Submitted:
13 August 2024
Posted:
13 August 2024
You are already at the latest version
Abstract
Background: Eosinophilic granulomatosis with polyangiitis (EGPA) is an extremely rare type of vasculitis characterized by inflammation within small blood vessels or tissues that may cause damage to the lungs, heart, kidneys, and other organs. Here, we present a rare case of EGPA with cardiac involvement that presented with acute heart failure.
Clinical findings: A 44-year-old woman with a history of bronchial asthma and sinusitis pre-sented with fever, shortness of breath, fatigue, unintentional weight loss, and polyarthritis. Physical examination revealed a fine basal crepitation and mononeuritis multiplex.
Diagnosis: The peripheral film revealed dimorphic features, an elevated eosinophil count, low hemoglobin (Hb), and abnormally high lactate dehydrogenase levels. The cardiac enzyme levels were elevated upon admission. Cardiac magnetic resonance imaging (CMR) revealed global hypokinesia and features suggestive of myocarditis. Echocardiography showed a low ejection fraction of 25%. Thus, the patient diagnosed with EPGA and myocarditis presented with acute heart failure.
Interventions: The patient was administered high-dose corticosteroids (intravenous bolus methylprednisolone 500 mg for three days, followed by 1 mg/kg of prednisolone) and cyclo-phosphamide 750 mg intravenously.
Outcome: After one months, the patient showed a marked improvement in clinical and labora-tory parameters. The ejection fraction improved to 30-40%, the eosinophil count returned to normal, and the hemolytic anemia resolved. The patient was sent home with mycophenolate mofetil 1 g twice a day as maintenance therapy.
Conclusion:
Patients with EGPA have a higher morbidity and mortality rate when they have cardiac in-volvement. The pathophysiological mechanism of cardiac involvement in EGPA warrants con-sideration of immunosuppressive therapy in addition to standard heart failure treatment.
Keywords:
Eosinophilia
; myocarditis
; vasculitis
; autoimmune haemolytic anaemia
; heart failure
1. Introduction
Eosinophilic granulomatosis with polyangiitis (EGPA), which was previously referred to as Churg-Strauss syndrome (CSS), is a subtype of small-to medium-sized vessel anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Necrotizing granulomatous inflammation, high concentration of eosinophils, and involvement of the respiratory tract are hallmarks of this condition [1].
The eosinophilic phase of the disease exhibits eosinophilic organ infiltration (heart, lungs, and gastrointestinal tract) as the primary pathological finding, and is usually associated with ANCA-negative disease. The vasculitis phase is characterized by purpura, peripheral neuropathy, glomerulonephritis, and constitutional symptoms. ANCA-positive patients were more likely to have vasculitis features [2,3].
Asthma usually precedes vasculitis symptoms (mean 9.3 + 10.8 years). Approximately 90–100% of patients are affected, and they have distinct features compared to other patients with asthma. It is typically late-onset asthma, manifesting in adults between the ages of 30 and 40. Allergy-related upper respiratory tract symptoms, such as allergic rhinitis, chronic sinusitis (70–90%), and nasal polyps, frequently coexist with asthma in patients with EGPA. Additionally, eosinophilic infiltration affects the lungs, and 70% of patients present with peripheral reticulonodular or consolidative pulmonary opacities on chest radiography. Pleural effusion and hilar or mediastinal lymphadenopathy are the two less common thoracic symptoms of EGPA [3].
The prevalence of cardiac involvement ranges from 16.0% to 29.0% in different studies [20], and eosinophilia and its cytotoxicity are the primary factors contributing to heart damage caused by EGPA. These patients have a higher eosinophil count at diagnosis, higher disease activity, negative ANCA, and higher C-reactive protein levels [3,4].
EGPA is associated with an increased mortality risk and worsened prognosis in the presence of cardiac involvement. The primary cause of first-year mortality and overall mortality in EGPA is cardiomyopathy, which accounts for nearly one-third of the deaths. [5]. It is recommended that all patients undergo periodic electrocardiography and echocardiography to detect any asymptomatic cardiac involvement early. Cardiac magnetic resonance (CMR) imaging is the gold standard method for diagnosing cardiomyopathies and assessing the staging of myocarditis [4].
Clinical manifestations of EGPA cardiomyopathy may be reversible with treatment despite its severe nature in rare cases, emphasizing the significance of promptly detecting and managing cardiac lesions [1,4].
It is extremely uncommon for autoimmune hemolytic anemia (AIHA) to be caused by warm-reacting IgM autoantibodies. Although there are no reports linking warm IgM autoantibodies to EGPA, they have been linked to immune disorders, such as immune thrombocytopenic purpura, severe combined immunodeficiency, and Sjogren's syndrome. The prognosis of AIHA caused by warm IgM autoantibodies is very poor, and the condition is typically resistant to standard treatments for IgG-mediated hemolytic anemia. A highly positive direct antiglobulin test (DAT) is generally linked to severe AIHA, and consequently, significant anemia [6,7].
