
Submitted:
16 August 2024
Posted:
20 August 2024
You are already at the latest version
Abstract
Goldenhar Syndrome (GS), also known as oculoauriculovertebral spectrum (OVAS), is a rare congenital condition characterized by impaired development of structures such as ears, eyes, nose, palate, lip, mandible, maxilla, and teeth. The etiopathogenesis is multifactorial and dependent on genetic and environmental factors, yet the syndrome has many unknowns. We report a case of a 31-year-old male diagnosed clinically with GS as an infant with an absent left ear, hearing loss, absent left lung lobe, reduced growth of left upper limb, decreased size of left face, absent left kidney, and a missing left thumb, who was seen in the genetic clinic for prenatal genetic counseling. A microarray comparative genome hybridization (aCGH) was performed for copy number abnormalities and targeted NGS panels were performed for hereditary hearing loss and limb/digital malformations. aCGH revealed a 471kb loss at 16p12.2 and a mosaic 7.3Mb gain at 19p12q12. Metaphase FISH confirmed the orientation of 19p12q12 gain as small supernumerary ring chromosome 19. NGS revealed multiple variants (MYH14 c.4001C>T, PDE1C c.8C>T, ADGRV1 c.15502A>G, CDH23 c.8772C>A, MYO3A c.484G>A, ADAMTS10 c.506G>A, CHUK c.1294C>T, CSPP1 c.1972A>G, and TCTN2 c.160G>A). Due to the heterogeneous clinical manifestation and genetic heterogeneity of GS, a comprehensive genetic testing strategy is needed for timely diagnosis and better patient care.
Keywords:
Goldenhar Syndrome
; small supernumerary marker chromosome (sSMC)
; mosaic ring chromosome 19
; 16p12.2 deletions
; chromosomal microarray
; comparative genomic hybridization
; genetic testing
1. Introduction
Goldenhar Syndrome (GS) – oculoauriculovertebral spectrum (OAVS)- is a rare condition that is present at birth. The occurrence ranges from 1 in 3500 to 1 in 5600, and there is a higher prevalence among males compared to females, with a ratio of 3 to 2 [1-4]. It is a unilateral, or bilateral deformity characterized most by impaired development of the eyes, ears, lip, tongue, palate, mandible, maxilla, and teeth [1]. This craniofacial syndrome is also known as 1st and 2nd Branchial Arch Syndrome as it is produced by neural crest migration disturbances resulting in abnormal development of the first and second branchial pouches and occlusion of placental vessels. [4, 5]. These patients have ocular symptoms, auricular symptoms, craniofacial deformities, skeletal abnormalities, and internal organ abnormalities [4, 6, 7, 8, 9]. Due to maxillary and/or mandibular hypoplasia, nearly all GS patients reveal some degree of hemifacial macrosomia (HFM). The majority of patients also suffer from some degree of hearing loss; hence GS patients require an audiological assessment [10]. Additionally, in children with GS, it is also possible to see speech disorders, autistic behaviors, short stature, and delayed psychomotor development [11].
The etiology and pathogenesis of the Goldenhar syndrome are influenced by a combination of genetic and environmental factors, although numerous aspects of the condition remain unexplored. In patients with phenotypic characteristics of GS, chromosomal anomalies support a genetic basis. Beleza-Meireles A, et al. published a review of the aCGH copy number variation loci reported in patients with GS and their associated phenotypes [10]. These anomalies include deletions, duplications, and chromosomal rearrangements. Deletion 5p15 (5p15.33-pter), deletions 22q11.2, X chromosome aneuploidies, duplication 14q23.1, mosaicism of trisomy 7, 9, 22 are some of many examples of published chromosomal anomalies in patients with phenotypic characteristics of GS [10]. Additionally, single nucleotide variants (SNVs) by NGS or WES have also been identified in Goldenhar patients. Tingaud-Sequeira A, et al. listed several genes frequently involved in the pathogenesis of GS [12], including SF3B2, MYT1, VWA1, ZIC3, EYA3 and ZYG11B. Even though some common molecular markers have been identified, it is expected that the new variants may be identified in GS patients due to heterogenous clinical manifestation and multiple overlapping syndromes.
Small supernumerary marker chromosomes (sSMCs) are structural anomalies whose origins cannot be determined through conventional cytogenetics alone, necessitating molecular approaches. Notably, 70% of sSMCs are of de novo origin, 20% are inherited maternally, and 10% are inherited paternally [13, 14]. The phenotypes associated with sSMCs are extremely variable, from normal to severely abnormal depending on its origin and nature [15]. Approximately 70% of individuals carrying small supernumerary marker chromosomes (sSMCs) exhibit no phenotypic abnormalities. Conversely, the remaining 30% manifest developmental delays, intellectual disabilities, mixed gonadal dysgenesis (MGS), or infertility, with the specific outcome contingent upon the origin of the sSMC [13]. Many of these sSMCs have been reported before the era of DNA microarray technology and thus the nature of these sSMCs were not fully identified.
Our detailed literature review suggests that no GS patient with either 16p12.2 deletion or mosaic ring 19 chromosome has been reported yet. Multiple congenital anomalies and overlapping phenotypes make it difficult for clinicians and healthcare providers to diagnose these patients appropriately without the knowledge of the relationship between phenotype and genotype. It is recommended that the analysis of chromosomal rearrangements using chromosomal microarrays, molecular cytogenetic, and molecular testing should be performed in order to comprehend the complexity of the phenotypes in known syndromes, followed by genetic counseling of the patients and families.
2. Case study and Methods
The proband, a 31-year-old single male from non-consanguineous parents of Italian descent consulted a private ambulatory clinic in Strong Memorial Hospital. He was interested in having children in the future and wished to speak to a geneticist regarding risk to future children.
2.1. Case Presentation
Proband was born after a pregnancy of 34 weeks. The mother experienced excessive bleeding and skull fusion issues during labor and has a history of 4 miscarriages during early pregnancy. He was diagnosed with Goldenhar syndrome as an infant with an absent left ear, absent lung lobe on the left side, reduced growth of left upper limb, decreased size of left face, absent left kidney, and a missing left thumb. His active problem list includes acute gastric volvulus, Goldenhar Syndrome, thrombocytopenia, splenomegaly, testicular cancer, systemic lupus erythematosus, cough, anxiety, long-term use of Plaquenil, latent hypermetropia of both eyes, amblyopia in the left eye, long term use of immunosuppressant medication. He had multiple surgical interventions, such as abdominal surgery, hand surgery, exploration of undescended testis, inguinal hernia repair x2, vesicoureteroreflux surgery, and tonsillectomy. He had no reported history of fine/gross motor delay or learning disability. Facial asymmetry was observed on clinical examination with the right being more dominant than the left, down slanting palpebral fissures and microtia on the left side, and surgically repaired cleft palate. In his upper limb, he was noted to have a left radial anomaly with a short forearm and absent thumb. Cardiac issues include bicuspid aortic valve, premature ventricular contractions, left bundle branch block, and history of pericarditis associated with Lupus. Due to the complex clinical picture, the patient was referred for genetic testing.
