
Submitted:
04 August 2024
Posted:
05 August 2024
You are already at the latest version
Abstract
Fibromyalgia (FM), classified by ICD-11 with code MG30.0, is a chronic debilitating disease characterized by wide-spread pain, fatigue, cognitive impairment, sleep and intestinal alterations, among other. FM affects a large proportion of the world-wide population, with increased prevalence among women. The lack of understanding of its etiology and pathophysiology hampers the development of effective treatments. Our group had developed a manual therapy (MT) pressure-controlled custom manual protocol on FM showing hyperalgesia/allodynia, fatigue and patient’s quality of life benefits in a cohort of 38 FM cases (NCT04174300). With the aim of understanding the therapeutic molecular mechanisms triggered by MT, this study interrogated PBMC transcriptomes from FM participants of this clinical trial using RNAseq and RT-qPCR technologies. The results showed that the salt-induced kinase SIK-1 was consistently downregulated by MT in FM, correlating with improvement of patient symptoms. In addition, the study compared the findings in a non-FM control cohort subjected to the same MT protocol evidencing that the changes in SIK1 with MT only occurred in individuals with FM. This positions SIK-1 as a potential biomarker to monitor response to MT, and as a therapeutic target of FM, to be further explored by continuation studies.
Keywords:
fibromyalgia
; myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS)
; pressure point threshold (PPT)
; physiotherapy
; manual therapy (MT)
; NCT04174300
; SIK1
1. Introduction
The latest version of the international classification of diseases (ICD-11) adopted by the WHO on May 2019 [1], classifies fibromyalgia (FM) as a multifactorial chronic primary widespread pain syndrome (code MG30.0) presenting diffuse pain in at least 4 of 5 body regions, anxiety, depression and overall functional disability [1].
Diagnosis is based on clinical criteria defined by the ACR (American College of Rheumatology) 1990 case definition with revisions [2,3]. Appropriate diagnosis should ensure pain is not directly attributable to a nociceptive process but consistent with nociplastic pain [1], caused by poorly understood mechanisms involving ongoing inflammation and general tissue damage, rather than local nerve damage (neuropathic pain) [4]. Additional symptoms include non-restorative sleep, fatigue, cognitive impairment and intestinal problems, overlapping with symptoms present in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) [5,6]. Presentation peaks between 20-55 years with marked increased prevalence in women [7,8]. Epidemiology reports vary across countries and regions with worldwide impact showing 2.7% of the general population and 3.7% in the Valencian Community of Spain studied here [7,8,9].
Because FM etiology and pathophysiology remain unknown, current treatments are directed to palliate symptoms, often leading to polypharmacy [10] and further health deterioration.
Clinical guidelines on non-pharmacological therapies include passive therapies such as hyperbaric oxygen therapy, repetitive transcranial magnetic stimulation, and other, including manual therapy (MT). Positive effect of physiotherapy (MT) on pain, physical capacity and quality of life have been repeatedly reported [11]. Our group had developed a MT pressure-controlled custom manual protocol on FM showing hyperalgesia/allodynia, fatigue and patient’s quality of life benefits in a cohort of 38 FM cases [12]. The registered clinical trial (NCT04174300) also built a biobanked collection of blood samples taken at different points of the treatment which were analyzed in this study to help understand the molecular mechanisms behind patients´ response to MT.
2. Results
2.1. Study Design, Demographics and Phenotyping
This prospective observational study evaluated changes in the immune system in response to treatment by measuring gene expression levels of PBMCs before and after eight sessions of controlled manual therapy (MT) (two weekly), as detailed in Methods, on FM patients (n=38) (NCT04174300 clinical trial) [12], and on non-FM volunteers (n=12). To assess participant baseline status and to monitor response to therapy questionnaire scores were obtained with the fibromyalgia impact questionnaire (FIQ) [13,14], the multi-fatigue inventory (MFI) [15] and the SF-36 quality of life instrument (Likert scale) [16]. In addition, pressure point thresholds (PPTs) at baseline and after treatment were measured in the FM, as previously described [12], as well as in the non-FM “control” cohort.
2.1.1. Demographics of Participants by Study Cohort
Patient cohort included 38 FM patients (35 females and 3 males) who fulfilled 1990 and/or 2010 ACR criteria [2,3], 50% (19/38) of them presenting comorbid ME/CFS according to Canadian and/or International diagnostic criteria [5,6], as previously described [12]. Average age for the FM cohort was 55.6 ± 7.2 years (range 43–71), and time from primary FM diagnosis over 3 years, 10.3 ± 7.5 years (range 3–21). A representative subcohort of 6 participants composed entirely by women, with an average age of 54 years ± 8.44 and a range of between 43 and 69 years, was selected for PBMC RNAseq analysis to evaluate the effects of the MT program on the immune system of FM (see section 2.2. for details).
On another side, a non-FM matched cohort of 12 female participants with an average age of 52.33 ± 6.2 years (range 43-61) was subjected to the same MT protocol towards investigating whether gene expression changes were specific for FM or, if by contrast, it was a generic response to MT. In this “control” non-FM cohort only 3 participants had a diagnosed pathology, assuming 25% of the sample. Pathologies were diabetes in one participant (8.33%), and osteoarthritis in 2 participants (16.66%), none having ever received an FM or ME/CFS diagnostic.
2.1.2. Participant Phenotyping
As mentioned in the Study design section above, all participants were finely phenotyped with the use of FIQ [13,14], MFI [15] and SF-36 (Likert scale) [16] validated questionnaires. As shown on Table 1, participating patients presented severe FM (Total FIQ>59), moderate fatigue with most domains being above the non-FM cohort with the exception of General Fatigue, and quality of life (SF-36) was much superior (>50 in all domains) on the non-FM cohort than the FM. No major baseline (pretreatment) differences were found between the subcohort of FM (n=6) subjected to RNAseq analysis, and the previously described complete FM cohort (n=38) [12], while the non-FM cohort statistically differed from the complete, as well as the sequenced sub-cohort. Individual participant scores available on Supplementary Table S1.
In addition, comparison of baseline PPTs showed that Low cervical points were the most sensitive while Gluteus, Trochanters and Knees were the most resistant to pain in both FM cohorts, with increased sensitivity in the Trapezius right point for the RNAseq FM subcohort (n=6) (Table 2). As expected, non-FM participants showed marked differences of resistance for pressure-triggered pain (Table 2). Individual participant PPT values available on Supplementary Table S2.
2.2. Differential Gene Expression in PBMCs of FM with Therapy
Differential gene expression in PBMCs of FM with MT, was assessed by RNAseq analysis of total RNA prepared from a representative sub-cohort of FM patients (discovery phase) before and after the treatment (n=12 paired samples from six patients). The results showed overexpression of 72 transcripts and underexpression of 256 transcripts (p<0.05) (Figure 1, and Supplementary Table S3).
At the individual level significant changes with treatment varied across participants, with upregulation of at least 22 genes and downregulation of at least 42 (see Supplementary Figure S1 for individual volcano plots and Supplementary Table S4 for RNAseq DE analysis at the individual level).
2.3. Gene Enrichment and Pathway Analysis with MT in the Immune Ssytem of FM
To understand the biological significance of the genes differentially expressed (DE) with MT in PBMCs of FM, we performed enrichment analysis using Gene ontology (GO) knowledgebase (http://geneontology.org) [17,18] (Figure 2, left panel), and the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (https://www.genome.jp/kegg/pathway.html ) (Figure 2, right panel), as detailed in Methods.
The findings included response to stress and infectious processes, with involvement of cytokine, chemokine, MAPK and NFkB signaling processes (Figure 2 and Supplementary Table S5).
2.4. RT-qPCR Validation of Protein-Coding Genes Differentially Expressed in Response to MT in FM
In order to validate top relevant functions among the 328 differentially expressed (DE) transcripts with treatment (p<0.05), now in the complete FM cohort of (n=38), we selected only protein-coding RNAs (Supplementary Table S4, Protein-coding tab), which included 123 DE mRNAs (34 upregulated and 89 downregulated with MT). Then, we evaluated DE at the individual level, in at least 50% of the samples (n≥3, p<0.05) (Table 3), which reduced the list to 10 genes being overexpressed and 14 underexpressed by MT, as top candidate effectors of the therapy. Individual DE data is provided on Supplementary Table S4 and summarized on Table 3 (Indiv.pval<0.05).
GO and KEGG analysis, was then reassessed for these top 24 DE genes representative of individual response to MT in at least 50% of the FM cases, finding that response to bacteria and chemokine signaling were among top cell functions affected by MT (Figure 3).
To orthogonally validate DE RNAseq genes by the alternative RT-qPCR method, primer sets were designed for three randomly selected DE from the upregulated group (CD3E, CX3CR1 and HBEGF) and another three for the downregulated (EGR2, EREG and SIK1) (Supplementary Table S6). These primer sets, together with a set to detect the house-keeping GAPDH gene were then used for technical validation of the RNAseq results in our FM subcohort (n=6), as well as in the complete FM cohort (n=38) (population validation of our RNAseq data).
The upper panel of Figure 4 illustrates individual relative DE values of the protein-coding genes selected for RT-qPCR validation (p<0.05), according to RNAseq individual data (Supplementary Table S4), while the lower panel of Figure 3 shows the results obtained by the alternative RT-qPCR approach, performed to validate RNAseq results in the FM subcohort (n=6). As observed, only SIK1 downregulation could be validated, while HBEGF significance showed opposite tendency (downregulation) to the upregulation obtained by RNAseq. None of the four remaining selected genes appeared significantly changed with MT by RT-qPCR (Figure 3, lower panel).
However, RT-qPCR analysis of the complete FM cohort (n=38) confirmed upregulation of CX3CR1 and down regulation of all three selected genes among the found downregulated by RNAseq. HBEGF, again showed a significant change towards downregulation with MT in contrast to the upregulation detected by RNAseq analysis (Table 3). No significant change with MT was found for CD3E by RT-qPCR (Figure 5, upper panel).
To investigate whether the DE with MT were exclusive of FM or their change with MT corresponded to a generalized response, DE was also measured in PBMCs from non-FM matched volunteers subjected to the same 8 session MT treatment program (n=12). As can be observed on the lower panel of Figure 5, while upregulation of CX3CR1 seemed also upregulated and HBEGF appeared downregulated by MT (interpreted as potential general responders to MT), all the three genes downregulated in FM by MT did not show significant changes, interestingly suggesting that these genes may constitute sensors of the response to MT therapy in FM.
