
Submitted:
19 July 2024
Posted:
22 July 2024
You are already at the latest version
Abstract
Due to its application as an anti-cancer drug, pazopanib (1) has attracted the interest of many researchers and several studies on pazopanib synthesis have been reported over the years. This paper provides a novel route for synthesizing N,2,3-trimethyl-2H-indazol-6-amine (5) which is a crucial building block in the synthesis of pazopanib from 3-methyl-6-nitro-1H-indazole (6). By alternating between the reduction and two methylation steps compound 5 was obtained in comparable yield (55.0%) to what has been reported (54%). Noteworthy, the last step of N2-methylation also yielded N,N,2,3-tetramethyl-2H-indazol-6-amine (5’) as a new compound ever reported. Furthermore, the data presented in this paper can serve as instrumental for future research aimed at further refining the synthesis process of pazopanib.
Keywords:
-methyl-6-nitro-1H-indazole
; pazopanib
; N
; 2
; 3-tetramethyl-2H-indazol-6-amine
; 3-trimethyl-2H-indazol-6-amine
1. Introduction
Pazopanib hydrochloride, marketed under the brand name VotrientTM, is an oral anti-cancer drug that inhibits the activity of various kinase enzymes. In 2009, it was approved by the United States Food and Drug Administration (FDA) for the treatment of soft tissue sarcoma progression and renal cell carcinoma [1]. The drug works by blocking vascular endothelial growth factor receptors (VEGFR-1, -2, and-3), and also platelet endothelial growth factor receptors (PDGFR–α and -β) which leads to the inhibition of angiogenesis and subsequent tumour growth [2].
