
Submitted:
24 July 2024
Posted:
25 July 2024
You are already at the latest version
Abstract
In recent years, bacterial resistance to conventional antibiotics has become a major concern in the medical field. The global misuse of antibiotics in clinics, personal use, and agriculture has accelerated this resistance, making infections increasingly difficult to treat and rendering new antibiotics ineffective more quickly. Finding new antibiotics is challenging due to the complexity of bacterial mechanisms, high costs and low financial incentives for the development of new molecular scaffolds, and stringent regulatory requirements. Additionally, innovation has slowed, with many new antibiotics being modifications of existing drugs rather than entirely new classes. Antimicrobial peptides (AMPs) are a valid alternative to small molecule antibiotics offering several advantages, including broad-spectrum activity and a lower likelihood of inducing resistance due to their multifaceted mechanisms of action. However, AMPs face challenges such as stability issues in physiological conditions, potential toxicity to human cells, high production costs, and difficulties in large-scale manufacturing. A reliable strategy to overcome the drawbacks associate with the use of small molecule antibiotics and AMPs is the combination therapy, namely the simultaneous co-administration of two or more antibiotics or the synthesis of covalently linked conjugates. This review is intended to cover and critically analyze the design, synthetic strategies, antibacterial activity and, when possible, the cytotoxicity of antibiotic-AMPs conjugates.
Keywords:
antimicrobial peptides
; antibiotics
; ‐lactams
; vancomycin
; aminoglycosides
; chemical conjugation
; combination therapy
1. Introduction
Antibiotics are a class of drugs, being among the highest prescribed drugs worldwide, specifically designed to combat bacterial infections by either killing bacteria or inhibiting their growth [1]. Discovered in the early 20th century, antibiotics revolutionized medicine and have saved countless lives by effectively treating previously lethal infections [2,3]. These medications work through various mechanisms, such as inhibiting or disrupting cell wall synthesis, protein production, DNA replication in bacteria, or interfering with metabolic pathways [4,5]. Despite their critical role in healthcare, the overuse and misuse of antibiotics have led to a growing issue of antibiotic resistance, where bacteria evolve mechanisms to evade the effects of these drugs [6,7,8]. Moreover, the fact that most antibacterial agents in use today are derived from natural products synthesized by bacteria or fungi to defend against bacterial competitors further facilitated the development of antimicrobial resistance (AMR). This resistance poses a significant challenge to global health, necessitating ongoing research and the development of new antibiotics and alternative treatments which are not advancing as fast as resistance [9]. Indeed, it is projected that by 2050 the annual fatalities could exceed 10 million unless appropriate measures are implemented, and research and development efforts are intensified to address AMR [10].
The most urgent need for new antibiotics is dictated to combat resistant strains, especially Gram-negative bacteria P. aeruginosa and E. coli, which have a second polar outer membrane and numerous efflux pumps that make them less susceptible to drug intervention, defined as critical pathogens and to a slightly lesser extent methicillin-resistant S. aureus (MRSA), defined as high priority [11]. Despite extensive screening efforts with traditional drug-like libraries, very few non-natural product-derived antibacterial agents have been discovered [12,13]. It has become increasingly apparent that the physicochemical properties required to evade these bacterial defenses differ from those needed for traditional drugs, since antibiotics are generally more polar and larger than drugs targeting other conditions [14]. Having evolved over millennia to target bacteria, antibiotic natural products inherently possess the necessary attributes to overcome bacterial defenses and have thus been successfully used for decades as starting points for semi-synthetically derived next-generation antibiotics [15]. However, finding new antibiotics is challenging not only because it is very difficult to find new scaffolds that are both effective and safe [16,17], being bacteria able to rapidly develop resistance to new antibiotics through mutations and horizontal gene transfer, often rendering new drugs ineffective shortly after they are introduced [6], but also due to economic and regulatory factors which hinder the development of new antibiotics. Developing a new antibiotic is expensive, often costing upwards of a billion dollars, which includes costs for discovery, preclinical testing, clinical trials, and regulatory approval [18]. Furthermore, antibiotics are typically used for short durations, unlike treatments for chronic diseases, resulting in lower financial returns and the pharmaceutical market prioritizes drugs with higher profitability, such as those for chronic conditions or lifestyle diseases, leading to underinvestment in antibiotics [19]. To compound the issue, new antibiotics are often held in reserve to delay resistance, further limiting sales. Moreover, regulatory agencies require extensive testing to ensure safety and efficacy, which can be particularly challenging for antibiotics due to the need for novel approaches and considerations of resistance [20]. Additionally, continuous post-approval surveillance for resistance and effectiveness in real-world use is necessary, adding to the complexity and cost of antibiotic development [21]. Nonetheless, some new antibiotics have been launched in the market during the last twenty-year demonstrating that the quest to fight AMR with small molecules is not at a death end [22].
One strategy to avoid the challenging search for new scaffolds and to chase an old antibiotics renaissance is to administer two or more antibiotics simultaneously to obtain a synergistic effect in the so-called combination therapy [23]. This approach can enhance the efficacy of treatment by attacking pathogens through different mechanisms, reducing the likelihood of resistance development. By combining drugs with complementary actions, combination therapy can achieve synergistic effects, lower required dosages, and minimize toxicity. Additionally, it can target a broader spectrum of pathogens, including multi-drug resistant strains [24]. However, the strategy may not always work in vivo because of the different pharmacokinetics of the antibiotics [25]. Differences in the absorption, distribution, metabolism, and excretion of the drugs can lead to suboptimal levels of one or more components, reducing overall efficacy and increasing the risk of side effects. Moreover, there is also a risk of synergistic toxicity, where the combined drugs produce greater adverse effects than expected. This can limit the dosage and effectiveness of the treatment.
Alternatively, combination therapy involving conjugates is an innovative approach to fight AMR. This strategy involves linking an antimicrobial agent with another molecule, such as a drug, peptide, or targeting ligand, to enhance its effectiveness and specificity [26,27]. Conjugates have shown promise in preclinical studies and are being explored for their potential to target a wide range of pathogens, including multi-drug-resistant bacteria [28]. Particularly interesting are the conjugates between conventional antibiotics and antimicrobial peptides (AMPs) because they possess several advantages compared to conjugates between small molecule antibiotics. Apart from an enhanced activity due to a synergistic effect and/or broader spectrum of activity, AMPs can significantly enhance the effectiveness of antibiotics through improved penetration and targeted delivery. Indeed, AMPs facilitate the penetration of antibiotics into bacterial cells by interacting, disrupting, or translocating cell membranes, thereby increasing the intracellular concentration of the antibiotic [29]. Furthermore, conjugation can enhance the specificity of antibiotics towards bacterial cells over host cells, reducing off-target effects and toxicity [30]. Moreover, the increased potency of antibiotic-AMP conjugates can lead to dose sparing, requiring lower dosages of each component and thus reducing potential side effects and toxicity [31]. Additionally, with improved targeting and lower dosages, the risk of adverse effects is minimized, making treatments safer for patients [32]. Lastly, the conjugates can be designed to incorporate a variety of AMPs and antibiotics with suitable linkers, allowing for tailored treatments against specific pathogens and resistance profiles.
This review aims to describe the rationale behind the design of the antibiotics-AMP conjugates appeared in literature [33], encompassing de facto AMPs and cell-penetrating peptides (CPPs), the strategies used for their synthesis, and briefly discuss their activity against the bacterial targets. This review is not intended to give a full overview of the different classes of antibiotics, AMPs and their mechanisms of action, which will be only summarized in the following two sections for the sake of a better comprehension of the main topic. The readers interested in a more detailed discussion on the features of the single class of antibiotics or AMPs can refer to the reviews cited therein.
2. Small Molecule Antibiotics
Small molecule antibiotics, both unconsciously and consciously, have been used in various forms for thousands of years. [34]. The identification of salvarsan as anti-syphilis drug in 1909 [35] followed by the serendipitous discovery of broad-spectrum antimicrobial penicillin by Fleming in 1928 [36] opened an incredibly prolific era during which different small molecule antimicrobials have been developed and marketed. More specifically, we witnessed two decades referred to as “the golden age of discovery” of antibiotics during which different classes of natural antibiotics have been isolated from natural sources and identified, followed by a second “golden age of medicinal chemistry” where successive generations of the natural scaffolds have been developed by chemical modification [37]. Both natural and synthetic antimicrobials can be classified according to the chemical core structure and mechanism of actions (Figure 1).
Sulfonamides are an important class of antibiotic drugs with a broad spectrum of activity, highly effective against gram-positive bacteria and some gram-negative bacteria, which were serendipitously discovered form the chemical dyes industry [3]. Sulfonamides act as competitive antagonists and structural analogues of p-aminobenzoic acid (PABA) in the synthesis of folic acid, which is essential for bacterial DNA production [38]. Thanks to chemical modification, they have witnessed 80 years of continuous use, not only as antibiotics but also as diuretics, antidiabetic, antiarrhythmic and COX2 inhibitors [39]. With the advent of penicillin and the increasing antibiotic resistance, the demand of new sulfonamides witnessed a decrease. Indeed, after the discovery of penicillin, β-lactam antibiotics encompassing penicillin and following generation cephalosporins, carbapenems, penems and monobactams (Figure 1) became probably the most popular class of antibiotics revolutionizing the treatment of infectious diseases [40]. Their action depends on the structure of the constrained β-lactam four membered ring common to all the generations which renders the amide bond prone to react with alcohols, and in particular with the side chain of serines belonging to the so-called penicillin-binding peptides responsible for the cell wall synthesis [41,42]. However, over the past 60 years, resistance to penicillins mostly due to the microbial production of β-lactamases but also to conformational changes in penicillin-binding proteins, permeability changes in the outer membrane; and activation of efflux pumps, has been steadily increasing so much that there are serious concerns that β-lactams may soon become ineffective against deadly bacterial infections [43,44,45].
Discovered in the 1940s, tetracyclines are a family of antibiotics that inhibit protein synthesis by preventing the attachment of aminoacyl-tRNA to the ribosomal acceptor (A) site [46]. Tetracyclines are broad-spectrum agents, effective against a wide range of gram-positive and gram-negative bacteria. Their favorable antimicrobial properties and lack of major adverse side effects have led to extensive use in treating infections in both humans and animals. However, during the two decades between 70s and 80s tetracyclines became a second choice in the treatments because of the development of growing availability of other antibiotics and acquired resistance mainly due to two major mechanisms such as efflux pumps and ribosomal protection proteins. Chemical modification, mostly performed on the D cycle of the scaffold, led to the development of three different generations of tetracyclines, the last one encompassing eravacycline, omadacycline, and tigecycline, which were approved within the past 15 years, representing a new era in the use these antibiotics (Figure 1) [47].
Also the era of aminoglycoside antibiotics started in the 1940s when streptomycin was discovered [48]. Aminoglycosides are a class of natural and semi-synthetic polyamino sugars most of them having in common a central cyclohexane ring referred to as 2-deoxystraptamine (2-DOS) to which different aminosugars are bonded through glycosidic linkage (Figure 1) [49]. Aminoglycosides have enjoyed widespread application against many types of Gram-positive and Gram-negative pathogens, being still widely used worldwide, due to their ability to interact mainly thorough electrostatic interaction but also through hydrogen bond networking with the A-site of the 16S ribosomal RNA and to the 50S ribosomal subunit perturbing the “proof-reading” process that ensure protein translation and inhibiting translocation and ribosome recycling respectively [50,51,52]. During the time, pathogens have developed three different mechanisms of bacterial resistance. The principal mechanism involves the inactivation of aminoglycosides by a family of enzymes referred to as aminoglycoside-modifying enzymes (AMEs) expressed by resistant strains which are able to acetylate the amino groups or phosphorylate the hydroxy functions. Resistance can also arise through mutations in the ribosomal target or, increasingly, through the modification of the ribosome by ribosomal methyltransferase enzymes. Lastly, the alteration of the bacterial cell wall by acquired lipid modifications that repel highly polar aminoglycosides improving impermeability and the activation of efflux pumps result in a lower concentration of aminoglycosides in the bacterial cells [53]. To overcome and fight antibacterial resistance, many derivatives of aminoglycosides have been synthetized starting from the natural scaffolds [54,55]. Amphiphilic aminoglycosides, aminoglycoside heteroconjugates, aminoglycoside homo- and heterodimers, as well as conformationally-restricted aminoglycosides have been synthetized and tested providing a better knowledge on their mechanism of action and paving the way for new applications of aminoglycosides as antifungal, anticancer and gene/drug delivery vectors [56,57].
Macrolide antibiotics, renowned for their high efficacy against Gram-positive bacteria and safety, have been extensively used in clinical settings for over 50 years since the first antibiotic, pikromycin was isolated in 1950 [58]. Natural macrolide antibiotics share a common structural feature, i.e. a macrocyclic lactone of different sizes (from 12 to 15 members) bearing one or more amino- or deoxy-sugar (Figure 1), being erythromycin the most popular [59]. As members of the largest class of antibiotics, they are particularly effective in treating upper and lower respiratory tract infections by reversible binding to the 23S rRNA at or near the peptidyl transferase center thus inhibiting the synthesis of bacterial proteins [60]. However, despite their excellent antibacterial activity, macrolides often suffer from poor bioavailability, unpredictable pharmacokinetics, and low stability in the acidic environment of the stomach. These limitations along with the emerging antibacterial resistance prompted early efforts to develop new derivatives with enhanced properties raising a growing interest toward the synthesis of next generations of macrolide antibiotics. The second generation of macrolide antibiotics are semisynthetic derivatives, mostly derived from erythromycin [61,62] whereas a third generation has been prepared through a fully synthetic platform technology by assembling a left-hand component and a right-hand component through reductive amination and subsequent macrolactonization [63].
During the same period, the first glycopeptide vancomycin was isolated and readily commercialized by Eli Lilly in 1958 paving the way to the glycopeptide antibiotics era overall to fight Staphylococcus aureus (S. Aureus), a microorganism able to elude most of the classic antibiotics [64]. Glycopeptide antibiotics are a class of heptapeptides which are sub-classified according to the identity of amino acids in position 1 and 2 (Figure 1) [65]. Biochemical studies indicate that vancomycin and other glycopeptides inhibit peptidoglycan synthesis by forming a stoichiometric 1:1 complex with the peptidoglycan precursor UDP-N-acetylmuramylpentapeptide through five hydrogen bonds with the acyl-D-ala-D-ala moiety, eventually inhibiting the transglycosylase enzyme and transpeptidase enzyme reactions fundamental for the synthesis of the rigid cell wall peptidoglycan [66]. Prior to 1984, the glycopeptide class included only few members beyond vancomycin, teicoplanin, ristocetin, and avoparcin. However, due to administration issues and side effects and with the acknowledgment of the threat posed by antibiotic resistance, the class of glycopeptides swelled to include thousands of natural and semi-synthetic compounds possessing multiple mechanism of action. The semisynthetic approaches to designing glycopeptide second generation can be broadly categorized into three strategies, namely 1) the functionalization of the functional groups on the outer shell of the parent peptides, 2) modification of the amino acids through disassembling and reassembling of the peptidic scaffold, and 3) dimerizing or trimerizing the glycopeptide through covalent bonding [65,67], being the first one probably the most exploited [68].
At the end of the “golden age of discovery” of antibiotics the first quinolone, nalidixic acid, was isolated and suddenly introduced into the clinic [69]. However, quinolones became the most often prescribed antibiotics in the world to treat a range of microbial diseases in humans only in the 80s when the second generation of this class of antibiotics was synthetized [70]. The common structural features of such compounds are a bicyclic skeleton, a carboxylic acid, and a keto group at positions 3 and 4, respectively, which are necessary for their pharmacological activity (Figure 1) [71]. Quinolones act as DNA synthesis inhibitors because they are able to bind bacterial enzymes DNA gyrase and DNA topoisomerase IV, which are enzymes that play fundamental roles in most nucleic acid processes, and convert them in cellular toxins [72]. Like the other class of antibiotics, also the targets of quinolones have developed antibiotic resistance mainly through three different mechanism: 1) target-mediated resistance due to the modification on gyrase and topoisomerase IV structures, 2) plasmid-mediated resistance due to the presence of enzymes able to acetylate the free nitrogen on the quinolone scaffold and the generation of efflux pumps, and 3) chromosome-mediated resistance though the under expression of porins and over expression of efflux pumps leading a decrease of concentration of the drugs in the cell. Due to the relative simplicity of the scaffold and to their wide spectrum anti-infective efficacy, quinolones have witnessed a lot of interest in the medicinal chemistry field in the search of novel derivatives [73].
3. Antimicrobial Peptides (AMPs)
Since the isolation of gramicidin A and B in 1930s AMPs have emerged as promising candidates against antimicrobial resistance due to their broad-spectrum activity and multiple/unique modes of action [74]. AMPs, also known as host defense peptides, are a class of naturally occurring molecules that play a crucial role in the innate immune systems of animals, plants, insects, and microorganisms [75]. They show activity against Gram-positive and Gram-negative bacteria and other pathogens, such as fungi, viruses, and parasites [76,77]. Moreover, they can also exert potent antibiofilm activity against multiresistant bacteria [78]. Currently, more than 60 peptides have been approved by the FDA and more than 400 are in clinical trials, but only seven have reached the market, used mainly as topical medications and, in cases involving serious infections, as injectables [79,80].
Structurally, AMPs are small peptides, from 12 to 50 amino acids in length and typically share several common physicochemical features, namely amphipathicity and the capacity to form stable secondary motifs (α-helical, β-sheet, mixed and cyclic structures) which are responsible for their biological activity [81]. Amphipathicity contributes significantly to the AMPs ability to selectively target microbial membranes. In fact, their hydrophilic region, primarily composed of cationic residues (Lys and Arg) plays a fundamental role in the initial binding to negatively charged components on bacterial membranes through electrostatic interactions. Unlike mammalian cell membranes, bacterial membranes are rich in negatively charged components, such as phospholipids in the peptidoglycan cell wall of Gram-positive bacteria, and lipopolysaccharide in the outer membrane of Gram-negative. Once bound, AMPs exert their antimicrobial effects mainly through two different mechanisms: bactericidal (membrane disruption causing cell lysis) and bacteriostatic (metabolic processes interference through nucleic acids binding and modulation of bacteria essential functions) [82,83]
The membrane-targeting mechanisms of AMPs can be described through three different models: the barrel-stave, the toroidal pore and the carpet. In the barrel-stave model, AMPs insert into the lipid bilayer of the microbial membrane, aligning themselves perpendicularly to the plane of the membrane. The peptides aggregate to form a pore or channel through the membrane, resembling the staves of a barrel. The formation of these transmembrane channels leads to the uncontrolled leakage of ions and other small molecules, disrupting cellular homeostasis and ultimately causing cell death [84]. Similar to the barrel-stave model, AMPs in the toroidal pore model insert into the membrane. However, unlike barrel-stave pores, toroidal pores cause the lipid monolayers to bend continuously through the pore, resulting in a toroidal (doughnut-like) structure. This bending disrupts the integrity of the membrane, allowing ions and other molecules to pass through the pore, leading to cell death [85]. In the carpet model, AMPs align parallel to the membrane surface, covering it like a carpet. The peptides interact with the lipid head groups, leading to a destabilization of the membrane. As the concentration of peptides increases, this destabilization causes the membrane to disintegrate in a detergent-like manner, resulting in cell lysis.
Initially, the bactericidal effects of AMPs were attributed only to membrane-active mechanisms. However, it has now been recognized that many AMPs target essential cellular components and functions, leading to bacterial death. These AMPs translocate into the cell membrane without disturbing it and inhibit critical cellular processes by interacting with intracellular targets. To date, several mechanisms have been identified, including the inhibition of protein and nucleic acid synthesis, as well as the degradation of enzymes and proteins [86]. Notably, some AMPs exhibit multiple modes of action, killing bacteria by both disrupting their membranes and interacting with intracellular targets [87]. One well-known intracellular target is genomic DNA. AMPs can bind to DNA, affecting the expression of related genes and inhibiting the synthesis of essential macromolecules. This interaction can also lead to the degradation of DNA, further hampering bacterial survival [88]. The ribosome, a key component of the translation machinery, is another significant target for AMPs. By interacting with 70S ribosomes, AMPs can inhibit protein synthesis. This interaction is facilitated by multiple hydrogen bonds and stacking interactions, effectively halting bacterial growth and replication [89]. Intracellular enzymes and organelles can also be targets for AMPs. By inhibiting or degrading essential enzymes, AMPs disrupt vital metabolic processes within bacterial cells. This inhibition can result in the accumulation of toxic intermediates or the depletion of necessary substrates, leading to cell death [90].
4. Antibiotic-AMP Conjugates
The synergistic effects of antimicrobial peptides with conventional antibiotics represent a promising strategy to enhance antibacterial efficacy and combat antibiotic resistance. In fact, by combining different mechanisms of action, this approach can overcome bacterial defenses and reduce the likelihood of resistance development [91]. One primary mechanism by which AMPs enhance the efficacy of conventional antibiotics is through membrane disruption. Many AMPs destabilize bacterial membranes, increasing permeability and allowing antibiotics to penetrate more easily. This increased uptake can significantly enhance the antibacterial activity of antibiotics that typically have limited access to intracellular targets. This synergistic effect, that can be obtained by physical coadministration of the antibiotics or by the synthesis of covalently linked conjugates, is particularly relevant for antibiotics which require access to the bacterial inner compartments to exert their effects [92,93].
Antibiotic-AMP conjugates are synthetized by anchoring conventional antibiotic to an AMP or CPP through a suitable bifunctional linker. The peptide has two points of attachment, the N-terminus or the C- terminus, even if the selective use of a side chain could be, in theory, taken into consideration. In general, the linker can be classified as a stable covalent linker or cleavable stimuli-responsive linker. With cleavable stimuli-responsive linkers the AMP and the antibiotic could each act independently upon entering the bacterial cell, targeting their respective sites. Conversely, if the conjugate molecules remain intact, they function as a single, multimodal antibacterial compound. This allows them to bind to and affect their targets simultaneously, with dynamics that may differ from those of the individual components. Probably, the most problematic task for the synthesis of these conjugates is to find the right point of attachment on the antibiotic, since they usually possess many reactive functional groups in their scaffolds. Accordingly, the following sections of this chapter are organized considering the antibiotic scaffold.
4.1. Vanconomycin-AMP Conjugates
Vancomycin, exhibiting one of the strongest bindings known for low-molecular-weight organic compounds with the D-Ala-D-Ala motif of the cell wall precursor lipid II, was initially considered the drug devoted to treat antibiotic resistant bacteria being immune to the development of resistance [65,66,67]. However, 30 years after its discovery different vancomycin-resistant strains, such as Enterococcus faecium (VRE), vancomycin-intermediate and resistant Staphylococcus aureus (VISA and VRSA) have been observed for which the discovery and development of novel antibiotics are urgently needed. In this contest, since clinical resistance to vancomycin took a lot of time to arise, the modification of this glycopeptide could be a successful strategy. From a synthetic chemical perspective, the selective functionalization of vancomycin could seem very difficult due to the presence of many functional group. However, it has been shown that there are for functional groups, referred to as point of attachments, which can be exploited for the selective functionalization due to their unicity or particular reactivity (Figure 2). First, the carboxylic acid at the C-terminus of the peptide sequence is the only carboxy functional group present in the vancomycin scaffold, thus can be selectively reacted with amines upon worthy activation. Also the amino functional group at the N-terminus can be selectively coupled with activated carboxylic acids but only when it is not methylated (R = H, norvancomycin). Indeed, in vancomycin (R = Me) there is a more reactive amino group in the glycosyl moiety (vancosamine) that is more reactive in the presence of activated carboxylic acids because less sterically congested. Finally, the fourth point of attachment is the resorcinol aromatic carbon in ortho position of the two hydroxy groups that, being very electron rich, readily undergo electrophilic aromatic substitution with iminium salts in situ produced by reaction of formaldehyde and primary amines.
