
Submitted:
17 August 2024
Posted:
20 August 2024
You are already at the latest version
Abstract
Thoracic aortic and arterial aneurysms are cardiovascular conditions associated with life-threatening endpoints including dissection and rupture. The genetic basis underlying the development these disease states is an evolving field, and the reporting of variants associated with aneurysms in patients are essential for advancing screening and diagnostic tools. Among all patients in our program who underwent clinical genetic screening (N=145) for arterial aneurysm and dissection disorders, we identified three patients with single nucleotide variants in the TNXB gene. This gene has known roles in collagen fibrillogenesis and classic Ehlers Danlos Syndrome. This is one of the first reports to associate this gene with aortopathy/arteriopathy and advances the literature relating extracellular matrix genes to arterial aneurysm development.
Keywords:
TNXB
; aortic aneurysm
; carotid aneurysm
; cerebral aneurysm
; extracellular matrix
; genetics
; single nucleotide variants
1. Introduction
Aneurysms occur secondary to the weakening of blood vessels, resulting in dilatation of all three layers of the arterial wall. Aneurysms may form in any blood vessel, including the carotid and cerebral arteries, but are most frequently found in the aorta [1]. Thoracic aortic aneurysms (TAAs) are a relatively common clinical cardiovascular finding with an estimated prevalence of 6 persons per 100,000 per year. TAAs vary in symptoms, with most being clinically asymptomatic until dissection or rupture occurs [2]. These complications are potentially fatal, and 95% of all cases remain undetected until these problems present [3]. Aneurysms of the ascending thoracic aorta are typically due to cystic medial degeneration, histologically appearing as degeneration of elastic fibers and dropout of smooth muscle cells. This leads to the characteristic aortic wall weakening and subsequent aortic dilatation [4].
Genetic influence has been heavily researched in TAA development, and as many as 20% of impacted individuals have a first-degree relative with a TAA [5]. Further, this 20% approximation is thought to be an underestimation due to the lack of routine imaging in family members. For many seemingly isolated TAA cases, the proband may be the first in a potential line of inheritance [6]. Although significant progress has been made in determining the genetic risk factors for TAA development, further research is required to improve our risk stratification and diagnostic capabilities.
There are three known categories of TAAs; sporadic, familial non-syndromic, and familial syndromic [7]. Sporadic TAAs are secondary to de novo gene variants and are primarily associated with hypertension and aging [8]. Syndromic TAAs, responsible for roughly 20% of cases, includes aneurysms secondary to Marfan Syndrome (variants in the FBN1 gene), Ehlers-Danlos syndrome (variants in COL3A1, TGFßR1, or TGFßR2) or Loeys-Dietz Syndrome (variants in TGFßR1 or TGFßR2) [7]. Currently, 11 genes have been established as high risk for TAA formation in patients with or without syndromic features. Variants in these genes impact vascular smooth muscle contraction and/or metabolism, transforming growth factor ß formation, or extracellular matrix (ECM) biology. These genes include ACTA2, MYLK, COL3A1, PRKG1, SMAD3, FBN1, MYH11, TGFBR1, TGFB2, LOX, and TGFBR2 [9]. Familial non-syndromic variants are acquired in an autosomal dominant inheritance pattern [7].
2. Case Description
2.1. Case 1
A 57-year-old female with a prior medical history of hypertension and essential tremors controlled on low dose propranolol was referred to our practice for evaluation of an ascending aortic aneurysm of 42 mm found on computed tomography angiography (CTA). Our patient did not have any clinical cardiac symptoms. A transthoracic echocardiogram (TTE) revealed a structurally normal heart with an EF of 60-65%, no aortic valve insufficiency and a similar ascending aortic measurement of 42 mm. The patient was offered clinical genetic testing to exclude high-risk pathogenic variants that would impact longitudinal clinical management.
2.2. Case 2
A 48-year-old male with a prior medical history of hypertension and obesity was referred to our practice for evaluation of a 45 mm ascending aortic aneurysm found on CTA. The patient was symptom free. Our patient was offered clinical genetic testing to exclude high-risk pathogenic variants in a panel of genes associated with aneurysm development that would impact longitudinal clinical management.