2.2. Methods
DNA was isolated manually using QIAamp DNA Blood mini kit (Qiagen-51106), followed by array comparative genome hybridization to rule out any chromosomal gains and losses. DNA from the patient and normal male reference DNA was digested and labeled with an Agilent DNA labeling kit. Patient DNA was labeled with Cy5 and reference DNA with Cy3. Both DNA were then co-hybridized on SurePrint G3 ISCA Human CGH+SNP Microarray 4x180k arrays (Agilent- G4890A) for 40 hours at 64-68⁰C in a rotating oven. Array slides were then washed and scanned on SureScan DX Microarray Scanner (Agilent-G5761A). Array data was extracted using feature extraction software (Agilent, V12.1) and analysis was performed on CytoGenomics software (Agilent, V5.3.014). Genomic copy number changes were identified with the assistance of the Aberration Detection Method 2 algorithm with the sensitivity threshold set at 6.0 and minimum size of 250kb for deletion and duplication. Copy number changes identified in the samples were evaluated by using the UCSC Genome Browser website (http://genome.ucsc.edu) and the Database of Genomic Variants (http://projects.tcag.ca/variation). The array data was analyzed using the annotation GRCh37/hg19. The DECIPHER (http://decipher.sanger.ac.uk/) database was used to support genotype-phenotype correlation.
Peripheral blood samples were cultured using standard cytogenetic methods for 72 h with phytohemagglutinin (PHA) stimulation to yield metaphases. Fluorescence in situ hybridization (FISH) was performed with standard techniques using RP11-141O15 BAC probe (Spectrum Red, Empire Genomics, NY) at 16p12.2 and TelVysion 16p (Spectrum Green, Abbott Laboratories, Des Plaines, IL) at 16PTEL05 (control) confirming the deletion of 16p12.2. BAC probe RP11-359H18 (Spectrum Red, Empire Genomics, NY) at 19p12 and BAC probe RP11-714C4 (Spectrum Green, Empire Genomics, NY) at 19q11 were used to confirm the structural variation, ring chromosome 19.
Targeted next generation sequencing (NGS) was performed at Invitae and Prevention Genetics Reference labs for below panels;
-
Prevention Genetics Hereditary Hearing Loss and Deafness Panel: https://www.preventiongenetics.com/testInfo?val=Hereditary-Hearing-Loss-and-Deafness-PanelInvitae Limb and Digital Malformations Panel: https://www.invitae.com/us/providers/test-catalog/test-55010
3. Results
Microarray CGH analysis revealed a 471 kilobase loss at 16p12.2 from 21959891 to 22430592 base pairs encompassing the UQCRC2 gene (Figure 1) and a mosaic 7.3 megabase gain at 19p12-q12 from 22470583 to 29795821 base pairs, likely indicating mosaicism for a small supernumerary ring chromosome 19 (Figure 2).
Interphase and metaphase FISH confirmed the deletion 16p12.2 (Figure 3) and the duplication 19p12q12 (Figure 4a). In addition, metaphase FISH also confirmed the 19p12q12 duplication was the result of sSRC19 (Figure 4b). The level of mosaicism is approximately 90% on metaphases/reverse DAPI.
Variants identified by targeted NGS reported at the reference lab are listed in Table 1.
Segregation analysis on proband’s parents was not performed due to unavailability of parents.
4. Discussion
Although GS usually occurs sporadically with no family history, genetic predisposition has been proposed based on growing evidence from the literature [16-19]. Familial case reports of autosomal dominant or autosomal recessive inheritance, as well as evidence of a genetic association in two families and the presence of features of GS in patients with multiple chromosomal aberrations and gene imbalances, all of which suggest that GS has a genetic basis in some instances [10]. Segregation analysis performed in 311 members of the families of 74 probands with GS provided evidence for an autosomal dominant mode of inheritance with reduced penetrance [20]. Kaye et al. hypothesized that OAVS may represent one end of a phenotypic continuum produced by the segregation of a single gene [20]. In their research, Rollnick et al. analyzed pedigree data on 97 cases, 44 of whom had a family history of the same or similar anomaly [21]. The authors have reported that first-degree relatives were most often affected (35/433, 8%). Of 176 sibs tabulated, 11 (6%) were considered affected. The pattern of occurrence in many families suggested multifactorial determination, although other interpretations are possible [22]. Moreover, the authors observe a broad phenotypic spectrum within families, which has also been observed by others. They suggest that familial inheritance is more prevalent than originally thought. Phenotypes observed in familial OAVS cases do not differ from those in sporadic ones [10, 21, 22].
The presence of chromosomal anomalies in individuals exhibiting phenotypic characteristics within the GS provides additional evidence supporting a genetic basis for this syndrome. Some examples of published chromosomal anomalies in patients with phenotypic characteristics of GS includes the deletion at 5p15, documented in multiple patients displaying features of GS [23-25]. Microduplications on 14q23.1 were detected in two families exhibiting autosomal dominant Oculo-Auriculo-Vertebral Spectrum (OAVS) [26, 27]. Within one of these families, two first-degree relatives manifested clinical features of both OAVS and Branchio-oto-renal syndrome. This suggests that the 14q23.1 region could potentially contain candidate genes not only for OAVS but also for additional developmental disorders related to the first and second pharyngeal arch [26, 27]. Some patients with features of GS have been found to exhibit deletions in the 12p13.33 region, specifically involving the WNT5B gene; however, it’s noteworthy that not all individuals with GS characteristics display these deletions [28, 29]. Aberrations in the 22q region also have been frequently reported in individuals diagnosed with Goldenhar Syndrome [30-35]. Mosaicism for trisomy 22q has been also documented, suggesting that this genomic region could be a good candidate for certain cases of Goldenhar Syndrome [36].
In the study of Melis et al. (2011), cytogenetics revealed a female karyotype with the presence of de-novo small supernumerary chromosome rings in 100% of cells examined in a 15-year-old female with selective cognitive impairment and tall stature due to chromosome 19 supernumerary ring [37]. In 2020, Li et al. performed Next Generation Sequencing to identify the chromosomal origins of sSMCs and correlate certain sSMCs with a clinical picture in 75 patients. This study underlines the importance of setting of an economical and efficient methods for clinical small supernumerary marker chromosome diagnosis in terms of identifying genotype-phenotype correlations and integrating genomic data into clinical care [13]. The phenotype linked to partial trisomy 19q is marked by facial dysmorphism, growth and mental retardation, macrocephaly, as well as heart malformation, along with anomalies affecting the genitourinary and gastrointestinal tracts. On the other hand, the phenotype associated with partial trisomy 19p is characterized by dysmorphic features, severe mental retardation, abnormalities in brain morphology, and anomalies affecting the fingers [38-40].
The duplicated region in our case mainly contains Zinc Finger (ZNF) genes ZNF98, ZNF99, ZNF91, and ZNF254 and UQCRFS1 gene. ZNF genes have important role both in tissue homeostasis and disease. With an estimated 500-600 members, zinc finger (ZNF) genes are one of the largest gene families in the human genome [41-43]. Several studies have demonstrated their crucial role in cell and developmental differentiation by encoding transcriptional regulators. They consist of zinc finger domains that can bind selectively to certain DNA or RNA and associate with proteins, thus being able to regulate gene expression at both the transcriptional and translational levels [44]. Biallelic variations in the UQCRFS1 gene are correlated with Mitochondrial Complex III Deficiency, Cardiomyopathy, and Alopecia Totalis [45]. Our case has bicuspid aortic valve, premature ventricular contractions, left bundle branch block, and history of pericarditis associated with Lupus as cardiovascular diseases, which may be associated with UQCRFS1 gene overexpression.
