
Submitted:
27 July 2024
Posted:
30 July 2024
You are already at the latest version
Abstract
Activating the cytosolic innate immune sensor cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway via natural and synthetic agonists holds great potential for priming robust antitumor immunity. However, the effectiveness of STING agonists is often impeded by the insufficient tumor accumulation, poor cellular entry, and rapid clearance, thus leading to difficulty of their clinic translation. Herein, we apply the human serum albumin as the single-molecule nanoreactor to encapsulate SR-717 (SH-NPs) for enhanced targeting delivery efficiency and biological potency. SH-NPs with rational size distribution exhibited superior serum stability, considerable cellular uptake and abundant tumor accumulation, thus increasing the STING signaling including phosphorylation of STING-associated proteins (TBK1, IRF3), and cytokines secretion inside tumor region. Subsequently, SH-NPs remarkably reshape the immunosuppressive tumor microenvironment into an immunogenic one that boosts antitumor T-cell immunity and improves the therapeutic efficacy of checkpoint blockade against murine tumors. We also validate SH-NPs in freshly isolated human renal tumor tissues to highlight their clinic translation capability. These findings pave the way towards targeted STING agonist delivery for reinvigorated immunotherapy of intractable cancers.
Keywords:
human serum albumin
; targeted delivery
; STING agonist
; immunotherapy
1. Introduction
Immunotherapy, emerging as a prominent strategy in cancer treatment, has garnered significant attention and research efforts [1,2,3]. Innovative cancer immunotherapies, exemplified by monoclonal antibodies targeting cytotoxic T-lymphocyte antigen 4 (CTLA-4) and programmed cell death protein 1/programmed cell death ligand 1 (PD-1/PD-L1) [4,5,6], have revolutionized the landscape of cancer therapy. Immune checkpoint inhibitors (ICI) circumvent inhibitory checkpoint molecules on tumor cells, thereby enhancing T-cell responsiveness and preventing immune exhaustion, thereby restoring immune surveillance [7,8,9]. Despite achieving clinical success, these therapies demonstrate considerable variability in response rates across patients [9,10]. For instance, the response rate to ipilimumab is only about 15%, with rarely more than 25% in patients receiving anti-PD-1/PD-L1 ICI therapy [12,13,14]. The efficacy of ICI appears correlated with pre-existing pro-inflammatory tumor microenvironments (TME), characterized by heightened immune cell infiltration or PD-L1 expression, while "immune-desert" or "cold" tumors exhibit limited responsiveness to ICI [15,16,17]. To maximize the efficacy of ICI therapy, "cold" tumors must be reprogrammed into an immunogenic and pro-inflammatory or "hot" phenotype, which can reinvigorate anti-tumor immunity. One promising approach involves upregulating the stimulator of interferon genes (cGAS-STING) pathway, a major component of the innate immune system involved in antiviral and anti-tumor immunity [18,19]. cGAS-STING senses cytoplasmic double-stranded DNA (dsDNA) and triggers a cascade of downstream signaling events in response to infection [20]. This pathway bridges innate and adaptive immunity, facilitating the activation and migration of dendritic cells (DCs) and subsequent initiation of cytotoxic T lymphocytes (CTLs) at tumor sites [21]. Thus, this pathway has the potential to overcome immune-suppressive environments in certain cancers, rendering patients more responsive to immune checkpoint inhibitor (ICI) therapy [22]. Activation of the innate immune response through exogenous stimulation with STING agonists, such as cyclic GMP-AMPP, has proven effective in enhancing the efficacy of immunotherapy [23]. However, STING agonists based on cyclic dinucleotides (CDNs) are small molecules with hydrophilic negative charges, making them prone to enzymatic degradation and limiting their activation of the STING pathway in target tissues [24,25] Non-nucleotide STING agonists like SR-717 have shown potential in overcoming the instability associated with cyclic dinucleotides (CDN)-based agonists [26]. Nevertheless, these drugs are hydrophobic, which contributes to their poor targeting and side effects, restricting their clinical utility [27]. Therefore, there is a critical need to develop new treatment strategies capable of improving drug solubility, targeting specificity, and reducing side effects.
Targeted delivery system has been widely utilized in biomedical research to address these challenges [28,29,30]. Many researchers have proposed various drug delivery strategies aimed at improving STING agonists utilization efficiency to enhance anti-tumor immune effects. For example, Zhou et al. used PMOF nanoparticles for light-triggered STING agonist SR-717 release, enhancing photodynamic-immune therapy [31]. Similarly, Lu et al. developed hollow manganese dioxide nanoparticles with STING agonist MSA-2 and CRISPR-Cas9/sg-PD-L1 plasmid for sustained release in tumors, activating the cGAS-STING pathway and reducing PD-L1 [32]. Additionally, a biomimetic cancer cell membrane-coated nano-vaccine delivery system (PLGA/STING@EPBM) effectively delivers STING agonists and tumor antigens to Clec9a+ DCs, showing significant anti-tumor synergy with radiotherapy [33]. These advancements in nanocarrier development effectively address the limitations of STING agonists in clinical applications caused by delivery barriers and adverse reactions. However, the complex preparation process and challenges in translation to clinical practice remain significant drawbacks.
In recent years, our research has focused on developing innovative protein-based nanomedicine delivery systems with excellent targeting capabilities, high drug loading efficiency, and improved safety, significantly enhancing the efficacy of tumor therapy [34,35]. Building on this foundation, we have also explored the use of human serum albumin as nanoreactors at the single-molecule level. By leveraging biomineralization principles, we have achieved controlled nucleation and crystallization of therapeutic agents within the protein nanocage, resulting in multifunctional protein nanoparticles with tunable dimensions. These nanoparticles hold significant promise for targeted cancer therapies, including chemotherapy, photothermal therapy, and photodynamic therapy [36,37,38]. Herein, we employ a biomineralization approach using a single-molecule albumin template to encapsulate the STING agonist SR717 (SH-NPs). These nanoparticles exhibited excellent tumor targeting ability with superior serum stability and efficient cellular uptake, thus considerably activating the STING signaling pathway, followed by relieving the immunosuppression and improving the immunogenetic feature of TME. Finally, SH-NPs boosts antitumor T-cell immunity and promotes the therapeutic efficacy of checkpoint blockade, which is further validated in freshly isolated human renal tumor tissues, highlighting their clinical translation potential. These findings pave the way towards targeted STING agonist delivery for reinvigorated immunotherapy of intractable cancers (Scheme 1).
2. Materials and Methods
2.1. Materials
SR717 was purchased from Selleck Co., Ltd. Human serum albumin (HSA) was obtained from Aladdin Reagent. RPMI 1640 medium, fetal bovine serum (FBS), trypsin EDTA solution and Penicillin-streptomycin solution were purchased from Gibco Life Technologies (California, USA). IL-4 protein and IFN-γ protein were purchased from Peprotech. Cy5.5 N-hydroxysuccinimide (NHS) ester and LPS proteins were purchased from Sigma. ELISA kits for IFN-β, CXCL-10, IL-6, and TNF-α were purchased from Lianke. Anti-Phospho-TBK1 antibody (CST, catalog number 5483S, dilution: 1:1000), anti-Phospho-IRF3 antibody (CST, catalog number 29047S, dilution: 1:1000), anti-TBK1 antibody (CST, catalog number 38066S, dilution: 1:1000), anti-IRF3 antibody (CST, catalog number 4550S, dilution: 1:1000), and anti-GAPDH antibody (Abclonal, catalog number AC033, dilution: 1:5000) were purchased from CST. Goat anti-rabbit IgG H&L secondary antibody (Abcam, catalog number ab6702, dilution: 1:5000) was purchased from Abcam. PE anti-mouse CD80 (BioLegend, catalog number 104708, dilution: 1:200), APC anti-mouse CD86 (BioLegend, catalog number 105012, dilution: 1:200), PE anti-mouse CD86 (BioLegend, catalog number 105007, dilution: 1:200), PE anti-mouse CD206 (BioLegend, catalog number 141706, dilution: 1:200) FITC anti-mouse CD45 (BioLegend, catalog number 103108, dilution: 1:200), APC anti-mouse CD3 (BioLegend, catalog number 100236, dilution: 1:200), PE anti-mouse CD8a (BioLegend, catalog number 100708, dilution: 1:200), PE anti-mouse CD335 (BioLegend, catalog number 137604, dilution: 1:200), FITC anti-mouse CD11c (BioLegend, catalog number 117306, dilution: 1:200), PE anti-mouse CD80 (BioLegend, catalog number 104708, dilution: 1:200), FITC anti-mouse/human CD11b (BioLegend, catalog number 101206, dilution: 1:200), APC anti-mouse F4/80 (BioLegend, catalog number 123116, dilution: 1:200), FITC anti- human CD14 (BioLegend, catalog number 982502, dilution: 1:200), APC anti-human CD68 (BioLegend, catalog number 333809, dilution: 1:200), PE anti- human CD86 (BioLegend, catalog number 374205, dilution: 1:200), and PE anti-human CD206 (BioLegend, catalog number 321105, dilution: 1:200) were purchased from Biolegend.
