
Submitted:
01 August 2024
Posted:
02 August 2024
You are already at the latest version
Abstract
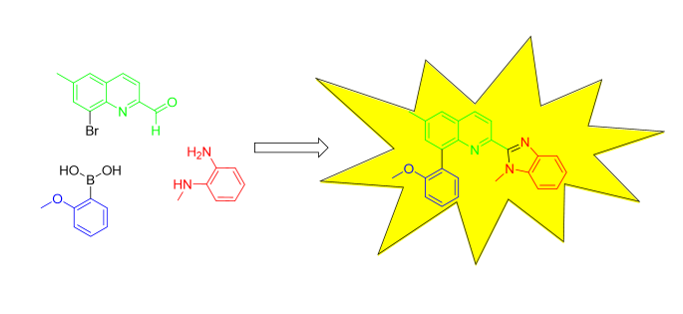
For very first time, we report the synthesis of 8-(2-methoxyphenyl)-6-methyl-2-(1-methyl-1H-benzo[d]imidazol-2-yl)quinoline 1. This was achieved in several steps, including usage of Suzuki reaction for functionalization of 2-(1H-benzo[d]imidazol-2-yl)quinoline moiety. The new compound exhibits blue fluorescence. Its structure was confirmed with 1D- and 2D-NMR spectroscopy, IR-spectroscopy, high-resolution mass spectrometry and X-ray analysis.
Keywords:
quinoline
; benzimidazole
; fluorescence
; single crystal diffraction
; Suzuki reaction
1. Introduction
Heterocyclic systems with 2-(1H-benzo[d]imidazol-2-yl)quinoline moiety can be intriguing for various fields of chemical research and application. This statement is based on several reasons - first, a great amount of quinoline derivatives exhibit biological activity and are used [1] as antimalarial, antiviral, antibacterial and anticancer pharmaceuticals. Second, benzimidazole derivatives are well known [2,3] for their analgesic, antiproliferative, antidiabetic, anticancer, anti-inflammatory and antioxidant activity. Third, 2-(1H-benzo[d]imidazol-2-yl)quinolines were studied [4,5] for cytotoxic, antimicrobial, insecticidal and herbicidal activity. Beyond their biological activity, 2-(1H-benzo[d]imidazol-2-yl)quinolines have been used as ligands [6,7,8], coordinated to transition metals.
Benzoimidazolylquinolines could be prepared by following several main synthetic strategies: from 1) quinaldines and benzene-1,2-diamine in the presence of mild oxidant (elemental suphur [4,9] or I2/DMSO [5,10] ), 2) carbaldehyde [11] or carboxylic acid [6,7] and benzene-1,2-diamine, and 3) trichalomethylquinoline and benzene-1,2-diamine [12]. A similar way could be applied for the synthesis of N-substituted in the benzimidazole system members of this class of compounds [8].
Herein, we report the synthesis of a new fluorescent compound, 8-(2-methoxyphenyl)-6-methyl-2-(1-methyl-1H-benzo[d]imidazol-2-yl)quinoline 1. This is the first benzoimidazolylquinoline representative with aromatic substituent in the quinoline system. Its structure and optical properties were also studied.
2. Results and Discussion
2.1. Synthesis
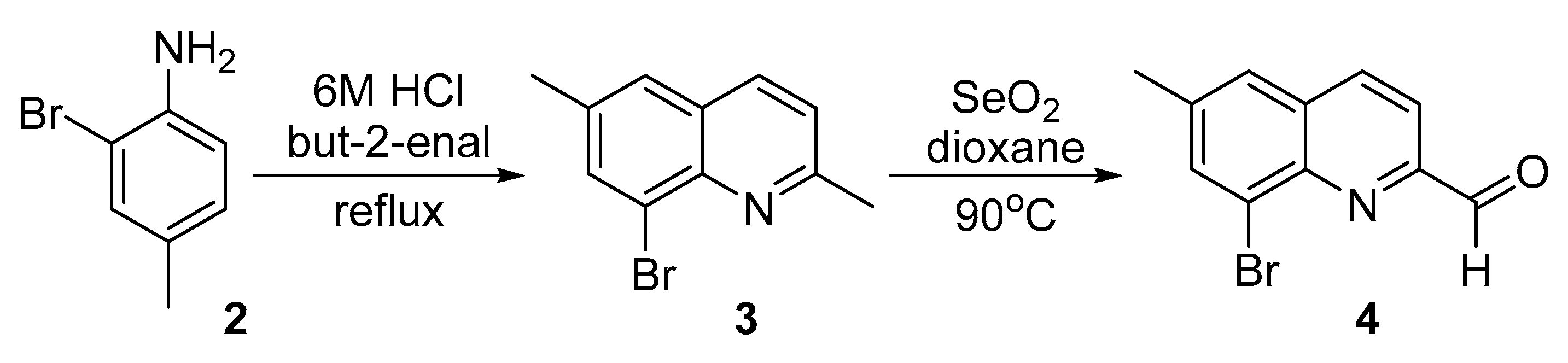
The synthesis of the new compound, 8-(2-methoxyphenyl)-6-methyl-2-(1-methyl-1H-benzo[d]imidazol-2-yl)quinoline 1, was achieved in several steps (Scheme 1, Scheme 2 and Scheme 3). First, 2-bromo-4-methylaniline 2 [13] was cyclized with but-2-enal (crotonaldehyde) to 8-bromo-2,6-dimethylquinoline 3 (Scheme 1). The crude product was purified via its ZnCl2-complex [14], which is insoluble in water and most organic solvents. This quinoline was reported previously [15], however a detailed procedure was not described. Later the isolated in pure form 8-bromo-2,6-dimethylquinoline 3 was oxidized with selenium dioxide to 8-bromo-6-methylquinoline-2-carbaldehyde 4, using procedure [16], previously applied for the 8-bromo-2-methylquinoline structure. It seems that these conditions are mild enough to oxidize the methyl group in 2-nd position, but to avoid the oxidation of the same group at 6-th position. Oxidation of the last one is possible, but at harsh reaction conditions [17]. The obtained aldehyde was previously noted in the literature [18], however again the synthetic procedure and compound’s physical properties were not reported.
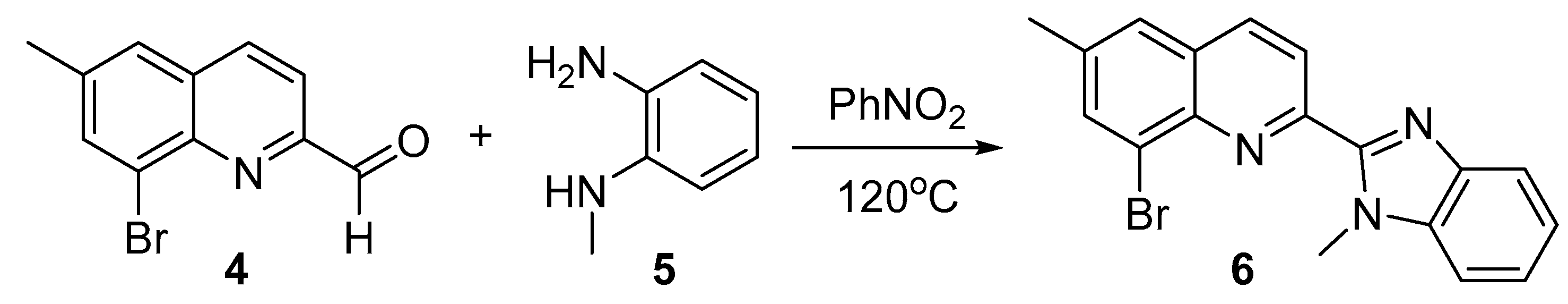
In the subsequent step (Scheme 2), 8-bromo-6-methylquinoline-2-carbaldehyde 4 was reacted with N1-methylbenzene-1,2-diamine 5 at high temperature in dry nitrobenzene media [19], serving at the same time as a solvent and as a mild oxidant reagent.
