
Submitted:
19 August 2024
Posted:
20 August 2024
You are already at the latest version
Abstract
This study evaluated changes over time in skeletal muscle atrophy, expressions of skeletal muscle anabolic and catabolic genes, and mitochondrial activity by skeletal muscle type in an adenine-induced chronic kidney disease (CKD) model. A CKD model was successfully established by feeding male Wistar rats a 0.75% adenine diet for 4 weeks starting at 8 weeks of age. Control and CKD groups were sacrificed at 12 and 20 weeks of age. Back muscles were analyzed histologically, and succinate dehydrogenase (SDH) staining was performed to evaluate mitochondrial activity. Gene expressions of myogenic determination gene number 1 and myogenin as indicators of muscle anabolism, atrogin-1 and muscle RING-finger protein-1 (MuRF1) as indicators of muscle catabolism, and peroxisome proliferator-activated receptor-γ coactivator-1-α as a marker of mitochondrial biogenesis were assessed. Type I and type II muscle cross-sectional areas (CSAs) were decreased at 12 weeks, but type I muscle CSA recovered at 20 weeks. SDH staining was lower in CKD than control rats at 12 weeks, but no significant difference was observed at 20 weeks. Increased expressions of myogenin, atrogin-1, and MuRF-1 were observed only at 12 weeks, but no differences were observed at 20 weeks. The adenine-induced CKD rat model appears to show changes in muscle atrophy over time.
Keywords:
chronic kidney disease
; sarcopenia
; animal model
; muscle atrophy
; mitochondria
1. Introduction
Chronic kidney disease (CKD) is widely recognized as a global public health problem [1], with an estimated global prevalence of 13.4%, which is expected to increase as the population ages [2]. In addition to decreased renal function and increased vascular calcification [3], CKD is associated with muscle wasting [4] and decreased physical function, which contribute to lower quality of life and increased mortality in patients with CKD [5]. Therefore, it is important to prevent and treat the skeletal muscle atrophy and decline in physical function associated with CKD. However, there are no established methods for the prevention or treatment of skeletal muscle atrophy or loss of physical function due to CKD.
Several factors are thought to contribute to skeletal muscle atrophy in CKD [6]. Most notably, an altered balance between catabolism and anabolism regulates skeletal muscle homeostasis [4]. Avin et al. reported that the muscle catabolism-related genes, muscle RING-finger protein-1 (MuRF-1) and atrogin-1, are increased in CKD rats [7]. In addition, it has recently been reported that mitochondrial dysfunction, which is thought to provide energy for physical function, is one of the factors contributing to the decline in physical function in CKD [8]. However, how skeletal muscle atrophy changes over time in CKD and how factors related to muscle catabolism and anabolism and mitochondrial activity change over time are unclear.
Several preclinical models of CKD have been used to study skeletal muscle atrophy in CKD, including nephrectomy [9], Cy/+ rats with polycystic kidney disease [7,10], diabetic kidney disease [11,12], and adenine-induced CKD [13,14]. Of these models, the adenine-induced CKD model has many advantages for use as a preclinical CKD model: it is minimally invasive, cost-effective, does not require surgery or postoperative care, and has a low mortality rate [15]. In the adenine-induced CKD model, atrophy of the extensor digitorum longus, soleus, and thigh muscles [13,15], decreased grip strength [16], and impaired physical function [17] have been reported. However, how skeletal muscle atrophy occurs over time in the adenine-induced CKD rat model, how the expressions of muscle anabolic and catabolic genes are altered, and how mitochondrial activity is changed are not clear. The elucidation of the temporal changes in these factors in adenine-induced CKD rat models, which are useful as CKD model rats, will be of great importance for future studies of the prevention and treatment of muscle atrophy in CKD. Therefore, the purpose of this study was to evaluate changes over time in skeletal muscle atrophy, expressions of skeletal muscle anabolic and catabolic genes, and mitochondrial activity according to skeletal muscle type in an adenine-induced CKD model.
2. Materials and Methods
2.1. Animal Model and Experimental Design
Eight-week-old, male Wistar rats (Charles River Laboratories Inc., Tokyo, Japan) were housed in a controlled environment (temperature 23 ± 2 °C, humidity 40 ± 20%) with a 12-h light-dark cycle with free access to water and rat food. The details were described in the previous study [18]. The control group consisted of rats that received a standard rodent chow (CE-7; Clea Japan, Tokyo, Japan) as their regular diet. The CKD group received a 0.75% adenine diet (Oriental Yeast Co., Ltd., Tokyo, Japan) from 8 to 12 weeks of age, followed by a standard rodent chow regular diet, thus inducing CKD. The 4-week treatment with the adenine diet was based on the previous study [19]. The results of blood biochemistry tests and renal histological findings in the adenine-induced CKD rat model used in this study were reported in our previously published paper [20]. The serum creatinine, phosphorus, and intact-parathyroid hormone (PTH) levels were elevated, and serum calcium levels were normal, indicating stage IV CKD, and there was enlargement of the urinary cavities and fibrosis of the renal interstitium in the CKD rats at 20 weeks [20]. The control and CKD groups were sacrificed at 12 and 20 weeks of age, respectively, with each group having seven rats at each time point, and the following parameters were evaluated. The animal experiments performed adhered to the protocols approved in advance by the Animal Care and Use Committee of our institute (approval number a-1-3070). Furthermore, all subsequent animal experiments were conducted in accordance with the Animal Care and Use Guidelines of our institute, which follow the guidelines for animal research prescribed by the National Research Council’s Guide for the Care and Use of Laboratory Animals.