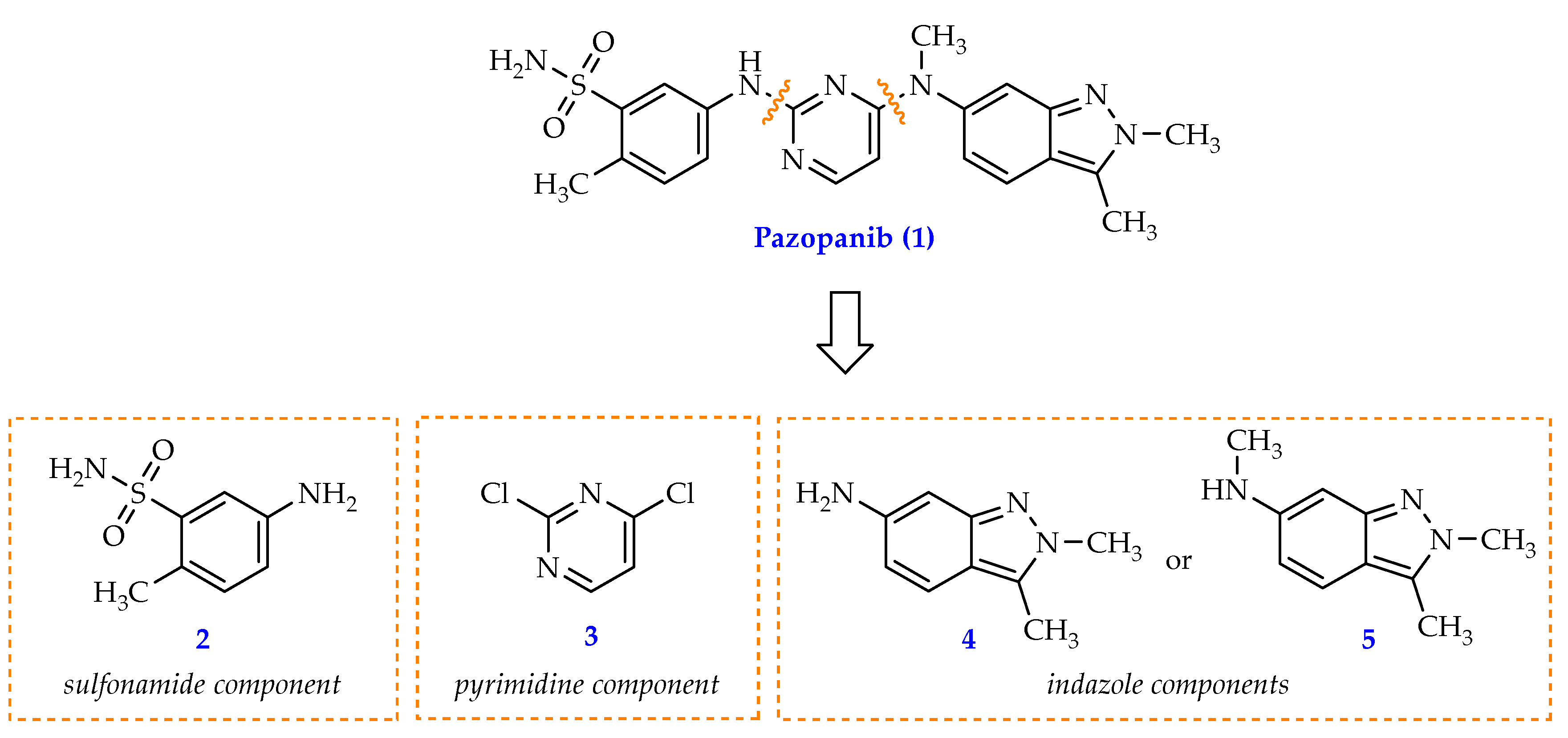
Pazopanib molecule can be divided into three parts, featuring as a sulfonamide, a pyrimidine, and an indazole component (Figure 1). These fragments can be separately synthesised and modified before being combined to offer the target compound (N,2,3-trimethyl-2H-indazol-6-amine, 5). In particular, the indazole component bears structural similarities to the adenine component of ATP, which enables the formation of hydrogen bonds within the tyrosine-kinase receptor’s binding pockets. As a result, this prevents ATP from binding to the receptor, and results in the inactivation of ATP-related intracellular reactions [3,4]. Due to its importance in expressing biological activity of pazopanib, the indazole fragment has received much attention with its synthesis having been extensively studied [5,6,7,8]
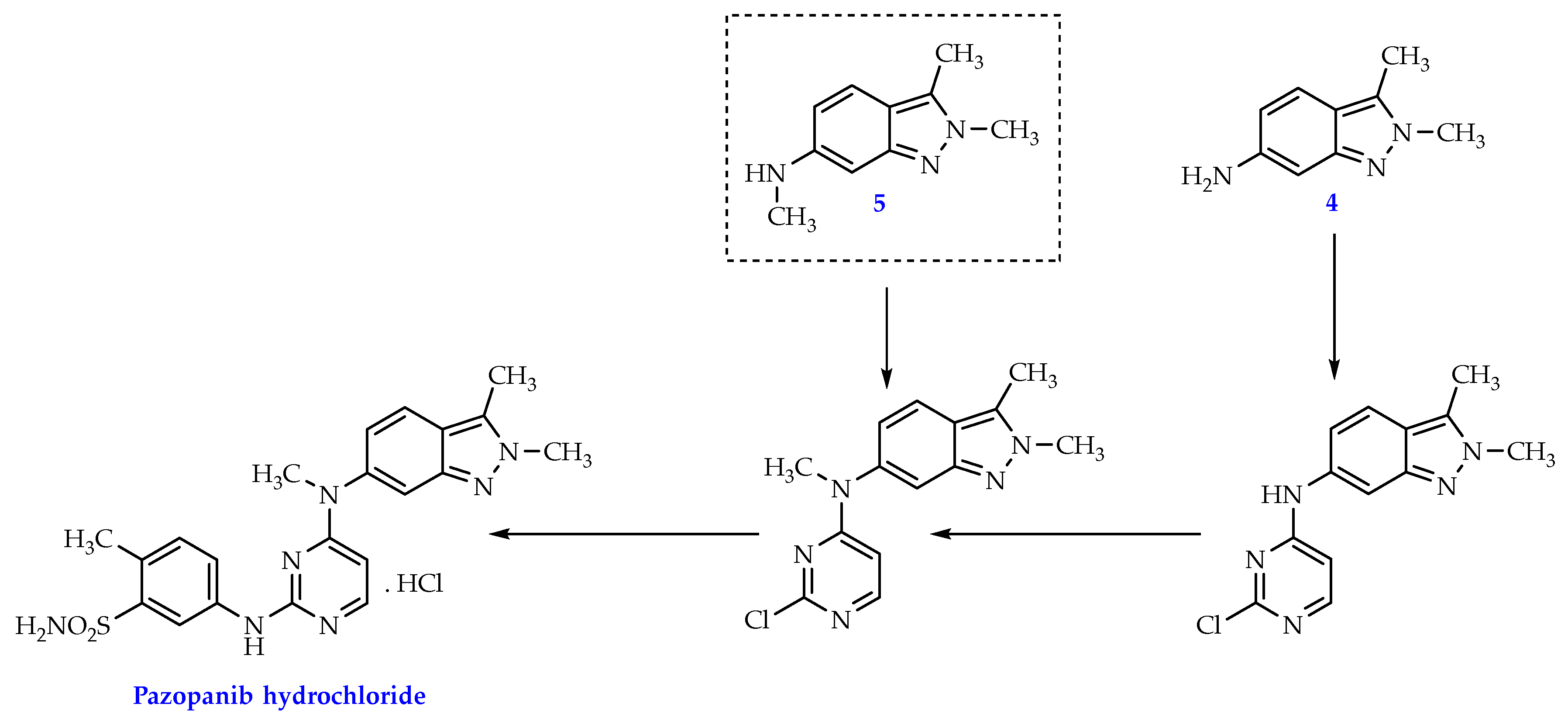
The synthesis of pazopanib can start from either indazole derivative 4 or 5 (Scheme 1) [5,6,7,8]. While compound 4 has the inherent instability of a free aniline and usually required a conversion to a stable HCl salt form, compound 5 posed to be a better candidate as it’s not only more stable but also doesn’t entail an extra N-methylation at the aniline group like compound 4 does (Scheme 1).
