Submitted:
20 July 2024
Posted:
22 July 2024
You are already at the latest version
Abstract
Facioscapulohumeral muscular dystrophy (FSHD) is an inherited myopathy, characterized by progressive and asymmetric muscle atrophy, primarily affecting muscles of the face, shoulder girdle, and upper arms before affecting muscles of the lower extremities with age and greater disease severity. FSHD is a disabling condition, and patients may also present with various extramuscular symptoms. FSHD is caused by the aberrant expression of double homeobox 4 (DUX4) in adult skeletal muscle, arising from compromised epigenetic repression of the D4Z4 array. DUX4 encodes the DUX4 protein, a transcription factor that activates myotoxic gene programs to produce the FSHD pathology. Therefore, sequence-specific oligonucleotides aimed at reducing DUX4 levels in patients is a compelling therapeutic approach, and one that has received considerable research interest over the last decade. This review aims to describe the current preclinical landscape of oligonucleotide therapies for FSHD. This includes outlining the mechanism of action of each therapy and summarizing the preclinical results obtained regarding their efficacy in cellular and/or murine disease models. The scope of this review is limited to oligonucleotide-based therapies that inhibit the DUX4 gene, mRNA, or protein in a way that does not involve gene editing.
Keywords:
facioscapulohumeral muscular dystrophy (FSHD)
; DUX4
; antisense oligonucleotides
; RNAi
; CRISPR
; DNA aptamers
; DNA decoys
; targeted therapy
; genetic therapy
1. Introduction
1.1. FSHD Overview
Facioscapulohumeral muscular dystrophy (FSHD, MIM: 158900 & 158901) is an autosomal dominant myopathy with a global incidence of approximately 1 in 8000–22000, making it one of the most common forms of muscular dystrophy worldwide [1,2,3]. FSHD is characterized by progressive muscle weakness and atrophy that develops in an asymmetric fashion, primarily affecting muscles of the face, shoulder girdle, and upper arms. Additional muscle groups can be affected with age, such as the ankle dorsiflexors and proximal leg muscles, resulting in obligate wheelchair use for approximately 20% of patients [1,4]. Some FSHD patients also experience extramuscular symptoms such as hearing loss, retinal vasculopathy, and/or cardiac conduction defects. FSHD is highly variable in terms of disease onset and severity [4]. There are no curative treatments available for FSHD, with current interventions limited to managing symptoms [5].
FSHD is a genetic condition with two distinct types. Patients can be classified as having either FSHD1 or FSHD2 depending on the genetic mechanism that results in the de-repression of the D4Z4 macrosatellite repeat array, located in the subtelomeric region 4q35 (Figure 1A). Healthy individuals have 11-100 3.3-kb D4Z4 repeat units. FSHD1, affecting 95% of patients, is caused by array contraction to ≤10 repeats [6]. FSHD2, affecting 5% of patients, is caused by mutation in genes involved in epigenetic methylation of the D4Z4 array (E.g., SMCHD1, DNMT3B, and LRIF1) [7,8,9]. Curiously, FSHD2 patients also tend to have fewer D4Z4 repeats than healthy individuals (12-16), demonstrating the complexity of this condition and how the distinction between FSHD1 and FSHD2 may not be as straightforward as initially thought. Regardless, the shared outcome in both types of FSHD is the loss of repressive methylation in the D4Z4 array. Therefore, clinical presentation is identical between FSHD1 and FSHD2 [4].
1.2. DUX4 Is the Central Cause of FSHD
FSHD is caused by the aberrant expression of double homeobox 4 (DUX4) protein in adult skeletal muscle, arising from the loss of repressive methylation in the D4Z4 array. The DUX4 gene encodes a transcription factor that is normally involved in zygotic genome activation during the 4-cell stage of early embryonic development [10,11]. Afterwards, DUX4 is epigenetically silenced in all adult tissues apart from limited expression of unrelated DUX4 isoforms in the testis and thymus [12,13]. Alternative splicing is known to occur for the DUX4 transcript, however, only mis-expression of the full-length isoform in muscle tissue is relevant to FSHD. Any mention hereafter of DUX4 mRNA is referring only to this full-length, pathogenic isoform.
Narrowing in on the genetic region from which FSHD arises, we return to the D4Z4 repeat array and the DUX4 gene therein. Within each 3.3-kb D4Z4 repeat unit is a retrogene containing exons 1 and 2 of the DUX4 open reading frame. A partial D4Z4 unit occurs after the most distal full unit, followed by the 3rd DUX4 exon (Figure 1B) [14]. Aberrant DUX4 expression occurs off this most distal D4Z4 unit in the FSHD-permissive 4qA haplotype. FSHD can only manifest in one of two major 4q allele variants: 4qA and 4qB. Unlike the non-permissive 4qB haplotype, the 4qA haplotype contains the pLAM region with a polyadenylation site (PAS), allowing transcription of a stable DUX4 mRNA when epigenetic repression is compromised in the D4Z4 array [1,14].
Following transcription of the DUX4 gene, stochastic, low-level DUX4 protein occurs in myofiber nuclei of the skeletal muscle. Inappropriate DUX4 protein in adult skeletal muscle is highly toxic, driving gene programs that result in oxidative stress, dysregulated transcript quality control, protein aggregation, inflammation, apoptosis, impaired myogenesis, and muscle atrophy (Figure 1B) [14,15,16]. DUX4 protein directly activates various genes including TRIM43, ZSCAN4, MBD3L2, WFDC3, PRAMEF1, RFPL2, and KHDC1 [17,18]. Disruption of these signaling pathways by reactivated DUX4 produces the FSHD pathology that we see in patients, manifesting primarily in the muscle tissue. In this review, we discuss targeted therapies aimed at removing or inhibiting this mis-expressed DUX4 protein, summarize the preclinical results obtained so far, and discuss further considerations for these treatment approaches.
2. Oligonucleotide Therapies Targeting DUX4
FSHD is a condition arising solely from the aberrant reactivation of a dormant gene: DUX4. Therefore, therapies that directly target aberrant DUX4 expression present a compelling treatment option. This review focuses on preclinical FSHD therapies that use a sequence-specific approach for targeting DUX4. This includes oligonucleotides with sequence complementarity to either the DUX4 gene, DUX4 mRNA, or DUX4 protein. This complementarity is used to inhibit DUX4 somewhere along its gene>mRNA>protein expression axis, thereby preventing DUX4 transactivation and the resulting FSHD pathology (Figure 2). Notably, the scope of this review is limited to only non-gene editing approaches that target DUX4.
The targeted oligonucleotide therapies discussed in this review are divided into three categories: Antisense Oligonucleotides (AOs), RNA interference (RNAi), and Other. These therapies have shown promising preclinical results in cellular and murine models of FSHD, primarily in their ability to lower DUX4 mRNA levels, reduce DUX4-target gene expression, and alleviate FSHD symptoms (Table 1, Table 2 and Table 3).
2.1. Antisense Oligonucleotides (AOs)
Antisense oligonucleotides (AOs) are synthetic, single-stranded nucleic acids that target a complementary mRNA molecule, dictated by Watson-Crick base pairing, to initiate post-transcriptional gene silencing. AOs were first identified in 1978 by Zamecnik and Stephenson, who found that complementary oligonucleotides inhibited translation of Rous sarcoma virus mRNA [19]. AOs bind to a target mRNA in a sequence-dependent manner and prevent its translation, thereby reducing the amount of target protein [20].
AOs are known to utilize various chemistries, a fact that makes them distinct from other oligonucleotide therapies. Modern AO drugs have chemical modifications to improve pharmacological properties like tolerability, target affinity, nuclease resistance, and intracellular uptake [21,22]. Commonly used AO chemistries involve modifying the phosphate backbone (PS, phosphorothioate; PMO, phosphorodiamidate morpholino oligomer) or ribose sugar (2’OMe, 2’-O-methyl; 2′-MOE, 2′-O-methoxyethyl; LNA, locked nucleic acid) [21]. In addition, peptide and fatty acid conjugate modifications have been used to facilitate delivery of DUX4-targeting AOs [23,24]. AOs can also be synthesized as gapmers: a chimeric molecule comprised of a central DNA region and a flanking region of modified RNA [25].
The formation of an AO-mRNA duplex results in (1) RNase-H mediated degradation of the target mRNA, or (2) steric blocking of the target mRNA (Figure 3A) [20,22,26]. Only gapmer AOs recruit RNase H to cleave target mRNA. Gapmer AOs produce a DNA:RNA substrate when bound to mRNA, recognizable by RNase H [27,28]. Steric blocking AOs inhibit proper mRNA translation, splicing and/or stability in an RNase H-independent manner [20,22,26]. Additionally, these steric blocking AOs can initiate further downstream degradation pathways, such as nonsense-mediated decay and no-go decay [29,30].