The resulting 8-bromo-6-methyl-2-(1-methyl-1H-benzo[d]imidazol-2-yl)quinoline 6 has not been published in the literature before. Moreover, it is the first heterocyclic system with 2-(1H-benzo[d]imidazol-2-yl)quinoline backbone, bearing bromine atom in the phenyl ring of the quinoline moiety. This structure is of a great importance because such type of halogens could be substituted with different organic groups by transition metal catalyzed reaction as we managed to do later in this research.
In the final step (Scheme 3), 8-bromo-6-methyl-2-(1-methyl-1H-benzo[d]imidazol-2-yl)quinoline 6 was used as a substrate in Pd(dppf)Cl2-catalyzed Suzuki reaction [20]. As its coupling partner 2-methoxyphenylboronic acid 7 was used.
After the performed Suzuki reaction (Scheme 3), 8-(2-methoxyphenyl)-6-methyl-2-(1-methyl-1H-benzo[d]imidazol-2-yl)quinoline 1 was isolated in pure form. It is a new compound and the first reported 8-phenyl substituted 2-(1H-benzo[d]imidazol-2-yl)quinoline. Its successful synthesis proves that the bromine atom in position 8-th is reactive in Suzuki reaction. On the other hand, this result demonstrates that Pd(dppf)Cl2 is suitable catalyst for functionalization of benzoimidazolylquinoline system with moderately sterically hindered boronic acids as 2-methoxyphenylboronic acid.
2.2. NMR Studies
All the structures of the obtained compounds were analyzed with NMR spectroscopy, 1H, 13C, COSY, HSQC and HMBC experiments were performed. See Supplementary Materials for copies of the NMR-spectra.
In the proton spectrum of 8-bromo-2,6-dimethylquinoline 3 two signals attributed to the two CH3-groups and four signals for the aromatic protons were observed (Figures S10 and S11). The signal of the CH3-group in the 2-nd position was observed in lower field, compared to the signal of the other methyl substituent.
In the proton spectrum of 8-bromo-6-methylquinoline-2-carbaldehyde 4 characteristic signal at 10.20 ppm was observed (Figures S17 and S18), related to the aldehyde group. The same substituent is responsible for the low fielded signal in the 13C-spectrum, observed at 193.46 ppm (Figures S19 and S20). In the proton spectrum (Figures S24 and S25) of 8-bromo-6-methyl-2-(1-methyl-1H-benzo[d]imidazol-2-yl)quinoline 6 signals for two methyl groups could be seen, respectively at 2.45 ppm for the CH3-group from the quinoline and at 4.51 ppm for the CH3-group from the benzoimidazolyl moiety. The most strongly shifted in low field are the signals in the 13C-spectrum of the compound correspond to the carbon atom in the second positions in the quinoline and benzoimidazolyl systems (Figures S26 and S27). A prove for the successful arylation by the Suzuki reaction could be seen from the proton spectra of 8-(2-methoxyphenyl)-6-methyl-2-(1-methyl-1H-benzo[d]imidazol-2-yl)quinoline 1 (Figures S1 and S2) and 8-bromo-6-methyl-2-(1-methyl-1H-benzo[d]imidazol-2-yl)quinoline 6 (Figures S24 and S25). By comparing the data, in the spectra of 1 additional signals for the alkyl protons and the aromatic system were determined.
2.3. Single Crystal Diffraction Studies
Suitable for single crystal x-ray diffraction measurements crystals were obtained by crystallization in acetonitrile. The titled compound 1 crystallizes in the triclinic P-1 space group with one molecule in the asymmetric. The molecular structure of the compound is shown in Figure 1 and the crystal data is summarized in Table 1. The CCDC deposition file can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033).
The crystal packing is shown in Figure 2 where weak intermolecular π – π stacking is observed between the two adjacent molecules in the unit cell (summarized in Table 2).
Crystal structure is additionally stabilized by intermolecular C – H ∙∙∙π interactions all with C – π distance less than 4 Å indicating relatively strong interactions. These interactions are summarized in Table 3 and shown in Figure S31.
2.4. Optical Properties
The recorded UV-Vis absorption spectra of the titled compound 1 in four different solvents are shown in Figure 3. The compound shows broad absorption peak in the range of 240 – 260 nm which can be attributed to π → π* transition due to the conjugated π-system. The broad emission band in the range of 270 – 320 nm is attributed to the presence of a lone pair (n → π*) of heteroatoms in the conjugated system (in this case nitrogen atoms). The last observed region of interest is between 325 – 375 nm involving three distinctive emission bands which are due to the extended conjugation system. Interestingly, these bands show solvatochromic effect and they are bathochromically shifted in respect to lowering the polarity of the solvent. Furthermore, the FWHM has lower values for the less polar solvents which is well expressed in pentane (the non-polar solvent of the four used).
The compound exhibits well defined fluorescence properties in the UV-blue region having a relatively large Stokes shift (λA - λF = 57 nm) when irradiated at λex = 250 nm (Figure 4). Different solvents had no effect on the fluorescence properties of the compound.
4. Materials and Methods
Melting points were determined on an SRS MPA120 EZ-Melt apparatus.
1H and 13C NMR spec-139 tra were recorded on a Bruker AVNEO 400 spectrometer (at 400 MHz for 1H and 100.6 140 MHz for 13C respectively. Chemical shifts are given in ppm. The NMR-spectra of all synthesized compounds could be found in the Supplementary Materials. Liquid chromatography mass spectrometry analysis (LC-HRAM) was carried out on Q Exactive® hybrid quadrupole-Orbitrap® mass spectrometer (ThermoScientific Co, Waltham, MA, USA) equipped with a HESI® (heated electrospray ionization) module, TurboFlow® Ultra High Performance Liquid Chromatography (UHPLC) system (ThermoScientific Co, Waltham, MA, USA) and HTC PAL® autosampler (CTC Analytics, Zwingen, Switzerland). The chromatographic separations of the analyzed compounds were achieved on Nucleoshell C18 (100 × 2.1 mm, 2.7 μm) analytical column (Macherey-Nagel, Düren, Germany). Full-scan mass spectra over the m/z range 100–600 were acquired in positive ion mode at resolution settings of 140,000. The used mass spectrometer operating parameters were: spray voltage—4.0 kV; capillary temperature—320 °C; probe heater temperature—300 °C; sheath gas flow rate 40 units; auxiliary gas flow 12 units; sweep gas 2 units (units refer to arbitrary values set by the Q Exactive Tune software); and S-Lens RF level of 50.00. Nitrogen was used for sample nebulization and collision gas in the HCD cell. All derivatives were quantified using 5 ppm mass tolerance filters to their theoretically calculated m/z values.