Furthermore, since our previous work in the context of this CT (NCT04174300) identified differences in response to MT among FM patients codiagnosed with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) [12], we reassessed our RT-qPCR results taking into account whether patients with FM had also received ME/CFS diagnostic or didn´t (Supplementary Table S1). The results indeed point out that the FM group with ME/CFs codiagnosis (n=19) do not seem to respond to MT by increasing their CX3CR1 levels, while DE of HBEGF and EGR2 appears more related to this group (Figure 6). EREG and SIK1 DE seem to specifically associate to both patient groups, without changes in the control non-FM group (Figure 5).
2.5. Correlation of of Genes Differentially Expressed in Response to MT with Patient Symptoms and Sesitivity to Pain (PPTs)
MT seems to provide improvement of patient symptoms to a certain extent, as shown by the changes detected for Overall and Total FIQ scores, as well as for the SF-36 “Bodily pain” domain for the complete FM group (n=38), as previously reported [12], changes not appreciated in the FM representative cohort (n=6), perhaps due to the small sample size, and are absent in non-FM cohort (n=12), as expected (Table 4). In the latter, improvement seems to rather associate to improved fatigue and mental health, perhaps relating to the improvement of their ailments, unrelated to FM.
On another side, the monitoring of PPT changes with MT, also had shown significant changes with treatment for the most sensitive “Low cervical” tender points (n=38) [12]. Again no differences were detected for the FM subcohort (n=6), and unexpectedly, thresholds changed with treatments for the non-FM cohort, in different anatomic locations (Table 5).
To determine if the DE genes may play a role in patient improvement of symptoms with MT, we evaluated the potential correlations between gene expression and symptom differences across our validated resuts. Figure 7, interestingly shows that low levels of SIK1 negatively correlate with higher scores for SF-36 “Bodily pain” domain, which indicates better health (reduced pain), supporting a potential role of the MT treatment in reducing pain in FM by decreasing SIK1 levels. Positive correlations of SIK1 levels with FIQ “Overall” and “Symptoms” domains also supports its potential participation in treatment-associated patient improvement. By contrast high MFI “Physical fatigue” domain scores associate with lower SIK1 levels which would indicate worsening of fatigue in FM with treatment (Figure 7, upper left panel). Importantly, no relevant correlations of SIK1 levels with questionnaire score changes after treatment were detected in the control group (Figure 7, upper right panel), supporting a specific role of SIK1 in FM symptom relief in FM with MT.
On another end, lower levels of SIK1 seem to correlate with increased threshold values in the lower cervical left tender point of FM patients (n=38) (Figure 7, lower left panel), indicating the changes of SIK1 may associate with improvement of patient allodynia. No correlations of changes in SIK1 with PPT value improvement were detected in the control non-FM group (n=12) (Figure 7, lower right pannel). Statistically significant correlations of PPT changes in other DE genes (CX3CR1 with low cervical right and EREG with gluteal left) were considered spurious since DE of CX3CR1 and EREG was found non-significant by RT-qPCR in this control group (Figure 5), and, therefore, their detailed analysis was not further pursued.
As differences in response to MT in patients having received ME/CFS co-diagnosis had been previously reported [12], we set to evaluate potential correlations between DE genes and symptoms, or between DE genes and PPT changes, with ME/CFS co-diagnosis. As shown on Figure 8 (upper left), patients not fulfilling diagnosis criteria for ME/CFS show a clear benefit of the MT program applied, mostly reproducing the results obtained with the full cohort. By contrast the subgroup of FM participants that had received diagnosis of ME/CFS as well, seemed to present a more reduced benefit of symptoms associating with decreased levels of SIK1 (Figure 8, upper right), with significant improvement of the Symptom domain of the FIQ questionnaire for low SIK1 levels only. Again, significant associations between symptoms and other DE genes different from SIK1 were no further pursued, as DE of the gene had not passed test of significance (e.g. CX3CR1 in the ME/CFS co-diagnosed group, or EGR2 in the FM subgroup not having received co-diagnosis of FM) (see Figure 6), or it showed lower correlation values (e.g. CX3CR1 with Mental health of the SF-36 questionnaire) (Figure 8, upper left).
With respect to changes in PPT values with reduced SIK1 levels, again, the FM subgroup not fulfilling ME/CFS diagnosis criteria, seems to behave as the complete FM group, indicating that the 50% of patients in the group (FM with ME/CFS diagnosis) were mostly irresponsive to MT, at least symptom-wise with SIK1 changes. As in former cases, statistically significant associations unrelated to SIK1 were not further examined, as the affected PPTs (e.g. great trochanters or lateral epicondyle humerus were not among the affected by MT in FM (Table 5).
3. Discussion
This study expands our previous knowledge on the improvement of FM symptoms by a self-designed controlled-pressure MT protocol (NCT04174300) [12] by evidencing molecular changes in the immune system of FM participants with MT. The study comprised two phases: a discovery phase of genome-wide transcriptomic profiling to detect changes in expression levels with MT in the immune system of a representative FM subcohort (n=6), and a validation phase, extending some of the findings to the complete cohort (n=38). This later validation phase also examined changes in expression levels with MT in the immune system of a non-FM control cohort (n=12) treated with the same self-designed controlled-pressure MT protocol as the FM cases [12]. The objective was to find out if the observed findings in the immune system with MT were specific to FM or, by contrast, corresponded with a general mechanism triggered by MT in all individuals. Although limitations associated to the selection process of a representative subcohort of FM and to the selection of DE genes with MT leave room for further findings, the results strikingly show that downregulation of SIK1 correlate with patient symptom improvement, particularly with some FIQ domains (“Symptoms” and “Overall”) as well as with the SF-36 “Bodily pain” subdomain, with the latter two domains having shown most improvement with MT [12]. It also shows and this correlation is specific for FM, as SIK1 levels do not seem to change with MT in the immune system of control non-FM participants.
SIK1, initially identified for their role in sodium sensing, belongs to the salt-inducible kinases (SIKs) family which includes three homologous serine-threonine kinases (SIK1, SIK2 and SIK3), that seem to regulate multiple aspects of human physiology in response to extracellular signals, including feeding/fasting metabolic responses, inflammation and immune responses, and sleep (circadian rhythms), among other [19]. Of the three kinases, only SIK1 expression is upregulated at the transcriptional level through a consensus CREB (cAMP response element) present in its promoter, as shown in myocytes and the suprachiasmatic nucleus of the brain (SCN) [19,20,21]. SIK activity regulates innate immunity responses by suppressing the production of the IL-10 anti-inflammatory cytokine in macrophages. In fact, pharmacological inhibition of SIK activity increases the levels of IL-10 while suppressing the levels of the proinflammatory IL-6, IL-12 and TNF-α after TLR (toll-like receptor) stimulation by LPS [20,22]. However, conflictive data regarding the production of proinflammatory cytokines and activation of the transcription factor NFκB with increased SIK activity exist [23,24].
The current intense research in the development of member-specific inhibitors of SIK activity [25,26,27] should eventually help to ascertain the precise attributes and contributions of each member of this family of proteins in particular cell and environmental scenarios, leading to the development of novel pharmacological treatments. For example, to increase the production of IL-10 in the gut Sundberg et al., screened a library of kinase inhibitors after challenging murine bone-marrow-derived dendritic cells (DCs) with the yeast cell wall preparation zymosan, finding that the protective effects involved SIK activity inhibition in a subpopulation of CD11c (+) CX3CR1(hi) cells isolated from murine gut tissue [25]. Thus, SIK activity seems relevant in still other immune system compartments, including mast cell IL-33 cytokine release [28], and even modulating the adaptive immunity through regulation of T-cell lineage commitment, differentiation and survival [29,30]. Although drastic SIK1 downregulation may not be desirable by its role in blood pressure, its tunning in certain cell types or in disease could constitute valued therapeutic options [23,31,32].
Whether SIK1 downregulation of transcript levels by MT are mediated through the conserved CREB element in its promoter or through alternative mechanisms seem important questions for future work in the field. Other possibilities worth exploring after this initial finding is the potential impact of MT on the muscle, blood pressure and cell metabolism, or on the circadian system through changes in SIK activity.
By contrast, CX3CR1 levels appeared significantly increased in both study groups (FM and non-FM individuals), indicating that MT triggers this change in all individuals, with exception of those FM patients co-diagnosed with ME/CFS. CX3CR1 is a G-protein coupled receptor and only binder of fractalkine (CX3CL1), present on a subset of immune cells, including monocytes and macrophages, as well as DCs, T helper (Th) 1, CD8+T effector/memory and γδ T lymphocytes, and NK cells [33]. Its main role in immune cells is to detect and migrate toward inflamed tissue, “crawling and patrolling” from blood vessel endothelium to different destinies according to fractalkine´s gradient, the objective being to initiate innate immune responses followed by adaptive responses [33,34]. In the brain it is mainly expressed in astrocytes and microglia regulating cellular communication between neurons, in addition to providing protection from the neurotoxicity induced by the HIV-1 envelope protein gp120 [35]. In the gut, CX3CR1-positive macrophages produce the IL-10 immunoregulatory cytokine [36], and lack of f CX3CR1 expression is associates with altered microbiome and impaired intestinal barrier [37]. Regulatory mechanisms of CX3CR1 expression and the implications of its overexpression are complex and require further research to understand their impact on health and disease. Why patients co-diagnosed with ME/CFS do not respond to MT with increased CX3CR1 levels is unknown at present, but seems to support differential response to MT, as previously shown [12].
Molecular differences between patients fulfilling only FM or also ME/CFS (co-diagnosis) have been found by our group [38] and by other [39], seemingly demanding a review of case definition for patients fulfilling both clinical criteria [38]. Our previous report of CT NCT04174300 [12], showed differences in response to MT between patients that had or had not received a co-diagnosis of ME/CFS. The results of this study further confirm differences across these two FM subgroups, not only for a lack of upregulation of CX3CR1 levels in response to MT, but also for the downregulation of EGR2, occurring only in the co-diagnosed group. EGR2 or early growth response 2 is a transcription factor with an essential epigenetic regulatory role (DNA methylation turnover) for the differentiation of human monocytes [40], and a novel regulator of senescence of fibroblast and epithelial cells [41]. Together with EGR3 it is needed for T and B cell development and activation [42].Whether MT preferential upregulation of EGR2 in patients with an ME/CFS status relates to increased EGR2 basal levels in these patients (as shown on Figure 6), coinciding with Dr. Kerr´s previous findings [43,44], and whether this relates to EBV infection history of the patient, seems as possibility to be further explored.
Finally, downregulation of EREG, also known as epiregulin, seems to discriminate responses to MT between both FM subgroups and the control. Being a soluble peptide hormone involved in inflammation and wound healing, upregulated by LPS induction and by stress of the endoplasmic reticulum [45], its downregulation by MT may relate with patient improvement. However, correlations with questionnaire scores did not detect such a link.
