
Submitted:
26 July 2024
Posted:
30 July 2024
You are already at the latest version
Abstract
Aim: Ovarian cancer is one of the most frequently diagnosed cancers in women in the world. Recognition of the differences between tumors in cancers and resistance to treatment in patients have made the treatment potential of chemotherapy, radiotherapy and immunotherapy controversial. Especially against drug resistance, laboratories have implemented the drug diversion system by putting old drugs into new forms. In this study, the cytotoxic and apoptotic effects of the combination of doxorubicin (DX) and thymoquinone (TQ) on ovarian adenocarcinoma cells (OVCAR3) were investigated.
Material Method: OVCAR3 cell line was cultured in RPMI medium containing 10% FBS, 1% penicillin/streptomycin. Cell viability and cell growth rate were determined by MTT viability test by combining doxorubicin and thymoquinone. Nucblue immunofluorescent staining was used to determine the mechanism of cell death at different doses of doxorubicin and thymoquinone compounds, qRT-PCR to determine the pathways underlying the apoptotic mechanism, and western blot methods to show protein levels.
Result: We found that the combination of DX and TQ reduced the viability of OVCAR3 cells (MTT assay), induced apoptosis (RAF, RAS, Bcl2, Bax by qRT-PCR) and increased RAF, RAS protein levels in TQ and DX-treated cells by western blot analysis.
Conclusion: The results obtained revealed for the first time that combining DX and TQ also had cytotoxic/apoptotic and anti-proliferative effects against ovarian adenocarcinoma cells.
Keywords:
Ovarian cancer
; apoptosis
; thymoquinone
; immunofluorescent staining
; western blot
Introduction
Cancer is a multifactorial disease in which certain normal cells in the body proliferate in an uncontrolled and anarchic manner. These cells evade normal differentiation mechanisms, regulate their proliferation, and resist programmed cell death [1]. Activation of oncogenes and/or inactivation of tumor suppressor genes results in uncontrolled cell cycle progression and inactivation of apoptotic mechanisms [2]. For a normal cell to become a cancer cell, it must accumulate certain precise changes in its physiology. Retention of proliferative signals, evasion of growth suppressors, resistance to cell death, infinite replicative potential, induction of angiogenesis, activation of invasion and metastasis, reprogramming of energy metabolism, evasion of immune system detection, inflammation favoring tumor growth, genomic instability and mutation [3]. The fifth leading cause of cancer-related deaths in women is ovarian cancer [4]. Ovarian cancer is classified according to the cell origin from which the tumor arises; It is divided into three groups: epithelial, germ cell and stromal. Several types of ovarian cancer, such as small cell carcinoma and sarcomas, which are extremely rare, have also been reported [5,6]. Epithelial ovarian cancer (EOC), which accounts for more than 85% of ovarian cancer cases, is responsible for the majority of ovarian cancer-related deaths. Most women with EOC are diagnosed with advanced, metastatic disease characterized by extensive peritoneal carcinomatosis and abdominal ascites. The incidence of death from ovarian cancer can be significantly reduced by developing new methods for early diagnosis and treatment of this fatal disease [7].
Rat sarcoma virus (RAS) proteins are intracellular guanine nucleotide-binding proteins (G proteins) that belong to the family of small GTPases. RAS proteins are generally involved in cell survival, cell cycle progression, cell polarity and movement, actin cytoskeleton organization, and vesicular and nuclear transport [8]. RAF is a proto-oncogene encoding a serine/threonine protein kinase, an effector protein downstream of RAS, and promotes cell proliferation and survival by transducing signals through mitogen-activated protein kinase. This pathway operates downstream of various receptor tyrosine kinases, such as EGFR, and is a key mediator of oncogenesis [9]. RAF mutations were found in 50 percent of malignant melanoma, 45 percent of papillary thyroid carcinoma, and 10 percent of colorectal cancer as well as ovarian, breast, and lung cancers [10]. Apoptosis, or programmed cell death, is a physiological process that involves cell shrinkage, chromatin condensation, protein cleavage, DNA breakage, and phagocytosis, among other morphological and biochemical changes. Apoptosis plays a very important role in the morphological and functional development of multicellular organisms and has a key role in the control of cancerous cells. The Bcl-2 family of proteins plays an important role in apoptosis. The Bcl-2 protein family includes anti-apoptotic and pro-apoptotic molecules [11].
Today, many plant-based treatment methods against cancer are applied as complementary treatments. Thymoquinone is found in the composition of Black Cumin, one of the plants used in this complementary treatment [12]. Despite the antineoplastic, antibacterial, immunostimulatory, anti-inflammatory and antioxidant properties of Thymoquinone, its mode of action has not been fully elucidated to date. However, Thymoquinone is reported to induce apoptosis in cancer cells [13]. It is also claimed that it inhibits DNA synthesis in cancer cell lines and, due to its immunostimulatory and antioxidant properties, reduces oxidative stress and inflammation in tumor cells, thus reducing malignant transformation and causing an increase in natural killer cell activities [14,15]. Although it has been shown by experimental models both in vitro and in vivo that thymoquinone and doxorubicin have an inhibitory effect on the growth and development of cancer cells, there is no study yet on whether these two agents direct ovarian adenocarcinoma cells to apoptosis. Therefore, this study aimed to investigate the effects of Thymoquinone on inducing apoptosis and proliferation of ovarian cancer cell lines in vitro.
Material Methods
Cell Culture
OVCAR3 cell line (NIH:OVCAR-3 (HTB-161™) was used in our laboratory. RPMI 1640 containing 10% Fetal Bovine Serum (Gibco), 2 mM L-glutamine, and 1% penicillin/streptomycin was added to make the medium for the cells. , were propagated as a monolayer culture in sterile culture dishes at 37°C in a humidified environment containing 5% CO and 95% air, and passaged with EDTA and trypsin began when the cells occupied 80–90% of the area of the cell culture dish in which they were grown. and the suspended cells were collected in a 15-milliliter centrifuge tube, then centrifuged in a centrifuge at 1000 rpm for 5 min. After the centrifuge was stopped completely, the supernatant was removed and the pellet was resuspended.
A hemocytometer is used to measure the number of cells in the suspension obtained by centrifugation. 10 μl of suspended cells were removed and then transferred to a 96-well plate. A 1:1 dilution was prepared by mixing 10 μl of cells with 10 μl of blue trypan. Cell count in the hemocytometer is determined according to the following equation:
Number of cells per ml = Number of viable cells counted x dilution factor x 104
Determination of IC50
In the study, stock solutions of Doxorubicin and Thymoquinone agents were prepared using pure ethanol. A stock solution of 5 mM for doxorubicin and 50 mM for thymoquinone was prepared. In the applications, the final concentration of the vehicle in the flasks or plate wells was reduced to 0.1%. To determine Doxorubicin and Thymoquinone IC50 doses, the OVCAR-3 cell line was planted in 96-well culture dishes with automatic multipipettes at 3x103 cells per well. At the end of one night (approximately 16 hours), doxorubicin was applied at 0.5-50 µM and thymoquinone 5-500 µM dose ranges in 9 different concentrations were incubated for 24, 48 and 72 hours.
MTT Assay
In MTT analysis, chemotherapy agent and control groups were designed to consist of 6 wells. Cell survival analysis was performed after incubation. For this, "Yellow tetrazolium MTT (3-(4, 5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide)" test solution, prepared at a dose of 5 mg/ml, was added to all wells at 20 µl/well. Then, the plates were incubated for 4 hours, and after incubation, 200 µl ultra puree DMSO (Merk, USA) was added to each well and incubated for 4 hours in the dark. At the end of this period, the plates were read spectrophotometrically at 492, 570 and 650 nm wavelengths. The value obtained from the control group was taken as 100% viability and the viability rate was determined comparatively. IC50 values for each cell line and chemotherapy agents in the control and experimental groups were calculated using probit analysis with the SPSS 20 statistical package program.
Metastasis Analysis
The wound healing assay, an experiment to study cell migration and cell-cell interaction, was performed to determine the effect of single and combined doses of TQ and DX agents in OVCAR-3 cells. For the wound healing experiment, cells were seeded in a 6-well plate to be 100% confluent. After observing the adhesion of the cells to the plate surface, the medium was removed. A scratch was made on the plate surface with the help of a 200 µl pipette tip. The purpose of the scratch is to create a cell-free area in order to observe cells migrating and closing the gap. Afterwards, it was washed again with PBS and dose groups were applied in the medium. At the 36th hour, when the control group scratch cells were completely closed, the study was terminated and photographed. The photographs taken were compared with the control group and analyzed.