The data set for single crystal x-ray diffraction analysis was collected using a Rigaku XtaLAB Synergy-S diffractometer with a microfocus sealed tube and a HyPix-6000HE Hybrid Photon Counting (HPC) detector. Monochromated MoKa radiation (l = 0.71073 Å) was used. Data was collected at 130(2) K and corrected for absorption effects using the multi-scan method. The structure was solved by direct methods using SHELXT [21] and was refined by full matrix least squares calculations on F2 (SHELXL2019 [22]) in the graphical user interface Shelxle [23].
All non-H-atoms were located in the electron density maps and refined anisotropically. C-bound H atoms were placed in positions of optimized geometry and treated as riding atoms. Their isotropic displacement parameters were coupled to the corresponding carrier atoms by a factor of 1.2 (CH) or 1.5 (CH3). The structure was refined as a two-component non-merohedral twin. Both components were found in the reciprocal lattice viewer of Crysalis-Pro (Rigaku). The structure was solved using the hklf4 file, whereas for the final refinement the hkfl5 was used (BASF 0.250(1). Component 2 was rotated by 179.8° around [0.85 0.42 0.32] in the reciprocal lattice.
The UV-VIS spectra of the titled compound 1 in different solvents (ethanol, pentane, acetonitrile, dichloromethane all of spectroscopic grade obtained by Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) were measured using Evolution 300 UV/VIS spectrometer (Thermo Scientific, Dreieich, Germany).
The fluorescence spectrum was recorded using Varian Cary Eclipse spectrometer (Agilent, Santa Clara, California, USA) equipped with a 150 W Xe flash lamp as an excitation source in acetonitrile solution.
The FT-IR analysis wеre carried out on a FT-IR Nicolet 6700-Thermo Scientific in KBr pellets.
N1-methylbenzene-1,2-diamine and 2-methoxyphenylboronic acid were purchased from Fluorochem and were used without further purification. All the other solvents and reagents were purchased from local suppliers. Nitrobenzene was dried by storing for one week over CaH2 and used without distillation.
Synthesis of 8-Bromo-2,6-dimethylquinoline 3
In a 100 mL round-bottom flask 2-bromo-4-methylaniline (22.300 g, 0.12 mol, 1 eqv) and 60 mL 6M HCl were mixed. The mixture was heated to reflux. Crotonaldehyde (9.250 g, 0.13 mol, 1.1 eqv) was added dropwise (for 25 minutes) to the boiling solution. The heating continued for 3.5 hours and the reaction mixture was cooled to room temperature. The solution was washed with diethyl ether (80 mL). The crude reaction mixture was transferred to a beaker and to it was added solution of ZnCl2 (16.360 g, 0.12 mol, 1 eqv) in minimal amount of water. The formed suspension was stirred for 30 min at room temperature, 30 min in an ice bath and then filtered. The yellow precipitate was transferred to a beaker with 50 mL ice water, stirred and filtered again. Zinc-quinoline complex was washed in the same manner, using 50 mL cold 3M HCl, 50 mL cold water, 100 mL isopropanol and finally 100 mL diethyl ether. The pale yellow crystalline solid has dried (18.1 g) and transferred to a beaker with 25 mL conc. aqueous NH3 and 50 mL ethyl acetate. The resulting emulsion was stirred for 5 min and the aqueous layer was extracted with dichloromethane. Combined organic layers were dried with Na2SO4 and the solvent was removed under reduced pressure, leaving pale yellow crystalline solid (11.667 g, 41%), m.p.= 92°-92.5°C (methanol), lit., [15] 96.0-97.0 °C (benzene).
1H NMR (500 MHz, CDCl3) δ 7.94 (d, J = 8.4 Hz, 1H, H4), 7.88 (d, J = 1.7 Hz, 1H, H7), 7.50 (s, 1H, H5), 7.30 (d, J = 8.4 Hz, 1H, H3), 2.81 (s, 3H, CH3-C2), 2.50 (s, 3H, CH3-C6). 13C NMR (126 MHz, CDCl3) δ 159.47 (4C2), 143.41 (4C8a), 136.09 (4C6), 135.94 (C4), 135.00 (C7), 127.65 (4C4a), 126.52 (C5), 123.71 (4C8), 122.83 (C3), 25.62 (CH3-C6), 21.10 (CH3-C2). HRMS (ESI) m/z calculated for [M+H]+ 236.00704, found 236.00693 (ppm: 0.47).
Synthesis of 8-Bromo-6-methylquinoline-2-carbaldehyde 4
In air, selenium dioxide (3.055 g, 27.5 mmol, 1.3 eqv) was dissolved in 110 mL dioxane. The resulting transparent colorless solution was heated at 90oC for 30 minutes. To the mixture 8-bromo-2,6-dimethylquinoline (5.000 g, 21.2 mmol, 1 eqv) was added and almost immediately the solution turned brown-red and dark precipitate was formed. The resulting suspension was heated at 90oC for 3.5 hours more, while monitored with TLC using alumina plates and mobile phase hexanes:dichloromethane=2:1 (on silica the starting quinoline and the respective carbaldehyde are indistinguishable). The reaction mixture was cooled to room temperature, filtered through celite pad and the volatiles were removed under reduced pressure. To the pale brown residue dichloromethane (150 mL) was added and the insoluble particles were filtered. The clear solution was transferred to a separatory funnel with 50mL saturated aqueous Na2CO3 solution. The water layer was extracted with dichloromethane and the combined organic extracts were dried with Na2SO4. After removal of the solvent, the crude product was purified by using column flash chromatography, mobile phase hexanes:dichloromethane from 3:1 to 1:1. The aldehyde was isolated as yellow crystals (4.75 g, 90%), m.p.=141°-142°C (dichloromethane). Rf=0.55 (hexanes:CH2Cl2=1:1) on silica.
1H NMR (500 MHz, CDCl3) δ 10.20 (s, 1H, CHO), 8.13 (d, J = 8.4 Hz, 1H, H4), 7.96 (d, J = 8.4 Hz, 1H, H3), 7.93 (d, J = 1.6 Hz, 1H, H7), 7.55 (s, 1H, H5), 2.49 (s, 3H, CH3). 13C NMR (126 MHz, CDCl3) δ 193.46 (CHO), 152.46 (4C2), 143.62 (4C8a), 140.34 (4C6), 137.28 (C4), 136.35 (C7), 131.30 (4C4a), 126.65 (C5), 125.65 (4C8), 118.16 (C3), 21.55 (CH3). HRMS (ESI) m/z calculated for [M+H]+ 249.98650, found 249.98619 (ppm: 1.24).