The fact that RTqPCR did not validate the increased levels of CD3Eor HBEGF with MT detected by RNAseq does not serve to refute its findings, as the methodological differences across methods, may indeed constitute the reason for the discrepancy.
On the question of what could the mechanisms that exert changes in the molecular profiles of immune cells by MT be, we are far from being able to give a detailed response. However, elucidation of the immunomodulatory effects of massage either by direct pressure/mechanotransduction, or by indirect pathways effected by MT such as cytokine, chemokine, or microRNA release, [46], sleep improvement [47], or other, are on their way.
4. Materials and Methods
4.1. Study Design and Intervention
Observational study consisting in the analysis of the molecular changes taken place in the circulating immune cells of FM patients (n=38) and non-FM patients (n=12) by a physiotherapy program of manual therapy. The program had consisted on eight sessions (twice weekly for four weeks) of a 25-min custom protocol including pressure maneuvers of about 4.5 N each of 10 out of the 18 FM tender points and surrounding areas (NCT04174300), as described in our previous publication [12]. This study also included a pilot interventional non-randomized single arm trial replicating the treatment program previously applied to FM, now in a matched non-FM cohort, as a control. Comparison across groups was performed to find out whether the MT program triggeers similar or distinct changes in FM vs non-FM individuals. Before vs after FIQ [13,14], MFI [15]and SF-36 [16] questionnaire scores, as well as PPT values of the 18-FM tender points were resgistered for all participants. The studies were approved by the Universidad Católica de Valencia San Vicente Mártir Ethics Committee with study codes UCV/2018-2019/076, and UCV/2020-2021/167, respectively. All participants signed an informed consent before they were included in the study. For the analysis of molecular changes in the immune system of FM whole transcriptome of a representative cohort of FM patients (n=6) was obtained before and after the program. Top differentially expressed genes were validated in the complete FM cohort (n=38) by RT-qPCR and studied in the non-FM cohort (n=12) for their comparison.
4.2. Total RNA Preparation and Quality Assesment
Total RNA was prepared from a previous collection of PBMC pellets (-150ºC) (≥106 cells) registered at the Institute of Health Carlos III National Biobank, Madrid, Spain (Ref. C.0006924) [12] with the RNeasy Mini Kit (Qiagen, cat. 74104) following manufacturer’s protocol. Cell lysis included addition of 1/100 β-mercaptoetanol (Sigma, cat. 63689) before 5 min vortex to favor lysis. Removal of contaminant DNA was done in column with the RNase-free DNase set (Qiagen, cat. 79254). Elution RNase free ddH2O was supplemented with RNasin (Promega, cat. N2118) to a final concentration of 0.4 U/µl before use. RNA yields and quality were obtained with an Agilent TapeStation 4200 (Agilent Technologies). Only samples with RIN≥7 were subjected to downstream analysis.
4.3. RNAseq
Upon ribosomal RNA depletion, 1 µg of RNA was used for whole transcriptome sequencing (Illumina NovaSeq 6000 PE150, 50M reads) (Novogene, Cambridge, UK). After removal of filtered to adapter sequences and low quality reads, sequences were aligned to the human GRCh38/hg38 genome using HISAT2 software [48]. Sequence assembly analysis were performed using Cufflinks, converted to BAM format and sorted and indexed with samtools [49]. Cuffdiff was used to calculate differential expression, expressed as FPKM (fragments per kilobase of exon per million mapped fragments) numbers for each sample. Differences were given as the absolute value log2Fold change of the ratio between the pretreated and the posttreatment samples >1, p-adjust<0.05. Heatmaps and volcano plots were generated with the CummeRbund R package (R version 4.2.1, cummeRbund 2.38.0) [50] and GraphPad Prism 8.0.2 software.
4.4. Enrichment Analysis
Gene Ontology (GO) enrichment analysis of differentially expressed genes was performed with Gene ontology (GO) knowledgebase (http://geneontology.org) [17,18], or with the GOseq R package [51]. After gene length bias correction, p values less than 0.05 were considered significantly enriched by differential expressed genes.
For pathway analysis of differentially expressed genes, Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of differentially expressed genes was done with the https://www.genome.jp/kegg/ online tool.
4.5. RT-qPCR Validation
Reverse transcription was performed using the High Capacity Complementary DNA (cDNA) Reverse Transcription Kit (Applied Biosystems, Waltham, MA, USA, cat. 4308228) with 1–2 μg of total RNA according to the manufacturer's guidelines. Gene relative expression was assesed by qPCR of triplicates per sample, with the primer sets on Supplementary TableS6. qPCR was performed with the PowerUP Sybr Green Master Mix (Applied Biosystems, cat. 100029283) on a Lightcycler LC480 instrument (Roche, Penzberg, Germany) and , with the following amplification conditions: a single preactivation cycle of the hotstart polymerase at 94ºC for 15 min, followed by 40 amplification cycles, each of which consisted of three steps: 95 ◦ C for 15 s, 54 ◦ C for 30 s and extension at 70 ◦ C for 30 s. Gene expression levels were normalized to GAPDH and quantified by the 2−∆∆Ct method [52].
4.6. Statistics
Continuous data are expressed as means ± SD (standard deviation) and range values. Statistical differences were assessed by paired t-tests for normal value distributions and either nonparametric Mann–Whitney or Wilcoxon analysis if values did not to follow a normal distribution. Normality was determined with the Shapiro–Wilk test. Differences between groups were considered significant when p ≤ 0.05. Analysis were conducted with Excel, the SPSS package 13.0 (SPSS Inc., Chicago, IL, USA) and R v4.2.1 [53]. Pearson correlations were evaluated with WGCNA R package [54]. Plots were drawn using the GraphPad Prism 5.0 program (San Diego, CA, USA) and the package ggplot2 [55].
5. Conclusions
In conclusion, the molecular data obtained by comparing the immune system transcriptome of FM before and after of a pressure-controlled MT protocol (NCT04174300) identifies downregulation of SIK1 as a prominent and specific target of MT in FM, associating with patient symptom improvement. In addition to its potential biomarker to monitor response to MT, SIK1 inhibitors combined with MT may result in improved treatments for FM. Research of SIK1-mediated mechanisms and MT complementary effects are needed for an improved understanding of SIK1´s role in FM and its potential function as mediator of MT therapeutics in FM.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Supplementary Table S1: Cohort demographics and validated Instruments (FIQ, MFI and SF-36);. Supplementary Table S2: Pressure Point Thresholds (PPTs); Supplementary Table S3: Differentially expressed mRNAs with MT treatment in FM, as determined by RNAseq; Supplementary Table S4: Individual differentially expressed genes for FM1-FM6; Supplementary Table S5: GO and KEGG enriched pathways with MT treatment in PBMCs from FM; Supplementary Table S6: Primer sets used in RT-qPCR validation of genes differentially expressed with MT.
Author Contributions
Conceptualization, JBF and EO; methodology, JBF and FJFV; software, JBF and KGO; validation, JBF and KGO; formal analysis, JBF and KGO; investigation, ; resources, X.X.; data curation, JBF and KGO; writing—original draft preparation, JBF, KGO and EO; writing—review and editing, ALL AUTHORS; visualization, JBF and KGO; supervision, MGE and EO; project administration, EO; funding acquisition, MGE and EO. All authors have read and agreed to the published version of the manuscript.
Funding
Please add: This research was funded by Universidad Católica de Valencia San Vicente Mártir, grant numbers 2019-266-001 to MGE and 2019-270-002 to EO. APC was funded by 2020-270-001 to EO.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board (or Ethics Committee) of UNIVERSIDAD CATÓLICA DE VALENCIA SAN VICENTE MÁRTIR ETHICS COMMITTEE (protocol code UCV/2018-2019/076, February 21st, 2019; and protocol code UCV2020-2021/167, December 22nd, 2021). All personal data were anonymized in fulfillment of Spanish data protection laws. All tasks were performed by collegiate professionals or qualified trained personnel.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
We encourage all authors of articles published in MDPI journals to share their research data. In this section, please provide details regarding where data supporting reported results can be found, including links to publicly archived datasets analyzed or generated during the study. Where no new data were created, or where data is unavailable due to privacy or ethical restrictions, a statement is still required. Suggested Data Availability Statements are available in section “MDPI Research Data Policies” at https://www.mdpi.com/ethics.