2.2. Cell Lines
RENCA (catalog number CL-0568), DC2.4 (catalog number SCC142) and RAW264.7 (catalog number K1673) were purchased from Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). These cell lines were cultured in RPMI 1640 Medium supplemented with 1% penicillin-streptomycin and 10% FBS under 5% CO2 at 37 °C in a humidified incubator.
2.3. Animals and Ethics Statement
BALB/c mice (female, 18 ± 2 g, 6-8 weeks), were purchased from Shanghai SLAC Animal Technology Co., Ltd. (Shanghai China). Mice were housed in an animal facility under constant environmental conditions (room temperature, 21 ± 1 °C; relative humidity, 40-70% and a 12 h light-dark cycle). All mice had access to food and water. All animal experiments were carried out following protocols approved by Laboratory Animal Center of Soochow University (No. ECSU-2019000179). In our experiment, the maximum tumor burden was 1500 mm3 in mice, which is lower than the maximal tumor burden permitted by Laboratory Animal Center of Soochow University.
2.4. Synthesis
For the preparation of SH-NPs, 1.0 mL SR-717 solution (1.0 mg mL-1) was added into 30.0 mg human serum albumin (HSA) dispersing in 3.0 mL deionized water under vigorous stirring together with adjusting the pH to 10. After complete dissolution, adjusted the pH to 6 and followed by reaction at 55 °C for 4 h. Finally, SH-NPs were purified through centrifugation and ultrafiltration (100 kDa MWCO, 1000 g, 20 min/each) for 5 times and stored in PBS solution (pH 7.4, 10.0 mM).
2.5. Characterization
The morphology of SH-NPs was observed by transmission electron microscopy (TEM) (HT7700, Hitachi). The hydrodynamic diameter and zeta potential were quantified via dynamic light scattering (DLS) (Zetasizer ZS90, Malvern). The concentration of SR717 was determined through the application of reversed-phase high-performance liquid chromatography (HPLC) (Agilent 1100, Agilent).
2.6. Drug Release
To investigate the drug release kinetics, SR717 was evaluated in free SR717 and SH-NPs using the dialysis method. A volume of 1.0 mL of the sample (0.2 mg mL−1 of SR717) was placed in dialysis bags with a molecular weight cut-off of 3.5 kDa. These drug-loaded dialysis bags were then submerged in pH 7.4 phosphate buffer, pH 5.0 acetate buffer, and pH 5.0 acetate buffer containing 10 μg mL−1 of CB, and subsequently agitated using an oscillator shaker at 37 °C. The concentration of SR717 in the buffer was measured at 0, 0.5, 1, 2, 4, 8, 12, and 24 h.
2.7. Cellular Uptakes and Endocytic Pathway
For cellular uptake of SH-NPs, DC2.4 cells were seeded in plates (1.0 × 106 cells/well) followed by the addition of free SR717 and SH-NPs (10.0 µM SR717) and further incubation for 12 and 24 h. Following incubation, the cells were collected for cell counting and disruption under ultrasonication, and HPLC was used to determine the SR717 amount. For the endocytic pathway, the inhibitors including 10.0 µg·mL−1 chlorpromazine, 100.0 µg·mL−1 amiloride, 5.0 µg·mL−1 nystatin were added into DC2.4 cells (1.0 × 106 cells/well) followed by 1 h incubation at 37 °C or 4 °C in serum-free RPMI 1640 medium. Then, SH-NPs (10.0 µM SR717) were added into the medium for 12 h incubation. Afterward, the cells were collected through trypsin treatment, centrifugation, and lysis under ultrasonication. Finally, the amount of SR717 was determined by the HPLC.
2.8. In Vitro Cytotoxicity
To evaluate the cytotoxicity, DC2.4 cells (1.0 × 104 cells/well) were incubated with free SR717 and SH-NPs at the concentration of 0, 1.25, 2.5, 5.0, 10.0 and 20.0 µM SR717 for 24 h. The MTT assay was applied to evaluate the cell viability.
2.9. Western Blot
DC2.4 cells were incubated with free SR717 and SH-NPs (10.0 µM SR717) for 24 h. Cells were collected to extract total proteins using Protein Extraction Kit from Beyotime. Protein concentration was determined using a BCA assay. Then, 20.0 µg of each sample's proteins were run on the 10% SDS-polyacrylamide gel and transferred to a PVDF membrane. The membrane was blocked with 5% bovine serum albumin for 2 h, followed by incubation with anti-Phospho-TBK1 antibody (CST, 5483S, dilution: 1:1000), anti-Phospho-IRF3 antibody (CST, 29047S, dilution: 1:1000), anti-TBK1 antibody (CST, 38066S, dilution: 1:1000), anti-IRF3 antibody (CST, 4550S, dilution: 1:1000), and anti-GAPDH antibody (Abclonal, AC033, dilution: 1:5000) overnight at 4 °C. The membrane was then incubated with goat anti-rabbit IgG H&L secondary antibody (Abcam, ab6702, dilution: 1:5000) followed by visualization with ECL using a detection system (GE healthcare). For the tumor tissue protein extraction, SH-NPs (30.0 mg kg−1) were intravenously injected into the BALB/c mice (female, 6-8 weeks) bearing RENCA tumors (n=3 mice per group). After 72 h, the mice were euthanized and the tumors were harvested. Tissues were lysed with 0.5% (v/v) CHAPS containing protease and phosphatase inhibitors.
2.10. DC Maturation
For DC stimulation experiments, DC2.4 cells were incubated with free SR717 and SH-NPs (10.0 µM SR717) for 24 h. Then DCs were stained with anti-CD86 PE and anti-CD80 APC, detected by flow cytometer to investigate their maturation via the surface expression of the costimulatory molecules CD86 and CD80. Additionally, the levels of IFN-β in the culture medium were measured by ELISA kits (Lianke).
2.11. Macrophage Polarization
For Macrophage polarization experiments, M1 macrophages were induced with 100.0 ng mL−1 of LPS and 20.0 ng mL−1 of IFN-γ, and M2 macrophages were induced with 20.0 ng mL−1 of IL-4. Polarized macrophage phenotypes were identified using flow cytometry. Subsequently, M2 macrophages were incubated with free SR717 and SH-NPs (10.0 µM SR717) for 24 h. Following incubation, RAW264.7 cells were stained with anti-CD206 PE and anti-CD86 PE, detected by flow cytometer to investigate their polarization via the surface expression of the costimulatory molecules CD206 and CD86.
2.12. Construction of RENCA Tumor Models
RENCA tumor cells (5.0 × 106 cells per mouse) were subcutaneously injected into the right back of the BALB/c mice (female, 6-8 weeks) to construct the above subcutaneous tumor models.
2.13. In Vivo Tumor Targeting
To evaluate the tumor targeting efficacy in RENCA tumors, Cy5-SH-NPs (30.0 mg kg−1 SR717) were intravenously injected into the mice bearing RENCA tumors (n=3 mice per group). Then fluorescence images were taken at intervals of 0, 2, 6, 12, 24, and 48 h, and analyzed using Living Imaging software.
2.14. In Vivo Biodistribution
To evaluate the biodistribution in RENCA tumors, free SR717 and SH-NPs (30.0 mg kg−1 SR717) were intravenously injected into the BALB/c mice (female, 6-8 weeks) bearing RENCA tumors (n=3 mice per group). Then, methanol was employed to extract SR717 from the heart, liver, spleen, lung, kidney, and tumor tissues at 12 h following injection. Finally, the amounts of SR717 were determined using HPLC.
2.15. In Vivo Antitumor Efficacy
To evaluate the antitumor efficacy, free SR717 and SH-NPs (30.0 mg kg−1 SR717) were intravenously injected into the mice bearing RENCA tumors (n=5 mice per group), followed by measurement of tumor volumes (V = 0.5 × L × W2, V, L, and W represent the tumor volume, long diameter, and short diameter, respectively.).
To evaluate the synergistic antitumor efficacy of SH-NPs and aPD-L1 against subcutaneous RENCA tumor models, SH-NPs (30.0 mg kg−1 SR717) and aPD-L1 (3.0 mg kg−1) were administered intravenously to mice with tumors measuring 75–100 mm³ (n=5 per group). Twenty-four hours after the initial injection, an additional dose of 3.0 mg/kg aPD-L1 was given intravenously. Tumor volumes were measured (V = 0.5 × L × W2, V, L, and W represent the tumor volume, long diameter, and short diameter, respectively.).
2.16. Cytokine Analysis
To evaluate the cytokine analysis, free SR717, SH-NPs (30.0 mg kg−1 SR717) and aPD-L1 (3.0 mg kg−1) were intravenously injected into the mice bearing RENCA tumors (n=3 mice per group). Afterward, 3.0 mg kg−1 aPD-L1 were intravenously injected after 24 h. The mice were euthanized and blood was collected at 3 days post-treatments, after which plasma was obtained by centrifugation. Then, the levels of IFN-β, CXCL-10, IL-6 and TNF-α were measured by ELISA kits (Lianke).
