
Submitted:
09 August 2024
Posted:
12 August 2024
You are already at the latest version
Abstract
Histone deacetylase inhibitors (HDACis) are being recognized as a potentially effective treatment approach for peripheral T-cell lymphoma (PTCL), which is a diverse group of aggressive lymphomas with a bleak prognosis. Recent evidence has shown that HDACi are effective in treating PTCL, especially in cases where the disease has relapsed or is resistant to other treatments, and there are few available options. Several clinical trials have demonstrated that HDACi, such as romidepsin and belinostat, can elicit long-lasting positive outcomes in individuals with PTCL, either when used alone or in conjunction with conventional chemotherapy. They exert their anti-tumor actions by regulating gene expression through inhibiting histone deacetylases, which results in the halting of cell division, programmed cell death, and the transformation of cancerous T-cell, as indicated by gene expression profile studies. Importantly, besides clinical trials, real-world evidence indicated that the utilization of HDACi presents a significant and beneficial treatment choice for PTCL. However, although HDACi showed potential effectiveness, they couldn’t cure most patients. Therefore, new combination with conventional drugs as well as new targeted agents are under investigation.

Keywords:
Peripheral T-cell lymphoma
; Histone deacetylase (HDAC)
; HDAC inhibitor (HDACi)
; targeted therapy
; epigenetic
; DNA mutation
; TET2
; DNMT3A
1. Introduction
Peripheral T-cell lymphomas (PTCLs) constitute a relatively rare and heterogeneous group of non-Hodgkin lymphomas (NHL) that arise from post thymic T-cells. According to the most recent classification of lymphoid neoplasms, PTCLs encompass a series of different entities that demonstrate various and intricate clinicopathologic features 1. The most commonly observed subtypes are the so-called PTCL not otherwise specified (PTCL NOS), the T-follicular helper related PTCL, angioimmunoblastic type (AITL) and the anaplastic large cell lymphomas (ALCL), anaplastic kinase-negative (ALK-) or positive (ALK+).
Overall, except for a few more indolent forms arising in the gastrointestinal tract, the skin or the bone marrow, the majority of PTCLs demonstrate a highly aggressive behavior2. Currently, the most commonly used first line chemotherapy is represented by CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) or CHOEP (cyclophosphamide, doxorubicin, vincristine, etoposide and prednisone). Some studies indicated that fit, chemosensitive patients may benefit from front line autologous stem cell transplant (ASCT)2. In contrast, despite the aggressive nature of this disease, allogeneic SCT is still considered an experimental option.
In patients with relapsed/refractory (R/R) peripheral T-cell lymphoma (PTCL), the median progression-free survival (PFS) and median overall survival (OS) were reported to be inferior to 5 and 6 months, respectively.2–4 Based on the poor outcomes, biologically meaningful targeted therapies are urgently warranted. In the last few years, a series of new agents has been approved in this setting, including pralatrexate, brentuximab vedotin (for CD30 positive cases)5 and histone deacetylase (HDAC) inhibitors (HDACi). As far as the latter are concerned, HDACs play a crucial role in the regulation of gene expression and HDACs can impede the growth of tumor cells by triggering the process of apoptosis, leading to cell death6.
In the following, the Authors summarize the rationale for HDACi in TCL as well as the main clinical evidence from clinical trials.
2. HDAC in Cellular Biology
The epigenetic regulation of gene expression by acetylation and deacetylation of histones represents an important mechanism involved in the cellular homeostasis, metabolism, division, and apoptosis. 7 Alterations in the finely balanced processes of acetylation (driven by histone acetyl transferases) and deacetylation (led by histone deacetylases) ultimately lead to abnormal DNA transcription, genomic instability and epigenetic disease8.
HDACs primarily regulate transcription by removing acetyl groups from the e-amino groups of histone tail lysine residues.9,10 This contributes to chromatin condensation, thus limiting the accessibility of transcription factors DNA. HDACs, as transcriptional corepressors, cause the nucleosome to compact, resulting in gene suppression. In addition to histone substrates, HDACs also regulate the stability and function of non-histone proteins by post-translational deacetylation. This process is another important regulation mechanism of both physiological and pathological cellular processes, including gene transcription, signal transmission, protein folding, autophagy, DNA repair, cell proliferation, and metabolism (Figure 1). 11–13 The non-histone substrates of HDACs include transcription factors that have been traditionally associated with various malignancies such as nuclear factor B (NF-B), TP53, GATA1, GATA2, STAT3, BCL6 and heat shock protein 90 (HSP90), regulatory proteins, but also chaperone proteins, structural proteins, and steroid receptors. 9,10.
In mammals, eighteen types of HDACs have been classified into four classes based on sequence patterns, cellular localization, tissue specialization, and enzymatic activity (Table 1).
| HDAC | Class | Cellular Localization | Substrate Specificity | Substrates | Function | Expression pattern | Associated diseases |
| HDAC1 | I | Nuclear | Histone proteins | Androgen receptor, SHP, TP53, MyoD, SMC4, E2F1, STAT3 | Gene regulation, cell cycle control | Ubiquitous | Cancer, neurodegenerative disorders |
| HDAC2 | I | Nuclear | Histone proteins | Glucocorticoid receptor, YY1, BCL6, STAT3 | Gene regulation, cell cycle control | Ubiquitous | Cancer, neurodegenerative disorders |
| HDAC3 | I | Nuclear, Cytoplasm, Membrane | Histone proteins | SHP, YY1, GATA1, RELA, STAT3, MEF2D | Gene regulation, cell cycle control | Ubiquitous | Cancer, metabolic diseases |
| HDAC4 | II A | Nuclear, Cytoplasmic | Histone and nonhistone | GCMA, GATA1, HP1 | Muscle differentiation, development | Tissue specific (heart, skeletal muscle, brain) | Muscular disorders, neurodegenerative diseases |
| HDAC5 | II A | Nuclear, Cytoplasmic | Histone and nonhistone | GCMA, SMAD7, HP1 | Muscle differentiation, development | Tissue specific (heart, skeletal muscle, brain) | Muscular disorders, neurodegenerative diseases |
| HDAC6 | IIB | Cytoplasmic | Cytoplasmic proteins | α-Tubulin, HSP90, SHP, SMAD7 | Aggresome formation, protein degradation | Tissue specific (heart, liver, kidney) | Neurodegenerative disorders, cancer |
| HDAC7 | II A | Nuclear | Histone and nonhistone | PLAG1, PLAG2 | Vascular development, immune response | Tissue specific (endothelium, heart, skeletal muscle, pancreas, placenta, thymys) | Cardiovascular diseases, cancer |
| HDAC8 | I | Nuclear | Histone proteins | - | Cell cycle | Ubiquitous | Cancer |
| HDAC9 | II A | Nuclear | Histone and nonhistone | - | Development, cardiac function | Tissue specific (brain, heart, skeletal muscle) | Cardiovascular diseases, cancer |
| HDAC10 | II B | Cytoplasmic, Nuclear | Cytoplasmic proteins | - | Cellular proliferation, apoptosis | Tissue specific (liver, spleen, kidney) | Cancer, neurodegenerative diseases |
| HDAC11 | IV | Nuclear | Histone proteins | - | Gene regulation | Tissue specific (brain, heart, kidney, testis) | Cancer, inflammatory diseases |
| SIRT1 | III | Nuclear, Cytoplasmic | Histone and nonhistone, NAD dependent | - | Metabolic, stress response | Ubiquitous | Aging related diseases, metabolism disorders |
| SIRT2 | III | Cytoplasmic, Nuclear | Histone and nonhistone, NAD dependent | - | Cell cycle, homeostasis | Ubiquitous | Metabolic diseases, cancer |
| SIRT3 | III | Mitochondrial | NAD dependent | - | Mitochondrial function, energy metabolism | Ubiquitous | Metabolic diseases, cancer |
| SIRT4 | III | Mitochondrial | NAD dependent | - | Metabolism | Tissue specific (pancreas) | Metabolic diseases, cancer |
| SIRT5 | III | Mitochondrial | NAD dependent | - | Metabolism | Ubiquitous | Metabolic diseases, cancer |
| SIRT6 | III | Nuclear | Histone and nonhistone, NAD dependent | - | DNA repair, genome stability | Ubiquitous | Aging related disease, cancer |
| SIRT7 | III | Nucleolar | Histone and nonhistone, NAD dependent | - | Ribosomal DNA transcription, cell growth | Ubiquitous | Cancer |
Class I includes HDAC1, 2, 3 and 8, are ubiquitously expressed in human tissues and are mainly located in the nucleus, regulating histone acetylation. Recently it has been discovered that HDAC8 is also extensively found in smooth muscles where it modulates contractility.14,15 HDAC1 and 2 are solely expressed at the nuclear lever, while HDAC3 can be found in the cytoplasm and also at the cell membrane level. However, during mitotic advancement, HDAC3 is exclusively localized on the mitotic spindle and preserves correct kinetochore-microtubule attachment and chromosome alignment16.
