Submitted:
02 August 2024
Posted:
06 August 2024
You are already at the latest version
Abstract
Capulus danieli, a distinct member of Capulidae, exhibits a unique ecological behaviour by attaching and drilling onto the pecten, distinguishing itself from other gastropods. Herein, we present the first characterisation of the mitochondrial genome of Capulus danieli. The results showed that the genome spans 15600 base pairs, with an A + T content of 71.12% and a G + C content of 28.88%. Within this genetic makeup, we identified 13 protein-coding genes, 22 transfer RNA genes, and 2 ribosomal RNA genes. Phylogenetic analysis revealed a close evolutionary relationship between Capulus danieli and members of the Ficoidea superfamily. The divergence time estimation suggested that Capulus danieli diverged approximately 52.29 million years ago. These findings significantly contribute to our understanding of the evolutionary history and genetic architecture of Capulus danieli, shedding light on its unique ecological adaptations to the marine environment.
Keywords:
Capulus danieli
; mitochondrial genome
; protein-coding genes
; divergence time estimation
1. Introduction
The family Capulidae belongs to the Phylum Mollusca, Subclass Caenogastropoda, Class Gastropoda, Order Littorinimorpha, and is a widely distributed marine gastropod worldwide. It comprises eighteen acknowledged genera, the preponderance of which exhibit a coiled form, although there exists a notable number of species resembling limpets [1], Among the uncoiled groups, Capulus Montfort, 1810 is the most widespread genus based on current taxonomy and includes at least 20 recent species [2], The gastropod Capulidae reached its highest level of limpetisation [3], Capulid shell plasticity is associated with a broad range of feeding ecology. Most coiled capulids exploit water currents generated by their hosts, which is obligate suspension feeding [4], Capulus danieli, a member of the family Capulidae, is a relatively large species within its genus, with diameters ranging from 20 to 30 mm [5], Characterised by its low, limpet-shaped form, this species possesses a unique shell structure with a protoconch composed of smooth, planispiral whorls, followed by a cap-shaped teleoconch featuring a large, horseshoe-shaped muscle scar [6].
Ecologically, Capulus danieli shows intriguing behaviours, notably its attachment and drilling onto the shells of pecten, reflecting a xenomorphic sculpture mirroring the form of its host [5], Despite its ecological significance, research on Capulus danieli remains limited.
Because most capulid species are rare and rarely collected alive, only three studies have attempted to produce a molecular phylogeny tree of the family Capulidae. [7], produced the first capulid phylogeny and investigated the larval ecology of Antarctic species. More recently, [8], proposed a new phylogenetic hypothesis using a taxonomic framework (six genera) to address the Indo-West Pacific diversity of Hyalorisia Dall, 1889. [1], also performed ancestral state reconstruction analysis on a time-calibrated phylogenetic tree within the family Capulidae and suggested that capulids evolved from a coiled suspension feeder lineage and that a shift to kleptoparasitism occurred in the family ancestor.
Here, we present the first characterisation of the mitochondrial genome of Capulus danieli to elucidate its gene function, phylogenetic relationships with limpet-like gastropods, and divergence time estimation. This study advances our understanding of Capulus danieli's evolutionary history and ecological adaptations, contributing valuable insights into the broader field of molluscan biology.
2. Materials and Methods
2.1. Sample Collection
Four specimens of Capulus danieli were collected in December 2022 from Yangjiang, Guangdong Province (21°85′N, 111°95′E). Although individual capture was not feasible, the specimens were discovered on the surface of shells of the economically significant scallop Amusium pleuronectes (Figure 1). Capulus specimens are relatively rare, with approximately one found in every 30 scallops. Morphological identification involved stripping fresh tissue from the shells, excising the digestive glands, and preserving the muscle tissue in anhydrous ethanol.
2.2. DNA Extraction, Library Preparation and Next Generation Sequence
The genomic DNA (gDNA) was extracted using the MagPure Bacterial DNA Kit from Magen (Guangzhou, China) and pre-grinding in liquid nitrogen. The concentration of the extracted DNA was tested using a Qubit dsDNA HS assay kit from Sangon (Shanghai, China), and its integrity was confirmed using 1% agarose gel electrophoresis. Subsequently, library preparation and next-generation sequencing were performed by Sangon Biotech (Shanghai) Co., Ltd. For the library preparation, 500 ng of the quantified DNA was randomly fragmented using Covaris (Woburn, USA). The Hieff NGS MaxUp II DNA Library Prep Kit for Illumina from YEASEN (Shanghai, China) was utilised for the following steps. The process included repairing the ends and adding a 3ʹ end A tail, followed by the ligation of adaptors using an enhancer and Fast T4 DNA ligase. Index primers were added through PCR, and the resulting amplified product (approximately 400 bp was selected using DNA selection beads. The concentration and size of the library were confirmed using the Qubit 4.0 (Thermo, Waltham, USA) and 2% agarose gel electrophoresis, respectively, and the libraries were pooled and loaded onto a Novaseq 6000 from Illumina (San Diego, USA) or DNBseq-T7 from BGI (Shenzhen, China) sequencer using a 2 × 150 bp paired-end sequence kit, following the manufacturer's instructions [9].
2.3. Sequence Assembly and Annotation of Mitochondrial Genome
Raw sequencing data of at least 6 GB were used for subsequent analyses. All raw reads were trimmed using Fastqc (version 0.11.2) [10]. SPAdes software (version 3.15) [11], was used to assemble the raw sequence reads into contigs. The complete mitochondrial genome was obtained by aligning the contigs against a reference mitochondrial genome, serving as the seed sequence for the software MITObim (version 1.9.1) with 30 iterations. An in-house genome annotation pipeline, utilizing NCBI-BLAST, was applied to annotate the protein-coding genes, tRNA, and rRNA genes. Subsequently, circular gene maps of Capulus danieli were generated using Circos software.
2.4. Sequence Analyses of Mitogenomes
The nucleotide composition of the entire mitogenome, protein-coding genes (PCGs), rRNA and tRNA genes, and A-T content were analysed using MEGA 7.0 [12]. Additionally, codon usage and relative synonymous codon usage (RSCU) of the PCGs were investigated. Base skew values were calculated using the formulas A-T skew = (A − T)/(A + T) and G-C skew = (G − C)/(G + C) [13,14]. Furthermore, DnaSP 6.0 [15,16], was selected to analyse the non-synonymous (Ka) and synonymous (Ks) substitution rates of mitogenomes in Capulus danieli to explore their evolutionary adaptation.
2.5. Systematic Analysis
We selected 25 species to construct a phylogenetic tree based on their complete mitochondrial genomes. The sequences of 23 species belonging to Haliotidae, Neritidae, Patellidae, Nacellidae, Calyptraeidae, Muricidae, Ficidae, Naticidae, Strombidae, Struthiolariids, and Xenophoridae in the Gastropoda class were obtained from the NCBI database (Table 1). Some species were selected based on their similarity in shell morphology to C. danieli [17]. Others were selected based on their close phylogenetic relationship with C. danieli as determined by the alignment of their mitochondrial genomes available at NCBI.