Total RNA Isolation
In the study, OVCAR-3 cells were incubated until the logarithmic phase. When the cells reached the logarithmic phase, Control, Doxorubicin IC50: 2.12 μM, Thymoquinone IC50: 62.9 μM doses were applied individually. RNA was isolated from the samples 48 hours after the agent treatment. During the isolation phase, Purelink RNA mini kit (Thermo, USA) was used and the kit protocol was followed. Accordingly, 1% mercaptoethanol was added to the lyszis solution included in the kit, the medium was removed, and 1 ml of this solution was placed in each 25-ml flask washed with D-PBS. These flasks were kept in a 37°C incubator for 20 minutes. At this stage, it was shaken by gentle shaking by hand every 5 minutes. Afterwards, the lysed cells in the flasks were collected in 2 ml Ependorph tubes and an equal volume of 1 ml of 70% ultrapure ethanol (Merk, USA) was added to them. This mixture was vortexed and then loaded into 700 µl volumes onto the columns provided in the kit. They were sequentially centrifuged at 12,000 g for 30 seconds and the RNAs were loaded onto the column, and then these columns were washed first with washing solution 1, then twice with washing solution 2, by centrifuging at 12,000 g for 30 seconds at each stage. After the washing process, the columns were centrifuged once at 12,000 g for 3 more minutes to dry them, then the columns were placed in new sterile 1.5 ml Ependorph tubes, 60 ul of the solution given in the kit was pipetted to the middle of the membrane in the column, and these columns were placed at 12,000 g. Pure RNAs were collected in an Ependorf tube by centrifugation for 1 minute. The purity of the collected RNAs was determined by Optizen NanoQ microvolume spectrophotometer (Mecasys, South Korea) and all were equalized with ultrapure water to 750 ng/10 µl.
cDNA Synthesis
cDNA synthesis was performed to enable the RNAs obtained after the synchronization process to be amplified by PCR. At this stage, High-Capacity cDNA Reverse Transcription Kit (Life Technologies, USA) was used, and according to the kit protocol, the enzyme, DNTP mix and random primers in the kit were mixed and pipetted into PCR tubes as 10 µl. Afterwards, the total RNA equal to 750 ng/10 µl in the previous section was placed into the same tubes. These tubes were incubated in the Applied Biosystems® ProFlex™ PCR System thermal cycler step 1 at 25 °C, 10 minutes; step 2 37 °C, 120 min; step 3 cDNA synthesis was performed using 85 °C, 5 min cycles. The obtained cDNAs were stored at -20 °C for ongoing studies.
Quantitative Real-Time PCR
In the study, the expression levels of RAS, RAF, Bcl2 and Bax genes treatment groups of OVCAR-3 cells were analyzed by qRT-PCR method. The primers of these genes are given below in the order 5'-3'.
RAS: F: ACAGAGAGTGGAGGATGCTTT, R: TTTCACACAGCCAGGAGTCTT
RAF : F: GGGAGCTTGGAAGACGATCAG, R: ACACGGATAGTGTTGCTTGTC
BCL-2: F: ATGTGTGTGGAGAGCGTCAA, R: ACAGTTCCACAAAGGCATCC
BAX: F: TTCATCCAGGATCGAGCAGA, R: GCAAAGTAGAAGGCAACG
β-Actin: F: CCTCTGAACCCTAAGGCCAAC, R: TGCCACAGGATTCCATACCC
GAPDH;F:CGGAGTCAACGGATTTGGTCGTAT, R: GCCTTCTCCATGGTGGTGAAGAC
cDNAs obtained by RNA isolation were used in gene expression. These cDNAs were performed in qRT-PCR in accordance with the Power Sybeer Green qPCR MasterMix (thermo, USA) protocol. In the study, cDNAs were amplified using the Applied Biosystems QuantStudio 5 Real-Time PCR device. Step 1 for the qRT-PCR reaction to take place: Enzyme activation: 95ºC-10 min; 2nd step: Denaturation: 95ºC-15s; Primer binding-Chain extension: 60ºC-1 min, Step 3: Melting curve: 95ºC-15s, 60ºC-1 min, 95ºC-15s. Ct values of the peaks obtained during the amplification process were used to determine gene expressions and gene expressions were calculated with the 2-∆∆Ct method. Endogenous control GAPDH (glyceraldehyde 3-phosphate dehydrogenase) and β-actin mRNA expressions were used as calibration and correction factors with the multiple control method.
Western Blot
In the study, OVCAR-3 cells were incubated until the logarithmic phase. When the cells reached the logarithmic phase, Control, Doxorubicin IC50: 2.12 μM, Thymoquinone IC50: 62.9 μM doses were applied individually. Protein was isolated from the samples 48 hours after the agent treatment. The medium was removed from the flasks and the collected cells were mixed with 2 μl of 500 μl RIPA lysis buffer. PMSF solution was homogenized with a mix containing 2 µl sodium orthovanadate solution and 2 µl protease inhibitor (RIPA Lysis Buffer System, sc-24948, Santa Cruz, USA) under cold conditions with a Daihan 15D tissue homogenizer (27,000 rpm). The homogenate was centrifuged at 14000 x g for 20 min. Protein amounts (approximately 1.2-1.6 mg/ml) were determined by the Protein A280 method using a nano spectrophotometer from Optizen Nano Q, Mecasys, Korea. Equation was performed with ultrapure water (Sigma, USA). 6.5 μl of the equalized proteins was placed on the strips on the cold block. 2.5 μl LDS Sample Buffer (4X) BoltTM and 1 μl Sample Reducing Agent (10X) BoltTM were added to them. The prepared samples were denatured in the PCR device at 80º for 12 minutes. After denaturation, the samples were kept in ice for a while and then placed on a cold block to cool down. Then, the proteins were loaded on NuPAGE® Bis-Tris polyacrylamide gel (10%) and electrophoresis was performed. Western Breze brand ready-made kits provided by Thermo Scientific company were used in the study. Blotting and transfer to the membrane was carried out with ready-made membranes and membranes with the iBlot 2 (Life Technologies) system. It was done using the kit and following the kit protocols. Proteins after blotting: B-Raf Antibody (OTI5A9) (Novus bio, CAT no: NBP1-47668), Ras Antibody (JF10-11) (Novusbio, CAT no: NBP2-67097), Beta actin Antibody (Invitrogen, CAT no: MA1 -140) specific primary antibodies, then the antibodies were labeled with appropriate secondaries and observed with the Micro ChemiDoc (DNR Bio-Imaging Systems Ltd, USA) gel imaging system. Band intensities were calculated using GelQuant software.
Protein-Protein Interaction (PPI) Analysis
PPI data were retrieved from the STRING database. The STRING database provides descriptions of protein-protein interactions (PPIs) as well as confidence intervals for data scores. A confidence score greater than or equal to 0.4 was chosen to construct the interaction network of proteins with target genes.
Enrichment Analysis
Data on the functional annotation of genes and the canonical pathways associated with the strong connections established with these proteins were obtained using the ShinyGO 0.80 program.
GO Functional Enrichment Analysis
Three types of Gene Ontologies (GO) were performed on possible target genes: cellular component (CC), biological process (BP) and molecular function (MF). The SRplot bioinformatics program was used to evaluate these data.
Statistical Analysis
The difference between the averages of cell viabilities determined by the MTT test and expression values obtained from qRT-PCR studies was determined by one-way ANOVA. The groups within which the averages fell were determined using the Tukey HSD test. Comparisons between two groups were determined by the independent sample t test or Mann Whitney U test, depending on the homogeneity of the data. Analyzes were performed with SPSS 20 (IBM, USA) program and p ≤ 0.05 was used.
Results
OVCAR-3 Findings
In the MTT test performed on OVCAR-3 cells by administering different doses of DX, the IC50 value could not be found because cell proliferation did not fall below 50% in 24-hour cell viability. However, after 2.5 µM, a statistically significant difference was detected compared to the control group. IC50 at the 48th hour was determined as 2.12 and IC50 at the 72nd hour was determined as 0.08 µM. A statistically significant difference was detected compared to the control group after 0.5 µM in 48 and 72 hours of DX treatment Figure 1.
In the TQ applied group, IC50 at 24 hours was determined as 97.9 µM, IC50 for 48 hours TQ was 62.9 µM and IC50 for 72 hours TQ was determined as 37.5 µM. A statistically significant difference was detected in comparison with the control group after 75 µM for 24-hour TQ, after 50 µM for 48-hour TQ application, and after 10 µM for 72-hour TQ teratment. The effects of TQ and DX according to varying dose ranges and time are seen in Figure 2.
Wound Healing Findings
In the wound healing model, the wound area of the control group closed 100% at the 36th hour, while the healing rate was approximately 50% in the DX-only group. While approximately 20% of the wound area was closed in TQ treatment alone, the healing rate was 10% in the TQ + DX treatment group. With combination therapy, almost complete prevention of metastasis was achieved (Figure 3).