2.2. Tissue Preparation
Back muscles were harvested and stored in liquid nitrogen for measurement of cross-sectional area (CSA) and succinate dehydrogenase (SDH) staining. Right tibialis anterior (TA) muscles were harvested and stored in RNAlater solution (Qiagen, Hilden, Germany) at 80 °C for real-time polymerase chain reaction (PCR) testing.
2.3. Histological Analysis of Muscle
Back muscles in the four groups were analyzed histologically. Samples were cut into 10-µm-thick transverse serial sections at the thickest part of the muscle belly, with the cryostat maintained at -18 °C. The sections were subjected to histochemical staining with adenosine triphosphatase (ATP) after preincubation at pH 10.6. When stained at this pH, type I muscle fibers stain light, and type II muscle fibers stain dark. To quantify the CSA of muscle fibers, five randomly selected fields were assessed, with 50 fibers measured per muscle, and the mean CSA for one muscle fiber was calculated, as previously described [18]. The CSA was assessed using an all-in-one fluorescence microscope (BZ-X800, KEYENCE, Osaka, Japan).
2.4. Mitochondrial Activity
In addition, succinate dehydrogenase (SDH) staining was performed to evaluate mitochondrial activity. The sections were incubated for 30 minutes with SDH (0.4 g sodium succinate, 0.04 g nitro-blue tetrazolium (NBT), 0.001 mg phenazine methosulfate) in 0.1 M Tris buffer at 37 °C, then extracted with 30–90% acetone and rinsed with distilled H2O, as previously reported [7]. The staining density was set to be divided into 256 levels (0–255), and the average intensity was calculated using the luminance measurement function of the all-in-one fluorescence microscope (BZ-X800, KEYENCE). For SDH staining intensity of muscle fibers, five randomly selected fields per muscle were evaluated, and the average staining intensity was calculated.
2.5. Gene Expression Analysis of Skeletal Muscle
The gene expressions of myogenic determination gene number 1 (MyoD) and myogenin as indicators of muscle anabolism, atrogin-1 and MuRF1 as indicators of muscle catabolism, and peroxisome proliferator-activated receptor-γ coactivator-1-α (PGC-1α) as a marker of mitochondrial biogenesis were assessed [20]. Tissue samples were pulverized using a homogenizer (MS-100R; Tomy, Tokyo, Japan). Total RNA was extracted from the tissue using TRIzol reagent (Life Technologies, Carlsbad, CA, USA) following the manufacturer’s protocol. The concentration of RNA was determined using a NanoDrop spectrophotometer ND-1000 (Thermo Fisher Scientific, Waltham, MA, USA). First-strand complementary DNA (cDNA) synthesis was carried out using the First-Strand cDNA Synthesis Kit (GE Healthcare, Milwaukee, WI, USA). Quantitative reverse-transcription PCR was performed using the Light Cycler 480 system (Roche, West Sussex, United Kingdom) according to the manufacturer’s instructions, with TaqMan probes specific for rat MyoD (TaqMan probe ID: Rn01457527_g1), myogenin (TaqMan probe ID: Rn01490689_g1), atrogin-1 (TaqMan probe ID: Rn00591730_m1), MuRF1 (TaqMan probe ID: Rn00590197_m1), and PGC-1α (TaqMan probe ID: Rn00580241_m1). Glyceraldehyde-3-phosphate dehydrogenase amplification was used as an internal control for sample normalization (TaqMan probe ID: Rn01775763_g1). The cycle number at which the amplification plot intersected the threshold (CT) was determined, and the ΔΔCT method was used to analyze the relative changes in gene expression.
2.6. Statistical Analyses
All data are presented as mean ± standard deviation (SD) values. Since the gene expression results deviated from the normal distribution, the nonparametric gene expression data were subjected to analysis using the Kruskal-Wallis test, followed by the Steel-Dwass method as a post hoc test. The Mann-Whitney U test was used to assess changes in CSA and SDH staining within the respective groups. All statistical analyses were conducted using EZR, a graphical user interface for R (The R Foundation for Statistical Computing, Vienna, Austria). Specifically, EZR is a modified version of R commander that incorporates frequently used statistical functions in the field of biostatistics [21]. Significance was defined as p < 0.05.
3. Results
3.1. Histological Findings and Cross-Sectional Area of Back Muscle
An ATP-stained histological section of back muscle is shown in Figure 1A. In CKD rats, muscle fibers of both type I and type II at 12 weeks and of only type II at 20 weeks were smaller than in controls. At 12 weeks, the CSAs of back muscle fibers of both type I (Figure 1B) and type II (Figure 1C) were significantly smaller in the CKD group than in the control group (p < 0.01). At 20 weeks, the CSA of only type II (Figure 1E) was significantly smaller in the CKD group than in the control group (p < 0.01), but there was no significant difference in type I (Figure 1D).