A significant proportion of patients with EGPA (42–76%) had nervous system involvement and were ANCA-positive. The most common symptom is mono-neuritis multiplex; however, the tibial, peroneal, median, and ulnar nerves are often affected [2,3]. Foot drop and symmetric polyneuropathy are common symptoms that worsen if the treatment is not initiated early. Up to 63% of patients describe pain, limb weakness, numbness, burning sensation, or other sensory disturbances as their initial symptoms. In contrast, only 5–29% of cases with neurological symptoms in EGPA are reported to involve the central nervous system (CNS). Ischemic cerebrovascular lesions accounted for 52%, which were caused by intracerebral and/or subarachnoid hemorrhage (24%), loss of visual acuity (33%), and cranial nerve palsies (21%) [3].
The diagnosis of EGPA is still difficult, in part because of the long-lasting nature of asthma and the need for long-term corticosteroids (CS), which can obscure other symptoms of the condition [3]. Currently, the new EGPA classification criteria endorsed by the European Alliance of Associations for Rheumatology (EULAR) and the American College of Rheumatology (ACR) are used to validate diagnoses. (Table 1) [1]. This case report analyzed and described the clinical characteristics and outcomes of EGPA patients with myocarditis and AIHA to improve the overall comprehensive understanding with useful information for clinical practice.
2. Case Description
A 44- year- old Saudi female, with a history of bronchial asthma, allergic rhinitis, and sinusitis was diagnosed at 18 years of age. She had experienced an attack of chest pain 27 days before presentation, for which she sought medical advice many times and received analgesia with partial response. Two days before admission, she presented to the emergency department as the chest pain became severe and more frequent and was associated with shortness of breath on mild to moderate exertion and bilateral lower limb swelling. She denied any other symptoms related to the cardiopulmonary system and had no history of cardiac disease. Her condition was accompanied by generalized fatigability, fever, and an unintentional weight loss of 3 kg. Furthermore, she described that the condition was associated with polyarthritis, as well as loss of sensation over the dorsum of the right foot and left thumb. However, she did not report any vasculitis or skin rash or gastrointestinal symptoms. Her regular medications included inhaled corticosteroids in addition to short-acting ꞵ agonist inhalers, as required.
Upon assessment, the patient was in a semi-sitting position and slightly pale. Her pulse 110/minutes, blood pressure was 105/70 mmHg, and Spo2 was 95% on room air. Cardiovascular examination showed tachycardia, normal S1 and S2, no murmur, and no added sounds. Chest examination revealed symmetrical air entry with bilateral fine basal crepitations. Abdominal examination results were normal, with no organomegaly. Musculoskeletal examination was unremarkable, apart from bilateral lower limb edema. Neurological examination revealed sensory loss in the right dorsal foot and the left thumb. Electrocardiography (ECG) showed sinus tachycardia, no ischemic changes, or other abnormalities. Chest radiography revealed pulmonary congestion. (Figure 1)
The patient was diagnosed with acute heart failure, admitted to a high dependency unit, and received oxygen and 80 mg intravenous (IV) furosemide as bolus dose followed by furosemide 40 mg intravenously for 12 hourly with 12 leads ECG monitoring and workup continued to look for the underlying cause with cardiology consultation.
Initial laboratory tests revealed a low hemoglobin = 9.3gm/dl, leukocytosis with a high eosinophil count (4 x109/l), and N = (0.2-0.5 x109/l). A peripheral smear revealed severe hemolytic anemia (dimorphic), neutrophil leukocytosis, and monocytosis. The direct antiglobulin test result was positive twice (DAT). There were high levels of inflammatory markers, erythrocyte sedimentation rate (109 mm/h), and C-reactive protein (96. Her cardiac markers were high: troponin, 2 ug/l; N, (0.01-0,02) ug/l; LDH = 1218, creatine kinase (CK)= 913, and creatine kinase-myocardial band (CKMB) = 192. Kidney function, liver function tests (LFT), and thyroid function tests (TFT) were normal. Virological screening and blood culture results were negative (Table 2). Transthoracic echocardiography revealed ventricular dysfunction with a low ejection fraction of 25%.
Given the extremely elevated eosinophil count, vasculitic pathology was thought to be the most likely diagnosis, and further investigations and rheumatologic work-up were initiated.
Cardiac MRI imaging suggested myocarditis. (Figure 2): Globally, hypokinemia occurs in the left ventricle with a mild to moderate reduction in systolic function. Focal delayed enhancement was observed along the anteroseptal wall of the midmyocardium. The inferolateral wall of the mid-myocardium also showed focal subendocardial enhancement. Pericardial fluid or thickening was not observed. The right side showed a small pleural effusion. No mediastinal lymph nodes were observed.
High-resolution computed tomography (HRCT) chest (Figure 3) showed a few peripheral and central faint patchy areas of ground glass densities in the lower segment of the right upper lobe, with no signs of lung masses, consolidation, or cavitation; there was also no pneumothorax; and no enlarged hilar or mediastinal lymph nodes. Impression: Ground glass densities in the right upper lobe, possibly indicative of an inflammatory or infectious process.
With a background history of asthma, eosinophilia, chest involvement in HRCT, and the presence of neuropathy, diagnosis of EGPA was considered, and further investigations were conducted. ANCA was negative. Urine analysis was negative for blood and red RBCs casts (Table 2).