Immunofluorescent Nucblue Staining Findings
Immunofluorescent Nucblue staining was performed to monitor the apoptotic process and to determine the apoptotic effects of the agents applied for treatment. While the cell morphology and nuclear structure were normal in the cells in the control group, a bright appearance was detected in the cells treated with TQ and DX as a result of the degradation of the nuclear structure. It was determined that the highest apoptosis occurred in the TQ+DX applied groups (Figure 4).
Western Blot Findings
When the RAS gene expression results from gelns, which have an important role in proliferation, were evaluated, no significant difference was observed between the control and treatment groups (Figure 5). While there was a significant change in the expression of the RAF gene between the control and treatment groups DX and TQ+DX, no significant difference was found between the control and TQ treatment groups (Figure 5). Again, when the expressions of genes regulating apoptosis were evaluated, Bcl2 showed a significant decrease in all groups compared to the control, while the highest decrease was seen only in DX and TQ + DX treatments. A statistically significant difference was detected in Bax gene expression between all treatment groups compared to the control. The highest increase was seen in the DX group, followed by TQ and DX+TQ, respectively (Figure 5).
Within the scope of the study, vehicle control, DX IC50: 2.12 μM, TQ IC50: 62.9 μM doses were applied to OVCAR-3 cells individually and in combination. RAS, and RAF protein levels were determined after 48 hours. All data were calculated as relative fold change in the OVCAR-3 cell series treated with Vehicle control according to target protein/actin = 1, and western blot analysis results are given in Figure 6. With DX treatment, the protein levels of RAS and RAF, which are regulators of proliferation and apoptosis, increased the most (Figure 6). In TQ treatment, while the RAS level decreased, the RAF level increased again. qRT-PCR and western blot results showed that TQ treatment could support DX treatment. It is recommended to evaluate it from synergistic and antagonistic aspects.
PPI Analysis
Predictions from STRING analysis were used to depict protein interactions. The visualization showed 11 nodes and 46 edges (Figure 7). Based on nodal degree, the following genes were identified as the top 10 central genes: BAD, BAX, BAK1, BCL2L1, BCL2L11, TP53, BIK, PMAP1, BECN1, BNIP3. These targets are hypothesized to be the primary targets in ovarian cancer of TQ.
KEGG Pathway Enrichment Analysis
KEGG pathway enrichment analysis of target genes was performed with Shiny 0.80 program. The findings showed that 123 genes were involved in the enrichment process and 75 pathways were cancer-related, exhibiting a significant correlation with target genes (p < 0.05). Basically apoptosis, platinum drug resistance, pancreatic cancer, chronic myeloid leukemia, colorectal cancer, tnf signaling pathway, small cell lung cancer, measles, hapatitis C, hepatitis B, the top 10 pathways that occur in diabetic complications are shown (Figure 8).
GO Functional Enrichment Analysis
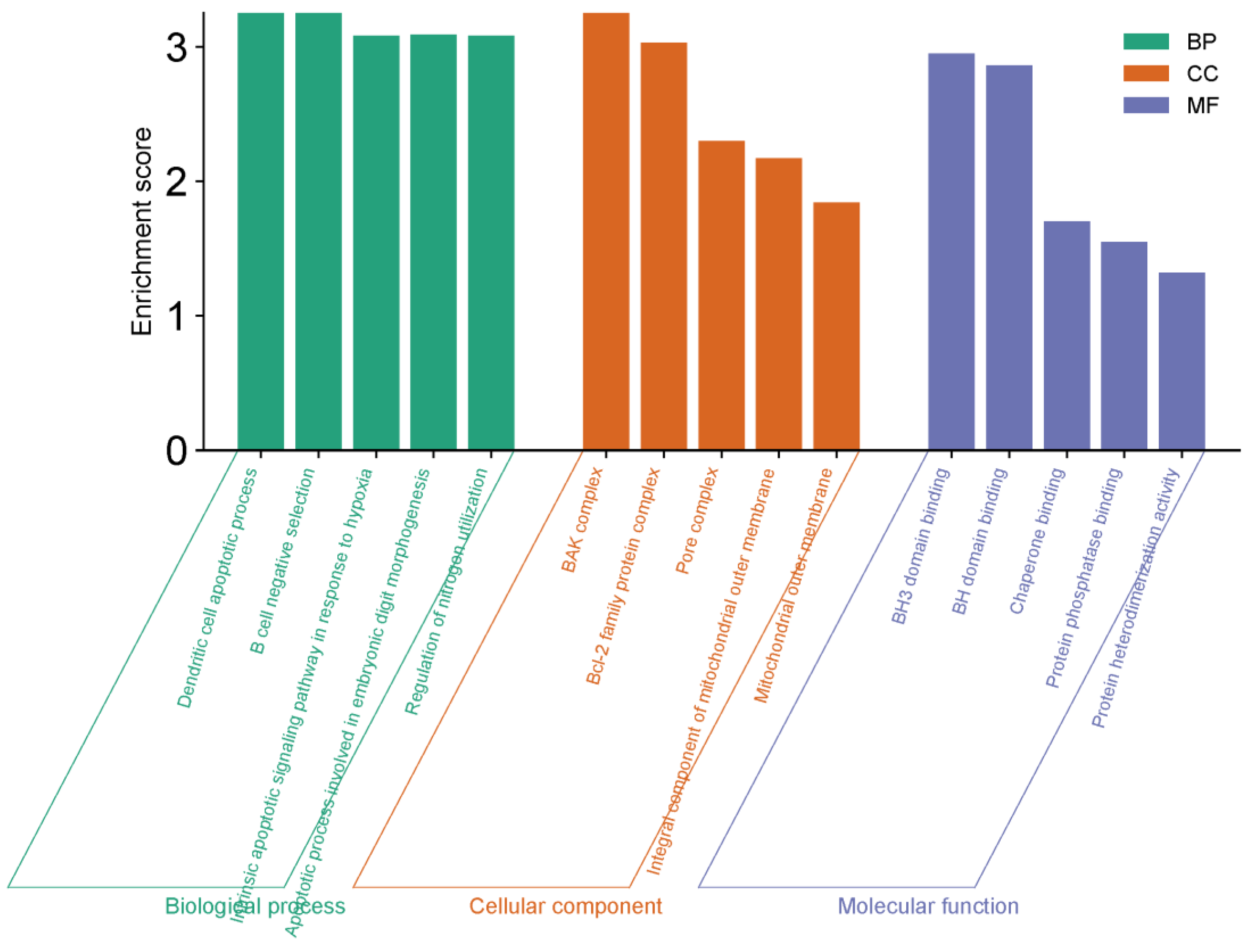
Analysis findings show only important functions (Figure 9). Target genes were found to be involved in various cellular components in the BP category, such as apoptotic process (Figure 9). In terms of cellular components, BAK complex, Bcl2- family complex. It was found that the MF category exhibited roles such as BH3 domain binding and Chaperone binding, protein phosphatase binding (Figure 9).
Discussion
In vitro studies have shown that the essential oils found in black cumin seeds have cytotoxic effects against different human cancer cell lines. Thymoquinone is also cytotoxic for human cancer cell lines such as colorectal, pancreatic adenocarcinoma, uterine sarcoma and leukemia [16,17]. In our study, in cell culture experiments, it was determined that all dilutions of thymoquinone up to 75 µM dilution were not cytotoxic on OVCAR3 cells when applied for 24 hours. However, it was reported that it inhibited the proliferation of OVCAR3 cells after this dose. Cytotoxicity also increased, especially with increasing dose-related time. The antitumor mechanisms of Nigella sativa have been shown in various studies [17]. Accordingly, depending on concentration, it has been reported that N. sativa extracts show antitumoral activity by inhibiting metastasis stimulating factors such as type 4 collegenase, metalloproteinase, angiogenic protein-fibroblastic growth factor, tissue type plasminogen activator, urokinase type plasminogen activator, plasminogen activator inhibitor type 1 and serineproteinase inhibitors [18,19,20]. To examine the effects of thymoquinone and doxorubicin, alone or in combination, on the migration of OVCAR3 cells, a wound healing assay was performed and the results are described above. IC50 values obtained by MTT test were used to observe the effect of drugs on cell migration. According to the results of the cell migration assay, doxorubicin, thymonquinone alone and in combination inhibited the migration of OVCAR3 cells in a time- and dose-dependent manner after 36 h. Finally, the greatest inhibition of OVCAR3 cell migration was determined in the combination treatment. Similar studies for thymoquinone have shown that it inhibits the migration of different cancer cells, but we did not find any studies for the effect of its combination with doxorubicin on ovarian cancer cells [21]. In this study, its reaction to 36 hours upon wound healing also revealed its metastatic role. It is also suggested that thymoquinone may have an antineoplastic effect by regulating antitumor immune responses [22].