As summarised in Table 1, numerous preclinical studies have demonstrated that AO therapies can effectively reduce the amount of DUX4 mRNA and DUX4 target gene expression both in vitro and in vivo [23,24,31,32,33,34,35,36,37,38,39,40,41,42]. Other measures were also used to evaluate AO treatment efficacy, such as DUX4 protein levels, muscle fibre health, and murine functional performance. Most groups used primary or immortalized myoblasts/myocytes (often differentiated into myotubes) as an in vitro FSHD model, and FLExDUX4 mice as an in vivo FSHD model [43,44,45]. Earlier studies used local (intramuscular) injection for in vivo AO treatment, while studies after 2021 evaluated systemic (intraperitoneal, subcutaneous) injection routes. Systemic injection, being more clinically viable, managed to yield a similarly efficient DUX4 knockdown compared to local injection. Regarding the DUX4 target site, the >10 studies produced since 2011 tend to target exons 2 and 3, often with an emphasis on the polyadenylations site (PAS), pre-mRNA cleavage sites, and/or splice sites therein (Figure 4).
2.2. RNA Interference (RNAi)
Like AOs, RNAi-based oligonucleotides act at the RNA level, binding to a target mRNA according to antisense sequence complementarity to initiate post-transcriptional gene silencing. Where these classifications differ is that RNAi-based oligonucleotides initiate the RNA interference (RNAi) pathway to knockdown target mRNAs. First defined by Fire and Mello in 1998, RNAi is a conserved, biological mechanism by which double-stranded RNA triggers the loss of homologous mRNA [46]. RNAi can be induced by miRNAs or siRNAs complementary to a mRNA transcript. DICER endonucleases cleave precursor molecules (pre-miRNA or dsRNA) to produce mature microRNA (miRNA) or small-interfering RNAs (siRNA), which then get loaded into the Argonaute (AGO) protein of the RNA-induced silencing complex (RISC). Using the guide strand, RISC targets a complementary mRNA transcript and induces translational inhibition, sequestration, and/or mRNA degradation (miRNA-RISC) or simply mRNA degradation (siRNA-RISC) (Figure 3B) [46,47].
The two types of RNAi-based oligonucleotide therapies for FSHD are miRNAs (natural or artificial) and siRNAs. Since 2011, several studies have shown that these RNAi-based oligos can knockdown DUX4 mRNA and reduce DUX4 transactivation, in addition to improving other markers of FSHD symptom reversal (E.g., DUX4 protein levels, muscle fibre health, murine functional performance, etc.) (Table 2) [31,32,48,49,50,51,52,53,54,55,56,57]. Notably, the amount of published work is less for RNAi-based approaches compared to AOs. These studies commonly use primary or immortalized myoblasts/myocytes (often differentiated into myotubes) as an in vitro FSHD model, and AAV-DUX4 mice as an in vivo FSHD model [43,44,58]. FLExDUX4 mice were also used as an in vivo model for testing RNAi therapies, but to a lesser extent [45]. Most studies used local (intramuscular) injection to evaluate preclinical efficacy in vivo, except for two groups with partially released findings using systemic (intravenous) injection [51,55]. All current preclinical studies opted for adeno-associated virus (AAV)-mediated delivery of siRNAs or miRNAs in vivo, except for the partially released findings from Avidity Biosciences, Inc. and Dyne Therapeutics, Inc., who each describe a proprietary anti-mTfR1 mAb conjugate for delivery [54,55,56,57]. Lastly, as summarized in Figure 4, these miRNAs and siRNAs target DUX4 mRNA at all 3 of its exons, especially exon 1. Other sites like upstream D4Z4 regions, intronic regions, and pre-mRNA cleavage sites have also been tested.
2.3. Other
Other non-gene editing, preclinical oligonucleotide therapies have also been investigated as potential treatments for FSHD (Table 3). Unlike AOs and RNAi therapies which target DUX4 mRNA, these oligos tend to target DUX4 expression at the gene or protein level (Figure 2). These approaches present further compelling options for treating FSHD, in addition to the antisense approaches previously discussed.
2.3.1. CRISPR/dCas9 Transcriptional Repression
Multiple research groups have explored CRISPR/dCas9-mediated transcriptional repression of the DUX4 gene as a targeted therapy for FSHD. This is a form of CRISPR inhibition (CRISPRi) which uses the sequence-specificity of the sgRNA-Cas9 complex to target the DUX4 promoter, but with a catalytically inactive, ‘dead’ Cas9 (dCas9) fused to a transcriptional repressor domain (TRD) [59]. This allows for specific re-silencing of the D4Z4 region, reducing DUX4 expression and the resulting FSHD pathology. While various TRDs have been used, most studies opted for the Krüppel-associated box (KRAB) domain. Evaluation of this treatment approach has been largely done in primary FSHD myoblasts, myocytes, or myotubes, with the only in vivo testing performed by Himeda et al. (2021) using AAV-delivery and local (intramuscular) injection in FLExDUX4 mice [60]. These studies have all reported reduction in DUX4 mRNA following treatment, as well as reduction in DUX4 target gene expression and/or increased H3K9 tri-methylation at the D4Z4 array [60,61,62,63,64]. Notably, rather than directly repressing the DUX4 gene, Himeda et al. (2018) used the CRISPR/dCas9-KRAB system to repress epigenetic activators of DUX4 (ASH1L, BRD2, KDM4C, SMARCA5). Similar reduction of DUX4 mRNA was observed for this approach [62].
Abstracts proposing other non-gene editing, CRISPR-based approaches have been recently published. Results are preliminary and not fully released, however. One group is developing CRISPR/Cas13-mediated cleavage of DUX4 mRNA, reporting effective DUX4 knockdown in vivo [65]. Another group suggests using CRISPR/Cas13-ADAR (adenosine deaminase acting on RNA)-mediated editing of DUX4 mRNA to create a C>U nonsense mutation [66]. No definitive results have been published at this time.
2.3.2. DNA Aptamers
Aptamers are single-stranded oligonucleotides that can bind to a specific protein or protein family thanks to their secondary and tertiary folding structure. The unique 3D conformation of an aptamer allows target interaction like that of an antigen and antibody [67]. Therefore, aptamers can be used to specifically target a protein of interest, and in the case of FSHD, bind to and inhibit DUX4. Klingler et al. (2020) designed DNA aptamers with high affinity to DUX4 protein [68]. While not evaluated, these DNA aptamers could be used to treat FSHD by sterically inhibiting DUX4 protein in skeletal muscle.
2.3.3. dsDNA Decoy Trapping
Mariot et al. (2020) demonstrated a unique approach to prevent DUX4 transactivation known as decoy trapping [69]. Decoy trapping uses double-stranded DNA fragments whose sequence corresponds to DUX4 binding motifs, akin to the DNA regions that DUX4 normally binds to as a transcription factor. By saturating the cellular environment with dsDNA decoy binding sites, DUX4 protein is trapped in a binding sink and unable to activate its normal target genes. Mariot et al. (2020) found that dsDNA treatment was able to reduce expression or downstream DUX4 target genes in vitro and in vivo [69].
2.3.4. U7-snRNA pre-mRNA Inhibition
Rashnonejad et al. (2021) describe a strategy to inhibit DUX4 mRNA expression using U7-small nuclear RNA (snRNA) antisense expression cassettes [70]. U7-snRNA is a part of the small nuclear ribonucleoprotein complex (snRNP), which is involved in 3’ end processing of histone pre-mRNAs in the nucleus. This therapeutic approach uses modified U7-snRNA with antisense sequence specificity to DUX4, capable of inhibiting pre-mRNA production or maturation. Rashnonejad et al. (2021) showed that these U7-snRNA expression cassettes, delivered by AAV, effectively reduced DUX4 mRNA, DUX4 protein, and DUX4 target gene expression in immortalized FSHD myotubes [70].
3. Further Considerations
Antisense therapies for FSHD, meaning AOs or RNAi drugs that target DUX4 mRNA, are the furthest along in preclinical development compared to other oligonucleotide approaches. Many AO and RNAi therapies have shown promising indications both in vitro and in vivo, as discussed previously. Therefore, discussion of certain advantages and disadvantages of DUX4-targeting oligonucleotide therapies will focus on antisense approaches only.
3.1. Advantages of Antisense Approaches
Antisense therapies are ideal for monogenic diseases that can be attributed to a single root cause. In the case of FSHD, this is aberrant DUX4 expression in adult skeletal muscle. DUX4 is an especially ideal therapeutic target for antisense therapies because it is practically absent in all adult tissues under normal, healthy circumstances, making concerns of unwanted DUX4 knockdown elsewhere in the body largely insignificant [12,13]. This, however, is not something that should be entirely ignored when evaluating candidate therapies for clinical trials. Overall, antisense therapies are highly specific and potent molecules with a relatively simple mechanism of action, often taking advantage of conserved cellular processes [20].
Compared to gene-editing approaches that prevent DUX4 expression, antisense therapies involve no changes to genomic DNA, acting only at the RNA level [20,26]. This makes them more acceptable from a regulatory standpoint, unburdened by the moral concern surrounding CRISPR/Cas9 editing of the human genome, even if for therapeutic purposes. For this reason, it may be fair to suggest that antisense therapies are a more clinically viable form of targeted, genetic therapy for FSHD.