The 16p12.2 deletion (previously termed as 16p12.1 deletion) observed in our case encompasses the EEF2K, POLR3E, UQCRC2, and CDR genes, this is a recurrent heterozygous deletion associated with variable phenotype and reduced penetrance. In most patients (~95.0%) this deletion is inherited from a parent, who may or may not have clinical traits linked to 16p12.2 recurrent deletion [46]. Although the clinical features of 16p12.2 deletion do not constitute a recognizable syndrome due to its varied expressivity and incomplete penetrance, it is associated with developmental delays, speech delays, cognitive impairment, mild dysmorphic facial features without a consistent pattern, sleep disturbance, epilepsy, cardiac/skeletal malformations, and a psychiatric and/or behavioral disorder commonly observed in probands. Based on the large study of 11873 cases, Girirajan et al. proposed ‘two-hit’ model to explain the severity and variability of 16p12.2/16p12.1 phenotypes [47]. Our case also supports the ‘two hit’ CNV model and is associated with severe phenotype due to the presence of large 7.3 Mb CNV on 19p12q12. It should be noted that most individuals with recurrent 16p12.2 deletions are identified by Chromosomal Microarray Analysis (CMA) performed in the context of evaluation for developmental delay, cognitive impairment, and/or autism spectrum disorder [46].
Targeted gene sequencing approach has been used in many studies to identify the candidate genes for GS [12,48]. Targeted NGS panels in our patient revealed multiple SNVs reported in patients with neurological, ocular, facial, sensorineural/ear, cardiac, skeletal and urogenital anomalies. Variants in MYH14, PDE1C, ADGRV1, CDH23, and MYO3A genes have been reported in both syndromic and non-syndromic patients with mild to complete hearing loss [49-56], which can be associated with hearing loss and ear anomalies present in our proband. Mutations in ADGRV1 and CDH23 are also reported in patients with ocular anomalies/ Vision loss [53, 57, 58]. Pulmonary, cardiac, and skeletal abnormalities in our case such as absent lung lobe, bicuspid aortic valve, facial asymmetry, and missing left thumb can be associated with ADAMTS10 variants [59-61]. In the studies of Khandelwal et al. [62] and Cadieux-Dion [63], CHUK gene variants have been associated with severe skin and orofacial phenotypes such as ankyloblepharon, ectodermal dysplasia, cleft lip/palate, ectrodactyly, syndactyly, hypogammaglobulinemia, and growth delay, most of these features are present in proband. CSPP1 and TCTN2 variants have been reported in Joubert/Meckel syndrome like phenotypes involving CNS, urogenital, skeletal and pulmonary systems [64-67]. Based on the genotype/phenotype correlation and literature review, it is highly anticipated that these SNVs contribute to the patient’s complex clinical manifestation as a result of multiple gene hits. Phenotypic/Genotypic correlation has been summarized in Table-2.

Table 2.
Phenotypic variation among the typical Goldenhar Syndrome, Proband, 16p12.2 deletion, sSRC19 and variants identified by targeted NGS.
Table 2.
Phenotypic variation among the typical Goldenhar Syndrome, Proband, 16p12.2 deletion, sSRC19 and variants identified by targeted NGS.
| Goldenhar Syndrome Phenotypes and their Prevalence [4,10] | Proband Phenotypes | 16p12.2 Deletion Phenotypes [46,47] | sSRC19 Phenotype [37] |
Possible Variant association and Genes |
|---|---|---|---|---|
| Head and Face Anomalies/Hemifacial microsomia (~83.5%) | + | - | + | + (CHUK, TCTN2) |
| Ocular Anomalies (23%) | + | - | - | + (ADGRV1, CDH23, ADAMTS10, CHUK, CSPP1) |
| Ear Anomalies/Sensorineural defects/Deafness (~55%) | + | + | - | + (MYH14, PDE1C, ADGRV1, CDH23, MYO3A) |
| Cardiac Anomalies & Congenital Heart Defects (~20.16%) | + | + | + | + (ADAMST10) |
| Skeletal Anomalies (~27%) | + | + | + | + (ADAMTS10, CHUK, TCTN2) |
| Urogenital Anomalies (~11.5%) | + | + | - | + (CHUK, TCTN2) |
| Pulmonary Anomalies (~8%) | + | - | - | + (CSPP1, ADAMTS10) |
| Gastrointestinal Anomalies (~7%) | + | - | - |
- |
| Psychiatric & Behavioral Anomalies | + | + | - | + (CSPP1, TCTN2) |
| Intellectual Disability | - | + | + | + (CSPP1, TCTN2) |
| Developmental Delay (~11.5%) | - | + | + | + (CSPP1, TCTN2) |
| Central Nervous System Anomalies (~10%) | - | - | + | + (CHUK, CSPP1, TCTN2) |
Due to complex and overlapping heterogeneous phenotypes of GS patients, clinicians diagnose these patients based on the principal anomalies, and many of the patients are not assessed for additional phenotypes. CMA and Exome Sequencing (ES) are recommended as the first-tier test by American College of Medical Genetics and Genomics (ACMG) for patients with neurodevelopmental disorders and/or multiple congenital anomalies [68,69]. The utilization of CMA has emerged as a pivotal diagnostic tool for the identification of genetic anomalies associated with additional or variable phenotypes in many syndromic patients. CMA at early ages can help in the proper diagnosis and management of patients. Additional Whole Exome Sequencing (WES) could identify pathogenic variants associated with the patient’s additional phenotypes. Genetic counseling also plays an important role in such cases by providing the support required to address the uncertainties families face, guidance for future pregnancies, and medical intervention as well.
5. Conclusions
In this case, we report 16p12.2 deletion, ring chromosome 19 mosaicism and multiple single nucleotide variants in an adult patient who was diagnosed with Goldenhar Syndrome in infancy. The phenotypes associated with sSMC are extremely variable, from normal to severely abnormal depending on its origin and nature. The correlation of specific sSMC with distinct clinical phenotype has been reported for the isochromosome 18p syndrome (OMIM: 614290), isochromosome 12p (Pallister-Killian) syndrome (OMIM: 601803), and derivative 22 chromosomes (cat-eye) syndromes (OMIM: 115470). The patient in this study visited the genetic clinic to ascertain risks for future pregnancies. In general, the risk for an abnormal phenotype in prenatally ascertained de novo cases with any sSMC are given as approximately 13%, 7% for sSMC from chromosomes 13, 14, 21 or 22 and 28% for non-acrocentric chromosomes. Because the carrier shows an abnormal phenotype, as in our case, there is a high risk for an abnormal phenotype associated with mosaic ring chromosome 19 in prenatal outcome.
Author Contributions
Conceptualization, M.A.I and B.D.D.; methodology, S.A., B.D.D.; software, S.A., B.D.D.; formal analysis S.A., B.D.D.; resources, M.A.I.; Data curation, S.A., B.D.D.; writing—original draft preparation, B.D.D.; writing—review and editing, B.D.D., S.A., M.A.I, B.Z.; supervision, M.A.I, B.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
University of Rochester, New York Research Subjects Review Board approval #RSRB00019926.
Informed Consent Statement
Patient consent was waived due to minimal risk.
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the article and its supplementary information files or from the authors upon request.
Conflicts of Interest
The authors declare no conflict of interest. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention. The use of trademarks is for identification purposes only and does not imply endorsement by the U.S. Department of Health and Human Services.
References
- Tidke SC, Vagha JD, Vagha K, Lohiya S, Hampe P. An Atypical Presentation of Goldenhar Syndrome With Seizures: A Rare Case Report. Cureus. 2023;15(10):e46627. Published 2023 Oct 7. [CrossRef]
- Tuna EB, Orino D, Ogawa K, et al. Craniofacial and dental characteristics of Goldenhar syndrome: a report of two cases. J Oral Sci. 2011;53(1):121-124. [CrossRef]
- Kulkarni V, Shah MD, Parikh A. Goldenhar syndrome (a case report). J Postgrad Med. 1985;31(3):177-179.