2.3. Case 3
A 61-year-old female was referred to us for evaluation of dizziness and near syncope. During our evaluation we confirmed pulsatile tinnitus and a recent cerebellar infarction with a subsequent CT angiographic imaging revealing a right paraclinoid internal carotid aneurysm. An invasive cerebral angiogram demonstrated a mildly lobulate saccular aneurysm projecting from the proximal right ophthalmic ICA segment measuring 4.1 x 5.2 mm with a 3.8 mm neck. She subsequently underwent a successful flow diversion embolization of the right ophthalmic ICA aneurysm utilizing a Surpass Evolve embolization device deployed between the mid right horizontal cavernous ICA segment to the distal right communicating ICA segment. All symptoms resolved and a follow-up cerebral angiogram six months later demonstrated a stable result. Our patient was offered clinical genetic testing to assess for the presence of high-risk, pathogenic variants that would influence our long-term surveillance and management.
3. Material and Methods
A commercial panel of 35 genes that have been reported as associated with extracellular matrix disorders were utilized to assess for pathogenic variants in patients. This panel of genes investigates variants in 35 genes including: “ZNF469, PRKG1, SLC2A10, PRDM5, BGN, MAT2A, SMAD3, NOTCH1, FOXE3, TGFB2, MED12, EFEMP2, COL3A1, COL5A1, ACTA2, SKI, FLNA, CHST14, FKBP14, MYH11, SMAD4, TGFBR2, FBN1, COL1A1, COL1A2, LOX, MYLK, PLOD1, TNXB, CBS, FBN2, TGFB3, MFAP5, COL5A2.” Genetic analysis is offered to every patient at our practice either through request or physician suggestion. To identify variants, Sanger and/or Next Generation sequencing is used. This identifies sequencing of coding domains, untranslated regions, and intronic domains. The NCBI sequence for TNXB was used for analysis of the gene throughout this report. Various methods including gross deletion and gross duplication were investigated to determine the gene copy number for covered exons and untranslated regions of the aforementioned genes. Bait capture was used to enrich coding exon sequences via “biotinylated oligonucleotide probes and subsequent polymerase chain reaction and sequencing and utilizing NCBI reference sequences [10].” Sanger sequencing was additionally used for those regions that were missing or for those with poor and not sufficient read depth coverage for reliable detection of variants [10]. Standard clinical genetic counseling was provided to our patients ad we assessed the population frequencies of discovered variants in ClinVar and GnomAD.
4. Genetic Results
4.1. Case 1
We identified a variant of unknown significance (VUS), p.T982I (c.2945C>T) in the TNXB gene. This variant is located in exon 6 of the gene and results in a C to T substitution at the nucleotide position 2945 (Figure 1). This results in a threonine at codon 982 being replaced by isoleucine, an amino acid that has relatively similar properties. Clinical significance is unclear, however, in silico analysis has predicted that the missense variant has a deleterious effect on protein structure and/or function [11]. No other pathogenic variant, VUS, gross deletions, or gross duplications were isolated in the 34 other genes analyzed.
4.2. Case 2
A heterozygous variant for c.4535_4552del18 (p.D1512_V1517del) VUS was isolated in coding exon 11 of the TNXB gene (Figure 2). This results from an in-frame deletion at nucleotide positions 4535 to 4552, resulting of a deletion of six amino acids (DGQPQV) between codons 1512 and 1517. This amino acid region is poorly conserved in vertebrate species, and the variant is predicted to be deleterious by in silico analysis.
4.3. Case 3
We identified a pathogenic variant, c.8510_8511delCT located on exon 24 of the TNXB gene (Figure 3). This results from a deletion of two nucleotides at positions 8510 to 8511, resulting in a translational frameshift with a predicted alternate stop codon (p.P2837Rfs*19). This is predicted to disrupt function by either mRNA decay or truncation resulting in a premature protein product. This variant is expected to cause autosomal recessive classic-like EDS.
4.4. Frequency of Variants of Unknown Significance
We analyzed each variant in ClinVar and GnomAD, which predicted the likelihood of tolerance for each individual variant. Although the precise frequency of each variant is not known due to the novelty of these variants, continued collection of data will allow for a more accurate prediction later.