3.2. Mitochondrial Activity Evaluated with SDH Staining
SDH-stained histological sections showed decreased SDH intensity in CKD rats than in control rats at 12 weeks, but not at 20 weeks (Figure 2A). Although SDH staining intensity was significantly lower in the CKD group than in the control group at 12 weeks (p < 0.05) (Figure 2B), there was no significant difference in staining intensity between the control and CKD groups at 20 weeks (Figure 2C).
3.3. Gene expression of Skeletal Muscle
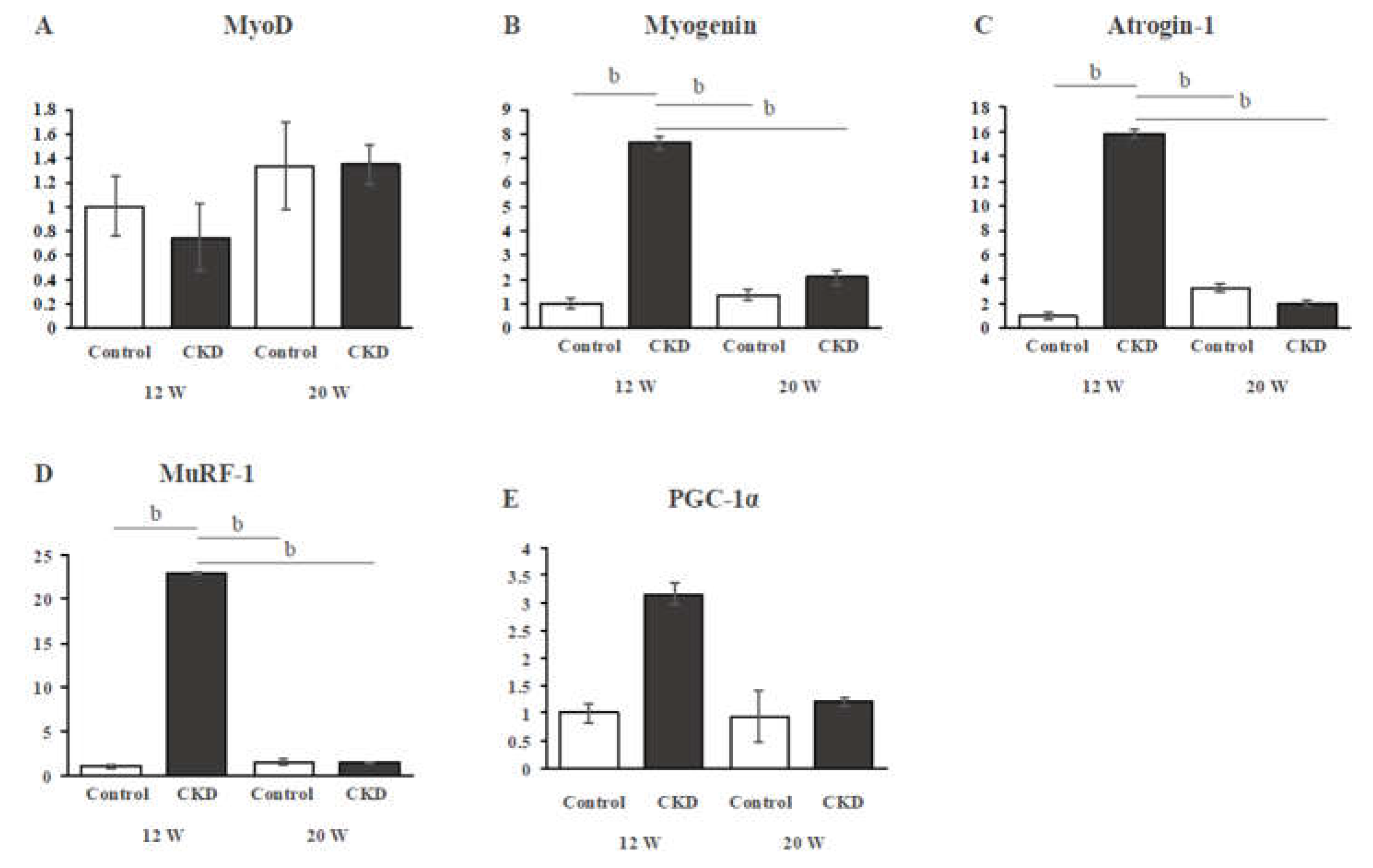
In TA muscle, gene expressions of MyoD were not significantly different between the control and CKD groups at both 12 and 20 weeks (Figure 3A). Muscle anabolic-related gene expression (myogenin) was significantly higher in the CKD group than in the control group at 12 weeks, but not at 20 weeks (Figure 3B). The mRNA expression levels of muscle catabolic-related genes, atrogin-1 and MuRF-1, were significantly higher in the CKD group than in the control group at 12 weeks (both P < 0.01), but not at 20 weeks (Figure 3C,D). The significantly increased expressions of myogenin, atrogin-1, and MuRF-1 in the CKD group at 12 weeks were significantly reduced in the CDK group at 20 weeks (all P < 0.01) (Figure 3B-D). Expression of PGC-1α mRNA showed a trend to being increased in the CKD group at 12 weeks, but there was no significant difference between the groups (Figure 3E).
