Submitted:
29 July 2024
Posted:
30 July 2024
You are already at the latest version
Abstract
Systemic inflammation and immunodeficiency are the key components of Cirrhosis Associated Immune Dysfunction (CAID); its severity is dynamic and progressive and is associated with the greater deterioration of the liver function. There are two different immune phenotypes: the low-grade systemic inflammation phenotype and the high-grade systemic inflammation phenotype. Each of these phenotypes is related with the function of liver cirrhosis; thus, the presence of high-grade inflammation is correlated with the severity of the hepatic insufficiency, the bacterial translocation, and the organic insufficiency, with which the risk of infections increases and the prognosis worsens. Bacterial translocation plays a relevant role in the persistent systemic inflammation in patients with cirrhosis; the prophylactic employment of antibiotics is useful for reducing events of infection and mortality.
Keywords:
liver cirrhosis
; cirrhosis associated immune dysfunction
; systemic inflammation
; bacterial translocation
1. Introduction
Liver cirrhosis is a morbid and multisystemic disease associated with frequent hospitalizations and high mortality, it represents 1.2 million deaths annually worldwide and is in 14th and 10th place among the principal causes of death worldwide and in the majority of developed countries, respectively [1]. 8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament
Cirrhosis is classically divided into two main periods: disease compensated cirrhosis (asymptomatic), and decompensated cirrhosis. The term acute decompensation (AD) defines the development of an acute complication (ascitis, variceal hemorrhage, hepatic encephalopathy, etc.); the first episode of AD signals the transition from compensated to decompensated cirrhosis. The course of decompensated cirrhosis is characterized by repeated episodes of AD; during these periods, patients are prone to developing bacterial infections. Finally, Acute-on-Chronic Liver Failure (ACLF) is characterized by the insufficiency of an organic system or of multiple organic systems, high short-term mortality, and always occurs within the context of AD. Systemic Inflammation (SI) is a well-recognized characteristic of decompensated cirrhosis [2]. 8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament Cirrhosis is a systemic disease that affects various organs and systems, a systemic disease that, including the immune system [3] 8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament, is considered an immunocompromised state that leads to a variety of infections, representing an approximate mortality of 30% [4]. 8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament The term Cirrhosis Associated Immune Dysfunction (CAID) is characterized by two important components: immunodeficiency, due to an altered response to pathogens, and systemic inflammation, as a consequence of a persistent and inadequate stimulation of the immune system; CAID should be considered a complication of cirrhosis of any etiology. [5] 8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament The immune dysfunction syndrome associated with cirrhosis is a multifactorial state that diminishes the patient’s capacity to eliminate bacteria, cytokines, and endotoxins from the circulation. The liver contains 90% of the reticuloendothelial cells (RE), such as the Kupffer sinusoidal endothelial cells, which are fundamental for the elimination of bacteria [6]. 8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament The most serious immune alterations are found in patients with Acute-on-Chronic Liver Failure (ACLF), a syndrome characterized by the acute decompensation of cirrhosis and hepatic and/or extra-hepatic organ failure with high mortality at the short term [7] 8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament.
Systemic Inflammation in Cirrhosis
The liver is a component of the innate immunological system. The molecular patterns associated with pathogens (PAMP) and the proinflammatory cytokines induce the sustained synthesis of acute-phase proteins. Therefore, the liver is a site of intense anabolic metabolism during SI. The inhibition of liver biotransformation in sepsis is induced by proinflammatory cytokines; this explains the presence of hyperbilirubinemia and jaundice [2]. 8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament Inflammation is a physiological response initially designed to restore homeostasis after different aggressions, such as bacterial infections or tissue lesions; similarly, it can be triggered by the recognition of distinctive bacteria-derived molecules (for example, Lipopolysaccharides [LPS]), known as PAMP. In the absence of infection, intracellular molecules released by damaged or dying cells, known as DAMP, could also activate systemic inflammation. DAMPs and PAMPs bind with PRR (Pattern Recognition Receptors) that are expressed in epithelial and in peripheral innate immune cells. PRR can be localized on the cellular surface (the type-4 Toll-like [TLP4]), the endolysosome (TLR9), or the cytosol (Nucleotide Oligomerization Dominion [NOD] 1) [3,8]. 8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament PAMPs released into the systemic circulation can also be translocated directly from the intestinal lumen; once translocated, the PAMPs interact with the PRRs in the gut-associated lymphoid tissue and the mesenteric lymph nodes; the latter leads to the expression of the genes codifying for the molecules responsible for the inflammation of the intestinal mucosa, and finally extends to the systemic circulation as well as to the peripheral organs [9]. The PAMPs-induced inflammatory response is indispensable for combating invasive bacteria; however, an excessive or chronic response can cause collateral tissue damage (immunopathology) and provoke a marked compensatory anti-inflammatory response that, in the last instance, leads to immune suppression and to a greater risk of secondary infections and with that, greater mortality; the TLR (principally TLR2 and TLR4) not only recognize PAMPs, but also DAMP, thus favoring the release of IL-1, IL-6, and TNFα [10]. (Figure A1)
8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament
Evidence of Systemic Inflammation
Cirrhosis is associated with SI evidenced by the increase in the count of white globules, neutrophils, activated circulating monocytes, plasma C-reactive Protein (PCR), proinflammatory cytokines, macrophage activation markers, and systemic oxidative stress [8]. The low-grade systemic inflammation is present in experimental models and in patients with compensated or decompensated cirrhosis without ACLF; this systemic inflammation is evidenced by the increase in the plasma concentration of acute-phase proteins (PCR and the LPS Binding Protein [LBP]), endothelial activation markers such as intercellular adhesion molecule 1 (ICAM1), the vascular cell adhesion protein 1 (VCAM1), the vascular endothelial growth factor (VEGF), von Willebrand Factor (vWF) P-selectin, nitrites, cytokines (TNF, IL-1β, IL-6, INFγ, and IL-17), and their soluble receptors [3]. 8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament
The dynamic nature of CAID is such that the intensity of the systemic inflammation increases as the cirrhosis progresses from the compensated to the decompensated stage. The mediators of adhesion and migration of leukocytes are particularly regulated in patients with ACLF, and curiously, some of these markers (VCAM1, ICAM1, granulocyte and macrophage colony-stimulating factor [GM-CSF]) are positively correlated with 3-month mortality in patients with ACLF. The extreme exacerbation of the SI in ACLF is exhibited by elevated levels of proinflammatory cytokines (TNF-α, IL-6, IL-8) and the anti-inflammatory cytokines (IL-10, IL-1RA), soluble markers of macrophage activation (sCD163 and the mannose receptor), CRP and leukocytes in plasma of patients with ACLF, there is a greater contribution of the innate response than the adaptative response to the SI [3,11]. 8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament
Mechanisms of Systemic Inflammation
8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament
There is the conversion of proinflammatory cytokines IL-1β and IL-18 into their active forms; the intracellular recognition of LPS is mediated by caspases 4 and 5, which triggers a type of programmed proinflammatory cellular death such as apoptosis. It has been demonstrated that IL-1α is an independent predictor of death in patients with ACLF and without previous clinical decompensations, while IL-1β is an independent predictor of death in those patients with previous episodes of ascites [12]. 8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament
Low-grade Systemic Inflammation
Bacterial translocation, defined as the passage of viable bacteria or bacterial subproducts (PAMPs, LPS) through the intestinal mucosa to the systemic circulation. [2] 8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament Intestinal permeability increases in patients with cirrhosis due to the presence of functional and anatomical alterations that affect the mucosal lining, the intercellular binding proteins, and the endothelial cells; in addition, excessive growth of bacteria and dysbiosis contribute to the migration of an increasing number of bacterial species and PAMPs of microbial origin from the intestinal lumen to the systemic circulation or the liver, following the lymphatic pathways and from the portal vein, respectively [13,14]. 8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament
The host and intestinal bacteria reside in a symbiotic state during health; any interruption of the intestinal homeostasis can cause quantitative and/or qualitative changes (dysbiosis) in the microbiota. Liver cirrhosis is associated as much with an excessive growth of intestinal bacteria as with dysbiosis, in that the native bacteria diminish and augment the potentially pathogenic bacteria; that is, there are fewer Bacteroidetes, but more Proteobacteria and Fusobacteria. Another common intestinal characteristic of cirrhosis is an altered intestinal barrier, which leads to pathological bacterial translocation [8, 15,16]. 8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament Hypergammaglobulinemia, which is common in decompensated cirrhosis, was considered a sign of escape of intestinal antigens into the systemic circulation and correlates with prognosis. Bacterial translocation and SI are chronically present in patients who are non-infected with decompensated cirrhosis [2]. 8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament
Infections represent a 4-fold increase in mortality among patients with cirrhosis; 30% of cases occur within the first month after the infectious process and 63% of patients die within 1 year. The Odds Ratio (OR) for dying in infected vs. non-infected patients is 3.75 (Confidence Interval [CI], 95% 2.12-4.23) [17]. Hospitalized patients with cirrhosis entertain a greater risk of developing infection, especially those with gastrointestinal (GI) bleeding. A 2001 study identified 34% (n = 150) bacterial infections in patients admitted with cirrhosis (89 were community-acquired and 61 were hospital-acquired), the urinary tract was most frequent site of infection (41%), 60% of cases at the time of admission already had an infectious process, and 40% occurred during hospitalization, the prevalence of bacterial peritonitis was 12%; however, it is noteworthy that 51% of bacterial peritonitis was found in asymptomatic patients; the main bacteria found were Gram-negative [18].
Since 1996, the effectiveness of antibiotic treatment was evaluated in the prevention of bacterial infections after hemorrhage in patients with cirrhosis; three groups were assigned: Group 1 included patients with Child-Pugh A or B without bleeding (without antibiotic prophylaxis); Group 2 were patients with Child-Pugh A or B and bleeding event as well as patients with Child-Pugh C with hemorrhage without antibiotic prophylaxis, and Group 3 included patients with Child-Pugh C with hemorrhage and antibiotic prophylaxis during 10 days. The incidence of bacterial infection was significantly higher in patients in group 2 than in those of group 1 (p 0.001), infections were more severe in group 2; however, when comparing mortality between groups 2 (23.5%) and 3 (13.3%) during the first 4 weeks, there was no significant difference [19]. 3916-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament
In the PREDICT study, a total 1,071 patients with AD were included; three groups of patients were identified, with the differential count of white globules and the PCR levels (as markers of inflammation); patients with pre-ACLF (n = 218) and who developed ACLF during the first hospitalization (20%) had a 3-month and 1 year mortality rate of 53.7% and 67.4%, respectively. Patients with unstable decompensated cirrhosis (n= 233) and who required >1 readmissions but without developing ACLF had lower mortality rates at 3 months (21%) and at 1 year (35.6%). Patients with stable decompensated cirrhosis (n = 620) who were not readmitted and who did not develop ACLF had a 1-year mortality rate of 9.5% [20]. 3916-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament
- High-grade Systemic Inflammation
Bacterial infections are the most common trigger of ACLF. [7] Chronic consumption of alcohol alters the intestinal barrier, contributing to dysbiosis, altering the tight junctions, reducing the mucous layer, and diminishing the production of antimicrobial peptides; in the context of ACLF, there is an increase of bacterial translocation. Sterile mechanisms triggered by the release of DAMPs by apoptotic hepatocytes are also implicated in alcohol-induced liver injury. Specifically, ethanol contributes to the release of the mitochondrial cytochrome c, which increases the expression of the Fas ligand, thus promoting hepatocyte apoptosis [21]. 8,16-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament
Immunological Role of the Resident Cells in the Liver
Within the liver, antigen-presenting cells (APC) (such as Kupffer cells, dendritic cells, and sinusoidal endothelial cells), T cells, B cells, natural killer (NK) cells, and monocytes control the local and systemic inflammatory response. Parenchymal cells, such as hepatocytes, play a role in surveillance, acting as APC by expressing major histocompatibility complex (MHC) type I and II, and costimulatory molecules [22]. 3916-18 fueron significativamenta mortalidad.l uso profil funcinttorios; encontrando que la IL6 e IL-18 fueron significativament In a sterile injury, the release of DAMPs from necrotic cells triggers recruitment of innate immune cells. The critical balance between immune activation and tolerance is altered in cirrhosis, which is correlated with dysregulation and altered synthesis of key immune proteins [23].