One way to face vancomycin-resistant strains is to modulate the structure of the drug to increase membrane binding and selectivity eventually enhancing drug concentration at the target site [94]. Accordingly, a library of vancomycin derivatives, referred to as vancapticins 1, was designed by coupling the free carboxylic acid on the glycopeptide with different cationic peptides, mostly polilysines, having distinct lipophilic membrane-insertive elements (MIE) tethered at the N-terminus through two linkers, one of them built on a cleavable disulfide bond (Figure 3). Structure-activity relationship studies (SAR) revealed that vancapticins 1 possess enhanced membrane affinity, which boosts their effectiveness against MRSA and various other Gram-positive bacteria. Additionally, vancapticins 1 retain their potency against strains that are resistant to traditional glycopeptides.
The same strategy, namely tethering polycationic peptides to vancomycin to fight antibacterial resistance, have been exploited for the synthesis of two vancomycin-polyarginine conjugates [95]. Exploiting again the reactivity of the free carboxylic acid, vancomycin was tethered to the N-terminus of D-octaarginine (r8) through not cleavable aminohexanoic acid (Ahx) linker obtaining the conjugate 2 (Scheme 1A) which could have a stronger affinity for the surface of cell membrane and enhanced cell permeability facilitating the action of vancomycin in arresting the cell wall synthesis and giving to vancomycin the access to intracellular binding targets. Octaarginine R8 4 was prepared trough solid phase peptide synthesis (SPPS) and coupled with Cbz-Ahx-OH 3 in solution leading to the formation of r8-Ahx-r8 5 after hydrogenolysis of the Cbz protecting group (Scheme 1B). Intermediate 5 was finally coupled to vancomycin producing the final conjugate vacomycin-Ahx-r8 2, which resulted to be much more active than vancomycin by orders of magnitude against difficult-to-treat MRSA populations, such as biofilms and persister cell, maintaining comparable minimal inhibitory concentration (MIC) against vancomycin-resistant Gram-positive organisms such as VISA and VRE.
Following the same rational, different vancomycin-polyarginine conjugates were synthetized at four distinct point of attachments, namely the free carboxylic acid (VC), the carbon in orto position to the hydroxy groups of the resorcinarene ring (VR), the N-methylammino function on the leucine residue (VN), and the free amino function on the glycosyl frame (VV) (in Scheme 2A the structure of the most performant VN derivative FU002 6 is represented) [96]. Apart from the site of attachment, the structures of the conjugates are very similar, being composed of the vancomycin antibiotic 7, a heterobifunctional cross linker and hexaarginine tagged with Cys at the C-terminus 9. The lead candidate FU002 6 was prepared by site specific coupling of vancomycin with sulfo-succinimidyl 4-(N-maleimidomethyl)cyclohaxane-1-carboxylate (sulfo-SMCC) providing intermediate 8 which was clicked in solution with Cys-(Arg)6-NH2 9 affording FU002 6 (Scheme 2B). FU002 6 showed a remarkable increased activity against the most important types of vancomycin-resistant bacteria having additional mechanism of action beyond the interaction with the D-Ala-D-Ala moiety responsible for the cell-wall synthesis and superior pharmacokinetics.
With the aim to increase the cell-permeability of vancomycin but also to exploit the synergistic effect of two antibiotics belonging to different classes, Adams et al. conjugated vancomycin to amphiphilic AMPs Hectate (Hec) which is an amphiphilic peptide with a net positive charge and α-helix predominant conformation (Figure 4) [97]. Exploiting the reactivity of the free carboxylic acid on vancomycin and without the use of any linker, vancomycin and Hec were coupled in solution producing Van-Hec conjugate 10 [98]. The synergistic effect of vancomycin conjugate to Hec, differently from the two antibiotics alone cause the disruption of the bacterial cell wall integrity resulting very active against wild type MRSA and VRSA.
Inspired by the vancomycin conjugates showed before and considering that Gram-negative strains are challenging to fight due to the presence of the impermeable lipopolysaccharide (LPS)-rich outer membrane [99], a novel series of conjugates have been recently designed and synthetized by tethering vancomycin to antimicrobial LPS binding peptides which have been previously demonstrated exhibiting strong effect against Gram-negative bacteria [100]. Actually, a library of 80 conjugates, referred to as vancomycin-LPS binding peptide conjugates (VPCs) has been synthetized exploiting all the four points of attachment on the vancomycin scaffold, different chemical inert bifunctional linkers, comprising alkyl and PEG linkers, and a collection of 6 LPS binding peptides [101]. After a first generation of conjugates where short peptides were tethered through click azide-alkyne reaction which showed modest MICs against Gram-negative strains, even if better activity that vancomycin against E. faecium Gram-positive strain, a second generation of VPCs was prepared, the structure of the most active of which, conjugate VPC 11, is showed in Scheme 3A. For this second generation of conjugates a different chemical strategy was chosen, namely functionalization of the vancomycin core 7 with bifunctional linkers 12 leading to the formation of intermediate 13 having a maleimide moiety at the other end of the vancomycin point of attachment, which was clicked in solution with Cys-functionalized LPS binding peptides 14 previously prepared on SP (Scheme 3B). Some of these conjugates were further functionalized on a different vancomycin point of attachment with lipophilic tails to study the synergistic affect of the latter and LPS binding peptides when tethered to vancomycin scaffold. The results in terms of MICs of VPNs compared to vancomycin against Gram-positive and Gram-negative strains showed an increase of activity against VRE and Gram-negative strains such as AB1157 (E. Coli), A. baumannii, PA01 (P. Aeruginosa), and K. pneumonia, showing that modification with LPS binding peptides (and further lipophilic tails) alters the antimicrobial profile of vancomycin in the fighting Gram-negative bacteria.
4.2. lactams-AMP Conjugates
With the evolution of many microorganisms that developed resistance to β-lactam antibiotics manly due to the widespread diffusion of β-lactamase enzymes, a huge effort has been devoted, and it is still ongoing, by the scientific community in the quest of new derivatives which eventually lead to new generations of these antibiotics. Since the β-lactam ring, the main feature for their activity, is very labile, the vast majority of the β-lactams analogues arose from chemical modification of the pharmacophoric scaffold rather then total synthesis [40]. Actually, both the penicillin scaffold and cephalosporin scaffold posses two functional groups, namely the amino and the carboxylic acid groups, easily to be derivatized upon protection of the other (Figure 5). Moreover, cephalosporin possesses a further point of attachment consisting in the hydroxy group at the 3’ position.
One of the drawbacks that limits the use AMPs as therapeutic agents is their toxicity mainly caused by the presence of different cationic moieties. Indeed, it has been shown that after blocking the amino groups of polymyxin E as methane sulphonate the resulting prodrug can be used systematically [102]. Inspired by the prodrug concept, the first β-lactam-AMP conjugate was synthetized by linking cephalotin to D-Bac8c(Leu2,5), an enantiomeric derivative of AMP Bac8c where the two D-isoleucine amino acids in position 2 and 5 are substituted with D-leucine, producing a conjugate, cephalotin- D-Bac8c(Leu2,5) 15, which has reduced net positive charge due to the presence of a carboxylate (Scheme 4A) [103]. The point of attachment chosen was the hydroxy group on commercially available 7-aminocephalosporanic acid (7-ACA). Accordingly, 7-ACA 19 was first deacetylated to obtain the required free 3’-OH group by using mild tetrabutylammonium hydroxide (TBAOH) at low temperature to avoid the β-lactam amide hydrolysis, then reacted with thienylacetyl chloride producing the amide intermediate 20 (Scheme 4B). Next, after protection of the carboxylic acid as diphenylmethyl ester, the OH group was converted into the corresponding tetrachloroethyl carbamate 21, which was finally transformed in 22 by reaction with propargyl amine followed by the ester protecting group cleavage in acidic condition. Resin bound D-Bac8c(Leu2,5)-NH2 16 was prepared by standard SPPS according to the Fmoc/tBu protecting groups strategy and converted to D-Bac8c(Leu2,5)-N3 18 by diazotransfer reaction with imidalole-1-sulfonyl azide hydrochloride and cleavage from the resin. Finally, click reaction between 22 and 18 was performed in solution leading to the formation to the target cephalotin- D-Bac8c(Leu2,5) 15. The conjugate 15 is considered a prodrug since the carbamate moiety of the conjugate acts as cleavable linker when the β-lactam ring is hydrolyzed in the presence of β-lactamases, delivering the free peptide.
However, even if the final conjugate 15 could be potentially used in systemic therapies preventing toxicity issues, it resulted to have a slightly lower MIC than the parent free peptide against E. Coli and MRSA probably due to a lower uptake.
A second exploited point of conjugation on β-lactam antibiotics is the free amino function on the β-lactam ring. Accordingly, Wade et al. investigated the possibility to link the N-terminus of three cationic AMPs, namely MSI-78, CA(1-7)M(2-9)NH2 and des-Chex1-Arg20, to the amino function of 7-ACA 19 and cephalosporins precursor 7-aminodesacetoxycephalosporanic acid (7-ADCA) 29 directly on solid phase through glutaric acid linker, producing 6 AMP-β-lactam conjugates 23-28 (Scheme 5A) [104]. Accordingly, by protecting the amino function of 7ACA and 7-ADCA as NH-Boc carbamate and the carboxylic acid as Fmoc-ester they obtained intermediates 30 that were selectively Boc-deprotected generating derivatives 31 that can be readily used in SPS. The AMPs were grown on Rink resin and after Fmoc-deprotection of the last amino acid of 32, they were coupled with glutaric anhydride leading to the formation of resin-bound peptides 33 which were coupled with 31, cleaved form the resin producing 34 and finally deprotected at the carboxylic function of the β-lactams in solution affording conjugates 23-28. The activity of these conjugates was measured against different nosocomial pathogens and only in one case did the conjugate MSI-78-ACA-25 and MSI-78-ADCA 26 revealed synergistic effect against A. baumannii and MDR A. baumannii 156.
Another small library of 4 β-lactam antibiotics-AMPs conjugates was synthetized exploiting the free amino group on the β-lactam ring and a stimuli responsive disulfide linker [105]. The rationale behind the design of such conjugates is to exploit the ability AMPs to cross the inner and outer bacterial cell membrane of Gram-negative bacteria to help the β-lactam antibiotic to reach his targets after the cleavage of the disulfide linker in the periplasm and cytosol [106]. Accordingly, ampicillin (Amp), herein used as a model β-lactam antibiotic, was tethered at either the N- and C-terminus of two AMPs having different characteristics, namely membrane-disrupting magainin analogue 2P2-2 that was developed by the same group [107] and proline-rich oncocin which is able to cross the inner and outer membranes without membrane lysis [108], producing the four conjugates 35-38 represented in Scheme 6A. Amp 43 was functionalized with 3-(2-pyridyldithio)propionic acid N-succinimidyl ester (PDPS) to obtain intermediate 44 which was coupled in solution with the two AMPs which were previously synthetized through SPPS and tagged with the required Cys either at the N- and C-terminus, yielding the target Amp-AMP conjugates 35-38 having cleavable disulfide linker. The MIC values of the four conjugates, along with undecorated AMPs, physical mixtures of AMP + Amp, and conjugates built with non-cleavable thioether linker were evaluated against Gram-negative bacteria E. Coli BW 25113 and A. baumannii ATCC 19606 and Gram-positive Staphylococcus epidermidis ATCC 12228. Interestingly, derivative Amp-SS.9P2-2 37 showed significantly increased activity against Amp-resistant A. baumannii and no cytotoxicity against HEK cells, whereas the oncocin-conjugates did not show enhanced antimicrobial activity probably due a lower membrane permeability induced by the introduction of the Cys tag on the peptide sequence. It is worth noting that conjugate 37 is the first β-lactam-AMP conjugate that showed remarkably increased activity against ampicillin-resistant Gram-negative bacteria highlighting the efficiency of an approach based on tethering suitable antibiotics with ad hoc AMPs through a cleavable linker.
4.3. Aminoglycoside-AMP Conjugates
Aminoglycosides have a broad spectrum of activity against pathogenic bacteria, which has led to their extensive use and occasional misuse over the past seventy years, establishing them as a valuable class of antibiotics. However, their clinical effectiveness is hampered by the toxic side effects associated to their use, in particular nephrotoxicity and ototoxicity, and by evolution of different mechanisms of resistance developed by bacteria, encompassing the decrease in AGs uptake and emergence of aminoglycoside-modifying enzymes (AMEs) [53,55]. Much effort has been dedicated, and it is still ongoing, to the chemical modification of the natural aminoglycosides to face a renaissance of these antibiotics. As glycopeptide antibiotics, also the chemical functionalization of aminoglycosides could seem tricky at a first sight due to the presence of many identical functional groups, i.e. hydroxy groups and amino groups that have clearly the same reactivity. However, aminoglycosides have primary alcohols or amino groups bonded to primary carbons that could be exploited for a selective functionalization because less sterically congested (Figure 6).
The first aminoglycoside-AMP conjugate appeared in literature, i.e. Pentobra 45 (Scheme 7A) was designed to target persister bacterial cells and to combat anaerobic bacterium Propionibacterium Acnes (P. acnes) which are difficult to treat since charged antibiotics are not able to penetrate into the largely lipophilic sebaceous membrane [109]. To increase the bacterial permeability of tobramycin without missing its ribosomal activity, Pentobra 45 was designed by linking tobramycin to a short 12mer AMP with ability to selectively permeate bacterial membrane, through a succinyl linker [110,111]. Accordingly, after Boc-protection of the amino groups of tobramycin, the primary hydroxy group of 46 was selectively functionalized with succinic anhydride leading to the formation of intermediate 47 which was coupled in SP to the N-terminus of the AMP producing after cleavage from the resin Pentobra 45 (Scheme 7B). The conjugate showed high activity against E. coli and S. aureus persister cells and a wide range of P. acnes thanks to the synergic effects, namely membrane activity and inhibition of protein synthesis [110], along with no adverse effect and anti-inflammatory activity [111].
In the attempt to increase the potentiality of Pentobra 45 in terms of accumulation in bacteria by increasing membrane permeability and limiting the action of the efflux systems activated by the bacterial species, the same group synthetized a collection of four new kanamycin-AMP conjugates, MAAP02-05 50-53, where the peptide transporter sequence is modified according to sequence principles based on quantum mechanical models for membranes-permeating peptides (Scheme 8A) [112]. Probably due to the low yield obtained in the functionalization of tobramycin for the synthesis of Pentobra 45, a new synthetic pathway has been employed. Accordingly, the primary hydroxy group of Boc-protected tobramycin 46 was selectively transformed in tosylate upon treatment with tosyl chloride in pyridine and substituted by an azide leading to the formation of intermediate 54 which was clicked with Fmoc-NH protected propargyl-alanine affording conjugate 55 (Scheme 8B). The free carboxylic acid was used to anchor 55 to 2-cholotrytyl chloride resin (CTC) where the AMP peptide was grown through Fmoc-strategy providing after cleavage the synthesis of two 13-mer and two 12-mer tobramycin-AMP conjugates, referred to as MAAPCs 50-53. The MAAPCs demonstrated good selectivity for bacterial cell membranes over mammalian cell membranes and do not cause significant hemolysis of human red blood cells. They also exhibit superior antibacterial activity against actively growing Gram-negative E. coli compared to Gram-positive S. aureus. Among them, MAAPC05 53, along with pentobra 45, exhibit the highest inner membrane permeability, which correlates well with antimicrobial activity against persisters, showing much better activity than tobramycin alone.
With the same rational, namely to increase the ability of aminoglycosides to cross bacterial cell membranes, aminoglycoside-AMP conjugates were designed to fight bacterial pathogens encompassing MRSA, Salmonella, Mycobacterium, and Brucella, which are internalized within mammalian cells macrophages [113,114,115]. Since aminoglycosides along with other antibiotics are characterized by insufficient membrane permeability within macrophages and suffer drug efflux, kanamycin was tethered to a modified proline-rich cell penetrating peptide with intrinsic, nonmembrane lytic antimicrobial activity targeting intracellular pathogenic bacteria [116], through both a cleavable disulfide linker or non-cleavable alkyl linker, generating P14kanS 58 and P14kanC 59, respectively (Scheme 9A) [117]. Boc-protected kanamycin 60 was reacted with 4,4’-dithiobutyric acid or sebacic acid giving rise to the formation of mixtures of isomers form which compounds 61 and 62, respectively, were isolated and fully characterized by NMR spectroscopy. The obtained intermediates 61, 62 were coupled in SP to the N-terminus of the proline-rich AMP generating after cleavage from the resin the target conjugates P14kanS 58 and P14kanC 59, respectively (Scheme 9B). Very interestingly, P14kanS 58 was more potent than P14kanC 59, P14LRR AMP and non-covalent mixture of kanamycin and P14LRR against different Gram-negative and Gram-positive bacteria, encompassing intracellular pathogens. Since the conjugates did not lyse membranes, as demonstrated by monitoring β-galactosidases release form E.coli after addition of the conjugates, these results demonstrated the synergistic effect of the antibiotics which can operate when the disulfide bond is cleaved in the reductive environment inside the cell and kanamycin is released. Moreover, in a successive work, P14kanS 58 has proved to have potent antimicrobial activity against ESKAPE pathogens along with anti-inflammatory activity and a great ability to treat biofilms [118].
Another proline-rich antimicrobial peptide (PrAMP) is Bac7, and in particular the segments Bac7(1-16) and Bac7(1-35), the 16-mer and 35-mer N-terminal segments that showed comparable antimicrobial activity than the parent full peptide [119,120]. These functional fragments are referred to as bacteria-penetrating peptides (BPPs) since they cross the bacterial inner membrane via the SbmA transporter without permeabilizing the membrane at active concentrations to eventually interact with the target ribosome and inhibit protein synthesis. To target bacterial ribosomes with two distinct synergistic mechanisms, Bac7(1-16) and Bac7(1-35) fragments were tethered to tobramycin with a cleavable disulfide linker that would release the active components in the intracellular reductive environment (Scheme 10A) [121]. Resin bound Cys 66, which was obtained by anchoring FmocNH-Cys(Tr)-OH to the Rink resin, was Fmoc deprotected and coupled with succinyl Boc-trobamycin 47, obtained as described in Scheme 7B, leading to the formation of tobramycin-Cys conjugate 67 after cleavage from the resin (Scheme 10B). 67 was reacted with 2,2’-dithiopyridine to yield 68 that was submitted to conjugation with Bac7(1-15)[Cys16]NH2 and Bac7(1-35)[Cys36]NH2 producing the final hybrid antibiotics mTob-Bac7(1-15)[Cys16]NH2 64 and mTob-Bac7(1-35)[Cys36]NH2 65, respectively. The resulting conjugates showed activity against strains to which tobramycin and the Bac7 segments were inefficient, such as clinically isolated Gram-negative bacteria strains E.coli and P. aetuginosa and others Gram-negative species (A. baumanii and S. enteridis), proving that the conjugation strategy is rewarding even if the real mechanism of action is not yet clear.
The antibacterial activity of PrAMPs depends also on the propensity of such peptides to assume more stable secondary conformations that have been shown very important for their ability to permeate and destabilize the bacterial cell membrane. A common strategy to stabilize the secondary structure of peptides, other than the introduction of prolines in the sequence, is the peptide stappling [122], a technique that has been successfully exploited in the design of active stapled antimicrobial peptides (StAMPs) [123]. A peptide that has witnessed an improvement in terms of proteolytic stability and antibacterial activity thanks to the stabilization of its helical structure upon hydrocarbon stapling is anoplin [124]. Recently, anolplin and stapled anoplin have been tethered to amikacin and neomycin through both non-cleavable triazole linker and a cleavable disulfide linker generating a small library of two not cleavable neomycin-anoplin conjugates, namely Neo-anoplin 69 and Neo-anoplin[2-6] 70, two cleavable neomycin- anoplin conjugates, i.e. Neo-SS-anoplin 71 and Neo-SS-anoplin[2-6] 72 and two not-cleavable amikacin-anoplin conjugates, i.e. Amk-anoplin 73 and Amk-anoplin[2-6] 74, which structures are reported in Scheme 11A [125].
For the synthesis of these conjugates, the presence of only one primary hydroxy group on both the aminoglycosides neomycin and amikacin was exploited. As an example, neomycin was first Boc protected at the amino functions, then reacted with bulky triisopropylsulfonyl chloride (TIPS-Cl) to selectively transform the primary hydroxy group in sulfate which was transformed into the corresponding azide 75 by nucleophilic substitution (Scheme 11B). Azide 75 can be either clicked with anoplin or anoplin[2-6] functionalized at the N-terminus with dec-9-ynoic acid for the synthesis of the not-cleavable derivatives 69, 70, respectively, or reacted with thiourea followed by 2-mercaptopryridined to afford intermediate 76, which were reacted in solution to anoplin or anoplin[2-6] functionalized at the N-terminus with Cys to afford cleavable Neo-SS-anoplin 71 and Neo-SS-anoplin[2-6] 72, respectively. In this case, the conjugates obtained, regardless the nature of the linker and the structure of the AMP were only slightly more active, or as active as the corresponding components, showing no synergistic effect
The improvement of the uptake of aminoglycosides to make them able to fight intracellular bacterial infections has been explored also by combination with CPPs. In particular two peptides, α1H and α2H which are two α-helices responsible for the penetration ability of the bacterial effector protein YopM into eukaryotic cell [126], have been tethered to gentamycin trough a not-cleavable linker producing two conjugates, namely α1H-gentamycin 77 and α2H-gentamicin 78 along with a third conjugate synthetized by anchoring gentamycin to the well-known polyarginine Tat peptide, i.e. Tat-gentamycin 79 (Scheme 12A) [127]. Accordingly, by reacting gentamycin 80 with cross linker succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) the intermediate 81 was obtained as the main product (Scheme 12B). 81 was then clicked trough thiol-maleimide chemistry to Cys-modified Tat, α1H and α2H peptides leading to the formation of the final conjugates 77-79, respectively.
Both α1H, α2H and Tat peptides were able to promote cellular internalization of gentamycin since the corresponding conjugates 77-79 were active against multiple intramolecular Gram-negative pathogenic bacteria, such as E. coli K1, Salmonella enterica and Shigella flexneri.
4.4. Miscellanous
Apart for β-lactams, vancomycin and aminoglycosides, also other classes of antibiotics have been used to build antibiotic-AMP conjugates.