2.17. Antitumor Immune Response
Briefly, SH-NPs (30 mg kg−1 SR717) was administrated intravenously into mice bearing the RENCA tumor model (n = 3 mice per group). Afterward, 3.0 mg kg−1 aPD-L1 were intravenously injected 24 h later. To evaluate in vivo antitumor immune response, lymph nodes and tumors were harvested at 3 days post-treatments from BALB/c mice (female, 6-8 weeks) bearing subcutaneous and RENCA tumors. The samples were then processed through homogenization or digestion with enzymes in staining buffers to prepare single cell suspensions. For T-cell and NK cells analysis, tumor cells were stained with FITC anti-mouse CD45 (BioLegend, 103108, dilution: 1:200), APC anti-mouse CD3 (BioLegend, 100236, dilution: 1:200), PE anti-mouse CD8a (BioLegend, 100708, dilution: 1:200) and PE anti-mouse CD335 (BioLegend, 137604, dilution: 1:200) antibodies for 60 min followed by analysis of CD45+CD3+CD8+ CTLs and CD45+CD3−CD335+ NK cells. For DCs analysis, lymph nodes were stained with FITC anti-mouse CD11c (BioLegend, 117306, dilution: 1:200), PE anti-mouse CD80 (BioLegend, 104708, dilution: 1:200) and APC anti-mouse CD86 (BioLegend, 105012, dilution: 1:200) antibodies for 60 min followed by analysis of CD11c+CD80+CD86+ DCs.For TAMs analysis, tumor cells were stained with FITC anti-mouse/human CD11b (BioLegend, 101206, dilution: 1:200), APC anti-mouse F4/80 (BioLegend, 123116, dilution: 1:200), and PE anti-mouse CD206 (BioLegend, 141706, dilution: 1:200), PE anti-mouse CD86 (BioLegend, 105007, dilution: 1:200) antibodies for 60 min followed by analysis of CD11b+F4/80+CD86+ TAMs and CD11b+F4/80+CD206+ TAMs. These immune cells data were detected with flow cytometry (FACS Aria III, BD) and analyzed with FlowJo software (Version 10).
2.18. Evaluation of Antitumor Immune Response in Resected Human Tissues
Patients provided consent for the use of biospecimens in research, as approved by the Fourth Affiliated Hospital of Soochow University. Freshly resected human tissues (primary RCC) were rinsed and divided into several sections (1–5 mm3) using a scalpel. Subsequently, these sections were injected with free SR717 and SH-NPs (500 µg SR717) in a 5% glucose solution within 30 minutes of resection. Each tissue section was cultured in 0.5 mL RPMI 1640 medium (supplemented with 10% heat-inactivated human serum, 1% insulin-transferrin-selenium, 1% GlutaMAX, and 1% penicillin–streptomycin) in a 24-well plate for 24 h. For TAMs analysis, tumour tissues were first digested by 1 mg mL−1 collagenase IV and 0.2 mg mL−1 DNase I (Sigma-Aldrich) for 45 min at 37 °C, then passed through a 70 μm nylon cell strainer to obtain single cells. Then, tumor cells were stained with FITC anti- human CD14 (BioLegend, 982502, dilution: 1:200), APC anti-human CD68 (BioLegend, 333809, dilution: 1:200), PE anti- human CD86 (BioLegend, 374205, dilution: 1:200), and PE anti-human CD206 (BioLegend, 321105, dilution: 1:200) antibodies for 60 min followed by analysis of CD14+CD68+CD86+ TAMs and CD14+CD68+CD206+ TAMs. The levels of IFN-β in the culture medium were measured by ELISA kits (Lianke).
2.19. Statistics and Reproducibility
Statistical analysis was performed using Graphpdad Prism (Version 8.0.2). All in vitro and in vivo experiments were repeated at least three times and data were presented as mean ± SD. Group size was determined on the basis of the preliminary experiments results, and no statistical method was used to predetermine sample size. The indicated sample size (n) represents biological replicates. For all studies, samples were randomly divided into different experimental groups. Group allocation and outcome assessment were not performed in a blinded manner. No data were excluded from the analyses. The statistical significance between two groups was calculated by unpaired two-tailed Student’s t test. The statistical significance between multiple groups was calculated by one-way ANOVA with Tukey’s post hoc test. Survival was measured using the Kaplan-Meier method and statistical significance was calculated by log-rank test. P < 0.05 was considered statistically significant.
3. Results
3.1. Preparation and Characterization of SH-NPs
In a typical synthesis process, human serum albumin was applied as a single-molecule nanoreactor to encapsulate SR-717. First, SR-717 was dissolved in basic solutions and incubated with albumin under stirring, followed by adjusting the pH to 6 to allow the quick precipitation of SR-717 inside albumin nanocages. The obtained SH-NPs were then purified via ultracentrifuge and stored for further use. As shown in Figure 1a, the SH-NPs showed round-like morphology with the size of 5.6 ± 1.8 nm. Furthermore, the result from dynamic laser scanning revealed that the diameter of SH-NPs was calculated to be 31.3 ± 4.2 nm with the PDI value of 0.122 (Figure 1b), indicating the uniform size distribution. SH-NPs exhibited satisfactory stability in aqueous solutions (Figure 1c), as evidenced by the negligible variations in hydrodynamic size over the course of 7 days when stored at 4 °C. Also, the surface potential of SH-NPs was negative with the value of -23.4 ± 0.7 mV (Figure 1d), implying that the negative surface charge facilitates the formation of stable complexes with positively charged serum proteins, potentially extending the circulation time and enhancing the nanoparticles' ability to reach target tissues[39]. Afterwards, we further applied the dialysis method to investigate the drug release behavior of SH-NPs in the presence and absence of cathepsin B (CB). We found that the accumulative release amount of SR-717 from SH-NPs remained relatively low during 24 h at pH 7.4 solution or pH 5.0 solution (Figure 1f). By contrast, free SR-717 rapidly released with the amount reaching 90%. Furthermore, the release behavior of SH-NPs was accelerated in the presence of cathepsin B (Figure 1f), which is resulted from the responsive degradation of albumin.
3.2. STING Activation and Immune Activation of SH-NPs Inside Immune Cells
To unravel the STING activation efficiency of SH-NPs, we first evaluated their cellular uptake in DC cells. As exhibited in Supplementary Figure S1, SH-NPs showed time-dependent internalization into immune cells during 24 h incubation and the cellular uptake amount was significantly higher than that of free SR-717, which is ascribed to the clathrin-mediated endocytosis (Supplementary Figure S2). We then tested the cytotoxicity of SH-NPs and no obvious cell death was found during 24 h even at the high concentration of 20 μM (Supplementary Figure S3), indicating their superior safety and biocompatibility. Afterwards, we investigated phosphorylates TANK-binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3) that are closely associated with STING pathway activation. After incubation of SH-NPs at the concentration of 10 μM for 24 h, the level of p-TBK1 and p-IRF3 was considerably elevated and their expressions were 1.5 times and 1.7 times higher as compared with that of free SR-717, respectively (Figure 2a, b). Given that p-TBK1 and p-IRF3 initiates the downstream production of IFN-I proteins, we further conducted enzyme-linked immunosorbent assay (ELISA) to evaluate the secretion of IFN-I. Owing to the enhanced cellular uptake and increased expression of p-TBK1 and p-IRF3 proteins, SH-NPs aroused the highest production of IFN-I with the amount 2.2-fold and 1.4-fold increases as compared with that of control and free SR-717 groups (Figure 2c).
Next, we sought to investigate the immune activation of SH-NPs using DC and macrophages as model cells. Followed by the effective activation of STING pathway, SH-NPs remarkably increased the critical co-stimulation signal (CD80 and CD86) in the cell surface, indicating the maturation of DC cells for subsequent antigen presentation (Figure 2d). However, the DC cell incubated with free SR-717 failed to induce any maturation owing to the weak IFN-I production (Figure 2e). Additionally, surface expression of CD206 (a canonical marker of M2-polarized macrophages) was decreased on macrophages after treatment with SH-NPs (Figure 2f, g), while CD86 (a canonical marker of M1-polarized macrophages) was increased with 1.3 times as compared with that of free SR-717 (Figure 2h, i), suggesting repolarization or recruitment of macrophages with reduced immunosuppressive capacity. These data indicated that STING activation of SH-NPs would potently stimulate an immunogenic tumor immune microenvironment with relieved immunosuppression, which is highly for improving the cancer immunotherapeutic efficacy.
3.3. Ttumor Accumulation and Antitumor Efficacy of SH-NPs
To understand the in vivo performance of SH-NPs, we constructed the subcutaneous renal tumor bearing mice to evaluate the tumor targeting capability of SH-NPs. We first covalently conjugated a fluorescent dye (Cy5) to the albumin shell of SH-NPs for visualizing their in vivo biodistribution. As descripted in Figure 3a, SH-NPs gradually accumulated in the tumor region and reach the peak at 12 h post-injection with the 2.8 times fluorescence intensity as compared to free Cy5, suggesting the superior targeting ability for maximizing the antitumor efficacy. Furthermore, we utilized HPLC to quantitatively measure the biodistribution of SH-NPs in tumor and major organs including heart, liver, spleen, lung, kidney. In consistence with previous result, SH-NPs showed excellent tumor accumulation with the amount up to 7.9 ID%/g, being 2.2-fold increase as compared to free SR-717 (Figure 3c).
