
Submitted:
20 August 2024
Posted:
20 August 2024
You are already at the latest version
Abstract
Developing recombinant Saccharomyces cerevisiae strains capable of transporting and fermenting cellobiose directly is a promising strategy for second-generation ethanol production from lignocellulosic biomass. In this study we cloned and expressed in the CEN.PK2-1C strain an intracellular βglucosidase (SpBGL7) from Spathaspora passalidarum, and co-expressed the cellobiose transporter SiHXT2.4 from Scheffersomyces illinoinensis, and two putative transporters from Candida tropicalis (CtCBT1) and Meyerozyma guilliermondii (MgCBT2). While all three transporters allowed cell growth on cellobiose, only the MgCBT2 permease allowed cellobiose fermentation, although cellobiose consumption was stuck (incomplete). The analysis of the βglucosidase and transport activities revealed that the cells stopped to consume cellobiose due to a drop in the transport activity. Since ubiquitinylation of lysine residues at the N- or C-terminal domains of the permease are involved in the endocytosis and degradation of sugar transporters, we constructed truncated versions of the permease lacking lysine residues at the C-terminal domain (MgCBT2ΔC), and at both the C- and N-terminal domain (MgCBT2ΔNΔC), and co-expressed these permeases with the SpBGL7 βglucosidase in an industrial strain. While the strain harboring the MgCBT2ΔC transporter continued to produce stuck cellobiose fermentations as the wild-type MgCBT2 permease, the strain with the MgCBT2ΔNΔC permease was able to consume and ferment all the cellobiose present in the medium. Thus, our results highlight the importance of expressing cellobiose transporters lacking lysine at the N- and C-terminal domains for efficient cellobiose fermentation by recombinant S. cerevisiae.
Keywords:
cellobiose
; transporter
; β-glucosidase
; Spathaspora passalidarum
; Meyerozyma guilliermondii
; recombinant Saccharomyces cerevisiae
1. Introduction
The production and use of sustainable alternatives to fossil fuels has been growing for the last few decades, and it will certainly become a necessity in the near future due to the environmental impact of fossil fuels on the climate. Bioethanol is the primary liquid biofuel used in the USA and Brazil, responsible for approx. 85% of the global production [1,2,3]. This so-called first-generation (1G) bioethanol is made from food-based plant sugars from corn (starch) or sugarcane (sucrose). Although accounting for approx. 1% of the global fuel ethanol production, second-generation (2G) bioethanol is made from plant residues rich in lignocellulosic biomass [3,4,5]. This renewable biomass is composed of cellulose (a linear polymer of β-1,4 linked glucose molecules), hemicellulose (a branched and highly heterogeneous polymer containing both hexoses and pentoses), and lignin. The biochemical conversion of lignocellulosic biomass involves several steps, including pre-treatment, enzymatic hydrolysis and fermentation, being the efficient conversion of all the sugars available a major requirement for an economically feasible 2G bioethanol process [5,6,7,8].
An optimized enzyme blend (containing endoglucanases, exoglucanases, hemicellulases, pectinases, β-glucosidases, xylanases, β-xylosidases, laccases, etc.) is required for the efficient conversion of cellulose and hemicellulose polymers into sugar syrups to be fermented by yeasts [9,10]. To avoid cellobiose accumulation, a strong inhibitor of both cellobiohydrolases and endoglucanases [11,12,13,14], enzyme blends usually contain high levels of β-glucosidases, increasing 2G bioethanol production costs. Thus, a solution for efficient conversion of lignocellulose to ethanol is the use of microorganisms capable of efficient cellobiose fermentation. Many yeasts can secrete β-glucosidases, and cellobiose is hydrolyzed to glucose in the extracellular environment, followed by glucose uptake and fermentation [15,16,17,18]. Other yeasts species have intracellular β-glucosidases, and thus cellobiose needs to be transported into the cell by cellodextrin permeases to allow its fermentation [19,20,21,22,23]. Some few yeasts may even have both pathways, extracellular hydrolysis besides transport and intracellular hydrolysis of cellobiose [24,25].