Synthesis of 8-Bromo-6-methyl-2-(1-methyl-1H-benzo[d]imidazol-2-yl)quinoline 6
In air, 8-bromo-6-methylquinoline-2-carbaldehyde (0.310 g, 1.24 mmol. 1 eqv) and N1-methylbenzene-1,2-diamine (0.151 g, 1.24 mmol. 1 eqv) were dissolved in 5 mL dry nitrobenzene. The solution was heated at 120oC for 22 hours, cooled to room temperature and directly introduced in silica loaded flash chromatography column. The product was isolated in the form of brown solid (0.383 g, 88%), m.p.= 194°-195°C (ethyl acetate). Rf=0.50 (CH2Cl2:ethyl acetate=8:1) on silica.
1H NMR (500 MHz, CDCl3) δ 8.54 (d, J = 8.6 Hz, 1H, H3-quinoline), 8.07 (d, J = 8.6 Hz, 1H, H4-quinoline), 7.84 (d, J = 1.5 Hz, 1H, H7-quinoline), 7.79 (d, J = 7.8 Hz, 1H, H4-benzoimidazolyl), 7.47 (s, J = 8.5 Hz, 1H, H5-quinoline), 7.40 (d, J = 8.0 Hz, 1H, H7-benzoimidazolyl), 7.29 (t, J = 7.2 Hz, 1H, H6-benzoimidazolyl), 7.25 (t, J = 7.8 Hz, 1H, H5-benzoimidazolyl), 4.51 (s, 3H, CH3-benzoimidazolyl), 2.45 (s, 3H, CH3-quinoline). 13C NMR (126 MHz, CDCl3) δ 150.30 (4C2-quinoline), 149.45 (4C2-benzoimidazolyl), 142.92 (4C8а-quinoline), 142.62 (4C3а-benzoimidazolyl), 138.03 (C6-quinoline), 137.69 (4C7а-benzoimidazolyl), 136.39 (C4-quinoline), 135.41 (C7-quinoline), 128.89 (4C4а-quinoline), 126.49 (C5-quinoline), 124.98 (4C8-quinoline), 123.78 (C6-benzoimidazolyl), 122.74 (C5-benzoimidazolyl), 122.37 (C3-quinoline), 120.25 (C4-benzoimidazolyl), 110.11 (C7-benzoimidazolyl), 33.85 (CH3-benzoimidazolyl), 21.38 (CH3-quinoline). HRMS (ESI) m/z calculated for [M+H]+ 352.04530, found 352.04438 (ppm: 2.61).
Synthesis of 8-(2-Methoxyphenyl)-6-methyl-2-(1-methyl-1H-benzo[d]imidazol-2-yl)quinoline 1
In inert atmosphere, 8-bromo-6-methyl-2-(1-methyl-1H-benzo[d]imidazol-2-yl)quinoline (0.176 g, 0.5 mmol, 1 eqv), 2-methoxyphenylboronic acid (0.091 g, 0.6 mmol, 1.2 eqv), K3PO4.7H2O (0.508 g, 1.5 mmol, 3 eqv) and Pd(dppf)Cl2 (0.0183 g, 25 µmol, 5 mol %) were suspended in mixture of 5 mL THF and 2 ml water. The resulting biphasic system was stirred at 70oC for 19 hours, cooled to room temperature and diluted with 50 mL ethyl acetate. The ethyl acetate solution was dried with Na2SO4 and the volatiles were removed under reduced pressure. The dark residue was purified using flash column chromatography on silica, eluting with mobile phase hexanes:ethyl acetate from 8:1 to 2:1. The resulting compound was recrystallized from ethyl acetate. The desired product was isolated in pure form in the form of pale-yellow solid (0.120 g, 63%), m.p.= 193°-195°C (ethyl acetate). Rf=0.44 (hexanes:ethyl acetate=2:1) on silica.
1H NMR (500 MHz, CDCl3) δ 8.46 (d, J = 8.6 Hz, 1H, H3-quinoline), 8.12 (d, J = 8.6 Hz, 1H, H4-quinoline), 7.77 – 7.71 (m, 1H, H4-N-benzoimidazolyl), 7.55 (s, 1H, H5-quinoline), 7.48 (d, J = 1.8 Hz, 1H, H7-quinoline), 7.34 (td, J = 8.3, 1.7 Hz, 1H, H4-2-methoxyphenyl), 7.26 (dd, J = 7.4, 1.7 Hz, 1H, H6-2-methoxyphenyl), 7.25 – 7.17 (m, 3H, H5,H6 and H7-N-benzoimidazolyl), 6.99 (td, J = 7.4, 0.8 Hz, 1H, H5-2-methoxyphenyl), 6.94 (d, J = 8.3 Hz, 1H, H3-2-methoxyphenyl), 3.75 (s, 3H, CH3-N-benzoimidazolyl), 3.57 (s, 3H, CH3-2-methoxyphenyl), 2.51 (s, 3H, CH3-quinoline). 13C NMR (126 MHz, CDCl3) δ 157.39 (4C2-2-methoxyphenyl), 150.24 (4C2-benzoimidazolyl), 148.96 (4C2-quinoline), 144.58 (4C8a-quinoline), 142.58 (4C7a-benzoimidazolyl), 138.54 (4C1-2-methoxyphenyl), 137.50 (4C3a-benzoimidazolyl), 136.91 (4C6-quinoline), 135.99 (C4-quinoline), 132.96 (C7-quinoline), 131.77 (C6-2-methoxyphenyl), 129.56 (4C8-quinoline), 128.78 (C4-2-methoxyphenyl), 127.79 (4C4a-quinoline), 126.32 (C5-quinoline), 123.32 (C6-benzoimidazolyl), 122.46 (C5-benzoimidazolyl), 121.32 (C3-quinoline), 120.22 (C5-2-methoxyphenyl), 120.07 (C4-benzoimidazolyl), 110.59 (C3-2-methoxyphenyl), 109.80 (C7-benzoimidazolyl), 55.59 (CH3-2-methoxyphenyl), 32.56 (CH3-N-benzoimidazolyl), 21.80 (CH3-quinoline). HRMS (ESI) m/z calculated for [M+H]+ 380.17633, found 380.17574 (ppm: 1.55). FT-IR (KBr/cm-1): 2933m, 1911w, 1728w, 1598s, 1474w, 1435s, 1393w, 1238s, 1183w, 924w, 874s, 740s (Figure S32).
5. Conclusions
The single crystal diffraction analysis shows the presence of weak π – π stacking between the two adjacent molecules in the same unit cell and relatively strong C-H∙∙∙π interaction between the molecules in neighboring unit cells. The material is a candidate for the construction a blue emitting organic light emiting diod due to his fluorescent properties.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figures S1–S32. 1H, 13C, COSY, HSQC, HMBC NMR spectra of all compounds, UV, fluorescent and IR spectra of compound 1.