Consequences of Systemic Inflammation
Proinflammatory cytokines stimulate, to an even greater degree, the endothelial production of nitric oxide (NO) and reactive oxygen species (ROS), worsening peripheral vasodilatation, portal hypertension, and cardiac dysfunction; the magnitude of the systolic dysfunction is related not only with the severity of the systemic circulatory dysfunction, but also with the markers of systemic inflammation and bacterial translocation [24]. It has been observed that patients with a stable disease course present low-grade inflammation, which improves later, while those with an unstable disease course and who require hospital readmission present persistent low-grade inflammation, with severe portal hypertension and 1-year mortality of 9.1% and 35%, respectively; patients with pre-ACLF exhibit a higher degree of inflammation upon hospital admission, which confers on them a mortality of 67.4% [3]. The hypothesis has been proposed that high-grade SI leads to organ failure by causing direct tissue damage (immunopathology) or as the result of an energy imbalance (immunometabolism). SI promotes ischemic acute renal tubular necrosis, caused by capillary leukocytes infiltration, microthrombosis, apoptosis, and mitochondrial injury, which substantially increases the risk of acute renal insufficiency in ACLF. The term “immunometabolism” refers to the energy imbalance that a metabolically active immune system causes in cells of other tissues, which contributes to organ dysfunction [25].
The degree of inflammation parallels both the presence of hepatic encephalopathy and the severity of hepatic, circulatory, and renal dysfunction. It has been studied and established that infections, particularly pneumonia and sepsis without a specific focus, increase the mortality of patients with cirrhosis and hepatic encephalopathy [7, 8, 26].
Immunodeficiency and Cirrhosis
The presence of abnormalities of immune system cells compromise their effector function and cause immune paralysis, including functional defects, the expansion of immune inhibitors, or reduced expression of costimulatory molecules [27]. Immunodeficiency is a dynamic characteristic of CAID that begins in compensated cirrhosis, increases with the progression of cirrhosis through the decompensated stage, and that reaches its maximal point in ACLF. Immunodeficiency in cirrhosis is the consequence of two main factors: structural distortion of the hepatic parenchyma, which compromises its surveillance function, and functional deterioration of circulating immune cells as an inadequate immune response to the triggering event [28].
Progressive Loss of Tolerance in Cirrhosis
Under physiological conditions, the “proinflammatory” hepatic environment (due to persistent exposure of low levels of gut-derived antigen) is altered by tolerance mechanisms that aim to decrease the capacity of APC to activate lymphocytes, a progressive deterioration of tolerance to antigen recognition in cirrhosis, in this manner conditioning the augmented proinflammatory response and that contributing to chronic inflammation. Monocytes from patients with cirrhosis exhibit hyperproduction of TNF after stimulation with LPS, due to the alteration in the IL-1R response (inducible suppressor of TNF); in the same manner, a defective production has been observed of the anti-inflammatory cytokine IL-10 by the Kupffer cells; in advanced cirrhosis, limited production of proinflammatory cytokines through stimulation with LPS [29].
Increased Release of DAMPs from Injured Hepatocytes
Chronic liver disease, of any etiology, progressively induces damage and death of hepatocytes. It has been proposed that the subsequent release of intracellular DAMPs contributes to sterile inflammation [30]. Alcohol consumption promotes inflammation and exerts a harmful effect on immune cells, alcohol alters both innate and adaptive responses; similarly, dietary factors (fructose, saturated fats, trans-fats, or cholesterol) trigger inflammation by means of lipotoxic effects, mitochondrial dysfunction, oxidative and endoplasmic reticulum stress, and sterile cellular death mechanisms [31].
Structural Distortion in Cirrhosis
The progression of cirrhosis alters the architecture of the liver, replacing the parenchymal and non-parenchymal cells with scar tissue, deposition of extracellular matrix, and sinusoidal capillarization renders difficult the surveillance function of the resident APC; the activity of the mononuclear phagocytic system is decreased in patients with cirrhosis, which is associated with a higher risk of bacterial infections and mortality. Furthermore, intrahepatic shunts through vascularized septa prevent systemic and portal bacteria from being filtered and eliminated by Kupffer cells [32]. Associated with this, the loss of hepatocytes diminishes the synthesis of proteins and immune receptors (complement components, soluble PRRs such as LPS binding protein and soluble CD14), albumin and acute phase protein. Serum concentrations of C3 and C4, as well as the hemolytic activity of complement are reduced in patients with decompensated cirrhosis compared to controls; these alterations predispose to infectious processes and confer higher mortality in patients with alcoholic cirrhosis [34].
Damage to the Circulating Immune Cells
CAID implies not only local but also systemic alterations that affect circulating innate and adaptive immune cells. Damage to circulating immune cells is evidenced by hyperactivation and up regulation of cellular activation and a dysfunctional effector response.

Table A1.
Damage to the immune cells in cirrhosis [3].
| Immune cells | Decompensated cirrhosis | ACLF |
| Monocytes | ⇑ HLA-DR expression ⇑ TNF production ⇑ CD14+CD16+ ⇓ capacity of the APC Altered phagocytes Altered chemiotaxis Production of OH+ Defective production of Fc |
⇓ HLA-DR ⇑ producción IL-10 ⇓ producción TNF Altered phagocytosis ⇑MERKT inhibits the activity of TLR and the proinflammatory cytokines inhibit the activity of NF-kB |
| Neutrophils | Altered phagocytosis Reduced chemiotaxis Re⇑ ROS |
⇑Expression of CXCR1 and CXCR2 that favor apoptosis and necrosis |
| Lymphocytes |
TCD4+ ⇓cytolytic activity of the NK cells |
Immunodeficiency is exemplified by alterations in APCs (i.e., monocytes and neutrophils) and reduced phagocytic capacity; however, it is more pronounced in patients with greater deterioration of liver function. Immunodeficiency in ACLF is characterized by decreased HLA-DR expression and impaired TNF production in response to LPS by monocytes, seriously altered phagocytic and oxidative capacity (by monocytes and neutrophils), elevated levels of MERKT+ monocytes, and by monocytes that produce anti-inflammatory cytokines (IL-10) [35].
Compensatory Anti-Inflammatory Response Syndrome (CARS)
Compensatory anti-inflammatory response syndrome (CARS) is characterized by an increase in lymphocyte apoptosis and anergy, a diminution in cytokine production and HLA expression following monocyte stimulation, and positive regulation of anti-inflammatory cytokines; it was originally described in critically ill patients with sepsis and trauma [36]. The magnitude of this compensatory response predicts a poor result in patients with ACLF, as was observed in a cohort of 51 patients with a diagnosis of ACLF and decompensated cirrhosis where the prognostic value of IL-10 levels on admission and its evolution during the early phase of treatment in the intensive care unit was investigated, in comparison with proinflammatory cytokines (IL-6 and TNF-α). It was found that it is not only the degree of SI, as reflected by the levels of IL-6 and TNF-α, that determines the outcome, but also the magnitude of the anti-inflammatory response. In patients with ACLF, IL-10 levels are higher and correlated with poor prognosis (p <0.001). In addition to IL-10, another finding is that together with the International Normalized Ratio (INR) (OR 1.7), at the moment of admission, they predict unfavorable clinical outcomes [37]. IL-10 constitutes an important part of CARS, which is closely related with the functional deactivation of monocytes, immunoparesis and predisposition to recurrent and opportunistic infections [38].
Dysfunction of Immune Cells Due to Metabolic Anomalies of Cirrhosis
In healthy individuals, albumin can bind in serum to proinflammatory molecules as well as to immunosuppressive mediators such as prostaglandin E2 (PGE), whose serum concentrations are markedly elevated; in patients with cirrhosis, in addition to presenting low albumin concentrations, they present alterations in their immune function [39]. Low serum albumin levels (<30 mg/dl) are the main cause of immunosuppression in acute decompensation and end-stage liver disease, they present elevated concentrations of circulating PGE2 secondary to increased production which in combination with hypoalbuminemia, drives innate immune dysfunction, resulting in vulnerability to infection. Human albumin infusion reduces circulating levels of PGE2 and attenuates suppressed secretion of proinflammatory cytokines from macrophages [40].
In a prospective multicenter study that included a total of 13,796 patients from 1,265 intensive care units (ICU), 410 of them patients had a diagnosis of cirrhosis; the hospital mortality rate due to an infectious process was 42%, compared to 24% in patients without cirrhosis with an infectious process (p <0.001) [41].
Intestinal—Liver Axis and the Intestinal Immune System
The gut—liver axis plays an important role in the pathogenesis of CAID. The intestinal and vascular epithelial barrier, the intestinal microbiota, the liver, and the immune system establish complex interactions in order to maintain tolerance to inoffensive stimuli and at the same time orchestrate an effective response against bacterial pathogens. However, cirrhosis is accompanied by harmful changes in the intestine, including dysbiosis and increased intestinal permeability, these alterations promote bacterial translocation from the intestinal lumen to the systemic and portal circulation, which notably contributes to systemic inflammation. The portal vein transports antigens derived from the intestine to the liver, which in turn supplies bile acids, lipids, and antibodies through the bile returning to the intestine; this bidirectional communication is profoundly altered in cirrhosis due to damage to intestinal, epithelial, vascular, and immune intestinal barriers at different levels [3]. Gut-associated lymphoid tissue (GALT) represents the largest immune organ in the human body and constitutes the first defense against antigens and pathogens derived from the intestine. GALT is composed of Peyer’s patches, intestinal lymphoid follicles, intraepithelial lymphocytes, and mesenteric lymph nodes [42].
In particular, the intestinal macrophages are effective in killing translocated bacteria, while intestinal dendritic cells transport them alive to mesenteric lymph nodes for antigen presentation and modulation of the adaptive T-cell response (that is, the induction of tolerance or preparation). The GALT houses about 94% of CD4 T cells; intraepithelial lymphocytes, particularly the γδ subpopulation maintain a close relationship with intestinal epithelial cells, acting as essential mediators that balance host microbial homeostasis [4,43]. (Figure A2)
Patients with cirrhosis exhibit slower intestinal motility, which leads to an overgrowth of intestinal bacteria; this, together with the portosystemic shunt, permits the perpetuation of the bacteria and can condition cause bacteremia; this in turn causes greater oxidative damage due to the increase in endotoxins, proinflammatory cytokines, and Nitric Oxide (NO); it alters the structure and capacity of intestinal permeability [44]. Toxins derived from bacteria, such as peptidoglycans from Gram-positive bacteria or LPS from Gram-negative bacteria, bind to Toll receptors, which initiate the cellular signaling cascade and release of cytokines such as TNF-α, IL-6, and IL-1. In systemic inflammatory response syndrome (SIRS), anti-inflammatory cytokines (IL-10, IL-4, IL-13, and PGE2) balance proinflammatory cytokines, a process known as the “cytokine storm”, leading to excessive inflammation [45]. The severity of the liver disease determines the development of SIRS, and this in turn favors the presence of variceal hemorrhage, hepatic encephalopathy, and negatively affects survival [46].