One of the main substrates of HDAC1 is represented by the TP53 protein and is overexpressed in numerous malignancies such as pancreatic, lung and prostate cancer, as well as in lymphomas. HDAC2 is often associated with HDAC1 as part of co-repressor complexes such as SIN3, NURD, MiDAC and CoREST. 7 In addition, HDAC2 overexpression regulates the IFNƔ signaling and is involved in the PDL1 nuclear translocation15. Furthermore, the overexpression of BCL6 in various B cell lymphomas requires the activity of HDAC2 to suppress the activity of regulating proteins, thus promoting abnormal tumor growth17.
HDAC3 has been shown to play an essential role in the regulation of metabolism by increasing the oxidation of fatty acids. Moreover, HDAC3 is essential for the inflammatory response in host defense against bacterial infection, as it amplifies TNF-mediated NF-kB activation.18 In addition to its effect on soft muscle contraction, HDAC8 has also been shown to have an anti-apoptotic effect by suppressing the transcription of the proapoptotic protein BCL2-modifying factor and is overexpressed in various adult and pediatric cancers. 19
Class II HDACs were further classified into IIa and IIb subclasses due to differences in sequence homology and domain organization9. Subclass IIa (HDAC4, 5, 7, and 9) exhibits signal-dependent nucleocytoplasmic shuttling as well as tissue-specific expression.9,20 Subclass IIb (HDAC6 and 10) contains two catalytic HDAC domains and is cytoplasmically localized in a restricted number of tissue types.9 HDAC6 is widely known for deacetylating particular cytosolic non-histone substrates involved in tumor genesis, progression, and metastasis. Tubulin, cortactin, peroxiredoxin, HSP90, and heat shock transcription factor-1 (HSF1) are examples of frequent substrates.21 In contrast to HDAC6, which is an acetyl lysine deacetylase, HDAC10 is a N8-acetylspermidine deacetylase and has been linked to dysregulated polyamine metabolism and neoplastic disorders such as colon cancer, prostate cancer, and neuroblastoma.22 HDAC10 enhances cell survival via autophagy in response to chemotherapeutic treatments and pathogen infection, according to several studies.22,23 As a result, inhibiting HDAC10 autophagy may be a unique method for sustaining cancer chemotherapeutic cytotoxicity, particularly in the treatment of advanced-stage neuroblastoma.22
HDACs class III are structurally distinct compared to class I and II and are collectively called Sirtuins. They are generally involved in metabolism regulation and, unlike other HDAc classes, the deacetylase activity of Sirtuins is NAD dependent and consequently on the cellular NAD/NADH ratio. Currently, 7 HDACs class III have been described and the roles they play range from normal cellular homeostasis, to aging, inflammation and oxidative stress.24,25
Because of a unique sequence pattern, HDAC11 is the sole class IV HDAC.9,26 HDAC11 exhibits deacetylase activity as well as more efficient defatty-acylase activity, both of which are important in lipid metabolism.26 HDAC11 was discovered to be a protective prognostic factor in particular cancers (e.g., kidney renal clear cell carcinoma and rectum adenocarcinoma) in a pan-cancer study.27 In addition, HDAC11 has emerged as a promising therapeutic target for chronic metabolic disorders28
3. HDACi as Anti-Cancer Treatment
HDACs are commonly overexpressed in malignant cells and the use of inhibitors can cause proliferation arrest, differentiation, and apoptosis in many cancers.29 Over five decades have passed since the emergence of the first HDAC inhibitor as a therapeutic agent. HDAC inhibitors, such as valproic acid, have first been used in neurology and psychiatry as mood stabilizers and anti-epileptics, but their efficacy started being tested in malignant disorders shortly afterwards. Sodium butyrate and Trichostatin A (TSA), another one of the classic and representative HDAC inhibitors, are among the first compounds used experimentally on cancer cells.30,31 Sodium butyrate, a natural short-chain fatty acid produced by bacteria in the colon was the first compound that sparked the interest in using HDAC inhibitors in the treatment of cancer due to the success in a limited number of acute leukemia cases.34 TSA, derived from a streptomycete metabolic product, led to the differentiation of leukemia cell lines as well as neoplastic cell apoptosis, however the poor biodisponibility made it impossible to use in vivo.
Shortly after, a number of prodrugs have been developed, but the action of these compounds was non-specific, had a short effect, harbored substantial side effects, and obtaining an effective inhibitory concentrations in vivo was often unattainable.35 The interest in HDAC inhibitors led however in the past 2 decades to the development of more effective and less toxic compounds such as SAHA (vorinostat), Depsipeptides (such as romidepsin), and Entinostat (MS-275), These drugs have shown efficacy both in vitro and in vivo and both vorinostat and romidepsin were granted FDA approval for human use.
Many lines of evidence suggest that histone hypo-acetylation suppresses the expression of tumor suppressor genes.36 In vitro and in vivo phase I and II clinical studies, small molecular HDAC inhibitors (HDACI) are highly efficient in up-regulating tumor suppressor gene expression, decreasing tumor growth, and triggering programmed cell death.34,37 The anticancer mechanism of HDAC inhibitor is based on three main mechanisms 1) cell cycle arrest and apoptosis; 2) inducing cell differentiations; and 3) prevention of angiogenesis. 34
4. Biological Rationale for HDACi Use in Peripheral T-cell Lymphomas
The dysregulation of acetylation and deacetylation processes is a prevalent factor contributing to genomic instability and aberrant gene expression in the context of cancer. PTCLs are not exempt from this phenomenon38. Within the realm of malignancies, PTCLs included, HDACs assume a significant role in governing downstream gene expression networks and signaling pathways, primarily through their deacetylation activity on non-histone substrates, such as transcription factors and signaling mediators.