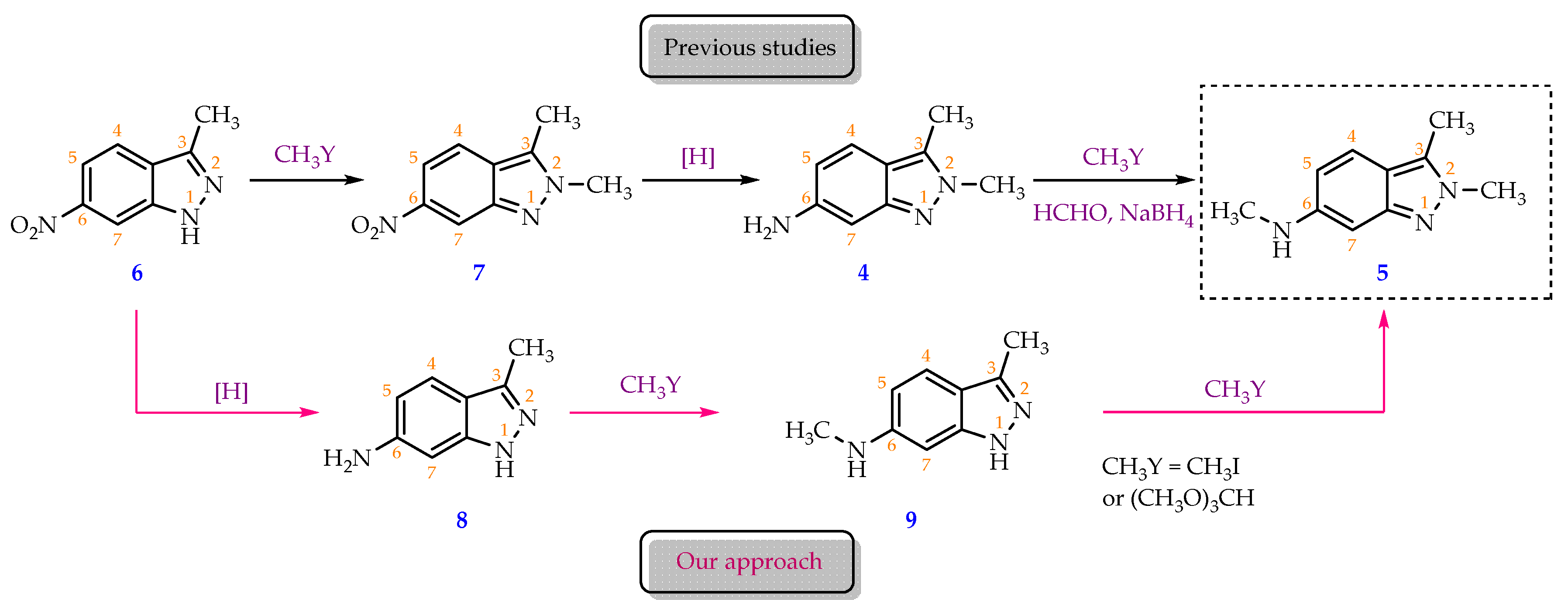
Theoretically, there are two possible routes to synthesise compound 5 from compound 6. The first one involved a selective N2-methylation on the indazole ring, followed by the reduction of nitro group and completed by an Eschweiler–Clarke methylation at the amine group (Scheme 2). This has been reported by Mei et al., who cleverly combined the last two steps in one synthetic sequence to afford compound 5 with the overall yield around 54% [6]. In this synthetic route, while the combined nitro reduction and methylation yielded 85.8%, the N2-methylation’s yield was limited at 63% which was comparable to previously reported data [5]. This was due to the intrinsic tautomerism of the indazole ring in which methylating reagent can generate either N1- or N2-methylation product [6,8].
The second route is our proposal which has not been reported elsewhere. We proposed to have the nitro reduction completed before the methylations at –NH2 and N2 positions, respectively (Scheme 2). Products at each step were purified and characterized by IR, NMR, MS, TLC and melting points which can serve as a handy database for research on synthesising pazopanib derivatives.
2. Results and Discussion
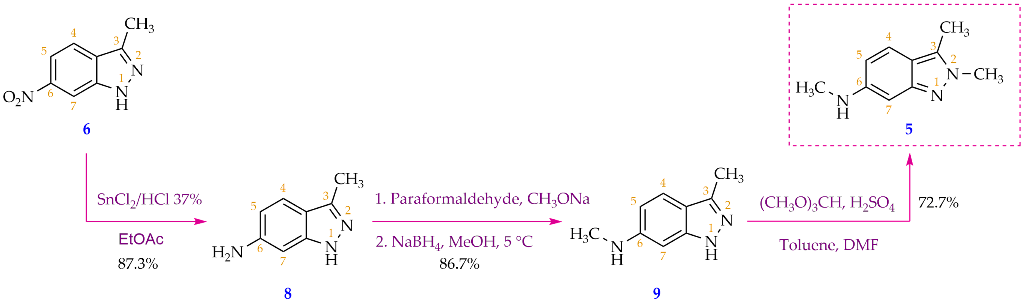
The target compound 5 was prepared in three separate steps from 3-methyl-6-nitro-1H-indazole (6), with an overall yield of 55.0%. Reaction conditions were modified from related references [5,7] (Scheme 3).
2.1. Process for 3-methyl-1H-indazol-6-amine (8)
The first step was the reduction of C6-nitro group by tin chloride in concentrated HCl. Due to its low cost and simpler handling, ethyl acetate (EA)was used as the sole solvent for both reaction and extraction instead of a combination of 2-methoxyethyl ether and diethyl ether as previously reported [5]. In the acidic condition, the freshly formed aniline group is readily protonated to result in a stable and water soluble HCl salt. Adjusting the pH to 9 by saturated Na2CO3 solution neutralized the reaction acidity and returned the free aniline which was extracted in EA layer leaving other by-products (SnO, Sn(OH)2, and NaCl) in the aqueous layer. To obtain the pure product without column chromatography, the extracted product in EA was further reacted with HCl solution to get the product back into aqueous HCl solution which was then separated from EA. Adjusting the pH of the resulting aqueous solution to basic range facilitated the precipitation of a pure product in aniline form which was filtered off and dried as a yellow crystalline solid (87.3%). In this nitro reduction step by SnCl2/HCl (conc), the molar ratio between SnCl2 and compound 6 was determined to be beneficial at 4:1 due to low chemical consumption and comparable efficiency when compared with other ratios examined (Table 1). Similar nitro reduction has been reported with the yield up to 92% [5], however, the product was isolated as a HCl salt, not a free aniline product as observed in our experiment.