Author Contributions
Conceptualization, R.L. and J.Z.; methodology, M.I., J.Z., M.T., V.L., B.M. and R.L.; software, M.T., R.L.; validation, M.I., J.Z., M.T., V.L., B.M. and R.L.; formal analysis, R.L., M.T.and J.Z.; investigation, M.I., J.Z., M.T., V.L., B.M. and R.L.; resources, R.L.,J.Z. and M.T.; data curation, R.L.,J.Z. and M.T.; writing—original draft preparation, R.L.,M.I.,J.Z. and M.T.; writing—review and editing, R.L.,J.Z. and M.T.; visualization, R.L.,J.Z. and M.T.; supervision, R.L. and J.Z.; project administration, J.Z.; funding acquisition, J.Z. All authors have read and agreed to the published version of the manuscript.
Funding
Please add: This research was funded by BG Fund for Scientific Investigations (project no. KP-06-N39/2019).
Data Availability Statement
Additional research data can be obtained from the corresponding author.
Acknowledgments
Instrumentation and technical assistance for this work were provided by the Service Center X-ray Diffraction, with financial support from Saarland University and German Science Foundation (project number INST 256/582-1.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Matada, B.S.; Pattanashettar, R.; Yernale, N.G. A comprehensive review on the biological interest of quinoline and its derivatives. Bioorg. Med. Chem. 2021, 32, 115973–115998. [Google Scholar] [CrossRef]
- Mohammed, L.A.; Farhan, M.A.; Dadoosh, S.A.; Alheety, M.A.; Majeed, A.H.; Mahmood, A.S.; Mahmoud, Z.H. A Review on Benzimidazole Heterocyclic Compounds: Synthesis and Their Medicinal Activity Applications. SynOpen 2023, 7, 652–673. [Google Scholar]
- Ebenezer, O.; Oyetunde-Joshua, F.; Omotoso, O.D.; Shapi, M. Benzimidazole and its derivatives: Recent Advances (2020–2022). Results Chem. 2023, 3, 100925–100955. [Google Scholar] [CrossRef]
- Hisano, T.; Ichikawa, M.; Tsumoto, K.; Tasaki, M. Synthesis of Benzoxazoles, Benzothiazoles and Benzimidazoles and Evaluation of Their Antifungal, Insecticidal and Herbicidal Activities. Chem. Pharm. Bull. 1982, 30, 2996–3004. [Google Scholar] [CrossRef]
- Baig, M.F.; Shaik, S.P.; Nayak, V.L.; Alarifi, A.; Kamal, A. Iodine-catalyzed Csp3-H functionalization of methylhetarenes: One-pot synthesis and cytotoxic evaluation of heteroarenyl-benzimidazoles and benzothiazole. BMCL, 2017, 27, 4039–4043. [Google Scholar] [CrossRef]
- Dayan, O.; Tercan, M.; Özdemir, N. Syntheses and molecular structures of novel Ru(II) complexes with bidentate benzimidazole based ligands and their catalytic efficiency for oxidation of benzyl alcohol. J. Mol. Struct. 2016, 1123, 35–43. [Google Scholar] [CrossRef]
- Sahki, F.A.; Messaaadia, L.; Merazig, H.; Chibani, A.; Bouraiou, A.; Bouacida, S. Synthesis, X-ray structure and theoretical investigation of 2-(2’-quinolyl)benzimidazole metal complexes. J. Chem. Sci. 2017, 129, 21–29. [Google Scholar] [CrossRef]
- Wei, X.; Jiang, Y.; Cui, X.; Li, Y.; Wang, H.; Qi, X. 2-Benzoimidazol-8-alkylquinolinylnickel(II) complexes as efficient catalysts for ethylene oligomerization and vinyl polymerization of norbornene. J. Coord. Chem., 2015, 68, 3825–3838. [Google Scholar] [CrossRef]
- Hisano, T.; Hisano, K. Studies of Organosulphur Compounds. I. Reaction between Active Methyl Groups and o-Substituted Bifunctional Molecule in the Presence of Sulfur. Yakugaku Zasshi 1971, 91, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Yaragorla, S.; Babu, P.V. Oxidative Csp3-H functionalization of 2-methylazaarenes: A practical synthesis of 2-azaarenyl-benzimidazoles and benzothiazoles. Tetrahedron Lett. 2017, 58, 1879–1882. [Google Scholar] [CrossRef]
- Lee, J.S.; Song, I.; Warkad, S.D.; Seong, Y.G.; Shinde, P.B.; Song, K.; Nimse, S.B. Synthesis and evaluation of 2-Aryl-1H-benzo[d]imidazole derivatives as potential microtubule targeting agents. Drug Dev. Res. 2022, 83, 769–782. [Google Scholar] [PubMed]
- Tynebor, R.; Millings, E. Novel Method of Synthesizing Various Five Membered Heterocycles from an Aryl Tribromomethyl Group. Synth. Commun. 2013, 43, 1902–1908. [Google Scholar] [CrossRef]
- Johnson, J.R.; Sandborn, L.T. 3-Bromo-4-aminotoluene. Org. Synth. 1926, 6, 8–9. [Google Scholar]
- Leir, C.M. An Improvement in the Doebner-Miller Synthesis of Quinaldines. J. Org. Chem., 1977, 42, 911–913. [Google Scholar] [CrossRef]
- Garrod, R.E.; Jones, H.O.; Evans, P.E. Some quinoline and tetrahydroquinoline derivatives obtained from aldol bases. J. Chem. Soc., Trans., 1912, 101, 1389–1394. [Google Scholar] [CrossRef]
- Barsoum, D.N.; Kirinda, V.C.; Kang, B.J.; Kalow, A. Remote-Controlled Exchange Rates by Photoswitchable Internal Catalysis of Dynamic Covalent Bonds. J. Am. Chem. Soc. 2022, 144, 10168–10173. [Google Scholar] [CrossRef] [PubMed]
- Seynah, M.; Fernelius, W.C. Formazyl Complexes of the Quinoline Series. J. Org. Chem. 1957, 22, 217–219. [Google Scholar]
- Martínez-González, S.; Rodríguez-Arístegui, S.; Oliva, C.A.G.; Hernández, A.I.; Cantalapiedra, E.G.; Varela, C.; García, A.B.; Rabal, O.; Oyarzabal, J.; Bischoff, J.R.; Klett, J.; Albarrán, M.I.; Cebriá, A.; Ajenjo, N.; García-Serelde, B.; Gómez-Casero, E.; Cuadrado-Urbano, M.; Cebrián, D.; Blanco-Aparicio, C.; Pastor, J. Discovery of novel triazolo[4,3-b]pyridazin-3-yl-quinoline derivatives as PIM inhibitors. Eur. J. Med. Chem. 2019, 168, 87–109. [Google Scholar] [CrossRef]
- Kumar, G.; Paul, K.; Luxami, V. Deciphering the excited state intramolecular charge-coupled double proton transfer in an asymmetric quinoline–benzimidazole system. New J. Chem. 2020, 44, 12866–12874. [Google Scholar] [CrossRef]
- Saha, A.; Ghosh, A.; Guin, S.; Panda, S.; Mal, D.K.; Majumdar, A.; Akita, M.; Maiti, D. Substrate-Rhodium Cooperativity in Photoinduced ortho-Alkynylation of Arenes, Angew. Chem., Int. Ed. Engl. 2022, 61, e202210492. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT - Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr C Struct Chem 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt Graphical User Interface for SHELXL. J Appl Crystallogr 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Synthesis of 8-bromo-2,6-dimethylquinoline 3 and 8-bromo-6-methylquinoline-2-carbaldehyde 4.