Previous studies have revealed that HDACs are implicated in the regulation of a multitude of oncogenes essential for lymphomagenesis, such as BCLXL, BCL2, MYC, TCRβ, and NOTCH339. In addition, gene expression profiling (GEP) studies, followed by mutational analyses indicated that HDACi can be a rational approach for mature TCL.38,46,47 TET2 and DNMT3A gene mutations are common in the early stages of PTCL pathogenesis.48 HDAC1 and 2 mediate TET2 protein deacetylation and degradation via the ubiquitin-proteasome pathway.49 TET2 proteins can recruit HDACs to the PDL1 gene promoter in breast cancer cells to decrease transcription, which may be independent of DNA demethylation.50
As mentioned above the use of HDAC inhibitors in peripheral T cell lymphomas can exert their therapeutic effect by promoting cell cycle arrest and apoptosis, cell differentiation and inhibition of angiogenesis. Regarding the first mechanism, HDACi can prompt apoptosis by addressing aberrant signaling pathways governed by HDACs, arising from distorted gene expression or premature degradation of pro-apoptotic proteins.29 For instance, HDAC1, HDAC2, and HDAC3 play a regulatory role in restricting the transcription of STAT3 target genes within the JAK/STAT pathway, resulting in subsequent epigenetic silencing of tumor suppressor genes. Inhibition of these HDACs with small molecules such as can thus lead to cell growth arrest or apoptosis.9,40
Alterations in the TP53 gene and/or disruptions in the TP53 pathway are also involved in the dysregulation of cell cycle arrest and apoptosis and play a significant role in the development of numerous human cancers, including T cell lymphomas. Previous studies have shown that the deacetylation of TP53 by HDAC1, inhibiting its activity, reduces the expression of the p21 protein, and diminishes apoptotic signaling. The effects of HDAC inhibitors-induced acetylation increases TP53 stability as seen in previous mouse models but can also restore the response to therapy in TP53 deleted tumoral cells.40 In T cell lymphomas, TP53 mutations manifest in late stages and are indicative of poor prognosis.40 In the case of TP53 mutations, HDAC1 and 2 can still integrate their expression, and contributing to the sustained expression of aberrant TP53 in human and genetically defined murine pancreatic cancer cells.41 In this context, the inhibition of HDAC1 and 2 reduce the expression of mutant TP53 and improve the response to therapy, cell cycle arrest and apoptosis.41
The deleterious effects of the HDACs also include the HDAC3-induced NF-κB activation through the TNF expression in the TCR/CD3 pathway, which can be mitigated using small molecule inhibitors such as Vorinostat. 39,42 Panobinostat showed in this setting an increased acetylation of HSP90, repression of anti-apoptotic protein BCL2 expression and downregulation of mitogen-activated protein kinase pathway signaling. 43,44
Besides apoptosis, autophagy is another possible therapeutic mechanism of HDAC inhibitors in T-cell lymphomas and SAHA (vorinostat) promotes the autophagy process by inhibiting mTOR and augmenting the efficacy of the autophagic factor L3.
Another mechanism by which HDAC inhibitors can interfere with the T cell leukemogenesis is the alteration of cytokine signaling. Interleukins 2, 4, 7 and 15 are recognized as promoters of disease and the use of HDAC inhibitors interferes with the production of interleukins by the tumoral environment and malignant cells. For example, the use of arginine butyrate induced cell apoptosis in peripheral T cell lymphoma by altering the Il-2 environmental signaling.30
An additional important therapeutic effect of HDAC inhibitors is the modulation of the supporting tumoral microenvironment by inhibiting angiogenesis. Various studies have shown that the administration of HDAC inhibitors leads to a reduction in the expression of proangiogenic genes, including basic fibroblast growth factor, vascular endothelial growth factor, angiopoietin, and endothelial nitric oxide synthase and alter the pro-angiogenic signaling pathway45.
4.1. Rational for HDAC Inhibitors In Virus-Induced PTCL
The role of viral infection in the etiopathogeny of lymphomas has been extensively described. In concern to T cell lymphomas, the Ebstein-Barr virus (EBV) and Human T cell lymphotropic virus type 1 (HTLV-1) are recognized as the main viral drivers of malignant transformation.
EBV is a gamma-herpesvirus that is thought to play a role in the progression of several human cancers, including lymphoma, gastric carcinoma, and nasopharyngeal carcinoma.51 In the latest edition of the WHO classification, EBV-positive TCL represent a distinct group of entities including EBV-positive nodal T- and NK-cell lymphoma, Extranodal NK-/T-cell lymphoma.1 Furthermore, the EBV-positive T- and NK-cell lymphoid proliferations and lymphomas of childhood are listed, including 4 entities (Table 1). In PTCL, EBV was found at a frequency ranging from 20% in PTCL/NOS to 30-up to 100% in extranodal NK-T cell lymphoma nasal type and the association seems to determine a more dismal prognosis.52,53 In PTCLs EBV can infect either neoplastic T-cells (as in some PTCL/NOS and the above mentioned EBV-positive TCL) or bystander B cells, as in AITL, implying that EBV plays both a direct and an indirect role in PTCL pathogenesis42
Romidepsin, a Class I HDACi, stimulates the EBV lytic cycle and facilitates apoptosis when associated with with ganciclovir in Burkitt lymphoma by inhibiting HDAC1, 2, and 3 and upregulating p21.54 EBV latent protein LMP1 upregulates STAT5A and recruits HDAC1/2 to the CEBPA gene locus, which is implicated in neoplastic plasticity control and cellular dedifferentiation.55 Furthermore, HDACis (romidepsin and chidamide) have been shown to restore CEBPA expression and reverse cellular dedifferentiation in EBV+ NPC in vitro.55 However, in PTCL, the link between HDACs and oncogenic viruses other than EBV has not been elucidated yet.
HTLV-1 infection is associated with virtually all the cases of adult T-cell leukemia/lymphoma (ATLL) and represents the major driver of malignant transformation. In ATLL the oncoprotein Tax physically and functionally interacts with HDAC1.56 Interestingly, HDACi can regulate Tax expression and therefore induce viral protein expression, enhancing T-cytotoxic response toward HTLV1 infected cells.57 However, whether this can be useful as anti-ATLL treatment has to be demonstrated.
4.2. HDACi as Therapeutic Option in Mature T-cell Lymphomas
Based on early clinical results in some acute leukemias and B-cell lymphomas and the pre-clinical evidence of their potential efficacy, HDACi were extensively tested in PTCLS.
4.2.1. HDACi in Relapsed/Refractory PTCL Patients
The treatment of R/R PTCL is challenging as the treatment options are limited and the response to conventional chemotherapy is poor. Along with brentuximab vedotin and the dihydrofolate reductase inhibitor pralatrexate, the HDAC inhibitors represent the most prominent newly developed class of drugs in this category of patients. To date , four HDAC inhibitors have been approved for the treatment of relapsed PTCL - class I, II and IV inhibitors (Vorinostat (SAHA), Belinostat), the selective HDAC1, 2, 3 and 10 inhibitor chidamide and the selective class I inhibitor Romidepsin. Among these, only Romidepsin, Belinostat and Vorinostat have received FDA approval for the treatment of R/R PTCL.
The reported ORR of single-agent HDAC inhibitor therapy in this setting is 24% for Vorinostat , 25% for Romidepsin, 25.8% for Belinostat and 28% for Chidamine.58–60 However, real-world multicentric data reported response rates as high as 33% in the R/R PTCL population when treated with Romidepsin single-agent.61
One of the most promising combinations in this setting is represented by the association of hypomethylating agents to the HDAC inhibitors. The real-life experience for the combination of Romidepsin in combination with Azacytidine, reported an ORR of 84% with a CR rate of 61%, significantly higher than 54% / 38% reported in the clinical trial setting.62
A recent meta analysis assessed pooled data from 16 studies on HDACi for R/R PTCL.63 These studies overall included 662 relapsed/refractory patients and used HDAC inhibitors either in monotherapy or in therapeutic combinations with hypomethylating agents, conventional chemotherapy, proteasome inhibitors +/- lenalidomide.Most of the studies evaluated romidepsin that was previously granted FDA approval in 2007 for the treatment of previously treated PTCL. The overall response rate (ORR) was 37%; specifically, the ORR in the HDAC inhibitor-based combination therapy cohort was 45% (95% CI, 36-54%), whereas the ORR in the HDAC inhibitor monotherapy subgroup was 33% (95% CI, 27-38%), this difference being statistically significant (P = 0.02).63 There was no difference in terms of efficacy when different HDACi were used (romidepsin, belinostat, or chidamide). The overall pooled CR rate was 14% with the CR rate in the combination treatment group being 22% and the CR rate in the HDACi inhibitor monotherapy subgroup was 13% (P = 0.02). There was no statistical difference in the CR rate between the three types of HDAC inhibitors (P = 0.06)63
4.2.2. HDACi in First Line Treatment of PTCL Patients
Due to the relative scarcity of these lymphomas, there is no supporting data coming from randomized clinical trials for the initial therapeutic approach. However, it is generally accepted that the standard initial therapy consists of CHOP or CHOP-like regimens followed by autologous stem-cell transplantation in eligible patients. The first major breakthrough was the development of brentuximab vedotin in the treatment of PTCL. The ECHELON study proved the efficacy of the BV-CHP combination compared to the standard CHOP regimen (Table 2).