Inspired by the excellent tumor targeting capability, we further evaluated the antitumor efficacy of SH-NPs in mice bearing renal tumors. SH-NPs were intravenously injected at the dose of 30.0 mg/kg when the tumor volume reach 140-150 mm3, and the tumor volume were measured during 16 days (Figure 3e). As shown in Figure 3f-g, renal tumors receiving treatment of free SR-717 exhibited aggressive growth profile and all the mice died with 55 days. In contrast, SH-NPs significantly delayed the tumor growth, probably owing to the enhanced tumor targeting and subsequent STING pathway activation (Figure 3f). Considering the elevated PD-L1 expression after STING activation, we further applied anti-PD-L1 antibody for synergistic therapy. Combination of anti-PD-L1 and SH-NPs did not result in complete tumor eradication. Nonetheless, it is noteworthy that this synergistic treatment regimen conferred a significant survival benefit, with 60% of the mice surviving past 80 days. In contrast, monotherapy with either SH-NPs or anti-PD-L1 exerted only a limited therapeutic effect on renal tumor growth. (Figure 3f). These data implies that SH-NPs with remarkable tumor targeting would induce potent antitumor effect and could further improve the therapeutic efficacy of checkpoint blockade against murine renal cancers.
3.4. STING Activation and Immune Responses of SH-NPs in Mice Bearing Renal Tumor
To unravel the detailed mechanism of the antitumor efficacy in the mice receiving SH-NPs treatment, we carefully assessed the STING activation and immune responses after various treatment. First, the essential proteins including p-TBK1 and p-IRF3 were evaluated via western blotting. In the mice treated with free SR-717 or anti-PD-L1, p-TBK1 and p-IRF3 were slightly upregulated, while SH-NPs significantly elevated their expressions with 1.7-fold and 1.8-fold increases as compared to that of free SR-717 groups (Figure 4a), respectively, which is ascribed to the enhanced tumor targeting and cellular uptakes. As expected, much higher secretion of IFN-I was observed in the mice receiving SH-NPs treatment regardless of combination with anti-PD-L1 or not. Meanwhile, chemoattractants such as CXCL10 (Figure 4d), closely associated with T cell proliferation and function, along with cytokines including IL-6 (Figure 4e) and TNF-α (Figure 4f), were elevated post-treatment with SH-NPs, a result of the robust activation of the STING pathway. Then, we collected the tumors from the mice treated with various formulations and analyzed the population of various immune cells at 3 days after injection. First, SH-NPs induced higher tumor-infiltrating cytotoxic T cells marked by CD45+CD3+CD8+ T cells, being 2.1-fold and 1.8-fold increase as compared to those of free SR-717 and anti-PD-L1 (Figure 4g, i), respectively. Meanwhile, another injection of anti-PD-L1 would further elevate the infiltration of T cells probably due to the immune checkpoint blockade. As a typical phenotype of innate immune system, natural killer (NK) cells showed similar trend, in which free SR-717 or anti-PD-L1 failed to arouse any significant improvement, while the highest infiltration amount of NK cells was observed in the mice treated with SH-NPs/anti-PD-L1. In particular, the NK cell proportion increased by a substantial 3.4-fold relative to the free SR-717 treatment and by 2.7-fold compared to the anti-PD-L1 treatment alone, underscoring the synergistic impact of the combined therapeutic approach. (Figure 4h, Supplementary Figure S4a, c). Furthermore, we also picked out the lymph nodes and tested the maturation of DCs after STING activation. Obviously, 2.2-fold higher DCs maturation was observed in the mice receiving SH-NPs as compared to those of free SR-717 (Figure 4j, Supplementary Figure S4b, d), suggesting the targeted delivery of STING agonist is highly advantageous for immune activation. Notably, we also found the combination of SH-NPs and anti-PD-L1 would further increase the DCs maturation, reasonably due to the intrinsic PD-L1 expression on the surface of DCs[40].
Considering the immunosuppressive microenvironment as a huge obstacle against immunotherapy, we then assessed the population of CD11b+F4/80+CD206+ tumor-associated macrophages (TAMs possess M2-like character) and CD11b+F4/80+CD86+ tumor-associated macrophages (TAMs possess M1-like character), and found that SH-NPs treatment significantly upregulated the amount of M1-like TAMs and downregulated the amount M2-like TAMs with the 6.0-fold higher CD11b+F4/80+CD86+ TAMs to CD11b+F4/80+CD206+ TAMs ratio than that in other control groups(Figure 4k, l, Supplementary Figure S5), suggesting their shift to an antitumor phenotype after SH-NPs treatment.
3.5. STING Activation in Human Tissues
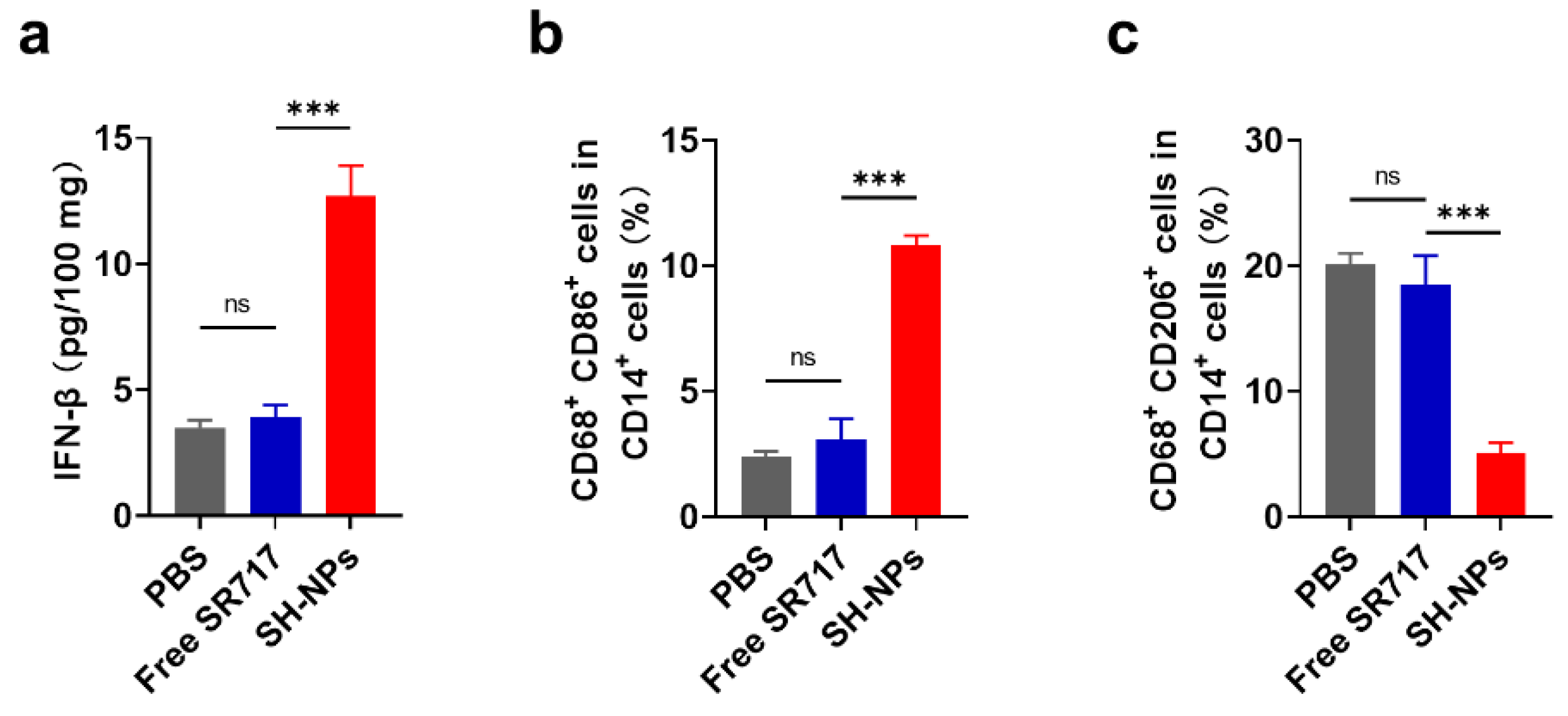
To examine the translational potential, we investigated the feasibility of STING activation in human tissues. We acquired freshly resected human tissues (primary RCC). We locally injected these tissues with free SR717 or SH-NPs, and evaluate the secretion of IFN-β. Free SR717 had a marginal effect on IFN-I expression over the control due to limited bioavailability. By contrast, SH-NPs aroused the highest production of IFN-I with the amount 3.6-fold and 3.2-fold increases as compared with that of control and free SR-717 groups (Figure 5a). Notably, M1 macrophage-associated markers was increased and the expression of M2 macrophage markers was decreased after treatment with SH-NPs compared with other groups (Fig. 5 b, c). This suggests that SH-NPs effectively activated STING in human RCC tissues, thereby significantly enhancing IFN-β secretion and modulating macrophage polarization.