Additionally, two bivalve species, Chlamys farreri and Mizuhopecten yessoensis, were included as outgroups, with GenBank accession numbers EF473269.1 and FJ595959.1, respectively. Prior to tree construction, PhyloSuite v1.2.3 [18,19], was utilised to extract the protein-coding genes (PCGs) from each sequence, followed by multiple sequence alignment using MAFFT [20,21]. The alignment results of the protein-coding gene sequences were optimised using MACSE [22], and Gblocks [23], were employed for sequence pruning. The extracted PCGs were concatenated into a comprehensive dataset, and ModelFinder [24,25], was used to partition the data and select the optimal evolutionary model. A phylogenetic tree was constructed using the MrBayes method [26].
2.6. Estimation of Differentiation Time of Capulidae
We used the 16S genes of 11 Capulidae species (Table 1) with the topological structure of the Bayesian tree as the framework to estimate the divergence time within the Capulidae family. We used BEAST v2.7.6 software [27], and a relaxed clock model for this analysis. The Yule process was used to determine the tree’s prior branch evolution rate.
We used the oldest confidently attributable species to the genus Capulus, i.e., Capulus onyxoides Cossmann, 1879, from the Ypresian period to incorporate fossil evidence into the analysis (Lower Eocene, 56–47.8 Ma) as a standard point. Similarly, we referenced Capulus (Hyalorisia) nettlesi Robinson, 1983† from the Upper Eocene period (41.2–33.9 Ma) for the genus Hyalorisia [1]. Markov chain Monte Carlo (MCMC) analysis was performed with 100 million generations sampled every 1000 generations. The TreeAnnotator v1.8.4 component of the BEAST software package was used to discard the first 25% of aging samples as burn-in. Convergence of the chain was confirmed using Tracer v.1.7, [28], ensuring effective sample size (ESS) values greater than 200. The resulting divergence time estimates were validated against Timetree fossil records, and the results were reported to verify the accuracy.We used TVBOT as a graphic beautification tool (https://www.chiplot.online/tvbot.html) [29].
3. Results
3.1. Distribution Characteristics of Capulus Danieli Mitochondria
The complete mitochondrial genome of Capulus danieli spanned 15,640 bp (GenBank accession number: NC 084349.1) and comprised a typical circular, closed, double-stranded molecule with a control region and 37 coding genes (Figure 2). Among these, there were 13 protein-coding genes (COX1, COX2, ATP8, ATP6, ND1, ND6, CYTB, ND4L, ND4, ND5, COX3, ND3, and ND2), including 3 cytochrome oxidase subunit genes, 7 NADH dehydrogenase subunit genes, 2 ATP synthase subunit genes, and 1 cytochrome b gene. Additionally, there were 22 tRNA genes (including, trnM, trnY, and trnC) and 2 rRNA genes (s-rRNA and l-rRNA).
The arrangement and location of genes in the mitochondrial genome of C. danieli were similar to those observed in most known neogastropods. The gene structure was densely packed with intervals and overlaps. The longest gene in the mitochondrial DNA was nad5 (1734 bp), whereas the shortest was trnQ (55 bp). Notably, the length of tRNA genes was generally less than 100 bp, with the anticodon positioned within the tRNA gene.
The mitochondrial genome of C. danieli contained 23 intergenic spacers with a combined length of 829 bp. The longest intergenic spacer was situated between trnF and COX3, spanning 473 bp, followed by the spacer between ATP6 and trnM, which was 77 bp in length. Three gene overlaps totalling 27 bp were observed, with the longest overlap occurring between ND5 and trnF (18 bp), followed by nad4l and nad4 (7 bp). Additionally, there were 11 regions with neither an overlap nor an interval.
All 13 protein-coding genes in the mitochondrial DNA of C. danieli contained initiation and termination codons. The initiation codon was ATG, whereas TAG was the termination codon for ND4L, ND4, and ND3. The remaining genes contained the TAA termination codon, except for ND2 and ND5, which featured an incomplete codon T (Table 2).
3.2. Base Content and Base Shift Degree
There were 4938, 6186, 2333, and 2183 A, T, G, and C bases of mtDNA in Capulus danieli, accounting for 31.57%, 39.55%, 14.92%, and 13.96% of the total bases, respectively. The A + T content was 71.12%, the G + C content was 28.88%, the AT-skew was −0.112, and the GC-skew was 0.033. The protein-coding gene length of Capulus daniel was 11247 bp, the tRNA length was 1439 bp, and the rRNA length was 2137 bp (Table 3). mtDNA showed AT base bias, and A + T was 70.03%, 71.72%, and 73.04% in the PCGs, tRNA, and rRNA genes, respectively.
In the mitochondrial DNA (mtDNA) of Capulus danieli, the number of A, T, G, and C bases was 4938, 6186, 2333, and 2183, respectively, constituting 31.57%, 39.55%, 14.92%, and 13.96% of the total bases. The A + T content was higher (71.12%) than the G + C content is lower (28.88%). The AT skew was −0.112, indicating a bias towards A over T, whereas the GC skew was 0.033. The length of the protein-coding gene (PCG) in Capulus danieli was 11,247 bp, with tRNA and rRNA lengths of 1439 and 2137 bp, respectively. mtDNA exhibited an AT base bias, with A + T content of 70.03%, 71.72%, and 73.04% in PCGs, tRNA, and rRNA genes, respectively. rRNA genes display the most significant bias towards A + T content (73.04%), exceeding G + C content (26.96%), with AT-skew at 0.083 and GC-skew at 0.139. Within protein-coding genes, the proportion of A bases was lower than that of T bases, with the largest difference observed in the AT base content percentages. In contrast, the difference in CG base content percentages is minimal, with AT-skew at −0.180 and GC-skew at 0.017 (Table 3). The control region segments typically exhibit a higher A + T content in mitochondrial gene sequences, making them prone to base mutations, thereby qualifying them as highly variable regions.
3.3. Mitochondrial Gene Codon Usage
In total, 3755 codons were present in the protein-coding genes (PCGs) of C. danieli. Among these, the UUA (L) codon exhibited the highest frequency of usage, occurring 351 times, followed by the relatively high-frequency UUU (F), AUU (I), and AUA (M) codons. The UAG codon was the least frequently used (Table 4). Analysis of the relative synonymous codon usage (RSCU) values for C. danieli revealed that TTA, TCT, CCT, and GCT were the top four preferred codons (Figure 3).
3.4. Selection Pressure Analysis
The Ka/Ks values for the 13 PCGs in Capulus danieli were below one (Figure 4), indicating that all 13 PCGs were subjected to purifying selection. As shown in Figure 4, the Ka value of cox1 was the lowest (0.002), whereas that of ND2 was the highest (0.256). Conversely, the Ks value of cox1 was the highest (11.49), whereas that of ND4L was the lowest (3.288). Although there were variations in the Ka and Ks values among the 13 PCGs, all Ka/Ks values were less than 1, with ND2 exhibiting the highest Ka/Ks value (0.055) and cox1 showing the lowest Ka/Ks value (Ka/Ks = 0.002). This indicated that atp8 had the highest evolutionary rate under purifying selection pressure, followed by ND4L (Ka/Ks = 0.042). Conversely, cox1 had the slowest evolutionary rate, suggesting that it was the most conserved during evolution.
3.5. Mitochondrial Dna Microsatellites
Microsatellites, also known as simple sequence repeats (SSRs), arise from DNA slippage and misalignment during DNA replication or repair or from unequal exchange between sister chromatids during mitosis or meiosis. These short, repetitive sequences are valuable for species-specific identification.