TQ belongs to a family of quinones that can undergo enzymatic or non-enzymatic redox cycling with semiquinone radicals to form superoxide anion radicals. It has proven its effectiveness against various diseases thanks to its many medical and pharmacological activities such as anti-inflammatory, antioxidant, hepatoprotector, neuroprotector, histone protein modulator, insecticidal effects, anti-ischemic, radioprotectors. TQ differentially activates a variety of molecular targets, and its effects are mediated by a variety of cellular mechanisms, including proliferation inhibition, induction of apoptosis, cell cycle disruption, production of reactive oxygen species (ROS), and inhibition of angiogenesis and cellular metastasis [23,24]. Thymoquinone also interferes with the structure of DNA. It targets cellular copper, which is found in chromatin and is strongly associated with DNA-based guanine, resulting in DNA oxidation and cancer cell death. It may also have an effect on DNA synthesis in cancer cells. It also inhibits the proliferation and migration of human non-small cell lung cancer by reducing ERK1/2 phosphorylation [17,25]. In addition to its inhibitory effect on cell proliferation and survival, TQ also promotes the apoptosis of cancer cells. According to numerous studies, TQ is thought to cause intrinsic apoptotic cell death by reducing the expression of the anti-apoptotic protein family BCL2 and increasing mitochondrial-dependent caspase activation [26]. TQ alone has demonstrated anticancer activity in various in vitro and in vivo studies, as well as in adjuvant therapy to prevent carcinogenesis or increase the effectiveness of conventional therapeutic techniques.
As a result, thymoquinone slows down reproduction in the ovarian cancer cell line, as in many different types of cancer, indicating that it is a strong chemical protector that protects DNA [27,28]. A similar situation has been observed for forestomach fibrosarcomas, colon, skin and liver tumors and has been suggested to be a potent chemoprotectant [29,30]. Ovarian cancer is an important health problem affecting women in all societies. The findings obtained in this study indicate that we can accelerate the apoptotic process and prevent cancer metastasis more quickly by applying TQ and DX or TQ and other chemotherapeutic agents together. Future studies in line with these findings are important. We think that thymoquinone should be proven as an important agent that can be used in the treatment of ovarian cancer through animal experiments.
Conclusion
In our study, it was shown that the proliferation, migration and invasion of OVCAR3 ovarian adenocarcinoma cells were inhibited by blocking the apoptosis signaling pathway with the combination of TQ and DX. The results of this study suggest that TQ may be a promising therapeutic agent for the prevention of proliferation and metastasis of ovarian cancer. However, further studies are needed to clarify the mechanism underlying these effects of TQ.
Funding
None.
Data Availability
All data supporting the findings of this study are available in public database from PubChem, STRING and within the paper.
Ethics Approval and Consent to Participate
Not Applicable.
Competing Interests
The authors declare that they have no competing interests.
References
- Chaudhry, G.E.; Akim, A.M.; Sung, Y.Y.; Muhammad, T.S.T. Cancer and Apoptosis. Methods Mol Biol. 2022, 2543, 191–210. [Google Scholar] [CrossRef] [PubMed]
- Dakal, T.C.; Dhabhai, B.; Pant, A.; Moar, K.; Chaudhary, K.; Yadav, V.; Ranga, V.; Sharma, N.K.; Kumar, A.; Maurya, P.K.; et al. Oncogenes and tumor suppressor genes: functions and roles in cancers. Medcomm 2024, 5, e582. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, H.; Wang, Y.; Hammarström, L.; Pan-Hammarström, Q. Hallmarks of Cancers: Primary Antibody Deficiency Versus Other Inborn Errors of Immunity. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Arora T, Mullangi S, Vadakekut ES, Lekkala MR. Epithelial Ovarian Cancer. 2024 May 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan. [PubMed]
- Boussios, S.; Zarkavelis, G.; Seraj, E.; Zerdes, I.; Tatsi, K.; Pentheroudakis, G. Non-epithelial Ovarian Cancer: Elucidating Uncommon Gynaecological Malignancies. Anticancer. Res. 2016, 36, 5031–5042. [Google Scholar] [CrossRef]
- Saani, I.; Raj, N.; Sood, R.; Ansari, S.; Mandviwala, H.A.; Sanchez, E.; Boussios, S. Clinical Challenges in the Management of Malignant Ovarian Germ Cell Tumours. Int. J. Environ. Res. Public Health 2023, 20, 6089. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA: A Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef] [PubMed]
- Unat, B. The Rat Sarcoma Virus (RAS) Family of Proteins in Sarcomas. Cureus 2024, 16, e57082. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK pathway for cancer therapy: from mechanism to clinical studies. Signal Transduct. Target. Ther. 2023, 8, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Chavda, J.; Bhatt, H. Systemic review on B-RafV600E mutation as potential therapeutic target for the treatment of cancer. Eur. J. Med. Chem. 2020, 206, 112675. [Google Scholar] [CrossRef]
- Kari, S.; Subramanian, K.; Altomonte, I.A.; Murugesan, A.; Yli-Harja, O.; Kandhavelu, M. Programmed cell death detection methods: a systematic review and a categorical comparison. Apoptosis 2022, 27, 482–508. [Google Scholar] [CrossRef]