In experimental models of cirrhosis, intestinal CD103+ dendritic cells are activated during liver decompensation (ascites), revealed by the expansion of the proinflammatory CD4+ dendritic cells subpopulation, the increase in the production of TNF, and the increase in phagocytosis and migration capacity. As cirrhosis progresses to the ascitic state, levels of intraepithelial and lamina propria lymphocytes (T helper cells, cytotoxic T cells, regulatory T cells, B cells, and NK cells) increase, T helper cells switch to a TH1 regulatory pattern with increased production of TNF and IFNγ in the lamina propria, with concomitant TH17 depletion. Interestingly, decontamination of the intestine leads to a redistribution of microbiota composition, a diminution in proinflammatory activation of mucosal immune cells, and a reduction in intestinal permeability and bacterial translocation, supporting the relevant role of the dysbiosis in intestinal inflammation in cirrhosis [47].
Macrophages show caspase1 activation and a marked increase in IL-1γ and IL-18 expression compared to circulating monocytes, suggesting inflammasome activation in ascites, facilitating an exacerbated inflammatory response, even in the absence of a primary signal [48]. Spontaneous bacterial peritonitis (SBP) is characterized by the disparity between the low microbial load in ascites and the intensity of the inflammatory response in the peritoneum, this latter exhibiting correlation with SI and the clinical outcomes, such as kidney failure. During SBP, CD206 levels in ascites, but not in serum, correlate with SI, peritoneal inflammation, and mortality; the massive release of proinflammatory cytokines during bacterial inflection contributes to systemic inflammation and organ failure [50].
Decontamination with non-absorbable antibiotics decreases vascular NO production, reduces inflammation and hemodynamic alterations in animal models and in humans with cirrhosis [51]. It has been observed that intestinal decontamination with antibiotics reduces endotoxemia and the severity of liver disease [3], due to that intestinal decontamination with antibiotics reestablishes immune surveillance in experimental cirrhosis. The intestinal microbiome can deplete the mucosal immune response capacity, the activation and function of intestinal dendritic cells in rats with cirrhosis and ascites, as well as its relationship with the bacterial translocation. Intestinal bacteria condition a different phenotypic and functional profile of the dendritic cells to produce effects that range from their activation and improvement of functions to exhaustion and tolerance [47].
Humoral Factors in Circulation
Endotoxins, prostaglandins, catecholamines, and products of cell death may contribute to CAID [40]. Norepinephrine has been demonstrated directly influence the microbiome, exerts detrimental effects on enterocytes, increases intestinal permeability, and contributes to the initiation and perpetuation of intestinal inflammation, observed in animal models as well as in humans models. Norepinephrine levels are known to be three times higher in ACLF than in acutely decompensated patients and strongly correlate with the severity of the systemic inflammatory response [52].
T helper cells play a crucial role in the induction and regulation of the adaptive immune response. Th cell lymphopenia is a common finding in patients with cirrhosis, generally attributed to splenic sequestration, as well as a reduction in the naïve Th cell compartment [53]. Lario et al. evaluated the number and distribution of circulating lymphocytes; compared with controls, patients with severe lymphopenia (p <0.001) with T-cell depletion (p <0.001) in peripheral blood showed a drastic reduction in the number of helper and cytotoxic T cells (p <0.001). The number of naive T cells was reduced 2.7-fold, while the number of memory Th cells was reduced 1.5-fold in patients with cirrhosis. The lymphopenic state of cirrhosis was associated with a redistribution of the peripheral Th cell compartment, characterized by a reduction in naïve Th cells, and an increase in memory Th cells, increased apoptosis and accumulation of splanchic Th-cells. The fundamental role that Th cells play in the regulating the immune response highlights the relevance of Th cells immunodeficiency in cirrhosis. Results reveal a severe reduction in the number of naïve CD31* Th cells in cirrhosis [54].
Elevated levels of TNF-α have been implicated in endothelial activation and hemodynamic disorders observed in cirrhosis [55]. LPS increase plasma levels of LBP, the main plasma protein responsible for transporting LPS to effector immune cells; decontamination of the intestine with norfloxacin diminishes enteric bacterial load, and therefore the risk of bacterial infections, diminishes circulating levels of TNF-α and LBP, and reduces hemodynamic abnormalities in patients with advanced cirrhosis. Not enough attention has been paid to alterations in immune effector cells responsible for modifying the humoral abnormalities and compromised immune function found in cirrhosis, as little is known about the immune cell source of TNF-α production in liver cirrhosis [56].
A prospective study was conducted that included 60 patients with alcoholic cirrhosis and a control population of 25 healthy subjects; the patients were aged between 25 and 70 years, with a diagnosis of cirrhosis, and at least 1 years of abstinence from alcohol; ascites were detected in 28 of 60 patients (46%). Serum LBP levels were above the threshold level of healthy controls in 11 of the 28 patients with ascites; the total number of monocytes is significantly increased in the peripheral blood of patients with ascites compared to that of patients without ascites and in healthy controls; the levels of LPS, LBP, sCD14, TNF-α, sTNFα-RI, and IL-6 were significantly increased in patients with ascites with normal LBP, patients without ascites and healthy controls; it was evident that the behavior of T cells is depleted, and that enteric bacteria play a relevant role in these abnormalities of cellular immunity [57].
LPS lead to the activation of the signaling pathways that induce an increase expression of the MHC class II and the costimulator molecule CD80 on the surface of monocytes, as well as an increase in serum LBP and CD14. The subtype of patients with cirrhosis and ascites revealed a higher number of monocytes and a higher expression of HLA-DR on the cell surface and, in particular, of the CD80 and sCD14 molecules, a pattern consistent with LPS signaling, as well as with the ability to modulate Th cells function [58].
Patients with acute decompensated liver cirrhosis have a reduced expression of HLA-DR antigen-presenting molecules on monocytes; this can also result in a decreased monocytes activation and cytokine secretion. In addition to dysfunction of the RE system, patients with cirrhosis demonstrate a diminution of neutrophil mobilization and phagocytic activity, a phenomenon that correlates with the severity of the liver disease [59]. Patients with cirrhosis have much lower levels of the immunoglobins IgM, IgG, and IgA in ascites, and the concentrations of C3, C4 and CH50 are significantly lower in serum as well as in ascites [60].
Most bacteria that cause serious infections must be phagocyted and eliminated by phagocytic cells; this process requires that the surface of the bacterial cell is first opsonized with IgG and/or with the third component of this process that requires complement (C3), with the fixation of the complement to the surface of the bacteria being the most important step in opsonization, and complement deficiencies are known to predispose to bacterial infections. Bruce et al. determined how opsonization activity in ascites correlates with the content of total protein (CH100) and complement (C3 and C4) in ascites; CH100 was measured in both ascites as well as in serum, observing that ascites caused by cirrhosis presents less opsonic activity than in ascites of other etiology [61].
Patients with cirrhosis have high levels of circulating endotoxins that inversely correlated with the liver failure (p <0.05); plasma levels of endotoxins and serum bilirubin are important factors for predicting short-term survival [62]. Protein C, high-density lipoproteins, and anti-inflammatory and anti-apoptotic factors are reduced in patients with cirrhosis; NO is increased in cirrhosis and it is known to contribute to oxidative stress and worsening vasodilatation in sepsis. Due to that endotoxemia improves the expression of inducible nitric oxide synthase (iNOS), the results suggest that circulating endotoxin in cirrhosis is responsible for the excessive synthesis and release of NO in the vasculature [63].
Infections in Cirrhosis
Gastrointestinal bleeding is associated with a higher incidence of infection, with approximately 17% to 45% of cases lead to an episode of SBP or bacteremia. The presence of infection increases the risk of early bleeding; therefore, the preferred prophylactic strategies are third-generation cephalosporins, both for Gram-negative and Gram-positive bacteria. In patients with advanced cirrhosis, it was found that 1 g of intravenous ceftriaxone during 7 days after bleeding was found to be effective in preventing bacterial infections than oral norfloxacin. Prokinetic agents can reduce dysmotility and bacterial translocation, prophylaxis with norfloxacin and cisapride significantly reduces the rate of SBP compared with norfloxacin alone [65].
The prevalence of SBP in hospitalized patients with cirrhosis and ascites ranges from 10% to 30%, approximately half of the cases occurring at the time of hospitalization and the other half of cases occurring during hospitalization [66]. The hospital mortality rate due to PBE is approximately 32%; renal failure develops in approximately one third of all patients with PBE and is a strong predictor of mortality during hospitalization [67]. The activation of the cytokine cascade and the production of NO in cirrhosis and SBP negatively impacts kidney function; therefore, the use of intravenous albumin (1.5 g/kg within 6 h of SBP followed by 1 g/kg on day 3) in conjunction with cefotaxime reduces the incidence of renal failure from 33% to 10% and the incidence of mortality from 29% to 10%. Albumin increases the mean arterial volume and binds to TNF-α and NO to counteract the inflammatory response due to the infectious process, eliminating toxins from the circulation [68].
In those patients without antibiotic prophylaxis, the SBP recurrence rate is 43% at 6 months, 69% at 1 year, and 74% at 2 years after the initial episode. Ginés et al. determined that 400 mg of norfloxacin orally per day reduces the recurrence of SBP from 68% to 20% [69].
It has been shown that in-hospital mortality is similar between patients with severe sepsis (32%) and ACLF (30%). The determination of CRP and procalcitonin (PCT) is higher in the sepsis group compared with patients with ACLF; the serum concentrations of IL-6 and IL-10 are considerably higher in patients with severe sepsis and different from those in subjects with ACLF or with stable cirrhosis. By contrast, CRP, PCT, and the average values of IL-6 and IL-10 are significantly higher in patients with ACLF than in patients with non-compensated cirrhosis [27].
As previously described, SI occurs in a setting attributable to the translocation of proinflammatory signals from the intestinal lumen to the systemic circulation and/or the release of DAMPs, that trigger proinflammatory mediators [70,71]. The direct deleterious effects of these proinflammatory mediators on organ microcirculation and homeostasis of cellular physiology can lead to organ failure [8]. At any stage of cirrhosis, patients can develop AD, but patients will only develop ACLF when systemic inflammation is widely activated [72].
Half of the CANONIC cohort developed ACLF without a precipitating event, which highlights that the triggers of ACLF are not completely known or that in some cases, the SI may be so high that it favors the development of ACLF without an external triggering. SI is a hallmark of ACLF; however, the dynamics of SI has not been described after successful AD treatment (that is, in re-compensation), and its role in the development of ACLF remains unclear [7, 73]. Inflammasome activation is a hallmark of the inflammatory response of the innate immune system leading to the release of IL-1α and IL-1β [74].
Monteiro et al. evaluated inflammasome activation, estimated by the proinflammatory cytokines IL-1α and IL-1β of compensated and recompensated patients and its role in the development of fatal ACLF. The hypothesis of this study was that SI is a prerequisite and necessary for the development of ACLF in patients with compensated and recompensated state. In total, 88 (35%) patients died, the most common cause of death was ACLF in 52 (58.5%) patients; in 63.5% of cases the trigger of ACLF was not identified. Compared with compensated patients, recompensated patients had significantly higher levels of MELD and CLIF-C AD (Hazard Ratio [HR] 1.11, p = 0.052), and lower levels of hemoglobin and albumin, while the recompensated patients had significantly higher rates of fatal ACLF and overall mortality compared to those with compensated patients. IL-1β levels were significantly higher and more frequently detectable in patients with ACLF development compared to those without development of ACLF, (69% vs 34%, p <0.05); patients with ACLF demonstrated significantly higher rates of IL-10 (60% vs. 33%) and IL-1β (16% vs. 8%) compared to patients without ACLF [75]. Evaluation of cytokine profiles in compensated and recompensated patients with cirrhosis could help identify patients at risk of developing fatal ACLF to a greater extent than MELD and CLIF-C AD. In some patients, plasma IL-6 levels show an increasing profile, with extremely high peaks, whose duration ranges between days and weeks; it is possible that these peaks are related with transitory episodes of massive PAMPs translocation [2].