Brain CT positive for ethmoid sinusitis. While nerve conduction study showed normal latency and amplitude and slow velocity of 41 m/s for the right tibial nerve, and peroneal nerve showed normal latency and amplitude with slow velocity at 39 and 40 m/s, and both ulnar nerves showed normal latency amplitude and slow velocity at 38 and 46 m/s (Table 3). Thus, the patient was diagnosed with EGPA, myocarditis, autoimmune hemolytic anemia, and peripheral neuropathy.
Treatment: The patient was administered high-dose corticosteroids (intravenous bolus methylprednisolone 500 mg for three days, followed by 1 mg/kg of prednisolone) and cyclophosphamide 750 mg intravenously according to the European Vasculitis Society (EUVAS) protocol [8]. Before starting immunosuppressive treatment, the patient was screened for latent tuberculosis (TB) and Interferon-Gamma Release Assays (IGRA), which were positive; therefore, she was started on prophylactic rifampicin 600 mg for four months.
After one month, the patient showed a marked improvement in clinical and laboratory parameters. Her respiratory symptoms and polyarthritis significantly improved, and her EF improved to 30-40%. The eosinophil count returned to normal, and the hemolytic anemia resolved.
The patient was sent home and scheduled for subsequent infusions of cyclophosphamide monthly for three months and periodic blood tests, during which the patient was improved clinically and laboratory tests were shifted to mycophenolate mofetil 1 g twice a day as maintenance therapy.
3. Discussion
This case report describes the clinical presentation of EGPA with myocarditis, AIHA, and multiple mononeuropathies in a background history of asthma and sinusitis. EGPA is a frequent systemic vasculitis affecting the heart, which is usually accompanied by eosinophilia and negative ANCA [1].
The young age of this female patient rendered her more prone to developing cardiac-related EGPA disease. Several researches show that EGPA patients with cardiac involvement started the disease earlier than those without (with mean ± SD: 38.4 ± 10.5 vs. 42.1 ± 15.9 years, respectively [4,9].
As our patient was female, this aligns with one study reporting that women were more likely than men to have cardiac involvement [10]. While some studies found that cardiac involvement is more frequent in men [4,11,12]. However, there was a greater number of female patients in the group with cardiac disease than in the group without cardiac involvement, suggesting that female patients in these studies had a greater incidence of cardiac disease.
The patient presented with atypical chest pain for three weeks, and received general nonspecific treatment until overt heart failure developed, which raised the suspicion of myocarditis as the underlying cause of her cardiac problem. Myocarditis, abrupt heart failure, as in this case, as well as coronary vasculitis, myocardial infarction, ventricular arrhythmias, and sudden cardiac death are frequently associated with EGPA [2,4]. Clinical heart failure was reported in a meta-analysis of 62 cases, with a common cardiac presentation in 51.6% and chest pain in 32.3% [11]. Lopes et al reported a patient who presented with fulminant eosinophilic myocarditis (13), while Sakurai. et al described a patient with severe EPGA associated with advanced atrioventricular block and cardiac arrest [14]. A fraction of patients will either show subtle presentation or no symptoms [1]. Dennert et al. [12] found that asymptomatic heart disease was more frequent than symptomatic heart disease but stated that all participants were in remission, in contrast to our patient who had an active disease. Consequently, in individuals with suspected EGPA, thorough examination of cardiac involvement is recommended.
Furthermore, patients with EGPA who experience cardiac involvement typically experience involvement in other organs, as it is evident from this case that patients with EGPA can present with cardiac, neurological, and hematological involvement [3,4,6,15]. Itagak et al 2023 reported cardiac involvement alone, as the patient exclusively had cardiac involvement as the only organ damage linked with EGPA, with the exception of sinusitis and asthma, which represent the prodromal phase of the disease [16],
Cardiac disease was confirmed by clinical presentation, ECG, cardiac biochemical markers, and cardiac images in this patient, which is consistent with the literature and other studies [2,4,11,17] Vasculitis rarely manifests itself as a major cardiac problem. Prior research has demonstrated that, although nerve involvement is common among individuals with EGPA, the heart and gastrointestinal systems are less commonly affected [9].
Cardiac involvement in patients with EGPA has a particularly extremely poor prognosis [12,17]. Comarmond et al. reported a mortality rate that was around four times higher than that of people without cardiac disease [18]. When EGPA-induced myocardial injury is suspected, it is crucial to perform multimodality imaging, such as cardiac magnetic resonance (CMR), particularly when endomyocardial biopsy (EMB) is negative or unavailable [13]. In this patient, the diagnosis was made based on CRM and prompt treatment was initiated earlier. This may have contributed to the patient's successful clinical outcome. Another factor that affected the prognosis in this case was the glucocorticoid treatment for bronchial asthma.
CMR imaging can help clarify the diagnosis and assess the degree of myocardial necrosis. However, EMB alone can provide conclusive diagnoses [3,4].
Furthermore, the case demonstrated laboratory data consistent with autoimmune hemolytic anemia based on positive direct Coombs and DAT tests, with no previous history of hemolysis. AIHA is a rare and unusual presentation of EGPA, and very few cases have reported this association [6].
