cell therapy.
Glioblastoma is the most common and aggressive malignant primary human brain cancer in adult patients, with a median overall survival from diagnosis of about 12-18 months. Glioblastoma is considered as one of the most aggressive and lethal among human malignancies and is resistant to every type of therapeutic strategy, including cytotoxic chemotherapy and molecular targeted therapies. Furthermore, glioblastomas are intrinsically resistant to T-cell based immunotherapies, including those based on PD1/PDL1 immune checkpoint blocking agents; thus, marked resistance to immunotherapy seems to be related to the paucity of T cells and to the immunosuppressive microenvironment present in these tumors, characterized by abnormal vascular niches and by the prominent infiltration of immunosuppressive macrophages.
Liu et al. have explored HER-2 expression in 43 primary glioblastoma cell lines and observed a significant positivity in 76% of cases; HER-2 expressed on glioblastoma cells is recognized by cytotoxic T cells [11]. Ramezani and coworkers have explored HER-2 expression by immunohistochemistry in 107 primary brain tumors and observed a higher frequency of positive tumors among high-grade astrocytomas (55%) than in low-grade astrocytomas (26%) [12]. Mineo and coworkers have compared the HER-2 expression in de novo glioblastoma (primary) and glioblastoma resulting from anaplastic transformation of low-grade glioma (secondary) and observed in the former ones a high HER-2 expression and in the latter ones a lower HER-2 expression [13]. High HER-2 expression was associated with shorter survival [13].
A preclinical study showed in an orthoptic murine xenograft model the efficacy of HER-2 specific CAR-T cells generated from 10 glioblastoma patients in mediating the killing of autologous glioblastoma cells, including the fraction of CD133-positive cancer stem cells [14].
In the clinical setting, the first patient treated with HER-2-targeted CAR-T cell therapy succumbed to death due to a cytokine storm; this patient (colon cancer) received the treatment with a CAR-T engineered with ScFv from high-affinity Trastuzumab antibody and co-stimulating domains CD28 and 41BB [15]. Given this severe event, a new CAR-T was redesigned including a ScFv from a low-affinity FRP5 antibody and different co-stimulatory domain inducing lower cytokine release in vivo; a study was carried out using these CAR-T cells in patients with progressive HER-2-positive glioblastoma, involving one or more infusions (up to 1x108 CAR-T cells), administered without prior lymphodepletion [16]. A total of 17 patients were enrolled and 16 were evaluable for response: 1 had partial response for more than 9 months, 7 had stable disease for 8 weeks to 29 months and 8 progressed after T-cell infusions. No dose-limiting toxic effects were reported [16]. The median OS post-treatment was 11.1 months and 24.5 months post-diagnosis; however, no conclusions can be drawn about a possible survival benefit [16].
Vitanza and coworkers have reported the preliminary results of the phase I BRAINChild-01 clinical trial involving locoregional infusion of HER-2-specific CAR-T cells in children and young patients with recurrent or refractory CNS tumors; The observations made in the first 3 treated patients showed no dose-limiting toxicities and evidence of local immune activation (with high concentrations of CXCL10 and CCL2 in the cerebrospinal fluid, thus supporting the clinical feasibility of this study [17].
Studies in an orthoptic xenograft model of HER-2+ breast cancer metastasis to the brain have shown an enhanced proliferative capacity and a reduced T-cell exhaustion phenotype of HER-2-CARs containing the CD28 co-stimulatory domain following regional intraventricular delivery of HER-2 CAR-T cells for the treatment of multifocal brain metastases [18].
Thus, two phase I clinical trials (NCT03389230 and NCRT03696030) have started the evaluation of these optimized HER-2-specific CAR-T cells among patients with HER-2-positive malignant gliomas and HER-2-positive metastases of breast cancers; other two studies are evaluating locoregional treatment of recurrent and/or refractory pediatric CNS tumors (NCT03500991 and NCT02442297).
Recent preclinical studies have reported the generation of HER-2-targeting CAR-T cells exerting a marked antitumor activity either through the development of a third-generation CAR-T cells or through the development of CAR-T cells with low-affinity HER-2CARs [19]. Furthermore, this study showed also that peritumoral intravenous CAR-T cell administration resulted in a better glioblastoma inhibition compared to intravenous administration [20].
2.2. EGFRvIII
Epidermal growth factor amplification is one of the most recurrent genetic alterations of glioblastoma and is present in more than 50% of primary glioblastomas and in less than 10% of glioblastomas evolving from recurrent low-grade gliomas; 30% to 50% of EGFR-amplified glioblastomas exhibit an in-frame deletion of exons 2 to 7, thus generating a truncated receptor with loss of extracellular ligand-binding domain, called EGFR variant III (EGFRvIII); EGFRvIII is expressed only in tumor cells and is associated with constitutive receptor signaling. Given this tumor-specific expression, EGFRvIII represents a therapeutic target in a part of glioblastoma patients, potentially suitable for CAR-T-mediated targeting.
The study of relevant genetic mouse models of glioblastomas identified distinct immune landscapes associated with expression of EGFR wild-type and mutant EGFRvIII; particularly, accumulation of polymorphonuclear myeloid-derived suppressor cells (PMN-MDSC) was mor pronounced in EGFRvIII-driven glioblastomas, association with resistance to PD-1 checkpoint blockade immunotherapy [21].
O’ Rourke et al. have performed a first-in-human study of intravenous delivery of a single-dose of autologous CAR-T cells targeting EGFRvIII in 10 recurrent glioblastoma patients [22]. Manufacturing and infusion of EGFRvIII-directed CAR-T cells was feasible and well-tolerated; no therapeutic responses were observed in these patients (only one patient had residual stable disease for over 18 months of follow-up); all patients displayed transient expansion of CART-EGFRvIII cells in peripheral blood and imaging studies suggested trafficking of CAR-T cells at the level of regions of active glioblastoma proliferation [22].
Goff et al. have investigated in a pilot phase I study the association of EGFRvIII-targeted CAR-T cell therapy with IL-2 infusion post-transfer, in R/R glioblastoma patients [23]. Cell infusion products ranged from 6.3x106 to 2.6x1010 anti-EGFRvIII CAR-T cells and were administered intravenously after lymhodepletion; median overall survival was 6.9 months, with two patients surviving over 1 year and a third patient was alive at 59 months [23]. Two patients experienced severe hypoxia and one of these patients succumbed to death after CAR-T cell infusion at the highest dose [23]. No objective responses were observed in the treated patients [23].
In order to reduce the risk of a tumor recurrence after anti-EGFRvIII CAR-T cell therapy by tumor cells expressing wild-type EGFR protein, Choi et al. have developed a peculiar strategy based on the use of bicistronic construct driving expression of a CAR specific for EGFRvIII, and a bispecific T-cell engager (BiTE) against EGFR [24]. Autologous T cells engineered with CARvIII-TEAM-E were intraventricularly reinfused to 3 patients with recurrent glioblastoma; none of these patients developed toxicities over grade 3 [25]. A dramatic radiographic tumor regression shortly after single infusion of CARvIII-TEAM-E CAR-T cells was observed in all three patients, but this response was transient in two of them, which correlated with limited persistence of CARvIII-TEAM-E cells [25]. The presence of CAR-T cells in the peripheral blood peaked 3 weeks after CAR-T cell infusion. The early responses observed in this study support future studies based on the intraventricular infusion of cell-based therapies to glioblastoma patients. However, the transient response observed in two of the three treated patients underscores the absolute need for additional research to improve and prolong in the time the efficacy of this therapy. Finally, this study supports the value of multitargeted CAR-T cell therapy in the treatment of brain tumors.
The study of the apheresis infusion products from the first trial of EGFRvIII-directed therapy showed that CAR-T cell therapy targeting EGFRvIII induced an upregulation of programmed death-ligand 1 (PD-L1) expression in tumor microenvironment; furthermore, the expression of PD-1 in CAR-T infusion products correlated with clinical response (PFS) [26]. These observations have supported a phase I trial (NCT03726515) exploring the concomitant administration intravenously of CAR-T-EGFRvIII cells with anti-PD-1 monoclonal antibody Pembrolizumab in 7 patients with newly diagnosed glioblastoma [27]. No limiting toxicities were observed; mPFS was 5.2 months and mOS 11.8 months; no objective responses were observed in these patients [27]. Comparison of tumor microenvironment in tumor specimens obtained before and after CAR-T cells therapy showed a consistent evolution of the infiltrating myeloid and T cells, with more exhausted, regulatory and interferon-stimulated T cells at relapse [27].