- Alsalahi, A.; Maarof, N.N.; Alshawsh, M.A.; Aljaberi, M.A.; Qasem, M.A.; Mahuob, A.; Badroon, N.A.; Mussa, E.A.; Hamat, R.A.; Abdallah, A.M. Immune stimulatory effect of Nigella sativa in healthy animal models: A systematic review and meta-analysis. Heliyon 2024, 10, e27390. [Google Scholar] [CrossRef] [PubMed]
- Kurowska, N.; Madej, M.; Strzalka-Mrozik, B. Thymoquinone: A Promising Therapeutic Agent for the Treatment of Colorectal Cancer. Curr. Issues Mol. Biol. 2023, 46, 121–139. [Google Scholar] [CrossRef] [PubMed]
- Adinew, G.M.; Messeha, S.S.; Taka, E.; Badisa, R.B.; Soliman, K.F.A. Anticancer Effects of Thymoquinone through the Antioxidant Activity, Upregulation of Nrf2, and Downregulation of PD-L1 in Triple-Negative Breast Cancer Cells. Nutrients 2022, 14, 4787. [Google Scholar] [CrossRef]
- AlDreini, S.; Fatfat, Z.; Ibrahim, N.A.; Fatfat, M.; Gali-Muhtasib, H.; Khalife, H. Thymoquinone enhances the antioxidant and anticancer activity of Lebanese propolis. World J. Clin. Oncol. 2023, 14, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Sheikhnia, F.; Rashidi, V.; Maghsoudi, H.; Majidinia, M. Potential anticancer properties and mechanisms of thymoquinone in colorectal cancer. Cancer Cell Int. 2023, 23, 1–15. [Google Scholar] [CrossRef]
- Almajali, B.; Al-Jamal, H.A.N.; Taib, W.R.W.; Ismail, I.; Johan, M.F.; Doolaanea, A.A.; Ibrahim, W.N. Thymoquinone, as a Novel Therapeutic Candidate of Cancers. Pharmaceuticals 2021, 14, 369. [Google Scholar] [CrossRef]
- Ibrahim, S.; Fahim, S.A.; Tadros, S.A.; Badary, O.A. Suppressive effects of thymoquinone on the initiation stage of diethylnitrosamine hepatocarcinogenesis in rats. J. Biochem. Mol. Toxicol. 2022, 36, e23078. [Google Scholar] [CrossRef] [PubMed]
- Al-Hayali, M.; Garces, A.; Stocks, M.; Collins, H.; Bradshaw, T.D. Concurrent Reactive Oxygen Species Generation and Aneuploidy Induction Contribute to Thymoquinone Anticancer Activity. Molecules 2021, 26, 5136. [Google Scholar] [CrossRef] [PubMed]
- Almatroodi, S.A.; Almatroudi, A.; Alsahli, M.A.; Khan, A.A.; Rahmani, A.H. Thymoquinone, an Active Compound of Nigella sativa: Role in Prevention and Treatment of Cancer. Curr. Pharm. Biotechnol. 2020, 21, 1028–1041. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fan, Y.; Huang, S.; Wang, G.; Han, R.; Lei, F.; Luo, A.; Jing, X.; Zhao, L.; Gu, S.; et al. Thymoquinone inhibits the metastasis of renal cell cancer cells by inducing autophagy via AMPK/mTOR signaling pathway. Cancer Sci. 2018, 109, 3865–3873. [Google Scholar] [CrossRef] [PubMed]
- Butnariu, M.; Quispe, C.; Herrera-Bravo, J.; Helon, P.; Kukula-Koch, W.; López, V.; Les, F.; Vergara, C.V.; Alarcón-Zapata, P.; Alarcón-Zapata, B.; et al. The effects of thymoquinone on pancreatic cancer: Evidence from preclinical studies. Biomed. Pharmacother. 2022, 153, 113364. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Mishra, R.K.; Vyawahare, A.; Kumar, A.; Rehman, M.U.; Qamar, W.; Khan, A.Q.; Khan, R. Thymoquinone (2-Isopropyl-5-methyl-1, 4-benzoquinone) as a chemopreventive/anticancer agent: Chemistry and biological effects. Saudi Pharm. J. 2019, 27, 1113–1126. [Google Scholar] [CrossRef] [PubMed]
- Zubair, H.; Khan, H.Y.; Sohail, A.; Azim, S.; Ullah, M.F.; Ahmad, A.; Sarkar, F.H.; Hadi, S.M. Redox cycling of endogenous copper by thymoquinone leads to ROS-mediated DNA breakage and consequent cell death: putative anticancer mechanism of antioxidants. Cell Death Dis. 2013, 4, e660–e660. [Google Scholar] [CrossRef]
- Khan, A.; Tania, M.; Fu, S.; Fu, J. Thymoquinone, as an anticancer molecule: from basic research to clinical investigation. Oncotarget 2017, 8, 51907–51919. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, Y.; Deng, H.; Liu, Y.; Lei, X.; He, P.; Dong, W. Thymoquinone inhibits the proliferation and invasion of esophageal cancer cells by disrupting theAKT/GSK-3β/Wnt signaling pathway viaPTENupregulation. Phytotherapy Res. 2020, 34, 3388–3399. [Google Scholar] [CrossRef] [PubMed]
- Garnique, A.; Rezende-Teixeira, P.; Machado-Santelli, G. Telomerase inhibitors TMPyP4 and thymoquinone decreased cell proliferation and induced cell death in the non-small cell lung cancer cell line LC-HK2, modifying the pattern of focal adhesion. Braz. J. Med Biol. Res. 2023, 56, e12897. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Kim, D.-H.; Ha, E.; Choi, S.M.; Choi, J.-S.; Chun, K.-S.; Joo, S.H. Thymoquinone induces apoptosis of human epidermoid carcinoma A431 cells through ROS-mediated suppression of STAT3. Chem. Interactions 2019, 312, 108799. [Google Scholar] [CrossRef] [PubMed]
- Al-Hayali, M.; Garces, A.; Stocks, M.; Collins, H.; Bradshaw, T.D. Concurrent Reactive Oxygen Species Generation and Aneuploidy Induction Contribute to Thymoquinone Anticancer Activity. Molecules 2021, 26, 5136. [Google Scholar] [CrossRef]
- Hossen, M.J.; Yang, W.S.; Kim, D.; Aravinthan, A.; Kim, J.-H.; Cho, J.Y. Thymoquinone: An IRAK1 inhibitor with in vivo and in vitro anti-inflammatory activities. Sci. Rep. 2017, 7, srep42995. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
% Cell viability obtained from MTT test in OVCAR-3 cell series after doxorubicin treatment and IC50 values calculated using probit analysis.
Figure 1.
% Cell viability obtained from MTT test in OVCAR-3 cell series after doxorubicin treatment and IC50 values calculated using probit analysis.
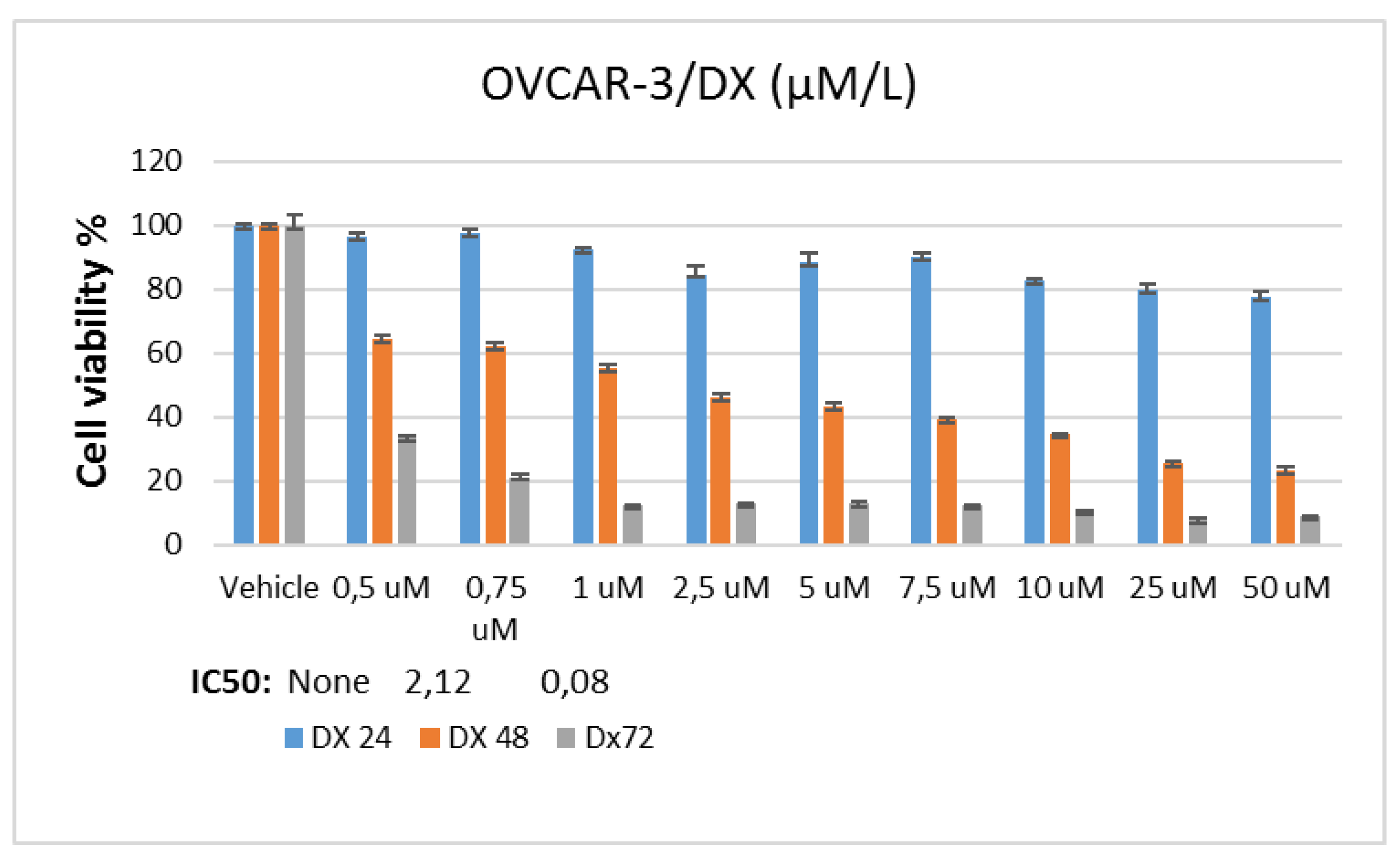
Figure 2.
% Cell viability obtained from MTT test in OVCAR-3 cell series after thymoquinone treatment and IC50 values calculated using probit analysis.
Figure 2.
% Cell viability obtained from MTT test in OVCAR-3 cell series after thymoquinone treatment and IC50 values calculated using probit analysis.