3.2. Disadvantages of Antisense Approaches
Efficient delivery to muscle tissue is a considerable challenge for antisense therapies, often hindering the clinical utility of oligonucleotides that otherwise demonstrate good preclinical efficacy. AOs and RNAi oligonucleotides are relatively large nucleic acids that tend to be negatively charged and hydrophilic [21]. Molecules with such properties do not readily pass through the plasma membrane. Furthermore, upon systemic injection, these molecules must avoid nuclease degradation, mononuclear phagocyte system entrapment, protein entrapment, and high renal clearance [71,72,73]. If these can be overcome, there remains the issue of inefficient cellular uptake, as these oligonucleotides are also prone to endosomal entrapment within the cell [71,72,73]. All this means that only a small percentage of injected drug becomes bioavailable to provide therapeutic benefit to a patient. However, various strategies to improve delivery are currently being explored for AOs and RNAi oligos, including chemical modification, delivery conjugates, and carrier molecules [71].
Persistence of therapeutic effect is another notable disadvantage of antisense therapies. Given that antisense therapies act on the mRNA, a transient and replenishable molecule, regular lifelong administrations may be necessary to offer long-term reversal of FSHD symptoms for patients. This problem is not shared by other proposed genetic therapies that would permanently inactivate the toxic DUX4 gene (E.g., CRISPR/Cas9 editing) such that multiple treatments are not needed.
3.3. Early-Stage Clinical Trials for Select FSHD Therapies
Two RNAi-based oligonucleotide therapies for FSHD are currently recruiting for Phase 1/2 clinical trials to evaluate the safety, tolerability, pharmacokinetics, pharmacodynamics, and efficacy in adult patients. First, ARO-DUX4, developed by Arrowhead Pharmaceuticals, is a DUX4-specific siRNA using an unspecified and proprietary delivery method (Phase 1/2 NCT06131983) [74,75]. Second, AOC 1020, developed by Avidity Biosciences, is a DUX4-specific siRNA using a proprietary anti-mTfR1 mAb delivery conjugate (Phase 1/2 FORTITUDE™ NCT0574792) [76,77]. Both therapies have previously demonstrated preclinical efficacy in cellular and murine models of FSHD [51,52,54,55].
4. Conclusions
Since DUX4 was identified as the central cause of FSHD, numerous targeted oligonucleotide therapies have been proposed, many of which have shown promising results in preclinical stages. However, despite DUX4 presenting itself as an ideal therapeutic target, there are still considerable challenges that may prevent these therapies from reaching clinical use and benefiting patients. Additionally, more progress towards fully characterizing FSHD is needed, as it remains an incredibly complicated condition with many unanswered questions. Further research into the molecular underpinnings of FSHD may offer additional therapeutic targets amenable to oligonucleotide therapies. Similarly, it is important to continue investigating the normal physiological role of DUX4 in the testis and thymus, as this is not fully understood and could impact decisions made when targeting DUX4 in skeletal muscle, possibly in terms of off-target effects. Another consideration would be the potential synergistic effect of combining multiple therapies together, particularly those that target DUX4 expression via different modes of action. This has not yet been attempted for FSHD. Overall, with a strong pipeline of candidate oligos from many different research groups, and two siRNA drugs entering early clinical trials, the future appears hopeful for a targeted treatment option for patients with FSHD.
Author Contributions
Conception and design, S.L.B.; literature review and writing—original draft preparation, S.L.B.; writing—review and editing, S.L.B. and T.Y.; supervision and funding acquisition, T.Y. All authors have read and agreed to the published version of the manuscript.
Funding
No specific grant support was received for this study. T.Y. is supported by the Muscular Dystrophy Canada, the Friends of Garrett Cumming Research Fund, the HM Toupin Neurological Science Research Fund, Canadian Institutes of Health Research (CIHR), the Canada Foundation for Innovation, Alberta Advanced Education and Technology, Alberta Innovates: Health Solutions (AIHS), Jesse’s Journey, and the Women and Children’s Health Research Institute (WCHRI), The Rare Disease Foundation, and the BC Children’s Hospital Foundation.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to acknowledge Saeed Anwar (Department of Medical Genetics, University of Alberta, Edmonton, AB, Canada) for providing advice and guidance throughout the writing of this review.
Conflicts of Interest
T.Y. is a co-founder and shareholder of OligomicsTx Inc., which aims to commercialize antisense technology. S.L.B. declares that this study was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
References
- Wang, L.H.; Tawil, R. Facioscapulohumeral Dystrophy. Curr Neurol Neurosci Rep 2016, 16, 66. [Google Scholar] [CrossRef] [PubMed]
- Mostacciuolo, M.; Pastorello, E.; Vazza, G.; Miorin, M.; Angelini, C.; Tomelleri, G.; Galluzzi, G.; Trevisan, C. Facioscapulohumeral Muscular Dystrophy: Epidemiological and Molecular Study in a North-east Italian Population Sample. Clinical Genetics 2009, 75, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Deenen, J.C.W.; Arnts, H.; Van Der Maarel, S.M.; Padberg, G.W.; Verschuuren, J.J.G.M.; Bakker, E.; Weinreich, S.S.; Verbeek, A.L.M.; Van Engelen, B.G.M. Population-Based Incidence and Prevalence of Facioscapulohumeral Dystrophy. Neurology 2014, 83, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Mul, K.; Lassche, S.; Voermans, N.C.; Padberg, G.W.; Horlings, C.G.; Van Engelen, B.G. What’s in a Name? The Clinical Features of Facioscapulohumeral Muscular Dystrophy. Pract Neurol 2016, 16, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.S.; Astudillo Moncayo, O.M.; Mosquera, J.; Muyolema Arce, V.E.; Gallegos, C.; Ortiz, J.F.; Andrade, A.F.; Oña, S.; Buj, M.J. Treatment of Facioscapulohumeral Muscular Dystrophy (FSHD): A Systematic Review. Cureus 2023. [Google Scholar] [CrossRef] [PubMed]
- Lemmers, R.J.L.F.; Van Der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camaño, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; Jan Van Ommen, G.; Padberg, G.W.; et al. A Unifying Genetic Model for Facioscapulohumeral Muscular Dystrophy. Science 2010, 329, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Lemmers, R.J.L.F.; Tawil, R.; Petek, L.M.; Balog, J.; Block, G.J.; Santen, G.W.E.; Amell, A.M.; Van Der Vliet, P.J.; Almomani, R.; Straasheijm, K.R.; et al. Digenic Inheritance of an SMCHD1 Mutation and an FSHD-Permissive D4Z4 Allele Causes Facioscapulohumeral Muscular Dystrophy Type 2. Nat Genet 2012, 44, 1370–1374. [Google Scholar] [CrossRef]
- Van den Boogaard, M.L.; Lemmers, R.J.L.F.; Balog, J.; Wohlgemuth, M.; Auranen, M.; Mitsuhashi, S.; van der Vliet, P.J.; Straasheijm, K.R.; van den Akker, R.F.P.; Kriek, M.; et al. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. The American Journal of Human Genetics 2016, 98, 1020–1029. [Google Scholar] [CrossRef]
- Hamanaka, K.; Šikrová, D.; Mitsuhashi, S.; Masuda, H.; Sekiguchi, Y.; Sugiyama, A.; Shibuya, K.; Lemmers, R.J.L.F.; Goossens, R.; Ogawa, M.; et al. Homozygous Nonsense Variant in LRIF1 Associated with Facioscapulohumeral Muscular Dystrophy. Neurology 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, P.G.; Doráis, J.A.; Grow, E.J.; Whiddon, J.L.; Lim, J.-W.; Wike, C.L.; Weaver, B.D.; Pflueger, C.; Emery, B.R.; Wilcox, A.L.; et al. Conserved Roles for Murine DUX and Human DUX4 in Activating Cleavage Stage Genes and MERVL/HERVL Retrotransposons. 2017.
