
Submitted:
28 July 2024
Posted:
31 July 2024
You are already at the latest version
Abstract
Cytologic diagnosis of extra‐adrenal paraganglioma presenting as a peripancreatic mass is challenging with a high rate of diagnostic error. We present a case of a peripancreatic mass identified by radiology as a gastrointestinal stromal tumor. Endoscopic ultrasound‐guided fine needle aspiration (FNA) of the mass showed a moderately cellular tumor composed of small to medium sized neoplastic cells with round to oval nuclei arranged singly and in loose clusters. The cells were positive for neuroendocrine markers (synaptophysin and chromogranin) and negative for CD117. A diagnosis of a neoplasm with a neuroendocrine tumor (NET) was made on FNA cytology. The subsequent surgical resection of the tumor revealed peripancreatic paraganglioma with immunohistochemistry positive for synaptophysin, chromogranin, and S100. The latter delineates the sustentacular cells. Although paraganglioma is a well recognized tumor, a detailed comparison of peripancreatic paraganglioma versus pancreatic/gastrointestinal NET is still lacking.
Keywords:
Paraganglioma
; Neuroendocrine neoplasm
; Synaptophysin
1. Introduction
Paraganglioma is a rare tumor arising from non-epithelial neural clusters from the chief cells. The annual incidence is 2–8 per 1 million people.[1] More than 40% of paragangliomas are associated with germline mutations.[2]Extra adrenal paragangliomas originate from the autonomic nervous system close to the gastric wall. Peripancreatic masses impose a great diagnostic challenge for both clinicians and pathologists. Non-specific symptoms plus endoscopic and radiological findings can result in huge diagnostic errors for these tumors.[3] Nearly 50% of cases are reported as NETs on fine-needle aspiration (FNA).[4]
2. Case Report
A 44-year-old hypertensive male presented with a complaint of abdominal pain for 3 months. He weighed 102 kgs with a body mass index of 29kg/mm2. On physical examination his abdomen was soft, non-tender, and non-distended.
The endoscopic ultrasound (EUS) showed a solid, hypoechoic, mild, vascular lesion with intact boarders measuring around 23 × 20 mm close to the gastric wall and anterior to the pancreatic head. The differential diagnosis of gastrointestinal stromal tumor versus neuroendocrine tumor was given.
The patient’s biochemical analysis was within normal limits. The chromogranin A level was 50.98 (normal value is <76.30); the gastrin level was 33.50 (normal range is 13–115 pg/mL); the 5-hydroxy indole acetic acid (5-HIAA) was 4.10 mg/24 hr (normal range is 2–8 mg/24 hr); and fluid amylase was 47U/L (normal range is 28–100).
The computed tomography and magnetic resonance imaging scans showed a solid, avidly enhancing nodule 2.2 cm in short axis—likely a lymph node just anterior to the pancreatic head.
The Ga-68 dotatate positron emission tomography scan reported that there was increased tracer uptake in the soft tissue nodule anterior to the pancreatic head (2.1 cm, SUV max 10.3).
EUS fine-needle aspiration cytology (FNAC) showed cellular smears with numerous abnormal cells appearing with a high nuclear–cytoplasmic ratio, salt and pepper chromatin with rare nuclear molding, and some markedly enlarged nuclei. Scant lymphocytes are seen in the background (Figure 1). Immunohistochemistry performed on the cell block showed tumor cells positive for CD56 and synaptophysin, and negative for CD117 and CD34. A diagnosis of neuroendocrine tumor was made and a resection was advised.
The patient went through laparoscopic resection and the resected nodule measured 27 × 25 × 20 mm with a homogenous yellow surface. Microscopically, a well-circumscribed tumor arranged in solid sheets and nests in a zellballen pattern with tumor cells showed abundant clear-to-granular cytoplasm, vesicular clumped chromatin, and small nucleoli. Also, spindle cells are seen at the periphery of the tumor nests. Mitoses were scarce (0–1/10 hpf). Immunohistochemistry S100 was positive with diffuse and mild cytoplasmic staining in chief cell and dense, strong staining in sustentacular cells (Figure 2). Synaptophysin, chromogranin, neuron-specific enolase, and CD99 were positive in chief cells. CD117 was patchy positive. The lesion was negative for CK7, AE1/3, CD10, CK19, and PAX8. A final diagnosis of paraganglioma low risk was made. The postoperative course was uneventful and the patient’s blood pressure returned to normal.