4. Discussion
We have developed HSA-based nanoparticles as a highly effective approach to deliver the STING agonist SR717, which significantly improved the cellular uptake, stability, and STING activation of SR717, leading to enhanced anti-tumor immune responses both in vitro and in vivo. These findings support the potential of SH-NPs as a promising delivery system for cancer immunotherapy. Previous studies have highlighted the challenges associated with the delivery of STING agonists, particularly regarding their stability and targeting specificity. Traditional STING agonists, such as CDNs, suffer from rapid degradation and limited efficacy due to poor tissue penetration and off-target effects[24,25]. Non-nucleotide STING agonists like SR-717 have shown potential but still face issues related to hydrophobicity and targeting[26,41]. Our approach using HSA-based nanoparticles addresses these challenges by enhancing the solubility and targeting efficiency of SR717. Our research reveal that SH-NPs not only improve the stability and cellular uptake of SR-717 but also significantly activate the STING pathway, as evidenced by the elevated levels of phosphorylated TBK1 and IRF3 (Figure 2a, b). This activation leads to the production of IFN-I proteins (Figure 2c), crucial for initiating anti-tumor immune responses. The enhanced STING activation further promotes the maturation of DCs and the polarization of macrophages towards a pro-inflammatory phenotype (Figure 2d, i). These immunological changes contribute to a more immunogenic tumor microenvironment, facilitating the recruitment and activation of CTLs and NK cells, which are essential for effective tumor eradication (Figure 4g, h). The in vivo studies in murine models demonstrated that SH-NPs significantly enhanced tumor targeting and retention of SR717, resulting in prolonged circulation time and higher tumor accumulation compared to free SR717 (Figure 3a). This targeted delivery translated into superior anti-tumor efficacy, with SH-NPs effectively delaying tumor growth and improving survival rates in renal tumor-bearing mice. The combination of SH-NPs with anti-PD-L1 antibodies further potentiated the anti-tumor effects, leading to significant tumor regression in some cases (Figure 3f). These results underscore the potential of SH-NPs in synergistic cancer immunotherapy.
In conclusion, this study demonstrates the potential of HSA-based nanoparticles for the targeted delivery of STING agonists, providing a promising strategy to enhance the efficacy of cancer immunotherapy. Our findings pave the way for the development of more effective and targeted therapeutic approaches, potentially transforming the landscape of cancer treatment.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: The interlized amount of free SR-717 or SH-NPs by dendritic cells (DCs) at different time points.; Figure S2: The cellular uptake amount of SR-717 after different treatments; Figure S3: Cell viability of DC2.4 cells treated with free SR717 or SH-NPs at various concentrations; Figure S4: Representative flow cytometric plots and percentages of CD45+CD3-CD335+ NK cells (a, c) in RCC tumors and matured dendritic cells (CD11c+CD80+CD86+ DCs) (b, d) inside tumor-draining lymph nodes; Figure S5: Representative flow cytometric plots and percentages of CD11b+F4/80+CD206+ TAMs (a, c) and CD11b+F4/80+CD86+ TAMs (b, d) inside RCC tumors.
Author Contributions
Conceptualization, T.Y. and G.S.; methodology, Z.M. and X.S.; software, X.S.; validation, Z.M., X.S. and A.X.; formal analysis, C.Y.; investigation, Z.M. and X.S.; resources, P.L.; data curation, X.S.; writing—original draft preparation, Y.L. and T.Y.; writing—review and editing, T.Y. and G.S.; visualization, X.S.; supervision, G.S.; project administration, Z.M.; funding acquisition, Z.M., T.Y. and G.S.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China (52373298), Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), Gusu Innovation and Entrepreneurship Leading Talent Plan (ZXL2022486), Project of Suzhou Science and Technology Development Program (SKJY2021029, SZM2023016) and Suzhou Industrial Park clinical medical expert team introduction project (0202140004).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee) of The Fourth Affiliated Hospital of Soochow University (241027). The animal study protocol was approved by the Institutional Review Board (or Ethics Committee) of Soochow University (202308A0335).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study. Written informed consent has been obtained from the patient(s) to publish this paper.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wang, C.; Zhang, S.J.A.A.N.M. Advantages of nanomedicine in cancer therapy: A review. ACS Appl. Nano Mater. 2023, 6, 22594–22610. [Google Scholar] [CrossRef]
- Kruger, S.; Ilmer, M.; Kobold, S.; Cadilha, B.L.; Endres, S.; Ormanns, S.; Schuebbe, G.; Renz, B.W.; D’Haese, J.G.; Schloesser, H.J.J. o. E.; et al. Advances in cancer immunotherapy 2019–latest trends. J. Exp. Clin. Cancer Res. 2019, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mathew, D.; Marmarelis, M.E.; Foley, C.; Bauml, J.M.; Ye, D.; Ghinnagow, R.; Ngiow, S.F.; Klapholz, M.; Jun, S.; Zhang, Z.J.S. Combined JAK inhibition and PD-1 immunotherapy for non–small cell lung cancer patients. Science 2024, 384, eadf1329. [Google Scholar] [CrossRef]
- Lyu, J.; Bai, L.; Li, Y.; Wang, X.; Xu, Z.; Ji, T.; Yang, H.; Song, Z.; Wang, Z.; Shang, Y.J.N.C. Plasma proteome profiling reveals dynamic of cholesterol marker after dual blocker therapy. Nat. Commun. 2024, 15, 3860. [Google Scholar] [CrossRef]
- Xu, Y.; Yan, J.; Tao, Y.; Qian, X.; Zhang, C.; Yin, L.; Gu, P.; Liu, Y.; Pan, Y.; Tang, R.J.S. Pituitary hormone α-MSH promotes tumor-induced myelopoiesis and immunosuppression. Science. 2022, 377, 1085–1091. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, P.J.T. i. p. s. Preserving the CTLA-4 checkpoint for safer and more effective cancer immunotherapy. Trends Pharmacol. Sci. 2020, 414, 412–418. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M.J.B. , The Journal of the American Society of Hematology. CTLA-4: a moving target in immunotherapy. J. Am. Soc. Hematol. 2018, 131, 58–67. [Google Scholar]
- Sharma, P.; Allison, J.P.J.S. The future of immune checkpoint therapy. J. Am. Soc. Hematol. 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- de Miguel, M.; Calvo, E.J.C. c. Clinical challenges of immune checkpoint inhibitors. Cancer Cell. 2020, 38, 326–333. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P.J.N.R.I. Dissecting the mechanisms of immune checkpoint therapy. Nat. Rev. Immunol. 2020, 20, 75–76. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M.J.C. c. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer cell. 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Siddiqui, B.A.; Anandhan, S.; Yadav, S.S.; Subudhi, S.K.; Gao, J.; Goswami, S.; Allison, J.P.J.C. d. The next decade of immune checkpoint therapy. Cancer Disco. 2021, 11, 838–857. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Minn, A.J.J.I. Combination cancer therapy with immune checkpoint blockade: mechanisms and strategies. Cancer Disco. 2018, 48, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M.J.N. r. c. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Bitton, K.; Michot, J.-M.; Barreau, E.; Lambotte, O.; Haigh, O.; Marabelle, A.; Voisin, A.-L.; Mateus, C.; Rémond, A.-L.; Couret, C.J.A.J. o. O. Prevalence and clinical patterns of ocular complications associated with anti-PD-1/PD-L1 anticancer immunotherapy. Am. J. Ophthalmol. 2019, 202, 109–117. [Google Scholar] [CrossRef]