2.2. Process for N,3-dimethyl-1H-indazol-6-amine (9) via Reductive Amination
The second step was a reductive amination between the aniline functional group (compound 8) and formaldehyde which resulted in compound 9 yielding at 86.7%. This reaction was performed in a basic condition due to the use of a weak organic or inorganic base as the catalyst (Table 2). We again adopted the purification method used in the synthesis of compound 8. We first protonated the product into a HCl salt which was selectively extracted into aqueous solution and leaving other organic matter in EA layer. This aqueous product solution was then separated, and pH adjusted to 9 (by Na2CO3) to neutralize the HCl salt facilitating the precipitation of the pure secondary amine product. This purification technique required no column chromatography as previously reported whilst significantly enhancing the yield of synthesizing compound 9 from 39% [7] to 86.7%.
In this synthesis of compound 9, we trialed several base catalysts and the results were presented in Table 2. Replacing the inorganic bases such as K2CO3 and Na2CO3 by an organic base such as potassium tert-butoxide (t-BuOK) and CH3ONa resulted in better efficiency and shorter reaction time (Table 2). While both organic bases (t-BuOK and CH3ONa) enabled comparable yields (79.4% vs 86.7%, Table 2), CH3ONa stood out in terms of cost when scaling up as it can freshly be made from Na and methanol in our lab. Additionally, we explored the optimal molar ratio between compound 8 and NaBH4. The shortest reaction time and highest efficiency were achieved with a molar ratio of 1:4 between compound 8 and the reducing agent NaBH4 (Table 3).
2.3. Process for N,2,3-trimethyl-2H-indazol-6-amine (5)
The synthesis of target compound 5 from 9 has never been reported before. It involved a selective methylation at N2 position on compound 9. A closely related reaction was reported for compound 6 using trimethyl orthoformate (TMOF) as the methylating reagent [6]. This reaction generated a mixture of N1- and N2-methylated products due to the tautomerism of the NH group in the indazole ring. It was found that in the presence of concentrated sulfuric acid, this reaction favoured the N2-methylated product [6]. However, in our experiment we found that beside the target N2-methylated product, a new by-product 5’ was formed and no N1-methylated product (5’’) was isolated (Scheme 4). Compound 5’, having not been reported in the literature, was found to contain a tertiary amine functional group attached on C6 of the indazole ring. This indicated that the methylating reagent also attacked the secondary amine of compound 9. This could be explained by the potent alkylating ability of TMOF [9] which readily attacked both the NH in the indazole and the secondary amine (Scheme 5). Although the possibility of generating multiple methylated products has limited the yield of the target product 5 at 72.7%, this was more efficient than similar N-methylation of a simpler compound previously reported (compound 6, 63%, [6]). Of note, compounds 5 and 5’ displayed very close Rf values (Rf ~ 0.30 in n-hexane/ethyl acetate, 3:7, and 0.70 in dichloromethane/methanol, 9:1), which prompted us to use our in-house preparative chromatography to purify them before characterization (yield: 27.6% for 5, and 21.3% for 5’). In this N-methylation reaction, sticking to concentrated H2SO4 as the sole catalyst was important, as changing it to either polyphosphoric acid or a mixture of p-toluenesulfonic acid (PTSA) and concentrated H2SO4 both resulted in unknown products whose Rf values were different from that of the target compound 5. Moreover, choosing the right ratio between TMOF and the starting material 9 was crucial. Using the ratio (~6.2:1) reported for a similar methylation reaction [6] led to more new spots including the aforementioned compound 5’, while reducing it to less than (4:1) returned unreactive starting material. As such, we utilized the ratio of (4:1) which enabled both the reaction completion and a cleaner TLC which prioritised the formation of target compound 5 at 72.6% yield.
We also trialed CH3I as an alternative methylating reagent accompanied by various base catalysts (CH3ONa, t-BuOK or K2CO3). However, their reaction mixtures displayed more unknown new spots on TLC when compared with that of TMOF, while the starting material (9) was still detectable. As a result, TMOF was chosen as the methylating reagent for our synthesis.
A probable mechanism for the N-methylation reaction using TMOF is suggested in Scheme 5. In acid media, a TMOF molecule was protonated into a methoxonium intermediate possessing an electrophilic H3Cδ+ center. This center then received a nucleophilic attack from the lone pair of electrons located on N2 position of the indazole ring to afford the target compound 5, and at the same time releasing a methanol and a methyl formate molecules as good leaving groups. When using a large amount of TMOF vs starting material 9 (6.2:1), this methylation process was continued on the newly formed compound 5 whose nucleophilic center was the lone pair of electrons locating on the secondary amine NH- connected to C6-indazole to afford a novel compound 5’ ever reported before [8,10].
3. Materials and Methods
3.1. General Information
3-Methyl-6-nitro-1H-indazole (compound 6) was synthesized in the lab. Trimethyl orthoformate (98.0 %) was purchased from Aladdin Company (China). Potassium carbonate (K2CO3) (99.0 %) and paraformaldehyde (≥ 95.0 %) were purchased from Guangdong Guanghua Sci-Tech, China. Ethyl acetate (99.5%) was purchased from Samchun, Korea. Sodium borohydride (NaBH4) (98.0 %), n-hexane (≥ 98.0 %), methanol (MeOH) (99.5 %), isopropanol (IPA) (99.7 %), anhydrous sodium sulfate, toluene (99.5 %), tin(II) chloride (SnCl2.2H2O) 98.0 % and N,N-dimethylformamide (DMF) (99,5 %) meeting analytical reagent (AR) purity standards were obtained from Xilong (China). All chemicals were used without further purification.