4. Discussion
In the present study, changes over time in skeletal muscle atrophy were examined in a CKD model of male Wistar rats fed a 0.75% adenine diet for 4 weeks starting at 8 weeks of age. At 20 weeks of age, only type II muscle CSA was decreased. SDH staining, which reflects mitochondrial activity, was lower in the CKD group than in the control group at 12 weeks of age, but there was no significant difference at 20 weeks of age. Regarding the expression of genes related to muscle differentiation, increased expressions of myogenin, a myogenic regulatory factor, and of MuRF-1 and atrogin-1, muscle catabolic markers, were found only at 12 weeks of age, early after the establishment of CKD, but at 20 weeks of age, there were no differences in their expressions.
In the present study, skeletal muscle atrophy in adenine-induced CKD model rats was found to change over time in different types of muscle fibers. Several previous studies involving CKD model rats have reported that skeletal muscle atrophy and changes in the molecular cellular mechanisms associated with muscle atrophy were different in adenine-induced CKD and 5/6 nephrectomy [7,15]. However, no studies have focused on these changes over time. The present study showed differences in the type of skeletal muscle that atrophies at 12 and 20 weeks of age, differences in gene expression, and differences in mitochondrial activity. These differences in gene expression and mitochondrial activity may account for the differences in the characteristics of muscle atrophy.
In the adenine-induced CKD rat model, the CSAs of both type I and type II skeletal muscles were significantly reduced compared to controls at 12 weeks of age, but only the CSA of type II skeletal muscle was significantly lower at 20 weeks of age, with no significant difference observed in type I skeletal muscle. In general, type I muscle fibers are more susceptible to atrophy caused by inactivity and denervation, whereas type II muscle fibers are more susceptible to aging, diabetes mellitus, chronic heart failure, and cancer [22,23]. It has been reported that fast-twitch muscle fibers tend to be decreased in CKD patients and CKD model rats [24]. Type II muscle fibers have a poorly distributed capillary network, which is thought to be due to the decreased hemoglobin concentration caused by worsening renal function due to CKD, resulting in tissue hypoxia [25]. Furthermore, it has been reported that a fast-to-slow fiber-type shift is observed with muscle atrophy, such as aging [23]. The present changes in muscle atrophy over time may indicate CKD-induced skeletal muscle atrophy and compensatory improvement in the CSA of slow type I muscle fibers.
In the adenine-induced CKD model rats, the mRNA expression levels of myogenin were significantly higher only at 12 weeks of age. However, the MyoD levels were not significantly different between the groups. MyoD and myogenin are important factors in muscle regeneration [26], and impaired skeletal muscle regeneration is involved in muscle atrophy. When skeletal muscle is injured, satellite cells initiate proliferation and differentiation, and the expressions of MyoD and myogenin increase [27]. MyoD expression is associated with satellite cell activation and proliferation, whereas myogenin reflects myoblast differentiation; a study examining skeletal muscle from Cy/+ rats reported that both MyoD and myogenin were upregulated [7], a result different from the present study. The increased expression of myogenin observed in the present study may reflect the process of muscle regeneration in an adenine-induced CKD rat model. Myogenin is involved in myoblast differentiation and acts to regulate myofiber maturation and size [28]. Among its functions, it has been suggested that it induces the fusion of skeletal muscle fibers, mainly affecting the formation of fast-twitch muscle fibers and muscle hypertrophy [29]. In the present study, myogenin expression was increased in 12-week-old CKD model rats. This may be a compensatory mechanism for the muscle wasting that occurs in the CKD rat model. In contrast, no significant difference was observed for MyoD, a myogenic marker.
It has been suggested that uremic toxin (UT) is also involved in muscle atrophy and weakness in CKD [30,31]. One in vitro study reported that different doses of uremic substances affect the myogenic process differently. Low doses of UT impair normal myogenic differentiation by promoting fibrotic and adipogenic differentiation of myoblasts, whereas high doses of UT impair myoblasts by inducing cell cycle arrest, disrupting their proliferation, and causing apoptosis [32]. MyoD is expressed at the muscle differentiation stage when satellite cells are activated before myoblast differentiation. Therefore, the difference in the expression of myogenic markers in the present study suggests that the model may have been exposed to high concentrations of uremic substances.
In the adenine-induced CKD rat model, SDH staining intensity was decreased only at 12 weeks. This result suggests that mitochondrial activity is reduced early after the model is created. It has been reported that mitochondrial dysfunction is associated with muscle weakness in CKD [33]. It has also been reported that CKD is preceded by a decrease in muscle strength and muscle mass and is associated with a decrease in muscle mitochondrial function [34]. In addition, type I muscle fibers that showed reduced CSA in this study were rich in mitochondria [35], and the reduction in type I muscle CSA early after model creation may be related to reduced mitochondrial activity.