3. Discussion
Authors Paragangliomas are often symptomatic and may cause sympathetic symptoms such as dry mouth, flushing of the face, racing heart, dilated pupils, constipation, fatigue, profuse sweating, headache, tremors, panic-like symptoms, and generalized weakness due to exhaustion.[4] However, the most classical presentation has the triad of headache, palpitations, and profuse sweating.
Paragangliomas are non-epithelial tumors arising from the neural crest, which is the chief cell of the sympathetic and parasympathetic chains. The majority of paraganglioma secreting hormones are known as functional paraganglioma; those that do not secrete are known as non-functional.[3,4] All paragangliomas have metastatic potential, so they are referred to as metastatic and non-metastatic instead of benign and malignant.[3,4]
Paragangliomas often present a diagnostic challenge due to the variety of locations and symptomatology. Diagnosing paragangliomas located in proximity to gastric and pancreatic regions presents a challenging task as there are many differential diagnoses, which pose diagnostic complexity.[4] FNAC is the most common preoperative diagnostic procedure in these locations. The morphologic overlap between paragangliomas and NETs is significant: the distinction can be confidently made only with immuno-peroxidase stains in cell block preparation. A definite diagnosis can be achieved through a combination of immunohistochemistry (IHC) stain using markers such as synaptophysin, chromogranin, S100, CD117, pan-CK, and GATA3 performed in cell block preparation. The paragangliomas are immunoreactive to S100 (on sustentacular cells), synaptophysin, chromogranin, and GATA3.5.[5]
Kapila et al.[6] studied that “the initial morphologic diagnosis of five of the seven paragangliomas were considered malignant (four undifferentiated and one adenocarcinoma).” These authors concluded that the major diagnostic dilemma was to differentiate the paraganglioma from neuroendocrine carcinoma.[6]
The study by Lanke et al.[7] mentioned “among 15 patients with pancreatic paragangliomas underwent EUS-FNA; six were correctly diagnosed as paragangliomas; six were misdiagnosed as NET; one had no diagnosis; one was diagnosed as spindle cell neoplasm; and one was diagnosed as pseudocyst.” These authors concluded that paragangliomas should be included in the differential diagnosis of peripancreatic/pancreatic masses and highlighted the difficulties in establishing an accurate preoperative diagnosis even after a second-round evaluation.[7]
The study by Nikas et al.[8] showed that the preoperative diagnosis of abdominal extra-adrenal paragangliomas with FNA or fine-needle biopsy is feasible. Diagnostic pitfalls exist but could be significantly avoided within a multidisciplinary setting and the application of IHC on the cytologic or histologic material.[8]
A significant proportion of paraganglioma has a genetic predisposition. Approximately 75% of paragangliomas are sporadic; the remaining 25% are hereditary.2 Mutations of the genes SDHA, SDHD, SDHB, RET, NF1, VHL, MAX, TMEM127, MEN 2A, and MEN 2B, have been associated with paragangliomas.[2,9,10] Genetic testing and counselling play a pivotal role in identifying at-risk individuals for management. Patients are offered genetic testing.[9,10]
Management of paragangliomas requires a multidisciplinary team approach. Treatment options include surgical resections and pharmacological control of catecholamines excess. In their experience, Omaima et al.[11] suggest that “treatment of paraganglioma and pheochromocytoma with octreotide might be of value in patients with paragangliomas and pheochromocytomas, having a having a quick and sustained treatment effect with minimal reported side effect”.[11]
In their study, Asa et al.[9] summarized that “clinical, biochemical, radiologic, and morphological features of pargangliomas are all important features that are required to properly structure a management plan for patients with such tumors. Clarity is assisted by using a comprehensive pathology reporting module that ensures collection and collation of the various components required for proper diagnosis.”