Another recent study explored the safety and the efficacy of intracellular administration of bivalent CAR-T cells engineered to target both EGFR and interleukin-13 receptor alpha 2 (IL13Rα2) in six recurrent glioblastoma patients [28]. All six treated patients had progressive multifocal disease at the time of treatment. In all six treated patients (3 with 1x107 cells and 3 with 2.5x107 cells) infusions of CAR-T-EGFR-IL13Rα2 cells were associated with early-onset neurotoxicity treated with dexamethasone and anti-IL1R antibody; radiologic evidence of tumor reduction was observed in all treated patients, but none reached criteria for an objective response [28]. A substantial CAR-T cell abundance and cytokine release was observed in all six treated patients [28]. The results of this study are still preliminary and need an evaluation after the completion of dose-escalation and a longer follow-up of patients.
Recently, a novel GCT102 CAR-T was developed, engineered with EGFRvIII-specific ScFv, targeting EGFRvIII with high affinity; in a xenograft model of human glioblastoma, GTC102 CAR-T cells efficiently killed tumor cells with decreased cytokine secretion [29]. In different preclinical glioblastoma models, GTC102 CAR-T cells displayed specificity for tumor cells expressing EGFRvIII, thus supporting future studies aiming to evaluate their activity in clinical setting [29].
2.3. Interleukin-13 Receptor Alpha2 (IL-13Rα2)
Two IL-13 receptor proteins were identified: (i) IL-13Rα1, a component of the signaling, heterodimeric high-affinity receptor for IL13- that is shared with IL-4; (ii) IL-13Rα2, a monomeric, IL-4-independent receptor, highly specific for IL-13 and not capable of binding IL-4. IL-13Rα1 and IL-13Rα2 are two members of the hematopoietic receptor superfamily exhibiting very low sequence homology. Human malignant glioma cells express high levels of IL-13Rα2, while this receptor chain is scarcely expressed on normal astrocytes [30]. IL-13Rα2 is a high-sffinity receptor of the inflammatory cytokine IL-13 and its preferential expression in many solid tumors, including malignant gliomas, supports its value as a potential attractive target for cancer immunotherapy [31].
Brown et al. have reported a first-in-human pilot safety and feasibility trial evaluating CAR-engineered, autologous primary human CD8+ lymphocytes targeting IL-13Rα2 (this CAR recognizes IL-13Rα2 via a membrane stethered IL-13 ligand mutated at a single site, E13Y, to reduce binding to the IL-13Rα1/IL-4Rα compelx and initiates cytolytic killing via an intracellular CD3ζ T-cell activating domain) [32]. In this pilot study, three patients with recurrent glioblastoma were treated with up to 12 local infusions (tumor intracavitary) of IL13(E13Y)-zetakine CD8+ CAR-T at a maximum dose of 108 CAR-T cells [32]. A transient anti-glioma effect was observed in two of these three patients [32]. The treatment was well tolerated, with manageable transient brain inflammation [32]. In 2016 it was reported a complete tumor regression observed in a patient with recurrent glioblastoma after treatment with IL-13Rα-CAR-T cells infused through two intracranial delivery routes (infusion into the resected tumor cavity, followed by infusions into ventricular system [33]. No toxic event of grade 3 or higher was observed [33].
Based on the remarkable clinical response observed in this patient to IL13Rα2-CAR-T cells, it was hypothesized that additional mechanisms could be involved beyond the direct effect of CAR-T cells on tumor targets. Thus, it was provided evidence that IL13Rα2 CAR-T cells exert effects on the endogenous immune system, mainly mediated through acute production of IFN-γ by CAR-T cells; IFN-γ mediates changes in the tumor microenvironment corresponding in an enrichment in activated memory or effector T cells in the lymphoid compartment and increase of activated myeloid cells and M1 type macrophages in the myeloid compartment [34]. These observations suggest a critical role for IFN-γ signaling to sustain a productive CAR-T cell therapy in glioblastoma patients and show that CAR-T cells can activate endogenous T-cell immunity, thus supporting a critical role for host innate and adaptive immunity for CAR-T cell therapy of glioblastoma.
In 2024, Brown and colleagues reported the results of the phase I trial based on IL13(E13Y)-zetochine CAR-T cells involving the enrollment of 65 patients with recurrent high-grade gliomas (in large part, recurrent glioblastomas); this trial evaluated three routes of locoregional CAR-T cell administration (intramural (ICT), intraventricular (ICV) and dual (ICT and ICV) and two manufacturing platforms and a final arm of treatment based on ICT/ICV and an optimized manufacturing procedure [35]. All routes of CAR-T cell delivery and infusion dose levels (from 2x106 to 200x106 CAR-T cells) were generally well tolerated, with grade 3 toxicities or above observed in 35% of patients; no dose-limiting treatment toxicities were observed for any patient [35]. The recommended dose for phase II was estimated to correspond to 200x106 CAR-T cells. 58 patients were evaluable for response and 50% of them displayed stable disease or better; 22% of patients achieved confirmed stable disease or better for ≥90 days, with 8/13 have relapsing high-grade glioma, grade 4; two patients achieved a partial response and one a complete response (all these three patients have IDH-mutated gliomas) [35]. A second patient achieved a complete response after additional CAR-T cell cycles off protocol. For glioblastoma patients, mOS was 7.7 months for all patients and 10.2 months for those with ICT/ICV and optimal CAR-T cell manufacturing [35]. CAR-T cell administration was associated with increased levels of inflammatory CNS cytokines; pretreatment intratumoral CD3 levels were positively associated with patient’s survival [35].
Brown and coworkers have performed a first-in-human study of locally administered, glocorticoid-resistant, allogeneic CAR-T cells administered to six patients with recurrent glioblastoma under treatment with dexamethasone to attenuate tumor-related neuro-edema [36]. Zinc finger nuclease-directed disruption of the glucocorticoid receptor gene was used for the generation of dexamethasone-resistant IL-13Rα2-targeted CAR-T cellks; these dexamethasone-resistant cells retained effector function in the presence of dexamethasone, without any rejection of the therapeutic allogeneic cells [36]. The treatment was well tolerated and showed in four of the six treated patients transient anti-tumor effects [36].
Stern and coworkers have identified two IL-13 variants (C4 and D7), bearing mutations that decrease binding addinity for IL-13Rα1 but did not markedly change affinity for IL-13R α2; in vivo biodistribution of CAR-T cells bearing these IL-13 variants were better able to traffic away from IL-13Rα1-positive lung tissue [37]. This study supports the use of CAR-T cells with IL-13 mutants selective for IL-13Rα2. In line with these observations, Kim et al. have reported the development of CAR-T cells with modified IL-13 (IL-13 was modified on the extracellular domain by substitution of amino acids with E13K, R66D, S69D and R109K) preferentially recognizing IL-13Rα2 and not IL-13Rα1 on malignant glioma cells; YYB-103 CAR-T cells exhibited selectivity for IL-!£Rα2-positive tumor cells and their intravenous infusion elicited inhibition of tumor growth in orthoptic models of human glioblastoma [38]. Furthermore, in a recent study, Loland and coworkers developed novel CAR-T cells with a scFv clone exhibiting high-affinity for IL-2Rα2 and high anti-tumor activity in models of human gliomas [39].
2.4. Disialogangloside (GD2)
GD2 is a carbohydrate-containing sphingolipid composed by a ceramide with two sialic residues attached three monosaccharide links. The intracellular synthesis of GD2 occurs in the Golgi apparatus through the action of two different glycosyltransferases. GD2 is a membrane tumor-associated antigen expressed by a wide range of tumors of neuroectodermal and epithelial origin, such as neuroblastoma, glioma, retinoblastoma, medulloblastoma, melanoma, small-cell lung cancer and other tumors. GD2 expression can be detected also on normal central and peripheral nervous system cells but its expression is markedly higher on tumor cells. Given these properties, GD2 is a potentially attractive target for immunotherapy of brain cancers.
Pediatric-type diffuse high-grade gliomas comprise four types of aggressive gliomas: diffuse midline glioma, H3K27 altered; hemispheric glioma, H3 G34 mutant; diffuse pediatric type high-grade glioma, H3 and IDH wild type; infant-type hemispheric glioma. Diffuse midline glioma (DMG), H3K37 altered, and diffuse hemispheric glioma, H3G4 mutant, are characterized by point mutations in histones, causing widespread epigenetic alterations [40]. DMG with H3K27 alterations grow in CNS midline structures and are associated with poor outcomes [40]. Some studies explored the targeting of GD2 in diffuse intrinsic pontine glioma (DIPG) and other diffuse midline gliomas (DMGs) with mutated H3KM27M, characterized by a high, uniform GD2 expression [41]. Anti-GD2 CAR-T cells incorporating a 4-1BB co-stimulatory domain showed killing of DMG cells in vitro and high antigen-dependent cytokine generation [41]. Furthermore, in patient-derived H3-K27M+ DMG orthoptic xenograft models, systemic administration of anti-GD2 CAR-T cells cleared engrafted tumor cells expressing low levels of GD2 [41].