S. cerevisiae is a favored platform for microbial engineering efforts to produce biofuels from cellulosic hydrolysates because of its industrial robustness and easy genetic manipulation, but it lacks the capacity to ferment cellobiose. The uptake and intracellular hydrolysis of cellobiose, an abundant mechanism for cellobiose utilization in filamentous fungi and also engineered in S. cerevisiae [26,27], is an interesting strategy to avoid the production of glucose in the medium and thus the competition between glucose and xylose for the HXT transporters of S. cerevisiae [28], which often considerable delays xylose consumption and the fermentation process [29,30,31,32,33]. Furthermore, complete enzymatic conversion of cellulose to glucose is problematic because high glucose concentrations inhibit both cellulases and β-glucosidases [10,11,12]. The first report showing the successful expression in S. cerevisiae of cellodextrin transporters and intracellular β-glucosidase was with genes from the cellulolytic fungi Neurospora crassa [26]. The authors expressed an intracellular β–glucosidase (encoded by the gh1-1 gene) and two transporters (CDT-1 and CDT-2 genes) that transport cellobiose and higher cellodextrins, as well as playing a critical role in hemicellulose sensing and utilization by N. crassa, while CDT-2 seems to also transport xylobiose and longer xylodextrins [26,34,35,36]. Another difference between these permeases is that CDT-1 is an active transporter, while CDT-2 seems to transport cellobiose by facilitated diffusion, although both transporters were reported to have very similar affinity to cellobiose [26,37].
There are dozens of other manuscripts reporting the use of the gh1-1 β-glucosidase and CDT-1 and CDT-2 transporters for cellobiose fermentation by several recombinant yeast, and while other fungal cellodextrin/xylodextrin permeases have been expressed in S. cerevisiae [38,39,40,41,42], unfortunately these transporters were characterized just by growth assays, with little information regarding kinetics of cellobiose transporters and their contribution to ethanol production during growth [27,43]. Regarding cellobiose transporters from yeasts, only two permeases (HXT2.4 from Sc. stipitis and Ls120451 from Lipomyces starkeyi) have been shown to allow cellobiose fermentation by recombinant S. cerevisiae when also expressing the gh1-1 β-glucosidase from N. crassa [44,45].
In the present work we have cloned an intracellular β-glucosidase (SpBGL7) from the cellobiose fermenting yeast Sp. passalidarum [46], and used a S. cerevisiae laboratory strain CEN.PK2-1C expressing this β-glucosidase to screen for cellobiose transporters from yeasts. The co-expression of the cellobiose transporter SiHXT2.4 from Sc. illinoinensis, and two putative transporters from C. tropicalis (CtCBT1) and M. guilliermondii (MgCBT2), allowed growth on cellobiose by the recombinant S. cerevisiae strains. However, only the MgCBT2 permease allowed cellobiose fermentation, although the consumption of cellobiose was stuck (incomplete). Since the incomplete consumption of cellobiose was the consequence of a drop in the transport activity by the cells, we analyzed this transporter in relation to the presence of lysine residues in the N- or C-terminal domains with potential for ubiquitinylation, and thus for being involved in downregulation of the transporter through endocytosis and vacuolar degradation [47,48]. The construction of a truncated version of the transporter lacking lysine residues at both the C- and N-terminal domain (MgCBT2ΔNΔC) allowed efficient fermentation of all the cellobiose present in the medium by an industrial fuel-ethanol yeast strain. The results obtained highlight the importance of removing lysine residues involved in endocytosis to allow efficient expression of heterologous sugar transporters in S. cerevisiae.