Acknowledgments
we are grateful to the collegiate nurse Ignacio Bonastre Ferez for his help with the phlebotomies, to Teresa Sánchez-Fito for technical support in the laboratory, to the Universidad Católica de Valencia Clinics personnel for their help with patient registration and appointment follow-ups, and to all volunteers that participated in the study.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Harrison, J.E.; Weber, S.; Jakob, R.; Chute, C.G. ICD-11: an international classification of diseases for the twenty-first century. BMC Med Inform Decis Mak. 2021, 21, 206. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wolfe, F.; Smythe, H.A.; Yunus, M.B.; Bennett, R.M.; Bombardier, C.; Goldenberg, D.L.; Tugwell, P.; Campbell, S.M.; Abeles, M.; Clark, P. , et al. The American College of Rheumatology 1990 Criteria for the Classification of Fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheum. 1990, 33, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, F.; Clauw, D.J.; Fitzcharles, M.A.; Goldenberg, D.L.; Katz, R.S.; Mease, P.; Russell, A.S.; Russell, I.J.; Winfield, J.B.; Yunus, M.B. The American College of Rheumatology preliminary diagnostic criteria for fibromyalgia and measurement of symptom severity. Arthritis Care Res 2010, 62, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Fitzcharles, M.A.; Cohen, S.P.; Clauw, D.J.; Littlejohn, G.; Usui, C.; Häuser, W. Nociplastic pain: towards an understanding of prevalent pain conditions. Lancet 2021, 397, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, B.M.; Jain, A.K.; De Meirleir, K.L.; Peterson, D.L.; Klimas, N.G.; Lerner, A.M.; van de Sande, M.I. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Clinical Working Case Definition, Diagnostic and Treatment Protocols. Journal Of Chronic Fatigue Syndrome 2003, 11, 7–115. [Google Scholar] [CrossRef]
- Carruthers, B.M.; van de Sande, M.I.; De Meirleir, K.L.; Klimas, N.G.; Broderick, G.; Mitchell, T.; Staines, D.; Powles, A.C.; Speight, N.; Vallings, R.; Bateman, L.; Baumgarten-Austrheim, B.; Bell, D.S.; Carlo-Stella, N.; Chia, J.; Darragh, A.; Jo, D.; Lewis, D.; Light, A.R.; Marshall-Gradisnik, S.; Mena, I.; Mikovits, J.A.; Miwa, K.; Murovska, M.; Pall, M.L.; Stevens, S. Myalgic encephalomyelitis: International Consensus Criteria. J Intern Med. 2011 Oct;270(4):327-38. doi: 10.1111/j.1365-2796.2011.02428.x. Epub 2011 Aug 22. Erratum in: J Intern Med. 2017 Oct;282(4):353. doi: 10.1111/joim.12658. PMID: 21777306; PMCID: PMC3427890. [CrossRef]
- Jones, G.T.; Atzeni, F.; Beasley, M.; Flüß, E.; Sarzi-Puttini, P.; Macfarlane, G.J. The prevalence of fibromyalgia in the general population: a comparison of the American College of Rheumatology 1990, 2010, and modified 2010 classification criteria. Arthritis Rheumatol. 2015, 67, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, L.P. Worldwide Epidemiology of Fibromyalgia. Curr Pain Headache Rep. 2013, 17, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cabo-Meseguer, A.; Cerdá-Olmedo, G.; Trillo-Mata, J.L. Fibromyalgia: Prevalence, epidemiologic profiles and economic costs. Med Clin 2017, 149, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Almenar-Pérez, E.; Sánchez-Fito, T.; Ovejero, T.; Nathanson, L.; Oltra, E. Impact of Polypharmacy on Candidate Biomarker miRNomes for the Diagnosis of Fibromyalgia and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Striking Back on Treatments. Pharmaceutics 2019, 11, 126. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carrasco-Vega, E.; Guiducci, S.; Nacci, F.; Bellando Randone, S.; Bevilacqua, C.; Gonzalez-Sanchez, M.; Barni, L. Efficacy of physiotherapy treatment in medium and long term in adults with fibromyalgia: an umbrella of systematic reviews. Clin Exp Rheumatol. 2024, 42, 1248–1261. [Google Scholar] [CrossRef] [PubMed]
- Falaguera-Vera, F.J.; Garcia-Escudero, M.; Bonastre-Férez, J.; Zacarés, M.; Oltra, E. Pressure Point Thresholds and ME/CFS Comorbidity as Indicators of Patient's Response to Manual Physiotherapy in Fibromyalgia. Int J Environ Res Public Health 2020, 17, 8044. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Burckhardt, C.S.; Clark, S.R.; Bennett, R.M. The fibromyalgia impact questionnaire: development and validation. J Rheumatol. 1991, 18, 728–733. [Google Scholar] [PubMed]
- Rivera, J.; González, T. The Fibromyalgia Impact Questionnaire: a validated Spanish version to assess the health status in women with fibromyalgia. Clin Exp Rheumatol. 2004, 22, 554–560. [Google Scholar] [PubMed]
- Smets, E.M.; Garssen, B.; Bonke, B.; De Haes, J.C. The Multidimensional Fatigue Inventory (MFI) psychometric qualities of an instrument to assess fatigue. J Psychosom Res. 1995, 39, 315–325. [Google Scholar] [CrossRef] [PubMed]
- McHorney, C.A.; Ware, J.E., Jr.; Raczek, A.E. The MOS 36-Item Short-Form Health Survey (SF-36): II. Psychometric and clinical tests of validity in measuring physical and mental health constructs. Med Care. 1993, 31, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; Harris, M.A.; Hill, D.P.; Issel-Tarver, L.; Kasarskis, A.; Lewis, S.; Matese, J.C.; Richardson, J.E.; Ringwald, M.; Rubin, G.M.; Sherlock, G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gene Ontology Consortium; Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; et al. The Gene Ontology knowledgebase in 2023. Genetics 2023, 224, iyad031. [CrossRef] [PubMed]
- Jagannath, A.; Taylor, L.; Ru, Y.; Wakaf, Z.; Akpobaro, K.; Vasudevan, S.; Foster, R.G. The multiple roles of salt-inducible kinases in regulating physiology. Physiol Rev. 2023, 103, 2231–2269. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jagannath, A.; Butler, R.; Godinho, S.I.; Couch, Y.; Brown, L.A.; Vasudevan, S.R.; Flanagan, K.C.; Anthony, D.; Churchill, G.C.; Wood, M.J.; Steiner, G.; Ebeling, M.; Hossbach, M.; Wettstein, J.G.; Duffield, G.E.; Gatti, S.; Hankins, M.W.; Foster, R.G.; Peirson, S.N. The CRTC1-SIK1 pathway regulates entrainment of the circadian clock. Cell 2013, 154, 1100–1111. [Google Scholar] [CrossRef]
- Stewart, R.; Akhmedov, D.; Robb, C.; Leiter, C.; Berdeaux, R. Regulation of SIK1 abundance and stability is critical for myogenesis. Proc Natl Acad Sci USA 2013, 110, 117–122. [Google Scholar] [CrossRef]
- Clark, K.; MacKenzie, K.F.; Petkevicius, K.; Kristariyanto, Y.; Zhang, J.; Choi, H.G.; Peggie, M.; Plater, L.; Pedrioli, P.G.; McIver, E.; Gray, N.S.; Arthur, J.S.; Cohen, P. Phosphorylation of CRTC3 by the salt-inducible kinases controls the interconversion of classically activated and regulatory macrophages. Proc Natl Acad Sci USA 2012, 109, 16986–16991. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yong Kim, S.; Jeong, S.; Chah, K.H.; Jung, E.; Baek, K.H.; Kim, S.T.; Shim, J.H.; Chun, E.; Lee, K.Y. Salt-inducible kinases 1 and 3 negatively regulate Toll-like receptor 4-mediated signal. Mol Endocrinol 2013, 27, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Sanosaka, M.; Fujimoto, M.; Ohkawara, T.; Nagatake, T.; Itoh, Y.; Kagawa, M.; Kumagai, A.; Fuchino, H.; Kunisawa, J.; Naka, T.; Takemori, H. Salt-inducible kinase 3 deficiency exacerbates lipopolysaccharide-induced endotoxin shock accompanied by increased levels of proinflammatory molecules in mice. Immunology 2015, 145, 268–278. [Google Scholar] [CrossRef]
- Sundberg, T.B.; Choi, H.G.; Song, J.H.; Russell, C.N.; Hussain, M.M.; Graham, D.B.; Khor, B.; Gagnon, J.; O'Connell, D.J.; Narayan, K.; Dančík, V.; Perez, J.R.; Reinecker, H.C.; Gray, N.S.; Schreiber, S.L.; Xavier, R.J.; Shamji, A.F. Small-molecule screening identifies inhibition of salt-inducible kinases as a therapeutic strategy to enhance immunoregulatory functions of dendritic cells. Proc Natl Acad Sci USA 2014, 111, 12468–12473. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ozanne, J.; Prescott, A.R.; Clark, K. The clinically approved drugs dasatinib and bosutinib induce anti-inflammatory macrophages by inhibiting the salt-inducible kinases. Biochem J. 2015, 465, 271–279. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Peixoto, C.; Joncour, A.; Temal-Laib, T.; Tirera, A.; Dos Santos, A.; Jary, H.; Bucher, D.; Laenen, W.; Pereira Fernandes, A.; Lavazais, S.; Delachaume, C.; Merciris, D.; Saccomani, C.; Drennan, M.; López-Ramos, M.; Wakselman, E.; Dupont, S.; Borgonovi, M.; Roca Magadan, C.; Monjardet, A.; Brys, R.; De Vos, S.; Andrews, M.; Jimenez, J.M.; Amantini, D.; Desroy, N. Discovery of Clinical Candidate GLPG3970: A Potent and Selective Dual SIK2/SIK3 Inhibitor for the Treatment of Autoimmune and Inflammatory Diseases. J Med Chem. 2024, 67, 5233–5258. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Darling, N.J.; Arthur, J.S.C.; Cohen, P. Salt-inducible kinases are required for the IL-33-dependent secretion of cytokines and chemokines in mast cells. J Biol Chem. 2021, 296, 100428. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nefla, M.; Darling, N.J.; van Gijsel Bonnello, M.; Cohen, P.; Arthur, J.S.C. Salt inducible kinases 2 and 3 are required for thymic T cell development. Sci Rep. 2021, 11, 21550. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Canté-Barrett, K.; Meijer, M.T.; Cordo', V.; Hagelaar, R.; Yang, W.; Yu, J.; Smits, W.K.; Nulle, M.E.; Jansen, J.P.; Pieters, R.; Yang, J.J.; Haigh, J.J.; Goossens, S.; Meijerink, J.P. MEF2C opposes Notch in lymphoid lineage decision and drives leukemia in the thymus. JCI Insight. 2022, 7, e150363. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, M.J.; Park, S.K.; Lee, J.H.; Jung, C.Y.; Sung, D.J.; Park, J.H.; Yoon, Y.S.; Park, J.; Park, K.G.; Song, D.K.; Cho, H.; Kim, S.T.; Koo, S.H. Salt-Inducible Kinase 1 Terminates cAMP Signaling by an Evolutionarily Conserved Negative-Feedback Loop in β-Cells. Diabetes 2015, 64, 3189–3202. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Huang, S.; Wu, X.; Feng, Y.; Shen, Y.; Zhao, Q.S.; Leng, Y. Activation of SIK1 by phanginin A inhibits hepatic gluconeogenesis by increasing PDE4 activity and suppressing the cAMP signaling pathway. Mol Metab. 2020, 41, 101045. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lee, M.; Lee, Y.; Song, J.; Lee, J.; Chang, S.Y. Tissue-specific Role of CX3CR1 Expressing Immune Cells and Their Relationships with Human Disease. Immune Netw. 2018, 18, e5. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, G.; Yu, H.; Liu, N.; Zhang, P.; Tang, Y.; Hu, Y.; Zhang, Y.; Pan, C.; Deng, H.; Wang, J.; Li, Q.; Tang, Z. Overexpression of CX3CR1 in Adipose-Derived Stem Cells Promotes Cell Migration and Functional Recovery After Experimental Intracerebral Hemorrhage. Front Neurosci. 2019, 13, 462. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Meucci, O.; Fatatis, A.; Simen, A.A.; Miller, R.J. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc Natl Acad Sci U S A. 2000 Jul 5;97(14):8075-80. doi: 10.1073/pnas.090017497. Erratum in: Proc Natl Acad Sci U S A 2001 Dec 18;98(26):15393. PMID: 10869418; PMCID: PMC16672.