The patient responded well to glucocorticoid therapy plus cyclophosphamide and continued maintenance treatment with 1 g g Mycophenolate mofetil twice a day. Patients with heart lesions and AIHA with EGPA require aggressive and prompt therapeutic approaches. This approach may enable the recovery of cardiac function and reduce the significant mortality associated with EGPA [19]. However, in patients with heart failure or mononeuritis multiplex, this treatment alone is ineffective [15]. Therefore, the patient continued maintenance treatment with 1 g of mycophenolate mofetil twice a day.
Early medical intervention can prevent potentially fatal episodes of heart disease and reduce the high mortality rate associated with EPGA-related myocarditis. Even in the absence of symptoms, a thorough cardiac evaluation should be carried out on an EPGA patient, as this will help avoid serious cardiac complications.
We report a case of myocarditis related to EPGA that responded well to standard corticosteroid-cyclophosphamide therapy; however, in some cases, this treatment is ineffective for EGPA patients who also have heart failure or mononeuritis multiplex [19]. Therefore, the patient continued maintenance treatment with 1 g of mycophenolate mofetil twice a day.
4. Conclusions
We report a case of myocarditis and AIHA related to EPGA that responded well to standard corticosteroid-cyclophosphamide therapy. As cardiac involvement substantially increases EGPA-related mortality and morbidity, early diagnosis and treatment can prevent patients from experiencing serious late-stage cardiac complications.
Author Contributions
Conceptualization, D.A. and F.A.; methodology, D.A.; software, N.A.; validation, D.A., F.A. and O.A.; formal analysis, O.A.; investigation, A.M. and A.K.; resources, D.A.; data curation, F.A.; writing—original draft preparation, O.A., D.A. and A.M.; writing—review and editing, N.A.; visualization, F.A.; supervision, D.A.; project administration, O.A., D.A. and A.M.; funding acquisition, F.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and bioethics standards of Saudi Arabia.
Informed Consent Statement
The patient received adequate information about the study and provided written informed consent in accordance with work center policies. The patient was informed that his case details would be kept anonymous and that the collected data would be used only for scientific purposes. The consent form is available upon request. The authors declare that no personal information is included in this article.
Data Availability Statement
Research data supporting this publication are available from the corresponding author (OA) and will be available upon request.
Acknowledgments
We acknowledge the participation of the patient in this study and his acceptance to use these information and images for publication.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Grayson, P.C., Ponte, C., Suppiah, R., Robson, J.C., Craven, A., Judge, A., Khalid, S., Hutchings, A., Luqmani, R.A., Watts, R.A. and Merkel, P.A. (2022). 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology Classification Criteria for Eosinophilic Granulomatosis with Polyangiitis. Annals of the Rheumatic Diseases, [online] 81(3), pp.309–314. [CrossRef]
- Emmi, G., Bettiol, A., Gelain, E., Bajema, I.M., Berti, A., Burns, S., Cid, M.C., Willem, J., Cottin, V., Durante, E., Holle, J., Mahr, A., Martinez, M., Chiara Marvisi, Mills, J.A., Sergey Moiseev, Moosig, F., Chetan Mukhtyar, Neumann, T. and Iacopo Olivotto (2023). Evidence-Based Guideline for the diagnosis and management of eosinophilic granulomatosis with polyangiitis. Nature Reviews Rheumatology, 19(6), pp.378–393. [CrossRef]
- Justyna Fijołek and Elżbieta Radzikowska (2023). Eosinophilic granulomatosis with polyangiitis – Advances in pathogenesis, diagnosis, and treatment. Frontiers in Medicine, 10. [CrossRef]
- Chen, Y., Guo, X., Zhou, J., Li, J., Wu, Q., Yang, H., Zhang, S., Fei, Y., Zhang, W., Zhao, Y. and Zhang, F., 2020. Cardiac involvement in eosinophilic granulomatosis with polyangiitis: a retrospective study in the Chinese population. Frontiers in medicine, 7, p.583944. [CrossRef]
- J. Vinit, Bielefeld, P., G. Müller, P. Pfitzenmeyer, Philippe Bonniaud, B. Lorcerie and J.-F. Besancenot (2010). Heart involvement in Churg–Strauss syndrome: Retrospective study in French Burgundy population in past 10years. European Journal of Internal Medicine, 21(4), pp.341–346. [CrossRef]
- Chao, M. P., Hong, J., Kunder, C., Lester, L., Schrier, S. L., & Majeti, R. (2014). Refractory warm IgM-mediated autoimmune hemolytic anemia associated with Churg-Strauss Syndrome responsive to eculizumab and rituximab. American Journal of Hematology, 90(1), 78. [CrossRef]
- Zanella, A., & Barcellini, W. (2014). Treatment of autoimmune hemolytic anemias. Haematologica, 99(10), 1547-1554. [CrossRef]
- Szczeklik, W. , Miszalski-Jamka, T., Mastalerz, L., Sokolowska, B., Dropinski, J., Banys, R., Hor, K.N., Mazur, W. and Musial, J. (2011). Multimodality Assessment of Cardiac Involvement in Churg-Strauss Syndrome Patients in Clinical Remission. Circulation Journal, 75(3), pp.649–655. [CrossRef]
- Neumann, T. , Manger, B., Schmid, M., Kroegel, C., Hansch, A., Kaiser, W.A., Reinhardt, D., Wolf, G., Hein, G., Mall, G., Schett, G. and Zwerina, J. (2009). Cardiac Involvement in Churg-Strauss Syndrome. Medicine, 88(4), pp.236–243. [CrossRef]
- Kanecki, K. , Nitsch-Osuch, A., Gorynski, P., Tarka, P. and Tyszko, P., 2017. Hospital morbidity database for epidemiological studies on Churg-Strauss syndrome. Respiratory System Diseases, pp.19-25. [CrossRef]
- Pakbaz, M. and Pakbaz, M., 2020. Cardiac involvement in eosinophilic granulomatosis with polyangiitis: a meta-analysis of 62 case reports. The Journal of Tehran University Heart Center, 15(1), p.18 . https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7360870/.