- Bogusiak K, Puch A, Arkuszewski P. Goldenhar syndrome: current perspectives. World J Pediatr. 2017;13(5):405-415. [CrossRef]
- Maldonado J, Revuelta Barbero JM, Rodas A, et al. Endoscopic Endonasal Odontoidectomy for Upper Cervical Spine and Brainstem Decompression in a Patient With Goldenhar Syndrome: 2-Dimensional Operative Video. Oper Neurosurg (Hagerstown). Published online November 23, 2023. [CrossRef]
- Lima Mde D, Marques YM, Alves Sde M Jr, Ortega KL, Soares MM, Magalhães MH. Distraction osteogenesis in Goldenhar Syndrome: case report and 8-year follow-up. Med Oral Patol Oral Cir Bucal. 2007;12(7):E528-E531. Published 2007 Nov 1.
- Jayaprakasan SK, Waheed MD, Batool S, Pimentel Campillo J, Nageye ME, Holder SS. Goldenhar Syndrome: An Atypical Presentation With Developmental and Speech Delay. Cureus. 2023;15(3):e36225. Published 2023 Mar 16. [CrossRef]
- Wilson GN. Cranial defects in the Goldenhar syndrome. Am J Med Genet. 1983;14(3):435-443. [CrossRef]
- Madiyal, A., Babu, S.G., Ajila, V., Madi, M., & Bhat, S. (2018). Goldenhar syndrome: Report of two cases with review of literature. CHRISMED Journal of Health and Research, 5, 67 - 71.
- Beleza-Meireles A, Clayton-Smith J, Saraiva JM, Tassabehji M. Oculo-auriculo-vertebral spectrum: a review of the literature and genetic update. J Med Genet. 2014;51(10):635-645. [CrossRef]
- Strömland K, Miller M, Sjögreen L, et al. Oculo-auriculo-vertebral spectrum: associated anomalies, functional deficits and possible developmental risk factors. Am J Med Genet A. 2007;143A(12):1317-1325. [CrossRef]
- Tingaud-Sequeira A, Trimouille A, Sagardoy T, Lacombe D, Rooryck C. Oculo-auriculo-vertebral spectrum: new genes and literature review on a complex disease. J Med Genet. 2022 May;59(5):417-427. Epub 2022 Feb 2. [CrossRef] [PubMed]
- Li T, Sang H, Chu G, et al. Genotype-phenotype correlation in 75 patients with small supernumerary marker chromosomes. Mol Cytogenet. 2020;13:30. Published 2020 Jul 14. [CrossRef]
- Jafari-Ghahfarokhi H, Moradi-Chaleshtori M, Liehr T, Hashemzadeh-Chaleshtori M, Teimori H, Ghasemi-Dehkordi P. Small supernumerary marker chromosomes and their correlation with specific syndromes. Adv Biomed Res. 2015;4:140. Published 2015 Jul 27. [CrossRef]
- Paoloni-Giacobino A, Morris MA, Dahoun SP. Prenatal supernumerary r(16) chromosome characterized by multiprobe FISH with normal pregnancy outcome. Prenat Diagn. 1998 Jul;18(7):751-2. [PubMed]
- Tasse C, Majewski F, Böhringer S, et al. A family with autosomal dominant oculo-auriculo-vertebral spectrum. Clin Dysmorphol. 2007;16(1):1-7. [CrossRef]
- Vendramini-Pittoli S, Kokitsu-Nakata NM. Oculoauriculovertebral spectrum: report of nine familial cases with evidence of autosomal dominant inheritance and review of the literature. Clin Dysmorphol. 2009;18(2):67-77. [CrossRef]
- Tsai FJ, Tsai CH. Autosomal dominant inherited oculo-auriculo-vertebral spectrum: report of one family. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi. 1993;34(1):27-31.
- Goodin K, Prucka S, Woolley AL, et al. Familial transmission of oculoauriculovertebral spectrum (Goldenhar syndrome) is not due to mutations in either EYA1 or SALL1. Am J Med Genet A. 2009;149A(3):535-538. [CrossRef]
- Kaye CI, Martin AO, Rollnick BR, et al. Oculoauriculovertebral anomaly: segregation analysis. Am J Med Genet. 1992;43(6):913-917. [CrossRef]
- Rollnick BR. Oculoauriculovertebral anomaly: variability and causal heterogeneity. Am J Med Genet Suppl. 1988;4:41-53. [CrossRef]
- Rollnick BR, Kaye CI, Nagatoshi K, Hauck W, Martin AO. Oculoauriculovertebral dysplasia and variants: phenotypic charac-teristics of 294 patients. Am J Med Genet. 1987;26(2):361-375. [CrossRef]
- Descartes M. Oculoauriculovertebral spectrum with 5p15.33-pter deletion. Clin Dysmorphol. 2006;15(3):153-154. [CrossRef]
- Josifova DJ, Patton MA, Marks K. Oculoauriculovertebral spectrum phenotype caused by an unbalanced t(5;8)(p15.31;p23.1) rearrangement. Clin Dysmorphol. 2004;13(3):151-153. [CrossRef]
- Ladekarl S. Combination of Goldenhar’s syndrome with the Cri-Du-Chat syndrome. Acta Ophthalmol (Copenh). 1968;46(3):605-610. [CrossRef]
- Ballesta-Martínez MJ, López-González V, Dulcet LA, Rodríguez-Santiago B, Garcia-Miñaúr S, Guillen-Navarro E. Autosomal dominant oculoauriculovertebral spectrum and 14q23.1 microduplication. Am J Med Genet A. 2013;161A(8):2030-2035. [CrossRef]
- Ou Z, Martin DM, Bedoyan JK, et al. Branchiootorenal syndrome and oculoauriculovertebral spectrum features associated with duplication of SIX1, SIX6, and OTX2 resulting from a complex chromosomal rearrangement. Am J Med Genet A. 2008;146A(19):2480-2489. [CrossRef]
- Rooryck C, Stef M, Burgelin I, et al. 2.3 Mb terminal deletion in 12p13.33 associated with oculoauriculovertebral spectrum and evaluation of WNT5B as a candidate gene. Eur J Med Genet. 2009;52(6):446-449. [CrossRef]
- Abdelmoity AT, Hall JJ, Bittel DC, Yu S. 1.39 Mb inherited interstitial deletion in 12p13.33 associated with developmental delay. Eur J Med Genet. 2011;54(2):198-203. [CrossRef]
- Herman GE, Greenberg F, Ledbetter DH. Multiple congenital anomaly/mental retardation (MCA/MR) syndrome with Goldenhar complex due to a terminal del(22q). Am J Med Genet. 1988;29(4):909-915. [CrossRef]
- Xu J, Fan YS, Siu VM. A child with features of Goldenhar syndrome and a novel 1.12 Mb deletion in 22q11.2 by cytogenetics and oligonucleotide array CGH: is this a candidate region for the syndrome?. Am J Med Genet A. 2008;146A(14):1886-1889. [CrossRef]