- Hadis, U.; Wahl, B.; Schulz, O.; Hardtke-Wolenski, M.; Schippers, A.; Wagner, N.; Müller, W.; Sparwasser, T.; Förster, R.; Pabst, O. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 2011, 34, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.M.; Bieghs, V.; Heymann, F.; Hu, W.; Dreymueller, D.; Liao, L.; Frissen, M.; Ludwig, A.; Gassler, N.; Pabst, O.; Latz, E.; Sellge, G.; Penders, J.; Tacke, F.; Trautwein, C. CX3CR1 is a gatekeeper for intestinal barrier integrity in mice: Limiting steatohepatitis by maintaining intestinal homeostasis. Hepatology 2015, 62, 1405–1416. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Orenga, K.; Martín-Martínez, E.; Nathanson, L.; Oltra, E. HERV activation segregates ME/CFS from fibromyalgia and defines a novel nosological entity for patients fulfilling both clinical criteria. bioRxiv 2023.10.05.56. [CrossRef]
- Nepotchatykh, E.; Caraus, I.; Elremaly, W.; Leveau, C.; Elbakry, M.; Godbout, C.; Rostami-Afshari, B.; Petre, D.; Khatami, N.; Franco, A.; Moreau, A. Circulating microRNA expression signatures accurately discriminate myalgic encephalomyelitis from fibromyalgia and comorbid conditions. Sci Rep. 2023, 13, 1896. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mendes, K.; Schmidhofer, S.; Minderjahn, J.; Glatz, D.; Kiesewetter, C.; Raithel, J.; Wimmer, J.; Gebhard, C.; Rehli, M. The epigenetic pioneer EGR2 initiates DNA demethylation in differentiating monocytes at both stable and transient binding sites. Nat Commun. 2021, 12, 1556. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tyler, E.J.; Gutierrez Del Arroyo, A.; Hughes, B.K.; Wallis, R.; Garbe, J.C.; Stampfer, M.R.; Koh, J.; Lowe, R.; Philpott, M.P.; Bishop, C.L. Early growth response 2 (EGR2) is a novel regulator of the senescence programme. Aging Cell 2021, 20, e13318. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, S.; Symonds, A.L.; Zhu, B.; Liu, M.; Raymond, M.V.; Miao, T.; Wang, P. Early growth response gene-2 (Egr-2) regulates the development of B and T cells. PLoS One 2011, 6, e18498. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kerr, J.R.; Petty, R.; Burke, B.; Gough, J.; Fear, D.; Sinclair, L.I.; Mattey, D.L.; Richards, S.C.; Montgomery, J.; Baldwin, D.A.; Kellam, P.; Harrison, T.J.; Griffin, G.E.; Main, J.; Enlander, D.; Nutt, D.J.; Holgate, S.T. Gene expression subtypes in patients with chronic fatigue syndrome/myalgic encephalomyelitis. J Infect Dis. 2008, 197, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J. Early Growth Response Gene Upregulation in Epstein-Barr Virus (EBV)-Associated Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Biomolecules 2020, 10, 1484. [Google Scholar] [CrossRef] [PubMed]
- Zhan, L.; Zheng, L.; Hosoi, T.; Okuma, Y.; Nomura, Y. Stress-induced neuroprotective effects of epiregulin and amphiregulin. PLoS One 2015, 10, e0118280. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Espejo, J.A.; García-Escudero, M.; Oltra, E. Unraveling the Molecular Determinants of Manual Therapy: An Approach to Integrative Therapeutics for the Treatment of Fibromyalgia and Chronic Fatigue Syndrome/Myalgic Encephalomyelitis. Int J Mol Sci. 2018, 19, 2673. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Navarro-Ledesma, S.; Hamed-Hamed, D.; Gonzalez-Muñoz, A.; Pruimboom, L. Impact of physical therapy techniques and common interventions on sleep quality in patients with chronic pain: A systematic review. Sleep Med Rev. 2024, 76, 101937. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kelley, L.G.C.T. cummeRbund; Bioconductor, 2017. Version 3.18, Bioconductor.
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biology 2010, 11, R14. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- R Core Team (2024). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. Available online: https://www.R-project.org/.
- Langfelder, P.; Horvath, S. WGCNA: an R package for weighted correlation network analysis. Bmc Bioinformatics 2008, 9, 559–559. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
Figure 1.
Volcano plot representation of differential gene expression in PBMCs of FM with therapy. Log2FoldChange values (X axis) are displayed with respect to -log10 of their p-values (Y axis), significance set at p<0.05.
Figure 1.
Volcano plot representation of differential gene expression in PBMCs of FM with therapy. Log2FoldChange values (X axis) are displayed with respect to -log10 of their p-values (Y axis), significance set at p<0.05.
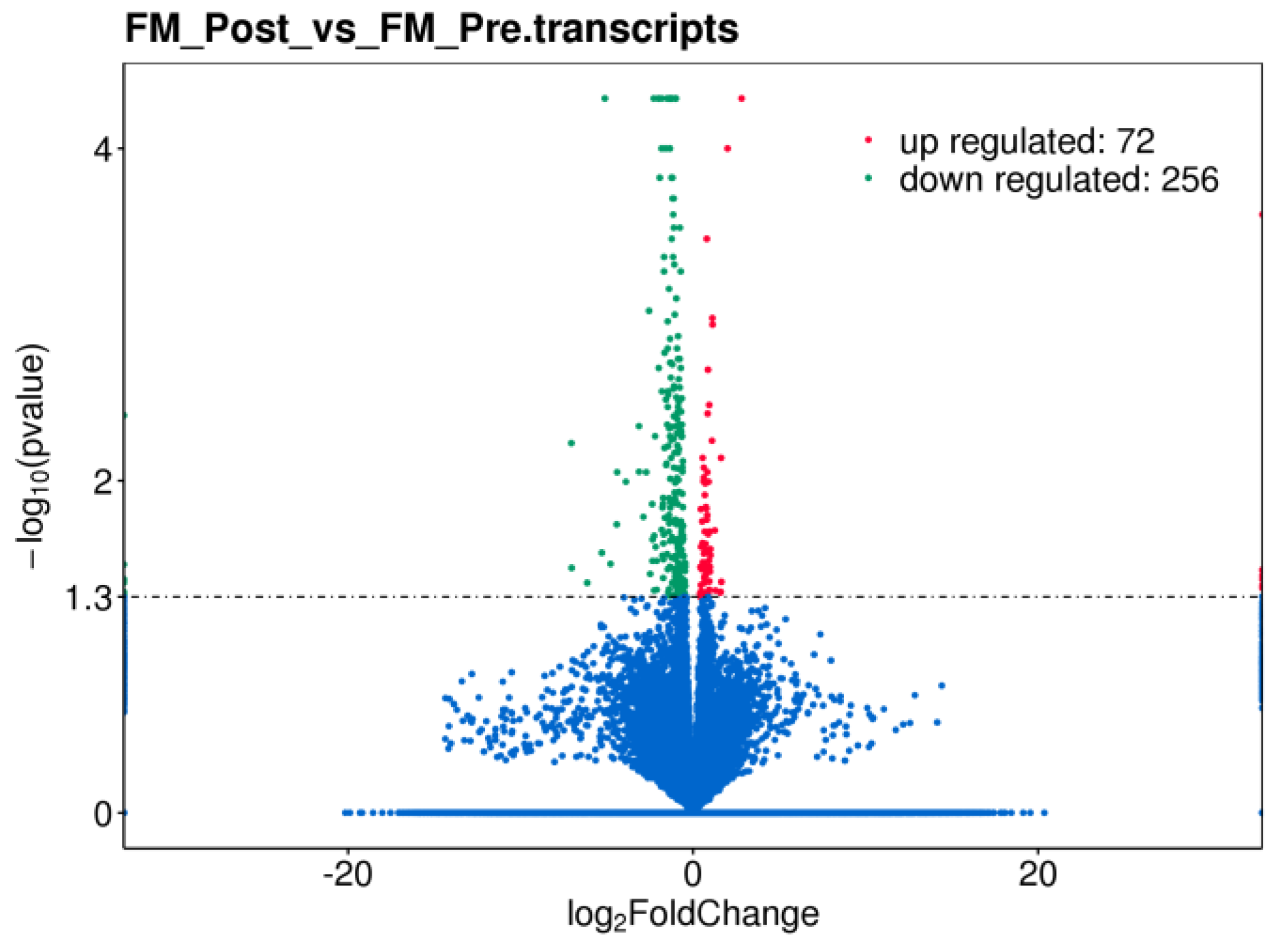
Figure 2.
GO (left) and KEGG (right) pathways targeted by MT in the immune system of FM. Function significance (color palette, padj<0.05) and approximate DE gene count in each pathway as indicated by dot thickness for each panel.
Figure 2.
GO (left) and KEGG (right) pathways targeted by MT in the immune system of FM. Function significance (color palette, padj<0.05) and approximate DE gene count in each pathway as indicated by dot thickness for each panel.
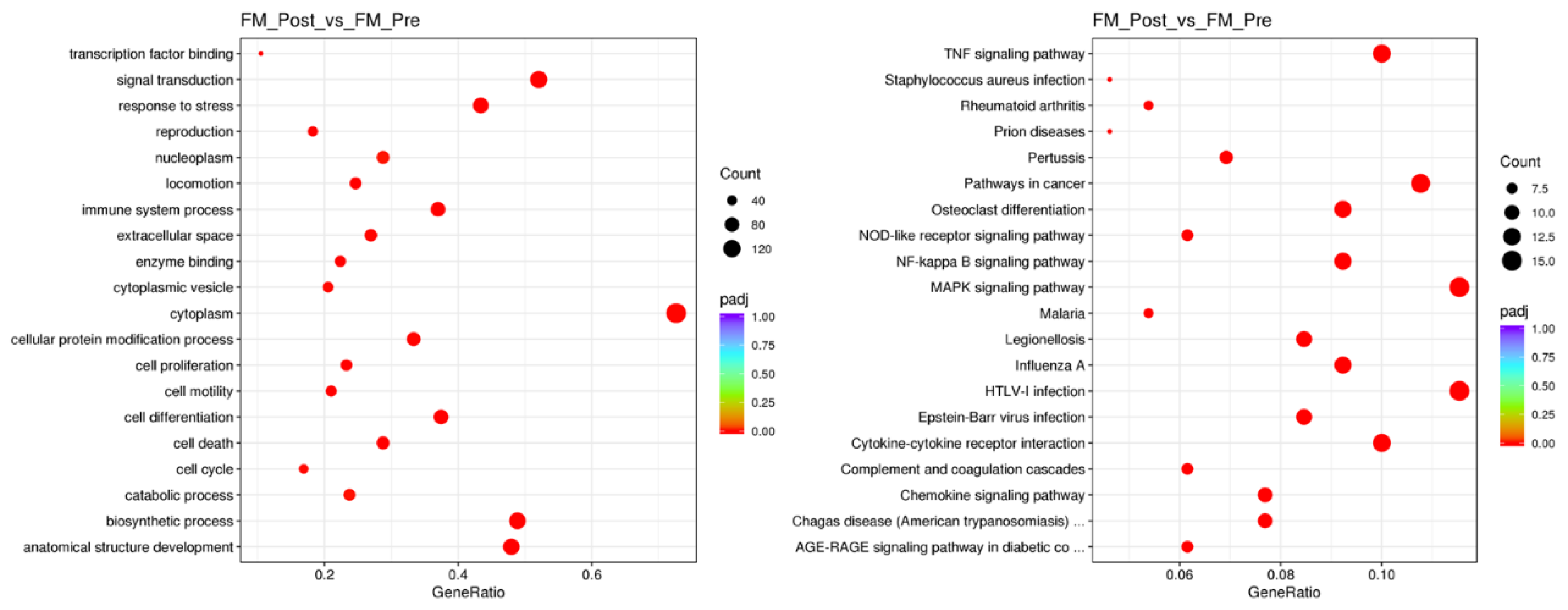
Figure 3.