- Ichihara, E.; Harada, D.; Inoue, K.; Sato, K.; Hosokawa, S.; Kishino, D.; Watanabe, K.; Ochi, N.; Oda, N.; Hara, N.J.L.C. The impact of body mass index on the efficacy of anti-PD-1/PD-L1 antibodies in patients with non-small cell lung cancer. Lung Cancer. 2020, 139, 140–145. [Google Scholar] [CrossRef] [PubMed]
- de Jong, F.C.; Rutten, V.C.; Zuiverloon, T.C.; Theodorescu, D.J.I. j. o. m. s. Improving anti-PD-1/PD-L1 therapy for localized bladder cancer. Int. J. Mol. Sc. 2021, 22, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Mosallanejad, K.; Kagan, J.C.J.I.; biology, c. Control of innate immunity by the cGAS-STING pathway. Immunol. Cell Biol. 2022, 100, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.-P.; Hornung, V.J.N. r. M. c. b. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A.J.N.R.I. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immuno. 2021, 21, 548–569. [Google Scholar] [CrossRef]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A.J.N.R.G. DNA sensing by the cGAS–STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Celias, D.P.; Hänggi, K.; Ruffell, B.J.C.R. Investigating the mechanisms involved in HMGB1-dependent DNA uptake and STING activation in dendritic cells. Cancer Res. 2023, 83, 678–678. [Google Scholar] [CrossRef]
- Umemura, K.; Kawamoto, Y.; Takahashi, Y.; Takakura, Y.J.M.P. Development of a Cytosolic DNA Sensor Agonist Using GALA Peptide-Conjugated DNA and Long Single-Stranded DNA. Mol. Pharmaceutics. 2024, 21, 1204–1213. [Google Scholar] [CrossRef] [PubMed]
- Morehouse, B.R.; Govande, A.A.; Millman, A.; Keszei, A.F.; Lowey, B.; Ofir, G.; Shao, S.; Sorek, R.; Kranzusch, P.J.J.N. STING cyclic dinucleotide sensing originated in bacteria. Nature. 2020, 586, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hou, X.; Du, S.; Xue, Y.; Yan, J.; Kang, D.D.; Zhong, Y.; Wang, C.; Deng, B.; McComb, D.W.J.N.N. Close the cancer–immunity cycle by integrating lipid nanoparticle–mRNA formulations and dendritic cell therapy. Nat. Nanotechnol. 2023, 18, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- Crunkhorn, S.J.N.R.I. Strengthening the sting of immunotherapy. Nat. Nanotechnol. 2020, 20, 589. [Google Scholar]
- Nguyen, P.H.D.; Jayasinghe, M.K.; Le, A.H.; Peng, B.; Le, M.T.J.A. n. Advances in drug delivery systems based on red blood cells and their membrane-derived nanoparticles. ACS nano. 2023, 17, 5187–5210. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Su, Y.-Y.; Jiang, X.-C.; Gao, J.-Q. J. D. D.; Research, T. Cell membrane-coated nanoparticles: a novel multifunctional biomimetic drug delivery system. Drug Deliv. Transl. Res. 2023, 13, 716–737. [Google Scholar] [CrossRef] [PubMed]
- Vyas, K.; Rathod, M.; Patel, M.M.J.N.N. , Biology; Medicine. Insight on nano drug delivery systems with targeted therapy in treatment of oral cancer. Nanomedicine: NBM. 2023, 49, 102662. [Google Scholar] [CrossRef]
- Senti, M.E.; Del Valle, L.G.; Schiffelers, R.M.J.A.D.D.R. mRNA delivery systems for cancer immunotherapy: lipid nanoparticles and beyond. Adv. Drug Deliv. 2024, 115190. [Google Scholar] [CrossRef]
- Zhou, Q.; Dutta, D.; Cao, Y.; Ge, Z.J.A. n. Oxidation-responsive PolyMOF nanoparticles for combination photodynamic-immunotherapy with enhanced STING activation. ACS Nano. 2023, 17, 9374–9387. [Google Scholar] [CrossRef]
- Lu, Q.; Chen, R.; Du, S.; Chen, C.; Pan, Y.; Luan, X.; Yang, J.; Zeng, F.; He, B.; Han, X.J.B. Activation of the cGAS-STING pathway combined with CRISPR-Cas9 gene editing triggering long-term immunotherapy. Biomaterials. 2022, 291, 121871. [Google Scholar] [CrossRef]
- Gou, S.; Liu, W.; Wang, S.; Chen, G.; Chen, Z.; Qiu, L.; Zhou, X.; Wu, Y.; Qi, Y.; Gao, Y.J.N.L. Engineered nanovaccine targeting Clec9a+ dendritic cells remarkably enhances the cancer immunotherapy effects of STING agonist. Nano Lett. 2021, 21, 9939–9950. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Wei, C.; Deng, Y.; Chen, H.; Shen, J.; Ke, H.J.J. o. M. C. B. Biomineralized iron oxide–polydopamine hybrid nanodots for contrast-enhanced T 1-weighted magnetic resonance imaging and photothermal tumor ablation. J. Mater. Chem. 2021, 9, 1781–1786. [Google Scholar]
- Li, M.; Wang, Y.; Li, T.; Zhang, J.; Wang, X.; Luo, J.; You, M.; Yang, T.; Deng, Y.; Yang, H.J.A.B. Albumin-templated platinum (II) sulfide nanodots for size-dependent cancer theranostics. Acta Biomater. 2023, 155, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, Y.; Zhu, J.; Zhang, F.; Xu, A. a.; Zhou, T.; Li, Y.; Liu, M.; Ke, H.; Yang, T.J.A.M. A pH-Activatable Copper-Biomineralized Proenzyme for Synergistic Chemodynamic/Chemo-Immunotherapy against Aggressive Cancers. Adv. Mater. 2023, 35, 2210201. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Liu, M.; Yang, T.; Luo, J.; Wei, C.; Shen, J.; Song, X.; Ke, H.; Sun, P.; Guo, M.J.J. o. C. R. Self-activated arsenic manganite nanohybrids for visible and synergistic thermo/immuno-arsenotherapy. J. Control. Release. 2022, 350, 761–776. [Google Scholar] [CrossRef]
- Liu, B.; Jiao, J.; Xu, W.; Zhang, M.; Cui, P.; Guo, Z.; Deng, Y.; Chen, H.; Sun, W.J.A.M. Highly Efficient Far-Red/NIR-Absorbing Neutral Ir (III) Complex Micelles for Potent Photodynamic/Photothermal Therapy. Adv. Mater. 2021, 33, 2100795. [Google Scholar] [CrossRef]
- Xue, X.; Qu, H.; Li, Y. Stimuli-responsive crosslinked nanomedicine for cancer treatment. Exploration. 2022, 2, 20210134. [Google Scholar] [CrossRef]
- Lucas, E.D.; Schafer, J.B.; Matsuda, J.; Kraus, M.; Burchill, M.A.; Tamburini, B.A.J. PD-L1 reverse signaling in dermal dendritic cells promotes dendritic cell migration required for skin immunity. Cell reports. 2020 33, 2, 108258. [Google Scholar]
- Lei, H.; Pei, Z.; Jiang, C.; et al. Recent progress of metal-based nanomaterials with anti-tumor biological effects for enhanced cancer therapy. Exploration. 2023, 3, 20220001. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
The synthesis process of using human serum albumin (HSA) as a single-molecule nanoreactor to encapsulate the STING agonist SR-717 and the subsequent process of immune activation in tumor immunotherapy.
Scheme 1.
The synthesis process of using human serum albumin (HSA) as a single-molecule nanoreactor to encapsulate the STING agonist SR-717 and the subsequent process of immune activation in tumor immunotherapy.
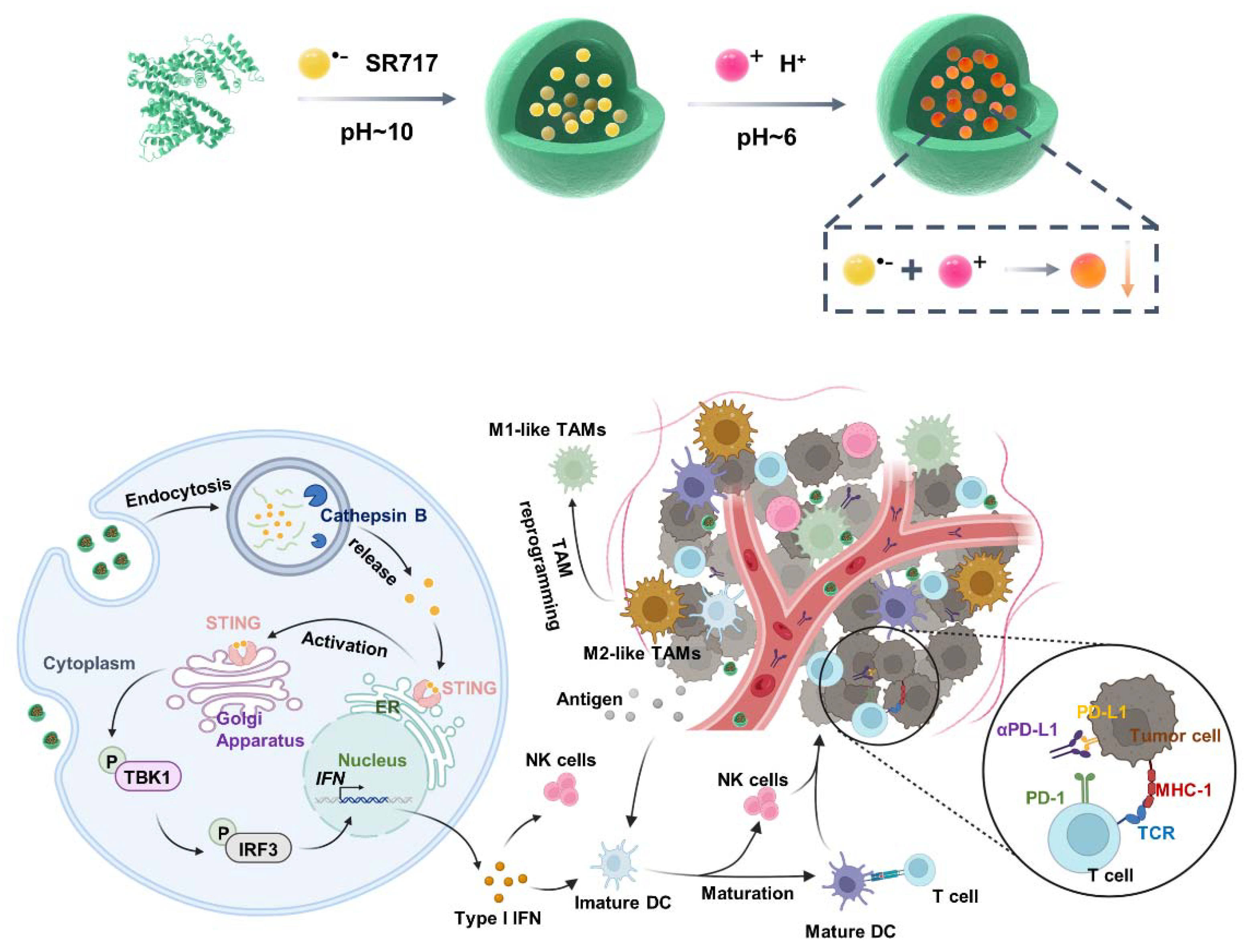
Figure 1.