Inspired by the observation that when fluoroquinolones are administered with AMPs the resulting cocktail showed a synergistic effect broadening the antibacterial spectrum of the antibiotics along with a decreasing therapeutic dose that would result in a lower adverse reactions [128,129], Toh et al. reasoned that a similar result could be obtained by linking levofloxacin to indolicin, an AMP with a broad spectrum of activity also against Gram-negative and Gram-positive bacteria [129], through a labile ester linkage or a more stable amide linker. The conjugation produced two AMP-levofloxacin conjugates, namely the prodrug levo-O-indolicidin 82 and the corresponding amide derivative levo-N-indolicidin 83, which ability to cross the outer membrane of bacteria could be higher than that of levofloxacin due to the present of the highly lipophobic peptide (Scheme 13A) [130]. The two conjugates 82, 83 were synthetized through SPPS by reacting the free carboxylic acid of levofloxacin 86 with the N-terminus of the peptide tagged with glycolic acid or Gly 84, 85 respectively (Scheme 13B). While the physical mixture of indolicin and levofloxacin was slightly more active compared to both antibiotics, in particular against B. subtilis ATCC 6633, the conjugates did not show the same effect.
Levofloxacin, along with another fluoroquinolone ciprofloxacin, was also tethered to a different AMP, namely HLopt2 which is an antimicrobial analogue of HLP-2 a segment of Lactoferrin with potent antimicrobial activity against both Gram-negative and Gram-positive bacteria [131]. The three conjugates were designed and synthetized to increase the permeability of the fluroquinolone antibiotics thanks to the ability of HLopt2 to destroy the bacterial cell trough pore formation mechanisms (Figure 8) [132]. All the conjugates were synthetized trough SPPS, LVX-HLopt2-NH2 87 by anchoring the carboxylic acid of levofloxacin to the N-terminus of HLopt2, CIP-CH2CO-HLopt2-NH2 88 by coupling the secondary amine of ciprofloxacin with the N-terminus of HLopt2 previously functionalized with bromoacetic acid, while CIP-Cys-SS-HLopt2-NH2 89, the only conjugate with a stimuli-responsive linker, by formation of a disulfide bond between Cys-modified ciprofloxacin and Cys residue linked to the N-terminus of HLopt2 [133]. Interestingly, all the conjugates showed increased activity along with low toxicity to mammalian cell and very low hemolytic activity, being CIP-Cys-SS-HLopt2-NH2 89 the most active against S. aures due to the reducing environment that trigged the disulfide bridge cleavage with the corresponding release of the two antibiotics.
Other than to increase the permeability of small molecule antibiotics, the conjugation strategy with AMP could be exploited to overcome the non-specificity of potent broad-spectrum antibiotics which suffer severe toxic side effects [134]. For instance, chloramphenicol (CAP) is one of the most effective broad-spectrum antimicrobial agents which clinical use was hampered by its high risk of bone marrow toxicity [135]. CAP, being lipid-soluble, diffuses through the bacterial cell membrane and reversibly binds to the L16 protein of the 50S subunit of bacterial ribosomes. This binding prevents the transfer of amino acids to growing peptide chains, likely by suppressing peptidyl transferase activity, thereby inhibiting peptide bond formation and subsequent protein synthesis. To overcome its no specificity, chloramphenicol was tethered to UBI29-41 which is a cationic AMP highly investigated for its capacity to bind bacteria with high affinity [136], through a not cleavable glutaric linker, leading to the formation of CAP- UBI29-41 90 (Scheme 14A) [137].
CAP 91 was reacted with glutaric anhydride yielding a mixture of products due to the indiscernible reactivity of the two hydroxy groups, from which intermediate 92 was isolated in around 50% yield. After that, 92 was coupled in solution with commercially available UBI29-41 yielding, after HPLC purification, CAP-UBI29-41 conjugate 90. Gratifyingly, in vitro studies demonstrated that CAP-UBI29-41 90 has enhanced antibacterial effects on S. aureus and E. coli., also showing significantly reduced toxicity to normal cells compared to CAP. Most importantly, this result was obtained also in bacteria-bearing mouse models, indicating that UBI29-41 is an ideal targeting ligand for constructing antibacterial agents for bacteria-targeting therapy.
Differently form CAP, selectivity is not a big issue for the use of macrolides, since their mechanism of action depends on their affinity to the so-called “macrolide binding site” which allows them to inhibit selectively translation in bacteria. However, this class of antibiotics suffers antibiotic resistance due to the ability of bacteria to modify the target binding site [59]. To fight antibacterial resistance, a huge body of work has been devoted to chemically modify the different scaffolds of macrolides. For instance, the modification of the 4’- and 4’’-hydroxyl groups of the mycaminose moiety of desmycosin (DES) leads to analogues able to fight antibacterial resistance [138]. With the same aim, DES was conjugated to fragments of oncocin, an AMP which activity depends on the interaction with binding site that overlaps with the binding site of macrolides [108,139]. DES-oncocin 93 (Scheme 15A) was synthetized starting from tylosin antibiotic 94 which was acetylated and hydrolyzed under acidic condition producing DES derivative 95 having the hydroxyl group in 4’ position unprotected, thus ready to be selectively functionalized (Scheme 15B) [140]. Accordingly, 95 was coupled with Boc-γ-aminobutyric acid (GABA) linker affording, after deprotection, intermediate 96 that was coupled with three different oncocin-fragments, the longer being Boc-Val-Asp(tBu)-Lys(Boc)-Pro-Pro-Tyr(tBu)-OH previously prepared through SPPS, yielding the target conjugate 93 after deprotection of all the side chain.
The resulting conjugates showed activity against some macrolide-resistant bacteria strains by binding to the E. coli 70S ribosome, thus inhibiting bacterial protein synthesis and suppressing bacterial growth.
5. Conclusions
Despite their widespread use and success in treating bacterial infections, conventional antibiotics face several significant challenges. One of the primary issues is the rapid development of bacterial resistance, which can render these drugs ineffective over time. The discovery and development of new antibiotics have slowed down significantly due to high costs, lengthy development times, and stringent regulatory requirements, making it difficult to keep pace with emerging resistant strains. AMPs are an alternative to small molecule antibiotics, offering several promising advantages in combating bacterial infections and antimicrobial resistance. One of their primary benefits is their broad-spectrum activity, allowing them to target a wide range of pathogens, including bacteria, fungi, and viruses. AMPs typically act rapidly with a mode of action that is less specific than traditional antibiotics reducing the likelihood of resistance development. Moreover, they can disrupt biofilms, which are protective layers formed by bacterial communities that conventional antibiotics often cannot penetrate. However, also AMPs suffer from some drawbacks, such as susceptibility to proteolytic degradation by enzymes in the human body, cytotoxicity towards human cells at higher concentrations, and poor pharmacokinetic properties necessitating frequent dosing or alternative administration routes, which can be less convenient for patients.
Combination therapy with old antibiotics and AMPs represent a promising frontier in the fight against AMR, overcoming most of the drawbacks associated with the use of the single components. This innovative therapy combines the potent, broad-spectrum activity of AMPs with the targeted efficacy of traditional antibiotics, resulting in enhanced antimicrobial effectiveness. The choice between AMPs-antibiotics conjugates and coadministration depends on various factors including the type of infection, the specific pathogens involved, patient characteristics, and available resources. Coadministration provides flexibility and simplicity but requires careful management to avoid issues with drug interactions and resistance. Conjugates offer the potential for highly targeted and effective treatment with lower resistance development, but they come with challenges related to complexity and cost. By leveraging the membrane active properties of AMPs, antibiotic-AMP conjugates facilitate improved antibiotic penetration and intracellular concentration, while also ensuring more specific targeting of bacterial cells over host cells. This dual-action approach not only increases the potency of the treatment but also allows for lower dosages, reducing potential side effects and minimizing the risk of developing resistance.
Although so far, no antibiotics-AMPs conjugates have successfully reached the marked, as research continues, with ongoing advancements in technology, innovations in peptide synthesis, delivery methods, and conjugation technologies, alongside a deeper understanding of bacterial biology and the mechanisms by which AMPs and small molecule antibiotics operate, antibiotic-AMP conjugates are emerging as a versatile and powerful strategy for addressing the global challenge of AMR.
Peptide synthesis technologies are evolving, allowing for the creation of more complex and stable AMPs with enhanced therapeutic properties. Advances in delivery methods are ensuring that these conjugates can be effectively transported to the target sites within the body, maximizing their efficacy while minimizing potential side effects. Additionally, new conjugation technologies are enabling the precise linkage of AMPs and antibiotics, enhancing their synergistic effects and overcoming bacterial defenses more effectively.
Our growing understanding of bacterial biology is crucial in this fight. Insights into bacterial resistance mechanisms, biofilm formation, and virulence factors are guiding the design of more effective conjugates. By targeting specific bacterial pathways and structures, these conjugates can disrupt the bacteria's ability to survive and replicate, even in the presence of traditional antibiotics. Furthermore, the mechanisms of action of AMPs and antibiotics are being elucidated in greater detail. This knowledge is instrumental in creating conjugates that can bypass resistance mechanisms, such as efflux pumps and enzymatic degradation, which bacteria commonly use to neutralize antibiotics. By combining the membrane-disrupting properties of AMPs with the intracellular targeting capabilities of antibiotics, these conjugates can deliver a one-two punch that bacteria find difficult to counteract. Additionally, the versatility of antibiotic-AMP conjugates lying in their potential to be tailored for specific infections and bacterial strains, can lead to more effective treatments with fewer side effects, as therapies can be designed to target only the pathogenic bacteria without harming the beneficial microbiota. This precision medicine approach not only enhances patient outcomes but also reduces the likelihood of resistance development.
Finally, economic and regulatory incentives must play a critical role in the development and deployment of antibiotic-AMP conjugates. Governments and health organizations are recognizing the urgent need to combat AMR and are providing funding, tax incentives, and streamlined regulatory pathways to accelerate the development of new antimicrobial agents. These incentives are crucial for encouraging pharmaceutical companies to invest in this high-risk, high-reward area of research and development.
In conclusion, the integration of cutting-edge technologies, a deepening understanding of bacterial biology, and supportive economic and regulatory frameworks are paving the way for antibiotic-AMP conjugates to become a cornerstone in the fight against AMR. These conjugates hold promise for revitalizing the efficacy of existing antibiotics and introducing new, potent antimicrobial therapies to safeguard public health for future generations.
Author Contributions
Conceptualization, M.C.B and A.V.; writing—original draft preparation, M.S. and A.V.; writing—review and editing, M.C.B, M.S, C.R.; visualization, A.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Theuretzbacher, U.; Gottwalt, S.; Beyer, P.; Butler, M.; Czaplewski, L.; Lienhardt, C.; Moja, L.; Paul, M.; Paulin, S.; Rex, J.H.; Silver, L.L.; Spigelman, M.; Thwaites, G.E.; Paccaud, J.P.; Harbarth, S. Analysis of the clinical antibacterial and antituberculosis pipeline. Lancet Infect Dis. 2019, 19, e40–e50. [Google Scholar] [CrossRef] [PubMed]
- Gould, K. Antibiotics: from prehistory to the present day. J. Antimocrob. Chemother. 2016, 71, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.B. Drugs that changed society: history and current status of the early antibiotics: salvarsan, sulfonamides, and b-lactams. Molecules 2021, 26, 6057. [Google Scholar] [CrossRef] [PubMed]
- Baran, A.; Kwiatkowska, A.; Potocki, L. Antibiotics and bacterial resistance – A short story of an endless arms race. Int. J. Mol. Sci. 2023, 24, 5777. [Google Scholar] [CrossRef]
- Halawa, E.M.; Fadel, M.; Al-Rabia, M.W.; Behairy, A.; Nouh, N.A.; Abdo, M.; Olga, R.; Fericean, L.; Atwa, A.M.; El-Nablaway, M.; Abdeen, A. Antibiotic action and resistance: updated review of mechanisms, spread, influencing factors, and alternative approaches for combating resistance. Front. Pharmacol. 2024, 14, 1305294. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Uddin, T.M.; Chakraborty, A.J.; Khusro, A.; Redwan Matin Zidan, B.M.; Mitra, S.; Emra, T.B.; Dhama, K.; Ripon, M.K.H.; Gajdács, M.; Sahibzada, M.U.K.; Hossain, M.J.; Koirala, N. Antibiotic resistance in microbes: History, mechanisms, therapeutic strategies and future prospects. J. Infect. Public Health 2021, 12, 1750–1766. [Google Scholar] [CrossRef] [PubMed]
- Darby, E.M.; Trampari, E.; Siasat, P.; Gaya, M.S.; Alav, I.; Webber, M.A.; Blair, J.M.A. Molecular mechanisms of antibiotic resistance revisited. Nat Rev Microbiol 2023, 21, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Kaye, K.S.; Pogue, J.M. Infections caused by resistant gram-negative bacteria: epidemiology and management. Pharmacotherapy 2015, 35, 949–962. [Google Scholar] [CrossRef]
- Cascioferro, S.; Parrino, B.; Carbone, D.; Pecoraro, C.; Diana, P. Novel strategies in the war against antibiotic resistance. Future Med. Chem. 2021, 13, 529–531. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; Ouellette, M.; Outterson, K.; Patel, J.; Cavaleri, M.; Cox, E.M.; Houchens, C.R.; Grayson, M.L.; Hansen, P.; Singh, N.; Theuretzbacher, U.; Magrini, N.; Aboderin, A.O.; Al-Abri, S.S.; Awang Jalil, N.; Benzonana, N.; Bhattacharya, S.; Brink, A.J.; Burkert, F.R.; Cars, O.; Cornaglia, G.; Dyar, O.J.; Friedrich, A.W.; Gales, A.C.; Gandra, S.; Giske, C.G.; Goff, D.A.; Goossens, H.; Gottlieb, T.; Guzman Blanco, M.; Hryniewicz, W.; Kattula, D.; Jinks, T.; Kanj, S.S.; Kerr, L.; Kieny, M.P.; Kim, Y.S.; Kozlov, R.S.; Labarca, J.; Laxminarayan, R.; Leder, K.; Leibovici, L.; Levy-Hara, G.; Littman, J.; Malhotra-Kumar, S.; Manchanda, V.; Moja, L.; Ndoye, B.; Pan, A.; Paterson, D.L.; Paul, M.; Qiu, H.; Ramon-Pardo, P.; Rodríguez-Baño, J.; Sanguinetti, M.; Sengupta, S.; Sharland, M.; Si-Mehand, M.; Silver, L.L.; Song, W.; Steinbakk, M.; Thomsen, J.; Thwaites, G.E.; van der Meer, J.W.; Van Kinh, N.; Vega, S.; Villegas, M.V.; Wechsler-Fördös, A.; Wertheim, H.F.L.; Wesangula, E.; Woodford, N.; Yilmaz, F.O.; Zorzet, A. Discovery, Research, and Development of New Antibiotics: The WHO Priority List of Antibiotic-Resistant Bacteria and Tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J.; Gwynn, M.N.; Holmes, D. J.; Pompliano, D.L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discovery 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Tommasi, R.; Brown, D.G.; Walkup, G.K.; Manchester, J.I.; Miller, A.A. ESKAPEing the labyrinth of antibacterial discovery. Nat. Rev. Drug Discovery 2015, 14, 529–42. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, R.; Moser, H.E. Physicochemical Properties of Antibacterial Compounds: Implications for Drug Discovery. J. Med. Chem. 2008, 51, 2871–2878. [Google Scholar] [CrossRef] [PubMed]
- Olsufyeva, E.N.; Yankovskaya, V.S. Main trends in the design of semi-synthetic antibiotics of a new generation. Russ. Chem. Rev. 2020, 89, 339–387. [Google Scholar] [CrossRef]
- Wright, G.D. Opportunities for natural products in 21st-century antibiotic discovery. Nat. Prod. Rep. 2017, 34, 694–701. [Google Scholar] [CrossRef]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef]
- Renwick, M.J.; Brogan, D.M.; Mossialos, E. A systematic review and critical assessment of incentive strategies for discovery and development of novel antibiotics. J. Antibiot. 2016, 69, 73–88. [Google Scholar] [CrossRef]
- Silver, L.L. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [Google Scholar] [CrossRef]
- Outterson, K.; Powers, J.H.; Daniel, G.W.; McClellan, M.B. Repairing the broken market for antibiotic innovation. Health Aff. 2015, 34, 277–285. [Google Scholar] [CrossRef]
- Ventola, C.L. The antibiotic resistance crisis: part 1: causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Shi, Z.; Zhang, J.; Tia, L.; Xin, L.; Liang, C.; Ren, X.; Li, M. A comprehensive overview of the antibiotics approved in the last two decades: retrospects and prospects. Molecules 2023, 28, 1762. [Google Scholar] [CrossRef] [PubMed]
- Coates, A.R.M.; Hu, Y.; Holt, J.; Yeh, P. Antibiotic combination therapy against resistant bacterial infections: synergy, rejuvenation, and resistance reduction. Expert Rev. Anti-Infect. Ther. 2020, 18, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, G.J.; Delgado, N.N.; Maharjan, R.; Cain, A.K. How antibiotics work together: molecular mechanisms behind combination therapy. Curr. Opin. Microbiol. 2020, 57, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.; Greenstein, T.; Colindres, R.V.; Aldridge, B.B. Leveraging laboratory and clinical studies to design effective antibiotic combination therapy. Curr. Opin. Microbiol. 2021, 64, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Alkhzem, A.H.; Woodman, T.J.; Blagbrought, S.I. Design and synthesis of hybrid compounds as novel drugs and medicines. RSC Adv. 2022, 12, 19470–19484. [Google Scholar] [CrossRef] [PubMed]
- Cavaco, M.; Castanho, M.A.R.B.; Neves, V. The Use of Antibody-Antibiotic Conjugates to Fight Bacterial Infections. Front. Microbiol. 2022, 13, 835677. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 17, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Raileanu, M.; Borlan, R.; Campu, A.; Janosi, L.; Turcu, I.; Focsan, M.; Bacalum, M. No country for old antibiotics! Antimicrobial peptides (AMP) as next-generation treatment for skin and soft tissue infection. Int. J. Pharm. 2023, 642, 123169. [Google Scholar] [CrossRef]
- Strempel, N.; Strehmel, J.; Overhage, J. Potential application of antimicrobial peptides in the treatment of bacterial biofilm infections. Curr. Pharm. Des. 2015, 21, 67–84. [Google Scholar] [CrossRef]
- David, A.A.; Park, S.E.; Parag, K.; Tiwari, R.K. Antibiotics-peptide conjugates: against multidrug-resistant bacterial pathogens. Curr. Top. Med. Chem. 2018, 18, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Le, C.-F.; Fang, C.-M.; Sekaran, S.D. Intracellular targeting mechanisms by antimicrobial peptides. Antimicrob. Agents Chemother. 2017, 61, e02340–16. [Google Scholar] [CrossRef]
- Hadjicharalambous, A.; Bournakas, N.; Newman, H.; Skynner, M.J.; Beswick, P. Antimicrobial and cell-penetrating peptides: understanding penetration for the design of novel conjugate antibiotics. Antibiotics 2022, 11, 1636. [Google Scholar] [CrossRef] [PubMed]
- Gould, K. Antibiotics: from prehistory to the present day. J. Antimicrob. Chemother. 2016, 71, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Gelpi, A.; Glberston, A.; Tucker, J.D. Magic bullet: Paul Ehrlich, Salvarsan and the birth of venereology. Sex. Transm. Infect. 2015, 91, 68–69. [Google Scholar] [CrossRef] [PubMed]
- Hare, R. New light on the history of penicillin. Med. Hist. 1982, 26, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Wencewicz, T.A. Prospects for new antibiotics: a molecule-centered perspective. J. Antibiot. 2014, 67, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Zessel, K.; Mohring, S.; Hamscher, G.; Kietzmann, M.; Stahl, J. Biocompatibility and antibacterial activity of photolytic products of sulfonamides. Chemosphere 2014, 100, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Azevedo-Barbosa, H.; Dias, D.F.; Franco, L.L.; Hawkes, J.A.; Carvalho, D.T. From Antibacterial to Antitumour Agents: A Brief Review on The Chemical and Medicinal Aspects of Sulfonamides. Mini-Rev. Med. Chem. 2020, 20, 2052–2066. [Google Scholar] [CrossRef]
- Lima, L.M.; Silva, B.N.M.; Barbosa, G.; Barreiro, E.J. β-Lactam antibiotics: An overview from a medicinal chemistry perspective. Eur. J. Med. Chem. 2020, 208, 112829. [Google Scholar] [CrossRef]
- Yocum, R.R.; Rasmussen, J.R.; Strominger, J.L. The mechanism of action of penicillin. Penicillin acylates the active site of Bacillus stearothermophilus D-alanine carboxypeptidase. J. Biol. Chem. 1980, 255, 3977e3986. [Google Scholar]