2. Materials and Methods
2.1. Strains, Media and Growth Conditions
The yeast strains and plasmids used in this study are listed in Table 1. The Escherichia coli DH5α strain (F’/ endA1hsdR17 (rK-mK+) glnV44 thi-1 recA1 gyrA (NaIr) relA1Δ (lacZYA-argF) U169 deoR Φ80dlac Δ(LacZ) M15) [49], was used for cloning and plasmid propagation, and was grown in Luria broth containing 1% tryptone, 0.5% yeast extract, 0.5% sodium chloride, pH 7.0, and 100 mg/L ampicillin when required (Sigma-Aldrich Brazil Ltda., São Paulo, SP, Brazil).
Yeasts were grown in rich YP medium (1% yeast extract, 2% Bacto peptone, Sigma-Aldrich), or in synthetic complete (YNB) medium (0.67% yeast nitrogen base without amino acids, supplemented with 1.92 g/L of yeast synthetic Drop-out media without uracil, or 1.82 g/L without uracil and tryptophan, Sigma-Aldrich), with 20 g/L glucose or cellobiose as carbon source. The pH of the medium was adjusted to pH 5.0 with HCl, and when required, 2% Bacto agar (Sigma-Aldrich) or 0.1 g/L nourseothricin (cloNAT, WERNER BioAgents GmbH, Jena, Germany) were added to the medium. The laboratory strains transformed with plasmids were pre-grown in YNB with glucose as carbon source, and used to inoculate new YNB medium containing 20 g/L cellobiose with an initial cell concentration of 0.1 optical density at 600 nm (A600nm), measured with a Cary 60 UV-VIS spectrophotometer (Agilent Technologies, Santa Clara, CA, USA). Growth was performed aerobically in cotton plugged Erlenmeyer flasks filled to 1/5 of the volume with medium at 28°C with 160 rpm orbital shaking. Cellular growth was followed by absorbance measurements at 600 nm (A600nm). For batch fermentations, cells were collected in the exponential phase of growth, centrifuged at 6,000 g for 5 min at 4°C, and washed twice with sterile water, and inoculated at a high cell density (~10 g dry cell weight/L) into a 25 mL flask containing 20 mL of rich YP medium containing 20 g/L cellobiose at 30°C, with shaking at 100 rpm. For the industrial strains, fermentations were also performed in rich YP medium containing 20 g/L glucose or cellobiose, or 20 g/L cellobiose plus 20 g/L xylose. Culture samples were harvested regularly, centrifuged (5,000 g, 1 min at 40C), and supernatants used for the quantification of substrates and fermentation products.
2.2. Molecular Biology Techniques
Standard procedures for DNA manipulation and analysis, as well as bacterial and yeast transformation, were employed [59,60]. Purification of plasmids, PCR products or Gibson Assembly products was performed using the QIAquick PCR Purification Kit (QIAGEN Antwerp, Belgium). The genomic DNA from the yeast strains was purified using a YeaStar Genomic DNA kit (Zymo Research, Irvine, CA, USA). For plasmid extraction, we used the manual mini-prep method [60], or using the commercial QIAprep Spin Miniprep Kit (QIAGEN). DNA fragments for cloning, sequencing, or transformations were PCR-amplified using Phusion High-Fidelity (Thermo Fisher Scientific Inc., Waltham, MA, USA) or PrimeSTAR® GXL DNA polymerases (Takara Bio Europe SAS, Saint-Germain-en-Laye, France). Purified plasmids, products of Gibson Assembly, or cells lysed by incubation at 100°C in 20 mM NaOH for 10 min (colony PCR) served as DNA templates.
Based on the genome of C. tropicalis and M. guilliermondii [61], Sc. stipitis [62] and Sp. passalidarum [63], primers were designed (Table 2) to amplify genes encoding a β-glucosidase from Sp. passalidarum (SpBGL7, NCBI Gene ID: 18871961, primers SpBGL7-F and SpBGL7-R), the cellobiose transporters HXT2.4 from Sc. illinoinensis (the HXT2.4 gene from Sc. stipitis has NCBI Gene ID: 4850978, primers HXT2.4-F and HXT2.4-R), CtCBT1 from C. tropicalis (NCBI Gene ID: 8298855, primers CtCBT1-F and CtCBT1-R), and MgCBT2 from M. guilliermondii (NCBI Gene ID: 5129179, primers MgCBT2-F and MgCBT2-R), introducing restriction sites (for BamHI, EcoRI, HindIII, SalI or XhoI enzymes) for cloning into multicopy shuttle vectors containing strong and constitutive promoters and terminators (pGPD-424 and pGPD-426, Table 1) as well as the TRP1 and URA3 genes used as selective marker.