This preclinical study has supported the development of a phase I clinical study (NCT 04196413) evaluating intravenous infusion of autologous anti-GD2 CAR-T cells (1x106 GD2 CAR-T cells per kg), followed by optional repeated intracerebroventricular (ICV) infusion of these CAR-T cells, in pediatric and young adult H3K27M-mutant DMG patients [42]. Radiographic and clinical benefit was observed in three out four patients, including a pronounced tumor reduction and neurologic improvement; the one non-responsive patient displayed elevated levels of immunosuppressive cytokines in cerebrospinal fluid, such as transforming growth factor-beta (TGF-β) [42]. Toxicity was largely related to the location of the tumor and was reversible with intensive supportive care; off-target toxicity was not observed. It is important to note that the addition of ICV dosing improved or stabilized clinical responses and was associated with increased levels of immunosuppressive myeloid cells in the CSF compared to IV infusions [42]. These initial observations hold promise for additional clinical trials in DMG patients involving the administration of GD2-CAR-T cells. In a second study presented at AACR 2022, the results on the first 13 patients enrolled in this study were reported: 4 treated at 1x106 GD2-CAR-T cells/Kg and 9 treated at 3x106 GD2-CAR-T cells/Kg [43]. Patients responding to the initial IV infusion of GD2-CAR-T cells ICV GD2-CAR-T cells every 4-8 weeks for a maximum of 12 doses [43]. Concerning the safety, three patients treated at 3x106 CAR-T cells/Kg experienced grade 4 cytokine release syndrome (CRS); furthermore, all treated patients displayed transient Tumor Inflammation-Associated Neurotoxicity [43]. 9/10 patients adequately assessed for benefit displayed radiographic and/or clinical benefit after CAR-T cell IV infusion; one participant, a 31-year-old with sDMG, has experienced a near-complete (> 95%) reduction in tumor volume and a 17-year-old with DIPG experienced a near-complete (> 98%) reduction in volume of a pontine tumor; ICV infusion was not associated with high-grade CRS; four subjects confirmed to receive ICV infusions on study and continued to display radiographic benefit at +11, +9.5, +8 and +7 months after enrollment (importantly, two of these four patients displayed a near-complete reduction (95-98%) in tumor volume) [43]. A more mature assessment of these patients showed that: (i) 1x105 GD2-CAR-T cells/kg by IV is the maximum tolerated dose; (ii) four patients displayed major tumor volumetric reductions (52%, 54%, )1% and 100%); (iii) one patient showed a complete response ongoing for >30 months since enrollment; (iv) eight patients displayed a clear neurological benefit as evidenced by an improvement of neurological deficits [44]. The 13 patients with DMG included in this study were explored for immunity-related changes observed during treatment with GD2-CAR-T cells [45]. GD2-CAR-T cell expansion following IV infusion was observed at the level of peripheral blood and persisted during ICV infusions but decreased over time; GD2-CAR-T cell expansion was observed also at the level of CSF after multiple repeated infusions [45]. Increased cytokine chemokine levels (such as IL-6 and IFN-γ) were present in the peripheral blood following IV GD2-CAR-T cell infusions, whereas chemokine/cytokine levels (such as CCL2 and CXCL9) were more pronounced in CSF following ICV CAR-T cell infusions [45]. Single cell analysis of CSF cells showed after IV CAR-T cell infusion an increase in CSF of regulatory T cells and suppressive myeloid cell populations compared to baseline; these immune suppressive cells were reduced following ICV infusions [45]. Given the positive results observed in these patients, two new arms of this study were launched to better assess safety and activity and to define the recommended phase II dose for ICV delivery of GD2-CAR-T cells without or with lymphodepletion. Furthermore, two additional phase I clinical trials (NCT 04099717, ACTRN 1262000675729) are evaluating DMG and DIPG patients as well as patients with other CNS tumors.
A recent phase I study explored the activity of GD2-CAR-T cells augmented with constitutive IL-7 receptor for treatment of high-grade pediatric gliomas [46]. The study was originated from the observation that CAR-T cell efficacy for brain tumors is constrained by the immunosuppressive microenvironment present in these tumors and characterized also by the limited availability of immunostimulatory cytokines; to overcome this hostile microenvironment it was developed an engineered IL-7 receptor (IL-/R) promoting constitutive signaling through Stat5 pathway and enhancing CAR-T cell survival, proliferation and function in murine and in vitro models [47]. Thus, to enhance T cell activity against GD2-positive brain tumors, Lin and coworkers have modified GHD2-directed CAR-T cells by introducing a constitutively active IL-7R [46]. Using these novel CAR-T cells, 11 pediatric patients (4-18 years) with DMG H3K27M-mutated or other recurrent GD2-expressing CNS tumors have been enrolled in a phase I study (NCT 04099797): 3 patients received treatment with GD2-CAR-T cells and 8 patients with CR7-GD2-CAR-T cells at two doses (1x107 cells/m2; 3x107/m2); all patients received standard chemo-radiotherapy before CAR-T cell infusions and lymphodepletion with fludarabine and cyclophosphamide before CAR-T cell infusions [44]. The CR7-GD2-CAR-T cell cohort developed grade 1 tumor inflammation-associated neurotoxicity in 88% of cases; CRS of grade 1 was observed in 75% of cases treated with CR7-GS2-CAR-Tcells [46]. Most of the enrolled patients had a diagnosis of DNG H3K27M-mutated, and 2 patients had recurrent medulloblastoma: The three patients treated with GD2-CAR-T cells did not display any response; patients receiving CR7-GD2-CAR-T cells exhibited a transient improvement of baseline neurologic deficits, 2/8 had a partial response and 7/8 remained eligible for additional treatment cycles [46].
Only one clinical study explored the safety and the efficacy of GD2-directed CAR-T cells in glioblastoma patients. This study evaluated 4SCAR-GD2 CAR-T cells in 8 glioblastoma patients; 4SCAR-GD2 CAR-T cells were obtained through transfection of T cells with 4SCAR-GD2 lentiviral vector constructed with DNA sequences of GD2 scFv, CD28 transmembrane and cytoplasmic domains, co-stimulatory 4-1BB intracellular TRAF binding domain, the CD3z chain intracellular domain, and an inducible suicide caspase 9 gene [48]. Eight patients with diagnosis if relapsed glioblastoma received the intravenous infusion of 2.5x106 4SCAR-GD2 T cells per Kg of body weight after lymphodepletion with fludarabine and cyclophosphamide; three of these patients with evidence of progressive disease and indication for surgery, received also an intracavitary infusion of 1x105 CAR-T 4SCAR-GD2 T cells per Kg of body weight [46]. The CAR-T cell treatment was well tolerated; 4SCAR-T cells expanded in vivo for 1-3 weeks and persisted at low frequency in peripheral blood; four patients displayed a partial response, lasting for 3 to 24 months; three patients had progressive disease and one stable disease [46]. Immunohistochemical studies showed that 4SCAR-T cell infusions induced partial antigen loss and immune cell activation at the level of tumor microenvironment [48].
Preclinical studies have shown the feasibility of efficient generation of GD2-targeting from autologous lymphocytes of glioblastoma patients endowed with a powerful and specific antitumor response against match ed primary glioblastoma cells [49]. Studies in orthoptic xenograft models of human glioblastoma have shown a significantly enhanced antitumor activity when GD2-targeting CAR-T cells were administered in association with Nivolumab [50] or using CAR-T cells manufactured with a retroviral vector encoding an interleukin-15 transgene alongside the GD2-specifric CAR [51].
GD2 is highly expressed in neuroblastomas and the treatment of high-risk neuroblastomas with anti-GD2 antibody, G_-CSF, IL-2 and isotretinoin improves overall survival compared to standard treatment with isotretinoin [52]. A first clinical study (NCT 02761915) evaluated the response of 12 children with R/R neuroblastoma to treatment with escalating doses of second-generation GD2-CAR-T cells; no radiological responses were observed and only three patients displayed regression of soft tissues and bone marrow disease [53]. Del Bufalo and coworkers reported the evaluation of GD2-CART01bcells in relapsed or refractory high-risk neuroblastoma patients [54]. In an academic, phase I/II study 27 children with relapsed or refractory (12 refractory, 14 relapsed and 1 in complete response), highly pretreated high-risk neuroblastoma were treated with autologous, third-generation GD2-CAR-T cells, expressing the inducible caspase 9 suicide gene [54]. Three dose levels were tested in the phase I (3, 6, and 10x106 cells (Kg of body weight); cytokine release syndrome occurred in 74% of patients and was mild in 20/21 patients; only one patient required activation of the suicide gene to obtain control of toxic events [52]. Six weeks after infusion of GD2-CAR-T01 cells, 33% of patients achieved a complete response and with a median follow-up of 1.7 years, a complete response was maintained in 5 of 9 of these responding patients; 30% of patients had a partial response; 19% had stable disease and 19% were resistant to treatment [54]. Patients with a low disease burden had significantly lower survival than those with high disease burden: at 3 years, overall survival was 67% vs 0%, respectively [54]. 11 patients received additional infusions of GD2-CAR-T cells, with 3 complete responses (2 consolidations and 1 complete response in a patient relapsing after first treatment) and 3 partial responses [54].