In this study, three SSRs were identified in the mitochondrial genome of Capulus danieli (Table 5). All these SSRs consist of single nucleotide repeats and belong to the A/T motif type.
3.6. Phylogenetic Relationship
The Bayesian inference (BI) method was used to build a tree of 13 PCGs sequences from 25 species, of which two species, Chlamys farreri and Mizuhopecten yessoensis, were used as outgroups (Figure 5). The phylogenetic tree contained 13 families, each of which formed a monophyletic branch. Previous studies have demonstrated that this is reasonable [30,31,32]. The posterior probability value of most branches was one, and the overall topology was stable. From the overall perspective of the phylogenetic tree, the following evolutionary relationships were observed: (Neritimorpha + Patellogastropoda) + Vetigastropoda + Caenogastropoda). From the phylogenetic relationship of gastropoda, it could be seen that Haliotidae in Vetigastropoda was located in the basal position of the phylogenetic tree and was the earliest branch of the Limpet-like shell. The gastropods that appeared after this were mainly divided into two major branches: (Neritimorpha + Patellogastropoda) and Caenogastropoda. Capulus danieli has the closest genetic relationship with Ficus, which also belongs to the Littorinimorph. Capulus danieli had a distant phylogenetic relationship with the limpet-like shell species Neritimorpha,Patellogastropoda, and Caenogastropoda, and was not within the same major branch. A Limpet-like shell appeared in multiple branches of the phylogenetic tree, indicating that it has evolved independently several times in Gastropoda. Statistically, limpet-like shells occur in at least 54 families of gastropods [17].
3.7. Divergence Time Estimation
We reconstructed a divergence time tree of Capulidae based on the analysis of the 16S gene sequence to explore its evolutionary process (Figure 6). According to the results, the origin of the Capulidae can be traced back to the end of the Callovian stage (Middle Jurassic) period about 155.25 million years ago. This finding provided us with an important time node for the origin of this family.
It began to evolve to the first limpet-like shell, which is reflected in the branch of Cryocapulus subcompressus, at 123.95 Mya (Early Cretaceous). Subsequently, the second evolution to the limpet-like shell, which dates back to about 82.65 million years ago (Late Cretaceous), resulted in a divergence between the cap-shaped genus Trichamathina and the spiral genus Torellia. About 66.44 million years ago (Late Cretaceous), there was a divergence between Hyalorisia and Capulus. The genus Trichamathina has a limpet-like shell type. It is mainly manifested in symmetrical and enlarged body snails but still retains some of the curling tower characteristics, which reveals its unique evolutionary path. About 66.44 million years ago (Late Cretaceous), there was a divergence between Hyalorisia and Capulus. Although there are differences in the taxonomic status of the two species, they show similar and highly capped shell-like characteristics, which further highlights the diversity and complexity of the evolution of the family. Finally, during the Eocene (Paleogene), about 52.29 to 36.03 million years ago, the differentiation of Capulidae reached its peak, and Capulidae gradually evolved a shell type similar to that of limpets. This period marked an important stage in the evolution of the family and provided valuable information about its evolution.
4. Discussion
The present study, leveraging mitochondrial protein-coding genes (PCGs), sheds light on the phylogenetic relationships within the Gastropoda class, particularly focusing on the Capulidae family. Our analysis, constrained by the absence of other Capulidae mitochondrial genomes in the NCBI public database, used the mitochondrial genome of Capulus danieli to establish a phylogenetic framework. The findings have significant implications for our understanding of the evolutionary history and morphological adaptations within this diverse group of marine gastropods. The system phylogenetic tree constructed in this study places Capulus danieli close to Ficus within the Order Littorinimorpha.
The recurrent emergence of the limpet-like shell within Capulidae, as observed in the Cryocapulus, Capulus + Hyalorisia, and Trichamathina lineages, underscores the complex evolutionary pressures that have shaped the diversity of shell forms in this family. Our results suggested that Capulus, which includes at least 20 extant limpet-like species [2], may be polyphyletic and have evolved through multiple independent lineages. This polyphyly indicated convergence in shell morphology, which is likely a response to similar ecological challenges or opportunities.
The molecular clock analysis estimated the origin of the Capulidae to be in the Middle Jurassic period, specifically during the Callovian stage, approximately 155.25 million years ago. [1], estimated that Capulidae originated 112.87 million years ago. This timeframe precedes the oldest known Capulidae fossil record from the Late Cretaceous-Campanian stage [33]. The discrepancy between molecular and fossil evidence underscores the need for further research to reconcile these divergent timelines and better understand the early evolutionary history of Capulidae.
The divergence time for Capulus danieli, dated to around 52.29 million years ago during the Palaeocene-Eocene transition, coinciding with a period of global warming and elevated sea surface temperatures [34]. The paleoenvironmental conditions of this era, characterised by transient eutrophication and ocean acidification [35,36,37], likely exerted selective pressures that favoured the adoption of a parasitic lifestyle in Capulus danieli. This ecological shift may have facilitated the transition to a limpet-like shell form, providing the species with enhanced access to nutrients via kleptoparasitising bivalves.
The current study was limited by the scarcity of mitochondrial genomic data for other Capulidae species, which restricted a comprehensive analysis of intrafamilial differences. Future studies should aim to fill this gap by sequencing additional mitochondrial genomes in this family. Additionally, ecological observations and molecular data are essential to validate the hypothesised adaptive transitions and provide a more nuanced understanding of the evolutionary trajectory of Capulidae.
5. Conclusions
In this study, we reported the characterisation of the mitochondrial genome of Capulus danieli, which is the first complete mitochondrial genome recorded in the family Capulidae. We analyzed the characteristics of the mitochondrial sequence of Capulus danieli, mainly including: AT skew, codon usage, selection pressure, microsatellites and elucidated the evolutionary relationship between Capulus danieli and limpet-like gastropod through phylogenetic analysis. Through the estimation of divergence time, the evolution period of Capulidae is estimated, and the process of its transformation into limpet-like shell is described.These findings significantly contribute to our understanding of the evolutionary history and genetic architecture of Capulus danieli, shedding light on its unique ecological adaptations to the marine environment.
Author Contributions
Yao-Yu Xie: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Investigation (equal); Software (equal);Validation (equal); Visualization (equal); Writing - original draft (equal). Yi-Da Han: Resources (equal). Jun-Xia Mao: Resources (equal). Xu-Bo Wang: Resources (equal). Zhen-Lin Hao: Resources (equal). Ying Tian: Project administration (equal); Supervision (equal); Writing - review & editing (equal).
Funding
This research was supported by the National Key Research and Development Program of China (2021YFB2600200); the Central Government Subsidy Project for Liaoning Fisheries (2023).
Institutional Review Board Statement
The experimental protocol was designed in accordance with the recommendations of the Regulations of the Laboratory Animal—Guideline for Ethical Review of Animal Welfare (National Standards of P. R. China, GB/T 35823—2018) and reviewed and approved by the animal care and use committee of Dalian Ocean University (DLOU-2023007).
Informed Consent Statement
Not applicable.
Acknowledgments
The authors are also grateful to the anonymous reviewers for the great elaboration of the manuscript through their critical reviewing and comments.