The melting point (M.p) was measured by using the capillary tube method with an SRS EZ-Melt apparatus (Stanford Research Systems, Sunnyvale, CA, USA). Mass spectra of compounds were recorded on an LC-MSD Trap SL machine, ionized by ESI (electrospray ionization) method. The FT-IR spectrum was recorded by a Shimadzu spectrometer (Kyoto, Japan). Nuclear magnetic resonance (1H, 13C, HSQC, HMBC, NOESY) experiments were measured on a Bruker Ascend spectrometer (Billerica, MA, USA) at 500 MHz for proton and 125 MHz for carbon-13 using CDCl3 as the solvent and tetramethylsilane (TMS) as an internal standard. The reaction mixtures were monitored, and the purity of the compounds was checked by thin-layer chromatography (TLC) on silica gel 60 F254 plates (Merck, Darmstadt, Germany).
3.2. Synthetic Procedure
3-Methyl-1H-indazol-6-amine (8)
In a 100 mL round-bottom flask, 3-methyl-6-nitro-1H-indazole (6) (2.00 g, 11.3 mmol, 1 eq) was dispersed into ethyl acetate (10 mL) at 0 °C, then slowly added tin(II) chloride dihydrate (10.19 g, 45.2 mmol, 4 eq). This was followed by dropwise addition of concentrated HCl (1 mL) over 1 min, keeping the reaction temperature below 10 °C with an ice bath. The ice bath was taken out when HCl addition was finished, and the mixture was stirred for three more hours. After the reaction was completed (as shown by TLC), 50 milliliters of ethyl acetate was added and then the reaction mixture was cooled to 5 °C. Reaction mixture was then neutralized by saturated Na2CO3 solution to a pH of 9. The organic phase was separated, and two more fractions of ethyl acetate (2 x 50 mL) were sequentially added to carry out the extraction two more times. Following that, the organic phase was combined and rinsed three times with distilled water (3 x 20 mL). The organic phase was then transferred to a conical flask, in which 10 mL of a 20% hydrochloric acid solution was gradually added, and the mixture was shaken to facilitate the HCl salt formation which was soluble in the aqueous phase. The latter was separated and cooled to 5 °C and saturated Na2CO3 solution was added in small portions under stirring to pH= 9, which enabled the formation of a precipitate. The precipitate was collected by vacuum filtration, washed three times with distilled water (3 x 5 mL), and dried to obtain 3-methyl-1H-indazol-6-amine (8) as a yellow-brown solid (1.45 g, 87.3% yield). M.p: 203.8-204.5 °C. TLC: Rf = 0.45 (n-hexane/ethyl acetate, 3:7). MS (ESI, MeOH), m/z: Calculated for C8H9N3 [M+H]+: 148.08, found: 147.9. FT-IR (KBr), vmax (cm-1): 3382, 3180 (N-H); 1640 (C=N); 1520 (C=C); 1H-NMR (500MHz, CDCl3), δ (ppm): 9.44 (s, 1H, N-NH); 7.43 (d, J = 8.5 Hz, 1H, H-4); 6.57-6.55 (m, 2H, H-5, H- 7); 3.84 (s, 2H, NH2); 2.50 (s, 3H, H-1’).13C-NMR (125MHz, CDCl3), δ (ppm): 146.1 (C-6); 143.5; (C-7a); 142.9 (C- 3); 121.0 (C-4); 116.8 (C-3a); 111.9 (C-5); 92.4 (C-7); 11.9 (C-1’).
N,3-Dimethyl-1H-indazol-6-amine (9)
In a 100 ml single neck round-bottom flask, 3-methyl-1H-indazol-6-amine (8) (2.00 g, 13.6 mmol, 1 equivalent) was dissolved in 20 ml of methanol. Then, CH3ONa (3.67 g, 67.9 mmol, 5 eq) and paraformaldehyde (2.04 g, 67.9 mmol, 5 eq) were added to the reaction vessel, respectively. The mixture was then refluxed for 5 minutes, and stirred at room temperature for 4 hours. After that, the reaction was cooled to 5 °C, and NaBH4 (2.06 g, 54.4 mmol, 4 eq) was slowly added under stirring for 5 minutes. The reaction mixture was refluxed for approximately 2 hours, and the solvent was removed by rotary evaporation. The residue was redistributed in water and washed three times with ethyl acetate (3x50 mL). The ethyl acetate extracts were combined and rinsed three times with purified water (3 x 30 mL). 20% HCl solution was added dropwise to the combined ethyl acetate layer under stirring until pH = 1. Subsequently, the aqueous phase was separated using a decanter. After the aqueous solution was cooled to room temperature, saturated Na2CO3 solution was gradually added under stirring until pH = 9, which enabled the precipitation of the base product (9). The precipitate was filtered using a Buchner filter funnel, collected, and dried under an infrared lamp. Compound 9 was obtained as a pink-white solid (1.90 g, 86.7 % yield). M.p: 215.6-217.3 °C. TLC: Rf = 0.60 (n-hexane/ethyl acetate, 3:7). MS (ESI, MeOH), m/z: Calculated for C9H11N3 [M+H]+: 162.10, found: 161.8. FT-IR (KBr), vmax (cm-1): 3338, 3221 (N-H); 2974, 2939, 2900 (C-H sp3), 1624 (C=N); 1589, 1510 (C=C); 1H-NMR (500 MHz, CDCl3), δ (ppm): 7.42 (d, J = 8.50 Hz, 1H, H-4); 6.52 (dd, J1 = 8.75 Hz, J2 = 1.75 Hz, 1H, H- 5); 6.43 (d, J = 1.50 Hz, 1H, H-7); 2.90 (s, 3H, H-2’); 2.52 (s, 3H, H-1’).13C-NMR (125 MHz, CDCl3), δ (ppm): 149.1 (C-7a); 143.3 (C-3); 120.7 (C- 4); 115.7 (C-3a); 111.5 (C-5); 87.9 (C-7); 30.8 (C-2’); 11.9 (C-1’).