- Waxman, D.J.; Strominger, J.L. Sequence of active site peptides from the penicillin-sensitive D-alanine carboxypeptidase of Bacillus subtilis. Mechanism of penicillin action and sequence homology to beta-lactamases. J. Biol. Chem. 1980, 255, 3964e3976. [Google Scholar]
- Amyes, S.G.B. Resistance to β-Lactams - The Permutations. Journal of Chemotherapy 2003, 15, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M. Can β-lactams be re-engineered to beat MRSA? CMI 2006, 12, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liao, Y.X.; Ding, T.; Ahn, J. Role of β-Lactamase Inhibitors as Potentiators in Antimicrobial Chemotherapy Targeting Gram-Negative Bacteria. Antibiotics 2023, 13, 260. [Google Scholar] [CrossRef] [PubMed]
- Chukwudi, C.U. rRNA binding sites and the molecular mechanism of action of the tetracyclines. Antimicrob. Agents Chemother. 2016, 60, 4433–4441. [Google Scholar] [CrossRef] [PubMed]
- LaPlante, K.L.; Dhand, A.; Wright, K.; Lauterio, M. Re-establishing the utility of tetracycline-class antibiotics for current challenges with antibiotic resistance. Annals of Medicine 2022, 54, 1686–1700. [Google Scholar] [CrossRef]
- Davies, J.; Wright, G.D. Bacterial resistance to aminoglycoside antibiotics. Trends Microbiol. 1997, 5, 234. [Google Scholar] [CrossRef] [PubMed]
- Houghton, J.L.; Green, K.D.; Chen, W.; Garneau-Tsodikova, S. The future of aminoglycosides: the end or renaissance? ChemBioChem 2010, 11, 880–902. [Google Scholar] [CrossRef]
- McCoy, L.S.; Xie, Y.; Tor, Y. Antibiotics that target protein synthesis. Wiley Interdiscip. Rev.: RNA. 2011, 2, 209–232. [Google Scholar] [CrossRef]
- Franҫois, B.; Russell, R.J.; Murray, J.B.; Aboul-ela, F.; Masquida, B.; Vicens, Q.; Westhof, E. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res. 2005, 33, 5677–5690. [Google Scholar] [CrossRef] [PubMed]
- Feldman, M.B.; Terry, D.S.; Altman, R.B.; Blanchard, S.C. Aminoglycoside activity observed on single pre-translocation ribosome complexes. Nat. Chem. Biol. 2010, 6, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Garneau-Tsodikova, S.; Labby, K.J. Mechanisms of resistance to aminoglycoside antibiotics: overview and perspectives. Med. Chem. Comm. 2016, 7, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Bera, S.; Mondal, D.; Palit, S.; Schweizer, F. Structural modifications of the neomycin class of aminoglycosides. Med. Chem. Comm. 2016, 7, 1499–1534. [Google Scholar] [CrossRef]
- Chandrika, N.T.; Garneau-Tsodikova, S. Comprehensive review of chemical strategies for the preparation of new aminoglycosides and their biological activities. Chem. Soc. Rev. 2018, 47, 1189–1249. [Google Scholar] [CrossRef]
- Fosso, M.Y.; Li, Y.; Garneau-Tsodikova, S. New trends in the use of aminoglycosides. Med. Chem. Commun. 2014, 5, 1075–1091. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, M.C.; Volonterio, A. Aminoglycosides: From Antibiotics to Building Blocks for the Synthesis and Development of Gene Delivery Vehicles. Antibiotics 2020, 9, 504. [Google Scholar] [CrossRef]
- Brockmann, H.; Hekel, W. Pikromycin, ein bitter schmeckendes antibioticum aus actinomyeceten. Chem Ber 1951, 84, 284–288. [Google Scholar] [CrossRef]
- Retsema, J.; Fu, W. Macrolides: Structures and microbial targets. Int. J. Antimicrob. Agents 2001, 18, S3–S10. [Google Scholar] [CrossRef]
- Jednačak, T.; Mikulandra, I.; Novak, P. Advanced Methods for Studying Structure and Interactions of Macrolide Antibiotics. Int. J. Mol. Sci. 2020, 21, 7799. [Google Scholar] [CrossRef]
- Dinos, P.G. The macrolide antibiotic renaissance. British Journal of Pharmacology 2017, 174, 2967–2983. [Google Scholar] [CrossRef] [PubMed]
- Undheim, K.; Scaffold Modifications in Erythromycin Macrolide Antibiotics. A Chemical Minireview. Molecules 2020, 25, 3941. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.G.; Clark, R.B. Discovery of Macrolide Antibiotics Effective against Multi-Drug Resistant Gram-Negative Pathogens. Acc. Chem. Res. 2021, 54, 1635–1645. [Google Scholar] [CrossRef] [PubMed]
- Blaskovich, M.A.T.; Hansford, K.A.; Butler, M.S.; Jia, Z.G.; Mark, A.E.; Cooper, M.A. Developments in Glycopeptide Antibiotics. ACS Infect. Dis. 2018, 4, 715–735. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Boddy, C.N.; Brase, S.; Winssinger, N. Chemistry, Biology, and Medicine of the Glycopeptide Antibiotics. Angew. Chem., Int. Ed. 1999, 38, 2096–2152. [Google Scholar] [CrossRef]
- Kahne, D.; Leimkuhler, C.; Lu, W.; Walsh, C. Glycopeptide and lipoglycopeptide antibiotics. Chem. Rev. 2005, 105, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Acharya, Y.; Dhanda, G.; Sarkara, P.; Haldar, J. Pursuit of next-generation glycopeptides: a journey with vancomycin. Chem. Commun. 2022, 58, 1881–1897. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Hansford, K.A.; Blaskovich, M.A.T.; Halai, R.; Cooper, M.A. Glycopeptide antibiotics: Back to the future. J. Antibiot. 2014, 67, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Lesher, G.Y.; Froelich, E.J.; Gruett, M.D.; Bailey, J.H.; Brundage, R.P. 1,8-Naphthyridine Derivatives: A New Class of Chemotherapeutic Agents. Journal of medicinal and pharmaceutical chemistry 1962, 91, 1063–1065. [Google Scholar] [CrossRef] [PubMed]
- Emmerson, A.M.; Jones, A.M. The quinolones: decades of development and use. J Antimicrob. Chemother. 2003, 51, 13–20. [Google Scholar] [CrossRef]
- Sharma, V.; Das, R.; Metha, D.K.; Sharma, D.; Aman, S.; Khan, M.U. Quinolone scaffolds as potential drug candidates against infectious microbes: a review. Mol. Divers. 2024. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of quinolone action and resistance. Biochem. 2014, 53, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Anuradha, *!!! REPLACE !!!*; Chandra, A.; Tanwar, T.; Sahu, S.K.; Mittal, A. Emerging quinoline- and quinolone-based antibiotics in the light of epidemics. Chem Biol Drug Des. 2022, 100, 765–785. [Google Scholar] [CrossRef] [PubMed]
- Bulet, P.; Stöcklin, R.; Menin, L. Anti-microbial peptides: from invertebrates to vertebrates. Immunol. Rev 2004, 198, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Felício, M.R.; Silva, O.N.; Gonçalves, S.; Santos, N.C.; Franco, O.L. Peptides with dual antimicrobial and anticancer activities. Front. Chem 2017, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, J.; Hart, R.J.; Soldano, A.; Chen, C.H.; Guha, S.; Hoffmann, J.P.; Wimley, W.C. Optimization of host cell-compatible, antimicrobial peptides effective against biofilms and clinical isolates of drug-resistant bacteria. ACS Infect. Dis. 2003, 9, 952–965. [Google Scholar] [CrossRef] [PubMed]
- Erdem Büyükkiraz, M.; Kesmen, Z. Antimicrobial peptides (AMPs): A promising class of antimicrobial compounds. J. App. Microbiol. 2022, 132(3), 1573–1596. [Google Scholar] [CrossRef]
- Chen, C.H.; Lu, T.K. Development and challenges of antimicrobial peptides for therapeutic applications. Antibiotics 2020, 9, 24. [Google Scholar] [CrossRef]
- Gan, B.H.; Gaynord, J.; Rowe, S.M.; Deingruber, T.; Spring, D.R. The multifaceted nature of antimicrobial peptides: Current synthetic chemistry approaches and future directions. Chem. Soc. Rev. 2021, 50, 7820–7880. [Google Scholar] [CrossRef] [PubMed]
- Madani, F.; Lindberg, S.; Langel, U.; Futaki, S.; Graslund, A. Mechanisms of cellular uptake of cell-penetrating peptides. Biophys. J. 2011, 414, 729. [Google Scholar] [CrossRef] [PubMed]
- Jenssen, H.; Hamill, P.; Hancock, R.E. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [PubMed]
- López-Meza, J.E.; Ochoa-Zarzosa, A.; Aguilar, J.A.; Loeza-Lara, P.D. Antimicrobial peptides: diversity and perspectives for their biomedical application. In Biomedical Engineering, Trends, Research and TechnoInc; 2011; pp. 275–304. [Google Scholar]
- Hazam, P.K.; Goyal, R.; Ramakrishnanet, V. Peptide based antimicrobials: design strategies and therapeutic potential. Progress in Biophysics and Molecular Biology 2018, 142, 10–22. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Benfield, A.H.; Henriques, S.T. Mode-of-Action of Antimicrobial Peptides: Membrane Disruption vs. Intracellular Mechanisms. Front. Med. Technol. 2020, 2, 610997. [Google Scholar] [CrossRef]
- Miao, J.; Zhou, X. Interaction between antimicrobial peptides and DNA: Mechanism of action and application in biotechnology. Biotechnol. Adv. 2016, 34, 808–820. [Google Scholar]
- Schenckbecher, K.; Meyer, A.; Ennifar, E. Antibiotics targeting the ribosome. Ribosome Structure and Function 2009, 1–21. [Google Scholar]
- Helmerhorst, E.J.; Troxler, R.F.; Oppenheim, F.G. The human salivary peptide histatin 5 exerts its antifungal activity through the formation of reactive oxygen species. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 14637–14642. [Google Scholar] [CrossRef]
- Regmi, S.; Yoon, S.C.; Yun, H.C.; Young, K.K.; Seung, S.C.; Jin, C.Y. Antimicrobial peptide from Bacillus subtilis CSB138: characterization, killing kinetics, and synergistic potency. Int. Microbiol. 2017, 20, 43–53. [Google Scholar]
- Pletzer, D.; Mansour, S.C.; Hancock, R.E. Synergy between conventional antibiotics and antimicrobial peptides in the treatment of chronic bacterial infections. J. Antibiot. 2018, 71, 87–98. [Google Scholar]
- Zhou, Y.; Peng, Y. Synergistic effect of clinically used antibiotics and peptide antibiotics against Gram-positive and Gram-negative bacteria. Exp. Ther. Med. 2013, 6, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Peetla, C.; Stine, A.; Labhasetwar, V. Biophysical interactions with model lipid membranes: applications in drug discovery and drug delivery. Mol. Pharm. 2009, 6, 1264–1276. [Google Scholar] [CrossRef] [PubMed]
- Blaskovich, M.A.T.; Hansford, K.A.; Gong, Y.; Butler, M.S.; Muldoon, C.; Huang, J.X.; Ramu, S.; Silva, A.B.; Cheng, M.; Kavanagh, A.M.; Ziora, Z.; Premraj, R.; Lindahl, F.; Bradford, T.A.; Lee, J.C.; Karoli, T.; Pelingon, R.; Edwards, D.J.; Amado, M.; Elliott, A.G.; Phetsang, W.; Daud, N.H.; Deecke, J.E.; Sidjabat, H.E.; Ramaologa, S.; Zuegg, J.; Betley, J.R.; Beevers, A.P.G.; Smith, R.A.G.; Roberts, J.A.; Paterson, D.L.; Cooper, M.A. Protein-inspired antibiotics active against vancomycin- and daptomycin-resistant bacteria. Nat Commun 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Umstätter, F.; Domhan, C.; Hertlein, T.; Ohlsen, K.; Mühlberg, E.; Kleist, C.; Zimmermann, S.; Beijer, B.; Klika, K.D.; Haberkorn, U.; Mier, W.; Uhl, P. Vancomycin Resistance Is Overcome by Conjugation of Polycationic Peptides. Angew. Chem. Int. Ed. 2020, 59, 8823–8827. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Muller, A.; Vuorenoja, S.; Tuominen, M.; et al. Use of Hecate chorionic gonadotropin beta conjugate in therapy of lutenizing hormone receptor expressing gonadal somatic cell tumors. Mol Cell Endocrinol. 2007, 269, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Jelinkova, P.; Splichal, Z.; Jimenez, A.M.; Haddad, Y.; Mazumdar, A.; Sur, V.P.; Milosavljevic, V.; Kopel, P.; Buchtelova, H.; Guran, R.; Zitka, O.; Richtera, L.; Hegerova, D.; Heger, Z.; Moulick, A.; Adam, V. Novel vancomycin–peptide conjugate as potent antibacterial agent against vancomycin-resistant Staphylococcus aureus. Infection and Drug Resistance 2018, 11, 1807–1817. [Google Scholar] [CrossRef] [PubMed]
- Meredith, T.C.; Aggarwal, P.; Mamat, U.; Lindner, B.; Woodard, R.W. Redefining the requisite lipopolysaccharide structure in Escherichia coli. ACS Chem. Bio. 2006, 1, 33–42. [Google Scholar] [CrossRef]
- Frecer, V.; Ho, B.; Ding, J.L. De. novo design of potent antimicrobial peptides. Antimicrob. Agents Chemother. 2004, 48, 3349–3357. [Google Scholar] [CrossRef]
- Shi, W.; Chen, F.; Zou, X.; Jiao, S.; Wang, S.; Hu, Y.; Lan, L.; Tang, F.; Huang, W. Design, synthesis, and antibacterial evaluation of vancomycin-LPS binding peptide conjugates. Bioorg. Chem. Med. Lett. 2021, 45, 128122. [Google Scholar] [CrossRef]
- Beveridge, E.G.; Martin, A.J. Sodium sulphomethyl derivatives of polymyxins. Br. J. Pharmacol. Chemother. 1967, 29, 125–135. [Google Scholar] [CrossRef]
- Desgranges, S.; Ruddle, C.C.; Burke, L.P.; McFadden, T.M.; O’Brien, J.E.; Fitzgerald-Hughes, D.; Humphreys, H.; Smyth, T.; Devocelle, M. b-Lactam-host defence peptide conjugates as antibiotic prodrug candidates targeting resistant bacteria. RCS Adv. 2012, 2, 2480–2492. [Google Scholar] [CrossRef]
- Li, W.; O’Briean-Simpson, N.M.; Holden, J.A.; Otvos, L.; Reynolds, E.C.; Separovic, F.; Hossian, M.A.; Wade, J.D. Covalent conjugation of cationic antimicrobial peptides with b-lactam antibiotic core. Peptide Science 2018, 110, e24059. [Google Scholar] [CrossRef]
- Yamauchi, R.; Kawano, K.; Yamaoka, Y.; Taniguchi, A.; Yano, Y.; Takasu, K.; Matsusaki, K. Development of antimicrobial peptide-antibiotic conjugates to improve the outer membrane permeability of antibiotics against Gram-negative bacteria. ACS Infect. Dis. 2022, 8, 2339–2347. [Google Scholar] [CrossRef]
- Ritz, D.; Beckwith, J. Roles of thiol-redox pathways in bacteria. Annu. Rev. Microbiol. 2001, 55, 21–48. [Google Scholar] [CrossRef]
- Azuma, E.; Choda, N.; Odaki, N.; Yano, Y.; Matsuzaki, K. Improvement of therapeutic index by the combination of enhanced peptide cationicity and proline introduction. ACS Infect. Dis. 2020, 6, 2271–2278. [Google Scholar] [CrossRef]
- Knappe, D.; Piantavigna, S.; Hansen, A.; Mechler, A.; Binas, A.; Nolte, O.; Martin, L.L.; Hoffmann, R. Oncocin (VDKPPYLPRPRPPRRIYNR-NH2): a novel antibacterial peptide optimized against gram-negative human pathogens. J. Med. Chem. 2010, 53, 5240–5247. [Google Scholar] [CrossRef]
- Williams, H.C.; Dellavalle, R.P.; Garner, S. Acne vulgaris. Lancet 2012, 379, 361–372. [Google Scholar] [CrossRef]
- Schmidt, N.W.; Deshayes, S.; Hawker, S.; Blacker, A.; Kasko, A.M.; Kim, J.; Wong, G.C.L. Engineering persister-specific antibiotics with synergistic antimicrobial functions. ACS Nano 2014, 8, 8786–8793. [Google Scholar] [CrossRef]
- Schmidt, N.W.; Aga, G.W.; Deshayes, S.; Yu, Y.; Blacker, A.; Champer, J.; Xian, W.; Kasko, A.M.; Kim, J.; Wong, G.C.L. Pentobra: A Potent Antibiotic with Multiple Layers of Selective Antimicrobial Mechanisms against Propionibacterium Acnes. J. Invest. Dermatol. 2015, 135, 1581–1589. [Google Scholar] [CrossRef]
- Schmidt, N.W.; Lis, M.; Zhao, K.; Lai, G.H.; Alexandrova, A.N.; Tew, G.N.; Wong, G.C.L. Molecular basis for nanoscopic membrane curvature generation from quantum mechanical models and synthetic transporter sequences. J. Am. Chem. Soc. 2012, 134, 19207–19216. [Google Scholar] [CrossRef]
- Deshayes, S.; Xian, W.; Schmidt, N.W.; Kordbacheh, S.; Lieng, J.; Wang, J.; Zarmer, S.; St. Germain, S.; Voyen, L.; Thulin, J.; Wong, G.C.L.; Kasko, A.M. Designing hybrid antibiotic peptide conjugates to cross bacterial membranes. Bioconjugate Chem. 2017, 28, 793–804. [Google Scholar] [CrossRef]
- Flannagan, R.S.; Cosío, G.; Grinstein, S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 2009, 7, 355–366. [Google Scholar]
- LaRock, D.L.; Chaudhary, A.; Miller, S.I. Salmonellae interactions with host processes. Nat. Rev. Microbiol. 2015, 13, 191–205. [Google Scholar] [CrossRef]
- Kuriakose, J.; Hernandez-Gordillo, V.; Nepal, M.; Brezden, A.; Pozzi, V.; Seleem, M.N.; Chmielewski, J. Targeting Intracellular Pathogenic Bacteria with Unnatural Proline-Rich Peptides: Coupling Antibacterial Activity with Macrophage Penetration. Angew. Chem., Int. Ed. 2013, 52, 9664–9667. [Google Scholar] [CrossRef]
- Brezden, A.; Mohamed, M.F.; Nepal, M.; Harwood, J.S.; Kuriakose, J.; Seleem, M.N.; Chmielewski, J. Dual Targeting of Intracellular Pathogenic Bacteria with a Cleavable Conjugate of Kanamycin and an Antibacterial Cell-Penetrating Peptide. J. Am. Chem. Soc. 2016, 138, 10945–10949. [Google Scholar] [CrossRef]
- Mohamed, M.F.; Brezden, A.; Mohammad, H.; Chmielewski, J.; Seleem, M.N. Targeting biofilms and persisters of ESKAPE pathogens with P14KanS, a kanamycin peptide conjugate. Biochim. Biophys. Acta 2017, 1861, 848–859. [Google Scholar] [CrossRef]
- Scocchi, M.; Tossi, A.; Gennaro, R. Proline-Rich Antimicrobial Peptides: Converging to a Non-Lytic Mechanism of Action. Cell. Mol. Life Sci. 2011, 68, 2317–2330. [Google Scholar] [CrossRef]
- Guida, F.; Benincasa, M.; Zahariev, S.; Scocchi, M.; Berti, F.; Gennaro, R.; Tossi, A. Effect of Size and N-Terminal Residue Characteristics on Bacterial Cell Penetration and Antibacterial Activity of the Proline-Rich Peptide Bac7. J. Med. Chem. 2015, 58, 1195–1204. [Google Scholar] [CrossRef]
- Gambato, S.; Bellotto, O.; Mardirossian, M.; Di Stasi, A.; Gennaro, R.; Pacor, S.; Caporale, A.; Berti, F.; Scocchi, M.; Tossi, A. Designing New Hybrid Antibiotics: Proline-Rich Antimicrobial Peptides Conjugated to the Aminoglycoside Tobramycin. Bioconjugate Chem. 2023, 34, 1212–1220. [Google Scholar] [CrossRef]
- Lau, Y.H.; De Andrade, P.; Wu, Y.; Spring, D.R. Peptide stapling techniques based on different macrocyclisation chemistries. Chem. Soc. Rev. 2015, 44, 91–102. [Google Scholar] [CrossRef]
- Lourenço, A.L.P.; Rios, T.B.; da Silva, A.P.; Franco, O.L.; Ramada, M.H.S. Peptide Stapling Applied to Antimicrobial Peptides. Antibiotics 2023, 12, 1400. [Google Scholar] [CrossRef]
- Wojciechowska, M.; Macyszyn, J.; Miszkiewicz, J.; Grzela, R.; Trylska, J. Stapled Anoplin as an Antibacterial Agent. Front. Microbiol. 2021, 12, 772038. [Google Scholar] [CrossRef]
- Macyszyn, J.; Burmistrz, M.; Mieczkowski, A.; Wojciechowska, M.; Trylska, J. ACS Omega 2023, 8, 19047–19056. [CrossRef]
- Rüter, C.; Buss, C.; Scharnert, J.; Heusipp, G.; Schmidt, M.A. A newly identified bacterial cell-penetrating peptide that reduces the transcription of pro-inflammatory cytokines. J Cell Sci 2010, 123, 2190–2198. [Google Scholar] [CrossRef]
- Gomarasca, M.; Martins, T.F.; Greune, L.; Hardwidge, P.R.; Schmidt, M.A.; Rüter, C. Bacterium-Derived Cell-Penetrating Peptides Deliver Gentamicin To Kill Intracellular Pathogens. Antimicrob. Agents Chemother. 2017, 61, e02545–16. [Google Scholar] [CrossRef]
- Choi, H.; Lee, D.G. Synergistic effect of antimicrobial peptide arenicin-1 in combination with antibiotics against pathogenic bacteria. Res., Microbiol. 2012, 163, 479–486. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Sun, Y.; Liu, Q.; Wang, X.; Li, Z.; Hao, J. In vitro synergistic activities of antimicrobial peptide brevinin 2CE with five kinds of antibiotics against multidrug-resistant clinical isolates. Curr. Microbiol. 2014, 68, 685–692. [Google Scholar] [CrossRef]
- Selsted, M.E.; Novotny, M.J.; Morris, W.L.; Tang, Y.Q.; Smith, W.; Cullor, J.S. Indolicidin, a novel bactericidal tridecapeptide amide from neutrophils. J. Biol. Chem. 1992, 267, 4292–4295. [Google Scholar] [CrossRef]
- Ghaffar, K.A.; Hussein, W.M.; Khalil, Z.G.; Capon, R.J.; Skwarczynski, M.; Toth, I. Levofloxacin and indolicidin for combination antimicrobial therapy. Curr. Drug Del. 2015, 12, 108–114. [Google Scholar] [CrossRef]
- Kondori, N.; Baltzer, L.; Dolphin, G.T.; Mattsby-Baltzer, I. Fungicidal Activity of Human Lactoferrin-Derived Peptides Based on the Antimicrobial Aβ Region. Int. J. Antimicrob. Agents 2011, 37, 51–57. [Google Scholar] [CrossRef]
- Ptaszyńska, N.; Gucwa, K.; Olkiewicz, K.; Łȩgowska, A.; Okońska, J.; Ruczyński, J.; Gitlin-Domagalska, A.; Dȩbowski, D.; Milewski, S.; Rolka, K. Antibiotic-Based Conjugates Containing Antimicrobial HLopt2Peptide: Design, Synthesis, Antimicrobial and Cytotoxic Activities. ACS Chem. Biol. 2019, 14, 2233–2242. [Google Scholar]
- Tang, E.N.; Nair, A.; Baker, D.W.; Hum, W.; Zhou, J. In vivo imaging of infection using a bacteria-targeting optical nanoprobe. J. Biomed. Nanotechnol. 2014, 10, 856–863. [Google Scholar] [CrossRef]
- Dinos, G.P.; Athanassopoulos, C.M.; Missiri, D.A.; Giannopoulou, P.C.; Vlachogiannis, I.A.; Papadopoulos, G.E.; Papaioannou, D.; Kalpaxis, D.L. Chloramphenicol Derivatives as Antibacterial and Anticancer Agents: Historic Problems and Current Solutions. Antibiotics 2016, 5, 20. [Google Scholar] [CrossRef]
- Saeed, S.; Zafar, J.; Khan, B.; Akhtar, A.; Qurieshi, S.; Fatima, S.; Ahmad, N.; Irfanullah, J. Utility of 99mTc-labelled antimicrobial peptide ubiquicidin (29−41) in the diagnosis of diabetic foot infection. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 737–743. [Google Scholar] [CrossRef]
- Chen, H.; Liu, C.; Chen, D.; Madrid, K.; Peng, S.; Dong, X.; Zhang, M.; Gu, Y. Bacteria-targeting conjugates based on antimicrobial peptide for bacteria diagnosis and therapy. Mol. Pharmaceutics 2015, 12, 2505–2516. [Google Scholar] [CrossRef]
- Phan, L.T.; Jian, T.; Chen, Z.; Qiu, Y.L.; Wang, Z.; Beach, T.; Polemeropoulos, A.; Sun Or, Y. Synthesis and Antibacterial Activity of a Novel Class of 4‘-Substituted 16-Membered Ring Macrolides Derived from Tylosin. J. Med. Chem. 2004, 47, 2965–2968. [Google Scholar] [CrossRef]
- Seefeldt, A.C.; Nguyen, F.; Antunes, S.; Pérébaskine, N.; Graf, M.; et al. The proline-rich antimicrobial peptide Onc112 inhibits translation by blocking and destabilizing the initiation complex. Nat. Struct. Mol. Biol. 2015, 22, 470–475. [Google Scholar] [CrossRef]
- Khairullina, Z.Z.; Makarov, G.I.; Tereshchenkov, A.G.; Buev, V.S.; Lukianov, D.A.; Polshakov, V.I.; Tashlitsky, V.N.; Osterman, I.A.; Sumbatyan, N.V. Conjugates of Desmycosin with Fragments of Antimicrobial Peptide Oncocin: Synthesis, Antibacterial Activity, Interaction with Ribosome. Biochemistry (Moscow) 2022, 87, 871–889. [Google Scholar] [CrossRef]
Figure 1.