Scheme 1.
Synthesis of 8-bromo-2,6-dimethylquinoline 3 and 8-bromo-6-methylquinoline-2-carbaldehyde 4.
Scheme 2.
Synthesis of 8-bromo-6-methyl-2-(1-methyl-1H-benzo[d]imidazol-2-yl)quinoline 6.
Scheme 3.
Synthesis of 8-(2-methoxyphenyl)-6-methyl-2-(1-methyl-1H-benzo[d]imidazol-2-yl)quinoline 1.
Scheme 3.
Synthesis of 8-(2-methoxyphenyl)-6-methyl-2-(1-methyl-1H-benzo[d]imidazol-2-yl)quinoline 1.
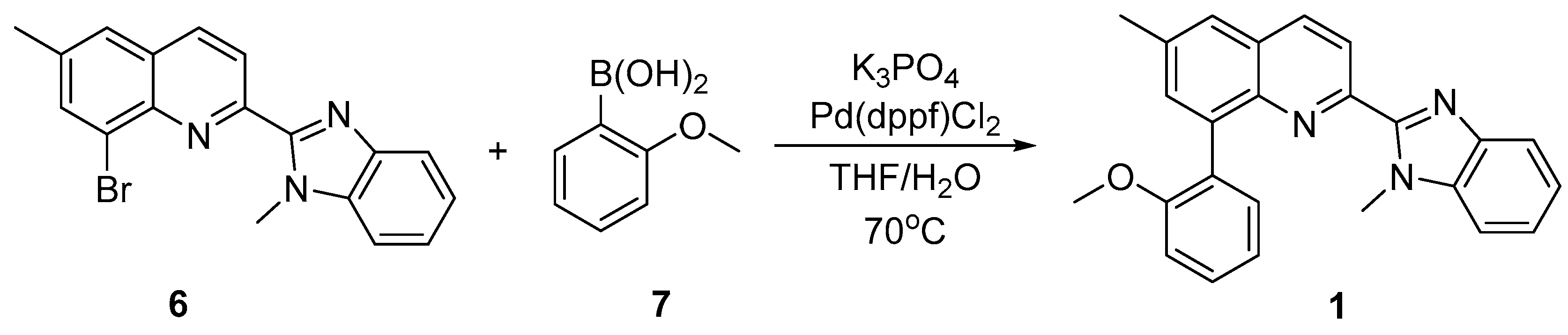
Figure 1.
Molecular structure of the titled compound. Thermal ellipsoids are at 50% level.
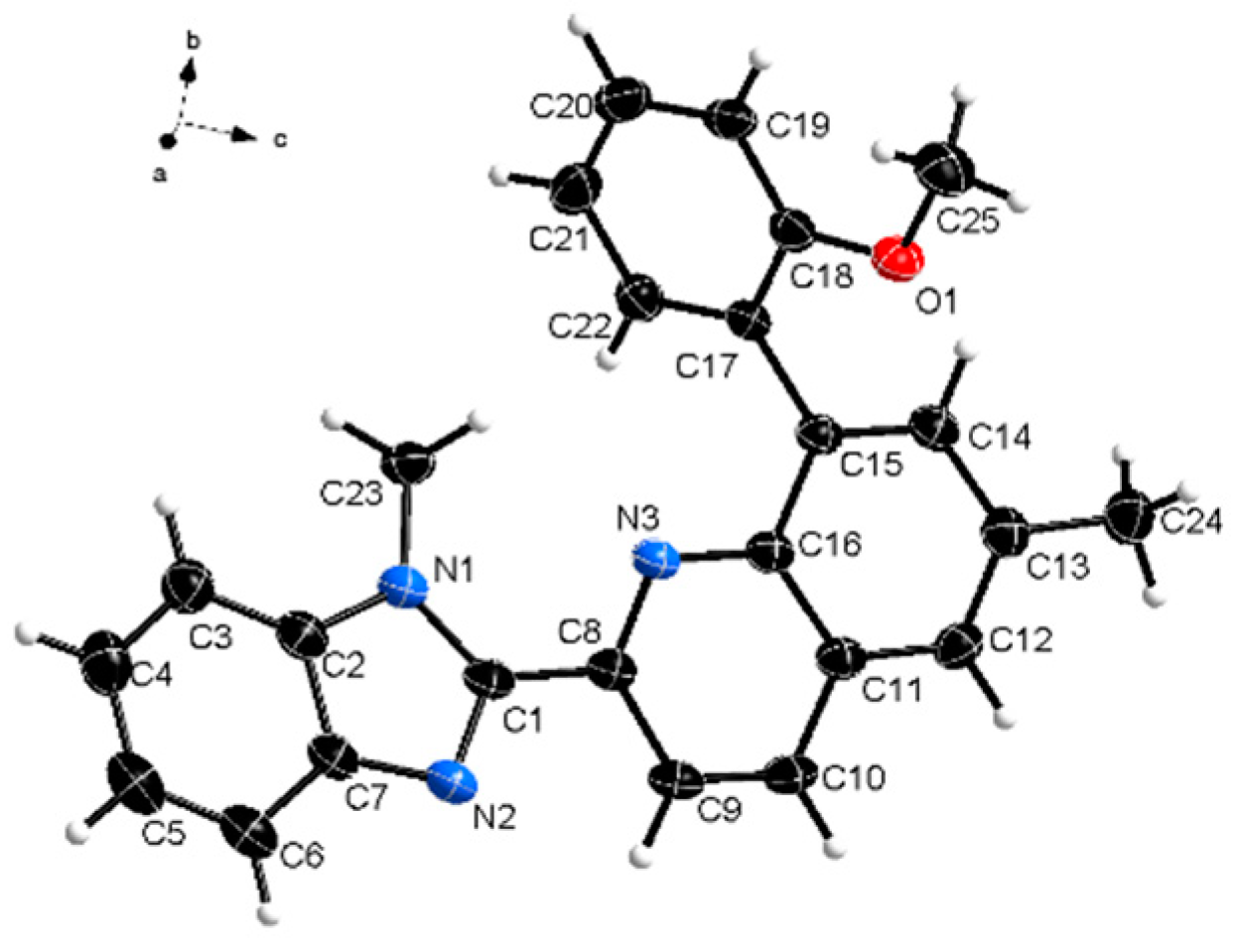
Figure 2.
Unit cell packing of the titled compound 1 with the π – π interactions showed with dashed green line. The thermal ellipsoids are at 50% and the hydrogen atoms are omitted for clarity.
Figure 2.
Unit cell packing of the titled compound 1 with the π – π interactions showed with dashed green line. The thermal ellipsoids are at 50% and the hydrogen atoms are omitted for clarity.