5. Discussion: TNXB
Identifying genetic variants responsible for the development of familial non-syndromic TAAs is necessary to further our understanding of TAA pathogenesis and to improve our risk-stratification and diagnostic tools. Variants in genes that impact the contraction of vascular smooth muscle, transforming growth factor beta (TGF-B) formation, or ECM adhesion have been isolated as major categories behind TAA development [9]. Understanding the genetic causes behind non-syndromic TAA development has continued to improve due to increased reporting of genetic variants in individuals diagnosed with aneurysms [6]. Variants in the TNXB gene, a key player in ECM structure and function, were isolated in three patients with aneurysms.
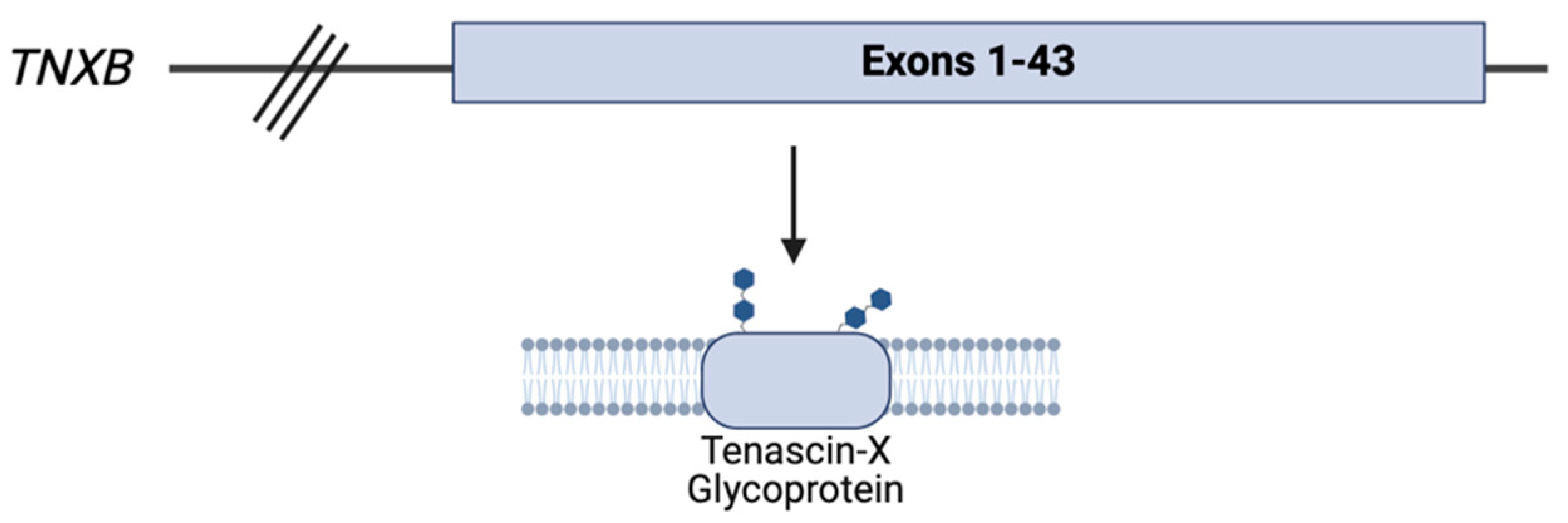
TNXB is located on chromosome 6p21.3 and contains 43 coding exons. Variants in TNXB are generally inherited in an autosomal recessive pattern and have been associated with classic-like Ehlers-Danlos Syndrome (EDS) [13]. TNXB haploinsufficiency has previously been identified in women with incomplete penetrance and is characterized by chronic musculoskeletal pain and hypermobile EDS [14].