HDAC inhibitor Romidepsin has been incorporated in the first line of therapy together with CHOP. However the combination failed to prove superiority compared to CHOP in terms of PFS, ORR and OS. 64 For the untreated patients, the generally used combination was a HDAC inhibitor plus the standard of care, CHOP combination chemotherapy. Two studies evaluated the combination of Romidepsin with CHOP either as phase I /II [30] or phase III clinical trial.64 Belinostat in combination with standard R-CHOP provided an ORR of 81% with a rate of CR of 71%.65 Consolidation with autologous stem cell transplant was not considered in these studies. Chidamide in combination with CHOP provided a 89.3% ORR with 57.1% attaining a CR.66
When combined with CHOEP, Chidamide conferred a 60.2% ORR, with a complete response rate of 40.7%. However, the addition of Etoposide did not offer a benefit in terms for the event free survival for the PCTL except for the ALK + ALCL.67
One study evaluated the safety and efficacy of Romidepsin in combination with the hypomethylating agent Azacytidine, a combination that proved to be highly efficacious in the treatment naive population with a ORR of 70% and a CR of 50%. The autologous stem cell transplant was allowed and 4 patients received the procedure, with 3 still being in remission at the time of the report (median PFS 20.8 months).68
The efficacy of HDACi-based combination therapy for untreated PTCL patients was recently revised in terms of ORR, CR rate, and PR rate in a meta analysis comprising a pooled total of 502 patients, from 7 clinical trials.63
The ORR turned out to be 72%. One of the seven studies used belinostat-based treatment and involved 21 patients, yielding an ORR of 86%. Three studies employed chidamide-based treatment and comprised a total of 224 patients, with a pooled ORR of 74%.66,67,69 Three studies with a total of 257 individuals used romidepsin-based treatment, resulting in a pooled ORR of 64%.68,70,71 In untreated PTCL patients, the research revealed a significant difference in ORR across the three HDACi-based combination therapy (P = 0.02).
The pooled CR rate was 44%. However, after subgroup analysis, the CR rate for the belinostat-based therapy was 67%, the pooled CR rete in the romidepsin-based therapy subgroup was 43% and for chidamide-based treatment it was 42% without statistically significant differences (P = 0.07).
4.2.3. HDACi Efficacy across PTCL Subtypes
Three of the studies on untreated PTCLs did include subtypes analyses.49,67,70 All 196 patients had an ORR of 68% (95% CI, 62-75%, fixed effect model). In the PTCL/NOS, AITL, and ALK-neg ALCL categories, the pooled ORR was 58%, 71%, and 76%, respectively. However, no significant difference (P = 0.17) was determined.63,60
The histological subtype analysis in R/R PTCL patients included eight investigations.59,62,64,68–70,72 Pooled together, the 418 patients had an ORR of 32%; the ORR in PTCL-NOS patients was 29%, in ALK-negative ALCL was 27% (95% CI, 16-38%), while in the AITL subgroup was 44%, strongly indicating a more efficient response compared to other subgroups (P = 0.01). The selective efficacy of HDAC inhibitors in AITL was further confirmed by real life retrospective data from a national Israeli study.61
5. Real-World Experience with HDAC Inhibitors in PTCL
To evaluate the real-world impact of HDACi in PTCL we conducted a systematic literature review. A comprehensive review using Scopus, PubMed, and EMBASE was conducted for English-language articles published from January 1, 2000, through November 30th, 2023. Key search terms included peripheral T cell lymphoma, Romidepsin, Chidamide, Vorinostat, Belinostat, real world, case report, HDAC inhibitors. Exclusion criteria included case reports with less than 3 patients, non-English texts. One thousand, fifty three papers have been identified out of which only 5 matched the inclusion criteria.
The first study by Shi et al.75 was conducted in China and involved 256 patients with PTCL, treated with Chidamide monotherapy. The overall response rate (ORR) was reported to be 39.06%. Notably, subgroup analyses within the study included patients with AITL and anaplastic lymphoma kinase-positive anaplastic large cell lymphoma (ALK+ ALCL). In AITL, Chidamide monotherapy demonstrated a higher ORR of 49.23%, emphasizing potential subtype-specific responses. For ALK+ ALCL, the ORR was even more promising at 66.67% when treated with Chidamide monotherapy, and 51.18% when Chidamide was combined with chemotherapy. Overall, the PFS for Chidamide monotherapy was 129 days (95% CI 82 to 194), vs. 152 days (95% CI 93 to 201) for the combination Chidamide+chemotherapy (P = 0.3266). In the monotherapy group, the AITL patients achieved a median PFS of 144.5 days, while the combination therapy delivered an impressive median PFS of 176 days.
When evaluating the efficacy of various chemotherapy regimens in association with the HDAC inhibitor, CHOP- like regimens resulted in the most effective, with a prolonged PFS of 172 days, while the platinum-containing regimens resulted in a PFS of only 119 days.
Regarding the safety profile, the most common adverse effects experienced by the monotherapy group are hematological (anemia, thrombocytopenia, neutropenia) and nausea/vomiting.
In a separate study by Shimony et al.76 conducted in Israel, Romidepsin monotherapy was administered to 42 PTCL patients, resulting in an ORR of 33% including a 12.5% rate of CR. The lower response rate suggests that the efficacy of HDAC inhibitors may vary among different agents although of note is that this population contained a relapsed-refractory category of patients that received a median of 2 prior lines of therapy (65% received 2 or 3 previous lines of therapy, range 1-5). However the duration of response exceeded 1 year (13.8 months) although the PFS 2.2 months. In alignment with the findings from the other studies, AITL subtype was associated with a longer EFS.
The most common side-effects were hematological, although one third of the patients required hospitalization due to infectious complications.
The investigation by Kalac et al.77 that included patients from the USA and Australia explored the combination of Romidepsin and azacitidine in 26 PTCL patients, including 3 newly-diagnosed patients, yielding an impressive ORR of 76.9% with a complete response rate (CRR) of 53%. As expected, the ATL patients benefited from a 69.5% ORR, with a CRR of 60.8%. Notably, this study included patients undergoing stem cell transplantation, indicating a potential role of ASCT transplantation in this treatment setting. From a mutational stempoint, the TET2, IDH and DNMT3A mutated cases benefited most from this combination therapy with an ORR of 77.7%.
Median PFS for the 26 patients was 13.3 months and the OS was not reached. Notably, then median PFS for patients undergoing stem cell transplant was not reached, while for the transplant-ineligible patients the PFS was 7.07 months.
Overall, the combination was well tolerated, the most important side effects including nausea, fatigue, rash, neutropenia, and thrombocytopenia.
Another significant contribution comes from Liu et al.78 in China, who conducted a large-scale study involving 725 PTCL patients. Chidamide monotherapy showed an ORR of 58.6%, while the combination with chemotherapy resulted in a higher response rate of 73.2%, suggesting a potential synergistic effect between HDAC inhibitors and chemotherapy. Furthermore, in AITL patients, Chidamide monotherapy and combination therapy achieved remarkable ORRs of 75.1%. The most common side effects for chidamide monotherapy included anemia, thrombocytopenia, neutropenia and fatigue in one third of the patients while fatigue was reported by almost 60% of the patients receiving chemotherapy -chidamide combination.
The study by Wei et al.79 in China retrospectively explored the efficacy of Chidamide in combination with CHOEP in newly diagnosed patients with PTCL, compared to standard CHOEP therapy. In this setting the ORR was 68.8%, with an impressive CRR of 56.3%. However, authors concluded that for patients that received CHOEP therapy did not achieve a statistically significant difference compared to Chidamide-CHOEP (ORR 54.8% versus 68.8%, p=0.256) although the CRR was double in the combination therapy compared to standard CHOEP (56.3% versus 25.8%, p=0.014). The median PFS for Chidamide-CHOEP was 12 months, and the OS was 57 months. 30% of the patients in the combination therapy group received stem cell transplant consolidation, and 36.4% Chidamide maintenance.