General structure of the main classes of antibiotics.
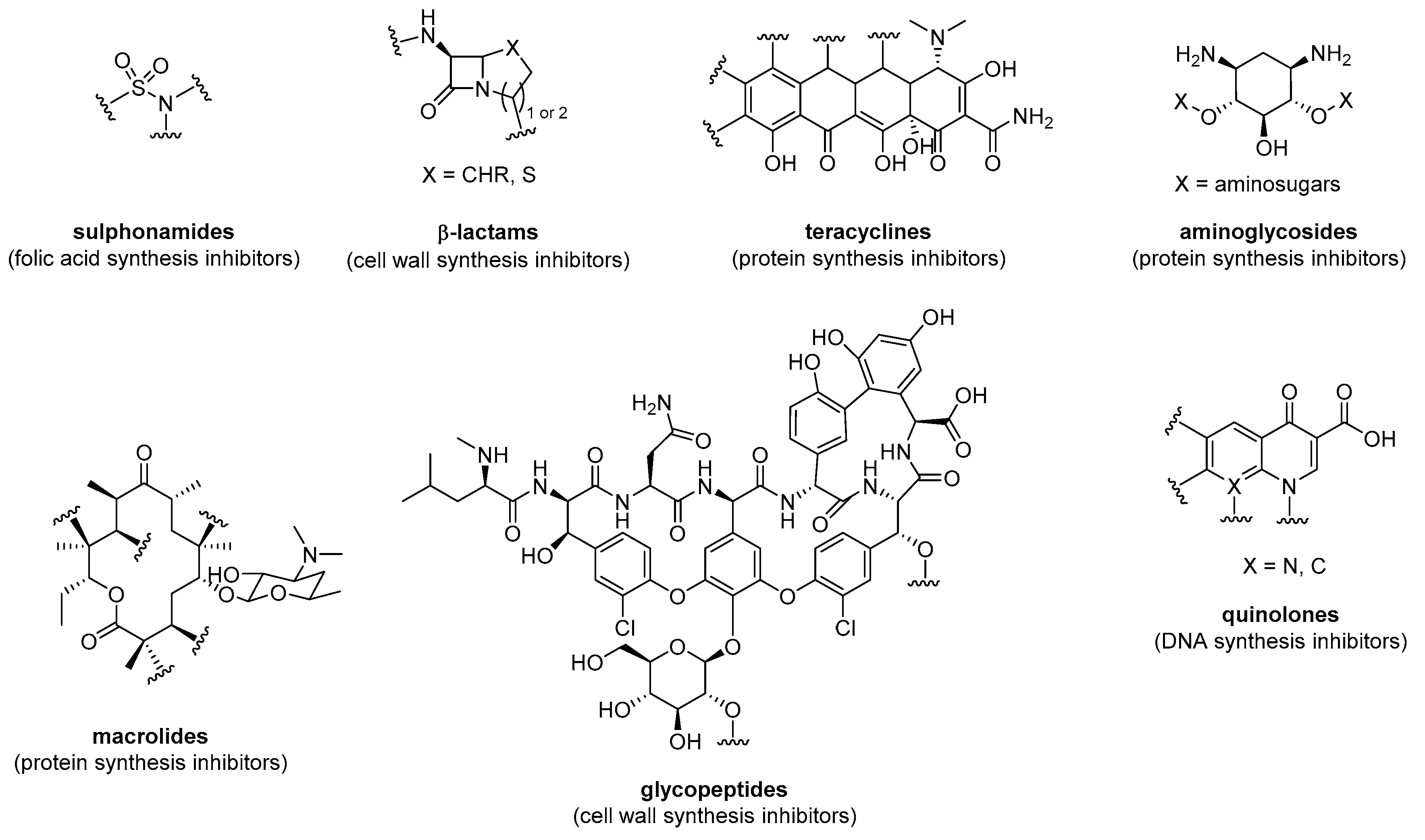
Figure 2.
Vancomycin’s points of attachment.
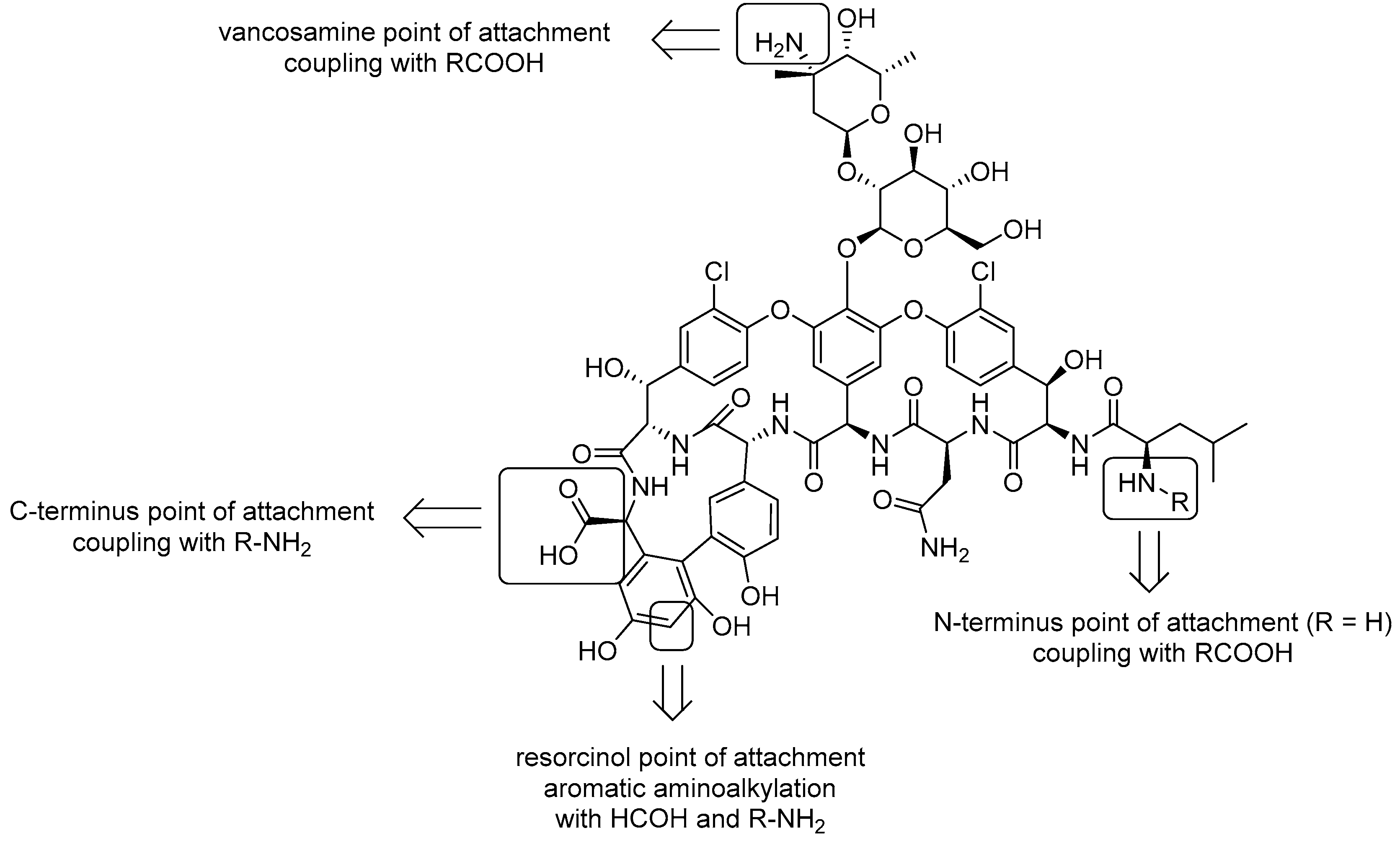
Figure 3.
Structure of vancapticins 1.
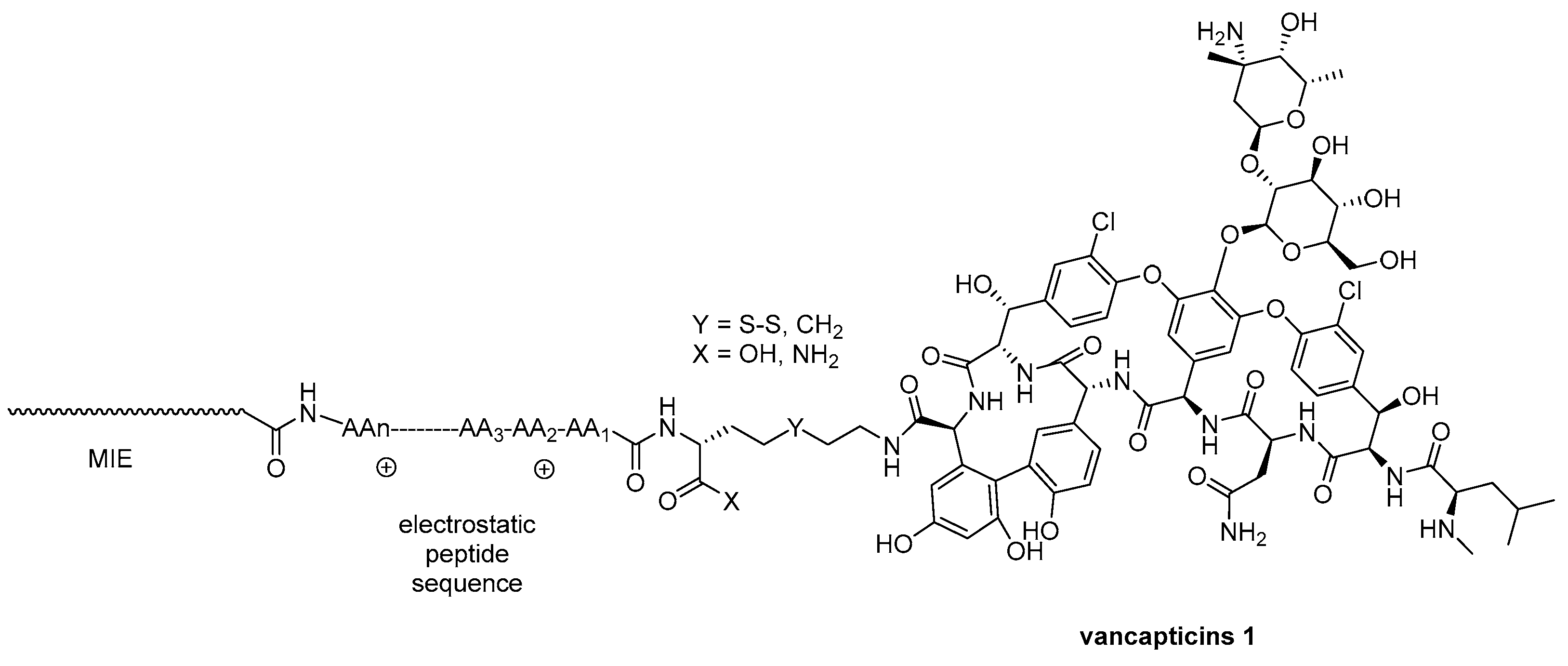
Scheme 1.
Structure of vancomycin-Ahx-r8 conjugate 2 (A) and its synthesis (B).
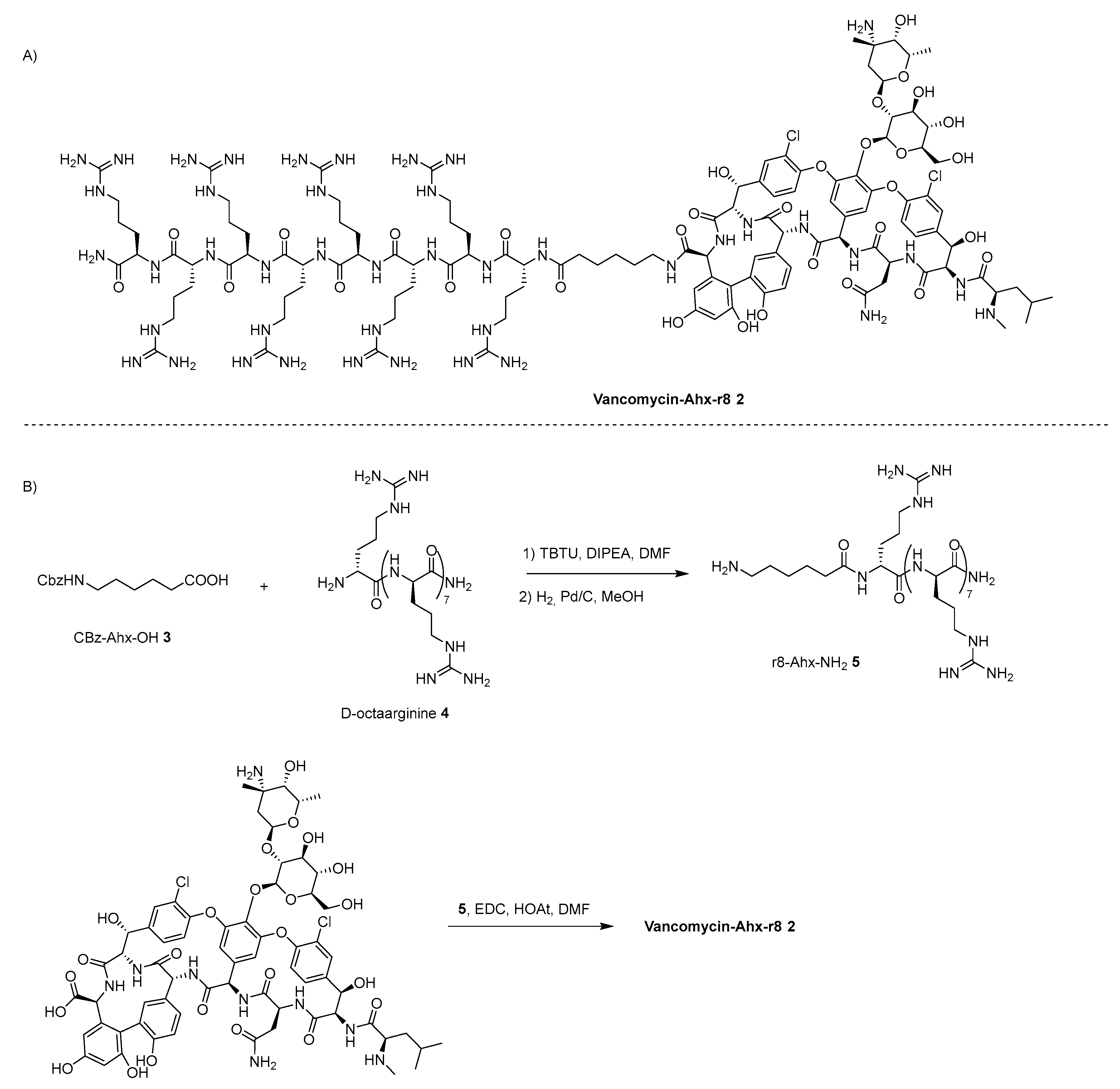
Scheme 2.
Structure of FU002 conjugate 6 (A) and its synthesis (B).
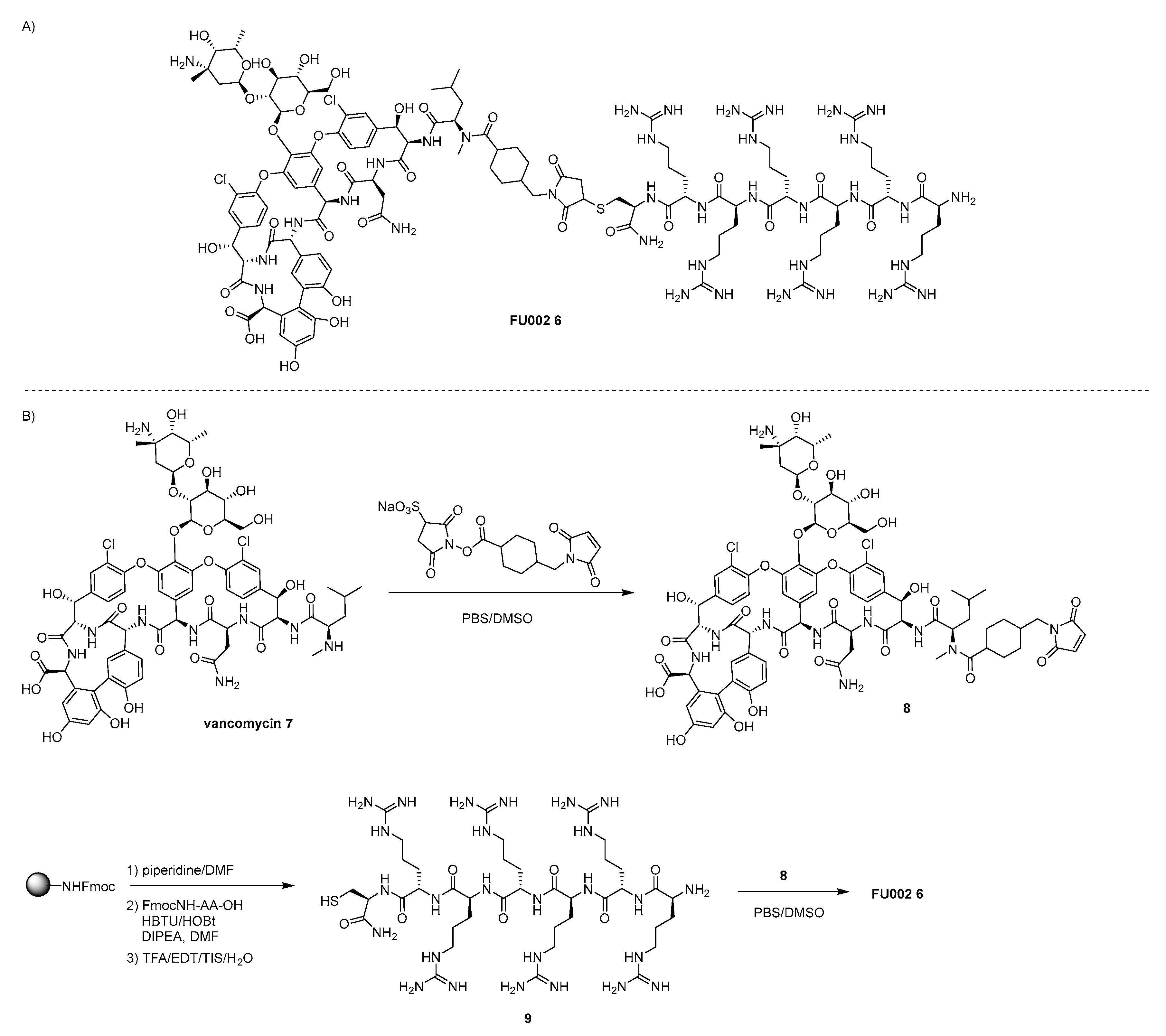
Figure 4.
Structure of Van-Hec 10.
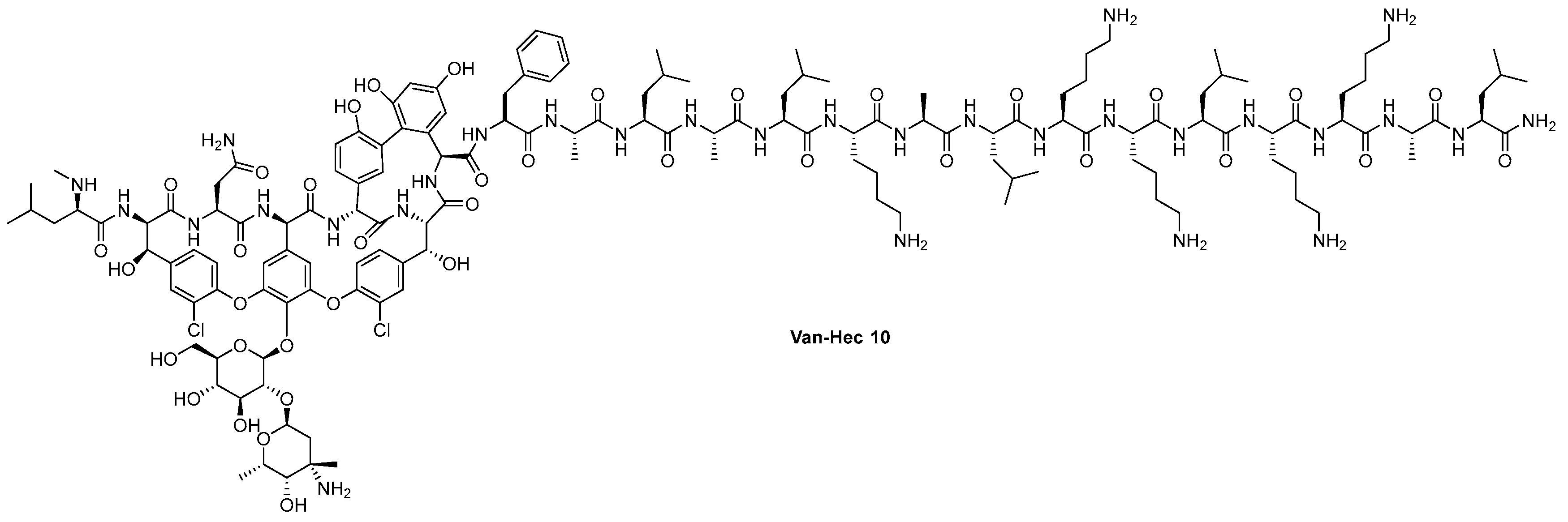
Scheme 3.
Structure of VPC conjugate 11 (A) and its synthesis (B).