In the observations carried out by Rolando et al., where 887 patients admitted to the hospital due to acute hepatic failure were included, the SI response was evaluated, which was present in 56% of the cases, regardless of whether the patients had or not bacterial infections. The prevalence of infections in patients with gastrointestinal hemorrhage is less than 2%. There is evidence that SI is involved in the development of ascites and renal failure; in fact, ascites and bacterial infections coincide in 30% of hospitalized patients with AD. Finally, as observed in sepsis, SI related with bacterial infections in decompensated cirrhosis may deteriorate in terms of liver failure to an even greater degree, may impair left ventricle contractility, and may reduce vascular resistance of the splenic and systemic systems. There are few clinical studies on the potential role of SI in gastrointestinal bleeding in cirrhosis; on the other hand, there are many experimental studies that indicate that SI can increase portal hypertension, and its severity correlates with the severity of portal hypertension in patients with cirrhosis. SI activates Toll-like receptors on hepatic stellate cells (HSC), making them sensitive to increased circulating levels or local release of vasoconstrictors (endothelin, norepinephrine, angiotensin II, leukotrienes, and thromboxane A2). The Kupffer cells are activated in the context of SI, which increases the production of proinflammatory cytokines and ROS; therefore, SI can induce an imbalance between vasoconstrictor and vasodilator mechanisms within the liver, leading to an increased vascular resistance [76].
SI Is the Common Mechanism for Major Complications and Organ Failure in AD
The CANONIC and PREDICT studies aimed to characterize ACLF syndrome on hospital admission (CANONIC study) and explore the critical periods before and within 3 months after admission (early follow-up period) in AD patients without ACLF (PREDICT study). Mortality rates at 3 months and one year after admission increased progressively and in parallel with the severity of AD. [8,9]
“Sterile” inflammation can derive from acute hepatic inflammatory processes; the spread of activated immune cells within the liver and DAMPs released by hepatocytes cause the systemic inflammatory response [77]. Preactivation of the innate immune system has been observed to be induced by an exaggerated inflammatory response to bacterial infections and other proinflammatory stimuli; in the same manner, it has been determined that the inflammasome is highly active in ascites in patients with cirrhosis without the presence of infection, explaining the exacerbated inflammatory response in the inflammasomes and that is associated with a higher degree of liver disease [78].
IL-6 is a sensitive marker of SI, the plasma level of IL-6 was normal on admission in only 40 (3.3%) patients among the 1,211 patients with AD included in the analysis; 37 of these patients showed elevated plasma levels of >2 of other SI markers (TNF-α, IL-8, IL-10, IL-1RA, and CPR). Of the 97 patients with compensated cirrhosis (patients without history of AD) included in the analysis, 48 (49.4%) showed normal plasma levels of IL-6 and IL-24 (24.7%). The SI correlates with the number of decompensations upon hospital admission, which is also a prognostic marker in AD [79, 80].
Patients with any precipitating factor represent 44% of patients with ACLF without AD; and 70% of ACLF patients with AD. Bacterial translocation is probably the precipitant of SI and AD in more than half of patients with ACLF-non-AD and in 30% of patients with ACLF-AD. The number, but not the type, of precipitants influences the severity of SI [81]. The clinical course of the disease is closely correlated with the evolution of SI [2].
Traditionally, it has been thought that SI causes dysfunction and organ failure through two different mechanisms that are not mutually exclusive. First, SI, by stimulating NO production in splanchnic arterioles, may accentuate preexisting systemic circulatory dysfunction, which results in increased inflammation, decreased effective arterial blood volume, and the overactivation of endogenous vasoconstrictor systems, resulting in organ hypoperfusion and subsequent impairment of organ function. In second place, SI may be associated with an activation of immune cells resulting in tissue damage and impairment of organ function [8]. A third mechanism has been suggested involving metabolic alterations associated with SI, which also plays a role in the development of organ dysfunction and failure in patients with cirrhosis [82].
Immunoparesis
Immunoparesis, as a mechanism of primary infection or the development of secondary infections, was described for the first time in patients with sepsis. [83] In the study by J. Fernández et al., 407 patients with ACLF were included, as well as 235 patients with AD; 152 patients (37%) had bacterial infection at the diagnosis of ACLF; 117 patients (46%) of the remaining 255 patients with ACLF developed bacterial infection during the 4-week follow-up. Serious infections (SBP, pneumonia, severe sepsis/shock, nosocomial infections) were more prevalent in patients with ACLF. Patients with ACLF and bacterial infections (either at diagnosis or during follow-up) showed a higher degree of SI at diagnosis, worse prognosis, and decreased 90-day survival compared to patients with ACLF without infection. Fungal infections developed in 9 patients with ACLF (2%) and in none with AD, they occurred mainly after the diagnosis of ACLF (78%), conferring a high mortality at 90 days (71%) [84]. The corresponding incidence of infections in the PREDICT study in patients with non-ACLF AD was 53% [81].
In patients with AD, the subset of circulating monocytes present characteristics of immune incompetence, these cells are present in patients with AD non-ACLF, and their number increases in patients with AD-ACLF. [85] In patients with decompensated cirrhosis, circulating neutrophils have differences in migration, bacteria recognition, and bacteria killing activities, including phagocytosis, phagocyte degranulation, as well as rapid production of large amounts of ROS [86].
Liver Failure and Acute Kidney Injury (AKI) in AD
In a retrospective study of four cohorts of patients treated with terlipressin and albumin in patients with cirrhosis and hepato-renal syndrome (HRS) type 1 as well as different degrees of ACFL, it was observed that the renal response to treatment depends largely on the degree of ACLF. Resolution of HRS was obtained in 60% of patients with ACLF-1, in 48% of patients with ACLF-2, and in only 29% of patients with ACLF-3; which could be predominantly related with renal inflammation (p <0.001 when comparing the three grades) [87].
Albumin Treatment Negatively Regulates Systemic Inflammation in Decompensated Patients with Cirrhosis
A recent study in patients with AD suggests that these beneficial effects are related to the negative regulation of SI, since the administration of albumin, both short and long term (12 weeks), in high doses, significantly reduces plasma levels of CPR and cytokines. A prospective study was carried out in which the long-term effect (12 weeks) of treatment at a low dose (1 g/kg of body weight every 2 weeks) and high-dose treatment (1.5 g/kg every week) of albumin on serum albumin levels, plasma renin, circulatory function, portal pressure, and plasma cytokine levels; the data were collected from 18 patients without bacterial infection but with decompensated cirrhosis (Pilot—PRECIOSA Study). Similarly, the effect of short-term treatment (1 week) with antibiotics alone vs. the combination of serum albumin plus antibiotics (1.5 g/kg on day 1 and 1 g/kg on day 3) on cytokine levels was evaluated in 78 patients with bacterial infections in a randomized controlled study (INFECIR-2). It was found that high dose of albumin for 12 weeks, but not the low dose of albumin, was associated with normalization of the serum albumin levels; improved the circulatory and left ventricular function, and reduced plasma cytokine levels (IL-6, granulocyte-stimulating factor, IL-1RA, and endothelial growth factor) without significant changes in portal pressure. The immunomodulatory effect of albumin was demonstrated in both studies (PRECIOSA and INFECIR-2) [88].
Sarcopenia in Cirrhosis
Sarcopenia is the progressive, widespread loss of skeletal muscle, mass, strength and function seen in up to 70% of patients with cirrhosis. Sarcopenia is relevant, because it leads to physical disability and functional impairment, low quality of life, poor prognosis, and increased mortality before and after liver transplantation [89,90]. Sarcopenia is more prevalent in patients with cirrhosis with low left ventricle mass [91]. Myokines are released by skeletal muscle in response to a contraction or degradation and they are relevant because they have multiple beneficial physiological functions, these include immunoregulation, they facilitate energy metabolism and the remodeling of cardiac architecture, in turn playing a role in improving insulin resistance and reducing systemic inflammation; due to all these functions, sarcopenia has a systemic impact. (Figure A3) Myostatin is a myokine responsible for inhibiting protein synthesis and regeneration; sarcopenia is the result of an imbalance between protein synthesis and catabolism [92].
Hyperammonemia plays a critical role in the development of sarcopenia, regulates the rise of myostatin levels and activates autophagia; elevated ammonia levels lead to impairment of the mTOR pathway through an increase mitochondrial dysfunction and ROS production; similarly, hyperammonemia affects muscle contractility and increases fatigue, this contributes to muscle dysfunction [93].
Physical disability and functional impairment, a result of sarcopenia, favors a sedentary lifestyle; this change in lifestyle plus reduction of energy expenditure can contribute to increase adipose tissue and obesity. Patients with cirrhosis can develop simultaneous loss of skeletal muscle and gain of adipose tissue, leading to sarcopenic obesity; this increase in adipose tissue observed in sarcopenia activates systemic inflammation [94]. (Figure A4)
Sarcopenia is associated with an increased risk of decompensation; it is associated with a 5-fold increased risk of mortality, regardless of MELD sodium score; it is also associated with a 2-fold increased risk of cancer, a higher risk of infection, and a higher mortality at 12 months after liver transplantation [95].
Cytokines and Their Correlation with Hepatic Encephalopathy (HE)
Hepatic Encephalopathy (HE) is a neuropsychiatric syndrome present in patients with chronic and acute liver disease that is associated with short survival in cirrhotic patients; therefore, cirrhotic patients who develop a first episode of acute hepatic encephalopathy should be considered potential candidates for liver transplantation, because the syndrome is associated with a poor prognosis [96, 97]. Minimal HE (MHE) is the first stage in the HE spectrum; inflammation has been proposed to plays a major role in cognitive impairment and to exacerbates the neuropsychological alterations induced by hyperammonemia [98].
Li W et al. studied 289 patients with liver cirrhosis of viral etiology; 3 groups were made: patients without HE (n =156); with MHE (n = 98), and with clinical HE (n = 213), in addition to including 88 healthy volunteers; the serum levels of IL-6, IFN-γ, and IL-17a were correlated with the presence of MHE (p <0.001) [99].
Shawcross et al. studied the role of ammonium and inflammation in MHE; 84 patients with cirrhosis were included, neuropsychological tests and serum determination of ammonia, leukocytes, CPR, nitrate/nitrite, IL-6, and amino acids, were performed before and after the induction of hyperammonemia through the administration of a solution that mimics the amino acid composition of hemoglobin (60) or placebo (24). It was observed that the presence and severity of MHE were independent of the severity of liver disease and ammonia concentration, but markers of inflammation were significantly higher in patients with MHE (IL-6: 36.83 ± 7.57 pg/mL; CPR: 35.6 ± 7.45, and leukocytes: 11.36 ± 137) vs. without MHE (IL-6: 20.1 ± 1.67 pg/mL, PCR: 16.94 ± 1.62, and leukocytes: 7.29 ± 0.42) [100]. Similarly, Montoliu C et al. studied in 55 patients with liver cirrhosis and 26 controls, the performance of the HE psychometric score and the critical blinking frequency, ammonia and IL-6 and IL-18 as inflammatory markers, these authors finding that IL-6 and IL-18 were significantly higher (2.5 times and 2.2 times, respectively) in patients with MHE; therefore, serum concentrations of IL-6 and IL-18 are considered to be useful to be useful for discriminating cirrhotic patients with and without MHE [100].