A second study reported the response of 10 patients with R/R neuroblastoma with progressive disease to 4SCAR-GD2 T cells; after CAR-T cell treatment, 6 patients displayed stable disease at 6 months and 4 of them remained with stable disease at 1 year and alive after 3-4 years of follow-up; the median OS was 25 months [55].
A third study evaluated the safety and the antitumor activity of GD2-NKT cells in neuroblastoma patients. Natural killer T cells (NKT) are a rare subset of T lymphocytes that coexpress a TCRα/β and several markers associated with NK cells. NKT cells differ from conventional T cells, in that their TCRs are much more limited in diversity (invariant NKT cells). Vα24-invariant NKT cells have antitumor properties that can be enhanced by CARs. Thus, Heczey have performed a first-in-human phase I trial evaluating autologous NKT cells co-expressing a GD2-specific CAR with IL-15 (GD2-CAR15-NKT) in 12 children with neuroblastoma [56]. A first initial report on this study assessed the feasibility of GD2-CAR15-NKT cells administration to 3 patients with neuroblastoma treated at dose level 1 (3x106 cells/m2) showing a good safety and a promising antitumor activity [57]. This initial study was then extended to 12 patients on four dose levels: 3x106, 1x107, 3x107 and 1x108 cells/m2; 8 patients received a single dose and 4 patients two infusions [56]. The treatment was well tolerated, and no dose-limiting toxicities were observed; one patient displayed grade 2 CRS [56]. After the first infusion of GD2-CAR15-NKT cells five patients had PD, four SD and three a PR; after the second infusion of GD2-CAR15-NKT cells, two patients had PD, one PR and one CR, maintained for 12 months [56]. Some markers correlated with response in these patients: the frequency of CD62L+ NKTs in CAR-T cell products was higher in responders; hyperexpression of BTG1 (antiproliferation factor 1) correlated with hyporesponsiveness of GD2-CAR15-NKT cells [56].
Very interestingly, the results of long-term survival of the first patients treated with GD2-CAR-T cells were recently reported. In 2008, Pule and coworkers engineered Epstein-Barr virus (EBV)-specific CTLs to express a CAR directed to GD2. In individuals with neuroblastoma, EBV-specific CTLs expressing a GD2-CAR survive better than T lymphocytes activated with anti-CD3 monoclonal antibody expressing a GD2-CAR but lacking virus specificity [58]. Infusion of these CAR-T cells was safe and was associated with tumor regression or necrosis in half of cases [58]. Two reports have evaluated the long-term clinical and immunological consequences of these GD2-CAR-T cells of first generation in 19 neuroblastoma patients: 8 in remission at infusion time and 11 with active disease at infusion; 6 weeks after infusion, the 8 patients in remission remained in remission, while the 11 patients with active disease displayed 3 PD, 2 SD, 1 PR, 3 CR, 2 tumor necrosis; after a follow-up of 3-4 years, 4 patients in remission at infusion remained in remission, while 4 relapsed, and 2 CR and 9 relapsing patients were observed among patients with active disease at infusion time [59]. A recent study reported the final results of this study with a follow-up of 13-18 years [60]. Of the 11 patients with active disease at time of infusion, 3 patients had CR and one PR; of the 3 patients with CR, one subsequently relapsed and two had sustained responses, one for 8 years and the other for 18 years [58]. Of the 8 patients in remission at the time of infusion, 5 were disease-free at the time of the last follow-up comprised between 13 and 14 years after infusion [60]. 12 of the 19 patients died between 2 months and 7 years post-infusion, all due to relapsed neuroblastoma. The long-term results of this study are consistent with the observations made by Del Bufalo et al [54] that patients with low tumor burden had significantly longer survival than those with a higher disease burden following GD2-CAR-T cell therapy.
A recent study showed that GD2 was expressed in a part of medulloblastomas, particularly in SHH (sonic hedgehog) and non-WNT/non-SHH group 4 subtypes [61]. Ciccone and coworkers have explored GD2 expression in a group of 52 medulloblastoma patients a reported a GD2 positivity in 82 of the analyzed samples, with a marked interspecimen heterogeneity, with an evrage of 50±36% of GD2-positive cells; the molecular medulloblastoma subtypes more positive were SHH, G4 and G3, while the WNT subtype was less GD2-positive [62]. The sensitivity of medulloblastoma cells to GD2-CAR-T cells identical to those used in the study of neuroblastoma patients [54] was explored, showing high levels of cell killing [62]. Importantly, the EZH2 inhibitor Tazemetostat induced a clear increase of GD2 expression on the surface of medulloblastoma cells, associated with a concomitant increase ot their sensitivity to the cytotoxic effects of GD2-CAR-T cells [60]. GD2-CAR-T cells exerted a marked in vivo antitumor effect in xenograft medulloblastoma models, potentiated by pretreatment with Tazemetostat; intravenously injected GD2-CAR-T cells were able to infiltrate medulloblastoma tissue [62]. Finally, the suicide gene present in the CAR vector was able, when drug-activated to promote GD2-CAR-T cell eradication [62]. These preclinical studies have supported the development of a phase I/II clinical study (NCT 05298995) for the evaluation of the safety and therapeutic efficacy of GD2-CAR-T cells in high-risk medulloblastoma patients [62].
2.5. B7-H3
B7-H3 (CD276) is an immunomodulatory protein that has emerged as an attractive target for cancer immunotherapy for its low expression in normal tissues but high expression in many solid tumors. Particularly, B7-H3 is highly expressed in pediatric solid tumors, including neuroblastoma and in many brain tumors, including medulloblastoma, high-grade glioma, DMG, ependymoma and atypical teratoid rhabdoid tumor (ATRRT); importantly, in these tumors, B7-H3 expression is high and comparable for its level of expression to that observed for GD2 [63]. B7-H3 targeted CAR-T cells display potent antitumor activity in patient-derived orthoptic xenografts of pediatric brain tumors [63].
B7-H3 mRNA and protein are also overexpressed in glioblastoma relative to normal brain in all glioblastoma subtypes; particularly, high expression of B7-H3 was observed in >75% of glioblastomas [64]. Furthermore, in preclinical studies, B7-H3 appeared to be a suitable CAR-T target for glioblastoma [64,65].
A recent study reported the results of the first-in-human clinical study (Brain Child-03) includes three distinct arms: arm A including localized recurrent/refractory CNS tumors; arm B including metastatic recurrent/refractory CNS tumors; arm C, including patients with DIPG enrolled at any time after radiotherapy. A first report concerned an interim analysis in the context of arm C [66]. The trial involved repeated weekly fixed doses (1x107 cells) of intraventricular infusions of B7-H3 CAR-T cells to three patients with recurrent DIPG. The three DIPG patients of this study received 10, 12 or 18 infusions of B7-H3-CAR-T cells; two patients showed no response to treatment and one patient displayed a partial response with improvement of neurologic deficits; two patients showed an extended survival [66]. Patients exhibited correlative evidence of local immune activation and persistence of B7-H3-CAR-T cells in cerebrospinal fluid [66]. A more extensive report of the arm C of the BrainChild study 03 was recently presented at the International Symposium on Pediatric Neuro Oncology (Philadelphia, 2024), including 21 patients with DIPG treated with multiple intraventricular infusions of B7-H3 CAR-T cells; these patients received a median of 7 doses of CAR-T cells [66]. 11 patients were enrolled after progression and survived 9.7 months after initial CAR-T cell infusions; 10 patients were enrolled before progression and survived 16.9 months; 3 patients were alive 3 years from diagnosis and two of them are still on protocol therapy [67]. The most common adverse events headache, nausea/vomiting, and fever [67].
A preclinical study supported the potential therapeutic value of B7-H3 targeting in atypical teratoid/rhabdoid tumors (ATRT), typically occurring in the CNS of children under 3 years of age [68]. ATRTs highly express B7-H3 and their targeting by B7-H3-CAR-T cells in ATRT xenograft cerebral models resulted in a consistent antitumor activity [68]. These observations strongly support the development of clinical studies implying targeting of ATRT, with B7-H3 CAR-T cells [68].