- De Iaco, A.; Planet, E.; Coluccio, A.; Verp, S.; Duc, J.; Trono, D. DUX-Family Transcription Factors Regulate Zygotic Genome Activation in Placental Mammals. Nat Genet 2017, 49, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Snider, L.; Geng, L.N.; Lemmers, R.J.L.F.; Kyba, M.; Ware, C.B.; Nelson, A.M.; Tawil, R.; Filippova, G.N.; Van Der Maarel, S.M.; Tapscott, S.J.; et al. Facioscapulohumeral Dystrophy: Incomplete Suppression of a Retrotransposed Gene. PLoS Genet 2010, 6, e1001181. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Chadwick, B.P. Influence of Repressive Histone and DNA Methylation upon D4Z4 Transcription in Non-Myogenic Cells. PLoS ONE 2016, 11, e0160022. [Google Scholar] [CrossRef] [PubMed]
- Himeda, C.L.; Jones, P.L. The Genetics and Epigenetics of Facioscapulohumeral Muscular Dystrophy. Annu. Rev. Genom. Hum. Genet. 2019, 20, 265–291. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Nguyen, Q.; Yokota, T. DUX4 Signalling in the Pathogenesis of Facioscapulohumeral Muscular Dystrophy. IJMS 2020, 21, 729. [Google Scholar] [CrossRef] [PubMed]
- Mocciaro, E.; Runfola, V.; Ghezzi, P.; Pannese, M.; Gabellini, D. DUX4 Role in Normal Physiology and in FSHD Muscular Dystrophy. Cells 2021, 10, 3322. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.N.; Yao, Z.; Snider, L.; Fong, A.P.; Cech, J.N.; Young, J.M.; van der Maarel, S.M.; Ruzzo, W.L.; Gentleman, R.C.; Tawil, R.; et al. DUX4 Activates Germline Genes, Retroelements, and Immune Mediators: Implications for Facioscapulohumeral Dystrophy. Developmental Cell 2012, 22, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Snider, L.; Balog, J.; Lemmers, R.J.L.F.; Van Der Maarel, S.M.; Tawil, R.; Tapscott, S.J. DUX4-Induced Gene Expression Is the Major Molecular Signature in FSHD Skeletal Muscle. Human Molecular Genetics 2014, 23, 5342–5352. [Google Scholar] [CrossRef]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous Sarcoma Viral RNA Translation by a Specific Oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. U.S.A. 1978, 75, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Dias, N.; Stein, C.A. Antisense Oligonucleotides: Basic Concepts and Mechanisms. Molecular Cancer Therapeutics 2002, 1, 347–355. [Google Scholar]
- Khvorova, A.; Watts, J.K. The Chemical Evolution of Oligonucleotide Therapies of Clinical Utility. Nat Biotechnol 2017, 35, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.J. Antisense Oligonucleotides. In Introduction to Basics of Pharmacology and Toxicology; Raj, G.M., Raveendran, R., Eds.; Springer Singapore: Singapore, 2019; pp. 407–411. ISBN 978-981-329-778-4. [Google Scholar]
- Ansseau, E.; Vanderplanck, C.; Wauters, A.; Harper, S.; Coppée, F.; Belayew, A. Antisense Oligonucleotides Used to Target the DUX4 mRNA as Therapeutic Approaches in FaciosScapuloHumeral Muscular Dystrophy (FSHD). Genes 2017, 8, 93. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, L.F.; Den Hamer, B.; Van Den Heuvel, A.; Franken, M.; Jackson, M.; Dwyer, C.A.; Tapscott, S.J.; Rigo, F.; Van Der Maarel, S.M.; De Greef, J.C. Systemic Delivery of a DUX4-Targeting Antisense Oligonucleotide to Treat Facioscapulohumeral Muscular Dystrophy. Molecular Therapy - Nucleic Acids 2021, 26, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Monia, B.P.; Lesnik, E.A.; Gonzalez, C.; Lima, W.F.; McGee, D.; Guinosso, C.J.; Kawasaki, A.M.; Cook, P.D.; Freier, S.M. Evaluation of 2‘-Modified Oligonucleotides Containing 2‘-Deoxy Gaps as Antisense Inhibitors of Gene Expression. Journal of Biological Chemistry 1993, 268, 14514–14522. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Baker, B.F.; Crooke, R.M.; Liang, X. Antisense Technology: An Overview and Prospectus. Nat Rev Drug Discov 2021, 20, 427–453. [Google Scholar] [CrossRef] [PubMed]
- Minshull, J.; Hunt, T. The Use of Single-Stranded DNA and RNase H to Promote Quantitative ‘Hybrid Arrest of Translation’ of mRNA/DNA Hybrids in Reticulocyte Lysate Cell-Free Translations. Nucl Acids Res 1986, 14, 6433–6451. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Oda, Y.; Iwai, S.; Inoue, H.; Ohtsuka, E.; Kanaya, S.; Kimura, S.; Katsuda, C.; Katayanagi, K.; Morikawa, K. How Does RNase H Recognize a DNA.RNA Hybrid? Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 11535–11539. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.J.; Norrbom, M.; Chun, S.; Bennett, C.F.; Rigo, F. Nonsense-Mediated Decay as a Terminating Mechanism for Antisense Oligonucleotides. Nucleic Acids Research 2014, 42, 5871–5879. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Nichols, J.G.; Hsu, C.-W.; Vickers, T.A.; Crooke, S.T. mRNA Levels Can Be Reduced by Antisense Oligonucleotides via No-Go Decay Pathway. Nucleic Acids Research 2019, 47, 6900–6916. [Google Scholar] [CrossRef] [PubMed]
- Vanderplanck, C.; Ansseau, E.; Charron, S.; Stricwant, N.; Tassin, A.; Laoudj-Chenivesse, D.; Wilton, S.D.; Coppée, F.; Belayew, A. The FSHD Atrophic Myotube Phenotype Is Caused by DUX4 Expression. PLoS ONE 2011, 6, e26820. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-W.; Snider, L.; Yao, Z.; Tawil, R.; Van Der Maarel, S.M.; Rigo, F.; Bennett, C.F.; Filippova, G.N.; Tapscott, S.J. DICER/AGO-Dependent Epigenetic Silencing of D4Z4 Repeats Enhanced by Exogenous siRNA Suggests Mechanisms and Therapies for FSHD. Hum. Mol. Genet. 2015, 24, 4817–4828. [Google Scholar] [CrossRef] [PubMed]
- Marsollier, A.-C.; Ciszewski, L.; Mariot, V.; Popplewell, L.; Voit, T.; Dickson, G.; Dumonceaux, J. Antisense Targeting of 3′ End Elements Involved in DUX4 mRNA Processing Is an Efficient Therapeutic Strategy for Facioscapulohumeral Dystrophy: A New Gene-Silencing Approach. Hum. Mol. Genet. 2016, 25, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; King, O.D.; Zhang, Y.; Clayton, N.P.; Spencer, C.; Wentworth, B.M.; Emerson, C.P.; Wagner, K.R. Morpholino-Mediated Knockdown of DUX4 Toward Facioscapulohumeral Muscular Dystrophy Therapeutics. Molecular Therapy 2016, 24, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Derenne, A.; Tassin, A.; Nguyen, T.H.; De Roeck, E.; Jenart, V.; Ansseau, E.; Belayew, A.; Coppée, F.; Declèves, A.-E.; Legrand, A. Induction of a Local Muscular Dystrophy Using Electroporation in Vivo: An Easy Tool for Screening Therapeutics. Sci Rep 2020, 10, 11301. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Maruyama, R.; Echigoya, Y.; Nguyen, Q.; Zhang, A.; Khawaja, H.; Sen Chandra, S.; Jones, T.; Jones, P.; Chen, Y.-W.; et al. Inhibition of DUX4 Expression with Antisense LNA Gapmers as a Therapy for Facioscapulohumeral Muscular Dystrophy. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 16509–16515. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Bittel, A.; Maruyama, R.; Echigoya, Y.; Nguyen, Q.; Huang, Y.; Dzierlega, K.; Zhang, A.; Chen, Y.-W.; Yokota, T. DUX4 Transcript Knockdown with Antisense 2′-O-Methoxyethyl Gapmers for the Treatment of Facioscapulohumeral Muscular Dystrophy. Molecular Therapy 2021, 29, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Falzarano, M.S.; Argenziano, M.; Marsollier, A.C.; Mariot, V.; Rossi, D.; Selvatici, R.; Dumonceaux, J.; Cavalli, R.; Ferlini, A. Chitosan-Shelled Nanobubbles Irreversibly Encapsulate Morpholino Conjugate Antisense Oligonucleotides and Are Ineffective for Phosphorodiamidate Morpholino-Mediated Gene Silencing of DUX4. Nucleic Acid Therapeutics 2021, 31, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Lu-Nguyen, N.; Malerba, A.; Herath, S.; Dickson, G.; Popplewell, L. Systemic Antisense Therapeutics Inhibiting DUX4 Expression Ameliorates FSHD-like Pathology in an FSHD Mouse Model. Human Molecular Genetics 2021, 30, 1398–1412. [Google Scholar] [CrossRef] [PubMed]
- Lu-Nguyen, N.; Malerba, A.; Antoni Pineda, M.; Dickson, G.; Popplewell, L. Improving Molecular and Histopathology in Diaphragm Muscle of the Double Transgenic ACTA1-MCM/FLExDUX4 Mouse Model of FSHD with Systemic Antisense Therapy. Human Gene Therapy 2022, 33, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Lu-Nguyen, N.; Dickson, G.; Malerba, A.; Popplewell, L. Long-Term Systemic Treatment of a Mouse Model Displaying Chronic FSHD-like Pathology with Antisense Therapeutics That Inhibit DUX4 Expression. Biomedicines 2022, 10, 1623. [Google Scholar] [CrossRef] [PubMed]
- Kakimoto, T.; Ogasawara, A.; Ishikawa, K.; Kurita, T.; Yoshida, K.; Harada, S.; Nonaka, T.; Inoue, Y.; Uchida, K.; Tateoka, T.; et al. A Systemically Administered Unconjugated Antisense Oligonucleotide Targeting DUX4 Improves Muscular Injury and Motor Function in FSHD Model Mice. Biomedicines 2023, 11, 2339. [Google Scholar] [CrossRef] [PubMed]