2. Conclusions
Alterations of the immune system are frequent findings in liver cirrhosis, from its initial stages to the development of ACLF; patients with cirrhosis develop an immune dysfunction favored both by the presence and persistence of systemic inflammation and by an immunodeficiency conditioned by alterations in their surveillance system against pathogens. Bacterial translocation is a determinant that favors the persistence of the inflammatory state and is a characteristic disorder in this condition; Likewise, it has been observed that the levels of certain cytokines play an important role in the development of hepatic encephalopathy. To date, we do not know if there is a characteristic inflammatory profile in the group of patients who frequently present clinical decompensation versus those patients who present only one decompensation during the course of their disease, or what causes one patient to decompensate and another not. Since, as mentioned, there are patients who develop decompensation or ACLF without a recognized triggering factor. We are currently carrying out a protocol with the intention of answering these questions.
Author Contributions
E.V.R.-N. and J.A.M.-G. conceived and designed the study; E.V.R.-N., K.S.-R., C.F.F.-F, K.P.-R. edited and wrote some portions of the paper, compiled the references, and analyzed the data; J.A.M.-G. and E.V.R.-N. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ren A, He W, Rao J, Ye D, Cheng P, Jian Q, Fu Z, Zhang X, Deng R, Gao Y, Ma Y. Dysregulation of innate cell types in the hepatic immune microenvironment of alcoholic liver cirrhosis. Front Immunol. 2023; 14: 1034356. [CrossRef]
- Albillos A, Martin-Mateos R, Van der Merwe S, Wiest R, Jalan R, Álvarez-Mon M. Cirrhosis-associated immune dysfunction. Nat Rev Gastroenterol Hepatol. 2022; 19(2): 112-134. [CrossRef]
- Bonnel AR, Bunchorntavakul C, Reddy KR. Immune dysfunction and infections in patients with cirrhosis. Clin Gastroenterol Hepatol. 2011; 9(9): 727-38. [CrossRef]
- Arvaniti V, D'Amico G, Fede G, Manousou P, Tsochatzis E, Pleguezuelo M, Burroughs AK. Infections in patients with cirrhosis increase mortality four-fold and should be used in determining prognosis. Gastroenterology. 2010; 139(4): 1246-56. [CrossRef]
- Borzio M, Salerno F, Piantoni L, Cazzaniga M, Angeli P, Bissoli F, Boccia S, Colloredo-Mels G, Corigliano P, Fornaciari G, Marenco G, Pistarà R, Salvagnini M, Sangiovanni A. Bacterial infection in patients with advanced cirrhosis: A multicentre prospective study. Dig Liver Dis. 2001; 33(1): 41-8. [CrossRef]
- Pauwels A, Mostefa-Kara N, Debenes B, Degoutte E, Lévy VG. Systemic antibiotic prophylaxis after gastrointestinal hemorrhage in cirrhotic patients with a high risk of infection. Hepatology. 1996; 24(4): 802-6. [CrossRef]
- Albillos A, Lario M, Álvarez-Mon M. Cirrhosis-associated immune dysfunction: Distinctive features and clinical relevance. J Hepatol. 2014; 61(6): 1385-96. [CrossRef]
- Moreau R, Jalan R, Gines P, Pavesi M, Angeli P, Cordoba J, Durand F, Gustot T, Saliba F, Domenicali M, Gerbes A, Wendon J, Alessandria C, Laleman W, Zeuzem S, Trebicka J, Bernardi M, Arroyo V; CANONIC Study Investigators of the EASL–CLIF Consortium. Acute-on-chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology. 2013; 144(7): 1426-37, 1437.e1-9. [CrossRef]
- Trebicka J, Fernandez J, Papp M, Caraceni P, Laleman W, Gambino C, Giovo I, Uschner FE, Jimenez C, Mookerjee R; PREDICT STUDY group of the EASL-CLIF Consortium. The PREDICT study uncovers three clinical courses of acutely decompensated cirrhosis that have distinct pathophysiology. J Hepatol. 2020; 73(4): 842-854. [CrossRef]
- Albillos A, de Gottardi A, Rescigno M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol. 2020; 72(3): 558-577. [CrossRef]
- Franco A, Barnaba V, Natali P, Balsano C, Musca A, Balsano F. Expression of class I and class II major histocompatibility complex antigens on human hepatocytes. Hepatology. 1988; 8(3): 449-54. [CrossRef]
- Gao B, Jeong WI, Tian Z. Liver: An organ with predominant innate immunity. Hepatology. 2008; 47(2): 729-36. [CrossRef]
- Mörbe UM, Jørgensen PB, Fenton TM, von Burg N, Riis LB, Spencer J, Agace WW. Human gut-associated lymphoid tissues (GALT); diversity, structure, and function. Mucosal Immunol. 2021; 14(4): 793-802. [CrossRef]
- MacPherson G, Milling S, Yrlid U, Cousins L, Turnbull E, Huang FP. Uptake of antigens from the intestine by dendritic cells. Ann N Y Acad Sci. 2004; 1029: 75-82. [CrossRef]
- Bernardi M, Moreau R, Angeli P, Schnabl B, Arroyo V. Mechanisms of decompensation and organ failure in cirrhosis: From peripheral arterial vasodilation to systemic inflammation hypothesis. J Hepatol. 2015; 63(5): 1272-84. [CrossRef]
- Bernardi M., Sriskandan S, Altmann DM. The immunology of sepsis. J Pathol. 2008; 214(2): 211-23. [CrossRef]
- Solé C, Solá E, Morales-Ruiz M, Fernández G, Huelin P, Graupera I, Moreira R, de Prada G, Ariza X, Pose E, Fabrellas N, Kalko SG, Jiménez W, Ginés P. Characterization of Inflammatory Response in Acute-on-Chronic Liver Failure and Relationship with Prognosis. Sci Rep. 2016; 6: 32341. [CrossRef]
- Monteiro S, Grandt J, Uschner FE, Kimer N, Madsen JL, Schierwagen R, Klein S, Welsch C, Schäfer L, Jansen C, Claria J, Alcaraz-Quiles J, Arroyo V, Moreau R, Fernandez J, Bendtsen F, Mehta G, Gluud LL, Møller S, Praktiknjo M, Trebicka J. Differential inflammasome activation predisposes to acute-on-chronic liver failure in human and experimental cirrhosis with and without previous decompensation. Gut. 2021; 70(2): 379-387. [CrossRef]
- Arroyo V, Angeli P, Moreau R, Jalan R, Clària J, Trebicka J, Fernández J, Gustot T, Caraceni P, Bernardi M; investigators from the EASL-CLIF Consortium, Grifols Chair and European Foundation for the Study of Chronic Liver Failure (EF-Clif). The systemic inflammation hypothesis: Towards a new paradigm of acute decompensation and multiorgan failure in cirrhosis. J Hepatol. 2021; 74(3): 670-685. [CrossRef]
- Wiest R, Albillos A, Trauner M, Bajaj JS, Jalan R. Targeting the gut-liver axis in liver disease. J Hepatol. 2017; 67(5): 1084-1103. [CrossRef]
- Gao B, Seki E, Brenner DA, Friedman S, Cohen JI, Nagy L, Szabo G, Zakhari S. Innate immunity in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2011; 300(4): G516-25. [CrossRef]
- Coant N, Simon-Rudler M, Gustot T, Fasseu M, Gandoura S, Ragot K, Abdel-Razek W, Thabut D, Lettéron P, Ogier-Denis E. Glycogen synthase kinase 3 involvement in the excessive proinflammatory response to LPS in patients with decompensated cirrhosis. J Hepatol. 2011; 55(4): 784-93. [CrossRef]
- Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. 2012; 143(5): 1158-1172. [CrossRef]
- Parlesak A, Schäfer C, Schütz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol. 2000; 32(5): 742-7. [CrossRef]
- Bellot P, García-Pagán JC, Francés R, Abraldes JG, Navasa M, Pérez-Mateo M, Such J, Bosch J. Bacterial DNA translocation is associated with systemic circulatory abnormalities and intrahepatic endothelial dysfunction in patients with cirrhosis. Hepatology. 2010; 52(6): 2044-52. [CrossRef]
- Ayres JS. Immunometabolism of infections. Nat Rev Immunol. 2020; 20(2): 79-80. [CrossRef]
- Romero-Gómez M, Montagnese S, Jalan R. Hepatic encephalopathy in patients with acute decompensation of cirrhosis and acute-on-chronic liver failure. J Hepatol. 2015; 62(2): 437-47. [CrossRef]
- Martin-Mateos R, Alvarez-Mon M, Albillos A. Dysfunctional Immune Response in Acute-on-Chronic Liver Failure: It Takes Two to Tango. Front Immunol. 2019; 10: 1-10. [CrossRef]
- Laleman W, Claria J, Van der Merwe S, Moreau R, Trebicka J. Systemic Inflammation and Acute-on-Chronic Liver Failure: Too Much, Not Enough. Can J Gastroenterol Hepatol. 2018; 2018: 1-10. [CrossRef]
- Papp M, Vitalis Z, Altorjay I, Tornai I, Udvardy M, Harsfalvi J, Vida A, Kappelmayer J, Lakatos PL, Antal-Szalmas P. Acute phase proteins in the diagnosis and prediction of cirrhosis associated bacterial infections. Liver Int. 2012; 32(4): 603-11. [CrossRef]
- Homann C, Varming K, Høgåsen K, Mollnes TE, Graudal N, Thomsen AC, Garred P. Acquired C3 deficiency in patients with alcoholic cirrhosis predisposes to infection and increased mortality. Gut. 1997; 40(4): 544-9. [CrossRef]
- Tritto G, Bechlis Z, Stadlbauer V, Davies N, Francés R, Shah N, Mookerjee RP, Such J, Jalan R. Evidence of neutrophil functional defect despite inflammation in stable cirrhosis. J Hepatol. 2011; 55(3): 574-581. [CrossRef]
- Ward NS, Casserly B, Ayala A. The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin Chest Med. 2008; 29(4): 617-25. [CrossRef]
- Berry PA, Antoniades CG, Hussain MJ, McPhail MJ, Bernal W, Vergani D, Wendon JA. Admission levels and early changes in serum interleukin-10 are predictive of poor outcome in acute liver failure and decompensated cirrhosis. Liver Int. 2010; 30(5): 733-40. [CrossRef]
- Bone RC, Grodzin CJ, Balk RA. Sepsis: A new hypothesis for pathogenesis of the disease process. Chest. 1997; 112(1): 235-43. [CrossRef]