Few studies have explored the safety and the antitumor activity of B7-H3 CAR-T cells in patients with glioblastoma and other high-grade gliomas. Tang et al. reported the case of a 56-year-old woman with recurrent glioblastoma treated with multiple intratumoral infusions of B7-H3 CAR-T cells and achieved a dramatic antitumor response lasting up to the first five cycles of CAR-T cell infusions; unfortunately, after these first five cycles, the patient displayed tumor recurrence [69]. The MAXIMUM Pharmaceuticals reported the results of the first 7 patients with high-grade glioma (5 glioblastoma and 2 DMG) treated with multiple intralumbar injections of 2x107 B7-H3-specific allogeneic universal CAR-T cells (B7H3 UCAR-T) [70]. The infusion of B7H3 UCAR-T cells was not associated with any toxic effect of grade 3 or higher and resulted in a significantly longer overall survival and a higher objective response rate than history data [70]. Zhang et al. reported the results of an additional phase I trial (NCT 05241392) involving 13 R/R glioblastoma patients treated with B7-H3-CAR-T cells (TX103), at three different dosage levels: 3pts at 3x107, 4pts at 6x107 and 6pts at 15x107 cells [71]. TX103 cells were administered biweekly, with five cycles as one course, intracavitary and/or intraventricularly through an Ommaya reservoir [71]. No dose-limiting toxicities were observed; some patients had grade 2 CRS; 2 patients displayed grade 3 neurologic events. At 12 months of follow-up, 83% of patients survived; mOS was 20.3 months; 2 of 3 patients from dose 2 level achieved a PR and a CR, respectively [71].
3. Conclusions
Studies carried out in these last least years with monospecific CAR-T cells targeting antigens specifically or preferentially expressed in tumor brain cells, such as tumor-specific mutation of EGFR (EGFRvIII) or GD2 or IL-13Rα2, have the capacity to target antigen-expressing tumor cells. However, CAR-T cells against single antigens are hampered by tumor brain cell heterogeneity, leading to immune escape and progression. These effects may be in part mitigated using CAR-T cells engineered with multitargeting capacity, as supported by the promising observations made by a pilot study using CAR-T cells targeting both ERGFRvIII and IL-13Rα2 in glioblastoma patients with recurrent disease.
The study of some pediatric tumors, such as relapsing/refractory neuroblastoma showed a good sensitivity to GD2-directed CAR-T cells, with a good potential therapeutic activity limited to patients with low tumor burden.
Furthermore, the study of GD2-apecific and B7-H3-specific CAR-t cells in pediatric MDSs showed a promising therapeutic activity limited to a minority of patients. At the moment, valuable strategies aiming to extend these responses to more patients remained undefined.
Author Contributions
GC and EP were involved in researching, writing and editing the manuscript. UT was involved in conceptualization, organization, researching and editing the manuscript. All authors have read and agree to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov 2013, 3, 388-398. [CrossRef]
- Roselli, E.; Faramand, R.; Davila, M.L. insight into next-generation CAR therapeutics: designing CAR T cells to improve clinical outcomes. J Clin Invest 2021, 131, e142030. [CrossRef]
- Ceja, M.A.; Khericha, M.; Harris, C.M.; Puig-Saus, C.; Chen Y.Y. CAR-T cell manufacturing: major process parameters and next-generation strategies. J Exp Med 2024, 221, e20230903. [CrossRef]
- Labanieh, L.; Mackall, C.I. CAR immune cells: design principles, resistance and the next generation. Nature 2023, 614, 635-648. [CrossRef]
- Savoldo, B.; Grover, N.; Dotti G. CAR T cells for hematological malignancies. J Clin Invest 2024, 134, e177160. [CrossRef]
- Khan, A.N.; Asija, S.; Pendhari, J.; Purwar R. CAR-T cell therapy in hematological malignancies: where are we now and are we heading for? Esur J Haematol 2024, 112, 112, 6-18.
- Albelda, S.M. CAR T cell therapy for patients with solid tumors: key lessons to learn and unlearn. Nat Rev Clin Oncol 2024, 21, 47-66.
- Amoròs-Perez, B.; Rivas-Pardo, B.; del Moral M.G.; Subiza, J.L.; Martinez-Naves, E. State of the art in CAR-T cell therapy for solid tumors: is there a sweeter future? Cells 2024, 13, 725. [CrossRef]
- Del Baldo, G.; Del Bufalo, F.; Pinacchio , C.; Carai, A.; Quintarelli, C.; De Angelis, B.; Merli, P.; Cacchione, A.; Locatelli, F.; Mastronuzzi A. The peculiar challenge of bringing CAR-T cells into the brain: perspective in the clinical application to the treatment of pediatric central nervous system tumors. Front Immunol 2023, 14, 1142597. [CrossRef]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang , X.; Chang, W.C.; Weng, L. Optimization of IL13Rα2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol Ther 2018, 26, 31-44.
- Liu, G.; Ying, H.; Zeng , G.; Wheelr, C.J.; Black, K.L.; Yu, J.S. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res 2004, 64, 4980-4986. [CrossRef]
- Ramezani, M.; Siami, S.; Rezaei, M.; Khazaei, S.; Sadeghi, M. An immunohistochemical study of HER2 expression in primary brain tumors. Biomedicine 2020, 10, 21-27. [CrossRef]
- Mineo, J.F.; Bordron, A.; Baroncini, M.; Maurage, C.A.; Ramirez, C.; Siminsky, R.M.; Brethou, C.; Hieu, P.D. Low HER2-expressing glioblastomas are often secondary to anaplastic transformation of low-grade glioma. J Neuroncol 2007, 85, 281-287. [CrossRef]
- Ahmed, N.; Salsman, S.V.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-sepcific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res 2010, 16: 474-485.
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Therapy 2010, 18: 843-851. [CrossRef]
- Ahmed, N.; Brawley, V.; Hedge, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol 2017, 3, 1094-1101.
- Vitanza, N.A.; Johnson, A.J.; Wilson, A.L.; Brown, C.; Yokoyama, J.K.; Kunkele, A.; Chang, C.A.; Rawlings-Rhea, S.; Huang, W.; Seidel, K.; et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: an interim analysis. Nat Med 2021, 27, 1544-1552. [CrossRef]
- Priceman, S.J.; Tilakawardane, D.; Jeang, B.; Aguilar, B.; Murad, J.P.; Park, A.K.; Chang, W.C.; Ostberg, J.R.; Neman, J.; Jandial, R.; et al. Regional delivery of chimeric antigen receptor-engineered T cells effectively targhets HER2+ breast cancer metastasis to the brain. Clin Cancer Res 2017, 24, 95-105.
- Li, X.; Zhao, L.; Li, W.; Gao, P.; Zhang, N. HER2-targeting CAR-T cells show highly efficient anti-tumor activity against glioblastoma both in vitro and in vivo. Genes & Immunity 2024, in press. [CrossRef]
- Shabaneh, T.B.; Stevens, A.R.; Stull, S.M.; Shimp, K.R.; Seaton, B.W.; Gad, E.A.; Jaeger-Ruckstuhl, C.A.; Simon, S.; Koehne, A.L.; Price, J.P.; et al. Systemically administered low-affinity HER2 CAR T cells mediate entitumor efficacy without toxicity. J Immunother Cancer 2024, 12, e008566.
- Yeo, A.T.; Shah, R.; Aliazis K.; Pal, R.; Xu, T.; Zhang, P.; Rawal, S.; Rose, C.M.; Varn, F.S.; Appleman, V.A.; et al. Driver mutations dictate the immunologic landscape and response to checkpoint immunotherapy of glioblastoma. Cancer Immunol Res 2023, 11, 629-645. [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.; Martinez-Lage M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directe CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Trans Med 2017, 9, eaaa0984.
- Goff, S.L.; Morgan, R.A.; Yang J.C.; Sherry R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.C.; Zheng, Z.; Xi, L.; et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother 2019, 42, 126-135. [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEWs circumvent antigen escape without detectable toxicity. Nat Biotechnol 2019, 37, 1049-1058.
- Choi, B.D.; Gerstner, E.R.; Frigault, M.J.; Leick, M.B.; Mount, C.W.; Balaj, L.; Nikiforow, S.; Carter, B.S.; Curry, W.T.; Gallagher, K.; et al. Intraventricular CARv3-TEAM-E T cells in recurrent glioblastoma. N Engl J Med 2024, 390, 1290-1298. [CrossRef]
- Tang, O.Y.; Tian, L.; Yoder, T.; Xu, R.; Kulikovskaya, I.; Gupta, M.; Malenhorst, J.J.; Lacey, S.F.; O’Rourke, D.M.; Binder, Z.A. PD1 expression in EGFRvIII-directed CAR T cell infusion product for glioblastoma is associated with clinical response. Front Immunol 2022, 13, 872756.
- Bagley, S.J.; Binder, Z.A.; Lamrani, L.; Marinari, E.; Desai, A.S.; Nasrallah, M.L.; Maloney, E.; Brem, S.; Lustig, R.A.; Kurtz, G.; et al. Repeated peripheral infusions of anti-EGFRvIII CAR T cells in combination with pembrolizumab show no efficacy in glioblastoma: a phase 1 trial. Nat Cancer 2024, 5, 517-531.