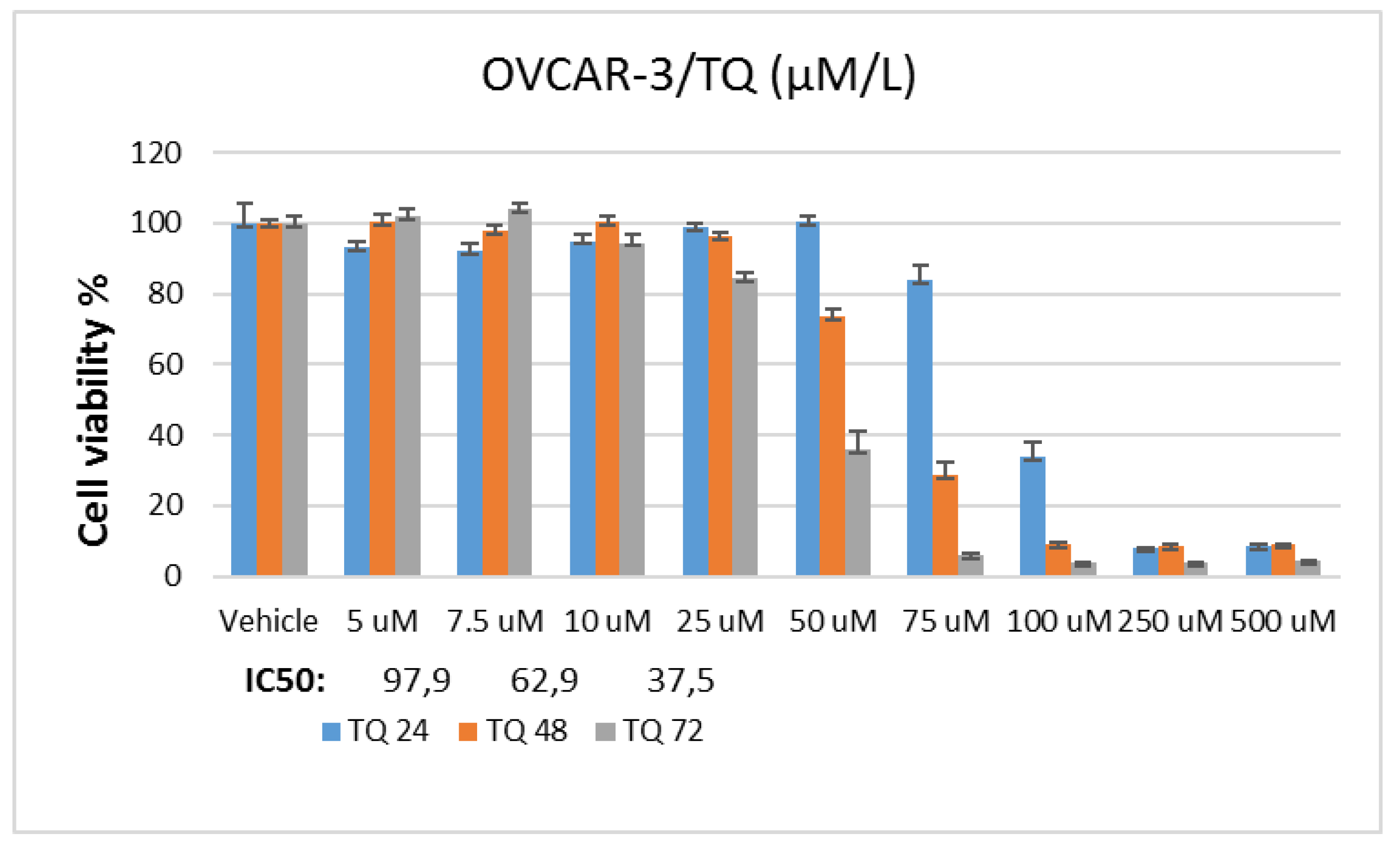
Figure 3.
Cell migration rate and % wound closure determined by wound healing assay (over 36 hours) in OVCAR-3 cell populations. n= 12, bars represent mean ± std error; * data are statistically different, calculated using independent samples t-test, p ≤ 0.001.
Figure 3.
Cell migration rate and % wound closure determined by wound healing assay (over 36 hours) in OVCAR-3 cell populations. n= 12, bars represent mean ± std error; * data are statistically different, calculated using independent samples t-test, p ≤ 0.001.
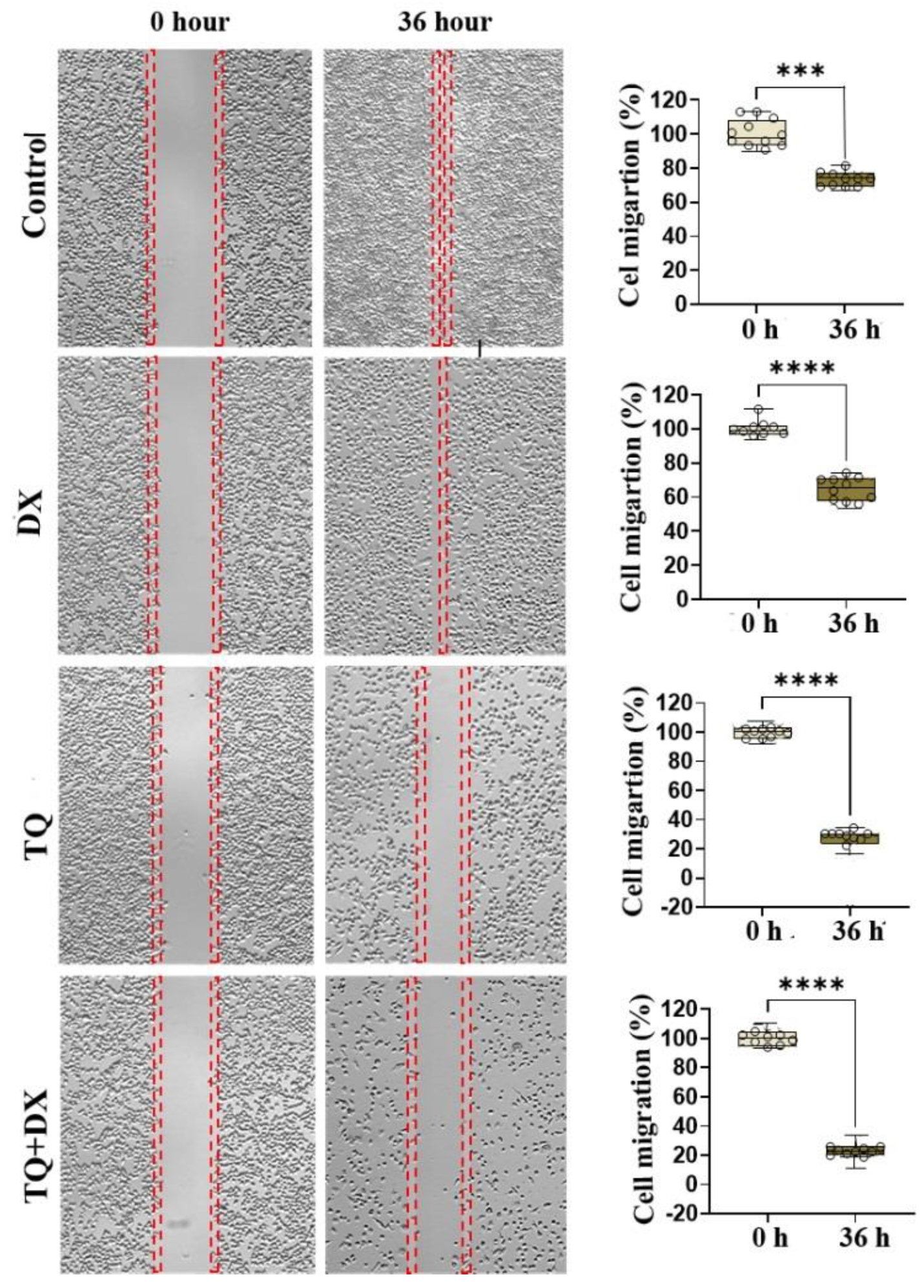
Figure 4.
Apoptotic body formation in OVCAR-3 cells treated with IC50 doses of treatment agents for 48 hours.
Figure 4.
Apoptotic body formation in OVCAR-3 cells treated with IC50 doses of treatment agents for 48 hours.
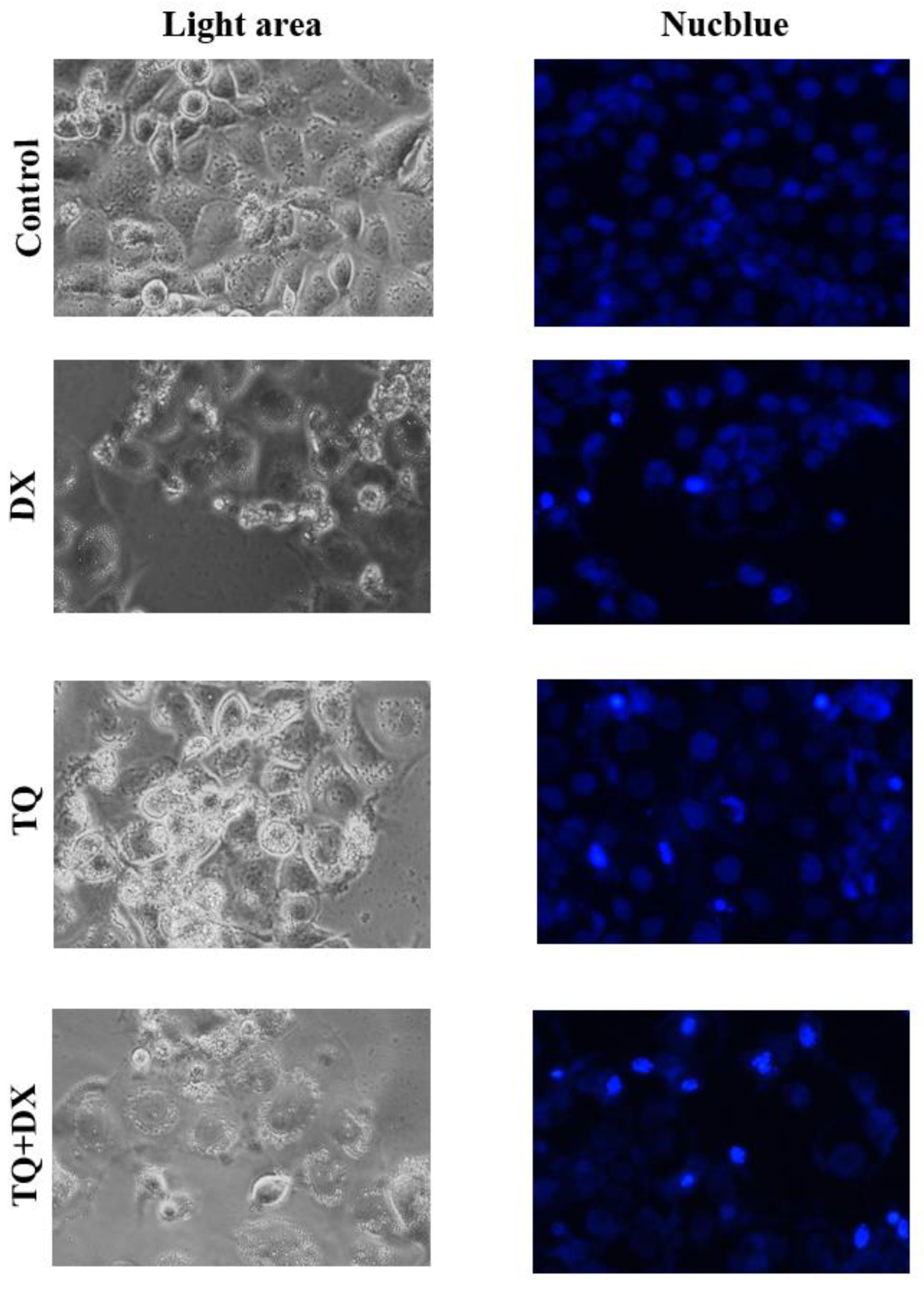
Figure 5.