N,2,3-Trimethyl-2H-indazol-6-amine (5)
30 mL of toluene, 3 mL of N,N-dimethylformamide (DMF) and 5.4 mL of trimethyl orthoformate (49.6 mmol; 4 eq) were charged to a 50 mL round-bottom flask under stirring, 0.5 mL of 98 % H2SO4 solution (8.7 mmol; 0.7 eq) was then dropwise added to the mixture which was stirred at 5 °C for 5 minutes before 2.00 g of N,3-dimethyl-1H-indazol-6-amine (9) (12.4 mmol; 1 eq) was added into the reaction vessel. The reaction mixture was then heated and stirred at 60 °C for 5 hours. Solvent was removed under reduced pressure and a thick, dark pink residue was obtained. 100 mL of distilled water was then added to this residue to form a clear, wine-red solution. The solution was washed with ethyl acetate (3 times x 120 mL/time) and the aqueous phase was separated. Saturated Na2CO3 solution was slowly added to the aqueous phase until the pH = 8. The resulting aqueous solution was washed with ethyl acetate (3x120mL). The ethyl acetate fractions were combined and then washed with distilled water (5x70mL) to remove DMF and water-soluble impurities. Finally, the organic phase was dried with sodium sulfate and solvent was removed via rotary evaporator to obtain a yellow powder (1.58 g, 72.7 % yield). M.p: 144.2-145.8 °C. TLC: Rf = 0.30 (n-hexane/ethyl acetate, 3:7); 0.65 (dichloromethane/methanol, 9:1). MS (ESI, MeOH), m/z: Calculated for C10H13N3 [M+H]+: 176.11, found: 175.9. FT-IR (KBr), vmax (cm-1): 3422 (N-H); 2982, 2904 (C-H sp3), 1645 (C=N); 1560, 1513 (C=C); 1H-NMR (500 MHz, CDCl3), δ (ppm): 7.28 (d, J = 9.00 Hz, 1H, H-4); 6.52 (s, 1H, H-7); 6.45 (dd, J1 = 8.75 Hz, J2 = 1.75 Hz, 1H, H-5); 3.97 (s, 3H, H-2’); 2.86 (s, 3H, H-3’); 2.49 (s, 3H, H-1’). 13C-NMR (125 MHz, CDCl3) δ (ppm): 149.6 (C-7a); 147.8 (C-6); 131.3 (C- 3); 119.9 (C-4); 115.5 (C-3a); 114.8 (C-5); 91.7 (C-7); 36.7 (C-3’); 30.8 (C-2’); 9.8 (C-1’).
N,N,2,3-Tetratramethyl-2H-indazol-6-amine (5’)
When using a larger ratio between TMOF: starting material 9 as (6.2:1), beside the target compound 5 (27.6%), a novel compound 5’ was isolated with 21.3% of yield.
Data for compound 5’: TLC Rf = 0.30 (n-hexane/ethyl acetate, 3:7); 0.70 (dichloromethane/methanol, 9:1). MS (ESI, MeOH), m/z: Calculated for C11H15N3 [M+H]+: 190.1, found: 189.9. FT-IR (KBr), vmax (cm-1): 2926 (C-H sp3); 1650 (C=N); 1559 (C=C). 1H-NMR (500 MHz, DMSO-d6), δ (ppm): 7.43 (d, J = 9.50 Hz, 1H, H-4); 6.77 (dd, J1 = 9.5 Hz, J2 = 1.50 Hz, 1H, H-5); 6.45 (d, J = 1.50 Hz,1H, H-7); 3.91 (s, 3H, H-2’); 2.88 (s, 6H, H-3’, H-4’), 2.50 (s, 3H, H-1’). 13C-NMR (125 MHz, DMSO-d6) δ (ppm): 149.6 (C-7a); 148.9 (C-6); 131.2 (C- 3); 120.5 (C-4); 115.4 (C-3a); 113.2 (C-5); 94.9 (C-7); 41.4 (C-3’,4’); 37.1 (C-2’); 9.8 (C-1’).
4. Conclusions
In conclusion, we have successfully developed a novel alternative approach to convert 3-methyl-6-nitro-1H-indazole (6) into N,2,3-trimethyl-2H-indazol-6-amine (5), an important intermediate in the synthesis of pazopanib. This approach involved a nitro reduction (87.3%), a reductive amination (86.7%), and a N-methylation at N2 position on the indazole ring (72.7 %) yielding an overall yield of the target compound 5 at 55.0 %. This yield was comparable to what has been reported (54%). Of note, we have optimized the reaction conditions so that column chromatography can be negated, which makes the synthesis suitable for upscaling. In addition, we discovered a new structure 5’ ever reported due to the potent methylating capability of TMOF, which facilitated multiple methylation on various nucleophilic centers on the indazole derivative. Products in each step were characterized using melting point determination, mass spectrometry, FT-IR spectroscopic analysis, as well as 1H-NMR, 13C-NMR, HSQC, HMBC, NOESY spectroscopy whose data can be found in the SI.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Spectral data of the compound 8, 9, 5, and 5’ are available online.