Top pathways targeted by MT in the immune system of FM, as determined by top representative DE genes. Function significance (color palette, padj<0.05) and approximate DE gene count in each pathway as indicated by dot thickness for each panel.
Figure 3.
Top pathways targeted by MT in the immune system of FM, as determined by top representative DE genes. Function significance (color palette, padj<0.05) and approximate DE gene count in each pathway as indicated by dot thickness for each panel.
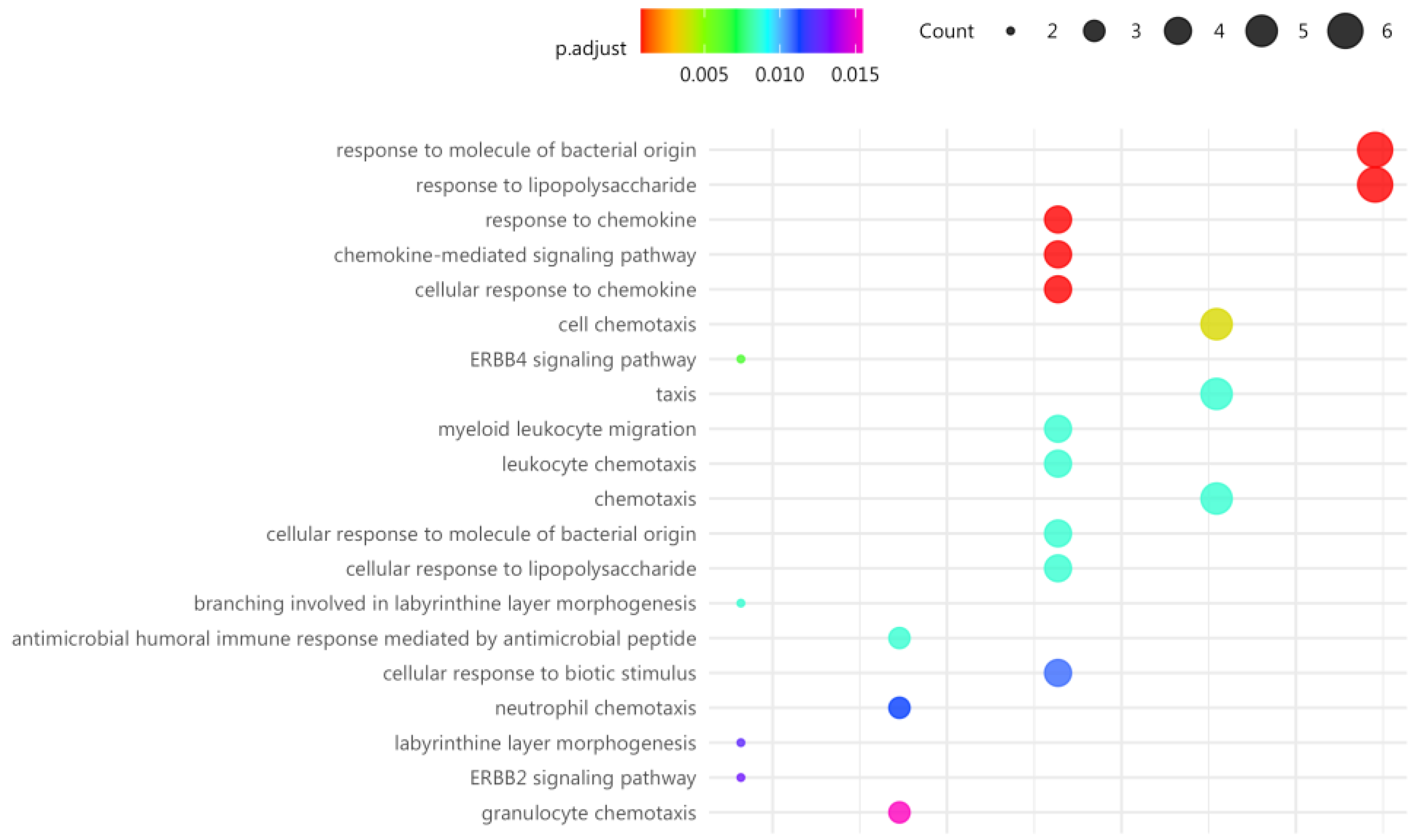
Figure 4.
Relative expression levels (Pre vs Post) of randomly selected coding genes DE with MT in a representative subcohort of FM patients at the individual level, as determined by RNAseq analysis (upper panel) (p<0.05, Supplementary Table S4), and as determined by RT-qPCR analysis (lower panel) (n=6), Wilcoxon test (p<0.05).
Figure 4.
Relative expression levels (Pre vs Post) of randomly selected coding genes DE with MT in a representative subcohort of FM patients at the individual level, as determined by RNAseq analysis (upper panel) (p<0.05, Supplementary Table S4), and as determined by RT-qPCR analysis (lower panel) (n=6), Wilcoxon test (p<0.05).
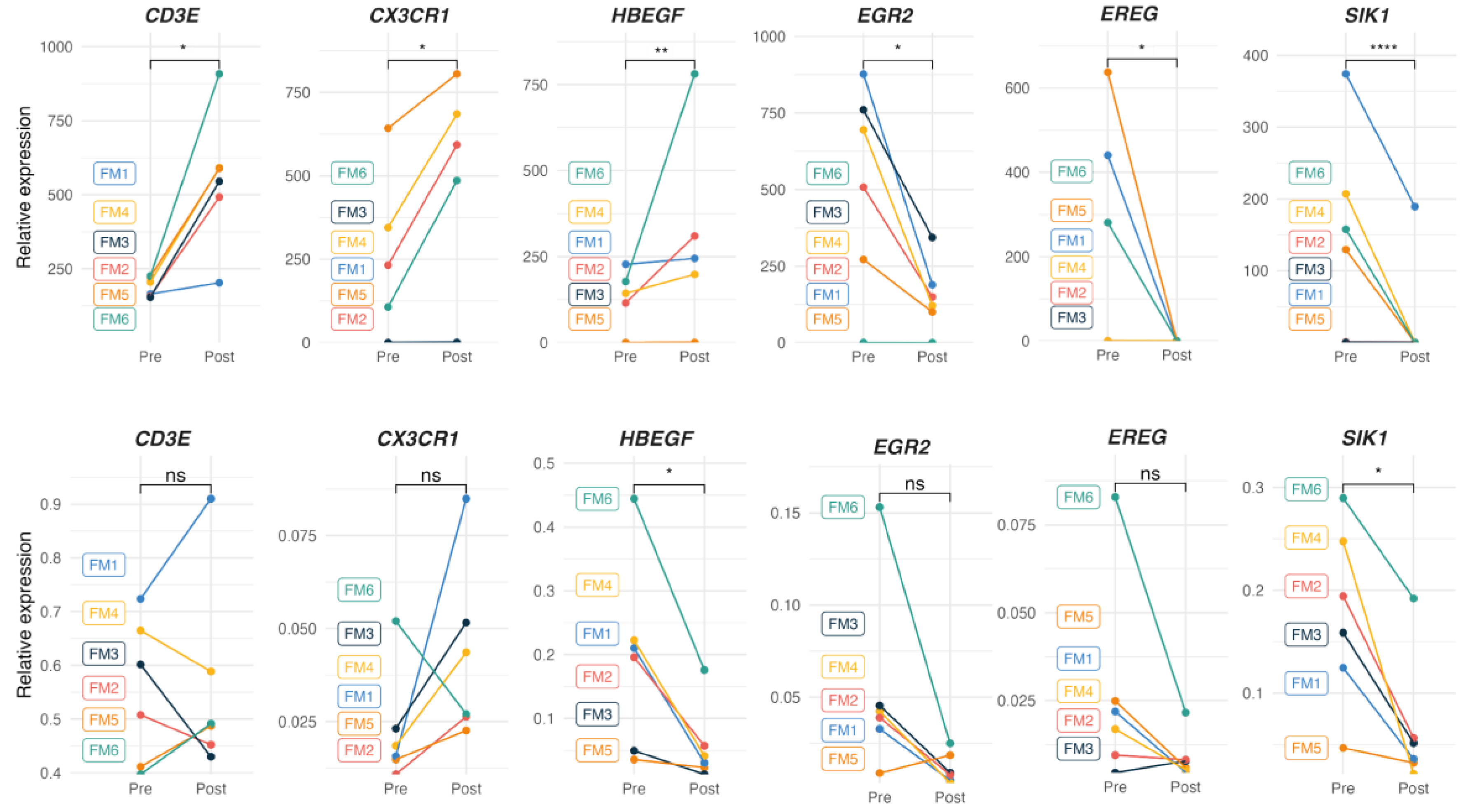
Figure 5.
DE expressed genes with MT on PBMCs from FM patients, as determined by RT-qPCR. Relative expression by ΔΔCt values upon GAPDH normalization for each sample (triplicates) in each study group (n=38 for the FM cohort in blue, upper panel; and n=12 for the non-FM control cohort in black, lower panel) are shown. Statistical paired two-Wilcoxon Test with Benjamin-Hochberg p-value correction. (*p<0.05, **p<0.01, ***p<0.001, ****p < 0.0001) was applied to assess significance of DE.
Figure 5.
DE expressed genes with MT on PBMCs from FM patients, as determined by RT-qPCR. Relative expression by ΔΔCt values upon GAPDH normalization for each sample (triplicates) in each study group (n=38 for the FM cohort in blue, upper panel; and n=12 for the non-FM control cohort in black, lower panel) are shown. Statistical paired two-Wilcoxon Test with Benjamin-Hochberg p-value correction. (*p<0.05, **p<0.01, ***p<0.001, ****p < 0.0001) was applied to assess significance of DE.