Data Availability Statement
The sequenced mitochondrial genome has been uploaded to NCBI (accession number of OR687630.1).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fassio, G.; Bouchet, P.; Lozouet, P.; Modica, M.V.; Russini, V.; Schiaparelli, S.; Oliverio, M. Becoming a limpet: An ‘intermittent limpetization’ process driven by host features in the kleptoparasitic gastropod family Capulidae. Mol. Phylogenet. Evol. 2020a, 155, 107014. [CrossRef]
- Worms. Available online: https://www.marinespecies.org/aphia.php?p=taxdetails&id=137729 (accessed on 24 july 2014).
- Simone, L.R.L. Main processes of body modification in gastropods: the limpetization. Malcopedia 2018, 1, 23–35.
- Iyengar, E.V. Suspension feeding and kleptoparasitism within the genus Trichotropis (Gastropoda: Capulidae). J. Mollus. Stud. 2007, 74, 55–62. [CrossRef]
- Orr, V. The drilling habit of Capulus danieli (Crosse) (Mollusca:Gastropoda). Veliger 1962, 5, 63–67.
- Ponder, W.F.; Lindberg, D.R. Towards a phylogeny of gastropod molluscs: an analysis using morphological characters. Zool. J. Linn. Soc. 1997, 119, 83–265. [CrossRef]
- Fassio, G.; Modica, M.V.; Alvaro, M.C.; Schiaparelli, S.; Oliverio, M.J.H. Developmental trade-offs in Southern Ocean mollusc kleptoparasitic species. Hydrobiologia 2015, 761, 121–141.
- Fassio, G.; Russini, V.; Buge, B.; Schiaparelli, S.; Modica, M.V.; Bouchet, P.; Oliverio, M. High cryptic diversity in the kleptoparasitic genus Hyalorisia Dall, 1889 (Littorinimorpha: Capulidae) with the description of nine new species from the Indo-West Pacific. J. Mollus. Stud. 2020b, 86, 401–421. [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884-i890. [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455-477. [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [CrossRef]
- Hassanin, A.; Léger, N.; Deutsch, J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst. Biol. 2005, 54, 277–298. [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [CrossRef]
- Rozas, J.; Rozas, R. DnaSP, DNA sequence polymorphism: An interactive program for estimating population genetics parameters from DNA sequence data. Comput. Appl. Biosci. 1995, 11, 621–625. [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25,1451–1452. [CrossRef]
- Vermeij, G.J. The limpet form in gastropods: evolution, distribution, and implications for the comparative study of history. Biol. J. Linn. Soc. 2017, 120, 22-37. [CrossRef]
- Zhang, D.; Gao, F.; Jakovlic, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [CrossRef]
- Xiang, C.; Gao, F.; Jakovlić, I.; Lei, H.; Hu, Y.; Zhang, H.; Zou, H.; Wang, G.; Zhang, D. Using PhyloSuite for Molecular Phylogeny and Tree-based Analyses. iMeta. 2023, 2, e87. [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Phylogenetics Evol. 2013, 30, 772–780. [CrossRef]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, W7–13. [CrossRef]
- Ranwez, V.; Douzery, E.J.P.; Cambon, C.; Chantret, N.; Delsuc, F. MACSE v2: toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol. Biol. Evol. 2018, 35, 2582–2584. [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564-577. [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017, 14, 587-589. [CrossRef]
- Shapiro, B.; Rambaut, A.; Drummond, A.J. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences, Mol. Biol. Evol. 2006, 23, 7–9. [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [CrossRef]
- Rambaut, A.; Drummond, A. J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [CrossRef]
- Peng, Y.Y.; Yan, H.H.; Guo, L.C.; Deng, C.; Wang, C.L.; Wang, Y.B.; Kang, L.P.; Zhou, P.P.; Yu, K.Q.; Dong, X.L.; et al. Referencegenome assemblies reveal the origin and evolution of allohexaploid oat. Nat. Genet. 2022, 54, 1248–1258. [CrossRef]
- Zhong, S.; Huang, L.; Huang, G.; Liu, Y.; Wang, W. The first complete mitochondrial genome of MAMMILLA from Mammilla mammata (Littorinimorpha: Naticidae). Mitochondrial DNA Part B 2020, 5, 96–7. [CrossRef]
- Qi, Y.; Zhong, Z.; Liu, X.; He, X.; Zhou, Y.; Zhang, L.; Chen, C.; Linse, K.; Qiu, JW.; Sun, J. Phylogenomic analyses reveal a single deep-water colonisation in Patellogastropoda. Mol. Biol. Evol. 2024, 190, 107968. [CrossRef]
- Ma, Y.; Zheng, B.; Li, J.; Meng, W.; Xu, K.; Ye, Y. Characterization of the complete mitochondrial genome of Desmaulus extinctorium (Littorinimorpha, Calyptraeoidea, Calyptraeidae) and molecular phylogeny of Littorinimorpha. PLoS One 2024, 19, e0301389. [CrossRef]
- Saul, L.R.; Squires, R.L. Cretaceous trichotropid gastropods from the Pacific slope of North America: possible pathways to calyptraeid morphology. Nautilus 2008, 122, 115–142.
- Tripati, A.; Zachos, J.C.; Bice, K.; Marincovich, L. Late Paleocene arcticcoastal climate inferred from molluscan stable and radiogenic isotope ratios. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2001, 17, 101–113. [CrossRef]
- Scheibner, C.; Speijer, R.P.; Marzouk, A.M. Turnover of largerforaminifera during the Paleocene-Eocene thermal aximumand paleoclimatic control on the evolution of platform ecosystems. Geology 2005, 33, 493–496. [CrossRef]
- Scheibner, C.; Speijer, R.P. Late Paleocene-Early Eocene Tethyan carbonate platform evolution: A response tolong and shortterm paleoclimatic change. Earth-Sci. Rev. 2008, 90, 71–102. [CrossRef]
- Alegret, L.; Ortiz, S. Global extinction event in benthic foraminifera across the Paleocene/Eocene boundary at the dababiyastratotype section. Micropaleontology 2007, 52, 433–447. [CrossRef]
Figure 1.
Capulus danieli and its host Amusium pleuronectes. A, The back view of Capulus danieli. B, The ventral view of Capulus danieli. C, The lateral view of Capulus danieli. D, Capulus danieli attached on Amusium pleuronectes. E, The shell of A. pleuronectes with drillhole from Capulus danieli.
Figure 1.
Capulus danieli and its host Amusium pleuronectes. A, The back view of Capulus danieli. B, The ventral view of Capulus danieli. C, The lateral view of Capulus danieli. D, Capulus danieli attached on Amusium pleuronectes. E, The shell of A. pleuronectes with drillhole from Capulus danieli.

Figure 2.
Gene map of the complete mitogenomes for Capulus danieli.