- Digilio MC, McDonald-McGinn DM, Heike C, et al. Three patients with oculo-auriculo-vertebral spectrum and microdeletion 22q11.2. Am J Med Genet A. 2009;149A(12):2860-2864. [CrossRef]
- Tan TY, Collins A, James PA, et al. Phenotypic variability of distal 22q11.2 copy number abnormalities. Am J Med Genet A. 2011;155A(7):1623-1633. [CrossRef]
- Quintero-Rivera F, Martinez-Agosto JA. Hemifacial microsomia in cat-eye syndrome: 22q11.1-q11.21 as candidate loci for facial symmetry. Am J Med Genet A. 2013;161A(8):1985-1991. [CrossRef]
- Torti EE, Braddock SR, Bernreuter K, Batanian JR. Oculo-auriculo-vertebral spectrum, cat eye, and distal 22q11 microdeletion syndromes: a unique double rearrangement. Am J Med Genet A. 2013;161A(8):1992-1998. [CrossRef]
- de Ravel TJ, Legius E, Brems H, Van Hoestenberghe R, Gillis PH, Fryns JP. Hemifacial microsomia in two patients further sup-porting chromosomal mosaicism as a causative factor. Clin Dysmorphol. 2001;10(4):263-267. [CrossRef]
- Melis D, Genesio R, Del Giudice E, et al. Selective cognitive impairment and tall stature due to chromosome 19 supernumerary ring. Clin Dysmorphol. 2012;21(1):27-32. [CrossRef]
- Novelli A, Ceccarini C, Bernardini L, et al. Pure trisomy 19p syndrome in an infant with an extra ring chromosome. Cytogenet Genome Res. 2005;111(2):182-185. [CrossRef]
- Vraneković J, Brajenović-Milić B, Modrusan-Mozetić Z, Babić I, Kapović M. Severe psychomotor retardation in a boy with a small supernumerary marker chromosome 19p. Cytogenet Genome Res. 2008;121(3-4):298-301. [CrossRef]
- Tercanli S, Hösli I, Berlinger A, Beyer R, Achermann J, Holzgreve W. Prenatal diagnosis of a partial trisomy 19q. Prenat Diagn. 2000;20(8):663-665. [CrossRef]
- Eichler EE, Hoffman SM, Adamson AA, et al. Complex beta-satellite repeat structures and the expansion of the zinc finger gene cluster in 19p12. Genome Res. 1998;8(8):791-808. [CrossRef]
- Hoovers JM, Mannens M, John R, et al. High-resolution localization of 69 potential human zinc finger protein genes: a number are clustered. Genomics. 1992;12(2):254-263. [CrossRef]
- Klug, A., Schwabe, J. W. R. Zinc fingers. FASEB J. 9, 597-604 (1995) . [CrossRef]
- Bu S, Lv Y, Liu Y, Qiao S, Wang H. Zinc Finger Proteins in Neuro-Related Diseases Progression. Front Neurosci. 2021;15:760567. Published 2021 Nov 18. [CrossRef]
- Gusic M, Schottmann G, Feichtinger RG, et al. Bi-Allelic UQCRFS1 Variants Are Associated with Mitochondrial Complex III De-ficiency, Cardiomyopathy, and Alopecia Totalis. Am J Hum Genet. 2020;106(1):102-111. [CrossRef]
- Girirajan S, Pizzo L, Moeschler J, et al. 16p12.2 Recurrent Deletion. 2015 Feb 26 [Updated 2018 Sep 13]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK274565/.
- Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, Vives L, Walsh T, McCarthy SE, Baker C, Mefford HC, Kidd JM, Browning SR, Browning BL, Dickel DE, Levy DL, Ballif BC, Platky K, Farber DM, Gowans GC, Wetherbee JJ, Asamoah A, Weaver DD, Mark PR, Dickerson J, Garg BP, Ellingwood SA, Smith R, Banks VC, Smith W, McDonald MT, Hoo JJ, French BN, Hudson C, Johnson JP, Ozmore JR, Moeschler JB, Surti U, Escobar LF, El-Khechen D, Gorski JL, Kussmann J, Salbert B, Lacassie Y, Biser A, McDonald-McGinn DM, Zackai EH, Deardorff MA, Shaikh TH, Haan E, Friend KL, Fichera M, Romano C, Gécz J, DeLisi LE, Sebat J, King MC, Shaffer LG, Eichler EE. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010 Mar;42(3):203-9. Epub 2010 Feb 14. [CrossRef] [PubMed] [PubMed Central]
- Zamariolli M, Colovati M, Moysés-Oliveira M, Nunes N, Caires Dos Santos L, Alvarez Perez AB, Bragagnolo S, Melaragno MI. Rare single-nucleotide variants in oculo-auriculo-vertebral spectrum (OAVS). Mol Genet Genomic Med 2019;7:e00959. [CrossRef]
- Choi BO, Kang SH, Hyun YS, Kanwal S, Park SW, Koo H, Kim SB, Choi YC, Yoo JH, Kim JW, Park KD, Choi KG, Kim SJ, Züchner S, Chung KW. A complex phenotype of peripheral neuropathy, myopathy, hoarseness, and hearing loss is linked to an autosomal dominant mutation in MYH14. Hum Mutat. 2011 Jun;32(6):669-77. Epub 2011 Apr 7. [CrossRef] [PubMed] [PubMed Central]
- Kim, B. J., Kim, A. R., Han, J. H., Lee, C., Oh, D. Y., & Choi, B. Y. (2017). Discovery of MYH14 as an important and unique deafness gene causing prelingually severe autosomal dominant nonsyndromic hearing loss. The Journal of Gene Medicine, 19(4). [CrossRef]
- Wang, L., Feng, Y., Yan, D., Qin, L., Grati, M., Mittal, R., Li, T., Sundhari, A. K., Liu, Y., Chapagain, P., Blanton, S. H., Liao, S., & Liu, X. (2018). A dominant variant in the PDE1C gene is associated with nonsyndromic hearing loss. Human Genetics, 137(6–7), 437–446. [CrossRef]
- Yoon, P. , Sumalde, A. , Ray, D. , Newton, S. , Cass, S. , Chan, K. & Santos-Cortez, R. (2020). Novel Variants in Hearing Loss Genes and Associations With Audiometric Thresholds in a Multi-ethnic Cohort of US Patients With Cochlear Implants. Otology & Neurotology, 41 (7), 978-985. [CrossRef]
- Pater, J. A., Green, J., O’Rielly, D. D., Griffin, A., Squires, J., Burt, T., Fernandez, S., Fernandez, B., Houston, J., Zhou, J., Roslin, N. M., & Young, T. L. (2019). Novel Usher syndrome pathogenic variants identified in cases with hearing and vision loss. BMC Medical Genetics, 20(1), 68–68. [CrossRef]