- Stadler, G.; Chen, J.C.; Wagner, K.; Robin, J.D.; Shay, J.W.; Emerson, C.P.; Wright, W.E. Establishment of Clonal Myogenic Cell Lines from Severely Affected Dystrophic Muscles - CDK4 Maintains the Myogenic Population. Skeletal Muscle 2011, 1, 12. [Google Scholar] [CrossRef] [PubMed]
- Krom, Y.D.; Dumonceaux, J.; Mamchaoui, K.; Den Hamer, B.; Mariot, V.; Negroni, E.; Geng, L.N.; Martin, N.; Tawil, R.; Tapscott, S.J.; et al. Generation of Isogenic D4Z4 Contracted and Noncontracted Immortal Muscle Cell Clones from a Mosaic Patient. The American Journal of Pathology 2012, 181, 1387–1401. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.; Jones, P.L. A Cre-Inducible DUX4 Transgenic Mouse Model for Investigating Facioscapulohumeral Muscular Dystrophy. PLoS ONE 2018, 13, e0192657. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and Specific Genetic Interference by Double-Stranded RNA In. Nature 1998, 391. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.C.; Doudna, J.A. Molecular Mechanisms of RNA Interference. Annu. Rev. Biophys. 2013, 42, 217–239. [Google Scholar] [CrossRef] [PubMed]
- Wallace, L.M.; Liu, J.; Domire, J.S.; Garwick-Coppens, S.E.; Guckes, S.M.; Mendell, J.R.; Flanigan, K.M.; Harper, S.Q. RNA Interference Inhibits DUX4-Induced Muscle Toxicity In Vivo: Implications for a Targeted FSHD Therapy. Molecular Therapy 2012, 20, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Wallace, L.M.; Saad, N.Y.; Pyne, N.K.; Fowler, A.M.; Eidahl, J.O.; Domire, J.S.; Griffin, D.A.; Herman, A.C.; Sahenk, Z.; Rodino-Klapac, L.R.; et al. Pre-Clinical Safety and Off-Target Studies to Support Translation of AAV-Mediated RNAi Therapy for FSHD. Molecular Therapy - Methods & Clinical Development 2018, 8, 121–130. [Google Scholar] [CrossRef]
- Saad, N.Y.; Al-Kharsan, M.; Garwick-Coppens, S.E.; Chermahini, G.A.; Harper, M.A.; Palo, A.; Boudreau, R.L.; Harper, S.Q. Human miRNA miR-675 Inhibits DUX4 Expression and May Be Exploited as a Potential Treatment for Facioscapulohumeral Muscular Dystrophy. Nat Commun 2021, 12, 7128. [Google Scholar] [CrossRef] [PubMed]
- Arrowhead Pharmaceuticals, Inc. Arrowhead Presents Preclinical Data on ARO-DUX4 at FSHD Society International Research Congress. 2021. Available online: https://ir.arrowheadpharma.com/news-releases/news-release-details/arrowhead-presents-preclinical-data-aro-dux4-fshd-society (accessed on 29 June 2024).
- Jagannathan, S.; De Greef, J.C.; Hayward, L.J.; Yokomori, K.; Gabellini, D.; Mul, K.; Sacconi, S.; Arjomand, J.; Kinoshita, J.; Harper, S.Q. Meeting Report: The 2021 FSHD International Research Congress. Skeletal Muscle 2022, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Mariot, V.; Sidlauskaite, E.; Gall, L.L.; Corbex, E.; Dumonceaux, J. VP. 61 An AAV-shRNA DUX4-Based Therapy to Treat Facioscapulohumeral Muscular Dystrophy (FSHD). Neuromuscular Disorders 2022, 32, S107. [Google Scholar] [CrossRef]
- Malecova, B.; Sala, D.; Melikian, G.M.; Erdogan, G.; Johns, R.; Young, J.; Ventre, E.; Gatto, S.; Onorato, M.; Pavlicek, A.; et al. DUX4 siRNA Optimization for the Development of an Antibody-Oligonucleotide Conjugate (AOC) for the Treatment of FSHD (P17-13.009). Neurology 2022, 98, 1776. [Google Scholar] [CrossRef]
- Avidity Biosciences, Inc. Targeting DUX4 for Silencing with AOC for the Treatment of FSHD. 2024. Available online: https://www.aviditybiosciences.com/wp-content/uploads/2024/03/MDA-2024-FSHD-RNASeq_v6.0_STC.pdf (accessed on 29 June 2024).
- Dyne Therapeutics, Inc. Dyne Therapeutics Presents New Preclinical Data for its Facioscapulohumeral Muscular Dystrophy Program During the FSHD Society International Research Congress. 2024. Available online: https://investors.dyne-tx.com/news-releases/news-release-details/dyne-therapeutics-presents-new-preclinical-data-its (accessed on 14 July 2024).
- Dyne Therapeutics, Inc. The FORCETM platform achieves robust and durable DUX4 suppression and functional benefit in FSHD mouse models. 2024. Available online: https://www.dyne-tx.com/wp-content/uploads/Natoli_FSHDIRC_June2024.pdf (accessed on 14 July 2024).
- Wallace, L.M.; Garwick, S.E.; Mei, W.; Belayew, A.; Coppee, F.; Ladner, K.J.; Guttridge, D.; Yang, J.; Harper, S.Q. DUX4, a Candidate Gene for Facioscapulohumeral Muscular Dystrophy, Causes P53-dependent Myopathy in Vivo. Annals of Neurology 2011, 69, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR Interference (CRISPRi) for Sequence-Specific Control of Gene Expression. Nat Protoc 2013, 8, 2180–2196. [Google Scholar] [CrossRef] [PubMed]
- Himeda, C.L.; Jones, T.I.; Jones, P.L. Targeted Epigenetic Repression by CRISPR/dSaCas9 Suppresses Pathogenic DUX4-Fl Expression in FSHD. Molecular Therapy - Methods & Clinical Development 2021, 20, 298–311. [Google Scholar] [CrossRef]
- Himeda, C.L.; Jones, T.I.; Jones, P.L. CRISPR/dCas9-Mediated Transcriptional Inhibition Ameliorates the Epigenetic Dysregulation at D4Z4 and Represses DUX4-Fl in FSH Muscular Dystrophy. Molecular Therapy 2016, 24, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Himeda, C.L.; Jones, T.I.; Virbasius, C.-M.; Zhu, L.J.; Green, M.R.; Jones, P.L. Identification of Epigenetic Regulators of DUX4-Fl for Targeted Therapy of Facioscapulohumeral Muscular Dystrophy. Molecular Therapy 2018, 26, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Chadwick, B.P. CRISPR Mediated Targeting of DUX4 Distal Regulatory Element Represses DUX4 Target Genes Dysregulated in Facioscapulohumeral Muscular Dystrophy. Sci Rep 2021, 11, 12598. [Google Scholar] [CrossRef] [PubMed]
- Sasaki-Honda, M.; Jonouchi, T.; Arai, M.; He, J.; Okita, K.; Sakurai, S.; Yamamoto, T.; Sakurai, H. Hit-and-Run Silencing of Endogenous DUX4 by Targeting DNA Hypomethylation on D4Z4 Repeats in Facioscapulohumeral Muscular Dystrophy. bioRxiv, 2022; (preprint). [Google Scholar]
- Rashnonejad, A.; Amini-Chermahini, G.; Taylor, N.; Fowler, A.; Kraus, E.; King, O.; Harper, S. FP. 29 AAV-CRISPR-Cas13 Gene Therapy for FSHD: DUX4 Gene Silencing Efficacy and Immune Responses to Cas13b Protein. Neuromuscular Disorders 2022, 32, S103–S104. [Google Scholar] [CrossRef]
- Saljoughian, N.; Rizzotto, L.; Sezgin, Y.; Faraji, H.; Wallace, L.; Kararoudi, M.N.; Palmieri, D.; Harper, S.P. 144 Developing Cas13-ADAR-Mediated DUX4 mRNA Editing as a Prospective Therapy for FSHD. Neuromuscular Disorders 2022, 32, S106. [Google Scholar] [CrossRef]
- Di Mauro, V.; Lauta, F.C.; Modica, J.; Appleton, S.L.; De Franciscis, V.; Catalucci, D. Diagnostic and Therapeutic Aptamers. JACC: Basic to Translational Science 2024, 9, 260–277. [Google Scholar] [CrossRef] [PubMed]
- Klingler, C.; Ashley, J.; Shi, K.; Stiefvater, A.; Kyba, M.; Sinnreich, M.; Aihara, H.; Kinter, J. DNA Aptamers against the DUX4 Protein Reveal Novel Therapeutic Implications for FSHD. FASEB j. 2020, 34, 4573–4590. [Google Scholar] [CrossRef] [PubMed]
- Mariot, V.; Joubert, R.; Marsollier, A.-C.; Hourdé, C.; Voit, T.; Dumonceaux, J. A Deoxyribonucleic Acid Decoy Trapping DUX4 for the Treatment of Facioscapulohumeral Muscular Dystrophy. Molecular Therapy - Nucleic Acids 2020, 22, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Rashnonejad, A.; Amini-Chermahini, G.; Taylor, N.K.; Wein, N.; Harper, S.Q. Designed U7 snRNAs Inhibit DUX4 Expression and Improve FSHD-Associated Outcomes in DUX4 Overexpressing Cells and FSHD Patient Myotubes. Molecular Therapy - Nucleic Acids 2021, 23, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in Oligonucleotide Drug Delivery. Nat Rev Drug Discov 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, M.; Ashizawa, A.T. The Challenges and Strategies of Antisense Oligonucleotide Drug Delivery. Biomedicines 2021, 9, 433. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Zhang, M.; Huang, Y. Three ‘E’ Challenges for siRNA Drug Development. Trends in Molecular Medicine 2024, 30, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Li, Y.; Yu, W.; Fu, Y.; Zhang, J.; Cao, H. Recent Progress of Small Interfering RNA Delivery on the Market and Clinical Stage. Mol. Pharmaceutics 2024, 21, 2081–2096. [Google Scholar] [CrossRef] [PubMed]
- Study of ARO-DUX4 in Adult Patients With Facioscapulohumeral Muscular Dystrophy Type 1. Available online: https://clinicaltrials.gov/study/NCT06131983 (accessed on 6 July 2024).