4. Conclusions
Paragangliomas can have many differential diagnoses. It is difficult to make a diagnosis on FNAC alone. A cell block from FNA with adequate IHC can help in differentiating paragangliomas from NETs. The presence of S100 positivity in sustentacular cells and GATA3 reactivity aid in confirming the paragangliomas as these markers are typically negative in NETs. However, the possibility of paragangliomas versus NETs always should be given in FNAC with a note as “A definite diagnosis cannot be concluded.”
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Written informed consent has been obtained from the patient(s) to publish this paper.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Acknowledgments
Nil.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- AAygun N, Uludag M. Pheochromocytoma and paraganglioma: from epidemiology to clinical findings. Sisli Etfal Hastan Tip Bul. 2020, 54, 159–68. [Google Scholar]
- Sarkadi, B.; Saskoi, E.; Butz, H.; Patocs, A. Genetics of Pheochromocytomas and Paragangliomas Determine the Therapeutical Approach. Int. J. Mol. Sci. 2022, 23, 1450. [Google Scholar] [CrossRef]
- Kimura, N.; Takayanagi, R.; Takizawa, N.; Itagaki, E.; Katabami, T.; Kakoi, N.; Rakugi, H.; Ikeda, Y.; Tanabe, A.; Nigawara, T.; et al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocrine-Related Cancer 2014, 21, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Brownlee RE, Shockley WW. Thyroid paraganglioma. Ann. Otol. Rhinol. Laryngol. 1992, 101, 293–9. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Fratini, G.; Sbrozzi-Vanni, A.; Giusti, A.; Manta, R.; Vignali, C.; Nesi, G.; Amorosi, A.; Cavazzana, A.; Arganini, M.; et al. Peripancreatic paraganglioma: Lesson from a round table. World J. Gastroenterol. 2022, 28, 2396–2402. [Google Scholar] [CrossRef]
- Kapila, K.; Tewari, M.C.; Verma, K. Paragangliomas--a diagnostic dilemma on fine needle aspirates. . 1993, 30, 152–7. [Google Scholar]
- Lanke, G.; Stewart, J.M.; Lee, J.H. Pancreatic paraganglioma diagnosed by endoscopic ultrasound-guided fine needle aspiration: A case report and review of literature. World J. Gastroenterol. 2021, 27, 6322–6331. [Google Scholar] [CrossRef]
- Nikas, I.P.; Ishak, A.; AlRawashdeh, M.M.; Klapsinou, E.; Sepsa, A.; Tzimas, G.N.; Panagiotakopoulos, D.; Papaioannou, D.; Salla, C. Preoperative Diagnosis of Abdominal Extra-Adrenal Paragangliomas with Fine-Needle Biopsy. Diagnostics 2022, 12, 1819. [Google Scholar] [CrossRef] [PubMed]
- Asa, S.L.; Ezzat, S.; Mete, O. The Diagnosis and Clinical Significance of Paragangliomas in Unusual Locations. J. Clin. Med. 2018, 7, 280. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, A.; Walsh, D.; Rodger, F.; Clark, G.; Martin, T.; Irving, R.; Sanna, M.; Yao, M.; Robledo, M.; Neumann, H.P.H.; et al. Profiling of Somatic Mutations in Phaeochromocytoma and Paraganglioma by Targeted Next Generation Sequencing Analysis. Int. J. Endocrinol. 2015, 2015, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Elshafie, O.T.; Khalil, A.C.B.; Alshaibi, M.A.; Itkin, B.L.; Ismail, B.M.; Woodhouse, N.J. Hypertensive Crisis in a Patient With a Functioning Mesenteric Paraganglioma: Dramatic Response to Octreotide Treatment. AACE Clin. Case Rep. 2023, 9, 149–152. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Fine needle aspiration cytology preparation shows sheets of cells arranged in nest and groups. The neoplastic cells show round to oval hyperchromatic nucleus (Giemsa stain image, 20×).
Figure 1.
Fine needle aspiration cytology preparation shows sheets of cells arranged in nest and groups. The neoplastic cells show round to oval hyperchromatic nucleus (Giemsa stain image, 20×).