- Usami, S., Isaka, Y., Miyagawa, M., & Nishio, S. (2022). Variants in CDH23 cause a broad spectrum of hearing loss: from non-syndromic to syndromic hearing loss as well as from congenital to age-related hearing loss. Human Genetics, 141(3–4), 903–914. [CrossRef]
- Doll, J., Hofrichter, M. A. H., Bahena, P., Heihoff, A., Segebarth, D., Müller, T., Dittrich, M., Haaf, T., & Vona, B. (2020). A novel missense variant in MYO3A is associated with autosomal dominant high-frequency hearing loss in a German family. Molecular Genetics & Genomic Medicine, 8(8), e1343-n/a. [CrossRef]
- Bueno, A. S., Nunes, K., Dias, A. M. M., Alves, L. U., Mendes, B. C. A., Sampaio-Silva, J., Smits, J., Yntema, H. G., Meyer, D., Lezirovitz, K., & Mingroni-Netto, R. C. (2022). Frequency and origin of the c.2090T>G p.(Leu697Trp) MYO3A variant associated with autosomal dominant hearing loss. European Journal of Human Genetics: EJHG, 30(1), 13–21. [CrossRef]
- González-Del Pozo, M., Fernández-Suárez, E., Martín-Sánchez, M., Bravo-Gil, N., Méndez-Vidal, C., Rodríguez-De La Rúa, E., Borrego, S., & Antiñolo, G. (2020). Unmasking Retinitis Pigmentosa complex cases by a whole genome sequencing algorithm based on open-access tools: Hidden recessive inheritance and potential oligogenic variants. Journal of Translational Medicine, 18(1), 73–73. [CrossRef]
- Brodie, K. D., Moore, A. T., Slavotinek, A. M., Meyer, A. K., Nadaraja, G. S., Conrad, D. E., Weinstein, J. E., & Chan, D. K. (2021). Genetic Testing Leading to Early Identification of Childhood Ocular Manifestations of Usher Syndrome. The Laryngoscope, 131(6), E2053–E2059. [CrossRef]
- Steinkellner, H., Etzler, J., Gogoll, L., Neesen, J., Stifter, E., Brandau, O., & Laccone, F. (2015). Identification and molecular characterisation of a homozygous missense mutation in the ADAMTS10 gene in a patient with Weill–Marchesani syndrome. European Journal of Human Genetics : EJHG, 23(9), 1186–1191. [CrossRef]
- Levitas, A., Aspit, L., Lowenthal, N., Shaki, D., Krymko, H., Slanovic, L., Yagev, R., & Parvari, R. (2023). A Novel Mutation in the ADAMTS10 Associated with Weil–Marchesani Syndrome with a Unique Presentation of Developed Membranes Causing Severe Stenosis of the Supra Pulmonic, Supramitral, and Subaortic Areas in the Heart. International Journal of Molecular Sciences, 24(10), 8864-. [CrossRef]
- Marzin P, Rondeau S, Alessandri J, et al. Weill-Marchesani syndrome: natural history and genotype-phenotype correlations from 18 news cases and review of literature Journal of Medical Genetics Published Online First: 21 September 2023. [CrossRef]
- Khandelwal, K. D., Ockeloen, C. W., Venselaar, H., Boulanger, C., Brichard, B., Sokal, E., Pfundt, R., Rinne, T., van Beusekom, E., Bloemen, M., Vriend, G., Revencu, N., Carels, C. E. L., van Bokhoven, H., & Zhou, H. (2017). Identification of a de novo variant in CHUK in a patient with an EEC/AEC syndrome-like phenotype and hypogammaglobulinemia. American Journal of Medical Genetics. Part A, 173(7), 1813–1820. [CrossRef]
- Cadieux-Dion, M., Safina, N. P., Engleman, K., Saunders, C., Repnikova, E., Raje, N., Canty, K., Farrow, E., Miller, N., Zellmer, L., & Thiffault, I. (2018). Novel heterozygous pathogenic variants in CHUK in a patient with AEC-like phenotype, immune deficiencies and 1q21.1 microdeletion syndrome: A case report. BMC Medical Genetics, 19(1), 41–41. [CrossRef]
- Wei, C., Zhang, H., Fu, M., Ye, J., & Yao, B. (2024). Novel compound heterozygous variants in the CSPP1 gene causes Joubert syndrome: case report and literature review of the CSPP1 gene’s pathogenic mechanism. Frontiers in Pediatrics, 12, 1305754-. [CrossRef]
- Ben-Omran, T., Alsulaiman, R., Kamel, H., Shaheen, R., & Alkuraya, F. S. (2015). Intrafamilial clinical heterogeneity of CSPP1-related ciliopathy. American Journal of Medical Genetics. Part A, 167A(10), 2478–2480. [CrossRef]
- Huppke, P., Wegener, E., Böhrer-Rabel, H., Bolz, H. J., Zoll, B., Gärtner, J., & Bergmann, C. (2015). Tectonic gene mutations in patients with Joubert syndrome. European Journal of Human Genetics : EJHG, 23(5), 616–620. [CrossRef]
- Fang, L., Wang, L., Yang, L., Xu, X., Pei, S., & Wu, D. (2023). Novel variants identified in five Chinese families with Joubert Syndrome: a case report. BMC Medical Genomics, 16(1), 1–221. [CrossRef]
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, Faucett WA, Feuk L, Friedman JM, Hamosh A, Jackson L, Kaminsky EB, Kok K, Krantz ID, Kuhn RM, Lee C, Ostell JM, Rosenberg C, Scherer SW, Spinner NB, Stavropoulos DJ, Tepperberg JH, Thorland EC, Vermeesch JR, Waggoner DJ, Watson MS, Martin CL, Ledbetter DH. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010 May 14;86(5):749-64. [CrossRef] [PubMed] [PubMed Central]
- Srivastava, S., Love-Nichols, J.A., Dies, K.A. et al. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med 21, 2413–2421 (2019). [CrossRef]
Figure 1.
aCGH showing 471 kb deletion in the short arm of chromosome 16 at p12.2 region (21959891-22430592). The deleted region is zoomed in on the right showing EEF2K, POLR3E, UQCRC2, and CDR genes.
Figure 1.
aCGH showing 471 kb deletion in the short arm of chromosome 16 at p12.2 region (21959891-22430592). The deleted region is zoomed in on the right showing EEF2K, POLR3E, UQCRC2, and CDR genes.
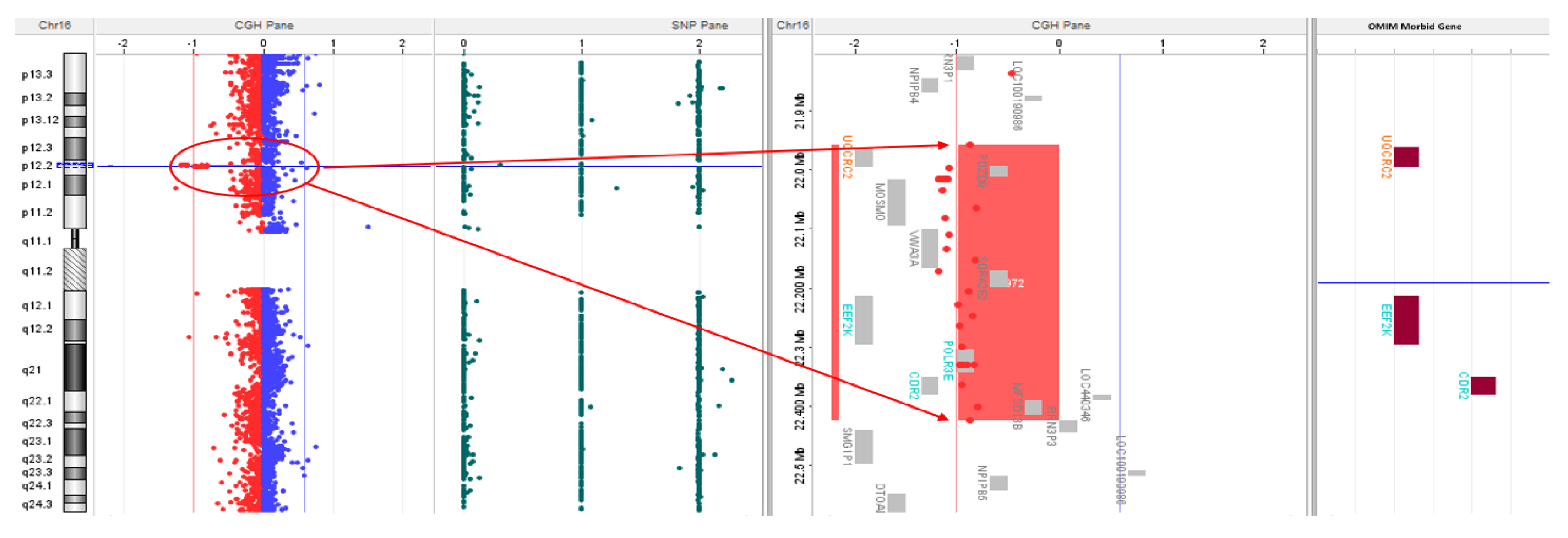
Figure 2.
aCGH showing 7.3 Mb duplication extending from 19p12-19q12 (22470583-29795821). The duplicated region is zoomed in on the right showing multiple ZNF genes and UQCRFS1 gene.