- Dennert RM, van Paassen P, Schalla S, Kuznetsova T, Alzand BS, Staessen JA, Velthuis S, Crijns HJ, Tervaert JW, Heymans S. Cardiac involvement in Churg-Strauss syndrome. Arthritis Rheum. 2010;62:627–634. [CrossRef]
- Lopes, P.M. , Rocha, B.M., Cunha, G.J., Ranchordas, S., Albuquerque, C., Ferreira, A.M., Aguiar, C., Trabulo, M., Neves, J.P. and Mendes, M., 2020. Fulminant eosinophilic myocarditis: a rare and life-threatening presentation of eosinophilic granulomatosis with polyangiitis. Case Reports, 2(5), pp.802-808. https://www.jacc.org/doi/abs/10.1016/j.jaccas.2020.01.
- Sakurai, Y., Oshikata, C., Katayama, T., Takagi, S., Kaneko, Y., Yo, K., Kaneko, T., Kubota, H., Matsubara, T., & Tsurikisawa, N. (2023). A case of eosinophilic polyangiitis with granulomatosis that evolved to cardiac arrest due to advanced atrioventricular block. Nagoya Journal of Medical Science, 85(1), 171-178. [CrossRef]
- Haas, C., Le Jeunne, C., Choubrac, P., Durand, H. and Hugues, F.C. (2001). [Churg-Strauss syndrome. Retrospective study of 20 cases]. Bulletin De l’Academie Nationale De Medecine, [online] 185(6), pp.1113–1130; discussion 1130-1133. Available at: https://pubmed.ncbi.nlm.nih.gov/11717829/.
- Itagaki, T. , Miura, T., Karasawa, S., Nomoto, F., Takamatsu, T., Sunohara, D., Komatsu, T., Tanaka, K., Mochidome, T., Kasai, T. and Ikeda, U., 2023. Eosinophilic granulomatosis with polyangiitis presenting with eosinophilic myocarditis as the only organ involvement. Journal of Cardiology Cases, 27(4), pp.172-175. [CrossRef]
- Hazebroek MR, Kemna MJ, Schalla S, Sanders-van Wijk S, Gerretsen SC, Dennert R, Merken J, Kuznetsova T, Staessen JA, Brunner-La Rocca HP, van Paassen P, Cohen Tervaert JW, Heymans S. Prevalence and prognostic relevance of cardiac involvement in ANCA-associated vasculitis: eosinophilic granulomatosis with polyangiitis and granulomatosis with polyangiitis. Int J Cardiol. 2015;199:170–179. [CrossRef]
- Comarmond, C. , Pagnoux, C., Khellaf, M., Cordier, J.F., Hamidou, M., Viallard, J.F., Maurier, F., Jouneau, S., Bienvenu, B., Puéchal, X. and Aumaître, O., 2013. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis & Rheumatism, 65(1), pp.270-281. [CrossRef]
- Sinico, R.A. , Di Toma, L., Maggiore, U., Bottero, P., Radice, A., Tosoni, C., Grasselli, C., Pavone, L., Gregorini, G., Monti, S., Frassi, M., Vecchio, F., Corace, C., Venegoni, E. and Buzio, C. (2005). Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome. Arthritis & Rheumatism, 52(9), pp.2926–2935. [CrossRef]
Figure 1.
Chest X Ray showing bilateral interstitial opacities consistence with pulmonary congestion.
Figure 1.
Chest X Ray showing bilateral interstitial opacities consistence with pulmonary congestion.