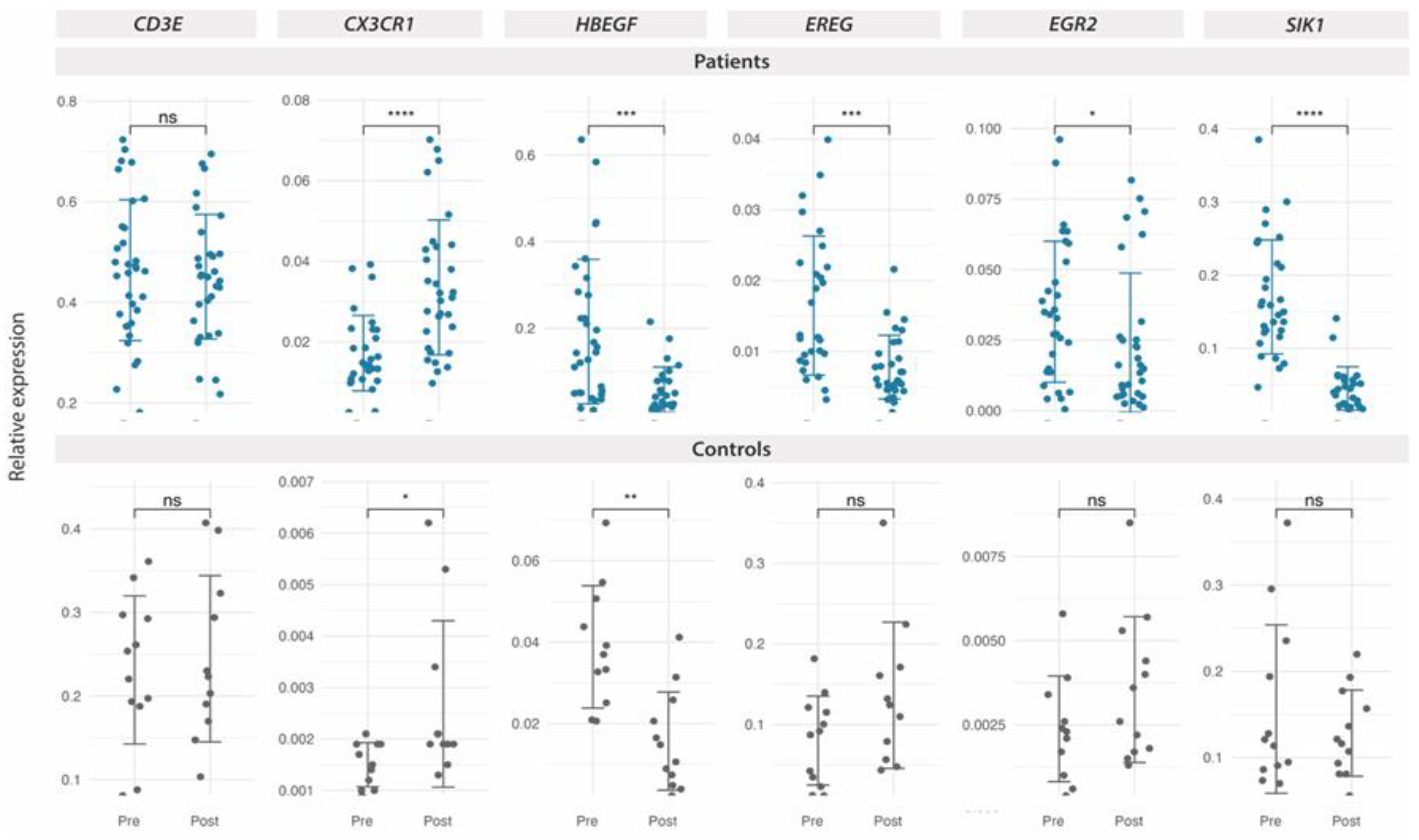
Figure 6.
DE expressed genes with MT on PBMCs from FM patients with or without ME/CFS co-diagnosis, as determined by RT-qPCR. Relative expression by ΔΔCt values upon GAPDH normalization for each sample (triplicates) in each study group (n=19 for the FM only group, dark blue, upper panel; and n=19 for the FM group with ME/CFS co-diagnosis, light blue, lower panel) are shown. Statistical paired two-Wilcoxon Test with Benjamin-Hochberg p-value correction. (*p<0.05, **p<0.01, ***p<0.001, ****p < 0.0001) was applied to assess significance of DE.
Figure 6.
DE expressed genes with MT on PBMCs from FM patients with or without ME/CFS co-diagnosis, as determined by RT-qPCR. Relative expression by ΔΔCt values upon GAPDH normalization for each sample (triplicates) in each study group (n=19 for the FM only group, dark blue, upper panel; and n=19 for the FM group with ME/CFS co-diagnosis, light blue, lower panel) are shown. Statistical paired two-Wilcoxon Test with Benjamin-Hochberg p-value correction. (*p<0.05, **p<0.01, ***p<0.001, ****p < 0.0001) was applied to assess significance of DE.
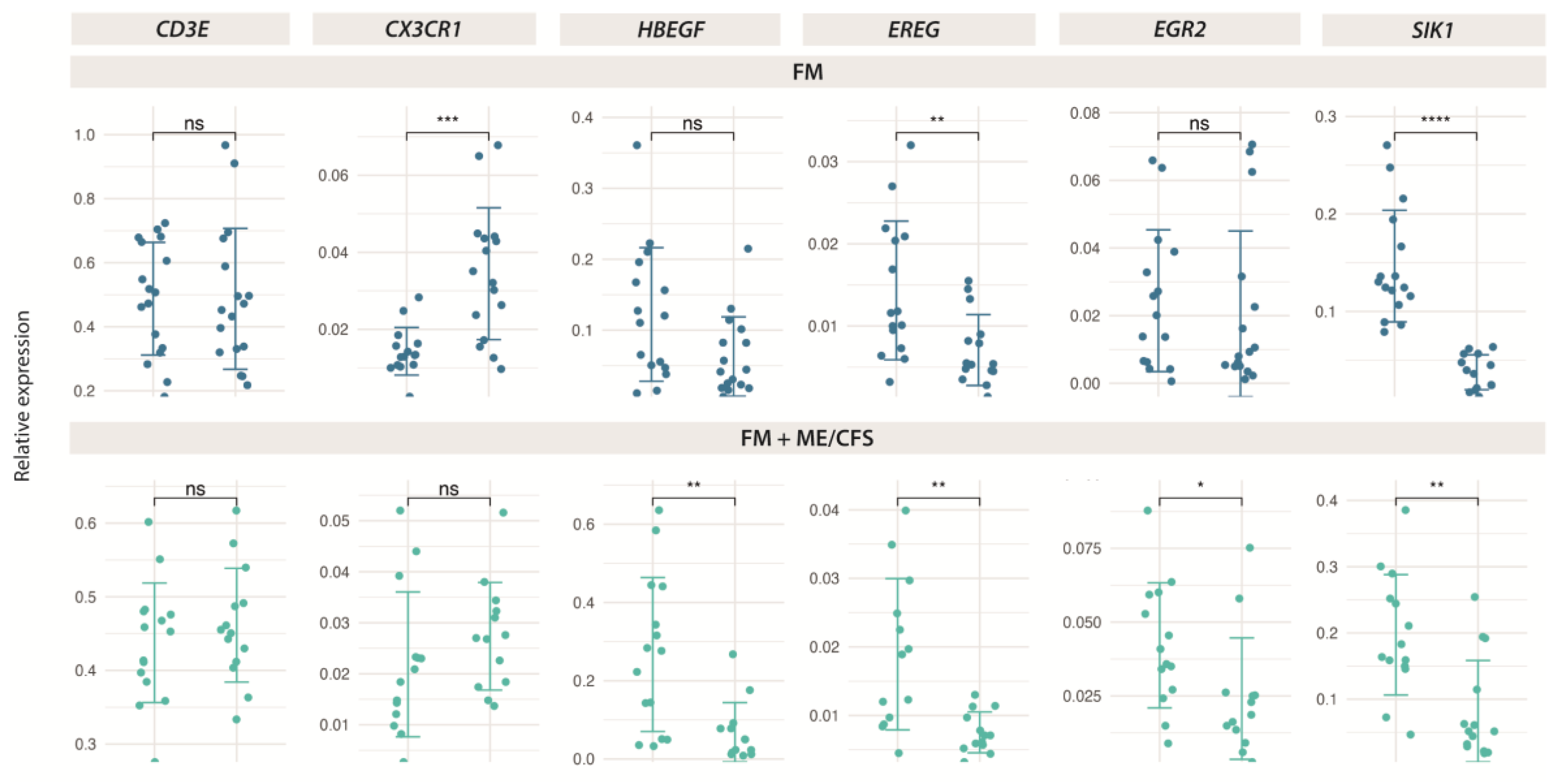
Figure 7.
Symptom improvement with validated DE genes with MT in FM (n=38) (upper left), and non-FM controls (n=12) (upper right), and correlation of PPT ratios (post-pre-) with DE genes in FM (n=38) (lower left), and non-FM controls (n=12) (lower right). Pearson correlation values and associated p-values (*, p<0.05; **, p<0.01; ***, p<0.001) between gene expression levels and symptom scores, or PPT ratios are shown.
Figure 7.
Symptom improvement with validated DE genes with MT in FM (n=38) (upper left), and non-FM controls (n=12) (upper right), and correlation of PPT ratios (post-pre-) with DE genes in FM (n=38) (lower left), and non-FM controls (n=12) (lower right). Pearson correlation values and associated p-values (*, p<0.05; **, p<0.01; ***, p<0.001) between gene expression levels and symptom scores, or PPT ratios are shown.