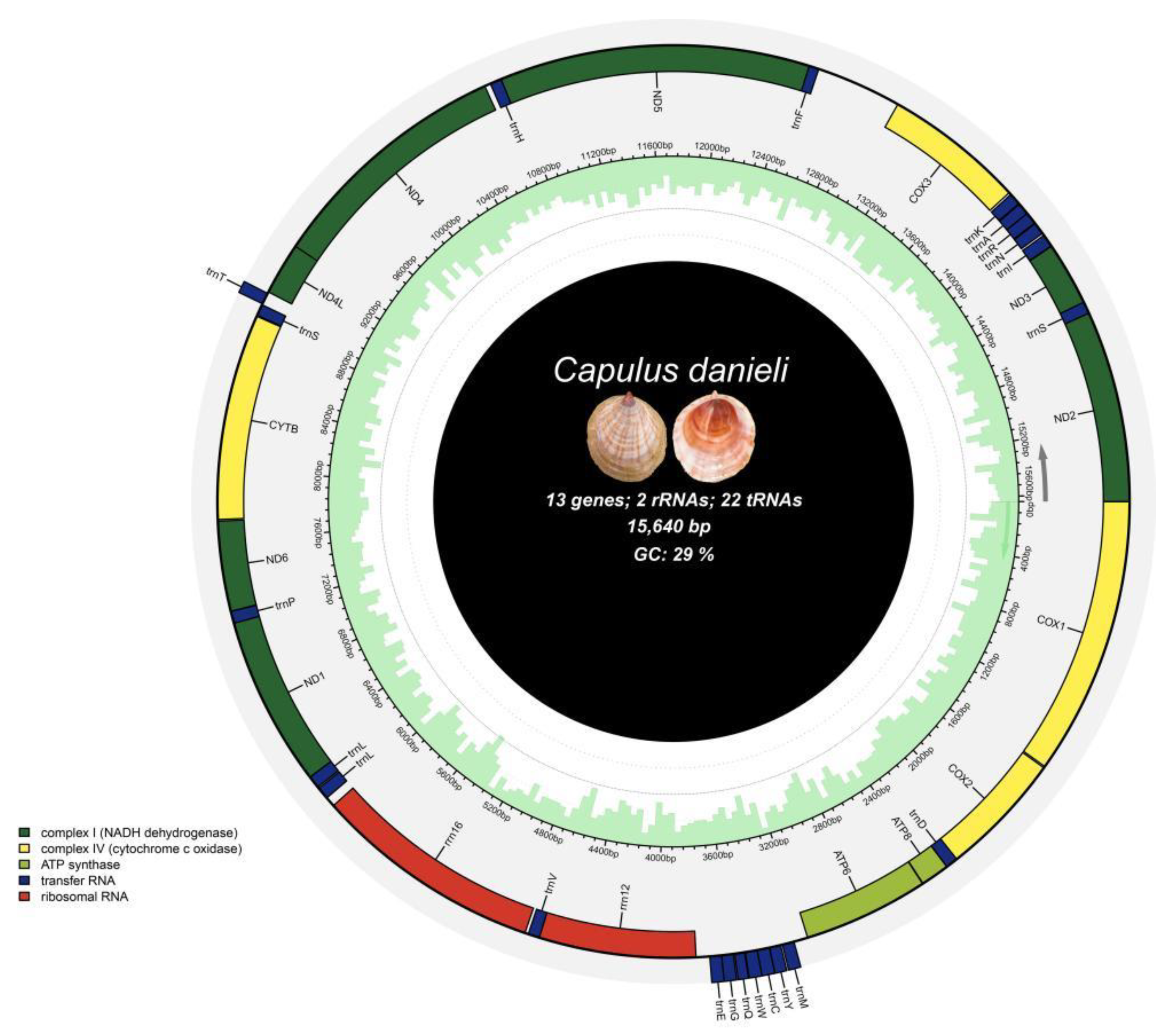
Figure 3.
The relative synonymous codon usage (RSCU) in the mitochondrial genomes of Capulus danieli.
Figure 3.
The relative synonymous codon usage (RSCU) in the mitochondrial genomes of Capulus danieli.
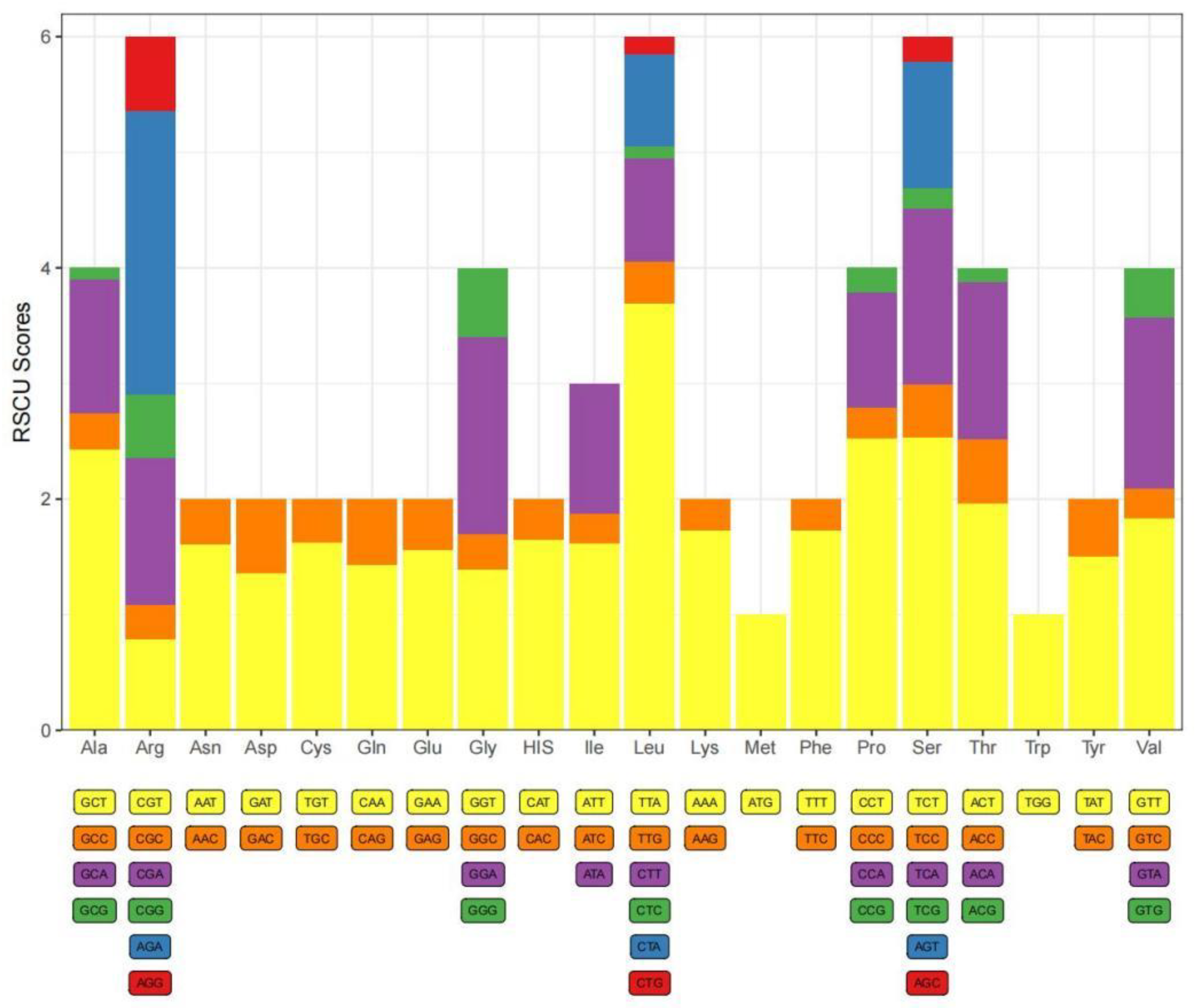
Figure 4.