- Jalan R, Schnurr K, Mookerjee RP, Sen S, Cheshire L, Hodges S, Muravsky V, Williams R, Matthes G, Davies NA. Alterations in the functional capacity of albumin in patients with decompensated cirrhosis are associated with increased mortality. Hepatology. 2009; 50(2): 555-64. [CrossRef]
- O'Brien AJ, Fullerton JN, Massey KA, Auld G, Sewell G, James S, Newson J, Karra E, Winstanley A, Alazawi W, Garcia-Martinez R, Cordoba J, Nicolaou A, Gilroy DW. Immunosuppression in acutely decompensated cirrhosis is mediated by prostaglandin E2. Nat Med. 2014; 20(5): 518-23. [CrossRef]
- Arvaniti V, D'Amico G, Fede G, Manousou P, Tsochatzis E, Pleguezuelo M, Burroughs AK. Infections in patients with cirrhosis increase mortality four-fold and should be used in determining prognosis. Gastroenterology. 2010; 139(4): 1246-56. [CrossRef]
- Gustot T, Felleiter P, Pickkers P, Sakr Y, Rello J, Velissaris D, Pierrakos C, Taccone FS, Sevcik P, Moreno C, Vincent JL; EPIC II Group of Investigators. Impact of infection on the prognosis of critically ill cirrhotic patients: Results from a large worldwide study. Liver Int. 2014; 34(10): 1496-503. [CrossRef]
- Muñoz L, José Borrero M, Ubeda M, Lario M, Díaz D, Francés R, Monserrat J, Pastor O, Aguado-Fraile E, Such J, Alvarez-Mon M, Albillos A. Interaction between intestinal dendritic cells and bacteria translocated from the gut in rats with cirrhosis. Hepatology. 2012; 56(5): 1861-9. [CrossRef]
- Lozano-Ruiz B, Bachiller V, García-Martínez I, Zapater P, Gómez-Hurtado I, Moratalla A, Giménez P, Bellot P, Francés R, Such J, González-Navajas JM. Absent in melanoma 2 triggers a heightened inflammasome response in ascitic fluid macrophages of patients with cirrhosis. J Hepatol. 2015; 62(1): 64-71. [CrossRef]
- Navasa M, Follo A, Filella X, Jiménez W, Francitorra A, Planas R, Rimola A, Arroyo V, Rodés J. Tumor necrosis factor and interleukin-6 in spontaneous bacterial peritonitis in cirrhosis: Relationship with the development of renal impairment and mortality. Hepatology. 1998; 27(5): 1227-32. [CrossRef]
- Stengel S, Quickert S, Lutz P, Ibidapo-Obe O, Steube A, Köse-Vogel N, Yarbakht M, Reuken PA, Busch M, Brandt A, Bergheim I, Deshmukh SD, Stallmach A, Bruns T. Peritoneal Level of CD206 Associates With Mortality and an Inflammatory Macrophage Phenotype in Patients With Decompensated Cirrhosis and Spontaneous Bacterial Peritonitis. Gastroenterology. 2020; 158(6): 1745-1761. [CrossRef]
- Rasaratnam B, Kaye D, Jennings G, Dudley F, Chin-Dusting J. The effect of selective intestinal decontamination on the hyperdynamic circulatory state in cirrhosis. A randomized trial. Ann Intern Med. 2003; 139(3): 186-93. [CrossRef]
- Mehta G, Mookerjee RP, Sharma V, Jalan R. Systemic inflammation is associated with increased intrahepatic resistance and mortality in alcohol-related acute-on-chronic liver failure. Liver Int. 2015; 35(3): 724-34. [CrossRef]
- McGovern BH, Golan Y, Lopez M, Pratt D, Lawton A, Moore G, Epstein M, Knox TA. The impact of cirrhosis on CD4+ T cell counts in HIV-seronegative patients. Clin Infect Dis. 2007; 44(3): 431-7. [CrossRef]
- Lario M, Muñoz L, Ubeda M, Borrero MJ, Martínez J, Monserrat J, Díaz D, Alvarez-Mon M, Albillos A. Defective thymopoiesis and poor peripheral homeostatic replenishment of T-helper cells cause T-cell lymphopenia in cirrhosis. J Hepatol. 2013; 59(4): 723-30. [CrossRef]
- Wiest R, Das S, Cadelina G, Garcia-Tsao G, Milstien S, Groszmann RJ. Bacterial translocation in cirrhotic rats stimulates eNOS-derived NO production and impairs mesenteric vascular contractility. J Clin Invest. 1999; 104(9): 1223-33. [CrossRef]
- Opal SM, Scannon PJ, Vincent JL, White M, Carroll SF, Palardy JE, Parejo NA, Pribble JP, Lemke JH. Relationship between plasma levels of lipopolysaccharide (LPS) and LPS-binding protein in patients with severe sepsis and septic shock. J Infect Dis. 1999; 180(5): 1584-9. [CrossRef]
- Albillos A, de la Hera A, González M, Moya JL, Calleja JL, Monserrat J, Ruiz-del-Arbol L, Alvarez-Mon M. Increased lipopolysaccharide binding protein in cirrhotic patients with marked immune and hemodynamic derangement. Hepatology. 2003; 37(1): 208-17. [CrossRef]
- Le Roy D, Di Padova F, Adachi Y, Glauser MP, Calandra T, Heumann D. Critical role of lipopolysaccharide-binding protein and CD14 in immune responses against gram-negative bacteria. J Immunol. 2001; 167(5): 2759-65. [CrossRef]
- Ghassemi S, Garcia-Tsao G. Prevention and treatment of infections in patients with cirrhosis. Best Pract Res Clin Gastroenterol. 2007; 21(1): 77-93. [CrossRef]
- Fiuza C, Salcedo M, Clemente G, Tellado JM. In vivo neutrophil dysfunction in cirrhotic patients with advanced liver disease. J Infect Dis. 2000; 182(2): 526-33. [CrossRef]
- Runyon BA, Morrissey RL, Hoefs JC, Wyle FA. Opsonic activity of human ascitic fluid: A potentially important protective mechanism against spontaneous bacterial peritonitis. Hepatology. 1985; 5(4): 634-7. [CrossRef]
- Bruce R, Richard M., John H., Frederic W. Opsonic Activity of Human Ascitic Fluid: A Potentially Important Protective Mechanism Against Spontaneous Bacterial Peritonitis. Hepatology. 1985; 5(4): 634-637.
- Wiest R, Garcia-Tsao G. Bacterial translocation (BT) in cirrhosis. Hepatology. 2005; 41(3): 422-33. [CrossRef]
- Gustot T, Durand F, Lebrec D, Vincent JL, Moreau R. Severe sepsis in cirrhosis. Hepatology. 2009; 50(6): 2022-33. Erratum in: Hepatology. 2010; 51(2): 725. [CrossRef]
- Cazzaniga M, Dionigi E, Gobbo G, Fioretti A, Monti V, Salerno F. The systemic inflammatory response syndrome in cirrhotic patients: Relationship with their in-hospital outcome. J Hepatol. 2009; 51(3): 475-82. [CrossRef]
- Chan CC, Hwang SJ, Lee FY, Wang SS, Chang FY, Li CP, Chu CJ, Lu RH, Lee SD. Prognostic value of plasma endotoxin levels in patients with cirrhosis. Scand J Gastroenterol. 1997; 32(9): 942-6. [CrossRef]
- Guarner C, Soriano G, Tomas A, Bulbena O, Novella MT, Balanzo J, Vilardell F, Mourelle M, Moncada S. Increased serum nitrite and nitrate levels in patients with cirrhosis: Relationship to endotoxemia. Hepatology. 1993; 18(5): 1139-43.
- Bernard B, Grangé JD, Khac EN, Amiot X, Opolon P, Poynard T. Antibiotic prophylaxis for the prevention of bacterial infections in cirrhotic patients with gastrointestinal bleeding: A meta-analysis. Hepatology. 1999; 29(6): 1655-61. [CrossRef]
- Sandhu BS, Gupta R, Sharma J, Singh J, Murthy NS, Sarin SK. Norfloxacin and cisapride combination decreases the incidence of spontaneous bacterial peritonitis in cirrhotic ascites. J Gastroenterol Hepatol. 2005; 20(4): 599-605. [CrossRef]
- Rimola A, García-Tsao G, Navasa M, Piddock LJ, Planas R, Bernard B, Inadomi JM. Diagnosis, treatment and prophylaxis of spontaneous bacterial peritonitis: A consensus document. International Ascites Club. J Hepatol. 2000; 32(1): 142-53. [CrossRef]
- Follo A, Llovet JM, Navasa M, Planas R, Forns X, Francitorra A, Rimola A, Gassull MA, Arroyo V, Rodés J. Renal impairment after spontaneous bacterial peritonitis in cirrhosis: Incidence, clinical course, predictive factors and prognosis. Hepatology. 1994; 20(6): 1495-501. [CrossRef]
- Sort P, Navasa M, Arroyo V, Aldeguer X, Planas R, Ruiz-del-Arbol L, Castells L, Vargas V, Soriano G, Guevara M, Ginès P, Rodés J. Effect of intravenous albumin on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis. N Engl J Med. 1999; 341(6): 403-9. [CrossRef]
- Ginés P, Rimola A, Planas R, Vargas V, Marco F, Almela M, Forné M, Miranda ML, Llach J, Salmerón JM; et al. Norfloxacin prevents spontaneous bacterial peritonitis recurrence in cirrhosis: Results of a double-blind, placebo-controlled trial. Hepatology. 1990; 12(4): 716-24. [CrossRef]
- Wasmuth HE, Kunz D, Yagmur E, Timmer-Stranghöner A, Vidacek D, Siewert E, Bach J, Geier A, Purucker EA, Gressner AM, Matern S, Lammert F. Patients with acute on chronic liver failure display "sepsis-like" immune paralysis. J Hepatol. 2005; 42(2): 195-201. [CrossRef]
- Úbeda M, Muñoz L, Borrero MJ, Díaz D, Francés R, Monserrat J, Lario M, Lledó L, Such J, Álvarez-Mon M, Albillos A. Critical role of the liver in the induction of systemic inflammation in rats with preascitic cirrhosis. Hepatology. 2010; 52(6): 2086-95. [CrossRef]
- Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008; 454(7203): 428-35. [CrossRef]
- Arroyo V, Moreau R, Jalan R, Ginès P; EASL-CLIF Consortium CANONIC Study. Acute-on-chronic liver failure: A new syndrome that will re-classify cirrhosis. J Hepatol. 2015; 62(1 Suppl): S131-43. [CrossRef]
- Piano S, Tonon M, Vettore E, Stanco M, Pilutti C, Romano A, Mareso S, Gambino C, Brocca A, Sticca A, Fasolato S, Angeli P. Incidence, predictors and outcomes of acute-on-chronic liver failure in outpatients with cirrhosis. J Hepatol. 2017; 67(6): 1177-1184. [CrossRef]
- Heneka MT, McManus RM, Latz E. Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci. 2018; 19(10): 610-621. [CrossRef]
- Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014; 146: 1513-1524. [CrossRef]
- Pascual S, Such J, Esteban A, Zapater P, Casellas JA, Aparicio JR; et al. Intestinal permeability is increased in patients with advanced cirrhosis. Hepatogastroenterology 2003; 50: 1482-1486.