To clone and overexpress modified versions of the MgCBT2 transporter, we amplified the MgCBT2 gene from the pGPD-426-MgCBT2 plasmid (Table 1) using specific primer pairs: MgCBT2ΔC-F and MgCBT2ΔC-R (Table 2) for generating a version of the gene that encodes a truncated permease in the C-terminal region (pGPD-426-MgCBT2ΔC plasmid, Table 1), and MgCBT2ΔNΔC-F and MgCBT2ΔC-R (Table 2) for generating a version that encodes a truncated permease in both N- and C-terminal regions (pGPD-426-MgCBT2ΔNΔC plasmid, Table 1). These primers ensured the retention of the ATG codon for the initial methionine and the TGA stop codon for protein synthesis termination. The gene encoding the transporter truncated in the C-terminal lacked base pairs 4 to 60, resulting in a protein lacking the first 19 amino acid residues after the initial methionine. The gene encoding the transporter truncated in both N- and C-terminal regions, in addition to having the same modification described above, also lacked the last 36 coding base pairs, resulting in a protein lacking the last 12 amino acid residues in addition to the first 19 after the initial methionine.
The pV1382 plasmid (Table 1) served as the platform for expressing the CRISPR-Cas9 system in S. cerevisiae [58]. The ARS208 and ARS1309 loci were chosen for integrating the overexpression modules of the SpBGL7 and MgCBT2 genes, respectively, based on the research by Reider Apel and colleagues [64]. After sequencing both regions of interest in the MP-C5H1 strain genome (Table 1), we identified 20 bp segments to serve as the crRNA recognition sites. These segments were required to be followed by a protospacer adjacent motif (PAM) sequence recognized by the CRISPR-Cas9 system ("NGG" in this case). For the ARS208 site, the selected sequence was "GTCCGCTAAACAAAAGATCT", followed by the PAM sequence "TGG", located approximately 325 base pairs upstream of the ARS208 locus. For the ARS1309 site, the chosen sequence was "CCTGTGGTGACTACGTATCC", followed by the PAM sequence "AGG", situated approximately 180 base pairs upstream of the ARS1309 locus.
Each DNA fragment responsible for crRNA transcription, specific to the sequences mentioned above, was cloned into pV1382 as described [58]. The vector pV1382 was treated with enzyme BsmBI (New England Biolabs, Leiden, The Netherlands), and the 5' ends of each DNA strand of the linearized plasmid were dephosphorylated using alkaline phosphatase (Quick CIP, New England Biolabs), followed by purification. The digested plasmid and the specific pair of oligonucleotides (sgRNA.ARS1309-F and sgRNA.ARS1309-R, Table 2) to target the CRISPR-Cas9 system to the ARS1309 locus, and sgRNA.ARS208-F and sgRNA.ARS208-R primers (Table 2) for the ARS208 locus, were incubated at 15°C for 16 hours in the presence of T4 DNA ligase (Thermo Fisher). The resulting plasmids were sequenced using the seq.p1382.sgRNA-F and seq.p1382.sgRNA-R primers (Table 2) to verify the correct insertion of the DNA fragments, yielding plasmids pV1382-ARS1309 and pV1382-ARS208 (Table 1).