In OVCAR-3 cell line, DX IC50: 2.12 μM, TQ IC50: 62.9 μM, relative fold increase values of RAS, RAF, Bcl2 and Bax gene expressions 48 hours after single and combined drug application (n=4 data mean±SH), * means are statistically different, one-way ANOVA, Tukey HSD test.
Figure 5.
In OVCAR-3 cell line, DX IC50: 2.12 μM, TQ IC50: 62.9 μM, relative fold increase values of RAS, RAF, Bcl2 and Bax gene expressions 48 hours after single and combined drug application (n=4 data mean±SH), * means are statistically different, one-way ANOVA, Tukey HSD test.
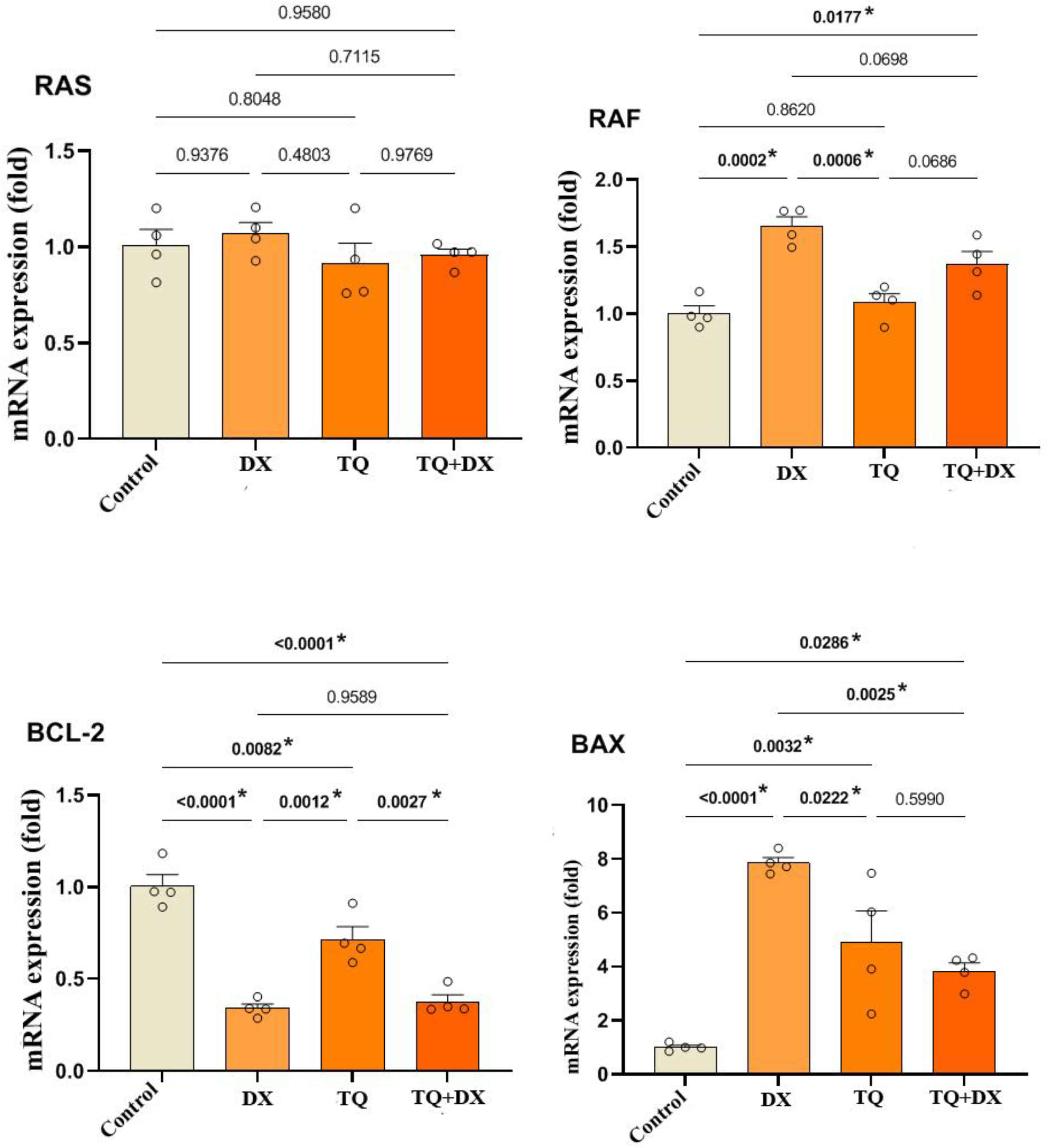
Figure 6.
RAF and RAS protein levels in OVCAR3 cells as a result of TQ and DX treatment with a 48-hour IC50 dose.
Figure 6.
RAF and RAS protein levels in OVCAR3 cells as a result of TQ and DX treatment with a 48-hour IC50 dose.
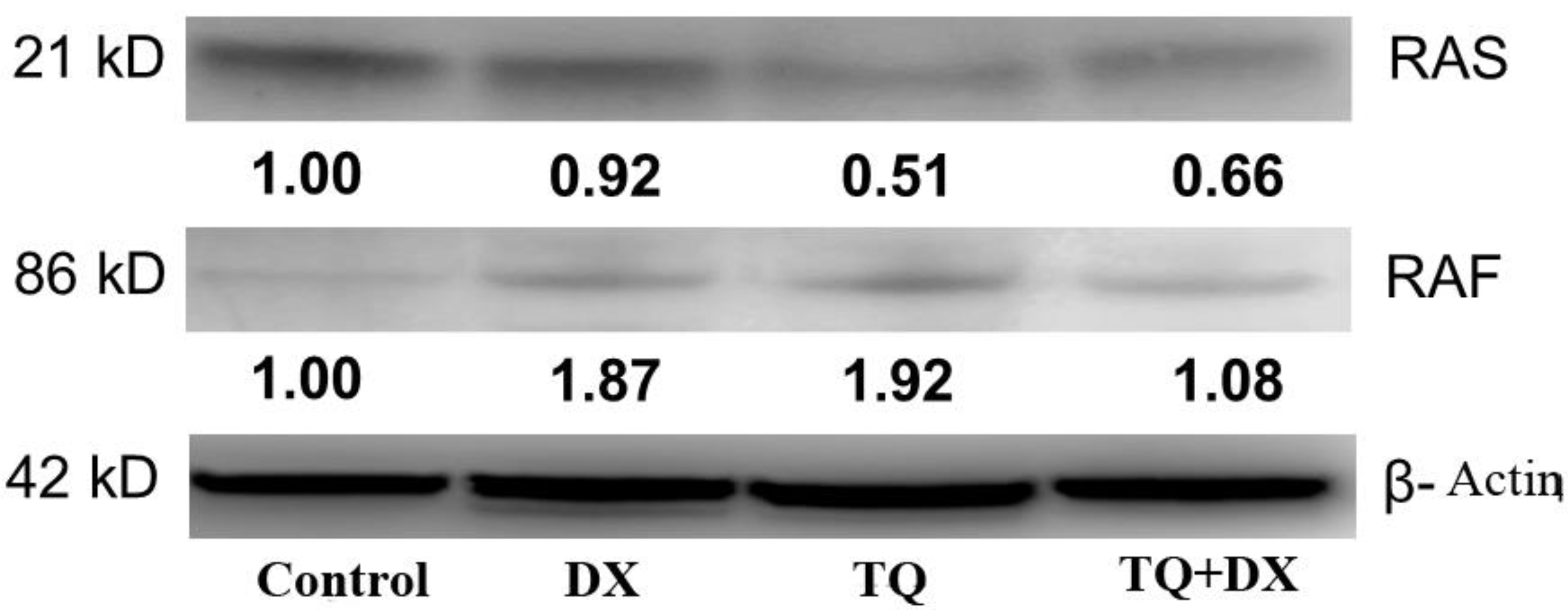
Figure 7.