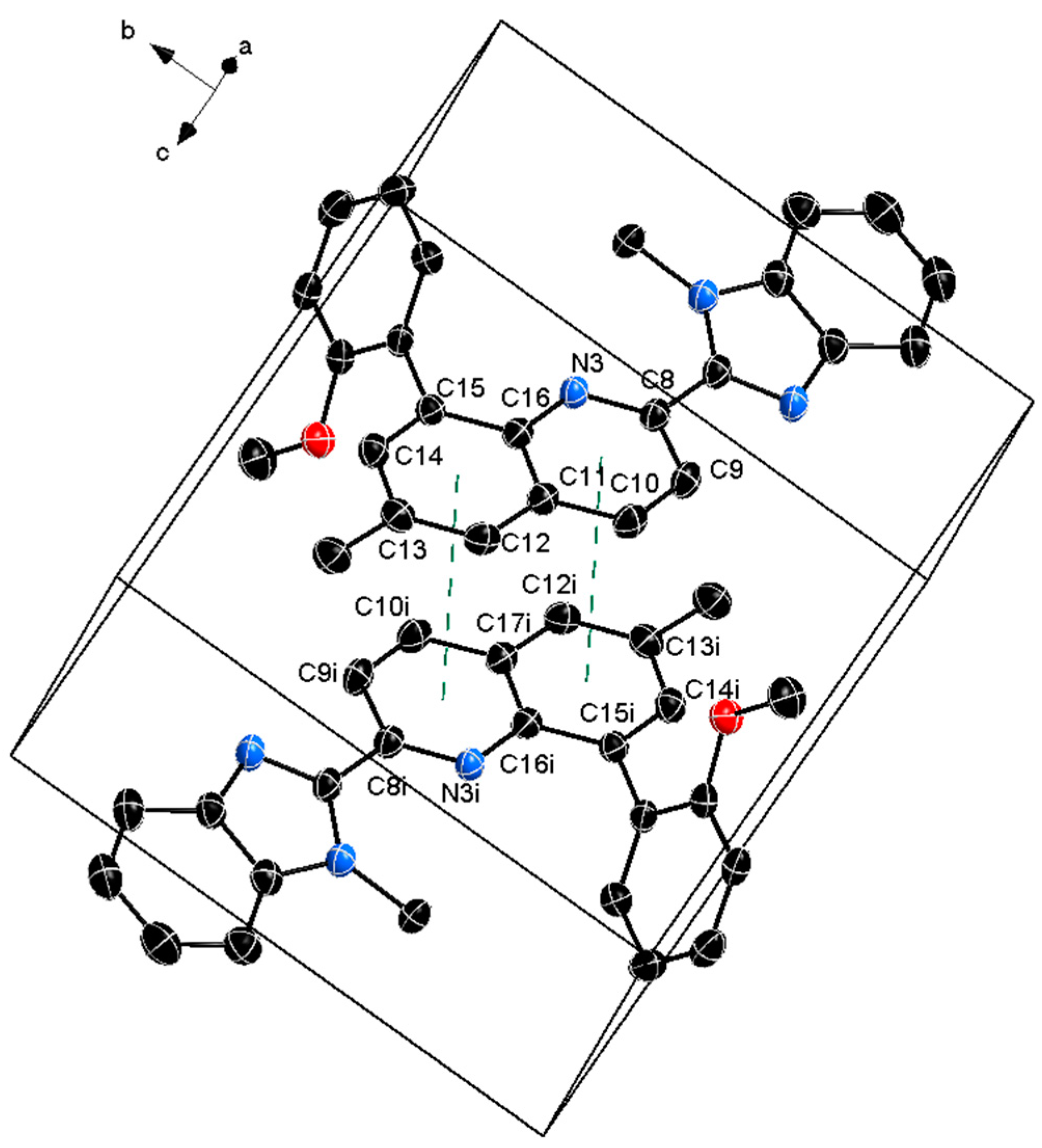
Figure 3.
UV-Vis absorption spectra of titled compound 1 in different solvents.
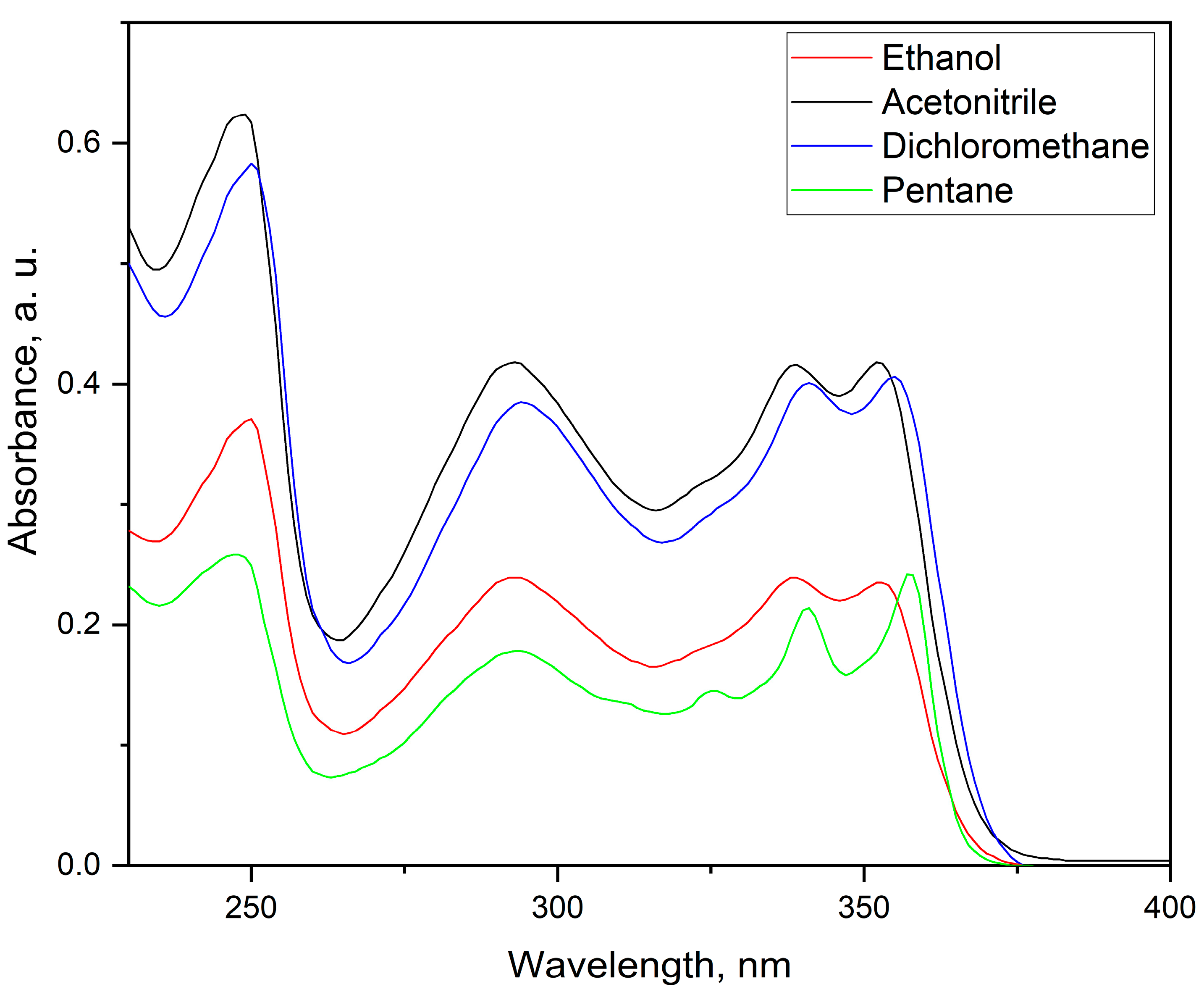
Figure 4.