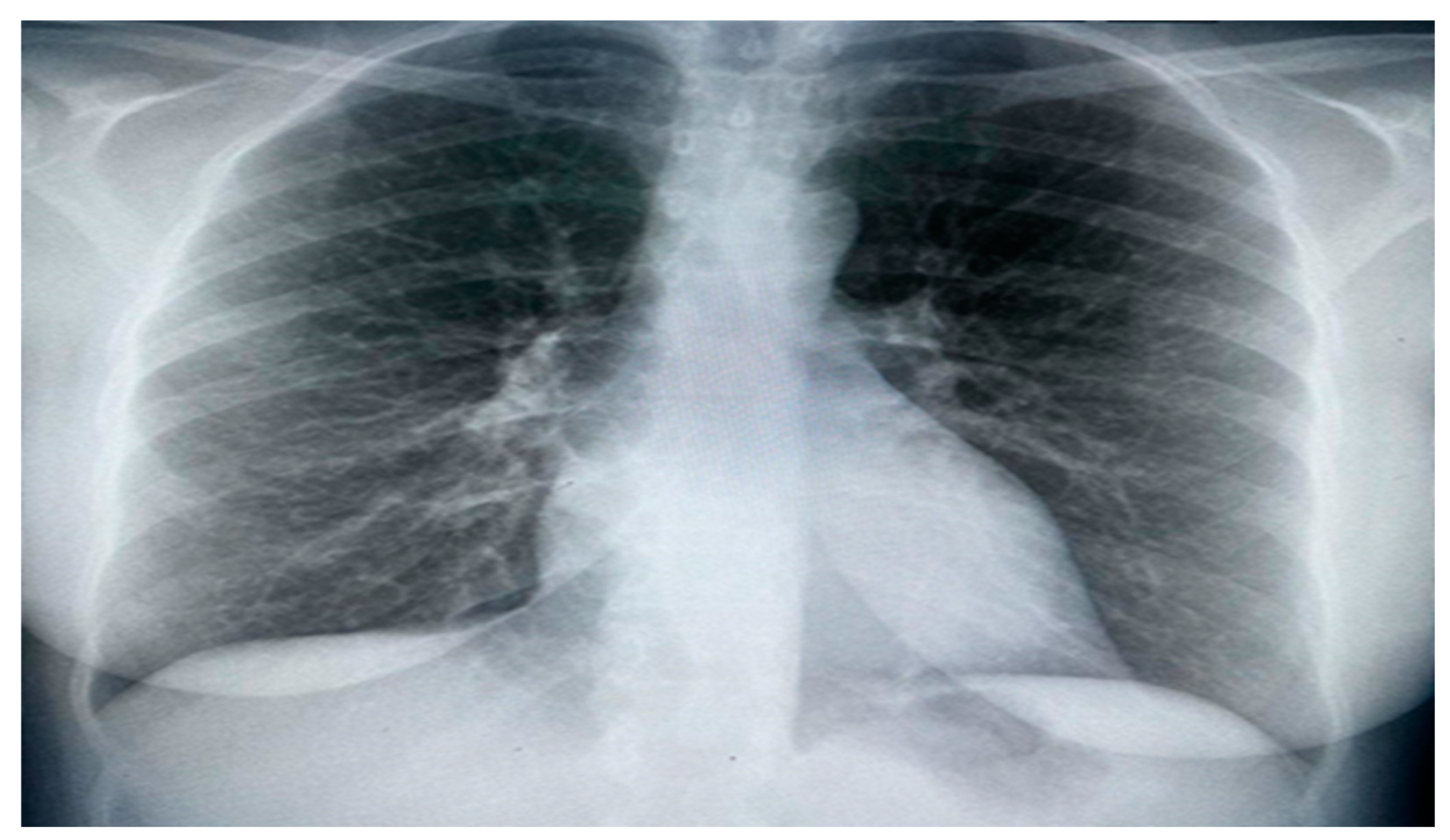
Figure 2.
Cardiac MRI There is non-territorial focal transmural delayed enhancement involving the antero-septal wall of the mid myocardium. Also there is focal sub endocardial enhancement of the inferior-lateral wall of the mid myocardium. Small right-sided pleural effusion is noted.
Figure 2.
Cardiac MRI There is non-territorial focal transmural delayed enhancement involving the antero-septal wall of the mid myocardium. Also there is focal sub endocardial enhancement of the inferior-lateral wall of the mid myocardium. Small right-sided pleural effusion is noted.