To assemble the PCR-amplified DNA fragments for constructing the repair and gene overexpression modules, we utilized Gibson Assembly® with the NEBuilder® HiFi DNA Assembly kit (New England Biolabs). Each PCR reaction employed a pair of primers (primer sequences can be provided upon request) designed with at least 20 base pairs at the 3' end that anneal to the beginning or end of the intended amplification region, and at least 20 nucleotides at the 5' end identical to the adjacent end of the DNA portion in the other DNA molecule intended for joining. For constructing the repair fragment for inserting the gene encoding each version of the MgCBT2 transporter, three initial fragments were joined: (I) a 515 bp DNA fragment identical to the region upstream of the cleavage site of the ARS1309 locus (5’ARS1309, obtained via colony PCR from the industrial strain MP-C5H1, Table 1); (II) a DNA fragment containing the PTDH3 promoter, the desired MgCBT2 version, and the TCYC1 terminator (obtained via PCR using pGPD-426-MgCBT2, pGPD-426-MgCBT2ΔC, or pGPD-426-MgCBT2ΔNΔC plasmids as templates, Table 1); and (III) a 624 bp DNA fragment identical to the region downstream of the cleavage site of the ARS1309 locus (3’ARS1309, obtained via colony PCR from the industrial strain MP-C5H1).
For constructing the repair fragment containing the SpBGL7 overexpression module, circular construction was chosen due to the low efficiency of Gibson Assembly in forming linear molecules from more than three distinct fragments. Circularization was achieved using the pMV vector (Table 1). The repair fragment was constructed by joining five distinct initial fragments: (I) a 695 bp DNA fragment identical to the region upstream of the cleavage site of the ARS208 locus (5’ARS208, obtained via colony PCR from strain MP-C5H1); (II) a DNA fragment containing 608 bp immediately upstream of the coding region of the TEF1 gene, corresponding to its promoter region (PTEF1, obtained via colony PCR from strain MP-C5H1); (III) a DNA fragment containing the SpBGL7 gene (obtained via PCR using pGPD-424-SpBGL2 plasmid as template); (IV) a DNA fragment containing the 428 bp immediately downstream of the coding region of the PGK1 gene, corresponding to its terminator region (TPGK1, obtained via colony PCR from strain MP-C5H1); and (V) a 742 bp DNA fragment identical to the region downstream of the cleavage site of the ARS208 locus (3’ARS208, obtained via colony PCR from strain MP-C5H1). We changed the constitutive promoter (PTEF1) and terminator (TPGK1) controlling the SpBGL7 gene to avoid any chromosomal instability with the promoter (PTDH3) and terminator (TCYC1) used for the MgCBT2 permeases. Each purified PCR fragment was incubated at 50°C for 60 minutes in the presence of NEBuilder® HiFi DNA Assembly reagent. After incubation, the resulting plasmid (pMV-SpBGL7, Table 1) was transformed into E. coli DH5α.
For the insertion of the SpBGL7 repair/overexpression module into the genome of the MP-C5H1 strain, transformations were performed using 300 ng of the purified pV1382-ARS208 plasmid and 10 µg of the repair/overexpression module amplified using primers ARS208-F and ARS208-R (Table 2) and the pMV-SpBGL7 plasmid as template (Table 1). Transformants were selected in YP-20 g/L glucose plates containing 0.1 g/L nourseothricin. Flipout of the pV1382-ARS208 plasmid was performed by overnight growth (twice) in nonselective liquid YP-20 g/L glucose medium. Drug-sensitive colonies, which had lost the plasmid, were identified by plating for single colonies on nonselective media and subsequent identification by replica plating to selective media. The correct insertion of the SpBGL7 module at ARS208 in the MP-B7 strain (Table 1) was confirmed by sequencing. The same concentrations of the pV1382-ARS1309 plasmid and the repair/overexpression modules containing the different MgCBT2 transporters (MgCBT2, MgCBT2ΔC or MgCBT2ΔNΔC), produced by Gibson Assembly, were used to transform strain MP-B7, yielding strains MP-B7-CBT2, MP-B7-CBT2ΔC and MP-B7-CBT2ΔNΔC, respectively (Table 1). All insertions at the ARS1309 locus were confirmed by sequencing.