The present study examined the changes in skeletal muscle atrophy over time in a rat model of adenine-induced CKD. This study has several limitations. First, it was not possible to confirm serum creatinine levels and other data in each group; thus, the relationship between blood data and muscle atrophy could not be evaluated. However, since the method used to create the model was the same as that in our previous study, which confirmed that creatinine levels worsened over time, we assumed that the early and late stages of CKD onset were compared in the present study. Second, nutritional status was not assessed. There is a trend toward weight loss when adenine is administered [36]. In our previous report of adenine-induced CKD rat models generated using the same protocol as in the present study [37], body weight was also decreased, suggesting that adenine administration may have contributed to the weight loss and associated deterioration in nutritional status.
5. Conclusions
The adenine-induced CKD rat model shows that type I and type II muscle CSAs were decreased at 12 weeks of age, early after adenine administration, but type I muscle CSA recovered at 20 weeks of age. In addition, increased expression of muscle catabolic markers and myogenin and decreased SDH staining intensity were observed at 12 weeks. These phenomena may be related to changes in muscle atrophy over time.
Author Contributions
Conceptualization, Y.K., K.N., and N.M.; methodology, Y.K. and K.N.; validation, K.O.; formal analysis, H.T., D.K., H.K., and Y.O.; investigation, K.O., H.T., H.K., Y.O., S.I., F.K., S.H., K.O., T.K., K.T., and M.W.; writing—original draft preparation, K.O.; writing—review and editing, Y.K., K.N., and N.M.; visualization, K.O.; supervision, N.M.; project administration, Y.K.; funding acquisition, N.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The animal study protocol was approved by the Ethics Committee of Akita University (a-1-3046/August 29, 2018).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data used to support the findings of this study are included within the article, and further information is available from the corresponding author upon request.
Acknowledgments
The authors would like to thank Ms. Kudo and Ms. Midorikawa for their support of our experiments.
Conflicts of Interest
The authors declare that they have no conflicts of interest regarding the publication of this paper.
References
- Levey, A.S.; Atkins, R. Chronic kidney disease as a global public health problem: approaches and initiatives - a position statement from Kidney Disease Improving Global Outcomes. Kidney Int. 2007, 72, 247–259. [Google Scholar] [CrossRef]
- Lv, J.C.; Zhang, L.X. Prevalence and Disease Burden of Chronic Kidney Disease. Adv Exp Med Biol. 2019, 1165, 3–15. [Google Scholar] [CrossRef]
- Palit, S.; Kendrick, J. Vascular calcification in chronic kidney disease: role of disordered mineral metabolism. Curr Pharm Des. 2014, 20, 5829–5833. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.C.; Huang, S.H. Muscle Wasting in Chronic Kidney Disease: Mechanism and Clinical Implications-A Narrative Review. Int J Mol Sci. 2022, 23, 6047. [Google Scholar] [CrossRef]
- Beddhu, S.; Baird, B.C. Physical activity and mortality in chronic kidney disease (NHANES III). Clin J Am Soc Nephrol. 2009, 4, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Watson, E.L.; Viana, J.L. The Effect of Resistance Exercise on Inflammatory and Myogenic Markers in Patients with Chronic Kidney Disease. Front Physiol. 2017, 8, 541. [Google Scholar] [CrossRef]
- Avin, K.G.; Chen, N.X. Skeletal Muscle Regeneration and Oxidative Stress Are Altered in Chronic Kidney Disease. PLoS One 2016, 11, e0159411. [Google Scholar] [CrossRef] [PubMed]
- Serrano, E.; Whitaker-Menezes, D. Uremic Myopathy and Mitochondrial Dysfunction in Kidney Disease. Int J Mol Sci. 2022, 23, 13515. [Google Scholar] [CrossRef]
- Flisiński, M.; Brymora, A. Influence of different stages of experimental chronic kidney disease on rats locomotor and postural skeletal muscles microcirculation. Ren Fail. 2008, 30, 443–451. [Google Scholar] [CrossRef]
- Organ, J.M.; Srisuwananukorn, A. Reduced skeletal muscle function is associated with decreased fiber cross-sectional area in the Cy/+ rat model of progressive kidney disease. Nephrol Dial Transplant. 2016, 31, 223–230. [Google Scholar] [CrossRef]
- Zhang, A.; Li, M. miRNA-23a/27a attenuates muscle atrophy and renal fibrosis through muscle-kidney crosstalk. J Cachexia Sarcopenia Muscle. 2018, 9, 755–770. [Google Scholar] [CrossRef]
- Mogi, M.; Kohara, K. Diabetic mice exhibited a peculiar alteration in body composition with exaggerated ectopic fat deposition after muscle injury due to anomalous cell differentiation. J Cachexia Sarcopenia Muscle. 2016, 7, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Mori, T. Metabolic alterations by indoxyl sulfate in skeletal muscle induce uremic sarcopenia in chronic kidney disease. Sci Rep. 2016, 6, 36618. [Google Scholar] [CrossRef]