Notably, a flat PFS curve was obtained in two circumstances: in patients receiving ASCT consolidation as well in the ones that benefited from chidamide maintenance.
In general, the C-CHOEP regimen exhibited good tolerance, with no significantly severe hematologic and non-hematologic toxicities observed when compared to the CHOEP regimen. Hematologic toxicities were the most frequently reported adverse events in both groups.
Lastly, Guo et al.80 investigated Chidamide maintenance therapy in 48 transplant-ineligible PTCL patients that received a first line induction with conventional chemotherapy. The study schema included a 2 cycle consolidation with chidamide + original induction therapy for patients achieving at least a PR after induction, or chidamide + conventional second line therapy as salvage therapy for patients not achieving a response. For patients who responded, the consolidation was followed by chidamide monotherapy maintenance. The HDAC inhibitor consolidation and maintenance demonstrated an impressive ORR of 93.8% with a CRR of 60.4%
AITL and ALCL (ALK + and -) subtypes presented with a staggering 100% ORR, and with CRR of 61.9% and 100% respectively (ALCL ALK+).
PFS showed no significant distinction between patients achieving CR and PR. In contrast, notable differences were evident when comparing patients undergoing first-line maintenance (40 cases) and salvage therapy (8 cases), with 1-year PFS rates of 80.8% and 46.9%, and 2-year PFS rates of 71.9% and 46.9%, respectively (P=0.012). The median PFS was not reached in the first-line maintenance group, while it was 10.1 months in the salvage therapy group.
The most common and severe side effects were hematological, however only one patient experienced infectious complications.
Although there is a marked heterogeneity between the reported outcomes in the identified studies, all of them reported superior ORR in the angioimmunoblastic T-cell lymphoma subtype suggesting that AITL may be particularly responsive to HDAC inhibitor therapy compared to other peripheral T cell lymphoma subtypes. This observation suggests a possible “Achille’s heel” of AITL that makes it more susceptible to the mechanisms of action of HDAC inhibitors. The main studies on real-wolrd experience with HDACi in PTCLs are summarized in Table 3.
5. Conclusions
The treatment of PTCL remains challenging as there is still a limited number of available therapeutic agents. In recent years, HDACs have emerged as new epigenetic therapeutic agents for both newly diagnosed as well as relapsed/refractory cases. For the latter case, this is particularly relevant since these patients represent an unmet medical need, as the widely used therapeutic approach is salvage chemotherapy and stem cell transplant for the eligible patients. The efficacy of HDAC inhibitors is proven in monotherapy as well as in therapeutic combinations, leading to robust response rates with manageable additional toxicity. In the R/R setting, the combination therapies based on HDACi can produce an impressive ORR of up to 45%. Despite the lack of large randomized trials, these response rates are unprecedented and represent a major argument in shifting the treatment paradigm.
The biological rationale for employing HDAC inhibitors in PTCLs is based on the existence of dysregulated acetylation and deacetylation processes, which contribute to genomic instability and aberrant gene expression, especially in the relapsed setting. The complex therapeutic mechanisms of HDAC inhibitors in PTCLs target cell cycle arrest and apoptosis, cell differentiation, inhibition of angiogenesis, and modulation of cytokine signaling. These mechanisms target key components of the complex molecular landscape of PTCLs, offering a comprehensive approach to hinder tumor growth and progression.
PTCL represents a heterogeneous group of lymphomas ranging from indolent to highly aggressive subtypes, however, the PTCL histology does not appear to be a major driver in outcome in the relapsed setting when treated with standard chemotherapy.73 It is interesting to note that, AITL, which is considered to have one of the poorest survival rates, appeared to be the most benefitting from HDACi based therapies, among nodal PTCL subtypes. Intriguingly, hypomethylating agents such as azacitidine and decitabine appeared to be also strikingly effective in R/R AITL, prompting new studies testing the duals epigenetic therapy combination in the first line as well as R/R setting.74
On the other hand, it is still hard to predict the subcategory of patients that would benefit most from the incorporation of HDAC inhibitors in the therapeutic scheme. The presence of single genetic lesions (including those affecting epigenetics) did not seem to correlate with clinical response and reliable predictive biomarkers are therefore lacking. We believe that identifying reliable biomarkers by introducing mutational analysis, gene and miRNA expression is a key step for future development of HDACi based therapies in the PTCL patients.
Author Contributions
Conceptualization, PPP.; methodology, PPP and RI; data curation, PPP and RI; writing—original draft preparation, PPP and RI; supervision, PPP.; project administration, PPP; funding acquisition, PPP. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Italian Ministry of Health (RC-2024-2790173 – Piccaluga), and PRIN 2022NXK38S (Dr. Irimia) and FIRB Futura 2011 RBFR12D1CB (Dr. Irimia).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Prof. Pier Paolo Piccaluga is currently affiliated to the University of Nairobi (Nairobi, Kenya), and the University of Botswana (Gaborone, Botswana).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Piccaluga PP, Khattab SS. A Comparison of the Fifth World Health Organization and the International Consensus Classifications of Mature T-Cell Lymphomas. Int J Mol Sci. 2023;24(18):14170. [CrossRef]
- D’Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093-3099. [CrossRef]
- Bellei M, Foss FM, Shustov AR, et al. The outcome of peripheral T-cell lymphoma patients failing first-line therapy: a report from the prospective, International T-Cell Project. Haematologica. 2018;103(7):1191-1197. [CrossRef]
- Mak V, Hamm J, Chhanabhai M, et al. Survival of patients with peripheral T-cell lymphoma after first relapse or progression: spectrum of disease and rare long-term survivors. J Clin Oncol. 2013;31(16):1970-1976. [CrossRef]
- Horwitz S, O’Connor OA, Pro B, et al. Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): a global, double-blind, randomised, phase 3 trial. Lancet. 2019;393(10168):229-240. [CrossRef]
- Jianfu Z, Jianzhao P, Jianzhao P, et al. Identification of Histone deacetylase (HDAC)-Associated Proteins with DNA-Programmed Affinity Labeling. Angew Chemie. 2020;132(40):17678-17685. Accessed November 19, 2023. https://jglobal.jst.go.jp/en/detail?JGLOBAL_ID=202002230005667956.