The TNXB gene encodes the tenascin-X (TNX) glycoprotein, a member of the tenascin family of ECM glycoproteins that is highly expressed in the cardiac, musculoskeletal, and dermal tissues. TNX, the largest glycoprotein in the tenascin family at 450 kDa, is a vital component of maintaining the structural function of the ECM through the regulation of collagen fibrillogenesis [13,15]
Deficiency of the TNX glycoprotein results in a recessive form of classical-like EDS. Affected individuals have generalized joint hypermobility, hyperextensible skin, and easy bruising. However, aside from these characteristic cEDS findings, patients impacted by TNX deficiency do not have the same atrophic scarring generally seen in cEDS. Other distinguishing findings include anomalies of the feet and hands, lower extremity edema in the absence of heart failure, muscle weakness, and axonal polyneuropathy. Notably, tissue fragility has been reported in these patients [16].
TNXB is strongly expressed in vascular walls and has been recognized as a potential biomarker for vascular disease [17]. Previously, an abstract has been published reporting associations of TNXB variants with aneurysms and/or dissections in 5 individuals [18], with no other reports published to date. As such, this report aims to contribute to the current literature to promote further mechanistic research behind TNXB and its potential association with TAA development.
Limitations to our report include the limited sample size and, thus, insufficient data to imply or investigate a causal role between TNXB and TAA development. This report instead aims to advance the current literature behind the gene and promote future mechanistic studies and investigation for a causal role with larger sample sizes.
6. Conclusion
TAAs are generally pathologies that are associated with potentially fatal complications including dissection and rupture. This report of three subjects adds to the current literature by describing a potential association between variants in TNXB and aneurysm development and advances the understanding of how disruptions in extracellular matrix proteins and their regulators may lead to vascular aneurysms. Detailed mechanistic research though larger clinical, translational, and transgenic animal model investigation will help elucidate the role of TNXB in aneurysm pathogenesis and doing so will help to advance diagnostic tools to more effectively risk stratify patients in this dangerous disease state.
Author Contributions
Conceptualization, M.S.; methodology, M.S.; images, P.M..; validation, M.S., P.M, G.B, K.S..; formal analysis, M.S., P.M, G.B, K.S.; investigation, M.S., P.M, G.B, K.S.; resources, M.S..; writing—original draft preparation, P.M, G.B, K.S.; writing—review and editing, M.S., P.M, G.B, K.S.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Potishman Foundation through a generalized research restricted grant to the Sathyamoorthy Laboratory at the Burnett School of Medicine at TCU. Funding number: Potishman Foundation: 02. The Foundation had no role in this research.
Institutional Review Board Statement
Due to the number of participants and each subjects’ provision of written informed consent, ethical review and approval were waived for this study per TCU IRB guidelines. These consents are on file at CCMS-FW.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to patient privacy considerations.
Acknowledgments
The authors wish to acknowledge staff at CCMS-FW for helping coordinate patient visits and consents. The authors recognize the neuro-interventional expertise and management of Dr. Matt Fiesta.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Salameh MJ, Black JH, Ratchford EV. Thoracic aortic aneurysm. Vascular Medicine. 2018;23(6):573-578. [CrossRef]
- Senser EM, Misra S, Henkin S. Thoracic Aortic Aneurysm. Cardiology Clinics. 2021;39(4):505-515. [CrossRef]
- Martin-Blazquez A, Heredero A, Aldamiz-Echevarria G, Martin-Lorenzo M, Alvarez-Llamas G. Non-syndromic thoracic aortic aneurysm: cellular and molecular insights. The Journal of Pathology. 2021;254(3):229-238. [CrossRef]
- Isselbacher EM. Thoracic and Abdominal Aortic Aneurysms. Circulation. 2005;111(6):816-828. [CrossRef]
- Isselbacher EM, Lino Cardenas CL, Lindsay ME. Hereditary Influence in Thoracic Aortic Aneurysm and Dissection. Circulation. 2016;133(24):2516-2528. [CrossRef]
- Ostberg N, Zafar M, Ziganshin B, Elefteriades J. The Genetics of Thoracic Aortic Aneurysms and Dissection: A Clinical Perspective. Biomolecules. 2020;10(2):182. [CrossRef]
- Rega S, Farina F, Bouhuis S, et al. Multi-omics in thoracic aortic aneurysm: the complex road to the simplification. Cell & Bioscience. 2023;13(1). [CrossRef]
- El-Hamamsy I, Yacoub MH. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nature Reviews Cardiology. 2009;6(12):771-786. [CrossRef]
- Pinard A, Jones GT, Milewicz DM. Genetics of Thoracic and Abdominal Aortic Diseases. Circulation Research. 2019;124(4):588-606. [CrossRef]
- Antolik C. TAADNext: Analyses of 35 Genes Associated with Thoracic Aortic Aneurysms and Dissections. Ambry Genetics; Aliso Viejo, CA, USA: 2022. pp. 1–6.