Author Contributions
V.H.N., T.T.C.B., V.G.N., and D.L.N. designed the experiments; T.T.C.B., H.L.L., T.T.L., and N.S.H.D. synthesized the compound and performed the optimization; T.T.C.B., V.H.N., H.L.L., T.N.N. analyzed spectroscopic data; T.T.C.B. and V.H.N. wrote the original draft preparation; V.H.N., H.D.N, N.T.T. edited the manuscript. All authors discussed the results, read, and approved the final version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The authors would like to thank Thai Nguyen University of Medicine and Pharmacy for the financial support and research facilities.
Data Availability Statement
The data presented in this study are available in this article and Supplementary Materials.
Acknowledgments
The authors would like to thank Hanoi University of Pharmacy for the financial support and research facilities.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- European Medicines Agency (2012), EPAR summary for the public, Votrient, Procedure No. EMEA/H/C/001141.
- Kumar, R.; Knick, V.B.; Rudolph, S.K.; Johnson, J.H.; Crosby, R.M.; Crouthamel, M.C.; Hopper, T.M.; Miller, C.G.; Harrington, L.E.; Onori, J.A.; et al. Pharmacokinetic-pharmacodynamic correlation from mouse to human with pazopanib, a multikinase angiogenesis inhibitor with potent antitumor and antiangiogenic activity. Mol Cancer Ther 2007, 6, 2012–2021. [Google Scholar] [CrossRef]
- Keisner, S.V.; Shah, S.R. Pazopanib: the newest tyrosine kinase inhibitor for the treatment of advanced or metastatic renal cell carcinoma. Drugs 2011, 71, 443–454. [Google Scholar] [CrossRef]
- McCormack, P.L. Pazopanib: a review of its use in the management of advanced renal cell carcinoma. Drugs 2014, 74, 1111–1125. [Google Scholar] [CrossRef]
- Harris, P.A.; Boloor, A.; Cheung, M.; Kumar, R.; Crosby, R.M.; Davis-Ward, R.G.; Epperly, A.H.; Hinkle, K.W.; Hunter, R.N., 3rd; Johnson, J.H.; et al. Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J Med Chem 2008, 51, 4632–4640. [Google Scholar] [CrossRef]
- Mei, Y.C.; Yang, B.W.; Chen, W.; Huang, D.D.; Li, Y.; Deng, X.; Liu, B.M.; Wang, J.J.; Qian, H.; Huang, W.L. A novel practical synthesis of pazopanib: An anticancer drug. Lett. Org. Chem. 2012, 9, 276–279. [Google Scholar]
- Boloor, A.; Cheung, M.; Davis, R.; Harris, P.A.; Hinkle, K.; Mook, R.A. Jr.; Stafford, J.A.; Veal, J.M. Pyrimidineamines as angiogenesis modulators. US7105530B2. 2006.