- Park SY, Kim JS. A short guide to histone deacetylases including recent progress on class II enzymes. Exp Mol Med 2020 522. 2020;52(2):204-212. [CrossRef]
- Gallinari P, Di Marco S, Jones P, Pallaoro M, Steinkühler C. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res 2007 173. 2007;17(3):195-211. [CrossRef]
- Chun, P. Histone deacetylase inhibitors in hematological malignancies and solid tumors. Arch Pharm Res. 2015;38(6):933-949. [CrossRef]
- Carpio LR, Bradley EW, McGee-Lawrence ME, et al. Histone deacetylase 3 supports endochondral bone formation by controlling cytokine signaling and matrix remodeling. Sci Signal. 2016;9(440):ra79. [CrossRef]
- Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol 2018 203. 2018;20(3):156-174. [CrossRef]
- New M, Olzscha H, La Thangue NB. HDAC inhibitor-based therapies: can we interpret the code? Mol Oncol. 2012;6(6):637-656. [CrossRef]
- Haery L, Thompson RC, Gilmore TD. Histone acetyltransferases and histone deacetylases in B- and T-cell development, physiology and malignancy. Genes Cancer. 2015;6(5-6):184. [CrossRef]
- Waltregny D, Glénisson W, Tran SL, et al. Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J. 2005;19(8):966-968. [CrossRef]
- Gao Y, Nihira NT, Bu X, et al. Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat Cell Biol. 2020;22(9):1064-1075. [CrossRef]
- Ishii S, Kurasawa Y, Wong J, Yu-Lee LY. Histone deacetylase 3 localizes to the mitotic spindle and is required for kinetochore-microtubule attachment. Proc Natl Acad Sci U S A. 2008;105(11):4179-4184. [CrossRef]
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 2006 59. 2006;5(9):769-784. [CrossRef]
- Yang L, Chen S, Xia J, et al. Histone deacetylase 3 facilitates TNFα-mediated NF-κB activation through suppressing CTSB induced RIP1 degradation and is required for host defense against bacterial infection. Cell Biosci. 2022;12(1):1-15. [CrossRef]
- Kang Y, Nian H, Rajendran P, et al. HDAC8 and STAT3 repress BMF gene activity in colon cancer cells. Cell Death Dis. 2014;5(10). [CrossRef]
- Edwards AJP, Pender SLF. Histone deacetylase inhibitors and their potential role in inflammatory bowel diseases. Biochem Soc Trans. 2011;39(4):1092-1095. [CrossRef]
- Pulya S, Amin SA, Adhikari N, Biswas S, Jha T, Ghosh B. HDAC6 as privileged target in drug discovery: A perspective. Pharmacol Res. 2021;163. [CrossRef]
- Hai Y, Shinsky SA, Porter NJ, Christianson DW. Histone deacetylase 10 structure and molecular function as a polyamine deacetylase. Nat Commun 2017 81. 2017;8(1):1-9. [CrossRef]
- Zhou W, Wang J, Wang X, et al. Degradation of HDAC10 by autophagy promotes IRF3-mediated antiviral innate immune responses. Sci Signal. 2022;15(765). [CrossRef]
- Wu QJ, Zhang TN, Chen HH, et al. The sirtuin family in health and disease. Signal Transduct Target Ther. 2022;7(1). [CrossRef]
- Dai Y, Faller D V. Transcription Regulation by Class III Histone Deacetylases (HDACs)-Sirtuins. Transl Oncogenomics. 2008;3(3):53-65. [CrossRef]
- Liu SS, Wu F, Jin YM, Chang WQ, Xu TM. HDAC11: a rising star in epigenetics. Biomed Pharmacother. 2020;131. [CrossRef]
- Li R, Wu X, Zhao P, Xue K, Li J. A pan-cancer analysis identifies HDAC11 as an immunological and prognostic biomarker. FASEB J. 2022;36(7). [CrossRef]
- Chen H, Xie C, Chen Q, Zhuang S. HDAC11, an emerging therapeutic target for metabolic disorders. Front Endocrinol (Lausanne). 2022;13:989305. [CrossRef]
- Achachi A, Florins A, Gillet N, et al. Valproate activates bovine leukemia virus gene expression, triggers apoptosis, and induces leukemia/lymphoma regression in vivo. Proc Natl Acad Sci U S A. 2005;102(29):10309-10314. [CrossRef]
- Shao RH, Tian X, Gorgun G, Urbano AG, Foss FM. Arginine butyrate increases the cytotoxicity of DAB389IL-2 in leukemia and lymphoma cells by upregulation of IL-2Rβ gene. Leuk Res. 2002;26(12):1077-1083. [CrossRef]
- [Effect of trichostatin A on histone acetylation level and apoptosis in HL-60 cells] - PubMed. Accessed November 19, 2023. https://pubmed.ncbi.nlm.nih.gov/15228659/.
- Liu T, Kuljaca S, Tee A, Marshall GM. Histone deacetylase inhibitors: multifunctional anticancer agents. Cancer Treat Rev. 2006;32(3):157-165. [CrossRef]
- Finnin MS, Donigian JR, Cohen A, et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401(6749):188-193. [CrossRef]
- Bi G, Jiang G. The Molecular Mechanism of HDAC Inhibitors in Anticancer Effects. Cell Mol Immunol. 2006;3(4):285-290.
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276(39):36734-36741. [CrossRef]
- The molecular mechanism of HDAC inhibitors in anticancer effects - PubMed. Accessed November 19, 2023. https://pubmed.ncbi.nlm.nih.gov/16978537/.
- Sakajiri S, Kumagai T, Kawamata N, Saitoh T, Said JW, Koeffler HP. Histone deacetylase inhibitors profoundly decrease proliferation of human lymphoid cancer cell lines. Exp Hematol. 2005;33(1):53-61. [CrossRef]
- Ji MM, Huang YH, Huang JY, et al. Histone modifier gene mutations in peripheral T-cell lymphoma not otherwise specified. Haematologica. 2018;103(4):679-687. [CrossRef]
- Wang P, Wang Z, Liu J. Role of HDACs in normal and malignant hematopoiesis. Mol Cancer 2020 191. 2020;19(1):1-21. [CrossRef]
- Yu X, Li H, Zhu M, et al. Involvement of p53 Acetylation in Growth Suppression of Cutaneous T-Cell Lymphomas Induced by HDAC Inhibition. J Invest Dermatol. 2020;140(10):2009-2022.e4. [CrossRef]
- Stojanovic N, Hassan Z, Wirth M, et al. HDAC1 and HDAC2 integrate the expression of p53 mutants in pancreatic cancer. Oncogene. 2017;36(13):1804-1815. [CrossRef]
- Pizzi M, Margolskee E, Inghirami G. Pathogenesis of Peripheral T Cell Lymphoma. Annu Rev Pathol. 2018;13:293-320. [CrossRef]
- Rosato RR, Grant S. Histone deacetylase inhibitors: insights into mechanisms of lethality. Expert Opin Ther Targets. 2005;9(4):809-824. [CrossRef]
- Taylor GP, Matsuoka M. Natural history of adult T-cell leukemia/lymphoma and approaches to therapy. Oncogene. 2005;24(39):6047-6057. [CrossRef]
- Li Y, Seto E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med. 2016;6(10). [CrossRef]
- Piccaluga PP, Agostinelli C, Califano A, et al. Gene expression analysis of peripheral T cell lymphoma, unspecified, reveals distinct profiles and new potential therapeutic targets. J Clin Invest. 2007;117(3):823-834. [CrossRef]
- Vega F, Amador C, Chadburn A, et al. Genetic profiling and biomarkers in peripheral T-cell lymphomas: current role in the diagnostic work-up. Mod Pathol. 2022;35(3):306-318. [CrossRef]
- Xie C, Li X, Zeng H, Qian W. Molecular insights into pathogenesis and targeted therapy of peripheral T cell lymphoma. Exp Hematol Oncol. 2020;9(1). [CrossRef]
- Wang Y, Zhang M, Song W, et al. Chidamide plus prednisone, etoposide, and thalidomide for untreated angioimmunoblastic T-cell lymphoma in a Chinese population: A multicenter phase II trial. Am J Hematol. 2022;97(5):623-629. [CrossRef]
- Shen Y, Liu L, Wang M, et al. TET2 Inhibits PD-L1 Gene Expression in Breast Cancer Cells through Histone Deacetylation. Cancers (Basel). 2021;13(9). [CrossRef]