Fluorescence spectrum of the titled compound.
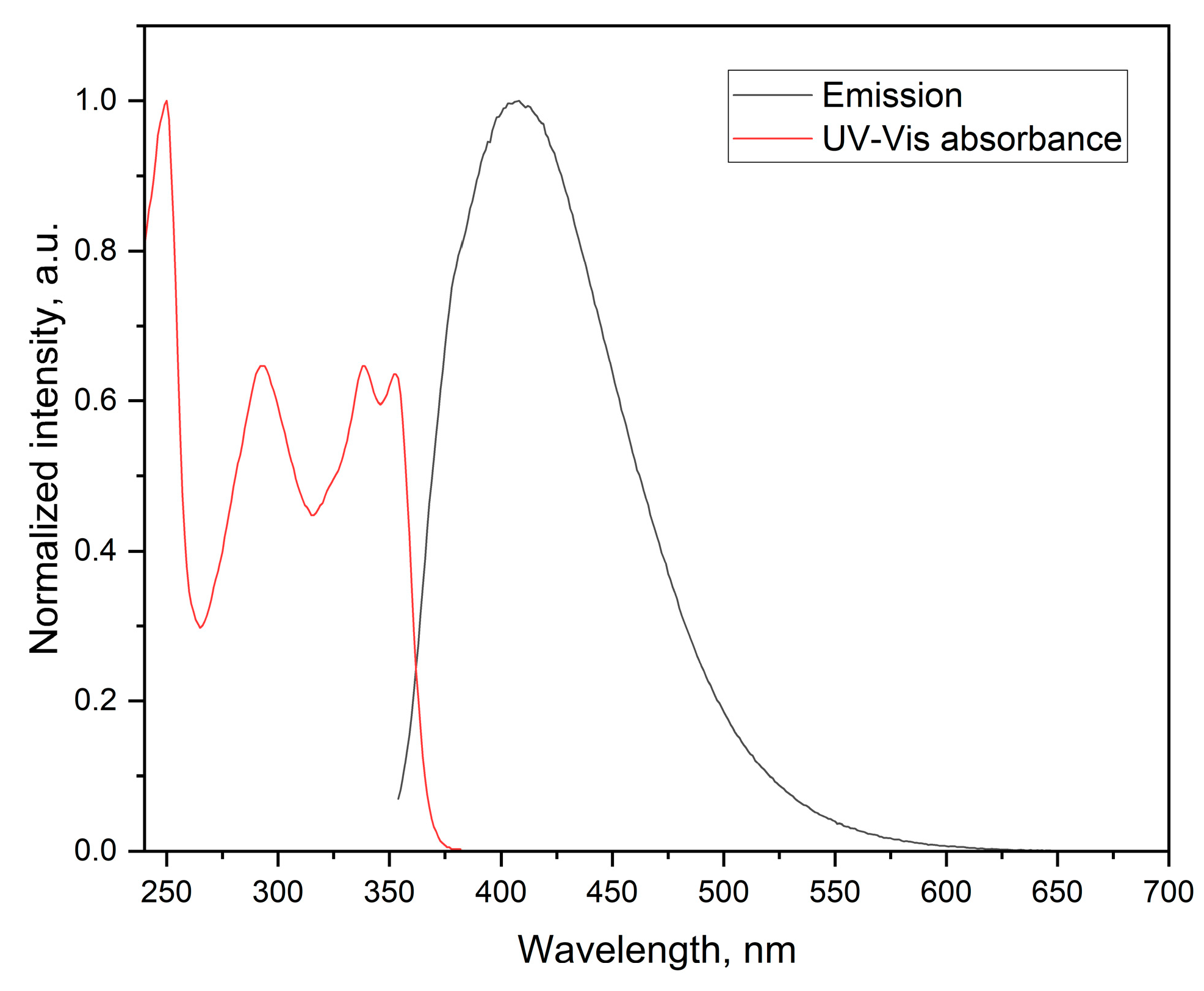

Table 1.
Experimental details of the single crystal diffraction.
| Crystal data | |
|---|---|
| CCDC numbers | 2374257 |
| Empirical formula | C25H21N3O |
| Molecular weight, (g/mol) | 379.45 |
| Crystal system, space group | Triclinic, P |
| Temperature (K) | 133(2) |
| a, b, c(Å) | 7.0467(5), 10.8556(7), 13.5823(9) |
| α, β, γ(°) | 79.469(6), 79.307(6), 71.684(6) |
| V, (Å3) | 960.56(12) |
| Z | 2 |
| Radiation type | Mo Kα |
| μ, (mm−1) | 0.082 |
| Crystal size (mm) | 0.200 x 0.080 x 0.060 |
| Data collection | |
| Diffractometer | Rigaku XtaLAB Synergy |
| Absorption correction | Spherical Harmonics implemented in SCALE3 ABSPACK scaling algorithm |
| Tmin, Tmax | 0.8728, 1.0000 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 4365, 4365, 3704 |
| (sin θ/λ)max, (Å−1) | 0.625 |
| Refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.0501, 0.1409, 1.066 |
| No. of reflections | 4365 |
| No. of restrains | 0 |
| No. of parameters | 266 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.169, -0.240 |
Table 2.
Observed π – π interactions where Cg2 and Cg4 are the centroids of the C11/C12 – C16 and N3 – C16, respectively. Cg2i and Cg4i are their respective counterparts generated by symmetry code (i) 1-x, 1-y, 1-z.
Table 2.
Observed π – π interactions where Cg2 and Cg4 are the centroids of the C11/C12 – C16 and N3 – C16, respectively. Cg2i and Cg4i are their respective counterparts generated by symmetry code (i) 1-x, 1-y, 1-z.
| π – π interaction | Cg – Cg, Å | α, o | Slippage, Å |
|---|---|---|---|
| Cg2 – Cg4i | 3.8224(14) | 0.58(11) | 1.639 |
| Cg2i – Cg4 | 3.8224(14) | 0.58(11) | 1.606 |
Table 3.
The intermolecular C – H ∙∙∙π interactions.
| Interaction | H∙∙∙π, Å | C∙∙∙ π, Å | C – H∙∙∙ π, o |
|---|---|---|---|
| C23 – H23B∙∙∙Cg3ii | 2.62 | 3.513(3) | 152 |
| C24 – H24A∙∙∙Cg5iii | 2.94 | 3.723(3) | 138 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