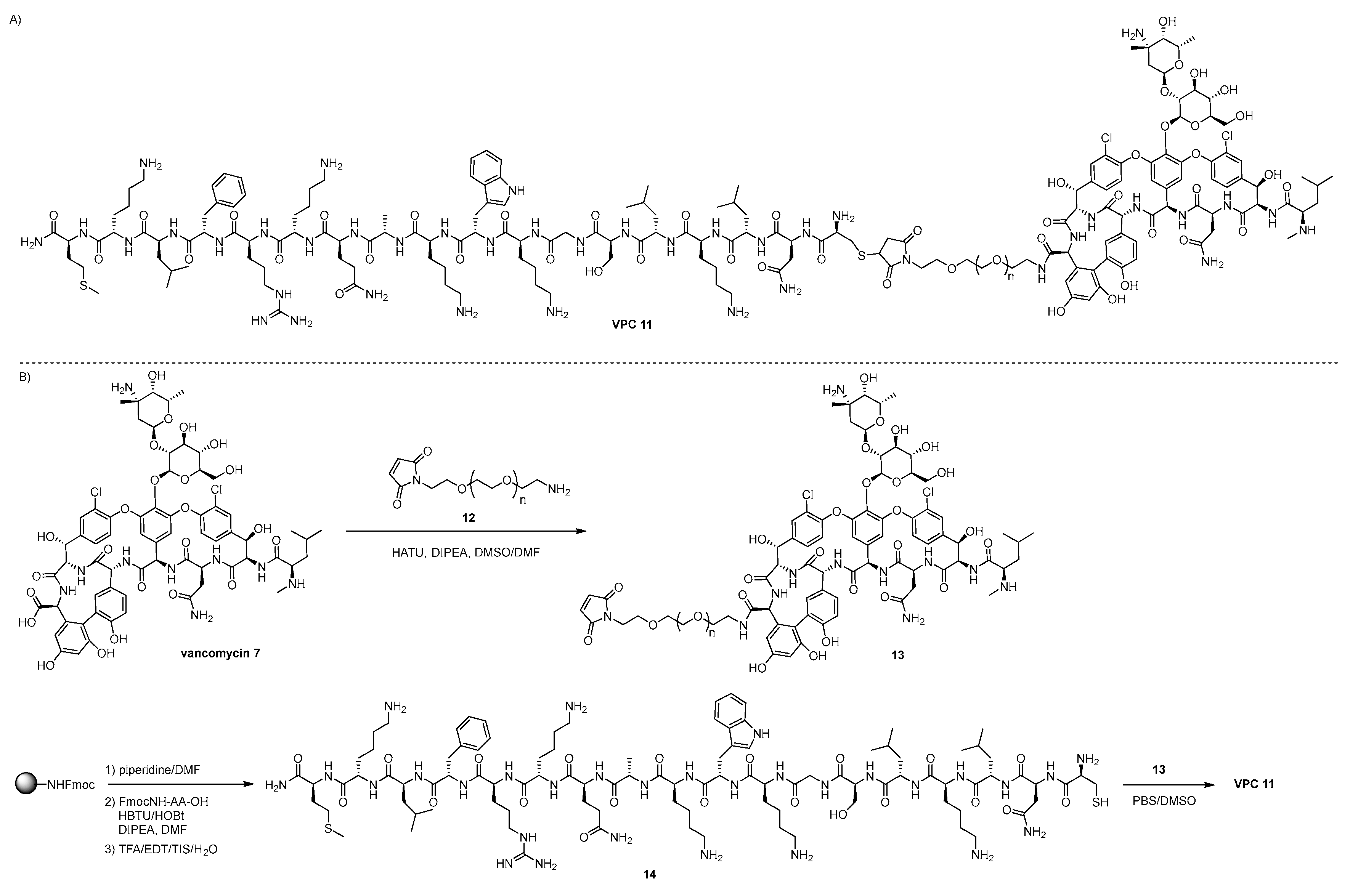
Figure 5.
β-lactam’s points of attachment.
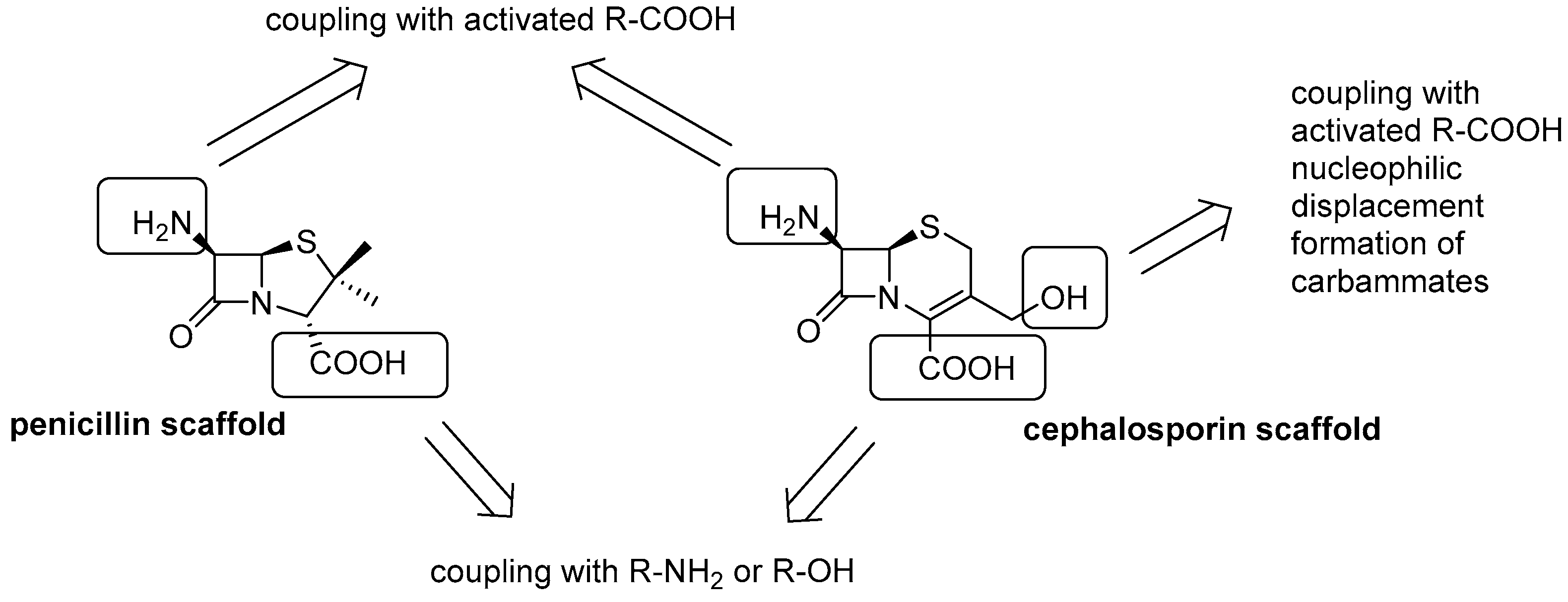
Scheme 4.
Structure of cephalotin- D-Bac8c(Leu2,5) conjugate 15 (A) and is synthesis (B).
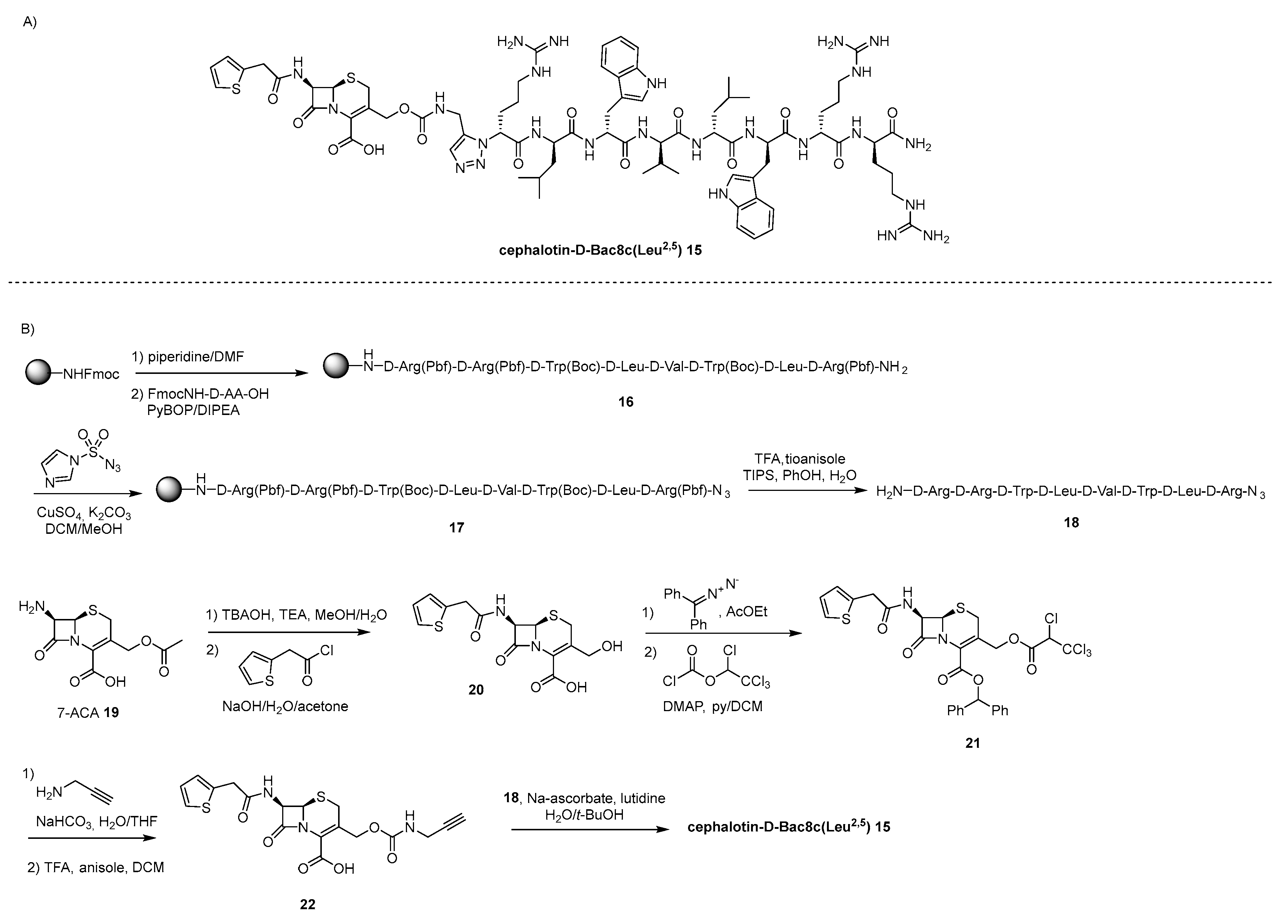
Scheme 5.
Structure of ACA- and ADCA-conjugates 23-28 (A) and their synthesis (B).
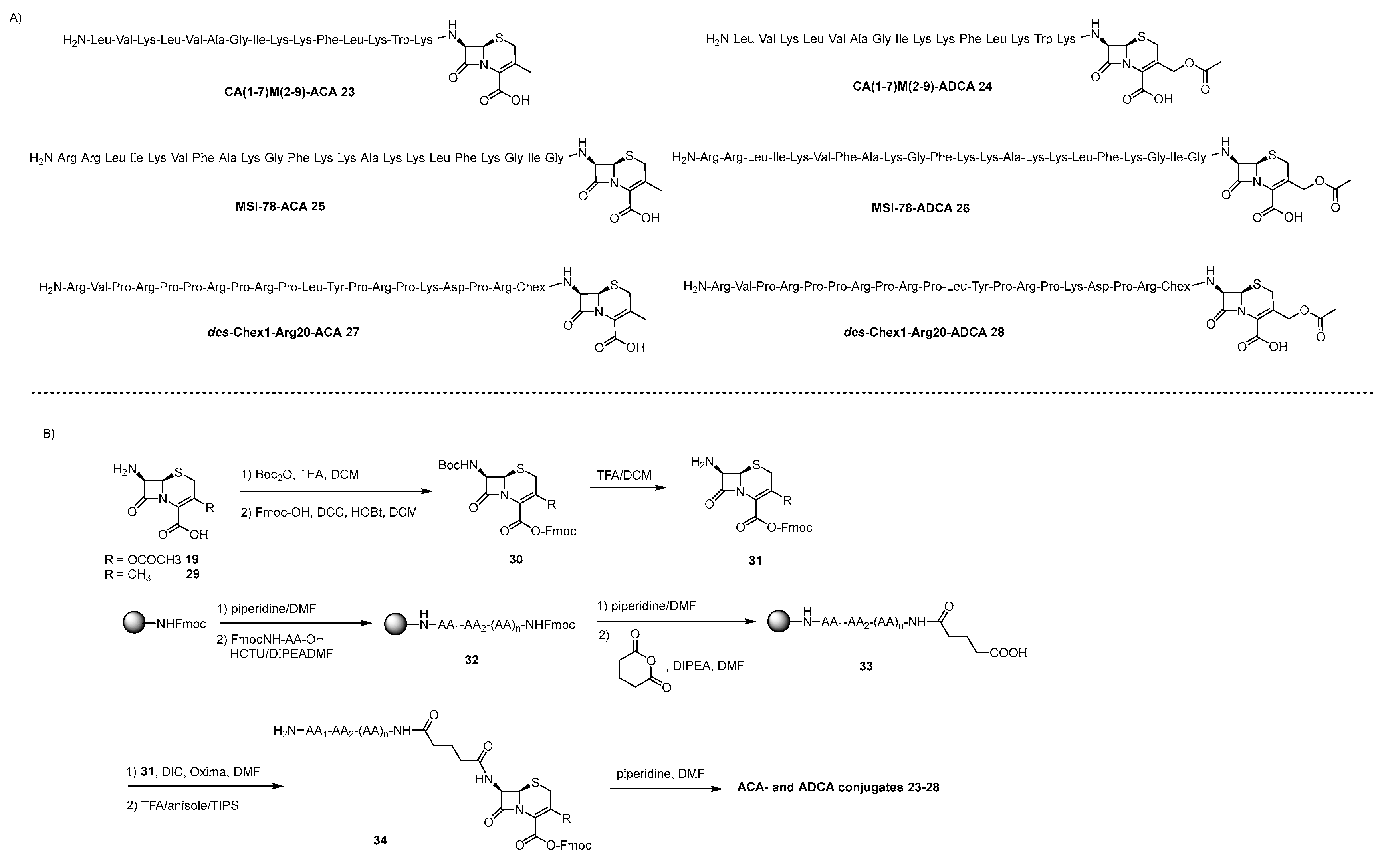
Scheme 6.
Structure of Amp-conjugates 35-38 (A) and their synthesis (B).
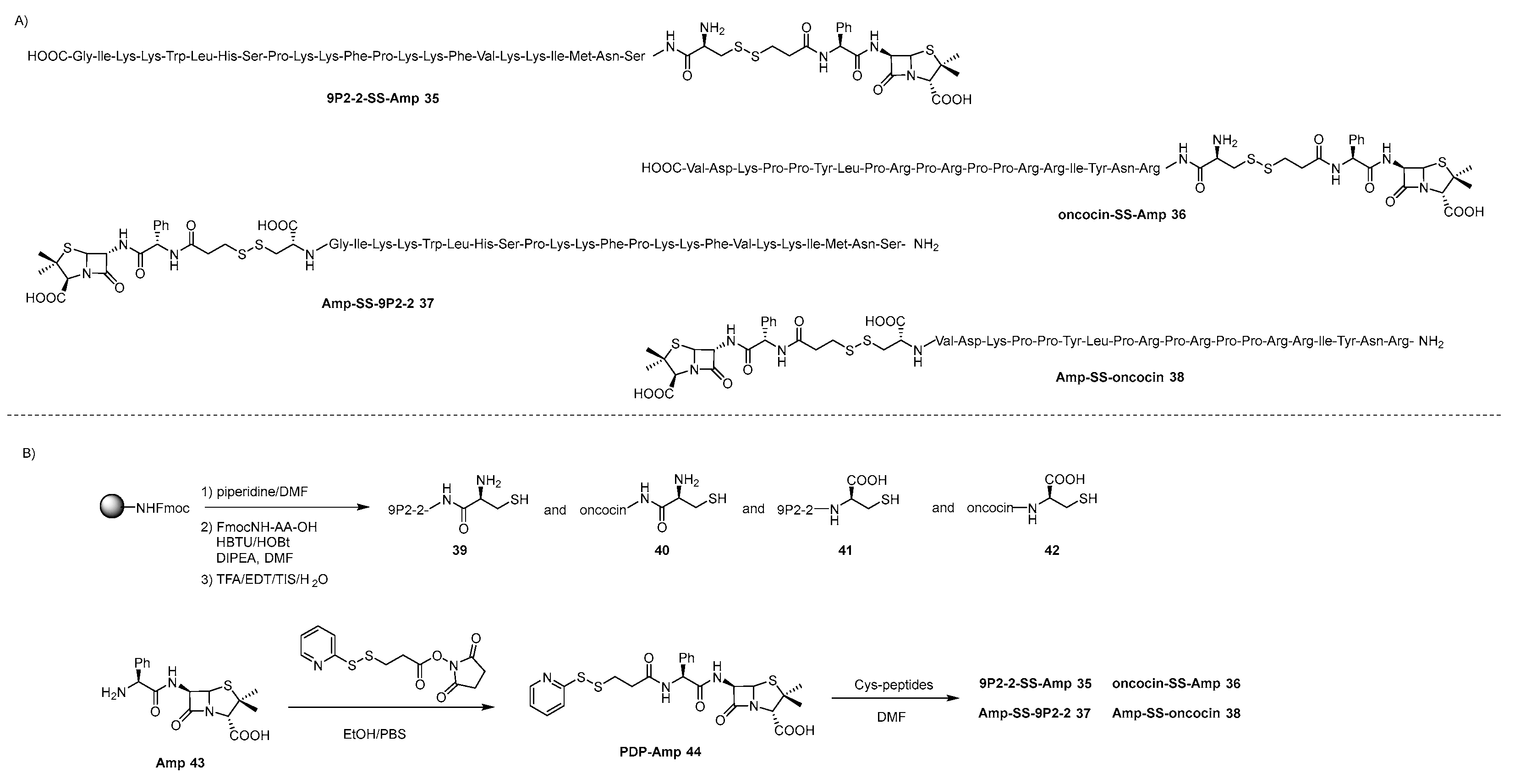
Figure 6.
Kanamycin and tobramycin selective functionalization.
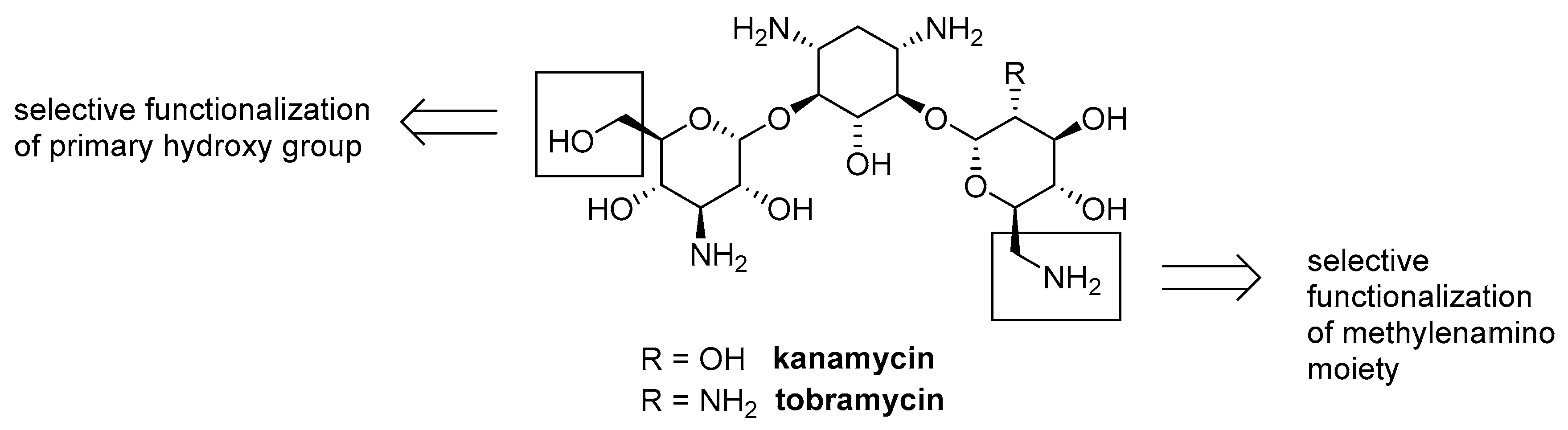
Scheme 7.
A) Structure of tobramycin-AMP conjugate Pentobra 45. B) Synthesis of Pentobra 45.
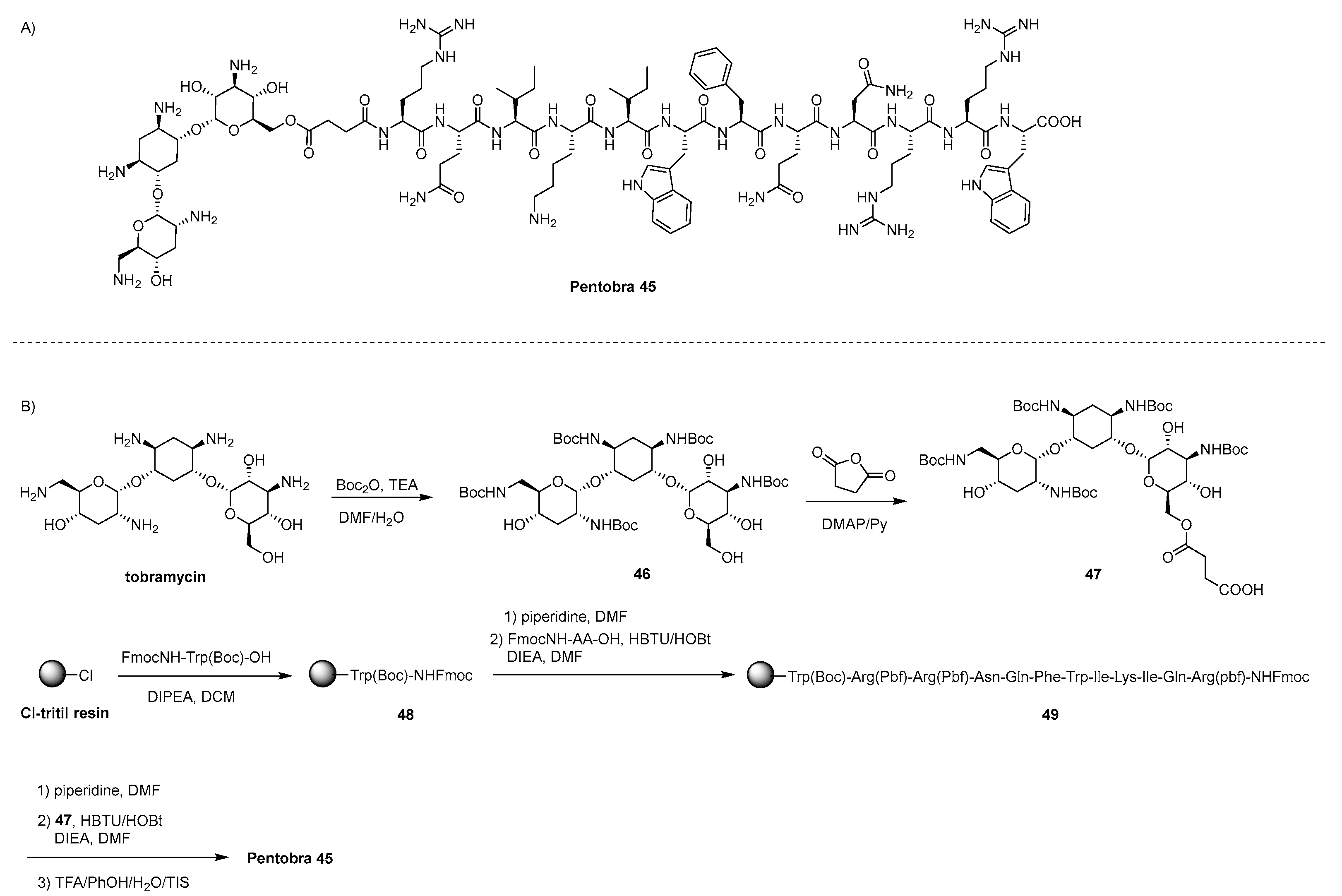
Scheme 8.