- Andres-Hernando, A.; Lanaspa, M.A. Obesity causes renal mitochondrial dysfunction and energy imbalance and accelerates chronic kidney disease in mice. Am J Physiol Renal Physiol. 2019, 317, F941–F948. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Anderson, E.M. Skeletal myopathy in CKD: a comparison of adenine-induced nephropathy and 5/6 nephrectomy models in mice. Am J Physiol Renal Physiol. 2021, 321, F106–F119. [Google Scholar] [CrossRef] [PubMed]
- Czaya, B.; Heitman, K. Hyperphosphatemia increases inflammation to exacerbate anemia and skeletal muscle wasting independently of FGF23-FGFR4 signaling. Elife. 2022, 11, e74782. [Google Scholar] [CrossRef]
- Moecke, D.M.P.; Martins, G.H.C. Aerobic Exercise Attenuates Kidney Injury, Improves Physical Performance, and Increases Antioxidant Defenses in Lungs of Adenine-Induced Chronic Kidney Disease Mice. Inflammation. 2022, 45, 1895–1910. [Google Scholar] [CrossRef]
- Kinoshita, H.; Miyakoshi, N. Effects of eldecalcitol on bone and skeletal muscles in glucocorticoid-treated rats. J Bone Miner Metab. 2016, 34, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Yokozawa, T.; Zheng, P.D. Animal model of adenine-induced chronic renal failure in rats. Nephron. 1986, 44, 230–234. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Martin-Sanchez, D. The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef]
- Kanda, Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013, 48, 452–458. [Google Scholar] [CrossRef]
- Wang, Y.; Pessin, J.E. Mechanisms for fiber-type specificity of skeletal muscle atrophy. Curr Opin Clin Nutr Metab Care. 2013, 16, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Ciciliot, S.; Rossi, A.C. Muscle type and fiber type specificity in muscle wasting. Int J Biochem Cell Biol. 2013, 45, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Sakkas, G.K.; Ball, D. Atrophy of non-locomotor muscle in patients with end-stage renal failure. Nephrol Dial Transplant. 2003, 18, 2074–2081. [Google Scholar] [CrossRef] [PubMed]
- Flisinski, M.; Brymora, A. Morphometric analysis of muscle fibre types in rat locomotor and postural skeletal muscles in different stages of chronic kidney disease. J Physiol Pharmacol. 2014, 65, 567–576. [Google Scholar]
- Hernández-Hernández, J.M.; García-González, E.G. The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin Cell Dev Biol. 2017, 72, 10–18. [Google Scholar] [CrossRef]
- Sabourin, L.A.; Girgis-Gabardo, A. Reduced differentiation potential of primary MyoD-/- myogenic cells derived from adult skeletal muscle. J Cell Biol. 1999, 144, 631–643. [Google Scholar] [CrossRef]
- Zammit, P.S. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin Cell Dev Biol. 2017, 72, 19–32. [Google Scholar] [CrossRef]
- Ganassi, M.; Badodi, S. Myogenin promotes myocyte fusion to balance fibre number and size. Nat Commun. 2018, 9, 4232. [Google Scholar] [CrossRef]
- Enoki, Y.; Watanabe, H. Indoxyl sulfate potentiates skeletal muscle atrophy by inducing the oxidative stress-mediated expression of myostatin and atrogin-1. Sci Rep. 2016, 6, 32084. [Google Scholar] [CrossRef]
- Enoki, Y.; Watanabe, H. Potential therapeutic interventions for chronic kidney disease-associated sarcopenia via indoxyl sulfate-induced mitochondrial dysfunction. J Cachexia Sarcopenia Muscle. 2017, 8, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Alcalde-Estévez, E.; Sosa, P. Uraemic toxins impair skeletal muscle regeneration by inhibiting myoblast proliferation, reducing myogenic differentiation, and promoting muscular fibrosis. Sci Rep. 2021, 11, 512. [Google Scholar] [CrossRef] [PubMed]
- Takemura, K.; Nishi, H. Mitochondrial Dysfunction in Kidney Disease and Uremic Sarcopenia. Front Physiol. 2020, 11, 565023. [Google Scholar] [CrossRef]
- Tamaki, M.; Miyashita, K. Chronic kidney disease reduces muscle mitochondria and exercise endurance and its exacerbation by dietary protein through inactivation of pyruvate dehydrogenase. Kidney Int. 2014, 85, 1330–1339. [Google Scholar] [CrossRef]
- Sanchez, B.; Li, J. Differentiation of the intracellular structure of slow- versus fast-twitch muscle fibers through evaluation of the dielectric properties of tissue. Phys Med Biol. 2014, 59, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Hayeeawaema, F.; Muangnil, P. A novel model of adenine-induced chronic kidney disease-associated gastrointestinal dysfunction in mice: The gut-kidney axis. Saudi J Biol Sci. 2023, 30, 103660. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Miyakoshi, N. Analysis of bone in adenine-induced chronic kidney disease model rats. Osteoporos Sarcopenia. 2021, 7, 121–126. [Google Scholar] [CrossRef]
Figure 1.