2.3. Enzymatic and Transport Activity Assays
The hydrolysis of p-nitrophenyl-β-D-glucopyranoside (pNPβG), cellobiose, or p-nitrophenyl-β-D-xylopyranoside (pNPβX) was determined using permeabilized yeast cells [65]. Approximately 50 μL of permeabilized cell suspension (at concentrations ranging from approximately 0.1 to 0.4 g/L) were added to 450 μL of 100 mM MOPS-NaOH, pH 6.8 buffer containing the desired amount of substrate, and incubated at 30°C for 10 minutes. The reaction was stopped by placing the tubes at 100°C for 3 minutes. Pre-boiled cells for 3 minutes were used as controls. We used final concentrations ranging from 0.05 to 10 mM of pNPβG and pNPβX, or 1 to 80 mM cellobiose as substrates. After the reaction, cells were centrifuged at 2600x g for 5 minutes, and the supernatant from assays using pNPβG and pNPβX were used to determine the enzymatic activity by measuring the concentration of p-nitrophenol released by substrate hydrolysis, at an absorbance of 400 nm. To determine cellobiose hydrolysis, the supernatant from the assays was used to measure the concentration of glucose formed using a commercial glucose oxidase-peroxidase kit (Glicose Pap Liquiform Labtest, Centerlab, Belo Horizonte, MG, Brazil). Activities are expressed as nmol of p-nitrophenol or glucose produced by (mg dry cell weight)-1 min-1. The values of Km and Vmax were determined through nonlinear regression applied to the Michaelis-Menten kinetic model using the GraphPad Prism v. 8.0 software (GraphPad Software, Boston, MA, USA).
The transport assays followed a colorimetric method originally developed for determination of α-glucoside (p-nitrophenyl-α-D-glucopyranoside) transport by yeast maltose permeases [66]. Cells were harvested from liquid culture, washed twice with chilled (4°C) sterile distilled water, and resuspended in 50 mM succinate-Tris pH 5.0 buffer to achieve a cell concentration of approximately 30 g/L. Aliquots of 50 μL of this cell suspension were transferred to Eppendorf tubes, and a volume of 50 μL of 10 mM pNPβG or pNPβX in the same buffer was added. The cells were incubated at 30°C for 10 minutes, during which the internalized substrate underwent hydrolysis due to the activity of the intracellular β-glucosidase. The reaction was stopped by incubating the tubes at 100°C for 3 minutes. Subsequently, 200 μL of 2 M NaHCO3 was added. Pre-boiled cells for 3 minutes were used as negative controls. Cells were centrifuged at 2600x g for 5 minutes, and the supernatant was used to determine the concentration of p-nitrophenol produced, measured by absorbance at 400 nm. The transport activities are expressed as nmol of pNPβG or pNPβX transported (p-nitrophenol produced) by (mg dry cell weight)-1 min-1.
2.4. Analytical Methods
Cellobiose and xylose substrates, and ethanol, xylitol, and glycerol products in medium samples were quantified by high performance liquid chromatography (Prominence HPLC system) equipped with a RID-20A refractive index detector (Shimadzu Co., Tokyo, Japan) using an Aminex HPX-87H column (Bio-Rad Laboratories, Hercules, CA, USA). The HPLC apparatus was operated at 50°C using 5 mM H2SO4 as mobile phase at a flow rate of 0.6 mL/min, and 10 μL injection volume.
2.5. Prediction of Lysine Residues in MgCBT2 Transporter with Ubiquitinylation Potential
To predict potential transmembrane domains (TMs) of the MgCBT2 transporter (NCBI GenBank ID: PP826654.1), we used PRALINE with the PSIPRED and HMMTOP methods [67]. The lysine residues with ubiquitinylation potential in the N- and C-terminal domains of the permease were determined with the BDM-PUB [68] and UbPred programs [69].