Figure 2.
aCGH showing 7.3 Mb duplication extending from 19p12-19q12 (22470583-29795821). The duplicated region is zoomed in on the right showing multiple ZNF genes and UQCRFS1 gene.
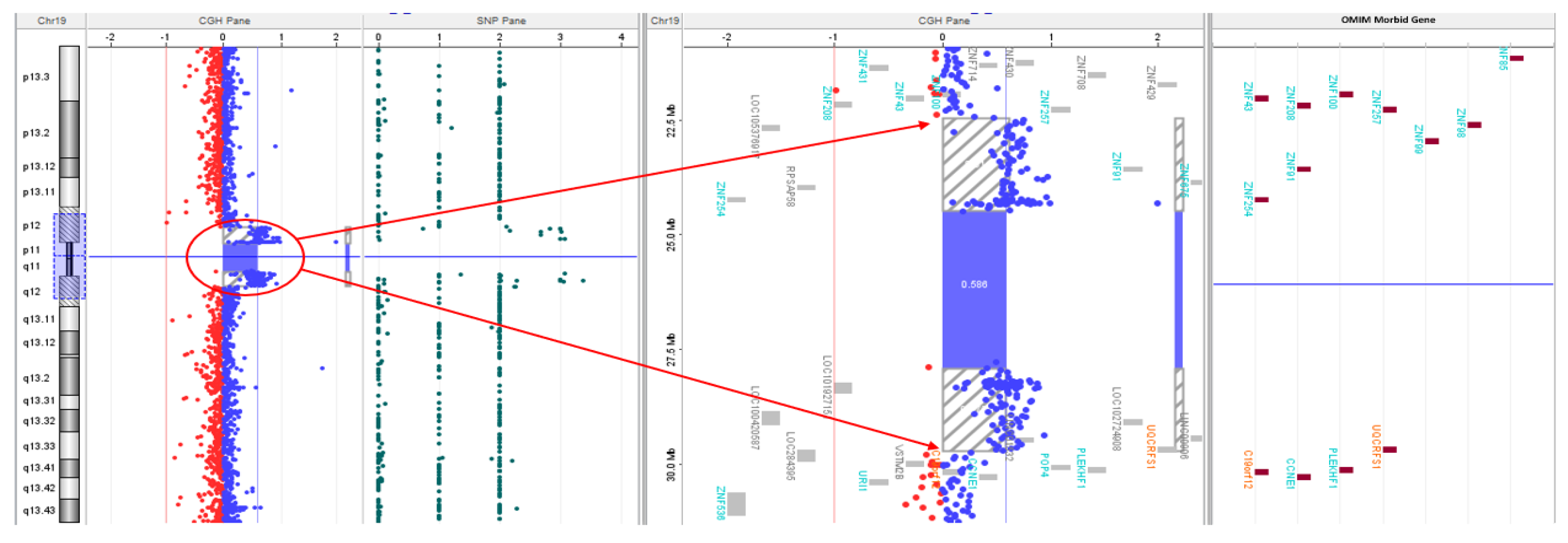
Figure 3.
FISH analysis using BAC probe RP11-141O15 (16p12.2p12.2)-spectrum red and TelVysion 16p-spectrum green shows one normal chromosome 16 (one red signal and one green signal) and a deletion of 16p12.2 chromosome (only one green signal) in proband’s metaphase.
Figure 3.
FISH analysis using BAC probe RP11-141O15 (16p12.2p12.2)-spectrum red and TelVysion 16p-spectrum green shows one normal chromosome 16 (one red signal and one green signal) and a deletion of 16p12.2 chromosome (only one green signal) in proband’s metaphase.
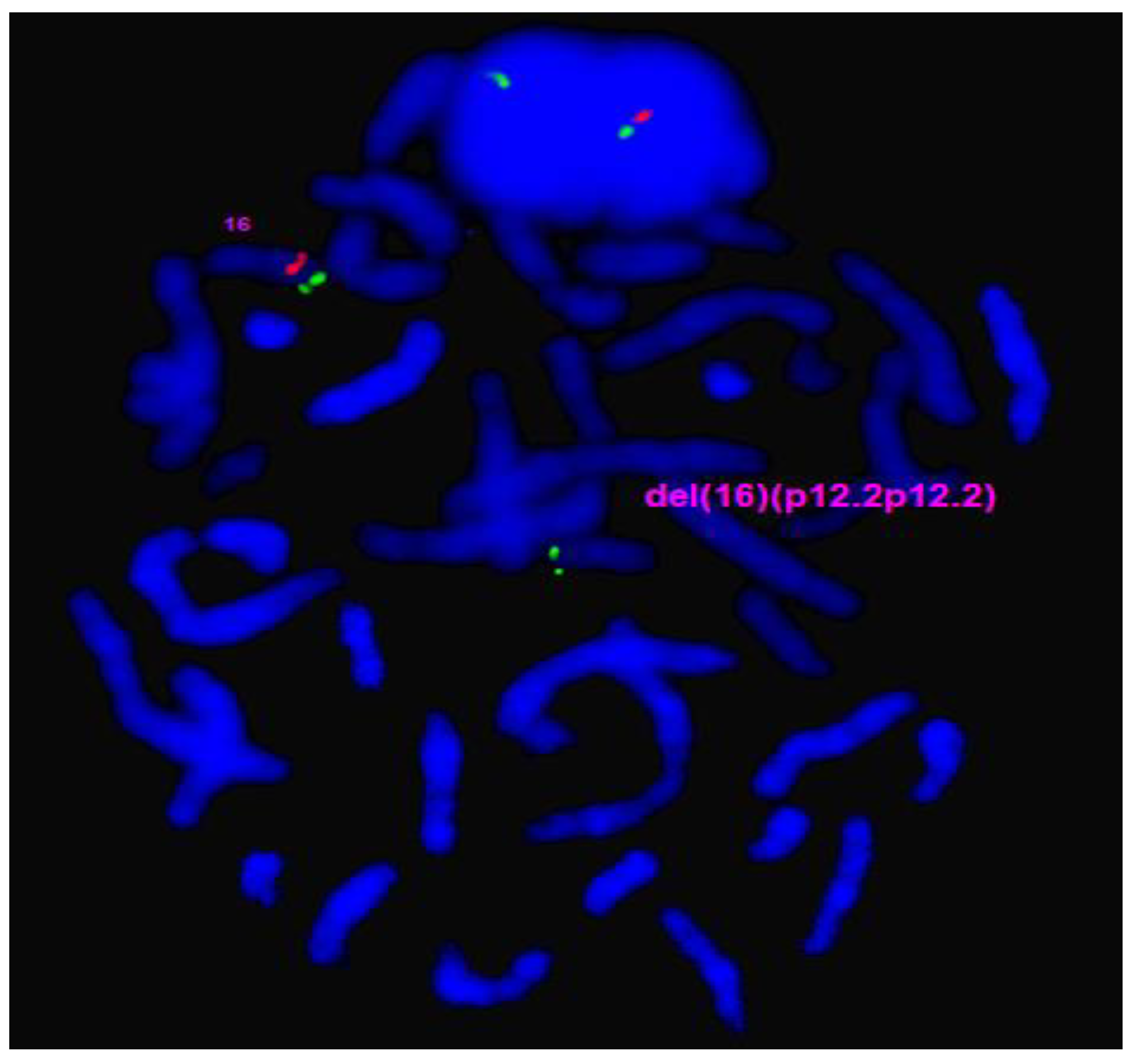
Figure 4.