- Gandoura S, Weiss E, Ratou PE, Fassen M, Gustot T, Lemoine F, Hurtado-Nedelec M, Hego C, Vadrot N, Elkrief L; et al. Gene and exon-expression profiling reveals an extensive LPS-induced response in immune cells in patients with cirrhosis. J Hepatol 2013; 58: 936-948. [CrossRef]
- Michelena J, Altamirano J, Abraldes JG, Affò S, Morales-Ibanez O, Sancho-Bru P; et al. Systemic inflammatory response and serum lipopolysaccharide levels predict multiple organ failure and death in alcoholic hepatitis. Hepatology 2015; 62(3): 762-72. [CrossRef]
- Lozano-Ruiz B, Bachiller V, García-Martinez I, Zapater P, Gomez-Hurtado I, Moratalla A, Giménez P, Bellot P, Francés R, Such J; et al. Absent in melanoma 2 triggers a heightened inflammasome response in ascitic fluid macrophages of patients with cirrhosis. J Hepatol 2015; 62: 64-71. [CrossRef]
- Gottardi A, Gronbaek H, Saliba F, Trautwein C, Özdogan OC, Francque S, Ryder S, Nahon P, Romero-Gomez M, Van Vlierberghe H; PREDICT STUDY group of the EASL-CLIF Consortium. The PREDICT study uncovers three clinical courses of acutely decompensated cirrhosis that have distinct pathophysiology. J Hepatol. 2020; 73(4): 842-854. [CrossRef]
- Clària J; Stauber R; Coenraad M; Moreau R; Jalan R; Pavesi M; Amorós À; Titos E; Alcaraz J; Oettl K. Systemic inflammation in decompensated cirrhosis: Characterization and role in acute on chronic liver failure. Hepatology. 2016; 64(4): 1249-1264. [CrossRef]
- Trebicka J, Fernandez J, Papp M, Caraceni P, Laleman W, Gambino C, Giovo I, Uschner FE, Jansen C, Jimenez C. PREDICT STUDY group of the EASL-CLIF CONSORTIUM. PREDICT identifies precipitating events associated with the clinical course of acutely decompensated cirrhosis. J Hepatol. 2021; 74(5): 1097-1108. [CrossRef]
- Moreau R, Clària J, Aguilar F, Fenaille F, Lozano JJ, Junot C, Colsch B, Caraceni P, Trebicka J, Pavesi M; et al. Blood metabolomics uncovers inflammation-associated mitochondrial dysfunction as a potential mechanism underlying ACLF. Journal of Hepatology. 2020; 72(4): 688-701. [CrossRef]
- Cohen J. The immunopathogenesis of sepsis. Nature. 2002; 420(6917): 885-91. [CrossRef]
- Fernández J, Acevedo J, Wiest R, Gustot T, Amorós A, Deulofeu C; et al. Bacterial and fungal infections in acute-on-chronic liver failure: Prevalence, characteristics and impact on prognosis. Gut 2018; 67(10): 1870–1880. [CrossRef]
- Clària J, Moreau R, Fenaille F, Amorós A, Junot C, Gronbaek H, Coenraad MJ, Pruvost A, Ghettas A, Chu-Van E; CANONIC Study Investigators of the EASL Clif Consortium, Grifols Chair and the European Foundation for the Study of Chronic Liver Failure (EF Clif). Orchestration of Tryptophan-Kynurenine Pathway, Acute Decompensation, and Acute-on-Chronic Liver Failure in Cirrhosis. Hepatology. 2019; 69(4): 1686-1701. [CrossRef]
- Bernsmeier C, van der Merwe S, Périanin A. Innate immune cells in cirrhosis. J Hepatol. 2020; 73(1): 186-201. [CrossRef]
- Piano S, Schmidt HH, Ariza X, Amoros A, Romano A, Hüsing-Kabar A, Solà E, Gerbes A, Bernardi M, Alessandria C; EASL CLIF Consortium, European Foundation for the Study of Chronic Liver Failure (EF Clif). Association Between Grade of Acute on Chronic Liver Failure and Response to Terlipressin and Albumin in Patients With Hepatorenal Syndrome. Clin Gastroenterol Hepatol. 2018; 16(11): 1792-1800. [CrossRef]
- Fernández J, Clària J, Amorós A, Aguilar F, Castro M, Casulleras M, Acevedo J, Duran-Güell M, Nuñez L, Costa M. Effects of Albumin Treatment on Systemic and Portal Hemodynamics and Systemic Inflammation in Patients With Decompensated Cirrhosis. Gastroenterology. 2019; 157(1): 149-162. [CrossRef]
- Bhanji R, Montaño-Loza A., Watt K. Sarcopenia in Cirrhosis: Loking Beyond the Skeletal Muscle Loss to See the Systemic Disease. Hepatology. 2019; 70(6): 2193-2203. [CrossRef]
- Cruz-Jentoft AJ, Baeyens JP, Bauer JM, Boirie Y, Cederholm T, Landi F, Martin FC, Michel JP, Rolland Y, Schneider SM, Topinková E, Vandewoude M, Zamboni M; European Working Group on Sarcopenia in Older People. Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Ageing. 2010; 39(4): 412-23. [CrossRef]
- Kazemi-Bajestani SM, Becher H, Ghosh S, Montano-Loza AJ, Baracos VE. Concurrent depletion of skeletal muscle, fat, and left ventricular mass in patients with cirrhosis of the liver. J Cachexia Sarcopenia Muscle. 2016; 7(1): 97-9. [CrossRef]
- Iizuka K, Machida T, Hirafuji M. Skeletal muscle is an endocrine organ. J Pharmacol Sci. 2014; 125(2): 125-31. [CrossRef]
- Dasarathy S, Merli M. Sarcopenia from mechanism to diagnosis and treatment in liver disease. J Hepatol. 2016; 65(6): 1232-1244. [CrossRef]
- Kim TN, Park MS, Lim KI, Choi HY, Yang SJ, Yoo HJ, Kang HJ, Song W, Choi H, Baik SH, Choi DS, Choi KM. Relationships between sarcopenic obesity and insulin resistance, inflammation, and vitamin D status: The Korean Sarcopenic Obesity Study. Clin Endocrinol (Oxf). 2013; 78(4): 525-32. [CrossRef]
- Kalafateli M, Mantzoukis K, Choi Yau Y, Mohammad AO, Arora S, Rodrigues S, de Vos M, Papadimitriou K, Thorburn D, O'Beirne J; et al. Malnutrition and sarcopenia predict post– liver transplantation outcomes independently of the Model for End-Stage Liver Disease score. J Cachexia Sarcopenia Muscle 2017; 8: 113-121. [CrossRef]
- Bustamante J, Rimola A, Ventura PJ, Navasa M, Cirera I, Reggiardo V, Rodés J. Prognostic significance of hepatic encephalopathy in patients with cirrhosis. J Hepatol. 1999; 30(5): 890-5. [CrossRef]
- Hui AY, Chan HL, Leung NW, Hung LC, Chan FK, Sung JJ. Survival and prognostic indicators in patients with hepatitis B virus-related cirrhosis after onset of hepatic decompensation. J Clin Gastroenterol. 2002; 34(5): 569-72. [CrossRef]
- Odeh M, Sabo E, Srugo I, Oliven A. Relationship between tumor necrosis factor-alpha and ammonia in patients with hepatic encephalopathy due to chronic liver failure. Ann Med. 2005; 37(8): 603-12. [CrossRef]
- Li W, Li N, Wang R, Li Q, Wu H. Interferon gamma, interleukin-6, and -17a levels were correlated with minimal hepatic encephalopathy in HBV patients. Hepatol Int. 2015; 9(2): 218-23. [CrossRef]
- Shawcross DL, Wright G, Olde Damink SW, Jalan R. Role of ammonia and inflammation in minimal hepatic encephalopathy. Metab Brain Dis. 2007; 22(1): 125-38. [CrossRef]
- Montoliu C, Piedrafita B, Serra MA, del Olmo JA, Urios A, Rodrigo JM, Felipo V. IL-6 and IL-18 in blood may discriminate cirrhotic patients with and without minimal hepatic encephalopathy. J Clin Gastroenterol. 2009; 43(3): 272-9. [CrossRef]
Figure A1.
Dysbiosis and increased intestinal permeability cause chronic inflammation in liver cirrhosis.
Figure A1.
Dysbiosis and increased intestinal permeability cause chronic inflammation in liver cirrhosis.
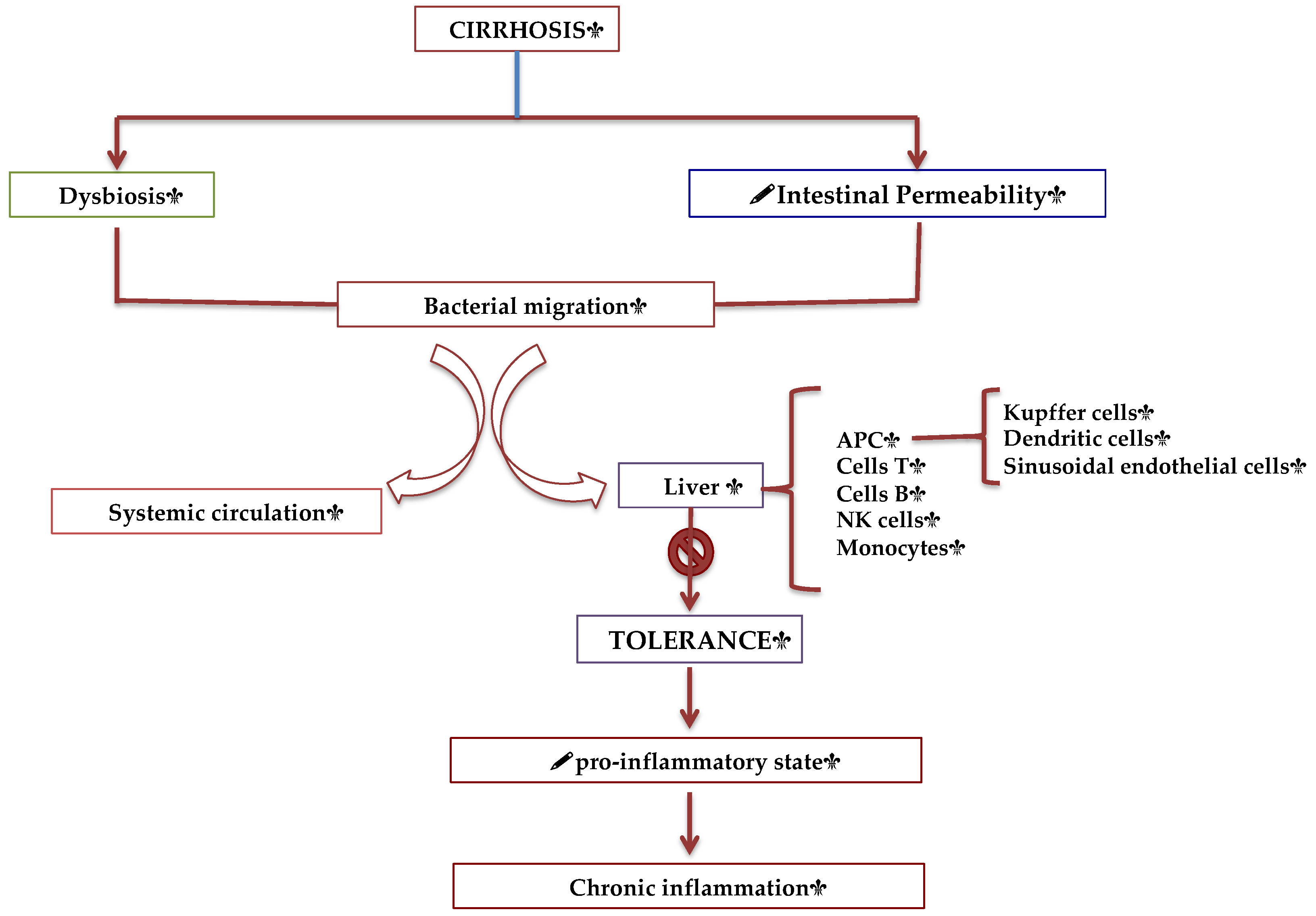
Figure A2.
Phatophysiology of cirrhosis-associated immune dysfunction. PAMPs y DAMPS bind to PRR for cytokine release.
Figure A2.
Phatophysiology of cirrhosis-associated immune dysfunction. PAMPs y DAMPS bind to PRR for cytokine release.