- VCV001215797.11 - ClinVar - NCBI. www.ncbi.nlm.nih.gov. Accessed May 27, 2024. https://www.ncbi.nlm.nih.gov/clinvar/variation/1215797/.
- Biorender. BioRender. Home. Published 2019. Accessed May 27, 2024. https://biorender.com.
- O’Connell M, Burrows NP, Van Vlijmen-Willems MJJ, Clark SM, Schalkwijk J. Tenascin-X deficiency and Ehlers-Danlos syndrome: a case report and review of the literature. British Journal of Dermatology. 2010;163(6):1340-1345. [CrossRef]
- Zweers MC, Bristow J, Steijlen PM, et al. Haploinsufficiency of TNXB Is Associated with Hypermobility Type of Ehlers-Danlos Syndrome. American Journal of Human Genetics. 2003;73(1):214-217. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1180584/.
- TNXB tenascin XB [Homo sapiens (human)] - Gene - NCBI. www.ncbi.nlm.nih.gov. Accessed May 27, 2024. https://www.ncbi.nlm.nih.gov/gene/7148#general-protein-info.
- van Dijk FS, Ghali N, Demirdas S, Baker D. TNXB-Related Classical-Like Ehlers-Danlos Syndrome. PubMed. Published 1993. Accessed February 21, 2024. https://www.ncbi.nlm.nih.gov/books/NBK584019/.
- Imanaka-Yoshida K, Matsumoto K. Multiple Roles of Tenascins in Homeostasis and Pathophysiology of Aorta. Annals of Vascular Diseases. 2018;11(2):169-180. [CrossRef]
- Arpita Neogi, Towne M, Dykas D, et al. Abstract 554: The Association Of Multiple Variants In The TNXB Gene With Vascular Aneurysms And Dissections. Arteriosclerosis, thrombosis, and vascular biology. 2022;42(Suppl_1). [CrossRef]
Figure 1.
A variant of unknown significance found in Exon 6 of the TNXB gene was isolated in case 1. This variant results in a C to T substitution at the 2945 nucleotide position, and a threonine to isoleucine substitution at codon 982 [12].
Figure 1.
A variant of unknown significance found in Exon 6 of the TNXB gene was isolated in case 1. This variant results in a C to T substitution at the 2945 nucleotide position, and a threonine to isoleucine substitution at codon 982 [12].
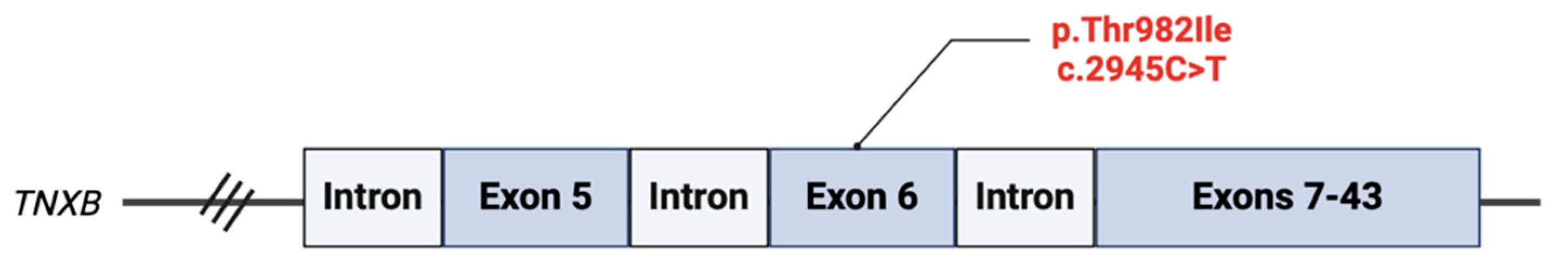
Figure 2.