Histological changes of the back muscles of control rats and CKD rats. (A) In CKD rats, both type I and type II cross-sectional areas (CSAs) are reduced compared to controls at 12 weeks; only type II CSA is reduced at 20 weeks. (B) CSA of Type I fibers in control and CKD rats at 12 weeks. (C) CSA of Type II fibers in control and CKD rats at 12 weeks. (D) CSA of Type I fibers in control and CKD rats at 20 weeks. (E) CSA of Type II fibers in control and CKD rats at 20 weeks. a: p < 0.05, b: p < 0.01 between control and CKD rats by the t-test.
Figure 1.
Histological changes of the back muscles of control rats and CKD rats. (A) In CKD rats, both type I and type II cross-sectional areas (CSAs) are reduced compared to controls at 12 weeks; only type II CSA is reduced at 20 weeks. (B) CSA of Type I fibers in control and CKD rats at 12 weeks. (C) CSA of Type II fibers in control and CKD rats at 12 weeks. (D) CSA of Type I fibers in control and CKD rats at 20 weeks. (E) CSA of Type II fibers in control and CKD rats at 20 weeks. a: p < 0.05, b: p < 0.01 between control and CKD rats by the t-test.
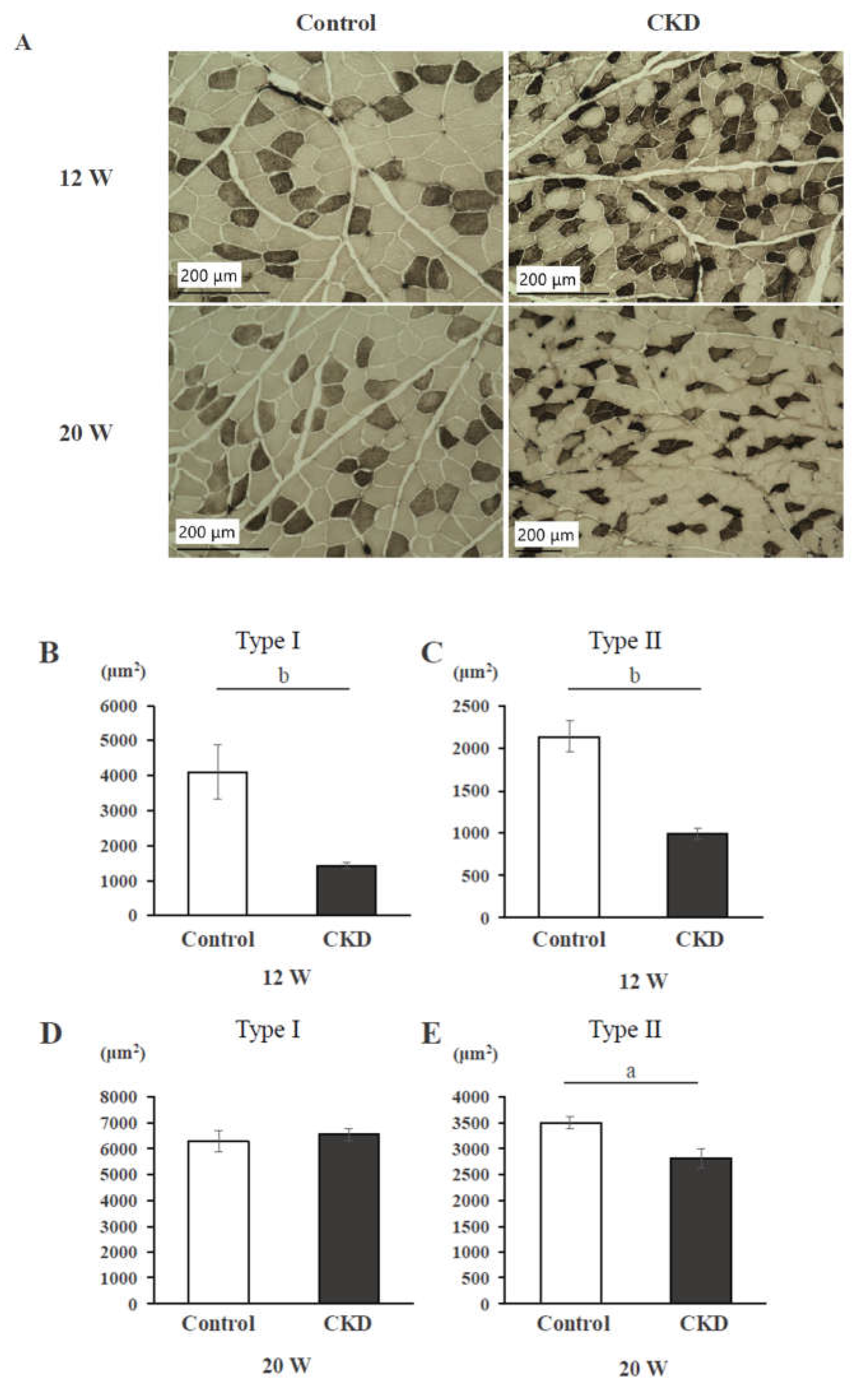
Figure 2.