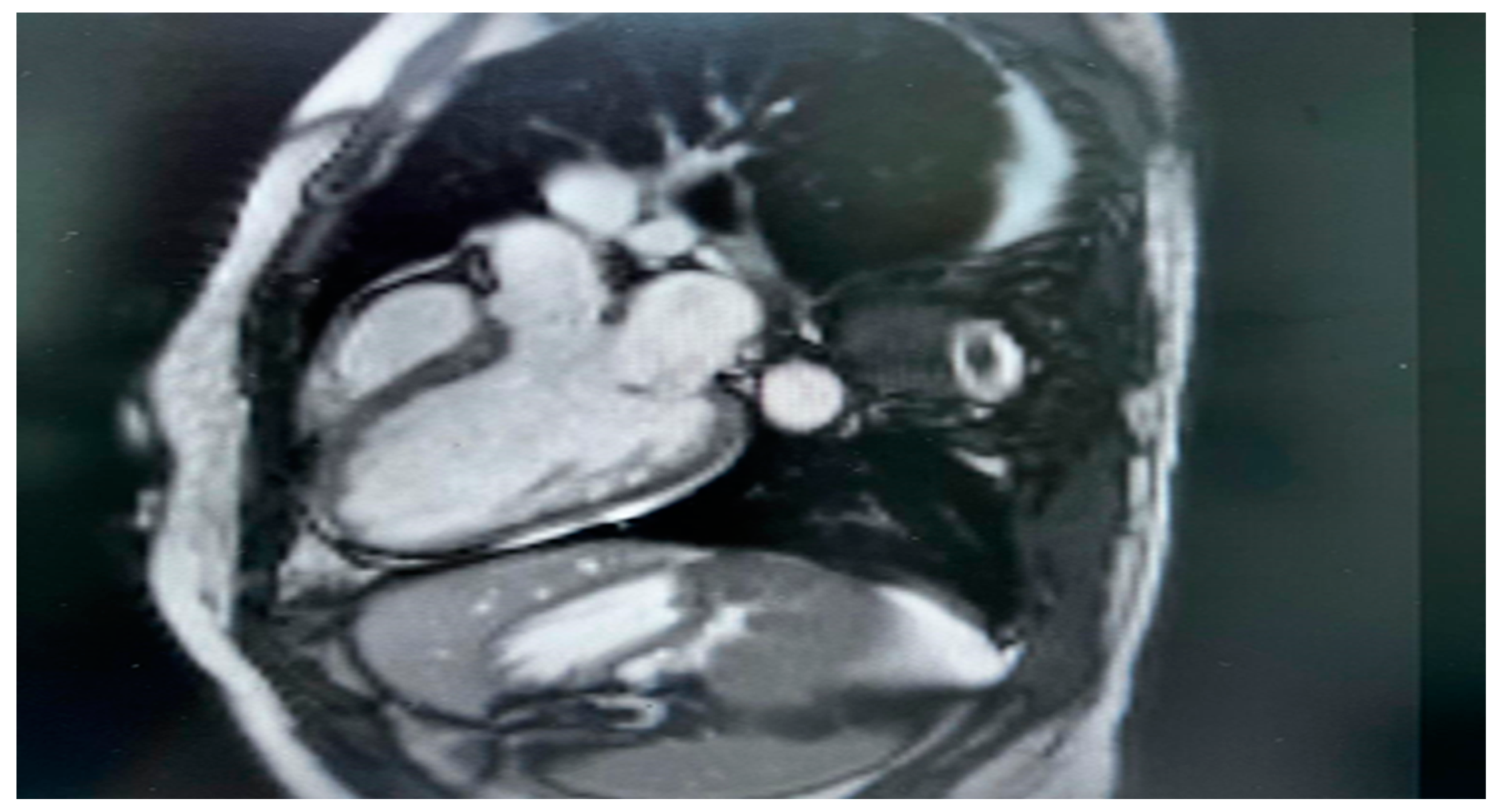
Figure 3.
High resolution computed tomography (HRCT) chest showing peripheral and central faint patchy areas of ground glass densities within the lower segment of right upper lobe.
Figure 3.
High resolution computed tomography (HRCT) chest showing peripheral and central faint patchy areas of ground glass densities within the lower segment of right upper lobe.
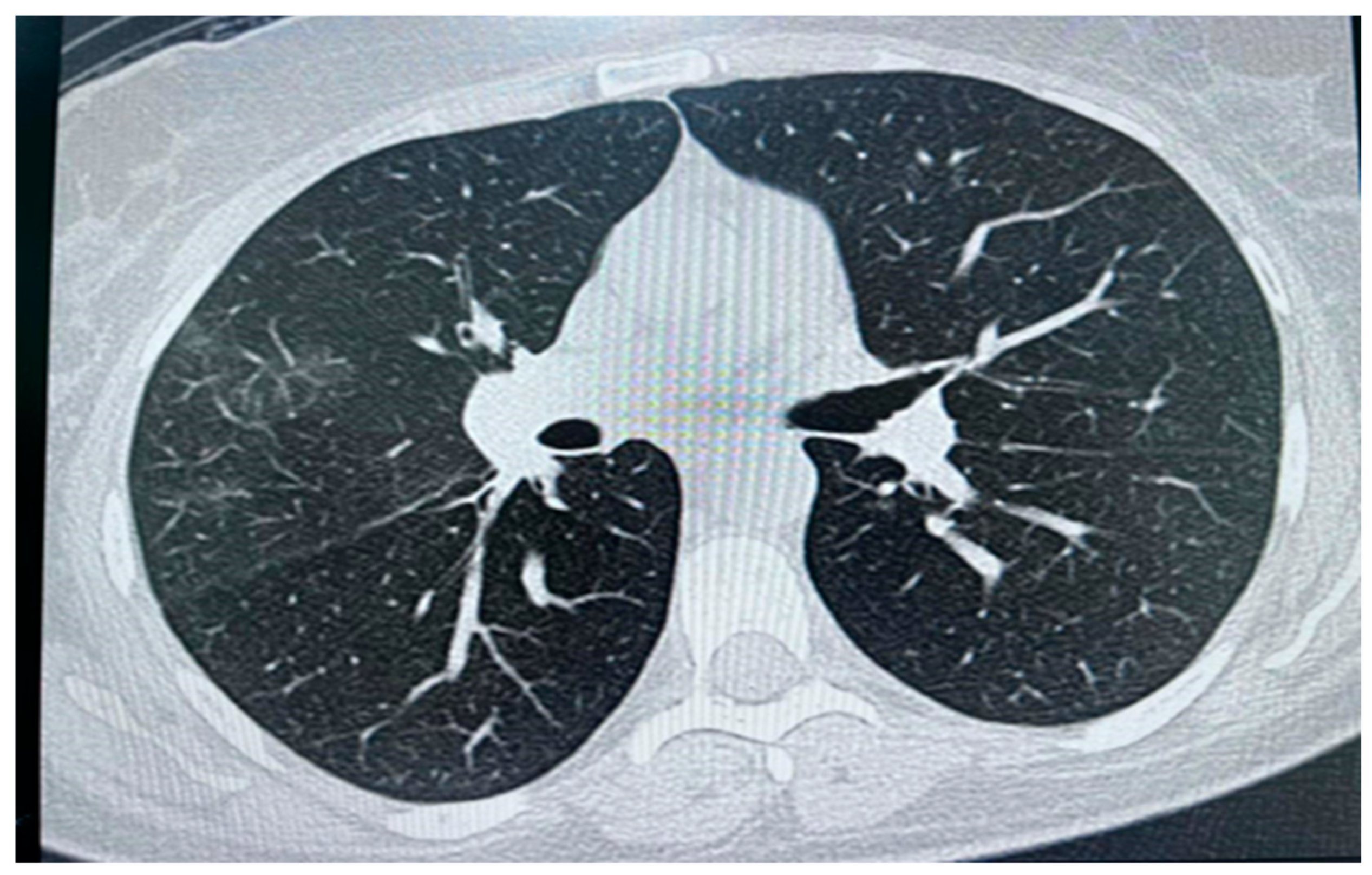

Table 1.
The American College of Rheumatology and the European Alliance of Associations for Rheumatology (2022) Classification criteria for EPGA.
Table 1.
The American College of Rheumatology and the European Alliance of Associations for Rheumatology (2022) Classification criteria for EPGA.
 |
Table 2.
Laboratory work-up.
| Investigations | Patient Results | Normal range |
| WBC1 | 17 x109/L | 4.5 - 11.0 × 109/L |
| Neutrophils | 8 x109/L | 2.5 - 6× 109/L |
| Lymphocytes | 3.5 x109/L | 1.0 – 4.8× 109/L |
| Eosinophil | 4 x109/L | 0.2- 0.5×109/L |
| Haemoglobin | 9.3g/dl | 12-16g/dl |
| Peripheral blood smear | Haemolytic anemia (dimorphic) of severe degree, Neutrophil leukocytosis and monocytosis. | |
| Creatinine | 71 µmol/L | 61.9 to 114.9 µmol/L |
| Hepatic profile | Normal | |
| ESR2: | 109 mm/hr | ≤ 20 mm/hr |
| CRP3 | 96 mg/dL | < 0.3 mg/dL |
| Hepatitis B surface antigen | Negative | |
| Hepatitis C antibody | Negative | |
| HIV4 | Negative | |
| LDH5 | 1218 units/L | 105 to 233 units/L |
| CK6 | 913 IU/L | 30 to 145 U/L |
| CKMB7 | 192 IU/L | 5 and 25 IU/L |
| Tropnin | 2ug/l | (0.01-0,02)ug/l |
| TFT8 | Normal | |
| Blood cultures | Negative | |
| DAT9 | Positive in two occations | |