- Nakhoul H, Lin Z, Wang X, Roberts C, Dong Y, Flemington E. High-Throughput Sequence Analysis of Peripheral T-Cell Lymphomas Indicates Subtype-Specific Viral Gene Expression Patterns and Immune Cell Microenvironments. mSphere. 2019;4(4). [CrossRef]
- Haverkos BM, Alpdogan O, Baiocchi RA, et al. Targeted therapy with nanatinostat and valganciclovir in recurrent EBV-positive lymphoid malignancies: a phase 1b/2 study. Blood Adv. 2023;7(20). [CrossRef]
- Kim TY, Min GJ, Jeon YW, et al. Impact of Epstein-Barr Virus on Peripheral T-Cell Lymphoma Not Otherwise Specified and Angioimmunoblastic T-Cell Lymphoma. Front Oncol. 2022;11. [CrossRef]
- Hui KF, Cheung AKL, Choi CK, et al. Inhibition of class I histone deacetylases by romidepsin potently induces Epstein-Barr virus lytic cycle and mediates enhanced cell death with ganciclovir. Int J cancer. 2016;138(1):125-136. [CrossRef]
- Xie J, Wang Z, Fan W, et al. Targeting cancer cell plasticity by HDAC inhibition to reverse EBV-induced dedifferentiation in nasopharyngeal carcinoma. Signal Transduct Target Ther. 2021;6(1). [CrossRef]
- Ego T, Ariumi Y, Shimotohno K. The interaction of HTLV-1 Tax with HDAC1 negatively regulates the viral gene expression. Oncogene. 2002;21(47):7241-7246. [CrossRef]
- Schnell AP, Kohrt S, Thoma-Kress AK. Latency Reversing Agents: Kick and Kill of HTLV-1? Int J Mol Sci 2021, Vol 22, Page 5545. 2021;22(11):5545. [CrossRef]
- Zhang Q, Wang S, Chen J, Yu Z. Histone Deacetylases (HDACs) Guided Novel Therapies for T-cell lymphomas. Int J Med Sci. 2019;16(3):424. [CrossRef]
- Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631-636. [CrossRef]
- O’Connor OA, Horwitz S, Masszi T, et al. Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J Clin Oncol. 2015;33(23):2492-2499. [CrossRef]
- Shimony S, Horowitz N, Ribakovsky E, et al. Romidepsin treatment for relapsed or refractory peripheral and cutaneous T-cell lymphoma: Real-life data from a national multicenter observational study. Hematol Oncol. 2019;37(5):569-577. [CrossRef]
- Kalac M, Jain S, Tam CS, et al. Real-world experience of combined treatment with azacitidine and romidepsin in patients with peripheral T-cell lymphoma. Blood Adv. 2023;7(14):3760-3763. [CrossRef]
- Yang P, Tao Y, Zhao A, et al. Efficacy and safety of histone deacetylase inhibitors in peripheral T-cell lymphoma: a systematic review and meta-analysis on prospective clinical trials. Front Oncol. 2023;13:1127112. [CrossRef]
- Bachy E, Camus V, Thieblemont C, et al. Final Analysis of the Ro-CHOP Phase III Study (Conducted by LYSA): Romidepsin Plus CHOP in Patients with Peripheral T-Cell Lymphoma. Blood. 2020;136(Supplement 1):32-33. [CrossRef]
- Johnston PB, Cashen AF, Nikolinakos PG, et al. Belinostat in combination with standard cyclophosphamide, doxorubicin, vincristine and prednisone as first-line treatment for patients with newly diagnosed peripheral T-cell lymphoma. Exp Hematol Oncol. 2021;10(1). [CrossRef]
- Gui L, Cao J, Ji D, et al. Chidamide combined with cyclophosphamide, doxorubicin, vincristine and prednisone in previously untreated patients with peripheral T-cell lymphoma. Chin J Cancer Res. 2021;33(5):616-626. [CrossRef]
- Zhang W, Su L, Liu L, et al. The combination of chidamide with the CHOEP regimen in previously untreated patients with peripheral T-cell lymphoma: a prospective, multicenter, single arm, phase 1b/2 study. Cancer Biol Med. 2021;18(3):841. [CrossRef]
- Falchi L, Ma H, Klein S, et al. Combined oral 5-azacytidine and romidepsin are highly effective in patients with PTCL: a multicenter phase 2 study. Blood. 2021;137(16):2161-2170. [CrossRef]
- Gui L, Cao J, Ji D, et al. Chidamide combined with cyclophosphamide, doxorubicin, vincristine and prednisone in previously untreated patients with peripheral T-cell lymphoma. Chinese J Cancer Res. 2021;33(5):616. [CrossRef]
- Dupuis J, Morschhauser F, Ghesquières H, et al. Combination of romidepsin with cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated patients with peripheral T-cell lymphoma: a non-randomised, phase 1b/2 study. Lancet Haematol. 2015;2(4):e160-e165. [CrossRef]
- Bachy E, Camus V, Thieblemont C, et al. Romidepsin Plus CHOP Versus CHOP in Patients With Previously Untreated Peripheral T-Cell Lymphoma: Results of the Ro-CHOP Phase III Study (Conducted by LYSA). J Clin Oncol. 2022;40(3):242-251. [CrossRef]
- Shaker S, Bernstein M, Momparler RL. Antineoplastic action of 5-aza-2′-deoxycytidine (Dacogen) and depsipeptide on Raji lymphoma cells. Oncol Rep. 2004;11(6):1253-1256. [CrossRef]
- Foster C, Kuruvilla J. Treatment approaches in relapsed or refractory peripheral T-cell lymphomas. F1000Research. 2020;9. [CrossRef]
- Zhang P, Zhang M. Epigenetic alterations and advancement of treatment in peripheral T-cell lymphoma. Clin Epigenetics 2020 121. 2020;12(1):1-17. [CrossRef]
- Shi Y, Jia B, Xu W, Li W, Liu T, Liu P, Zhao W, Zhang H, Sun X, Yang H, Zhang X, Jin J, Jin Z, Li Z, Qiu L, Dong M, Huang X, Luo Y, Wang X, Wang X, Wu J, Xu J, Yi P, Zhou J, He H, Liu L, Shen J, Tang X, Wang J, Yang J, Zeng Q, Zhang Z, Cai Z, Chen X, Ding K, Hou M, Huang H, Li X, Liang R, Liu Q, Song Y, Su H, Gao Y, Liu L, Luo J, Su L, Sun Z, Tan H, Wang H, Wang J, Wang S, Zhang H, Zhang X, Zhou D, Bai O, Wu G, Zhang L, Zhang Y. Chidamide in relapsed or refractory peripheral T cell lymphoma: a multicenter real-world study in China. J Hematol Oncol. 2017 Mar 15;10(1):69. [CrossRef] [PubMed]
- Shimony S, Horowitz N, Ribakovsky E, et al. Romidepsin treatment for relapsed or refractory peripheral and cutaneous T-cell lymphoma: Real-life data from a national multicenter observational study. Hematological Oncology. 2019; 37: 569–577. [CrossRef]
- Kalac M, Jain S, Tam CS, Xiao Z, Montanari F, Kanakry J, Huber BD, Goldfinger M, O'Connor OA, Marchi E. Real-world experience of combined treatment with azacitidine and romidepsin in patients with peripheral T-cell lymphoma. Blood Adv. 2023 Jul 25;7(14):3760-3763. [CrossRef] [PubMed]
- Liu W, Zhao D, Liu T, Niu T, Song Y, Xu W, Jin J, Cai Q, Huang H, Li Z, Hou M, Zhang H, Zhou J, Hu J, Shen J, Shi Y, Yang Y, Zhang L, Zhao W, Ding K, Qiu L, Tan H, Zhang Z, Liu L, Wang J, Xu B, Zhou H, Gao G, Xue H, Bai O, Feng R, Huang X, Yang H, Yan X, Zeng Q, Liu P, Li W, Mao M, Su H, Wang X, Xu J, Zhou D, Zhang H, Ma J, Shen Z, Zhu J. A Multi-Center, Real-World Study of Chidamide for Patients With Relapsed or Refractory Peripheral T-Cell Lymphomas in China. Front Oncol. 2021 Nov 4;11:750323. [CrossRef] [PubMed]
- Wei C, Zhao D, Zhang Y, Wang W, Zhou D, Zhang W. Long-time follow-up of patients with untreated peripheral T cell lymphoma following chidamide combined with cyclophosphamide, epirubicin, vindesine, prednisone, and etoposide therapy: a single-center propensity score-matching study. Clin Transl Oncol. 2023 Aug;25(8):2514-2522. [CrossRef] [PubMed]
- Guo W, Wang X, Li J, Yin X, Zhao Y, Tang Y, Wang A, Bai O. Chidamide Maintenance Therapy Following Induction Therapy in Patients With Peripheral T-Cell Lymphoma Who Are Ineligible for Autologous Stem Cell Transplantation: Case Series From China. Front Oncol. 2022 Jun 7;12:875469. [CrossRef] [PubMed]
Figure 1.