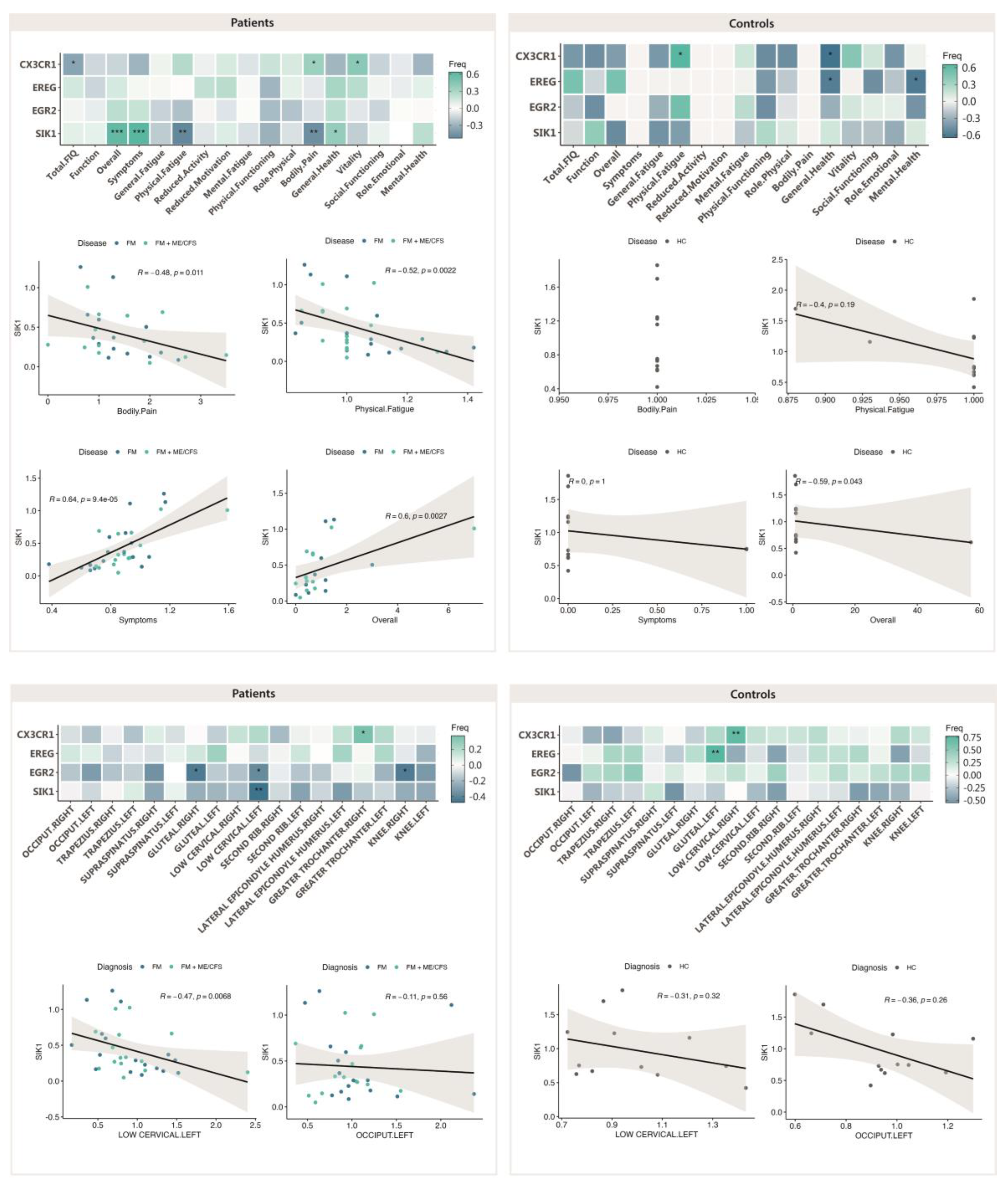
Figure 8.
Symptom improvement with validated DE genes with MT in FM (n=19) (upper left), and FM with co-diagnosis of ME/CFS (n=19) (upper right), and correlation of PPT ratios (post-pre-) with DE genes in FM (n=19) (lower left), and FM with co-diagnosis of ME/CFS (n=19) (lower right). Pearson correlation values and associated p-values (*, p<0.05; **, p<0.01; ***, p<0.001) between gene expression levels and symptom scores, or PPT ratios are shown.
Figure 8.
Symptom improvement with validated DE genes with MT in FM (n=19) (upper left), and FM with co-diagnosis of ME/CFS (n=19) (upper right), and correlation of PPT ratios (post-pre-) with DE genes in FM (n=19) (lower left), and FM with co-diagnosis of ME/CFS (n=19) (lower right). Pearson correlation values and associated p-values (*, p<0.05; **, p<0.01; ***, p<0.001) between gene expression levels and symptom scores, or PPT ratios are shown.

Table 1.
Participant baseline FIQ [13,14], MFI [15] and SF-36 (Likert scale) [16] questionnaire scores by cohort, as indicated.
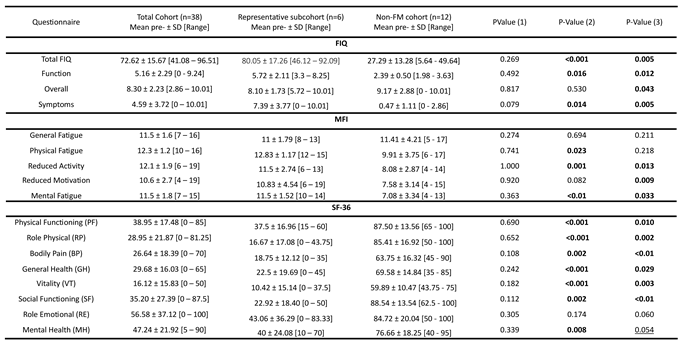
PValue (1) refers to the pvalues obtained by comparing the complete FM cohort (n=38) and the representative FM subcohort (n=6); pValue (2) refers to the p-values obtained by comparing the complete FM cohort (n=38) and the and non-FM cohort (n=12); and pValue (3) refers to the p-values obtained by comparing the representative FM subcohort (n=6) and non-FM cohort (n=12). Statistically significant differences (p≤0.05) appear bolded, tendencies (p≤0.1) are underlined.
Table 2.
Participant baseline PPTs by studied cohort, as indicated. Patient tender point sensitivity assessment, as determined by triplicate measurements in lbf with a FDIX Force Gage, ForceOne algometer (Wagner Instruments, Greenwich, CT, USA) [12] at baseline, by cohort, as indicated.
Table 2.
Participant baseline PPTs by studied cohort, as indicated. Patient tender point sensitivity assessment, as determined by triplicate measurements in lbf with a FDIX Force Gage, ForceOne algometer (Wagner Instruments, Greenwich, CT, USA) [12] at baseline, by cohort, as indicated.
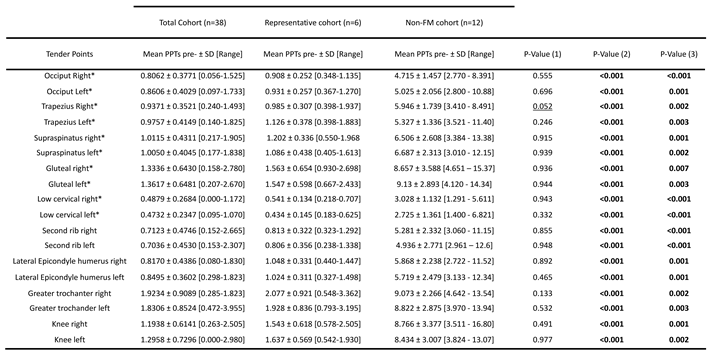
PValue (1) refers to the pvalues obtained by comparing the complete FM cohort (n=38) and the representative FM subcohort (n=6); pValue (2) refers to the p-values obtained by comparing the complete FM cohort (n=38) and the and non-FM cohort (n=12); and pValue (3) refers to the p-values obtained by comparing the representative FM subcohort (n=6) and non-FM cohort (n=12). Statistically significant differences (p≤0.05) appear bolded, tendencies (p≤0.1) are underlined.
Table 3.
DE expressed genes with MT on PBMCs from FM patients, as determined by RNAseq.
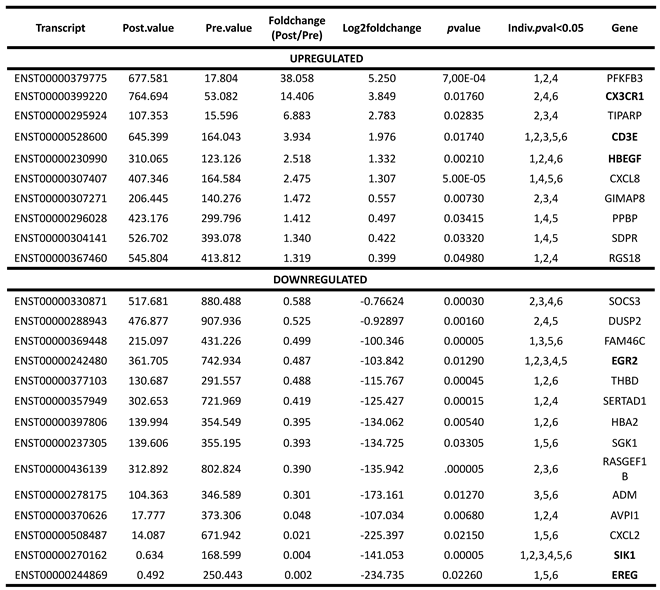
Numbers in the Indv.pval column indicate the participants that showed DE of the indicated genes with MT by RNAseq analysis, up or downregulated, as indicated. Genes randomly selected for RT-qPCR validation appear bolded.
Table 4.
Patient response to MT as evidenced by score differences in the standard, validated, FIQ, MFI and SF-36 instruments [13,14,15,16], by studied cohort. Significant differences (p≤0.05) are bolded.
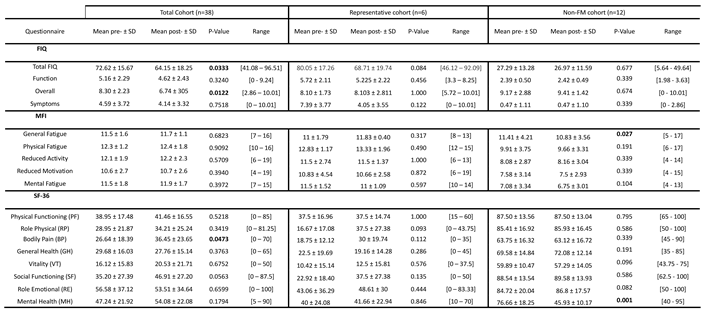
Table 5.
Patient response to MT as evidenced by PPT differences, by studied cohort.
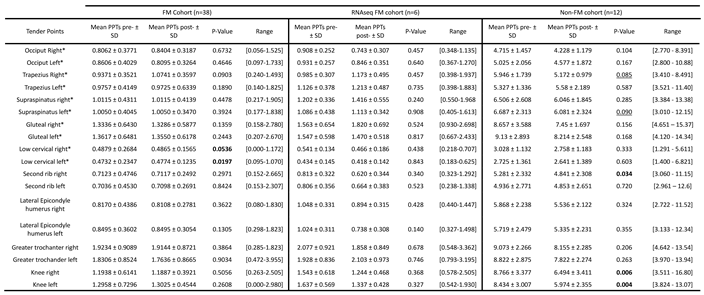
(*) Tender points in areas treated with manual therapy; PPT (Pressure Point Threshold); SD (Standard Deviation); pre- (pre-treatment); post- (post-treatment). Significant differences (p≤0.05) are bolded.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