PPI and interaction between various genes of ovarian cancer.
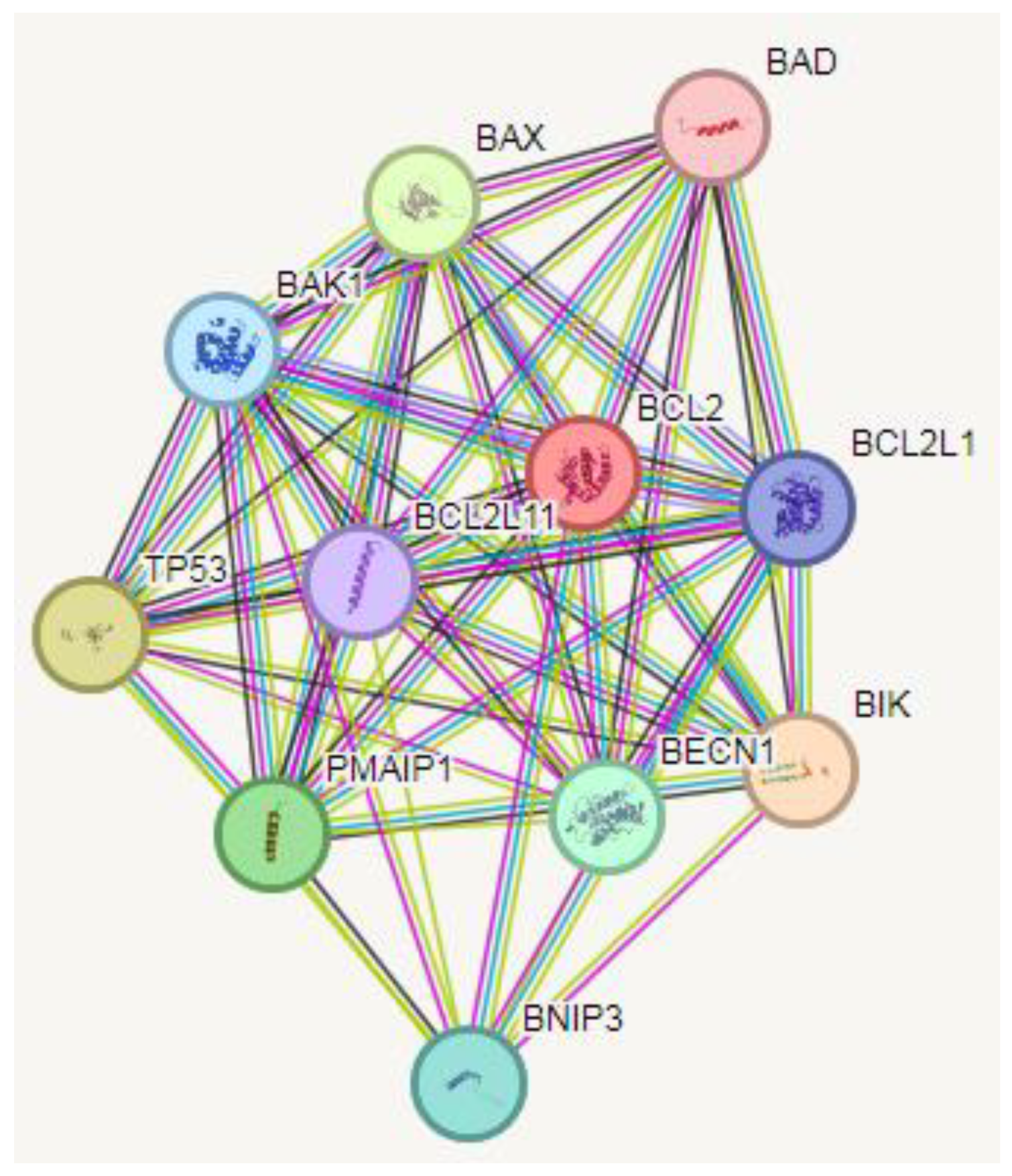
Figure 8.
Enrichment analysis for the 123 common compound targets.
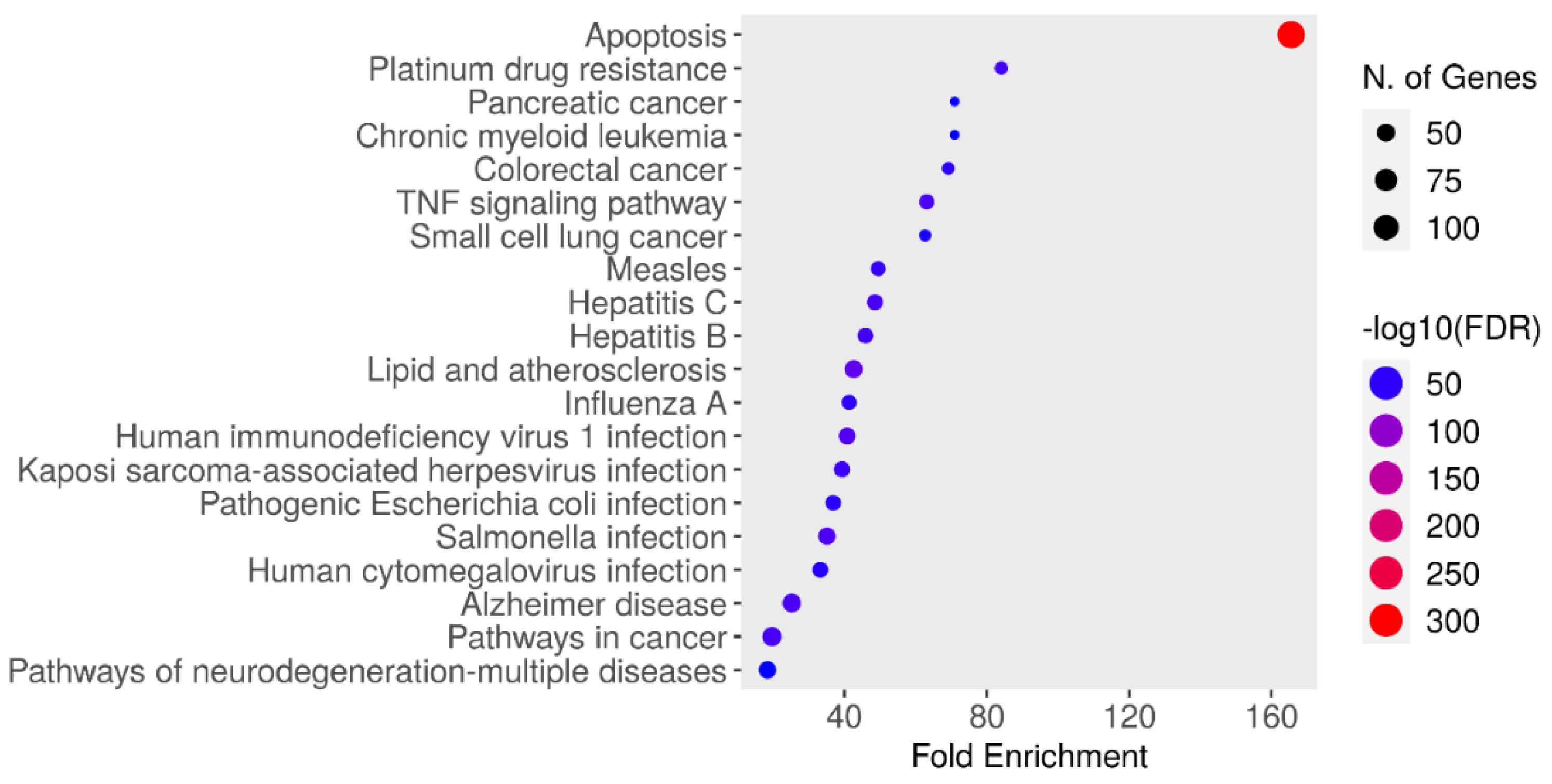
Figure 9.
GO (Biological process, molecular function, and cellular component) analysis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