A) Structure of tobramycin conjugates 50-53. B) Synthesis of MAAP05 53.
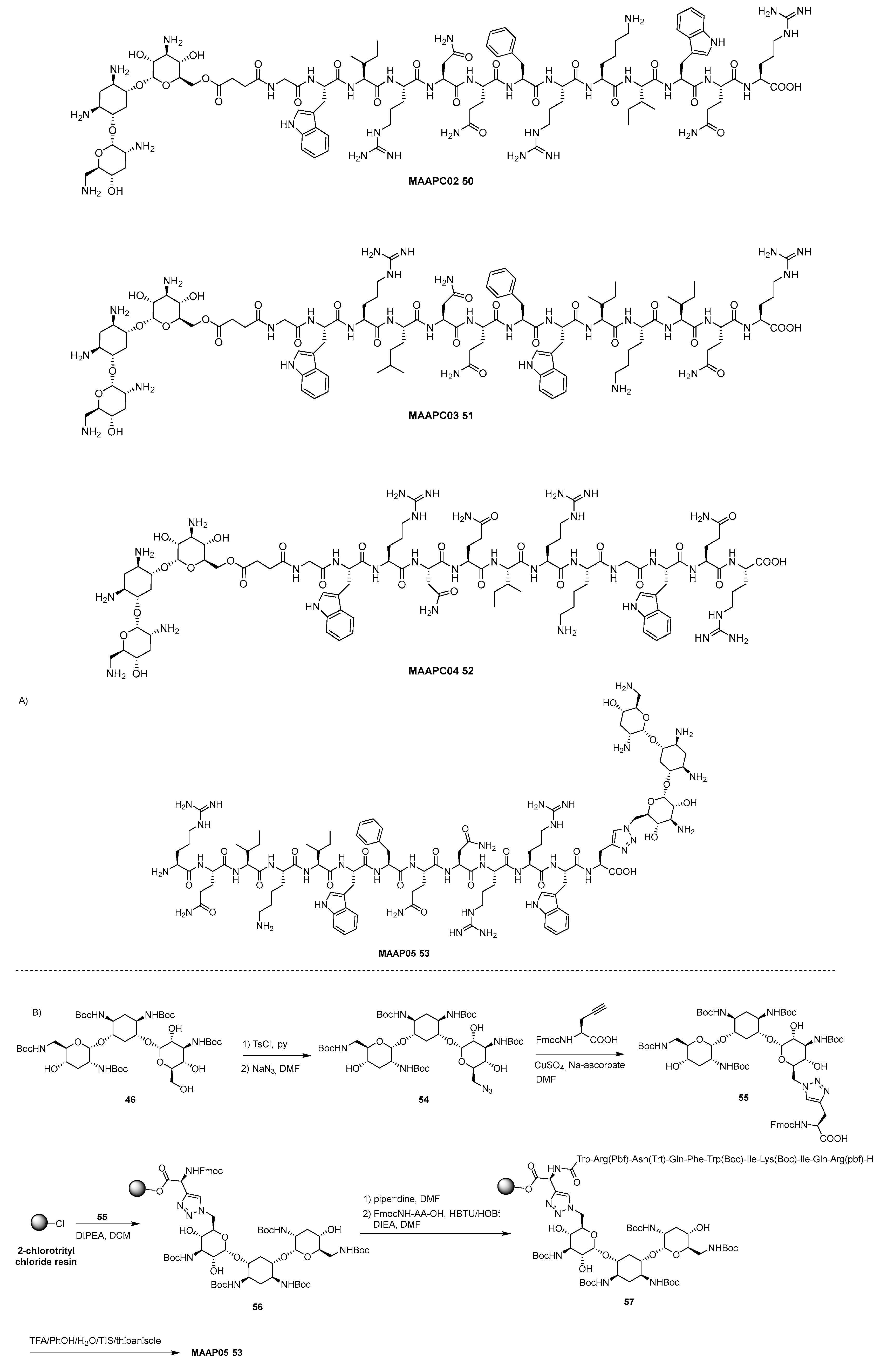
Scheme 9.
A) Structure of P14KanS 58 and P14KanC 59. B) Synthesis of P14KanS 58 and P14KanC 59.
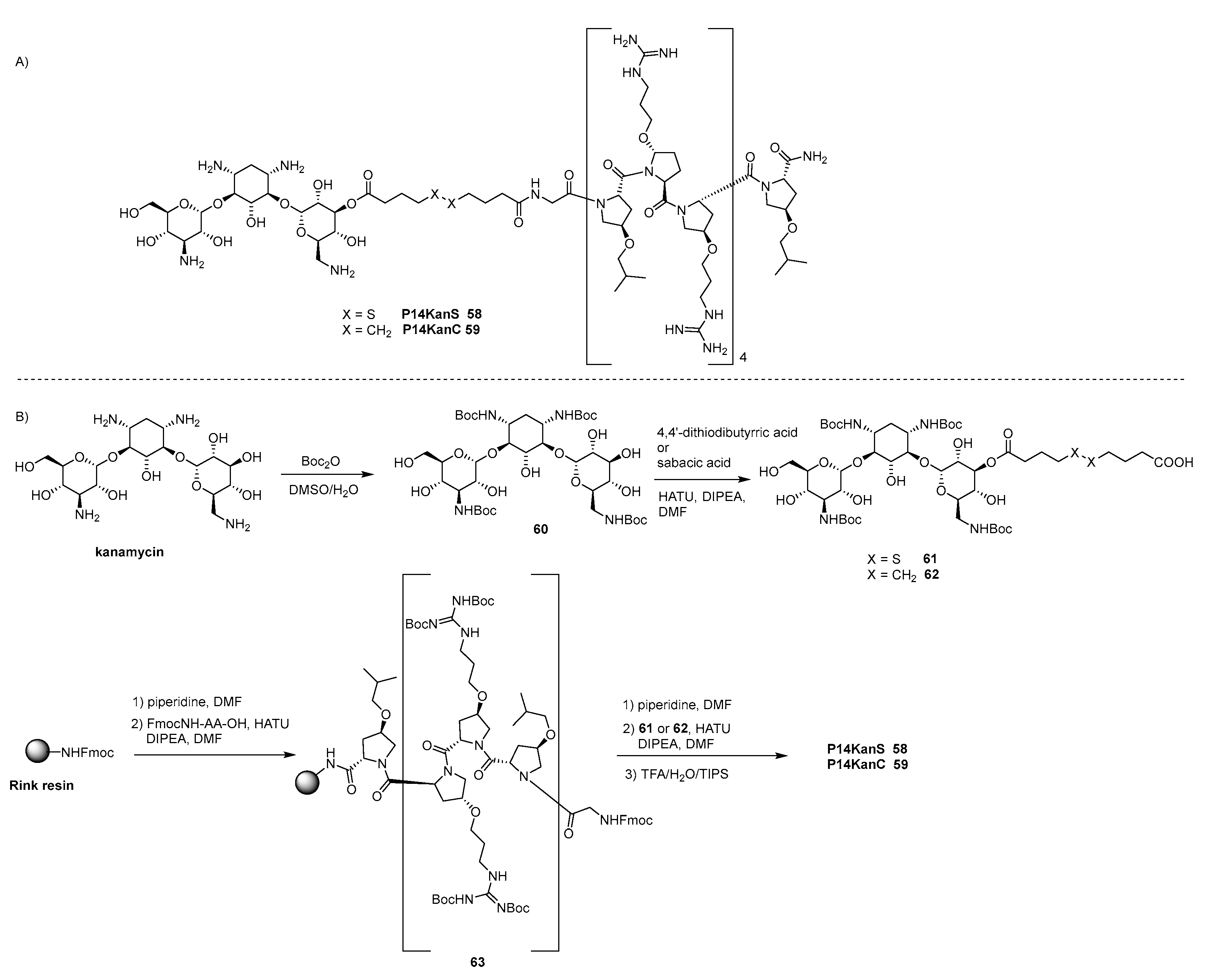
Scheme 10.
A) Structure of tobramycin-Bac7 conjugates 64, 65, and B) their synthesis.
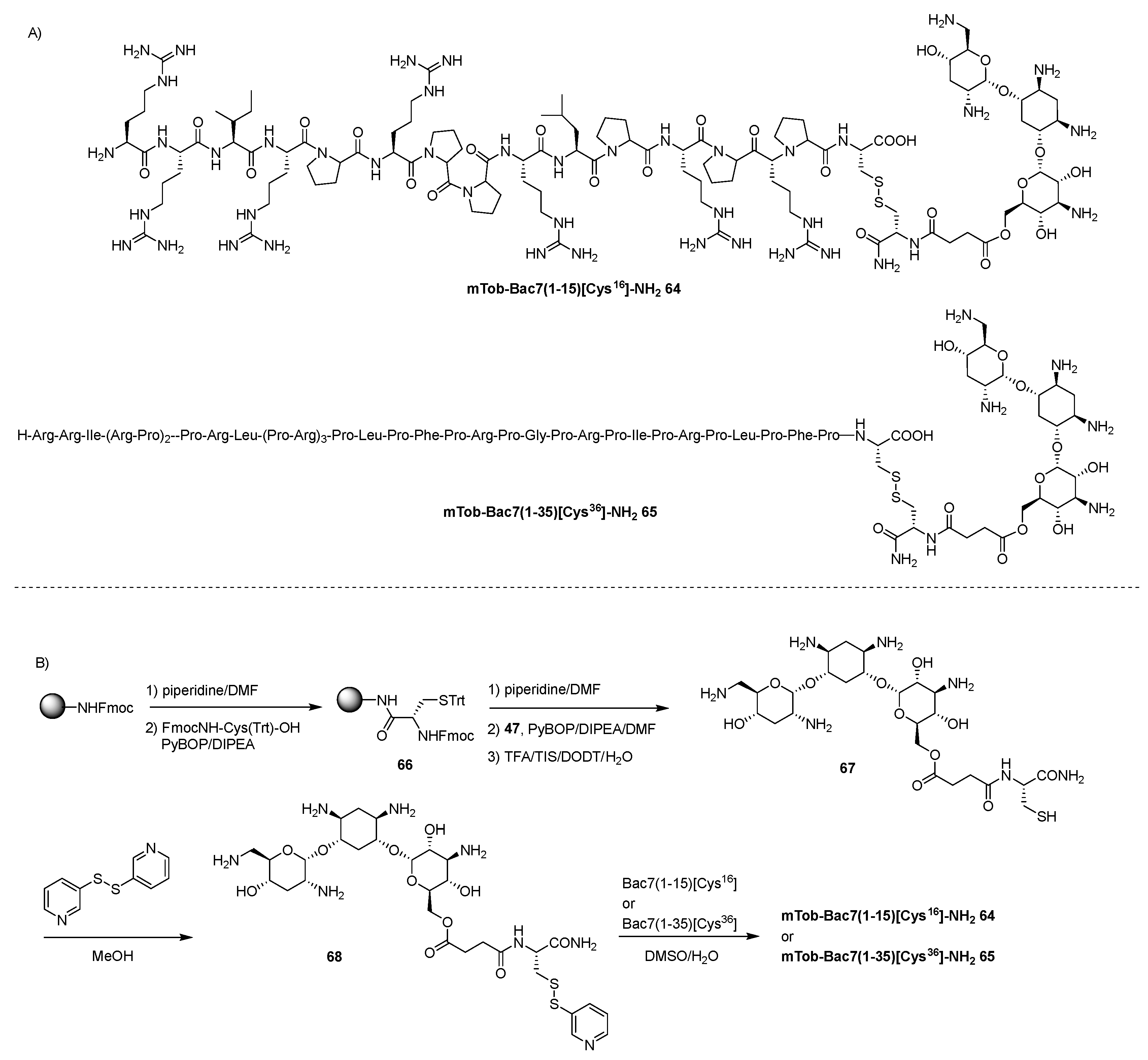
Figure 7.
Structure of Neo- and Amk-anoplin conjugates 69-74.
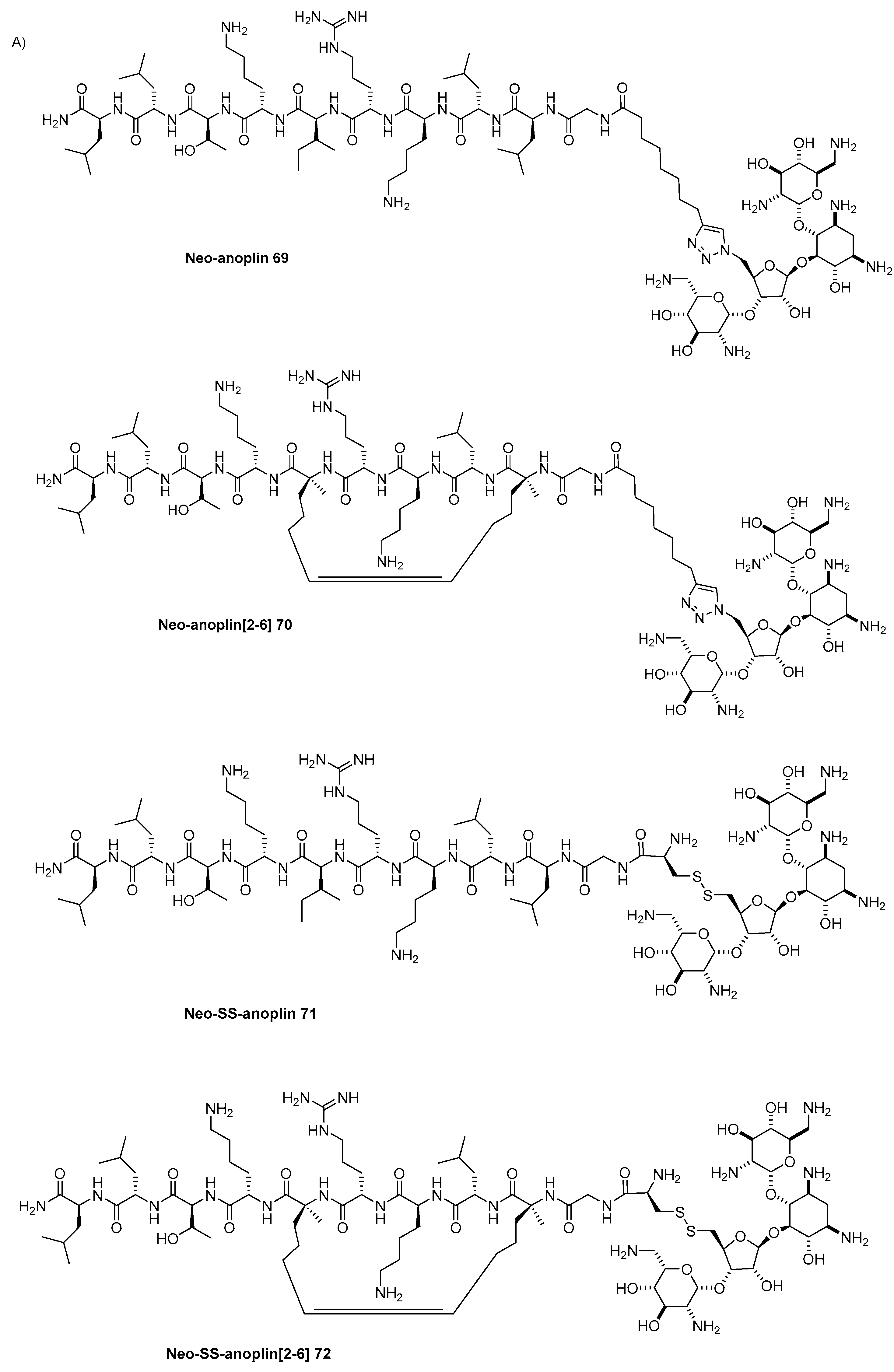
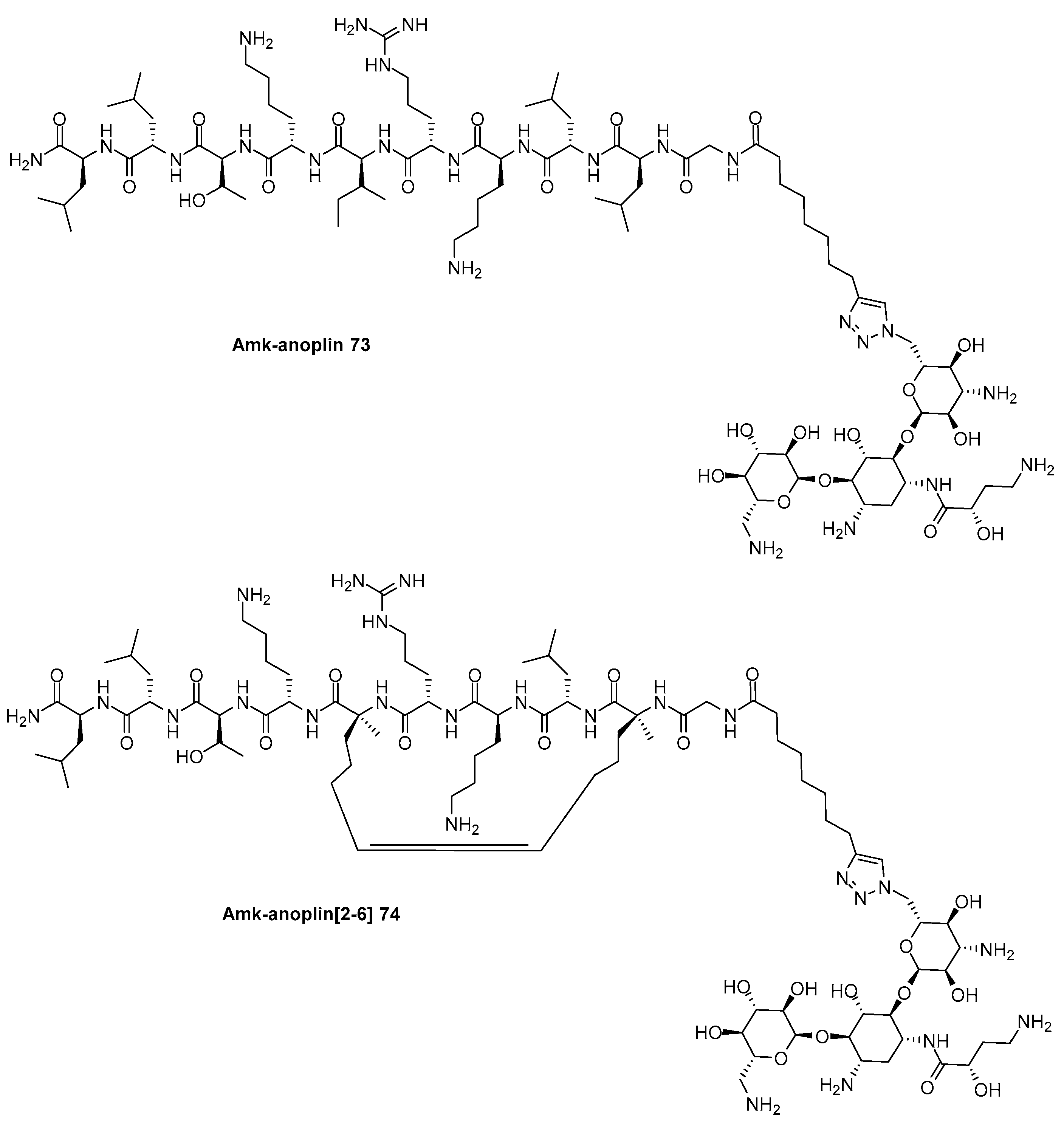
Scheme 11.
Synthesis of Neo-anoplin 69, Neo-anoplin[2-6] 70, and Neo-SS-anpolin 71.
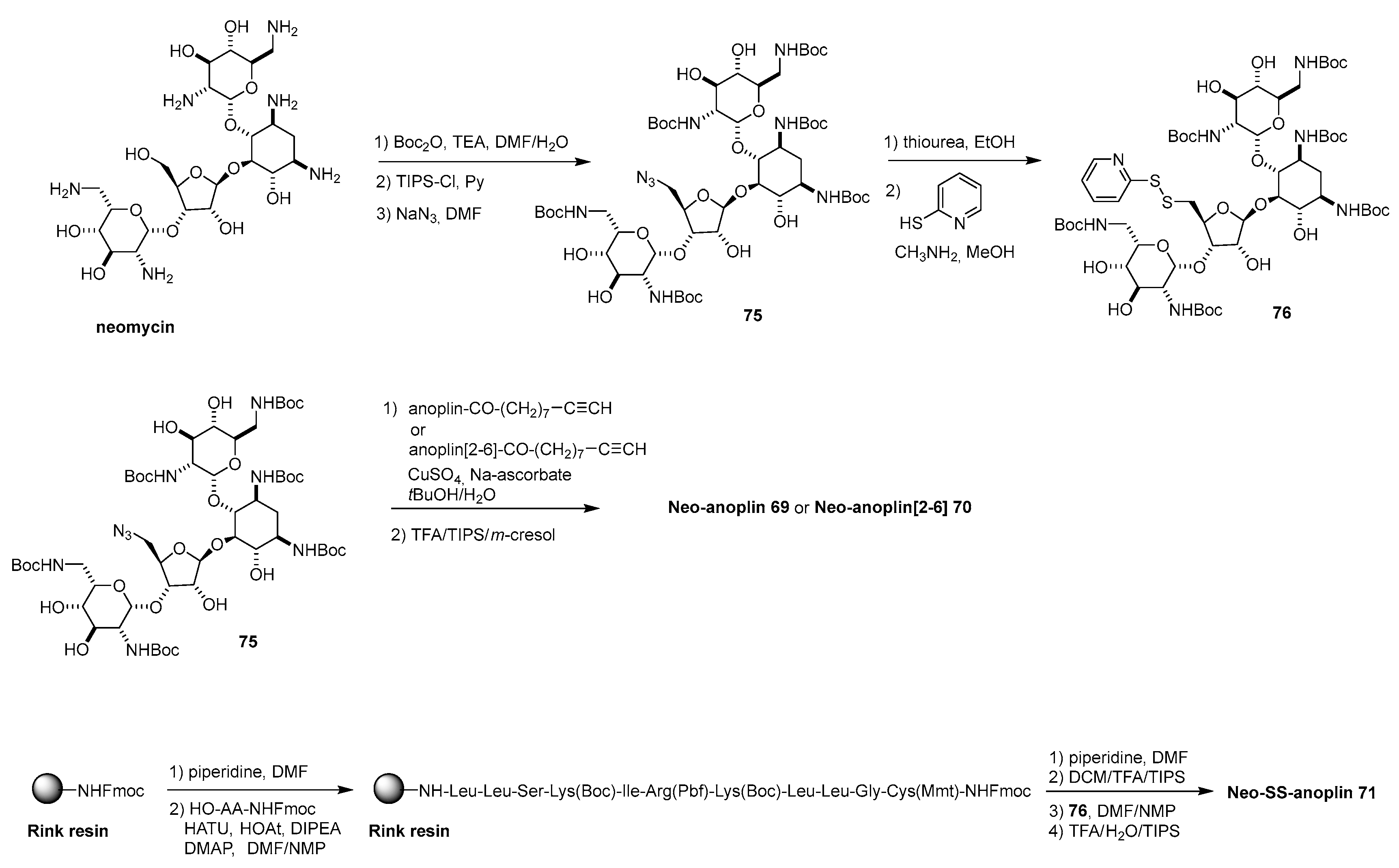
Scheme 12.