Selection pressure analysis of Capulus danieli ( Ka is non-synonymous substitution value, Ks is synonymous substitution value ).
Figure 4.
Selection pressure analysis of Capulus danieli ( Ka is non-synonymous substitution value, Ks is synonymous substitution value ).
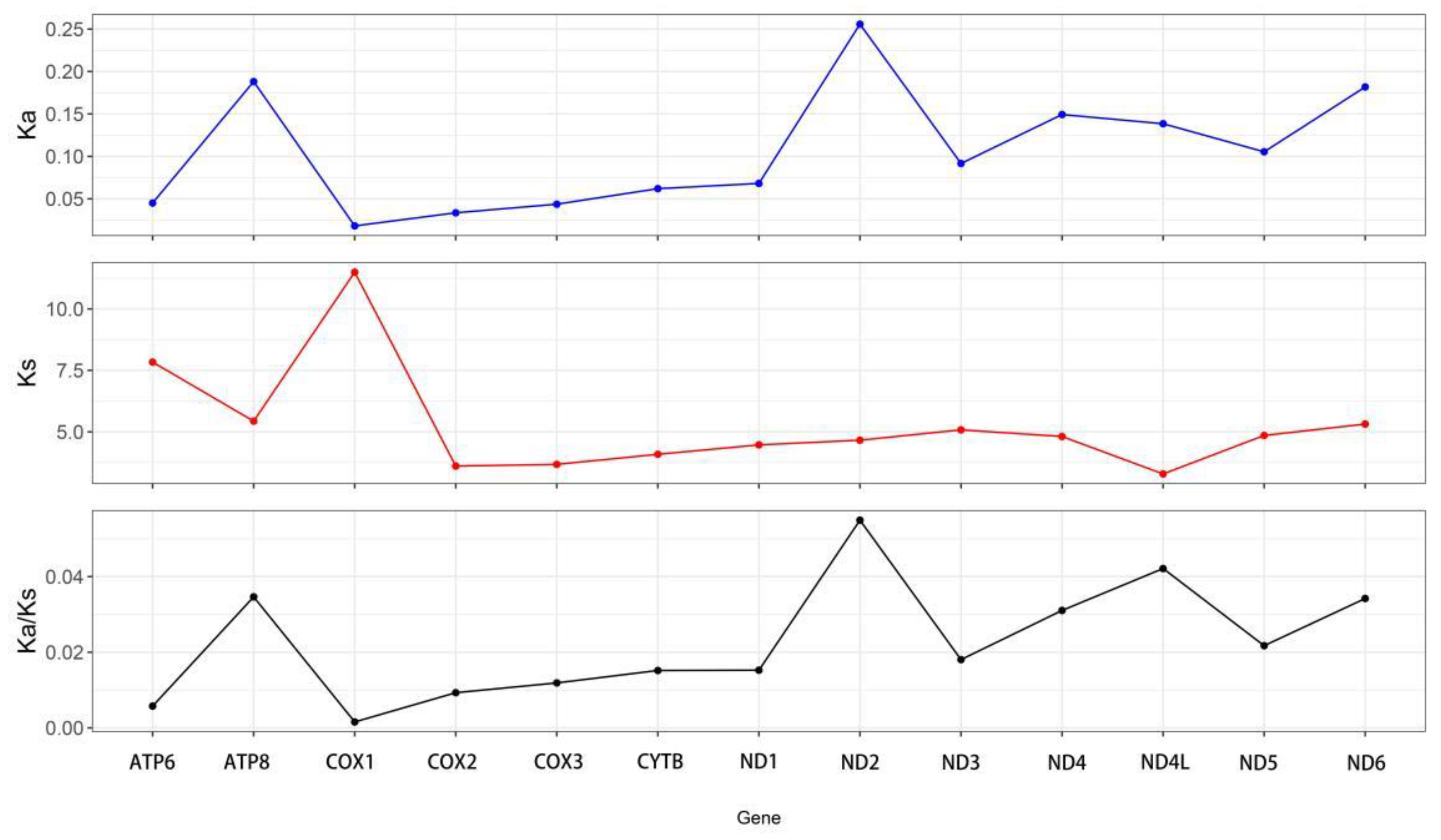
Figure 5.
Phylogenetic tree inferred using Bayesian inference methods based on concatenated sequences of 13 PCGs from 23 gastropod mitogenomes, The sequences of two bivalves were chosen as the outgroup.
Figure 5.
Phylogenetic tree inferred using Bayesian inference methods based on concatenated sequences of 13 PCGs from 23 gastropod mitogenomes, The sequences of two bivalves were chosen as the outgroup.
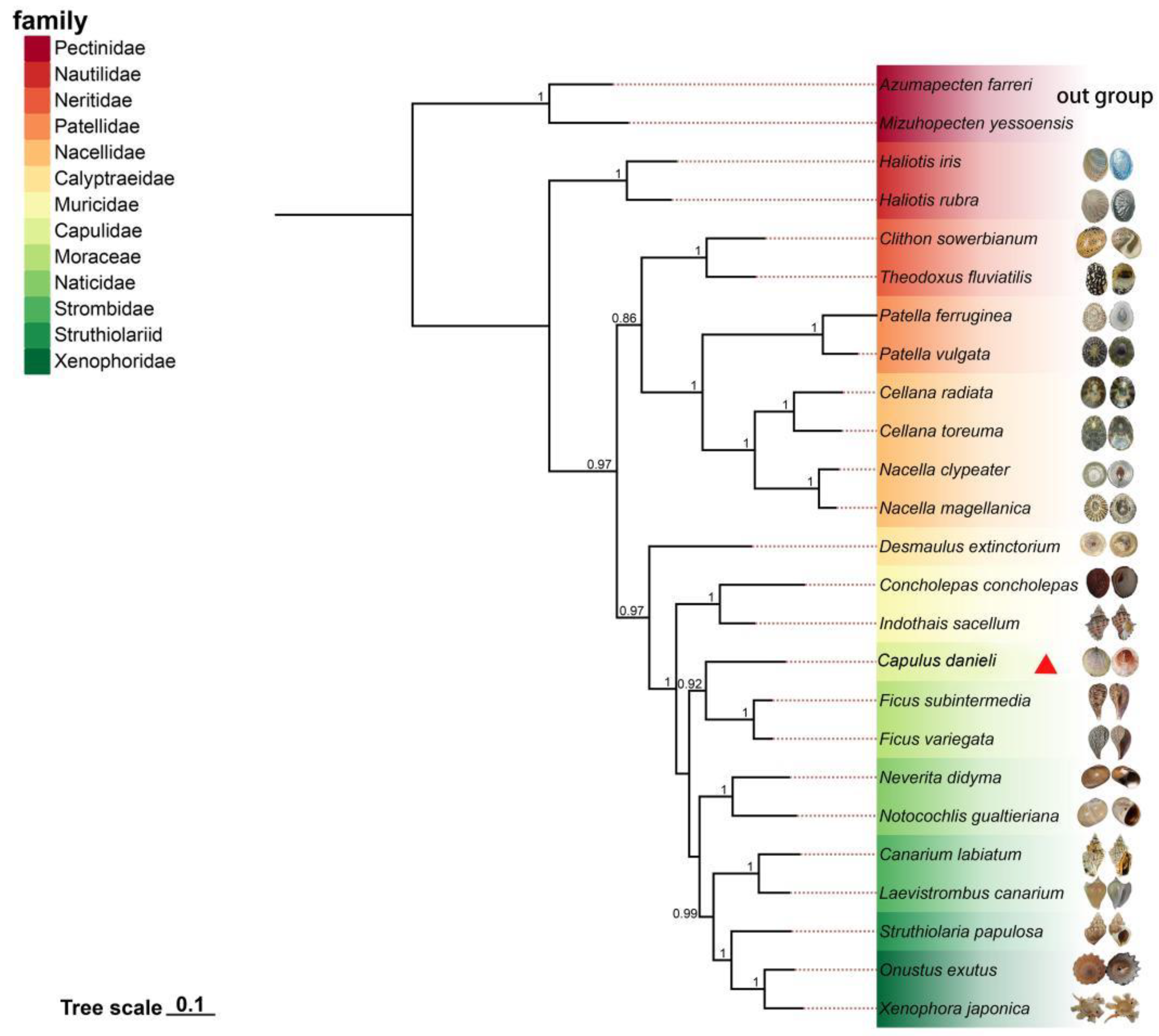
Figure 6.