- Halseth, A.; Ackermann, E.; Brandt, T.; Chen, C.; Cho, H.; Stahl, M.; DiTrapani, K.; Hughes, S.; Tawil, R.; Statland, J. P51 Phase 1/2 Study to Evaluate AOC 1020 for Adult Patients with Facioscapulohumeral Muscular Dystrophy: FORTITUDE Trial Design. Neuromuscular Disorders 2023, 33, S71. [Google Scholar] [CrossRef]
- Phase 1/2 Study of AOC 1020 in Adults With Facioscapulohumeral Muscular Dystrophy (FSHD) (FORTITUDE). Available online: https://clinicaltrials.gov/study/NCT05747924 (accessed on 6 July 2024).
Figure 1.
(A) Schematic representation of D4Z4 region in healthy individuals and FSHD patients. The D4Z4 macrosatellite tandem repeat array is found in the subtelomeric region 4q35. Each grey triangle indicates a 3.3 kb D4Z4 repeat unit, within each a DUX4 retrogene is contained. The 1st and 2nd DUX4 exons (blue boxes) occur in each full D4Z4 unit. A partial D4Z4 (grey trapezoid) occurs after the most distal full unit, followed by the 3rd DUX4 exon. Healthy individuals have 11-1000 repeats and full epigenetic repression (purple line). FSHD patients have fewer repeats and compromised epigenetic repression (purple dotted line) arising from one of two genetic changes indicated in red text. (B) Schematic representation of aberrant DUX4 expression from the most distal D4Z4 repeat within the FSHD-permissive 4qA haplotype. The 1st and 2nd DUX4 exons are within each D4Z4 unit. The 3rd DUX4 exon and PAS site are found directly downstream of the most distal D4Z4 unit. Compromised repression results in low-level DUX4 protein to exist within the skeletal muscle, perturbing downstream gene expression to cause the FSHD pathology (oxidative stress, apoptosis, impaired myogenesis, muscle atrophy, etc.).
Figure 1.
(A) Schematic representation of D4Z4 region in healthy individuals and FSHD patients. The D4Z4 macrosatellite tandem repeat array is found in the subtelomeric region 4q35. Each grey triangle indicates a 3.3 kb D4Z4 repeat unit, within each a DUX4 retrogene is contained. The 1st and 2nd DUX4 exons (blue boxes) occur in each full D4Z4 unit. A partial D4Z4 (grey trapezoid) occurs after the most distal full unit, followed by the 3rd DUX4 exon. Healthy individuals have 11-1000 repeats and full epigenetic repression (purple line). FSHD patients have fewer repeats and compromised epigenetic repression (purple dotted line) arising from one of two genetic changes indicated in red text. (B) Schematic representation of aberrant DUX4 expression from the most distal D4Z4 repeat within the FSHD-permissive 4qA haplotype. The 1st and 2nd DUX4 exons are within each D4Z4 unit. The 3rd DUX4 exon and PAS site are found directly downstream of the most distal D4Z4 unit. Compromised repression results in low-level DUX4 protein to exist within the skeletal muscle, perturbing downstream gene expression to cause the FSHD pathology (oxidative stress, apoptosis, impaired myogenesis, muscle atrophy, etc.).
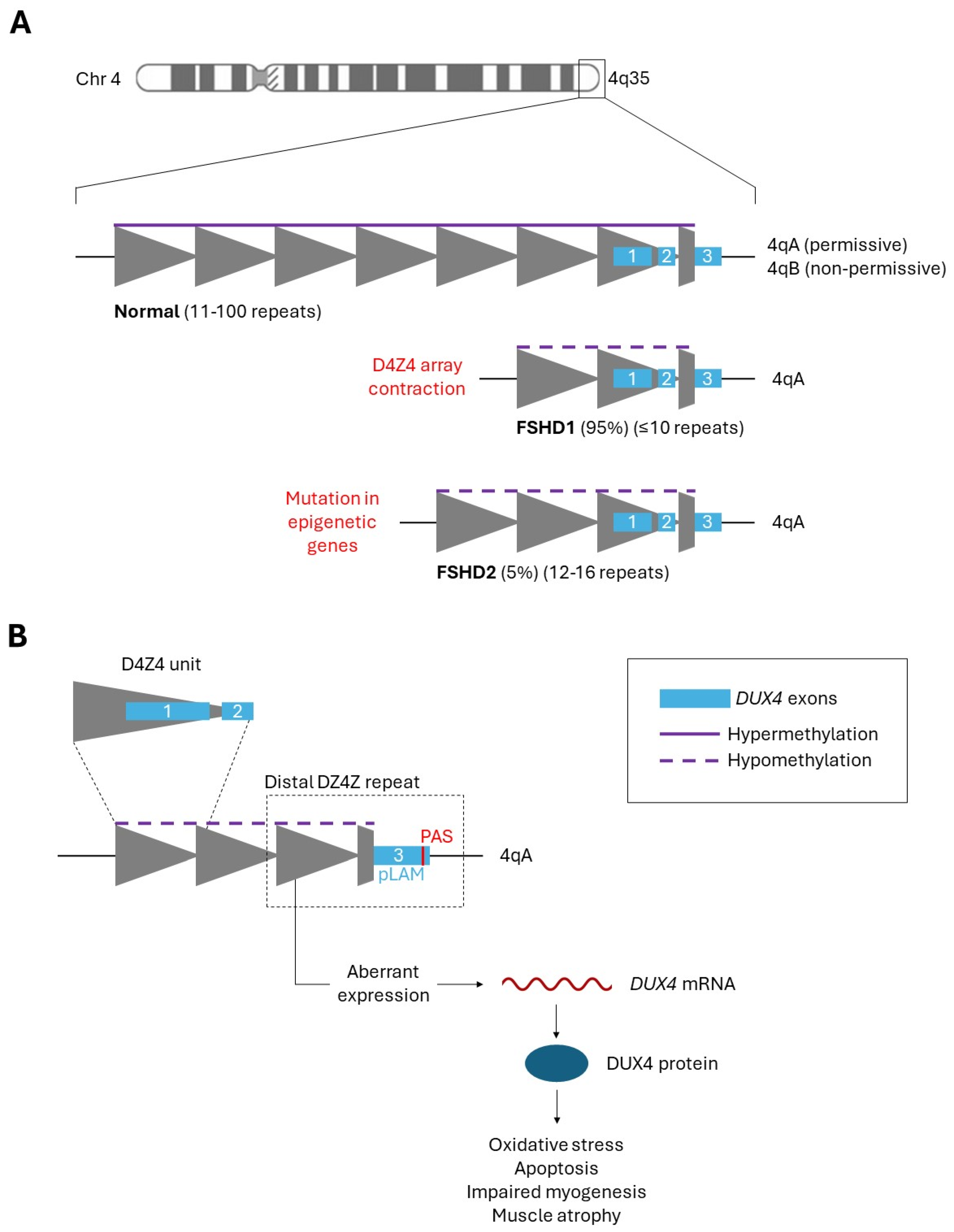
Figure 2.