- Bagley, S.J.; Logun, M.; Fraietta, J.A.; Wang, X.; Desai, A.S.; Bagley, L.J.; Nabavizadeh, A.; Jarocha, D.; Martins, R.; Maloney, F.; et al. Intratechal bivalent CART cell targeting EGFR and IL13Rα2 in recurrent glioblastoma: phase 1 trial interim results. Nat Med 2024, 30, 1320-1329.
- Abbott, R.C.; Iliopoulos, M.; Watson, K.A.; Arcucci, V.; Go, M.; Hughes-Parry, H.E.; Smith, P.; Call, M.J.; Cross, R.S.; Jenkins, M.R. Human EGFRvIII chimeric antigen receptor T cells demonstrate favorable safety profile and curative resonses in orthoptic glioblastoma. Clin Transl Immunol 2023, e1440.
- Joshi, B.H.; Plautz, G.E.; Puri, R.K. Interleukin-13 receptor α chain: a novel tumor-associated transmembrane protein in primary explants of human malignant gliomas. Cancer Res 2020, 60: 1168-1172.
- Knudson, K.M.; Hwang, S.; McCann, M.S.; Joshi, B.H.; Husain, S.R. Recent advances in IL-13Rα2-directed cancer immunotherapy. Front Immunol 2022, 13, 878365.
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res 2015, 21, 4062-4070.
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med 2016, 375, 2561-2569. [CrossRef]
- Alizadeh, D.; Wong, R.A.; Gholamin, S.; Maka, M.; Aftabizadeh, M.; Xang, W.; Pecoraro, J.R.; Jeppson, J.D.; Wang, D.; Aguilar, B.; et al. IFNγ is critical for CAR T cell-mediated myeloid activation and induction of endogenous immunity. Cancer Discov 2021; 11, 2248-2265.
- Brown, C.E.; Hibbard, J.C.; Alidazeh, D.; Blanchard, M.S.; Natri, H.M.; Wang, D.; Ostberg, J.R.; Aguilar, B.; Wagner, J.R.; Paul, J.A.; et al. Locoregional delivery of IL-13Rα2-targeting CAR-T cells in recurrent high-grade glioma: a phase 1 trial. Nat Med 2024, 30, 1001-1012.
- Brown, C.E.; Rodriguez, A.; Palmer, J.; Ostberg, J.R.; Naranjo, A.; Wagner, J.R.; Aguilar, B.; Starr, R.; Weng, L.; Synold, T.W.; et al. Off-the-shelf, steroid-resistant, IL13Rα2-specific CAR T cells for treatment of glioblastoma. Neuro-Oncol 2022, 24, 1318-1330.
- Stern, L.A.; Gholamin, S.; Moraga, I.; Yang, X.; Saravanakumar, S.; Cohen, J.R.; Cohen, J.R.; Starr, R.; Aguilar, B.; Salvary, V.; et al. Engineered IL13 variants direct specificity of IL13Rα2-targeted CAR T cell therapy. Proc Natl Acad Sci USA 2022, 119, e2112006119.
- Kim, K.; Gwak, H.S.; Han, N.; Hong, E.K.; Choi, B.K.; Lee, S.; Choi, S.; Park, Y.H.; Seok, J.H.; Jeon, Y.; et al. Chimeric antigen receptor T cells with modified interleukin-13 preferentially recognize IL13Rα2 and suppress malignant glioma: a preclinical study. Fron Immunol 2021, 12, 715000.
- Leland, P.; Degheidy, H.; Lea, A.; Bauer, S.R.; Puri, R.K.; Joshi, B.H. Identification and characterization of novel CAR-T cells to target IL13Rα2 positive human glioma in vitro and in vivo. Clin Transl Med 2024, 14, e1664.
- Weller, M.; Wen, P.Y.; Chang, S.M.; Dirven, L.; Lim, M.; Monje, M. Glioma Nat Rev Dis Prim 2024, 10, 33.
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rierberg, S.P.; et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-H27M+ diffuse midline gliomas. Nat Med 2018, 24, 572-579. [CrossRef]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Brasan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for K3K227M-mutated diffuse midline gliomas. Nature 2022, 603, 934-941. [CrossRef]
- Majzner, R.G.; Mahdi, J.; Ramakrishna, S.; Patel, S.; Chinna Samy, H.; Yeom, K.; Schultz, L.; Barsan, V.; Richards, R.; Conjar, C.; et al. Major tumor regressions in H3K27M-mutated diffuse midline glioma (DNG) following sequential intravenous (IV) and intracerebroventricular (ICV) delivery of GD2-CAR T cells. Cancer Res 2022, 81(suppl. 7), CT001.
- Monje, M.; Mahdi, J.; Majzner, R.; Yeom, K.; Schultz, L.M.; Richards, R.M.; Barsan, V.; Song, K.W.; Kamens, J.; Baggott, K.; et al. Sequential intravenous and intracerebroventricular GD2-CAR T-cell tharapy for H3K27M-mutated diffuse midline gliomas. medRxIV 2024, in press.
- Ramakrishna, S.; Good, Z.; Desai, M.; Zamler, D.; Mancusi, R.; Mahdi, J.; Majzner, R.; Schulz, L.; Richards, R.; Kamen, J.; et al. Immune signatures of GD2 CAR T cell activity in H3K27M+ diffuse midline glioma patients. Cancer Res 2023, 23 (suppl.7), 959. [CrossRef]
- Lin, F.Y.; Stuckert, A.; Tat, C.; White, M.; Ruggieri, L.; Zhang, H.; Mehta, B.; Lapteva, N.; Mei, Z.; Major, A.; et al. Phase I trial of GD2.CART cells augmented with constitutive interleukin-7 receptor for treatment of high-grade pediatric CNS tumors. J Clin Oncol 2024, in press. [CrossRef]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Oarihar, R.; et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov 2017, 7, 1238-1247. [CrossRef]
- Liy, Z.; Zhou, J.; Yang, X.; Liu, Y.; Zou, C.; Lv, W.; Chen, C.; Cheng, K.K.; Chen, T.; Chang, L.J.; et aL. Safety and antitumor activity of GD2-specific 4SCAR-T cells in patients with glioblastoma. Mol Cancer 2023, 22, 3. [CrossRef]
- Chiavelli, C.; Prapa, M.; Rovesti, C.; Slingardi, M.; Neri, G.; Pugliese, G.; Trudu, L.; Dall’Ora, M.; Golinelli, G.; Griseni, G.; et al. Autologous anti-GD2 CART cells efficiently target primay human glioblastoma. NPJ Precis Oncol 2024, 8, 26.
- Gargett, T.; Ebert, L.M.; Truong, N.; Kollis, P.M.; Sedivakova, K.; Yu, W.; Yeo, E.; Wittver, N.L.; Gliddon, B.L.; Tea, M.N.; et al. GD2-targheting CAR-T cells enhanced by transgenic IL-15 expression are an effective and clinically feasible therapy for glioblastoma. Immunother Cancer 2022, 10, e005187.
- Zhang, G.; Zhao, Y., Liu, Z.; Liu, W.; Wu, H.; Wang, X.; Chen, Z. GD2 CAR-T cells in combination with Nivolumab exhibit enhanced antitumor efficacy. Transl Oncol 2023, 32, 101663. [CrossRef]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 antibody with GM-CSF, interleukemi-2, and isotretinoin for neuroblastoma. N Eng J Med 2010, 363, 1324-1334. [CrossRef]
- Straathof, K.; Flutter, B.; Wallace, R.; Jain, N.; Loka, T.; Depani, S.; Wright, G.; Thomas, S.; Cheung, G.W.K.; Gileadi, T.; et al. Antitumor activity without on-target off-tumor toxicity of GD2-chimeric antigen receptor T cells in pateints with neuroblastoma, Sci Transl Med 2020, 12, eabd6169.
- Del Bufalo, F.; De Angelis, B.; Cornana, I.; Del Baldo, G.; De Ioris, M.A.; Serra, A.; Mastronuzzi, A.; Cefalo, M.G.; Pagliara, D.; Amicucci, M.; et al. GD2-CART001 for relapsed or refractory high-risk neuroblastoma. N Engl J Med 2023, 388, 1284-1295. [CrossRef]
- Yu, L.; Huang, L.; Lin, D.; Lai, X.; Wu, L.; Liao, X.; Liu, J.; Zeng, Y.; Liang, L.; Zhang, G.; et al. GD2-specific chimeric antigen receptor-modified T cells for the treatment of refractory and/or recurrent neuroblastoma in pediatric patients. J Cancer Res Clin Oncol 2022, 148, 2643-2652. [CrossRef]
- Heczey, A.; Xu, X.; Courtney, A.N.; Tian, G.; Barragan, G.A.; Guo, L.; Amador, C.M.; Ghatwai, N.; Rathi, P.; Wood, M.S.; et al. Anti-GD2 CAR-NKT celsl in erlapsed or refractory neuroblastoma: updated phase 1 trial interin results. Nat Med 2023, 29, 1379-1388.