| ANCA10 | Negative | |
| Urine analysis | Negative for RBCs11 casts |
1WBC; white blood count, 2 ESR; Erythrocyte sedimentation rate, 3 CRP; C- reactive protein, 4 HIV; Human immune deficiency virus, 5 LDH; Lactate dehydrogenase, 6 CK; Creatine Kinase, 7 CKMB; Creatine kinase myocardium bound, 8 TFT; Thyroid function test, 9 DAT; Direct antiglobulin test, 10 ANCA; anti-neutrophil cytoplasmic antibody, 11 RBC; Red blood cells.
Table 3.
Diagnostic Images and Nerve Conductive Study.
| Echo: EF[12] 25% |
|
CT[13] Brain: No acute territorial infarction. Preserved grey white matter differentiation. No intra cerebral focal lesion. No intracerebral hematoma or extra-axial collection. No shift of midline structures. Sulci, ventricles and basal cisterns are patent. Partially degraded images of posterior cranial fossa without suspicious lesion are seen. Orbits and mastoid air cells are unremarkable. No suspicious skull lesion. Mild ethmoid sinusitis is noted. |
|
Nerve conduction study: Tibial and comm. peroneal nerve bilateral NCS normal for median and ulnar nerve bilateral Right tibial nerve shows normal latency and amplitude and slow velocity 41 m/s. no response could be obtained from right common peroneal nerve Left tibial and left comm. peroneal nerve show normal latency, amplitude and slow velocity at 39 and 40 m/s. SNCS : no sensory response could be obtained from both median nerve Both ulnar nerve shows normal latency amplitude and slow velocity at 38 and 46 m/s. findings suggestive of myelinic neuropathy both lower limbs and diffuse sensory neuropathy. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