- Mei, Y.C.; Yang, B.W. The regioselective alkylation of some indazoles using trialkyl orthoformate. Indian J. Heterocycl. Chem. 2017, 27, 275–280. [Google Scholar]
- Frizzo, C.P. , Alkyl Orthoformate: A versatile reagent in organic synthesis. Synlett., 2009, 2009, 1019–1020. [Google Scholar] [CrossRef]
- Kim, D.J.; Oh, K.H.; Park, J.K. A general and direct synthesis of imidazolium ionic liquids using orthoesters. Green Chemistry, 2014, 16, 4098–4101. [Google Scholar] [CrossRef]
Figure 1.
Structural components (2, 3, 4, 5) of pazopanib (1)
Scheme 1.
Synthesis diagram of pazopanib hydrochloride from compound 4 or 5.
Scheme 2.
Approaches for preparing compound 5 from 3-methyl-6-nitro-1H-indazole (6)
Scheme 3.
Synthetic method of compound 5 starting from compound 6
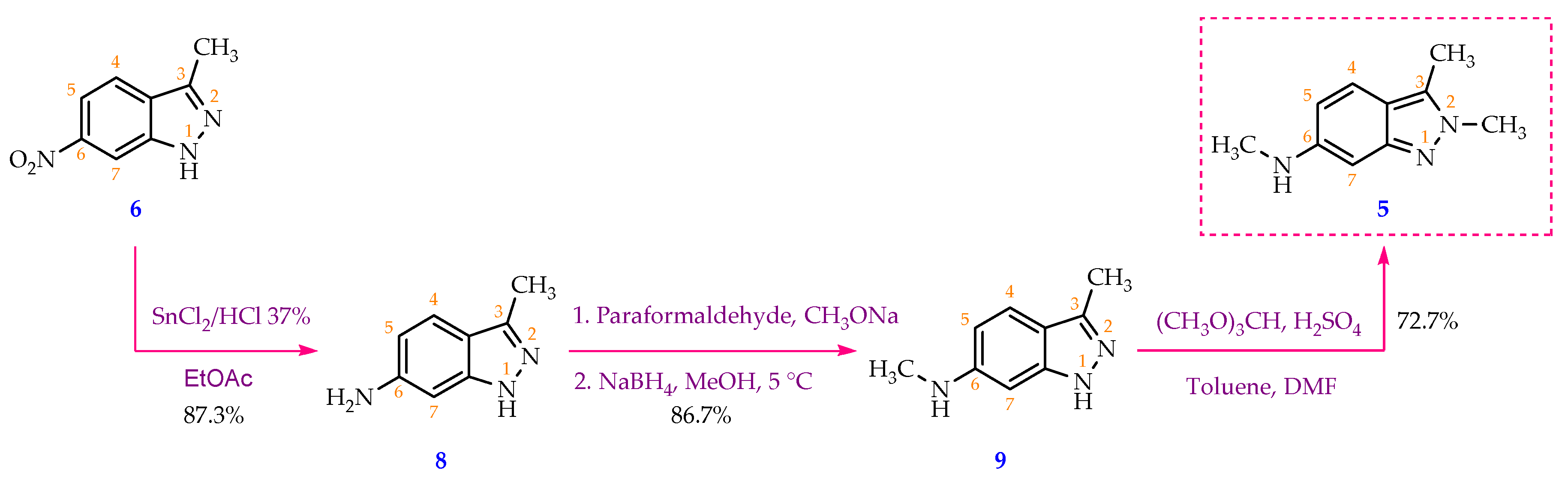
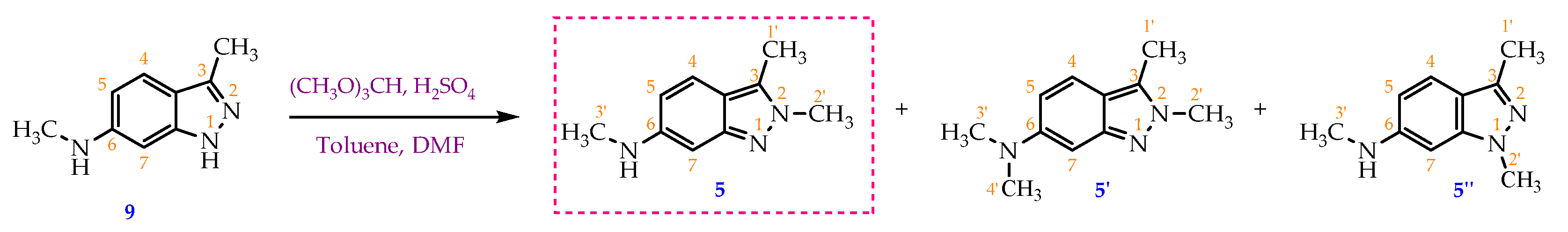
Scheme 4.
Methylation of compound 9 by TMOF (ratio of TMOF: starting material as 6.2:1) in concentrated H2SO4 generated a mixture of target compound 5 and by-product 5’ whose structure has not been reported in the literature. Compound 5’’ was not isolated.
Scheme 4.
Methylation of compound 9 by TMOF (ratio of TMOF: starting material as 6.2:1) in concentrated H2SO4 generated a mixture of target compound 5 and by-product 5’ whose structure has not been reported in the literature. Compound 5’’ was not isolated.
Scheme 5.
The probable mechanism for methylating compound 9 by TMOF in acidic condition.
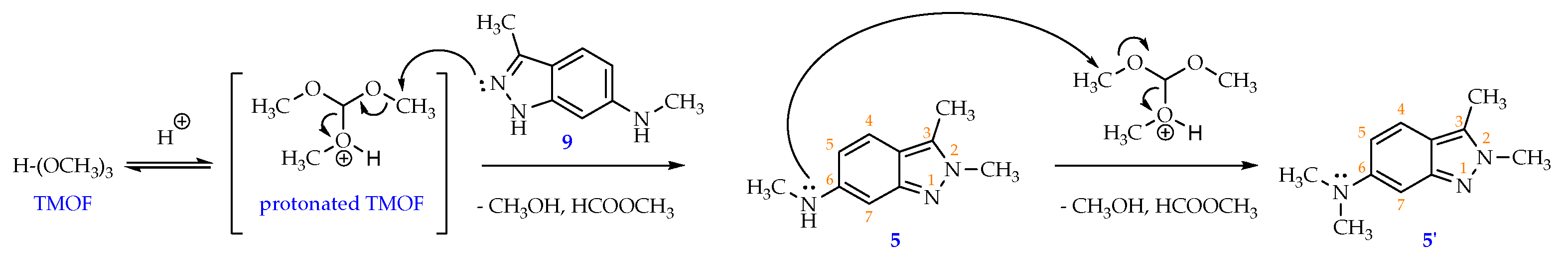

Table 1.
Various molar ratios between SnCl2 and compound 6 generated comparable yields of compound 8
Table 1.
Various molar ratios between SnCl2 and compound 6 generated comparable yields of compound 8
| Entry | Ratio (SnCl2 : compound 6) |
Time (h) | Product mass (g) | Yield (%) |
|---|---|---|---|---|
| 1 | 3 : 1 | 8 | 1.35 | 81.2 |
| 2 | 4 : 1 | 3 | 1.45 | 87.3 |
| 3 | 5 : 1 | 3 | 1.37 | 82.5 |
| 4 | 6 : 1 | 3 | 1.39 | 83.7 |
Table 2.
Organic and inorganic bases used as the catalysts in the synthesis of compound 9.
| Entry | Base catalyst | Time (h) | Product mass (g) | Yield (%) |
|---|---|---|---|---|
| 1 | CH3ONa | 6 | 1.90 | 86.7 |
| 2 | t-BuOK | 6 | 1.74 | 79.4 |
| 3 | K2CO3 | 10 | 1.43 | 65.3 |
| 4 | Na2CO3 | 10 | 1.46 | 66.6 |
Table 3.
The impact of the molar ratio between compound 8 and NaBH4 on product yield.
| Entry | Molar Ratio of Compound 8 : NaBH4 |
Time (h) | Yield (%) |
|---|---|---|---|
| 1 | 1:2 | 14 | 76.1 |
| 2 | 1:3 | 9 | 78.6 |
| 3 | 1:4 | 6 | 86.7 |
| 4 | 1:5 | 6 | 86.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