- Heczey, A.; Courtney, A.N.; Montalbano, A.; Robinson, S.; Liu, K.; Li, M.; Ghatway, N.; Dakhova, O.; Liu, B.; Raveh-Sadka, T.; et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: an interin analysis. Nat Med 2020, 26, 1686-1690.
- Pule, M.A.; Savoldo, B.; Meyrs, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 2008, 14, 1264-1270. [CrossRef]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050-6056. [CrossRef]
- Che-Hsing, L.; Sharma, S.; Heczey, A.A.; Steffin, D.; Louis. C.U.; Grilley, B.J.; Thakkar, S.G.; Wu, M.; Wang, T.; Rooney, C.M.; et al. Eighteen-year survival after GD2-direcetd chimeric antigen receptor-modified immune effector cell treatment for neuroblastoma. Res Sq 2024, rs.3.ers-4232549.
- Paret, C.; Ustjanzew, A.; Ersali, S.; Seidmann, L.; Jennemann, R.; Ziegler, N.; El Malki, K.; Russo, A.; Wingerter, A.; Ortmuller, F.; et al. GHD2 expression in medulloblastoma and neuroblastoma for personalized immunotherapy: a matter of subtype. Cancers 2022, 14, 6059.
- Ciccone, R.; Quintarelli, C.; Camera, A.; Pezzella, M.; Caruso, S.; Manni, S.; Ottaviani, A.; Guercio, M.; Del Bufalo, F.; Quadraccia, M.C.; et al. GD2-targeting CAR T-cell therapy for patients with GD2+ medulloblastoma. Clin Cancer Res 2024, 30, 2545-2557. [CrossRef]
- Haydar, D.; Houke, H.; Chiang, J.; Yi, Z.; Odé, Z.; Caldwell, K.; Zhu, X.; Mercer, K.S.; Stripay, J.L.; Shaw, T.I.; et al. Cell-surface antigen profiling of pediatric brain tumots: B7-H3 is consistently expressed and can be targeted via local or systemic CAR T-cell delivery. Neuro Oncology 2021, 23, 991-1101.
- Nehama, D.; Di Ianni, N.; Musio, S.; Du, H.; Patané, M.; Pollo, B.; Finocchiaro, G.; Park, J.; Dunn, D.E.; Edwards, D.S.; et al. B7-H3-redirected chimeric antigen receptor T cells target glioblastoma and neuropspheres. E BioMedicine 2019, 47, 33-43.
- Tang, X.; Zhao, S.; Zhang, Y.; Wang, Y.; Zhang, Z.; Yang, M.; Zhu, Y.; Zhang, G.; Guo, G.; Tong, A.; et al. B7-H3 as a novel CAR-T therapeutic target for glioblastoma. Mol Ther Oncolytics 2019, 14, 279-285. [CrossRef]
- Vitanza, N.A.; Wilson, A.L.; Huang, W.; Seidel, K.; Brown, C.; Gustafson, J.A.; Yokoyama, J.K.; Johnson, A.J.; Baxter, B.A.; Koning, R.W.; et al. Intraventricular b7-H3 CART cells for diffuse intrinsic pontine glioma: preliminary first-in-human bioactivity and safety. Cancer Discov 2023, 13, 114-131.
- Vitanza, N.A.; Ronsley, R.; Huang, W.; Seidel, K.; Thomsen, A.; Gust, J.; Hauptman, J.; Choe, M.; Crotty, E.; Leary, S.; et al. Intravenous B7-H3 CAR T cells for diffuse intrinsic pontine glioma: safety and efficacy report from the completed phase 1 trial BrainChild-03. Neuro Oncol 2024, 26 (suppl.4), TRLS-01.
- Theruvath, J.; Sotillo, E.; Mount, C.W.; Graef, C.M.; Delaidelli, A.; Heitzeneder, S.; Labanieh, L.; Dhingra, S.; Leruste, A.; Majzner, R.G.; et al. Locoregional administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat Med 2020, 26, 712-719. [CrossRef]
- Tang, X.; Wang, Y.; Huang, J.; Zhang, Z.; Liu, F.; Xu, J.; Guo, G.; Wang, W.; Tong, A.; Zhou, L. Administration of B7-H3-targeted chimeric antigen receptor-T cells induce regression of glioblastoma. Signal Transd Targetes Ther 2021, 6, 125. [CrossRef]
- Shang, X.; T-MAXIMUM Pharmaceutical. An exploratory clinical trial on intra-lumbar injection of B7-H3-specific allogeneic universal CAR-T cells in patients with recurrent high-grade gliomas. J Clin Oncol 2023, 141(suppl. 16), 2043.
- Zhang, Y.; Feng, R.; Chi, X.; Xian, N.; Chen, X.; Huang, N.; Zhang, Y.; Zhang, K.; Zhang, J.; Chen, L.; et al. Safety and efficacy of B7-H3 targeting CAR-T cell therapy for patients with recurrent GBM. J Clin Oncol 2024, 142 (suppl. 16), 2062. [CrossRef]
Figure 1.
Structure of different CAR generations. The core structure of a CAR involving components of the extracellular domain, the transmembrane domain and the intracellular domain. Evolution of CAR structure involves the passage from first generation CARs with only a signaling motif in the intracellular domain to second generation CARs containing one co-stimulatory molecule, to third generation with two co-stimulatory molecules and to the fourth generation with a cytokine inducer in addition to two co-stimulatory molecules.
Figure 1.
Structure of different CAR generations. The core structure of a CAR involving components of the extracellular domain, the transmembrane domain and the intracellular domain. Evolution of CAR structure involves the passage from first generation CARs with only a signaling motif in the intracellular domain to second generation CARs containing one co-stimulatory molecule, to third generation with two co-stimulatory molecules and to the fourth generation with a cytokine inducer in addition to two co-stimulatory molecules.
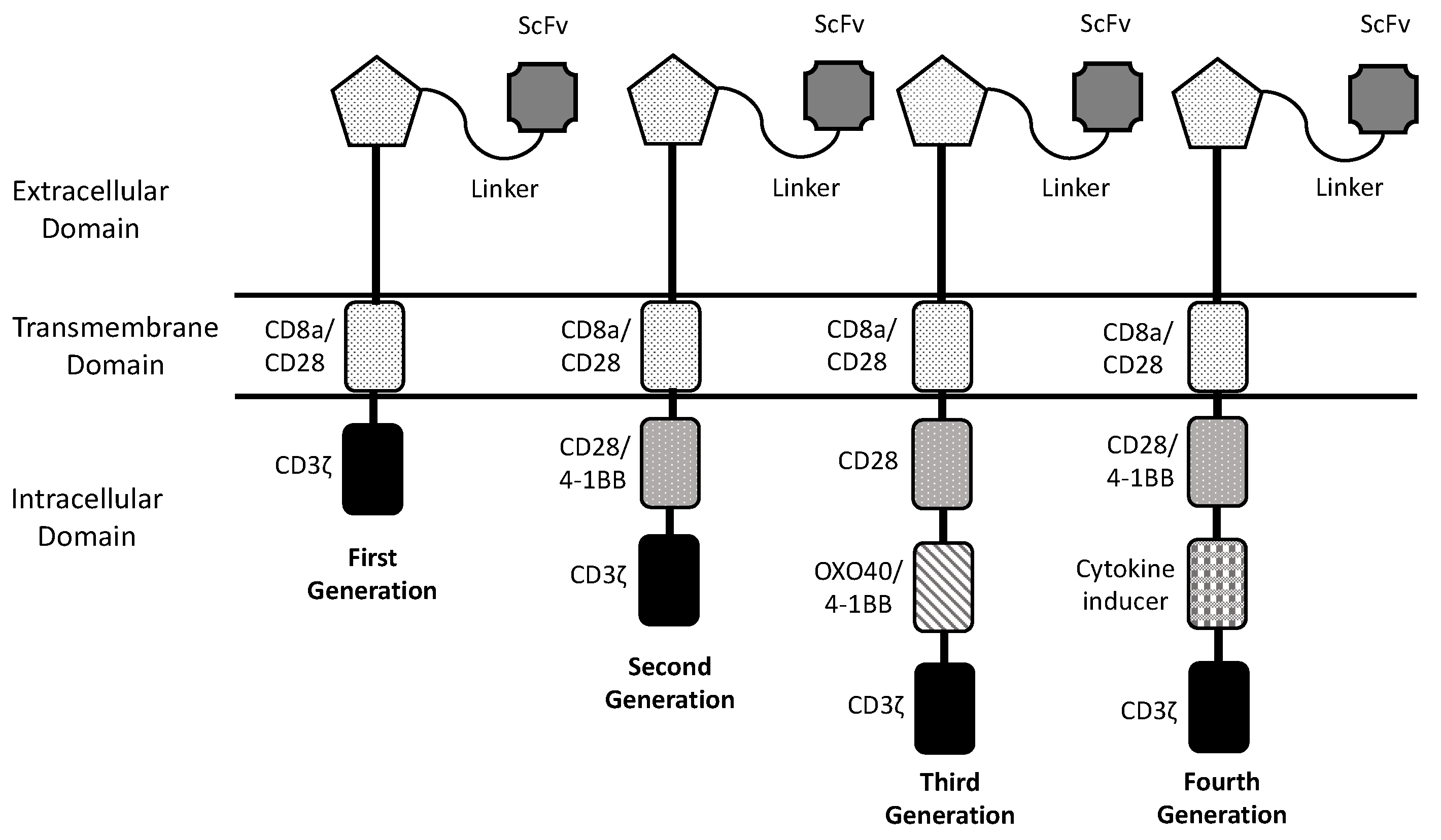

Table 1.