Characterization of SH-NPs. TEM image (a) and hydrodynamic size distribution (b) of SH-NPs; (c) Hydrodynamic size of SH-NPs stored at 4 °C for 7 days; (d) Zeta potential of SH-NPs; (f) Accumulative release of SR717 from free SR717 and SH-NPs in pH 7.4 phosphate buffer, pH 5.0 acetate buffer, and pH 5.0 acetate buffer containing 10 μg mL−1 of CB.
Figure 1.
Characterization of SH-NPs. TEM image (a) and hydrodynamic size distribution (b) of SH-NPs; (c) Hydrodynamic size of SH-NPs stored at 4 °C for 7 days; (d) Zeta potential of SH-NPs; (f) Accumulative release of SR717 from free SR717 and SH-NPs in pH 7.4 phosphate buffer, pH 5.0 acetate buffer, and pH 5.0 acetate buffer containing 10 μg mL−1 of CB.
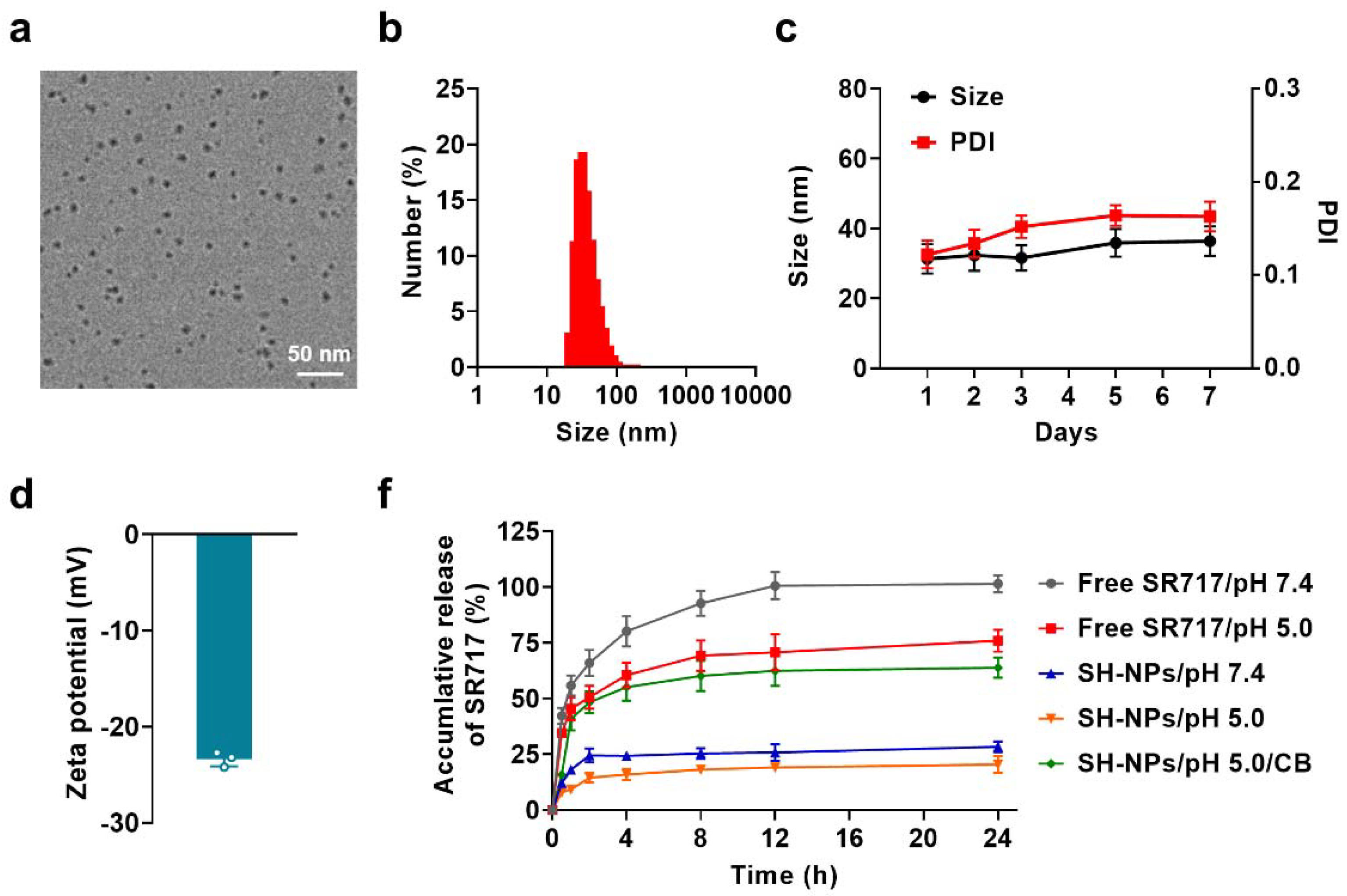
Figure 2.
Cellular behaviors of SH-NPs. Expression of downstream proteins of cGAS-STING signaling pathway including p-TBK1 and p-IRF3 (a) and statistics of protein relative gray value (b); (c) Concentration of IFN-β secreted by cells treated with free SR717 and SH-NPs; The expression of CD80 and CD86 on the surface of DC2.4 after administration (d) and statistics of maturation ratio (e); (f-i) Expression and the fluorescence intensity of CD206 (f,g) and CD86 (h,i) on the surface of macrophages after incubation with free SR717 and SH-NPs.
Figure 2.
Cellular behaviors of SH-NPs. Expression of downstream proteins of cGAS-STING signaling pathway including p-TBK1 and p-IRF3 (a) and statistics of protein relative gray value (b); (c) Concentration of IFN-β secreted by cells treated with free SR717 and SH-NPs; The expression of CD80 and CD86 on the surface of DC2.4 after administration (d) and statistics of maturation ratio (e); (f-i) Expression and the fluorescence intensity of CD206 (f,g) and CD86 (h,i) on the surface of macrophages after incubation with free SR717 and SH-NPs.
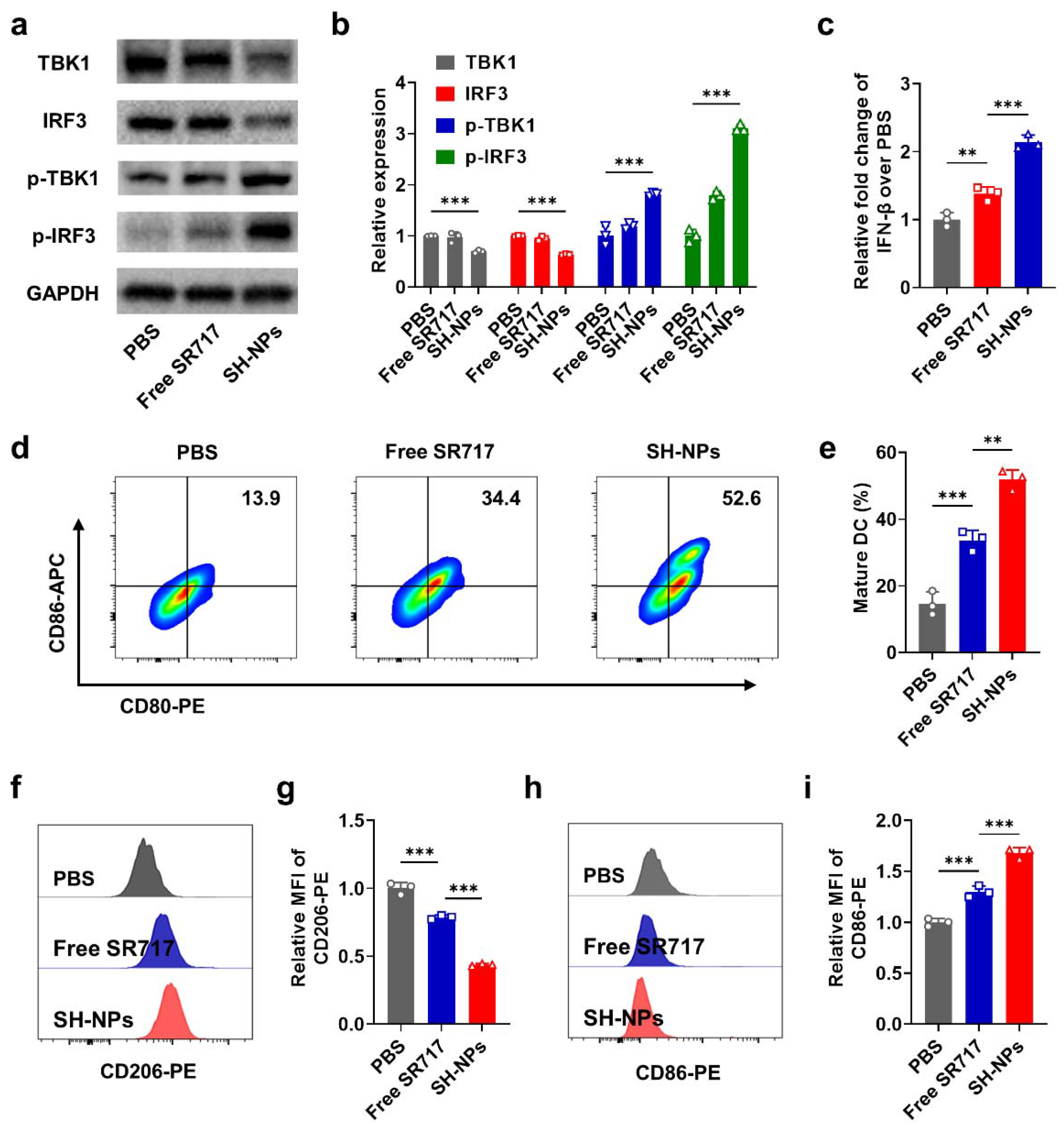
Figure 3.