Overview of oligonucleotide therapies for FSHD and where they inhibit DUX4 expression (gene, mRNA, or protein) to ameliorate FSHD symptoms. (1) sgRNA-dCas9-TRD targets the promoter or coding region of the DUX4 gene, resulting in transcriptional repression. (2) U7-snRNA alters the specificity of a small nuclear ribonucleoprotein complex (snRNP) to inhibit DUX4 pre-mRNA maturation. (3) Antisense oligonucleotides bind to DUX4 mRNA, causing steric blocking (and downstream effects) or RNase H-mediated degradation, depending on the AO type. (4) Various RNA molecules (miRNA, siRNA, etc.) degrade DUX4 mRNA through the RNA interference pathway. (5) Decoy dsDNA molecules have DUX4-binding motifs that trap DUX4 protein in a binding sink, inhibiting transactivation of downstream DUX4 targets. (6) DNA aptamers bind to DUX4 protein, inhibiting DUX4 activity through steric inhibition.
Figure 2.
Overview of oligonucleotide therapies for FSHD and where they inhibit DUX4 expression (gene, mRNA, or protein) to ameliorate FSHD symptoms. (1) sgRNA-dCas9-TRD targets the promoter or coding region of the DUX4 gene, resulting in transcriptional repression. (2) U7-snRNA alters the specificity of a small nuclear ribonucleoprotein complex (snRNP) to inhibit DUX4 pre-mRNA maturation. (3) Antisense oligonucleotides bind to DUX4 mRNA, causing steric blocking (and downstream effects) or RNase H-mediated degradation, depending on the AO type. (4) Various RNA molecules (miRNA, siRNA, etc.) degrade DUX4 mRNA through the RNA interference pathway. (5) Decoy dsDNA molecules have DUX4-binding motifs that trap DUX4 protein in a binding sink, inhibiting transactivation of downstream DUX4 targets. (6) DNA aptamers bind to DUX4 protein, inhibiting DUX4 activity through steric inhibition.

Figure 3.
Mechanism of action for (A) AO and (B) RNAi therapies. (A) Antisense oligonucleotides (AOs) can degrade target mRNA transcript by recruiting RNase H, or by inducing steric blocking. AO-mediated steric blocking of a target mRNA transcript can inhibit proper translation, splicing and/or stability. Further downstream degradation pathways can be initiated on steric blocked mRNA transcripts. (B) RNAi-based therapies (siRNAs, miRNAs, etc.) degrade target mRNA transcripts using the RNA interference (RNAi) pathway. RNAi can be induced by miRNAs or siRNAs complementary to a mRNA transcript. DICER processes the precursor molecules to produce miRNA or siRNAs, which then get loaded into the Argonaute (AGO) protein of the RNA-induced silencing complex (RISC). Using the guide strand, RISC targets a complementary mRNA transcript and induces translational inhibition, sequestration, and/or mRNA degradation (miRNA-RISC) or simply mRNA degradation (siRNA-RISC).
Figure 3.
Mechanism of action for (A) AO and (B) RNAi therapies. (A) Antisense oligonucleotides (AOs) can degrade target mRNA transcript by recruiting RNase H, or by inducing steric blocking. AO-mediated steric blocking of a target mRNA transcript can inhibit proper translation, splicing and/or stability. Further downstream degradation pathways can be initiated on steric blocked mRNA transcripts. (B) RNAi-based therapies (siRNAs, miRNAs, etc.) degrade target mRNA transcripts using the RNA interference (RNAi) pathway. RNAi can be induced by miRNAs or siRNAs complementary to a mRNA transcript. DICER processes the precursor molecules to produce miRNA or siRNAs, which then get loaded into the Argonaute (AGO) protein of the RNA-induced silencing complex (RISC). Using the guide strand, RISC targets a complementary mRNA transcript and induces translational inhibition, sequestration, and/or mRNA degradation (miRNA-RISC) or simply mRNA degradation (siRNA-RISC).
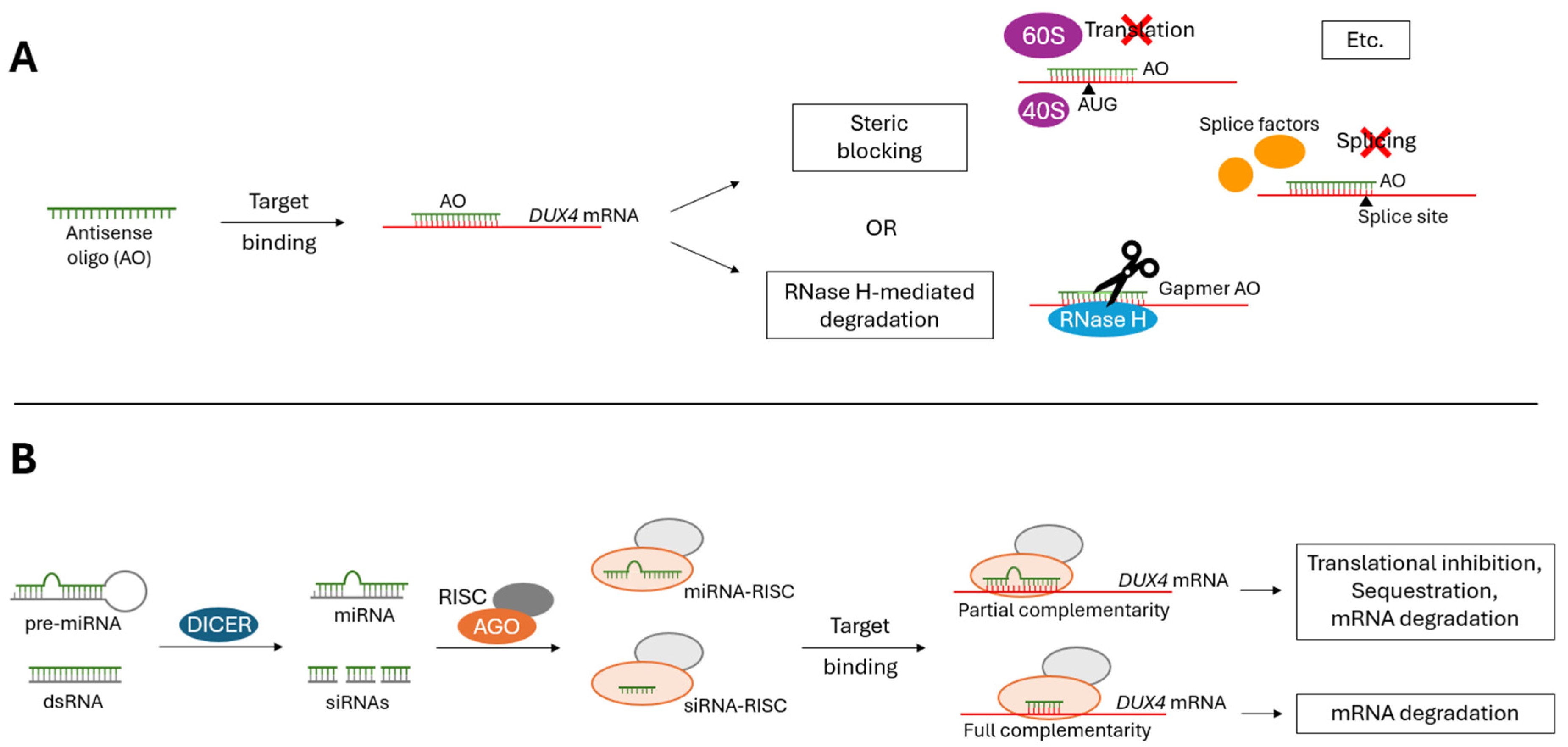
Figure 4.
Overview of oligonucleotide target sites on DUX4 mRNA. Orange (antisense oligonucleotides) and purple (RNAi) lines indicate oligo target sites. Partially overlapping target sites are simply represented as a continuous line. All attempted target sites are included, even oligos that showed poor indications in corresponding studies. Figure is not to scale and approximate oligo sites/sizes are shown. (Top) Schematic representation of DUX4 gene (downwards arrow indicates start codon; blue, open reading frame; boxes, exons; lines, introns; red line, polyadenylation signal). The distal D4Z4 unit and adjacent pLAM region are indicated by double-sided arrows. Note that the following groups used some or all the same oligonucleotides (indicated by *): Vanderplanck et al. (2011), Ansseau et al. (2017) and Derenne et al. (2020); Marsollier et al. (2016) and Chen et al. (2016); Marsollier et al. (2016) and Falzarano et al. (2021); Lu-Nguyen et al. (2021), Lu-Nguyen et al. (2022a) and Lu-Nguyen et al. (2022b); Wallace et al. (2012) and Wallace et al. (2018).
Figure 4.
Overview of oligonucleotide target sites on DUX4 mRNA. Orange (antisense oligonucleotides) and purple (RNAi) lines indicate oligo target sites. Partially overlapping target sites are simply represented as a continuous line. All attempted target sites are included, even oligos that showed poor indications in corresponding studies. Figure is not to scale and approximate oligo sites/sizes are shown. (Top) Schematic representation of DUX4 gene (downwards arrow indicates start codon; blue, open reading frame; boxes, exons; lines, introns; red line, polyadenylation signal). The distal D4Z4 unit and adjacent pLAM region are indicated by double-sided arrows. Note that the following groups used some or all the same oligonucleotides (indicated by *): Vanderplanck et al. (2011), Ansseau et al. (2017) and Derenne et al. (2020); Marsollier et al. (2016) and Chen et al. (2016); Marsollier et al. (2016) and Falzarano et al. (2021); Lu-Nguyen et al. (2021), Lu-Nguyen et al. (2022a) and Lu-Nguyen et al. (2022b); Wallace et al. (2012) and Wallace et al. (2018).