A heterozygous variant on exon 11, p.D1512_V1517del was found in case 2. This results in the deletion of six amino acids between codons 1512 and 1517 and was predicted to be deleterious by in silico analysis [12].
Figure 2.
A heterozygous variant on exon 11, p.D1512_V1517del was found in case 2. This results in the deletion of six amino acids between codons 1512 and 1517 and was predicted to be deleterious by in silico analysis [12].
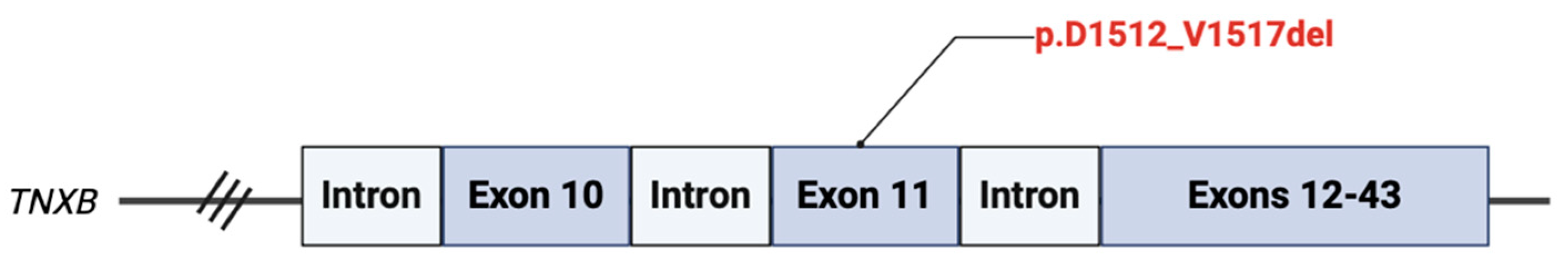
Figure 3.
A pathogenic variant, c.8510_8511delCT was located in case 3 resulting in a translational frameshift with a predicted alternate stop codon [12].
Figure 3.
A pathogenic variant, c.8510_8511delCT was located in case 3 resulting in a translational frameshift with a predicted alternate stop codon [12].
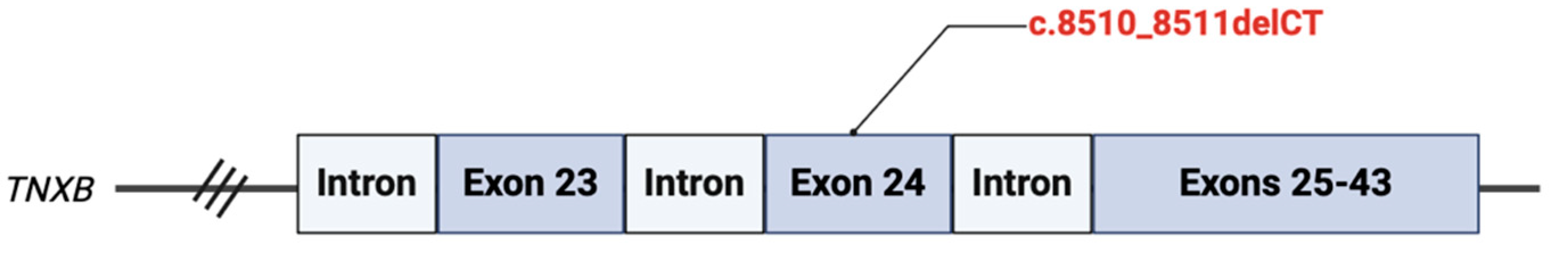
Figure 4.
The TNXB gene, containing 43 coding exons, is responsible for the production of the glycoprotein Tenascin-X (TNX), a member of the tenascin family of ECM glycoproteins. TNX serves as a vital component of the ECM through the regulation of collagen fibrillogenesis [12].
Figure 4.
The TNXB gene, containing 43 coding exons, is responsible for the production of the glycoprotein Tenascin-X (TNX), a member of the tenascin family of ECM glycoproteins. TNX serves as a vital component of the ECM through the regulation of collagen fibrillogenesis [12].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