Main clinical trials involving CAR-T cells in adult and pediatric nervous tissue tumors.
| Author | Trial and Phase | Target Antigen | Dose and Route of Administration |
Number of Patients (Age) |
Clinical Results | Adverse Events |
|---|---|---|---|---|---|---|
| Ahmed et al. 2017 | NCT 01109059 Phase I |
HER2 | 1x106 to 1x108 Intravenous without prior lymphodepletion |
Pediatric 7 (10-17 yr) Adult 10 (30-69 yr) Recurrent GBM |
1 partial response; 7 stable disease; 8 disease progression In adult pts: mOS 9.4 mo |
No severe adverse events related to treatment |
| O’Rourke et al. 2017 | NCT 02209376 Phase I |
EGFRvIII | 1.75x108 to 5x108 Intravenous after lymphodepletion |
11 Adult recurrent GBM |
1 SD; 10 no response Evidence of CAR-T cell trafficking to the tumor Reduction of target antigen |
No off-tumor toxicity No CRS |
| Goff et al. 2019 | NCT 01454596 Phase I |
EGFRvIII | 4pts 1x107 ; 3pts 1x108 5pts 1x109 ; 5pts 1x1010 Intravenous after lymphodepletion |
18 Adult recurrent GBM |
No objective response mPFS: 1.2 mo mOS: 6.9 mo |
Severe adverse events and dose-limiting toxicities in the group at 1x1010 CAR-T cells |
| Choi et al. 2024 | NCT 05660369 Phase I/Pilot |
EGFRvIII | Patient 1: 2 infusions of 10x106 CAR-T cells Patients 2 and 3: 1 infusion of 10x106 CAR-T cells |
3 Adult recurrent GBM |
All patients displayed rapid and dramatic radiographic tumor regression. In 2/3 pts, this response was transient | No dose-limiting toxicity No adverse events greater than 3 |
| Bagley et al. 2024 | NCT 03726515 Phase I |
EGFRvIII | 4.65x107 to 2x108 1-3 cycles of CAR-T cells 1 cycle Pembrolizumab |
7 (56-76 yr) Adult recurrent GBM |
No objective responses mPFS: 5.2 mo mOD: 11.8 mo |
No dose-limiting toxicity |
| Bagley et al. 2024 | NCT 05168423 Phase I |
EGFRvIII - IL-13Rα2 (bicistronic lentiviral vector) |
3 pts (1x107 cells/m2) 3 pts (2.5x107 cells/m2) Intrathecal |
6 (33-71 yr) Adult recurrent GBM |
Significant reduction of tumor size at MRI, but none with radiographic objective response | Low-grade CRS Early moderate-severe neurotoxicity |
| Brown et al. 2024 | NCT 02208362 Phase I |
IL-13Rα2 | 57pts From 2 to 200x106 IL13-CAR-T cells ICT or ICV or ICT and ICV infusions |
57pts (16-71 yr) 41pts GBM 2pts DMG 7pts gr4 Astrocytoma 7pts gr3 Glioma |
SD: 50% PR: 2pts CR: 2pts mOS: 7.7 mo GBM(all) mOS: 10.2 mo GBM(ICT and ICV) |
No-dose limiting toxicity 2pts grade3 neurologic events |
| Majzner et al. 2022 | NCT 04196413 Phase I |
GD2 | IV (1x106 cells/Kg) Optional ICV infusions in responding patients |
4 (5-25 yr) DMG H3K2M7-mutated |
75% patients clinical and radiographic response | Manageable toxicity On target, off-tumor toxicity not observed |
| Majzner et al. 2022 | NCT 04196413 Phase I |
GD2 | 4 pts IV (1x106 cells/Kg) 9 pts IV (3x106 cells/Kg) |
13 (2-30 yr) DMG H3K2M7-mutated |
90% pts clinical and radiographic response. 2 pts with complete response | Grade 4 CRS in 3 pts at 3x106 cells/Kg. Transient tumor inflammation neurotoxicity |
| Lin et al. 2024 | NCT 04099797 | GD2 – IL-7R | 3pts: IV GD2-CAR-T cells 1x107 cells/m2 Ppts: IV CR7-GD2-CAR-T cells 1x107 cells/m2 |
11 (4-18 yr) 8 DMG H3K27M-mut 2 recurrent MB 1 ATRT (atypical teratoid rhabdoid tumor) |
3 pts: GD2-CAR-T group: PD 8pts CR7-GD2-CAR-T group: 2pts PR; 6pts SD |
CRS grade 1 75% Tumor inflammation-associated toxicity grade 1 88% |
| Liu et al. 2023 | NCT 03170141 Phase I |
GD2 | 8pts IV: 3x107 - 2.1x108 3pts IC: 2.6x106 - 6.4x106 4SCAR-T cells |
8 (3-63 yr) Recurrent GBM |
3pts: PD; 4pts: PR; 1pt: SD mOS from diagnosis: 19.7 mo; mOS from infusions: 11.5 mo |
Grade 2 or 3 neurologic events: 2pts |
| Del Bufalo et al. 2023 | NCT 03373097 Phase I/II |
GD2 | 17pts single infusion; 11pts multiple infusions 3x106 , 6x106,10x106 GD2-CART01 cells/Kg |
27 (2.7-18.6 yr) Refractory/relapsed Neuroblastoma |
CR: 33%; PR: 30%; SD: 19%; NR: 19% At 3yr: OS 67% LDB vs 0% HDB; EFS 58% LDB vs 0% HDB |
No-dose limiting toxicities Grade 1-2 CRS: 70% Grade 3 CRS: 4% Neurologic toxicity grade 1-2: 22% |
| Heczey et al. 2023 | NCT 03294954 Phase I |
GD2 | 3pts 3x106, 3pts 1x107, 3pta 3x107, 3pts 1x108 GD2-CAR-NKT cells/m2 8pts single infusion, 4pts double infusion |
12 (2-12 yr) Refractory/relapsed Neuroblastoma |
After first infusion: PR: 3pta; SD: 4pts; PD: 5pts After second infusion: CR: 1pt; PR: 1pt; PD: 2pts. |
|
| Pule et al. 2008 Louise et al. 2011 Che-Hsing et al. 2024 |
NCT 00085930 Phase I |
GD2 | 19pts IV infusion 1.2x107 cells/m2 5x107 cells/m2 3.1x108 cells/m2 |
19pts with R/R Neuroblastoma 11 with active disease 8 in remission |
After a follow-up of 8-14 years survived: 5/8 in remission 2/11 with active disease |
No-dose limiting toxicities |
| Brown et al. 2024 | NCT 02208362 Phase I |
IL-13Rα2 | From 2x106 to 200x106 IL13-CAR-T cells ICT or ICV or ICT and ICV infusions |
57pts 41pts GBM 2pta DMG 7pts Astrocytoma 7pts Glioma |
SD: 50% PR: 2pts CR: 2pts mOS: 7.7mo (GBM); 10.2mo (GBM ICT and ICV) |
No-dose limiting toxicity 2pts with grade 3 neurologic events |
| Vitanza et al. 2024 | NCT 04185038 Phase I Brain Child 03- Arm C |
B7-H3 | ICV infusions of 10x107 B7-H3 CAR-T cells Multiple infusions (median 7) |
21 pediatric DIPG | mOS for pts after progression: 9.7mo, before progression: 16.9mo 3pts alive 3yrs from diagnosis |
No-dose limiting toxicity Common neurologic events: headache, nausea, vomiting, fever |
| Zhang et al. 2024 | NCT 05241392 Phase I |
B7-H3 | ICT or ICV infusions of B7-H3 CAR-T cells 3x107 cells (3pts) 6x107 cells (4pts) 15x107 cells (6pts) |
13 adult R/R GBM patients | At 12mo: 83% OS mOS: 20.3mo 1pt: PR 1pt: CR |
No-dose limiting toxicity 2pts neurologic events gr.3 some pts CRS gr2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