A) Structure of CPP-gentamicin conjugates 77-79, and B) their synthesis.
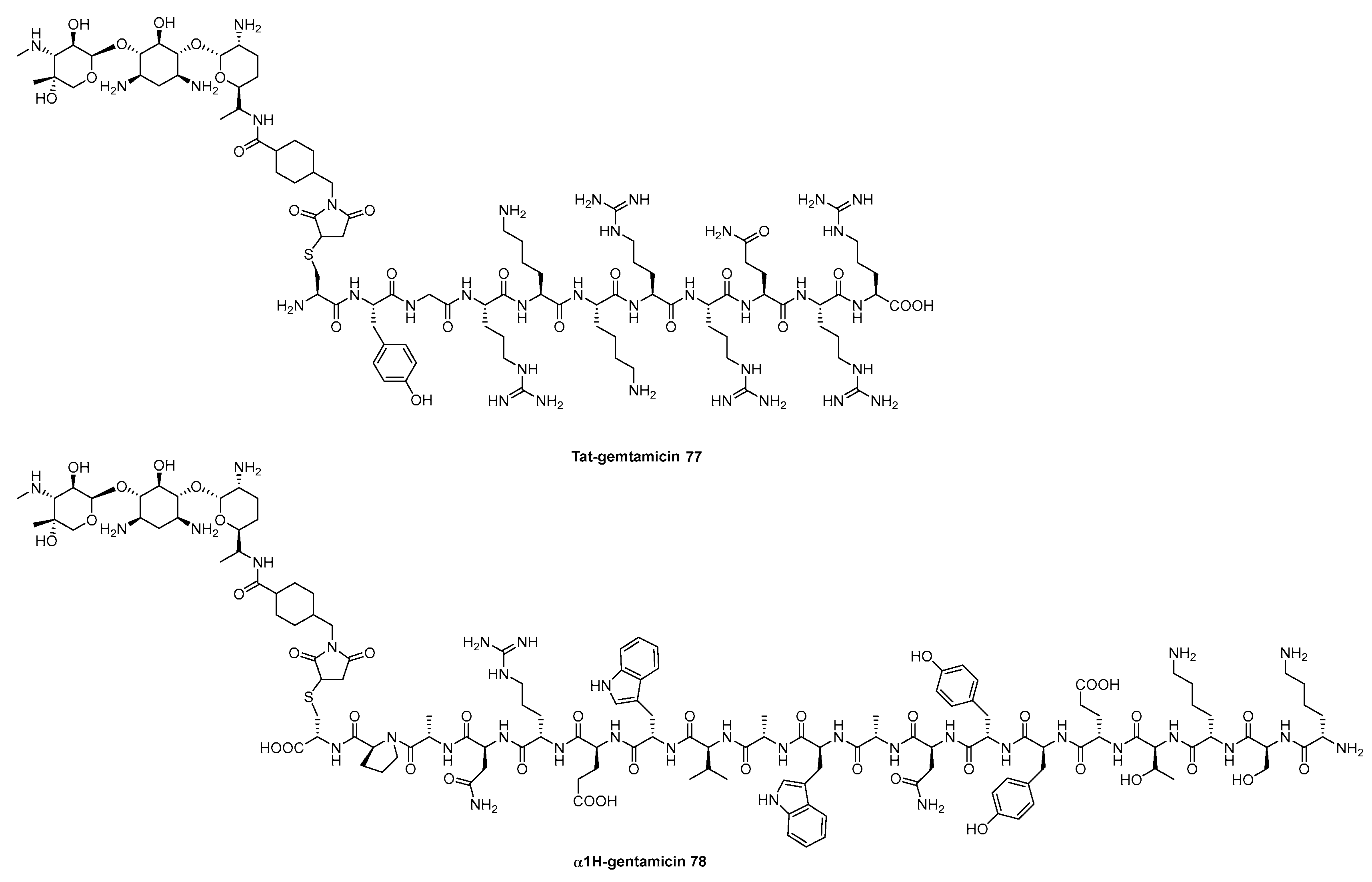
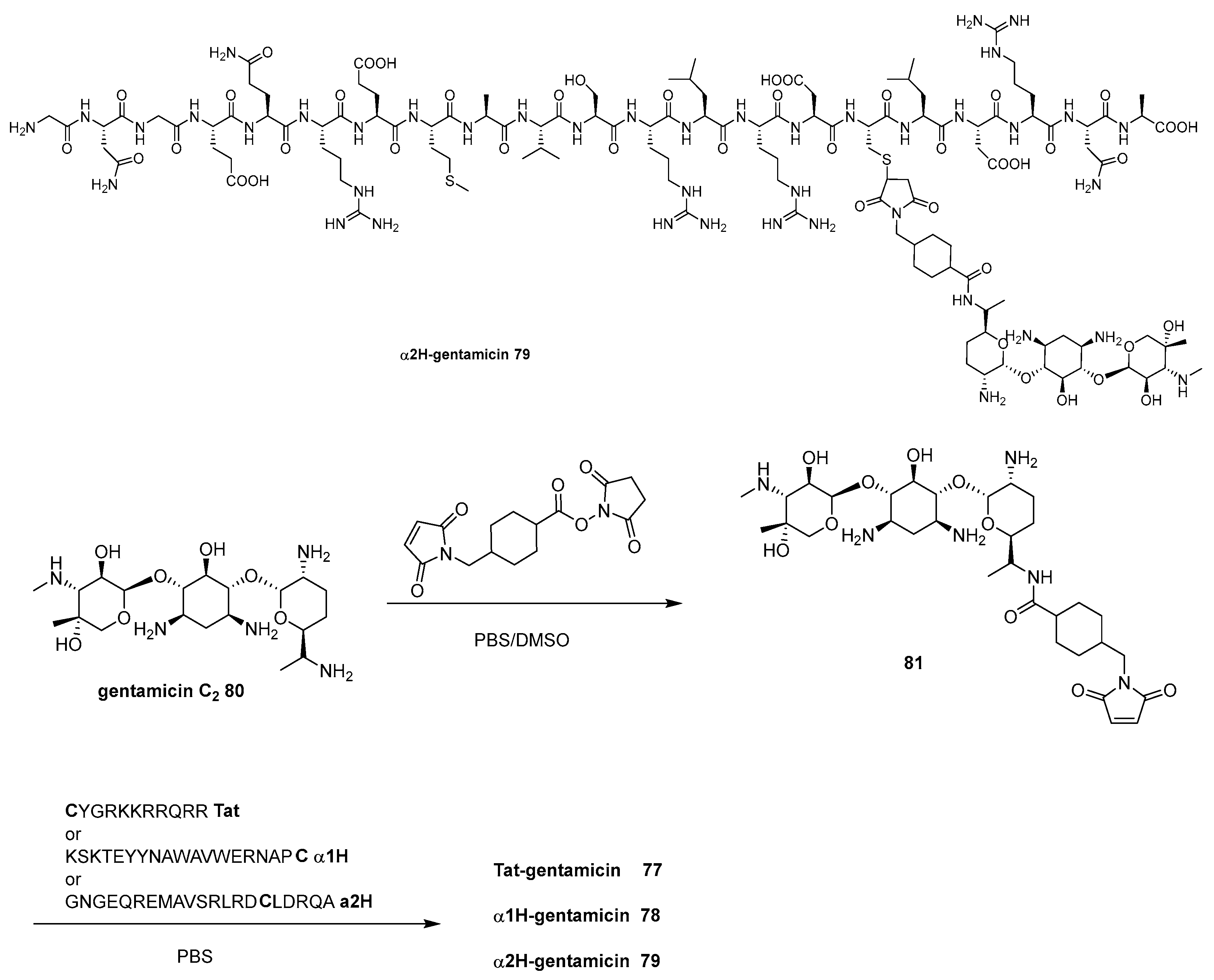
Scheme 13.
A) Structure levofloxacin-indolicidin conjugates 82, 83, and B) their synthesis.
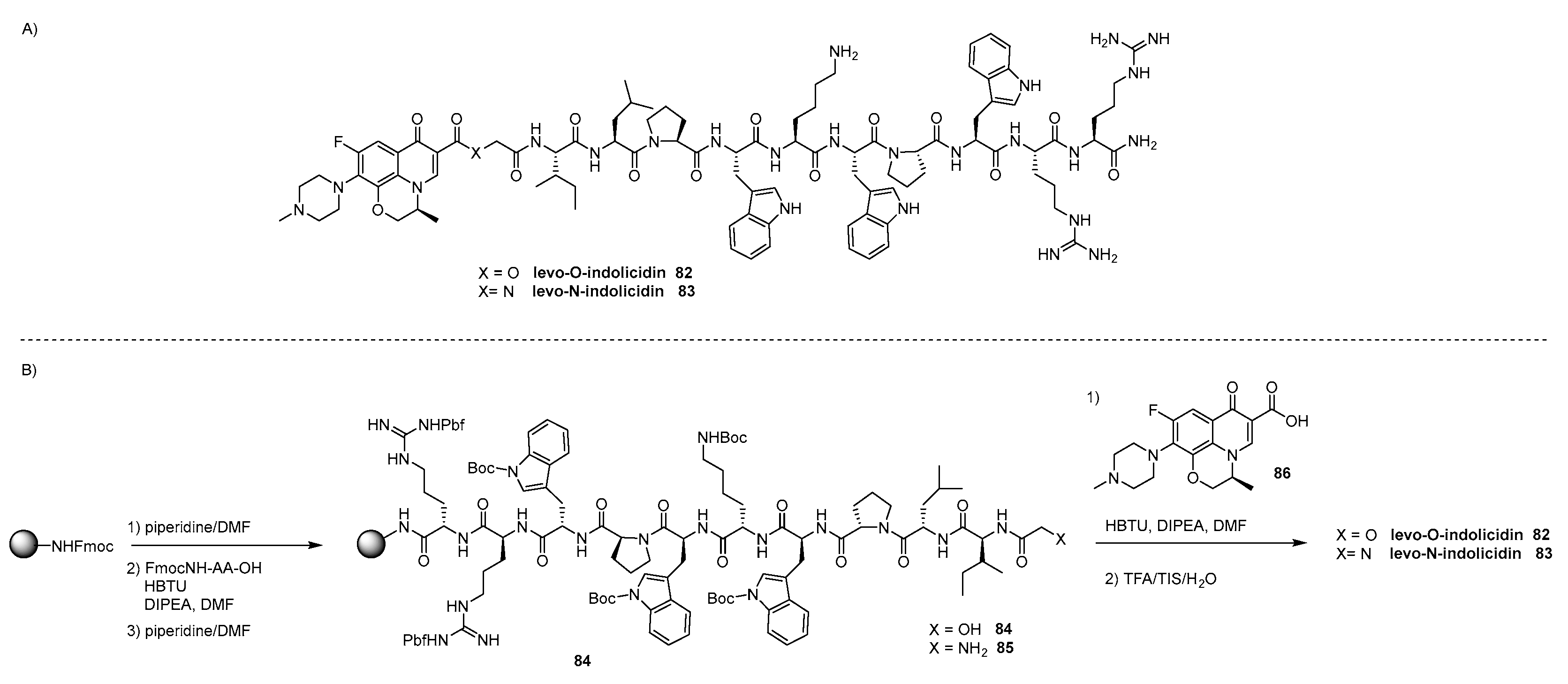
Figure 8.
Structure of HLopt2 conjugates 87-89.
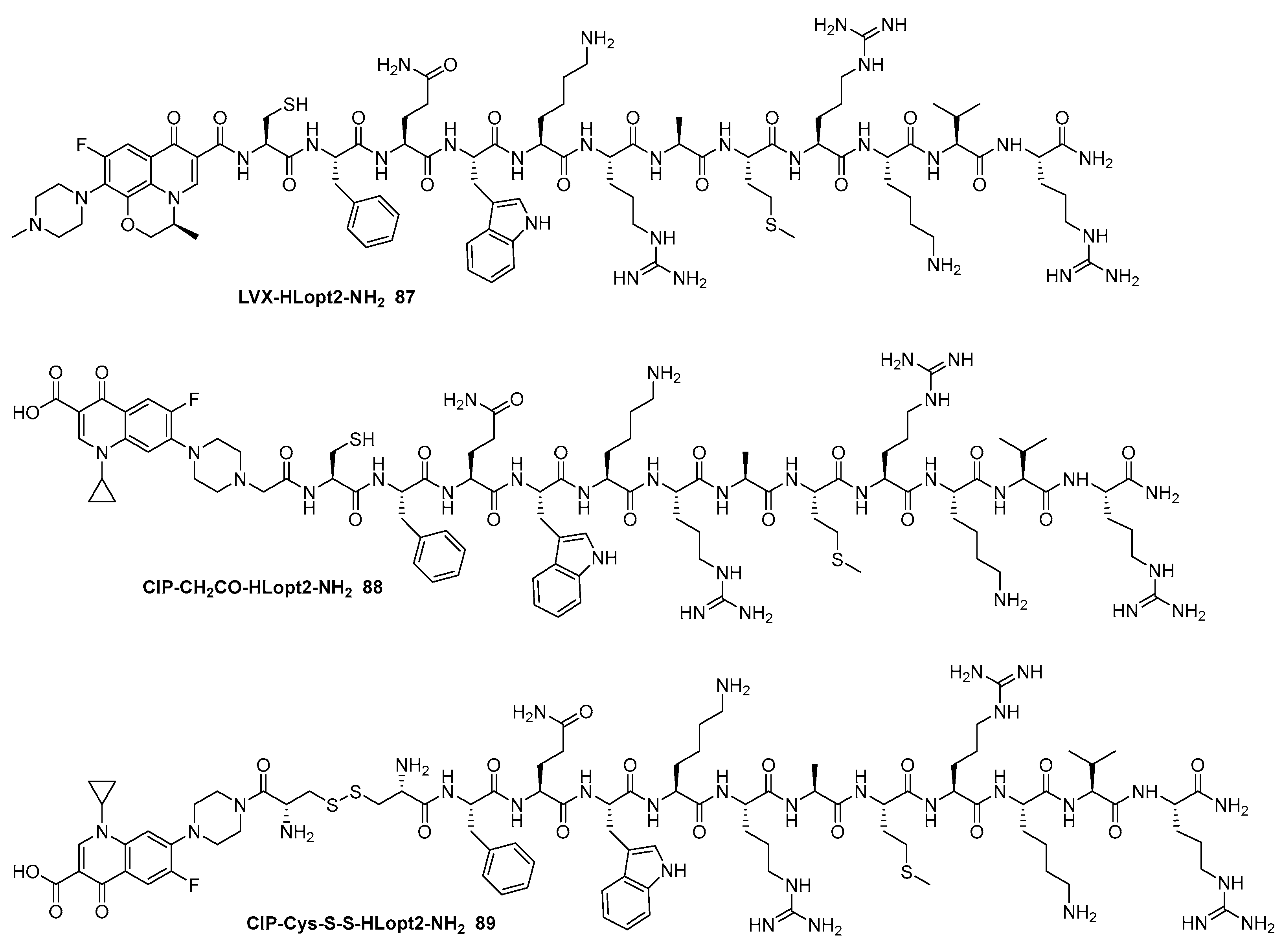
Scheme 14.
A) Structure of CAP-UBI29-41 90, and B) its synthesis.
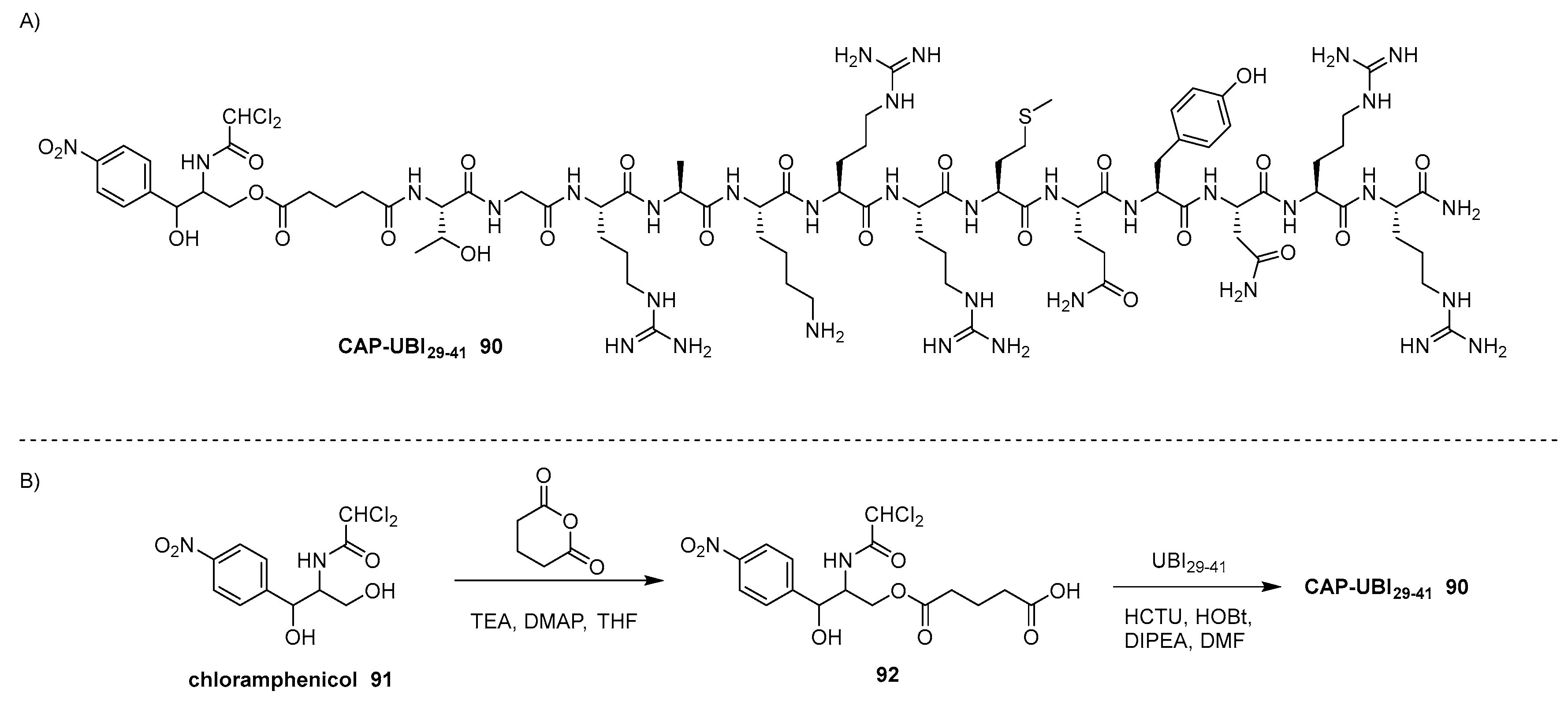
Scheme 15.
A) Structure of DES-oncocin 93, and B) its synthesis.
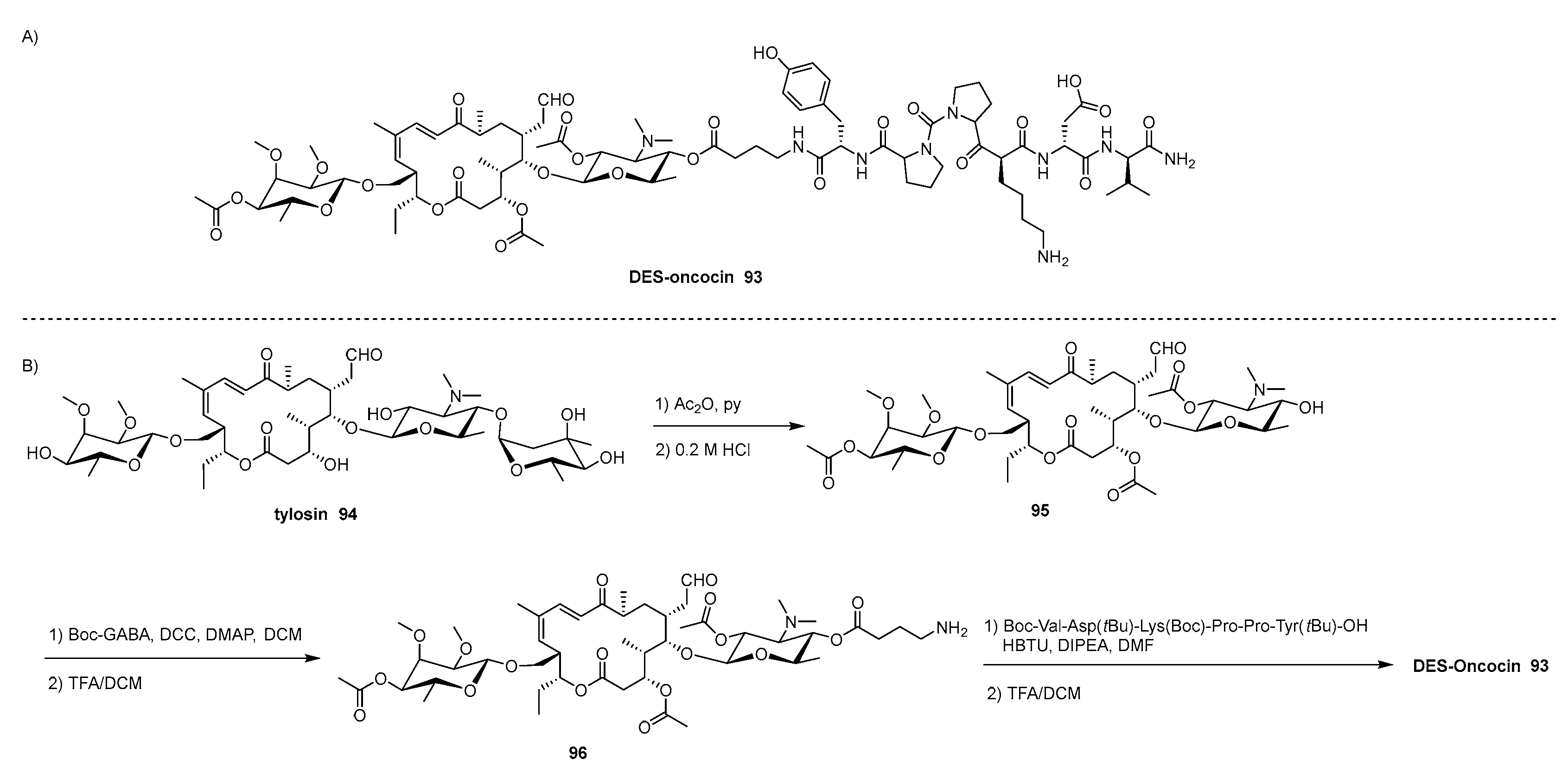
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