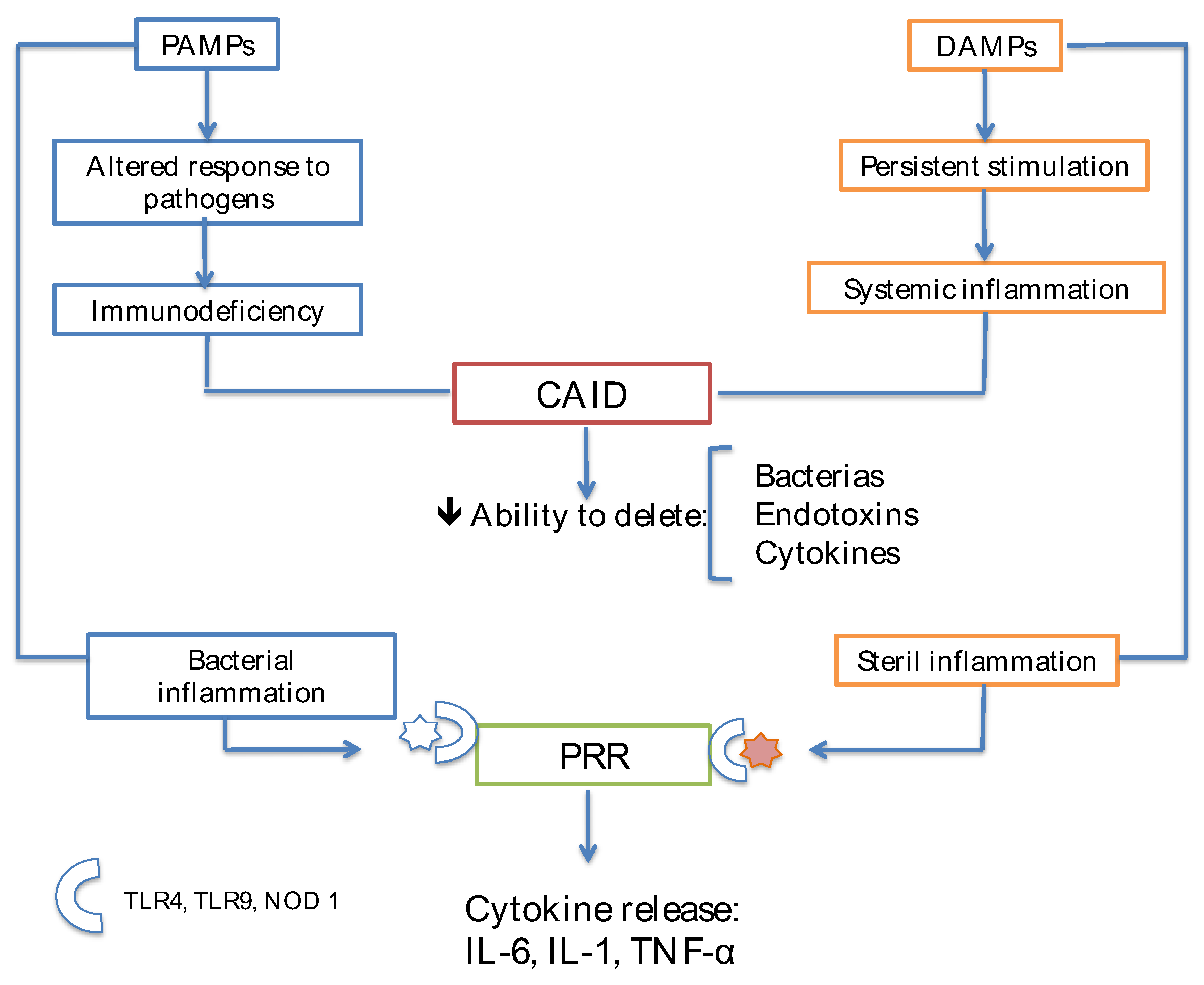
Figure A3.
Sarcopenia and Cirrhosis. Skeletal muscle has beneficial physiology functions, which deteriorate in liver cirrhosis.
Figure A3.
Sarcopenia and Cirrhosis. Skeletal muscle has beneficial physiology functions, which deteriorate in liver cirrhosis.
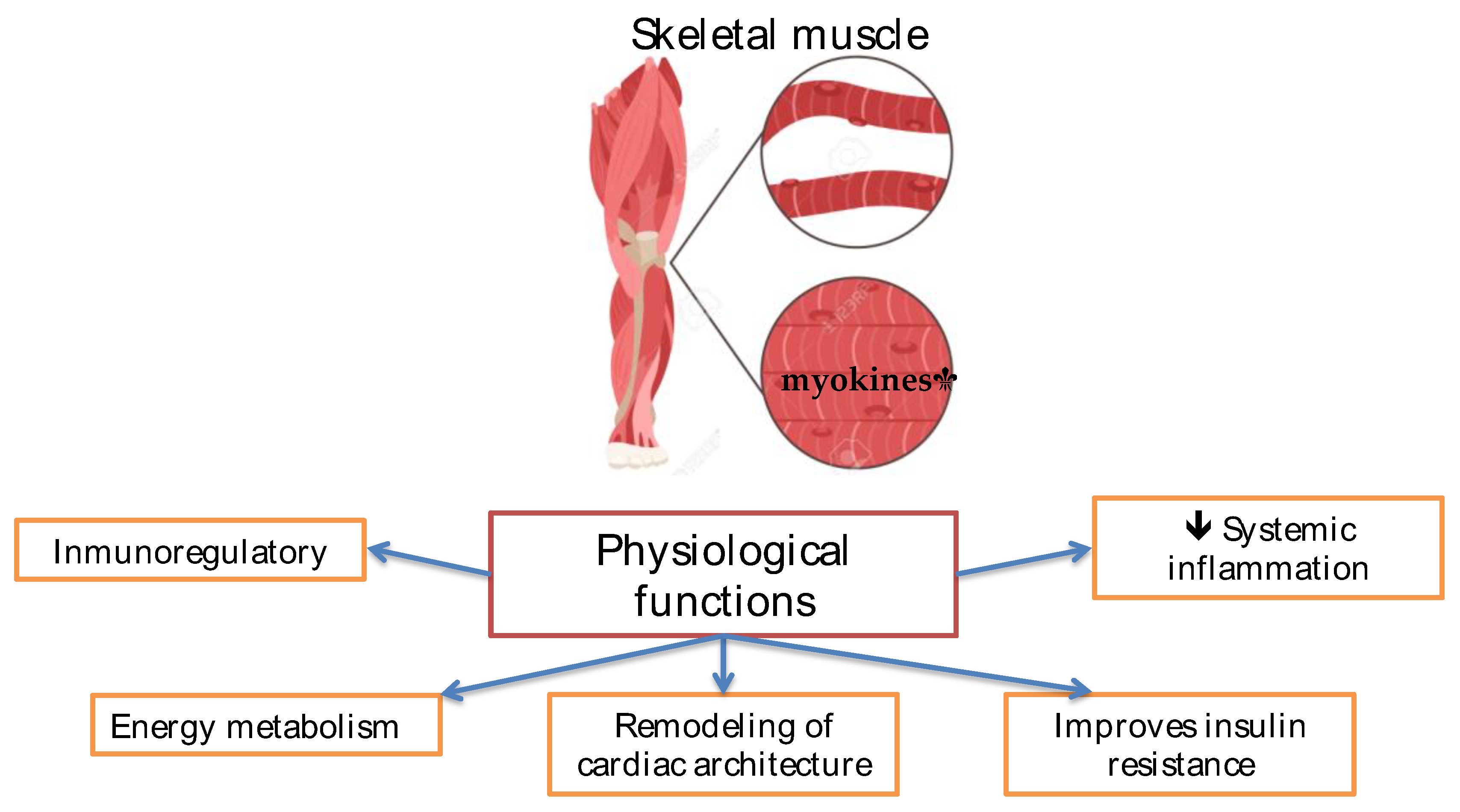
Figure A4.
Sarcopenic obesity in cirrhosis.
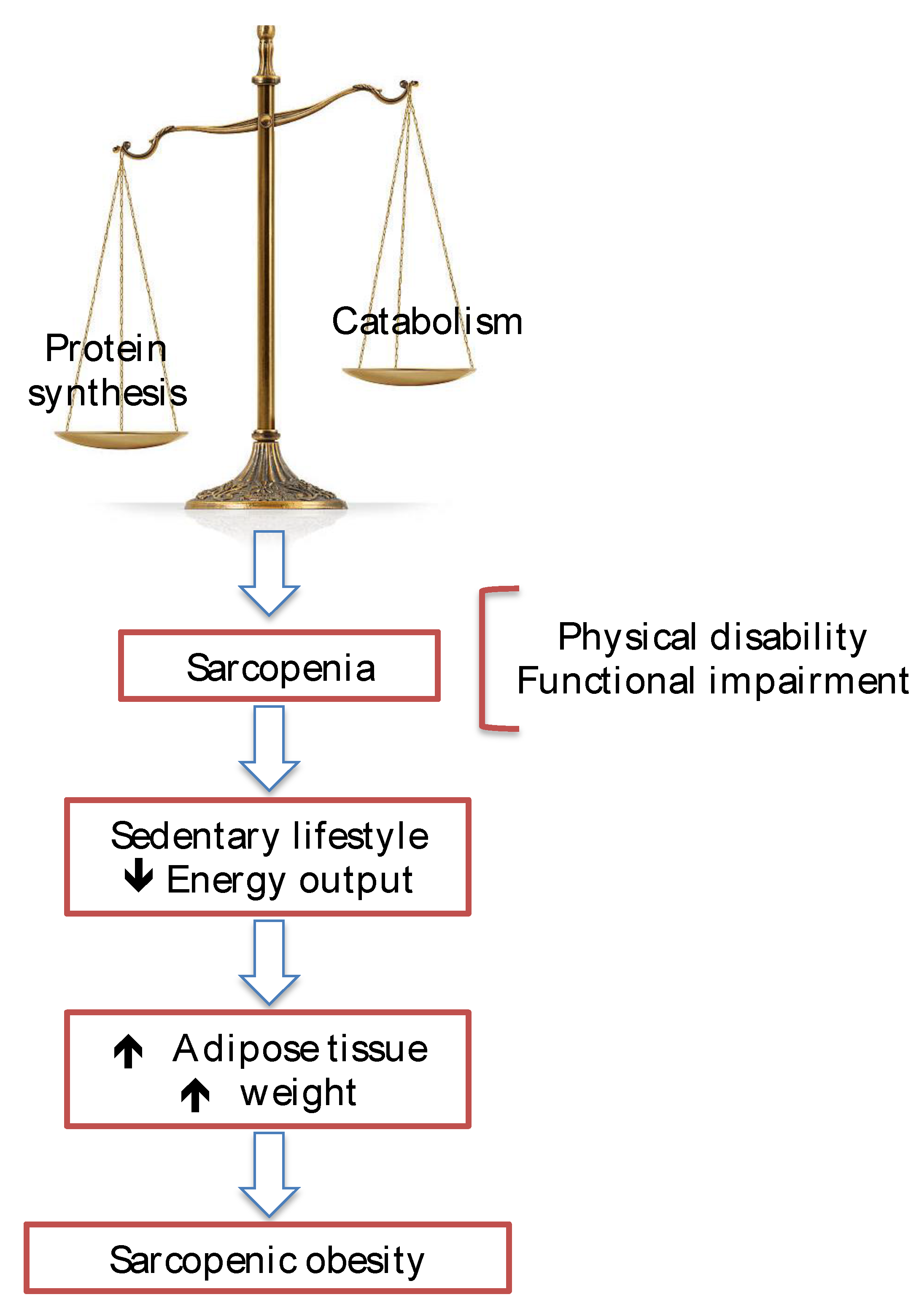
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