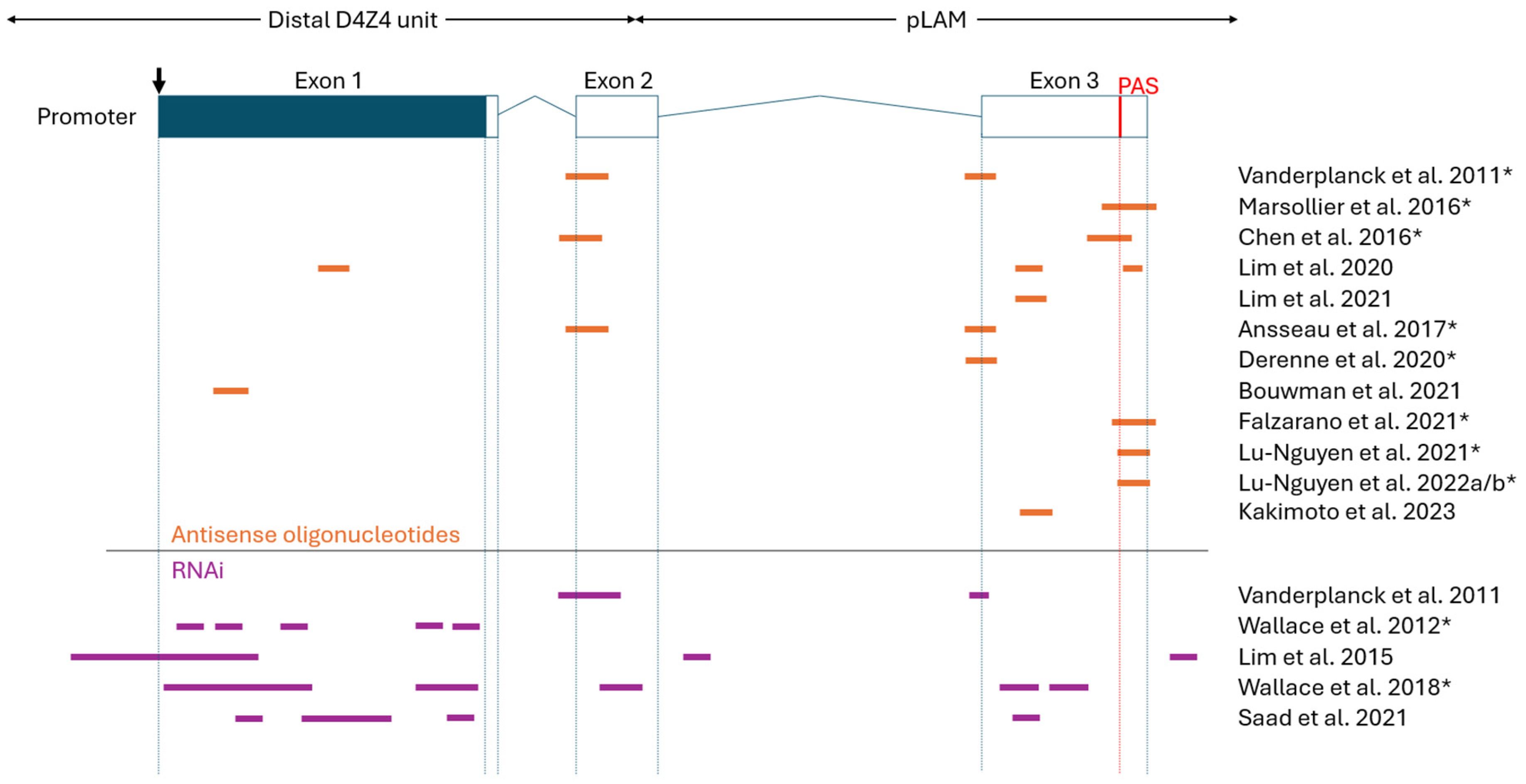

Table 1.
Overview of preclinical studies investigating antisense oligonucleotides to treat FSHD.
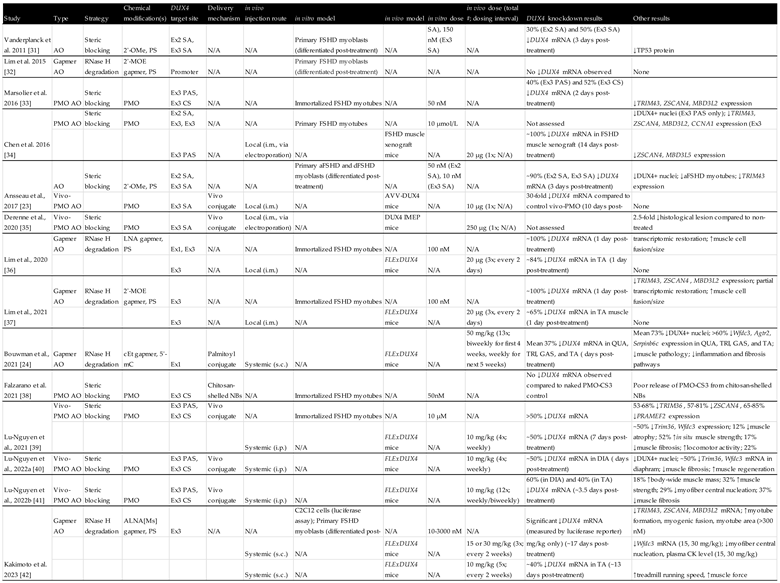
Table 2.
Overview of preclinical studies investigating RNAi-based oligonucleotides to treat FSHD.
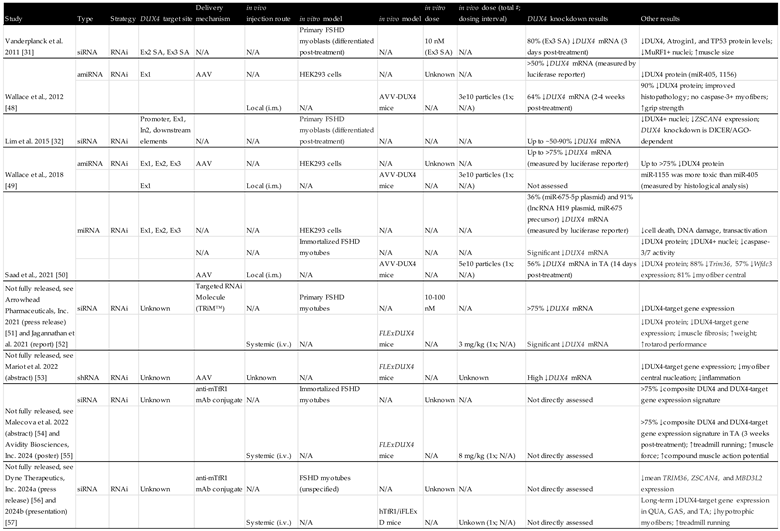
Table 3.
Overview of preclinical studies investigating other oligonucleotides to treat FSHD.

Table abbreviations: 2′-OMe, 2′-O-methyl; PS, phosphorothioated; PMO, phosphorodiamidate morpholino oligomer; LNA, locked nucleic acid; 2′-MOE, 2′-O-methoxyethyl; cEt, constrained ethyl; 5’-mC, 5’-methylcytosines; ALNA[MS], 2′-N-methanesulfonyl-2′-amino-locked nucleic acid; HEG, hexaethylene glycol; Ex, exon; In, intron; SA, splice acceptor; SD, splice donor; SE, splice enhancer; PAS, polyadenylation signal; CS, cleavage site; NBs, nanobubbles; AAV, adeno-associated virus; mAb, monoclonal antibody; i.m., intramuscular injection; s.c., subcutaneous injection; i.p., intraperitoneal injection; i.v., intravenous injection; QUA, quadriceps; TRI, triceps; GAS, gastrocnemius; TA, tibialis anterior; DIA, diaphram; iPSC, induced pluripotent stem cells; aFSHD, atrophic FSHD; dFSHD, disorganized FSHD; tEP, transient electroporation; IMEP, i.m. injection and electroporation of naked plasmid DNA; SELEX, systematic evolution of ligands by exponential enrichment; TR-FRET, Time-resolved fluorescence energy transfer; AGO, argonaute; mRNA, messenger RNA; siRNA, small interfering RNA; miRNA, microRNA; amiRNA, artificial microRNA; shRNA, short hairpin RNA; snRNA, small nuclear RNA; sgRNA, single guide RNA; CRISPR, clustered regularly interspaced short palindromic repeat; Cas, CRISPR-associated protein; dCas9, dead (endonuclease deficient) Cas9; KRAB, Krüppel-associated box; TRD, transcriptional repression domain; ADAR, adenosine deaminase acting on RNA; N/A, not applicable.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