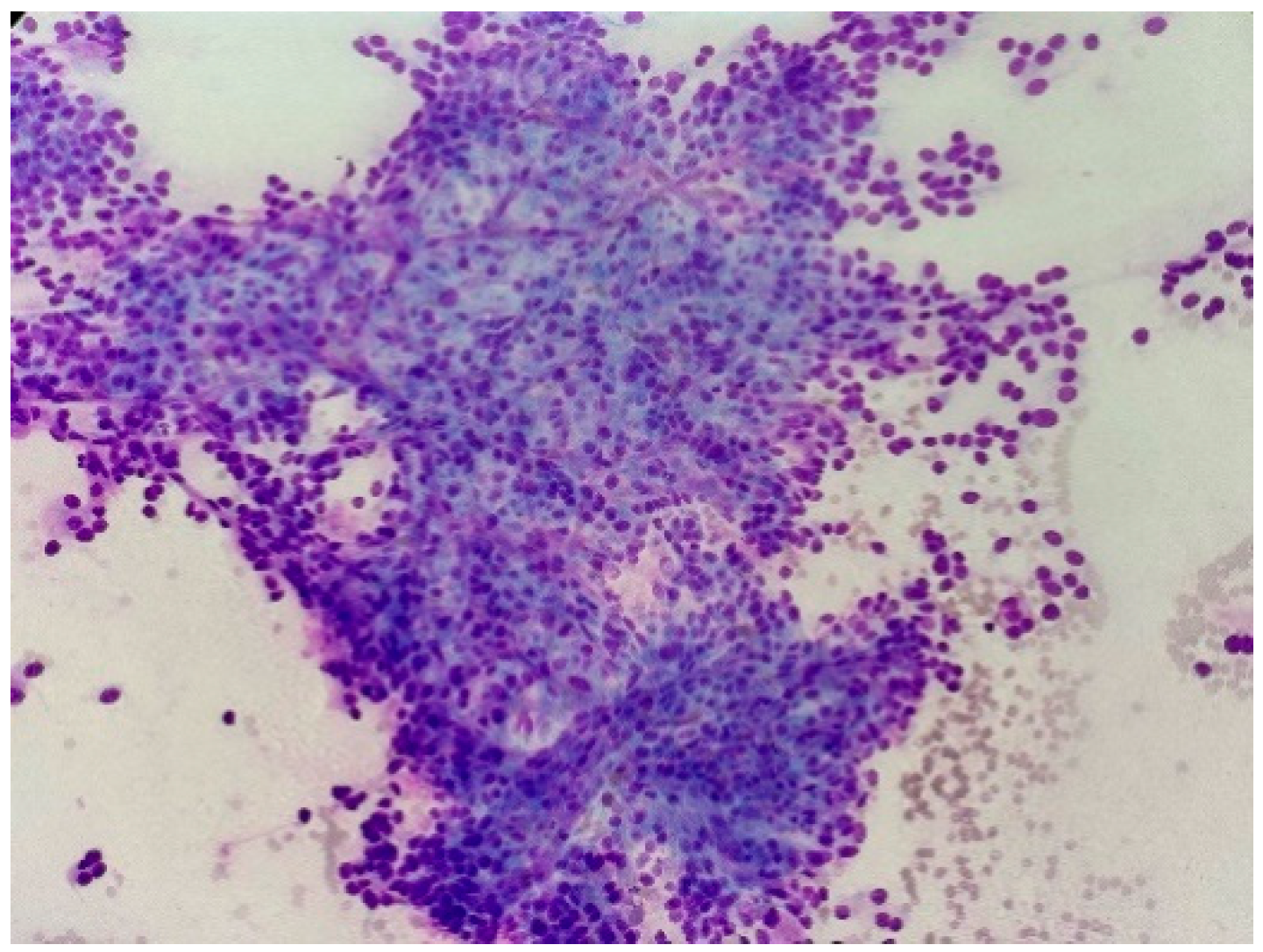
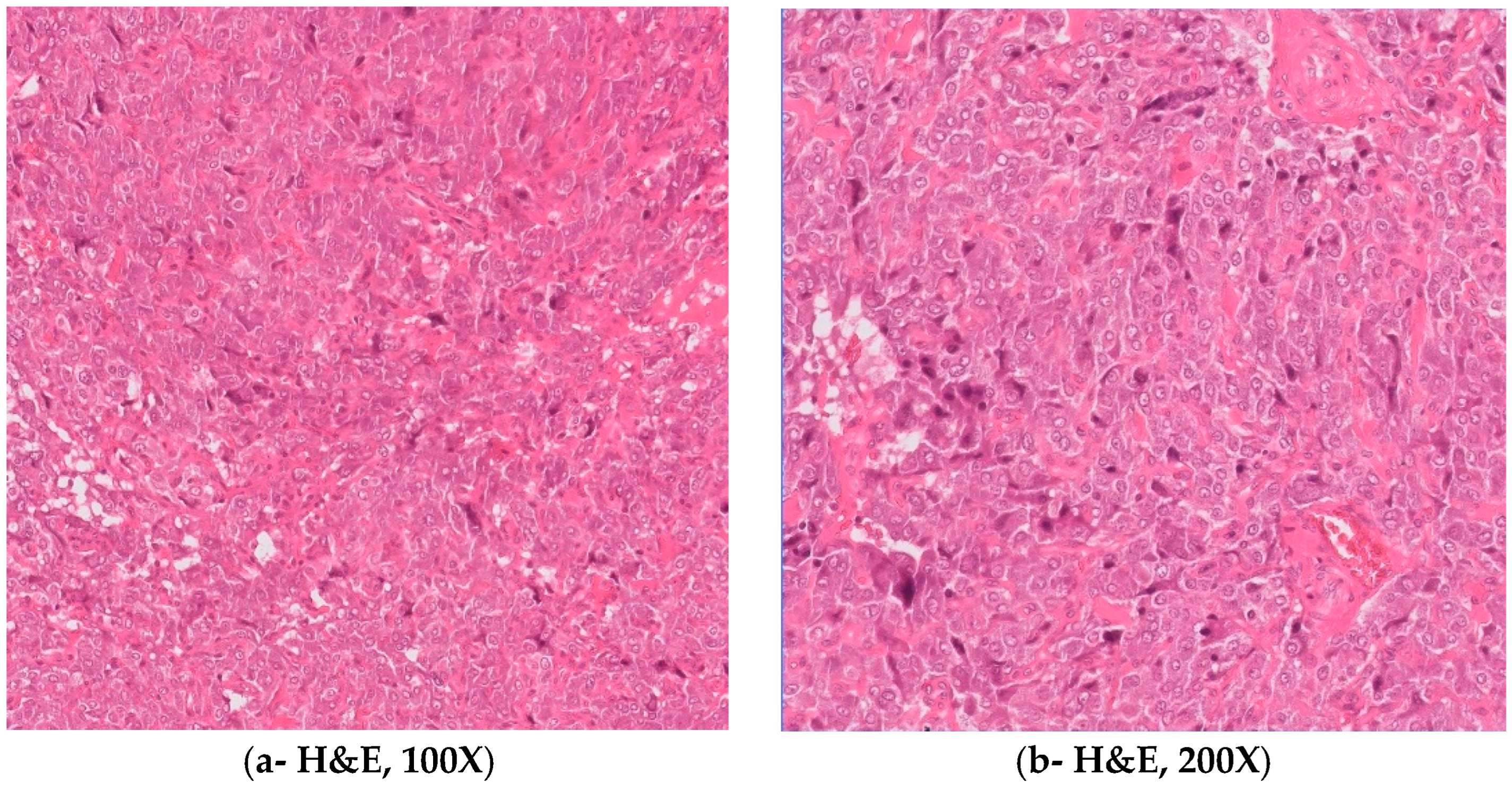
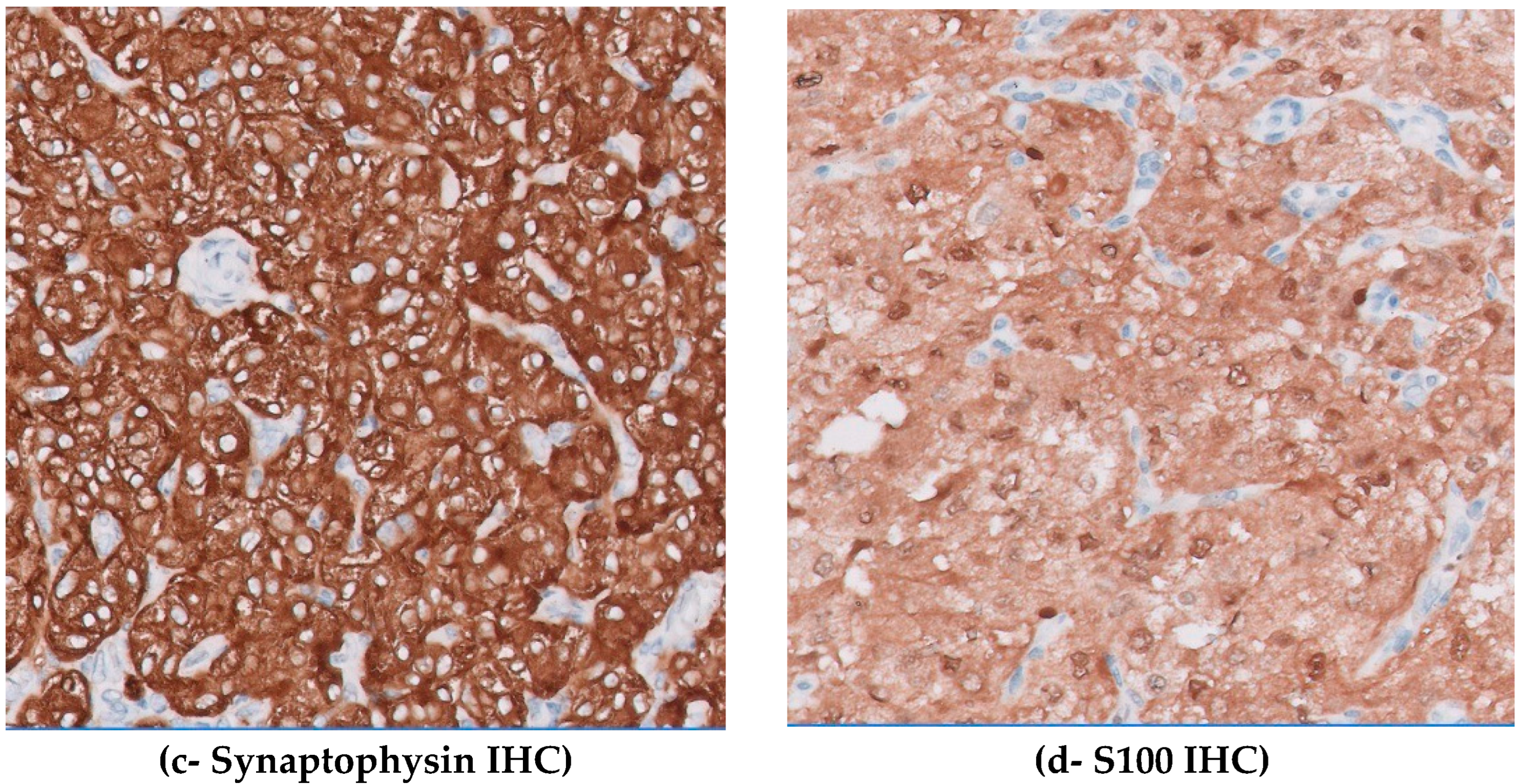
Figure 2.
(A) Microscopic H&E images show tumor cells arranged in nested and zellballen pattern with tumor cells showing abundant clear to granular cytoplasm having vesicular chromatin, clumped chromatin, and small nucleoli (A:100×; B: 200×). Spindle cells are also seen at the periphery of the tumor nests. (C) Immunohistochemistry synaptophysin is strongly positive in tumor cells (200×). (D) S100 is positive in sustentacular cells and faint cytoplasmic staining in chief cells is shown (200×). H&E: hematoxylin and eosin.
Figure 2.
(A) Microscopic H&E images show tumor cells arranged in nested and zellballen pattern with tumor cells showing abundant clear to granular cytoplasm having vesicular chromatin, clumped chromatin, and small nucleoli (A:100×; B: 200×). Spindle cells are also seen at the periphery of the tumor nests. (C) Immunohistochemistry synaptophysin is strongly positive in tumor cells (200×). (D) S100 is positive in sustentacular cells and faint cytoplasmic staining in chief cells is shown (200×). H&E: hematoxylin and eosin.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