Estimating the divergence time of Capulidae based on 11 16s sequences.bars indicate 95% highest posterior density intervals for node ages (Ma), and number at node the median (Ma).
Figure 6.
Estimating the divergence time of Capulidae based on 11 16s sequences.bars indicate 95% highest posterior density intervals for node ages (Ma), and number at node the median (Ma).
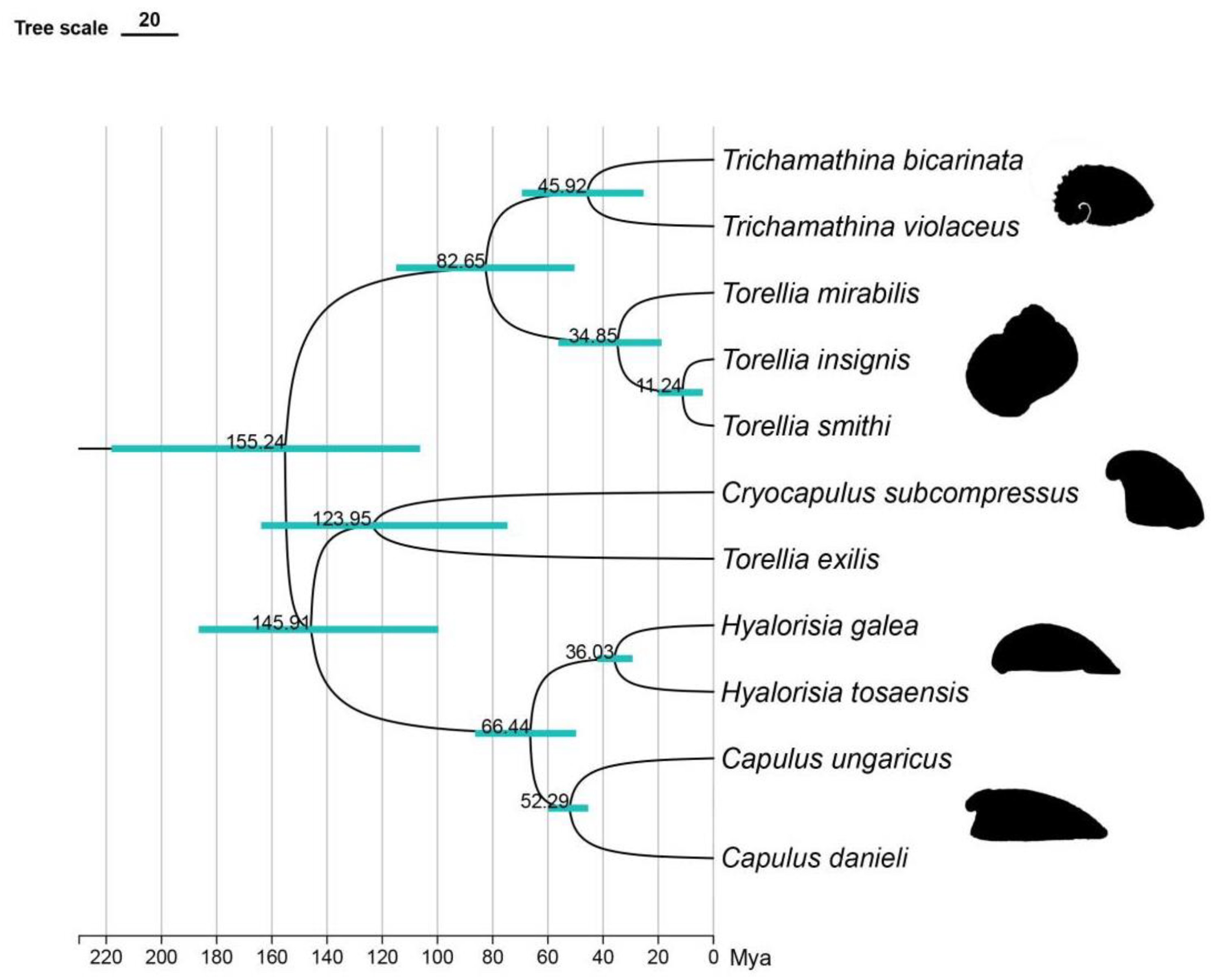

Table 1.
List of species analyzed in this study and their GenBank accession numbers.
| Classification(family) | Species name | Size(bp) | GenBank accession |
|---|---|---|---|
| Haliotidae | Haliotis iris | 17131 | KU310895.1 |
| Haliotis rubra | 16907 | AY588938.1 | |
| Neritidae | Clithon sowerbianum | 15919 | MT230542.1 |
| Theodoxus fluviatilis | 15667 | MT628587.1 | |
| Patellidae | Patella ferruginea | 14400 | MH916654.1 |
| Patella vulgata | 14808 | MH916653.1 | |
| Nacellidae | Cellana radiata | 16194 | MH916651.1 |
| Cellana toreuma | 16268 | ON018805.1 | |
| Nacella clypeater | 16742 | KT990124.1 | |
| Nacella magellanica | 16663 | KT990125.1 | |
| Calyptraeidae | Desmaulus extinctorium | 16608 | OQ511529.1 |
| Muricidae | Concholepas concholepas | 15495 | JQ446041.1 |
| Indothais sacellum | 15237 | NC 063938.1 | |
| Capulidae | Capulus danieli | 15640 | OR687630.1 |
| Capulus ungaricus | 525 | MT525803.1 | |
| Hyalorisia tosaensis | 405 | MT525849.1 | |
| Hyalorisia galea | 526 | MT525835.1 | |
| Cryocapulus subcompressus | 760 | KR364850.1 | |
| Torellia exilis | 529 | MT525847.1 | |
| Torellia smithi | 714 | KR364868.1 | |
| Torellia insignis | 769 | KR364865.1 | |
| Torellia mirabilis | 745 | KR364856.1 | |
| Trichamathina violaceus | 529 | MT525806.1 | |
| Trichamathina bicarinata | 529 | MT525846.1 | |
| Ficidae | Ficus subintermedia | 16255 | OR522697.1 |
| Ficus variegata | 15736 | NC 056153.1 | |
| Naticidae | Neverita didyma | 15629 | NC 046594.1 |
| Notocochlis qualtieriana | 15176 | NC 046705.1 | |
| Strombidae | Canarium labiatum | 15843 | NC 084213.1 |
| Laevistrombus canarium | 15626 | NC 053786.1 | |
| Struthiolariidae | Struthiolaria papulosa | 15475 | NC 059921.1 |
| Xenophoridae | Onustus exutus | 16043 | MK327366.1 |
| Xenophora japonica | 15684 | MW244823.1 |
Table 2.