SDH staining of back muscles. SDH staining intensity is significantly reduced in CKD rats compared to controls at 12 weeks, but there is no significant difference between the two groups at 20 weeks.
Figure 2.
SDH staining of back muscles. SDH staining intensity is significantly reduced in CKD rats compared to controls at 12 weeks, but there is no significant difference between the two groups at 20 weeks.
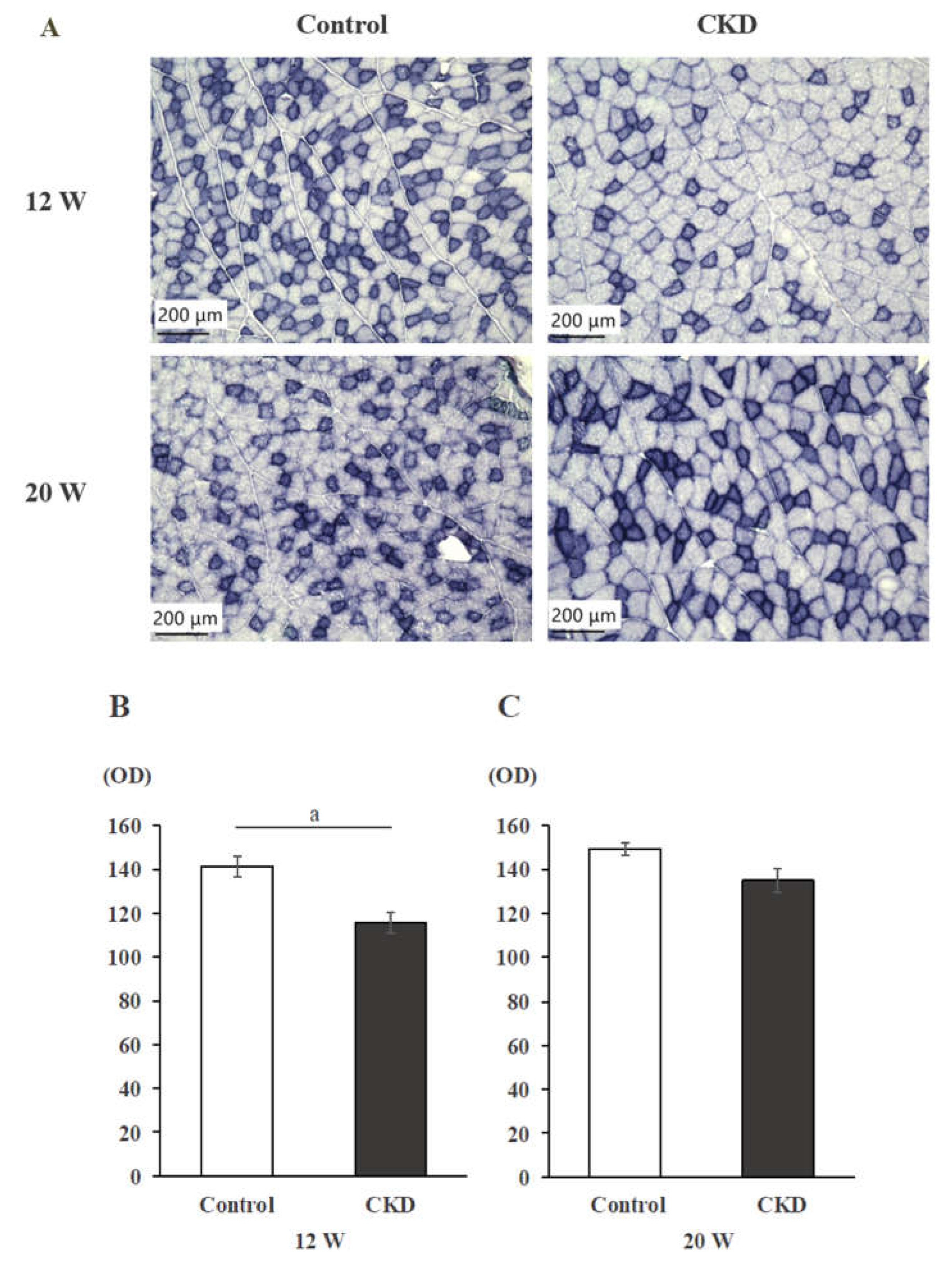
Figure 3.
Gene expressions of muscle anabolic, muscle catabolic, and mitochondrial biogenesis markers. Gene expression of MyoD shows no difference between the groups, whereas that of myogenin shows a significant increase in expression at 12 weeks in the CKD group. Both atrogin-1 and MuRF-1, muscle catabolic markers, are significantly upregulated at 12 weeks in the CKD group. There is no significant difference in gene expression of PGC-1α between the groups.
Figure 3.
Gene expressions of muscle anabolic, muscle catabolic, and mitochondrial biogenesis markers. Gene expression of MyoD shows no difference between the groups, whereas that of myogenin shows a significant increase in expression at 12 weeks in the CKD group. Both atrogin-1 and MuRF-1, muscle catabolic markers, are significantly upregulated at 12 weeks in the CKD group. There is no significant difference in gene expression of PGC-1α between the groups.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