The histone or non-histone substrates for and the integrated biological effects of HDACs. The acetylation of histone substrates modulates the chromatin structure to reduce the accessibility to transcriptional regulatory proteins and subsequent gene expression. For non-histone substrates, HDACs have an impact on their activity by acetylating. In general, HDACs contribute to proliferative effects (Adapted from Lu et al. Clinical Epigenetics (2023) 15:124 https://doi.org/10.1186/s13148-023-01531-8).
Figure 1.
The histone or non-histone substrates for and the integrated biological effects of HDACs. The acetylation of histone substrates modulates the chromatin structure to reduce the accessibility to transcriptional regulatory proteins and subsequent gene expression. For non-histone substrates, HDACs have an impact on their activity by acetylating. In general, HDACs contribute to proliferative effects (Adapted from Lu et al. Clinical Epigenetics (2023) 15:124 https://doi.org/10.1186/s13148-023-01531-8).
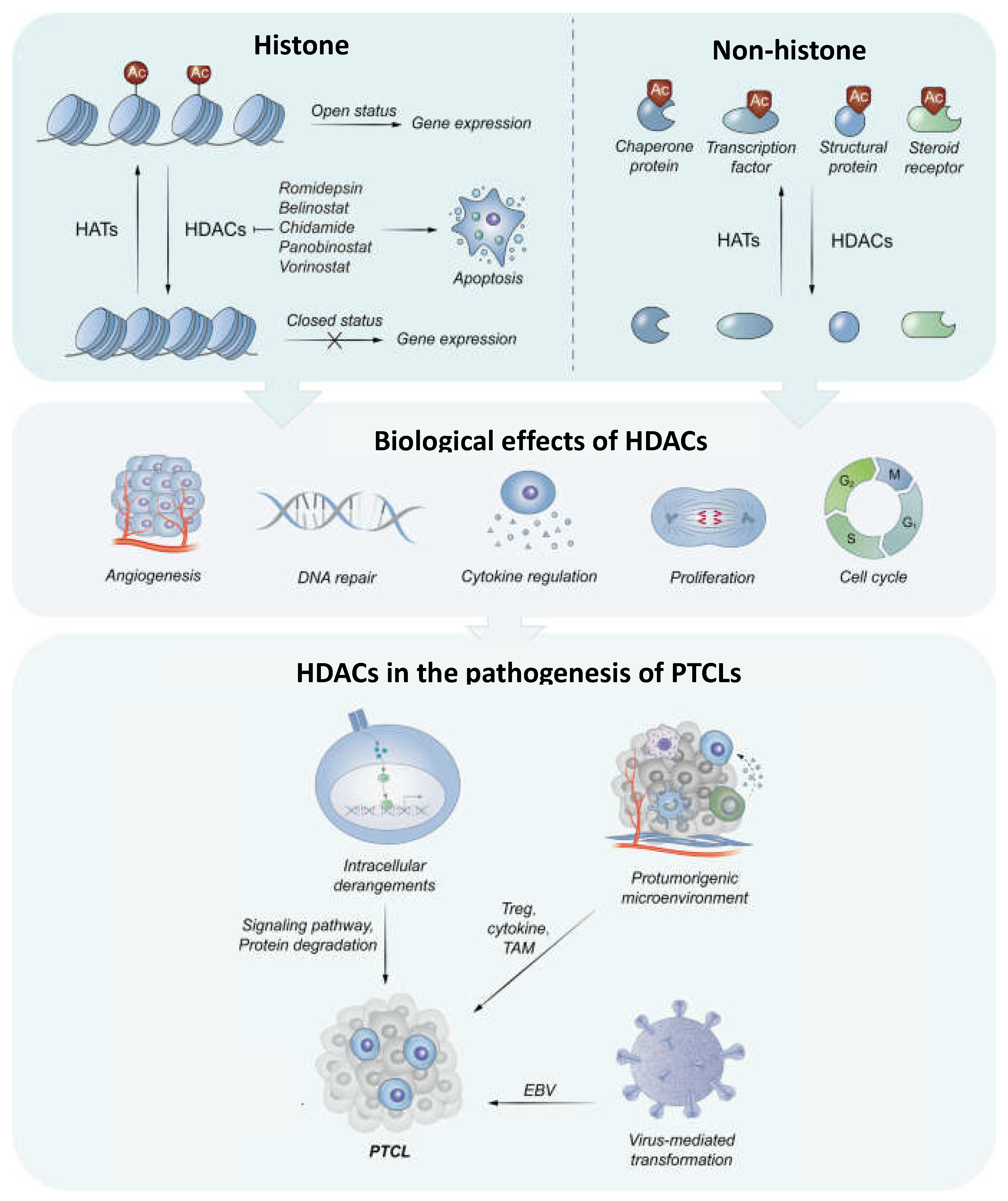

Table 2.
Clinical trials on HDACi in PTCLs.
| Study | Design | Tratment | ASCT permitted | Subtype efficacy |
Toxicity profile (grade ¾ events) |
|---|---|---|---|---|---|
| Dupuis 2015 | Phase I/II single-arm | Ro-CHOP | No | N/A | Cardiac toxicity Febrile neutropenia Hematologic toxicities |
| Bachy 2022 | Phase III, RCT | Ro-CHOP | No | AITL (PFS 19.5 mo vs 10.6 mo) |
>10% difference in >= grade 3 hematologic toxicities |
| Johnston2021 | Phase I, single arm | Bel-CHOP | No | Hematologic toxicity Febrile Neutropenia Nausea SAE rate 43% |
|
| Guy 2021 | Phase I, single arm | Chidamide-CHOP | No | N/A | Hematologic toxicity Vomiting |
| Wang 2022 | Phase II, single arm | Chidamide+ Prednison+ Etoposide + Thalidomide |
No | AITL (90.2%/ 54.9%) |
Hematologic toxicity |
| Zhang 2021 | Phase Ib/II, single arm | Chidamide + CHOEP | No | ALK+ AITL (65.9%/41.5%) | Hematologic toxicity |
| Falchi 2021 | Phase II, single arm | R-Azacytidine | Yes (4 patients / 3 in remission) | AITL (80%/60%) |
Hematologic toxicity |
Table 3.
Real-world experience with HDACi in PTCLs.
| Article | Country of experience | Number of patients | Subtype of PTCL | Therapeutic approach | Stem cell transplant included | Results ORR |
|---|---|---|---|---|---|---|
| Shi, Y75 | China | 256 | PTCL | Chidamide monotherapy | No | 39.06% |
| Shi, Y75 | China | 32 | AITL | Chidamide monotherapy | No | 49.23% |
| Shi, Y75 | China | 13 | ALK+ ALCL | Chidamide monotherapy | No | 66.67% |
| Shi, Y75 | China | 127 | ALK+ ALCL | Chidamide + Chemotherapy | No | 51.18% |
| Shimony, S76 | Israel | 42 | PTCL | Romidepsin monotherapy | No | 33% |
| Kalac, M77 | USA, Australia | 26 | PTCL | Romidepsin - azacitidine | Yes (1 Allo, 7 auto) | 76.9% |
| Kalac, M77 | USA, Australia | 19 | AITL | Romidepsin - azacitidine | Yes (1 Allo, 7 auto) | 69.5% |
| Liu, W78 | China | 261 | PTCL | Chidamine monotherapy | No | 58.6% |
| Liu, W78 | China | 287 | PTCL | Chidamide + Chemotherapy | No | 73.2% |
| Liu, W78 | China | 177 | AITL | Chidamine monotherapy/ Chidamide + Chemotherapy | No | 75.1% |
| Wei, C79 | China | 32 | PTCL | Chidamide + CHOEP | Yes | 68.8% |
| Guo, W80 | China | 48 | PTCL | Chidamide maintenance | No | 93.8% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