FISH analysis using BAC probe RP11-359H18 (19p12)-spectrum red and RP11-714C4 (19q12)- spectrum green show two normal chromosomes 19. (a) Two red and two green signals) and an arrow pointing to ring chromosome 19 (r19) with extra red and green signals in proband’s metaphase. (b) Ring chromosome confirmed on inverted DAPI.
Figure 4.
FISH analysis using BAC probe RP11-359H18 (19p12)-spectrum red and RP11-714C4 (19q12)- spectrum green show two normal chromosomes 19. (a) Two red and two green signals) and an arrow pointing to ring chromosome 19 (r19) with extra red and green signals in proband’s metaphase. (b) Ring chromosome confirmed on inverted DAPI.
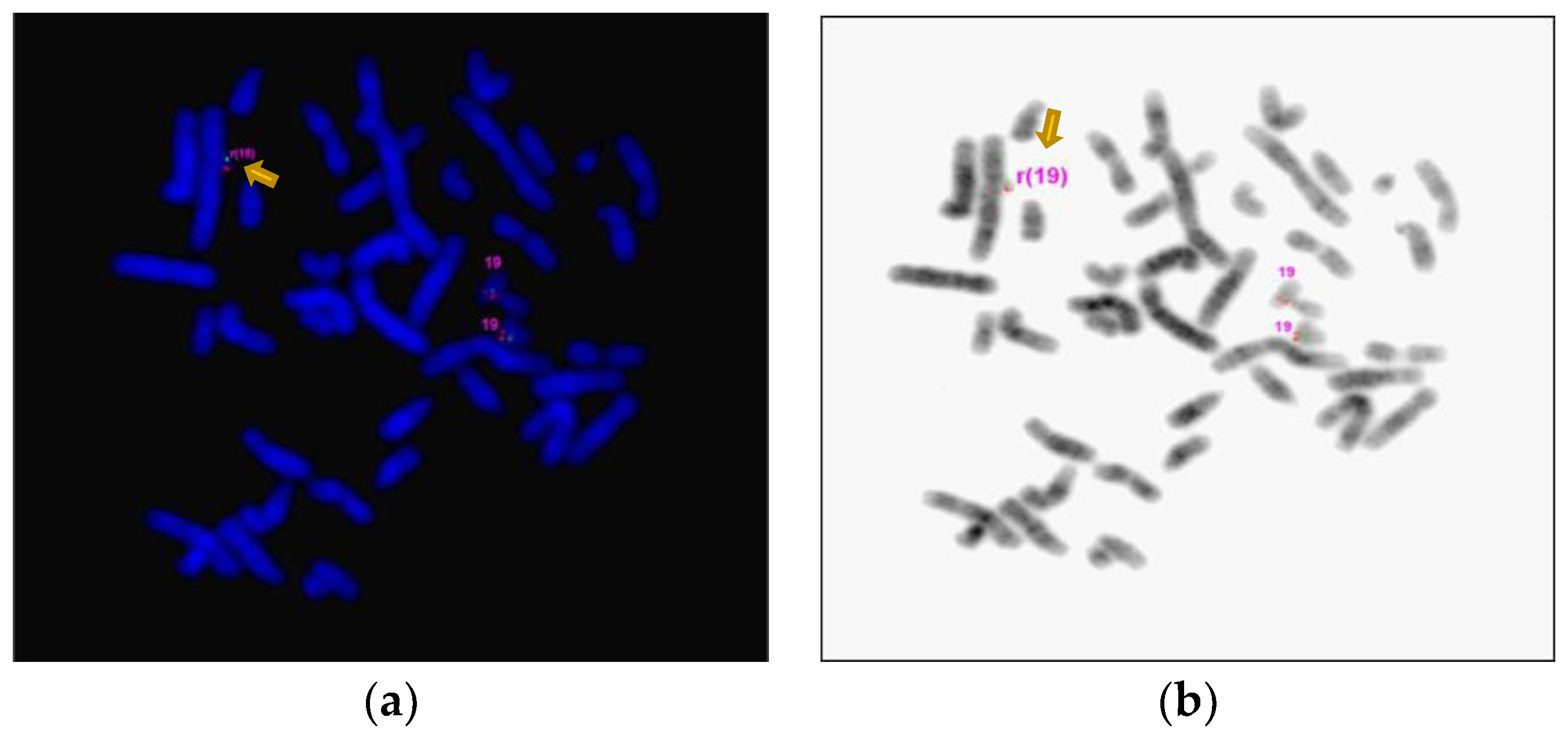
Table 1.
Variants identified by targeted Next Generation Sequencing (NGS) Panels. a) Hereditary Hearing Loss and Deafness Panel. b) Limb and Digital Malformation Panel:.
Table 1.
Variants identified by targeted Next Generation Sequencing (NGS) Panels. a) Hereditary Hearing Loss and Deafness Panel. b) Limb and Digital Malformation Panel:.
| Gene Transcript | Mode of Inheritance, OMIM ID | DNA variants, Predicted effects, Zygosity |
Highest Allele Frequency in a gnomAD Population |
|---|---|---|---|
|
MYH14, NM_001145809.1 |
Autosomal Dominant, 608568 |
c.4001C>T, p.Ala1334Val, Heterozygous |
0.0071% East Asian |
|
PDE1C, NM_001322059.1 |
Autosomal Dominant, 602987 |
c.8C>T, p.Ser3Leu, Heterozygous |
0.026% European (Non-Finnish) |
|
ADGRV1, NM_032119.3 |
Autosomal Recessive, 602851 |
c.15502A>G, p.Thr5168Ala, Heterozygous |
Not present |
|
CDH23, NM_022124.5 |
Autosomal Recessive, 605516 |
c.8772C>A, p.Ser2924Arg, Apparently Mosaic |
0.00089% European (Non-Finnish) |
|
MYO3A, NM_017433.4 |
Autosomal Recessive, 606808 |
c.484G>A, p.Gly162Ser, Heterozygous |
Not present |
| Gene Transcript |
Mode of Inheritance, OMIM ID |
DNA variants, Predicted effects, Zygosity |
Highest Allele Frequency in a gnomAD Population |
|
ADAMTS10, NM_030957.3 |
Autosomal Recessive, 608990 |
c.506G>A, p.Thr5168Ala, Heterozygous |
Not present |
|
CHUK, NM_001278.4 |
Autosomal Recessive, 600664 |
c.1294C>T, p.Ala1334Val, Heterozygous |
0.0017% European (Non-Finnish) |
|
CSPP1, NM_024790.6 |
Autosomal Recessive, 611654 |
c.1972A>G, p.Arg658Gly, Heterozygous |
0.092% (Admixed American) |
|
TCTN2, NM_024809.4 |
Autosomal Recessive, 613846 |
c.160G>A, p.Val54Met, Heterozygous |
0.0061% (African/African-American) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