Gene organization and annotation of Capulus danieli mitochondrial genome.
| Gene | Location | Length | amino acid | start codon | terminal codon | Intergenic nucleotide*(bp) | strand |
|---|---|---|---|---|---|---|---|
| COX1 | 1-1542 | 1542 | 514 | ATG | TAA | 8 | + |
| COX2 | 1551-2243 | 693 | 231 | ATG | TAA | -1 | + |
| trnD | 2242-2309 | 68 | 1 | + | |||
| ATP8 | 2311-2469 | 159 | 53 | ATG | TAA | 2 | + |
| ATP6 | 2472-3167 | 696 | 232 | ATG | TAA | 77 | + |
| trnM | 3245-3310 | 66 | 9 | - | |||
| trnY | 3320-3382 | 63 | 0 | - | |||
| trnC | 3383-3450 | 68 | 0 | - | |||
| trnW | 3451-3517 | 67 | 3 | - | |||
| trnQ | 3521-3575 | 55 | 7 | - | |||
| trnG | 3583-3647 | 65 | 0 | - | |||
| trnE | 3648-3712 | 65 | 74 | - | |||
| s-rRNA | 3787-4653 | 867 | 0 | + | |||
| trnV | 4654-4717 | 64 | 18 | + | |||
| l-rRNA | 4736-6005 | 1270 | 54 | + | |||
| trnL | 6060-6128 | 69 | 8 | + | |||
| trnL | 6137-6205 | 69 | 0 | + | |||
| ND1 | 6206-7147 | 942 | 314 | ATG | TAA | 0 | + |
| trnP | 7148-7214 | 67 | 1 | + | |||
| ND6 | 7216-7713 | 498 | 166 | ATG | TAA | 8 | + |
| CYTB | 7722-8861 | 1140 | 380 | ATG | TAA | 5 | + |
| trnS | 8867-8930 | 64 | 5 | + | |||
| trnT | 8936-8999 | 64 | 8 | - | |||
| ND4L | 9008-9304 | 297 | 99 | ATG | TAG | -7 | + |
| ND4 | 9298-10668 | 1371 | 457 | ATG | TAG | 34 | + |
| trnH | 10701-10764 | 64 | 0 | + | |||
| ND5 | 10765-12498 | 1734 | 578 | ATG | T | -18 | + |
| trnF | 12482-12537 | 56 | 473 | + | |||
| COX3 | 13010-13789 | 780 | 260 | ATG | TAA | 12 | + |
| trnK | 13802-13868 | 67 | 4 | + | |||
| trnA | 13873-13938 | 66 | 0 | + | |||
| trnR | 13939-14005 | 67 | 4 | + | |||
| trnN | 14010-14076 | 67 | 12 | + | |||
| trnI | 14089-14159 | 71 | 2 | + | |||
| ND3 | 14162-14515 | 354 | 118 | ATG | TAG | -2 | + |
| trnS | 14514-14580 | 67 | 0 | + | |||
| ND2 | 14581-15640 | 1060 | 353 | ATG | T | 0 | + |
Intergenic nucleotide*(bp): positive values indicated the interval sequence of adjacent genes, and negative values indicated the overlapping of adjacent genes.
Table 3.
Nucleotide composition of the mitochondrial genome of Capulus danieli.
| Item | length/bp Size | A% | T% | G% | C% | (A+T)% | (C+G)% | AT-skew | GC-skew |
|---|---|---|---|---|---|---|---|---|---|
| mtDNA | 15640 | 31.57 | 39.55 | 14.92 | 13.96 | 71.12 | 28.88 | -0.112 | 0.033 |
| PCGsgenes | 11247 | 28.71 | 41.32 | 15.23 | 14.73 | 70.03 | 29.96 | -0.180 | 0.017 |
| tRNA | 1439 | 36.28 | 35.44 | 15.77 | 12.51 | 71.72 | 28.28 | 0.012 | 0.115 |
| rRNA | 2137 | 39.54 | 33.50 | 15.35 | 11.61 | 73.04 | 26.96 | 0.083 | 0.139 |
Table 4.
The number of codons of protein-coding genes.
| Codon | Count | Codon | Count | Codon | Count | Codon | Count |
|---|---|---|---|---|---|---|---|
| UUU(F) | 290 | UCU(S) | 131 | UAU(Y) | 104 | UGU(C) | 35 |
| UUC(F) | 46 | UCC(S) | 25 | UAC(Y) | 33 | UGC(C) | 7 |
| UUA(L) | 351 | UCA(S) | 77 | UAA(*) | 9 | UGA(W) | 97 |
| UUG(L) | 37 | UCG(S) | 8 | UAG(*) | 3 | UGG(W) | 13 |
| CUU(L) | 86 | CCU(P) | 91 | CAU(H) | 68 | CGU(R) | 16 |
| CUC(L) | 10 | CCC(P) | 10 | CAC(H) | 16 | CGC(R) | 6 |
| CUA(L) | 81 | CCA(P) | 36 | CAA(Q) | 55 | CGA(R) | 27 |
| CUG(L) | 14 | CCG(P) | 7 | CAG(Q) | 21 | CGG(R) | 11 |
| AUU(I) | 265 | ACU(T) | 85 | AAU(N) | 109 | AGU(S) | 57 |
| AUC(I) | 44 | ACC(T) | 24 | AAC(N) | 23 | AGC(S) | 14 |
| AUA(M) | 188 | ACA(T) | 61 | AAA(K) | 86 | AGA(S) | 59 |
| AUG(M) | 38 | ACG(T) | 5 | AAG(K) | 13 | AGG(S) | 16 |
| GUU(V) | 104 | GCU(A) | 126 | GAU(D) | 51 | GGU(G) | 88 |
| GUC(V) | 15 | GCC(A) | 18 | GAC(D) | 23 | GGC(G) | 21 |
| GUA(V) | 88 | GCA(A) | 61 | GAA(E) | 70 | GGA(G) | 101 |
| GUG(V) | 25 | GCG(A) | 5 | GAG(E) | 17 | GGG(G) | 34 |
Note: * . Stop codon.
Table 5.
SSR in the mitochondrial sequence of Capulus danieli.
| Numbering | Gene | Initial position/bp | Termination position/bp | length/bp | SSR |
|---|---|---|---|---|---|
| 1 | COX | 188 | 197 | 10 | (T)10 |
| 2 | ND1 | 6955 | 6964 | 10 | (T)10 |
| 3 | COX3 | 12653 | 12667 | 15 | (A)15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