Tumor targeting and in vivo antitumor efficacy of SH-NPs. In vivo fluorescence imaging of renal tumor bearing mice receiving SH-NPs at different time points (a) and the fluorescence intensity of tumor region (b); (c) Tumor accumulation amount of SR717 in the renal tumor bearing mice at 12 h post-injection of SH-NPs; (e) Timeline schedule of treatment of renal tumor bearing mice; (f,g) Tumor growth profiles (f) and survival curve (g) of the mice bearing renal tumor treated with PBS, free SR717, aPD-L1, SH-NPs and SH-NPs/aPD-L1.
Figure 3.
Tumor targeting and in vivo antitumor efficacy of SH-NPs. In vivo fluorescence imaging of renal tumor bearing mice receiving SH-NPs at different time points (a) and the fluorescence intensity of tumor region (b); (c) Tumor accumulation amount of SR717 in the renal tumor bearing mice at 12 h post-injection of SH-NPs; (e) Timeline schedule of treatment of renal tumor bearing mice; (f,g) Tumor growth profiles (f) and survival curve (g) of the mice bearing renal tumor treated with PBS, free SR717, aPD-L1, SH-NPs and SH-NPs/aPD-L1.
Figure 4.
In vivo STING activation and immune responses. (a-b) Expression (a) and the relative protein gray value (b) of p-TBK1 and p-IRF3 in tumor tissue from the mice bearing renal tumor treated with PBS, free SR717, aPD-L1, SH-NPs and SH-NPs/aPD-L1 at 72 h post-injection; (c-f) Concentration of IFN-β (c), CXCL-10 (d), IL-6 (e), TNF-α(f) in tumor tissue from the mice bearing renal tumor treated with PBS, free SR717, aPD-L1, SH-NPs and SH-NPs/aPD-L1 at 72 h post-injection; (g-i) Quantification of tumor-infiltrating CD45+CD3+CD8+ CTLs (g,i) and CD45+CD3-CD335+ NK cells (h) in tumor tissue from the mice bearing renal tumor treated with PBS, free SR717, aPD-L1, SH-NPs and SH-NPs/aPD-L1 at 72 h post-injection; (j) Quantification of matured dendritic cells (CD11c+CD80+CD86+ DCs) inside tumor-draining lymph nodes from the mice bearing renal tumor treated with PBS, free SR717, aPD-L1, SH-NPs and SH-NPs/aPD-L1 at 72 h post-injection; (k, l) Quantification of CD11b+F4/80+CD206+ TAMs (k) and CD11b+F4/80+CD86+ TAMs (l) in tumor tissue from the mice bearing renal tumor treated with PBS, free SR717, aPD-L1, SH-NPs and SH-NPs/aPD-L1 at 72 h post-injection.
Figure 4.
In vivo STING activation and immune responses. (a-b) Expression (a) and the relative protein gray value (b) of p-TBK1 and p-IRF3 in tumor tissue from the mice bearing renal tumor treated with PBS, free SR717, aPD-L1, SH-NPs and SH-NPs/aPD-L1 at 72 h post-injection; (c-f) Concentration of IFN-β (c), CXCL-10 (d), IL-6 (e), TNF-α(f) in tumor tissue from the mice bearing renal tumor treated with PBS, free SR717, aPD-L1, SH-NPs and SH-NPs/aPD-L1 at 72 h post-injection; (g-i) Quantification of tumor-infiltrating CD45+CD3+CD8+ CTLs (g,i) and CD45+CD3-CD335+ NK cells (h) in tumor tissue from the mice bearing renal tumor treated with PBS, free SR717, aPD-L1, SH-NPs and SH-NPs/aPD-L1 at 72 h post-injection; (j) Quantification of matured dendritic cells (CD11c+CD80+CD86+ DCs) inside tumor-draining lymph nodes from the mice bearing renal tumor treated with PBS, free SR717, aPD-L1, SH-NPs and SH-NPs/aPD-L1 at 72 h post-injection; (k, l) Quantification of CD11b+F4/80+CD206+ TAMs (k) and CD11b+F4/80+CD86+ TAMs (l) in tumor tissue from the mice bearing renal tumor treated with PBS, free SR717, aPD-L1, SH-NPs and SH-NPs/aPD-L1 at 72 h post-injection.
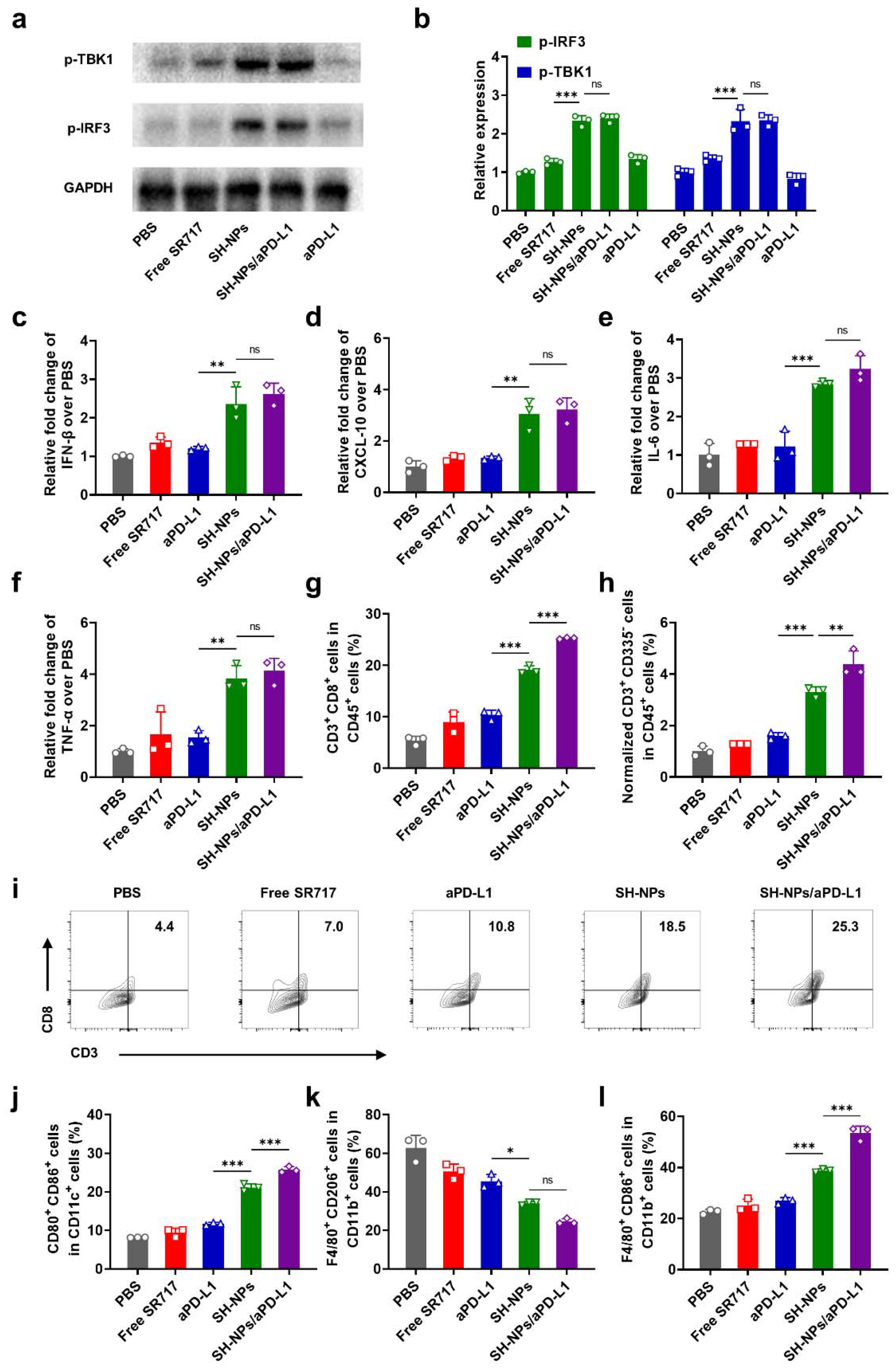
Figure 5.
STING activation in freshly isolated human renal tumor tissues. (a-c) Concentration of IFN-β CD14b+CD68+CD86+ TAMs (b) and CD14b+CD68+CD206+ TAMs (c) in human renal tumor tissues treated with intratumoral injection of PBS, free SR717 and SH-NPs.
Figure 5.
STING activation in freshly isolated human renal tumor tissues. (a-c) Concentration of IFN-β CD14b+CD68+CD86+ TAMs (b) and CD14b+CD68+CD206+ TAMs (c) in human renal tumor tissues treated with intratumoral injection of PBS, free SR717 and SH-NPs.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