3. Results
3.1. Cloning and Expression of an Intracellular Yeast β-Glucosidase in S. cerevisiae
Initially we used the amino acid sequence of the intracellular β–glucosidase encoded by the gh1-1 gene from N. crassa [26] to search for putative β–glucosidases from yeasts, but our analysis did not revealed a gene with significant identity. However, using the sequence of the β-glucosidase from the cellulolytic fungi Thielavia terrestris (TtBG gene, NCBI Gene ID: 11519932 [27]), revealed two genes with 33% (annotated as SpBGL2) and 29% (SpBGL7) of identity in the genome of the cellobiose-fermenting yeast Sp. passalidarum [46,63]. Attempts to clone the SpBGL2 gene from the Sp. passalidarum UFMG-CM-Y474 strain (Table 1) failed, but we succeeded in cloning the SpBGL7 β-glucosidase into the pGPD-424 plasmid (Table 1). When the pGPD-424-SpBGL7 plasmid was transformed into strain CEN.PK2-1C, the obtained strain (B7) could not use or grow on cellobiose (Figure 1), but we could clearly detect a intracellular β-glucosidase activity when we used permeabilized yeast cells, with a Km of 18.2 ± 3.9 mM and Vmax of 1,188 ± 85 nmol mg-1 min-1 with cellobiose, and higher affinity (Km of 0.8 ± 0.2 mM and Vmax of 5,368 ± 199 nmol mg-1 min-1) with pNPβG. This enzyme had also hydrolytic activity with pNPβX (a synthetic analog of xylobiose), with a Km of 1.0 ± 0.5 mM and Vmax of 1,088 ± 175 nmol mg-1 min-1.
3.2. Cloning and Expression of Yeast Sugar (Cellobiose) Transporters in S. cerevisiae
The S. cerevisiae strain B7, having high intracellular β-glucosidase activity but unable to grow on cellobiose, was used as platform to identifiy putative sugar transporters capable of mediating the uptake of cellobiose. First, and taking into account the close relationship between Sc. stipitis and Sc. illinoinensis [70], we used the genomic information of Sc. stipitis [63] to design primers and amplify the corresponding HXT2.4 cellobiose transporter [44] from Sc. illinoinensis. Primers HXT2.4-F and HXT2.4-R (Table 2) allowed the amplification of a 1.7-1.8 Kb DNA fragment from the genome of Sc. illinoinensis, which was cloned into the pGPD-426 plasmid (Table 1). We also used the amino acid sequence of the CDT-2 cellobiose transporter from N. crassa [26,34,36,37] to search for yeast putative cellobiose transporters, and found two sequences, one with 28% identity with CDT-2 present in the genome of the yeast C. tropicalis (and thus named CtCBT1), and another sequence with 27% identity in the genome of M. guilliermondii (named as MgCBT2). Using specific primers for these genes (Table 2), these putative cellobiose transporters were cloned into the pGPD-426 plasmid (Table 1). As can be seen in Figure 1, the co-expression in S. cerevisiae of the intracellular SpBGL7 β-glucosidase together with any of the three transporters (SiHXT2.4, CtCBT1 and MgCBT2) allowed cell growth on cellobiose, although cellobiose consumption and cell growth was lower in the strain expressing the SiHXT2.4 permease.
We next tested the capacity of these laboratory yeast strains to ferment cellobiose under microaerobic conditions (Figure 2). While the strain harboring the SiHXT2.4 permease consumed small amounts of cellobiose, and could not produce any ethanol, the strain with the CtCBT1 transporter also consumed very small amounts of cellobiose, and produced less than 1.5 g/L of ethanol from the sugar. The best results regarding cellobiose fermentation were with the strain expressing the SpBGL7 β-glucosidase together with the MgCBT2 permease, which consumed more than 16 g/L of cellobiose and produced almost 8 g/L of ethanol, although the fermentation was stuck (incomplete).
In order to better understand the reason for the cellobiose stuck fermentation by strain B7-CBT2 (pGPD-424-SpBGL7 + pGPD-426-MgCBT2, Table 1), we initially used pNPβG, a synthetic analog of cellobiose, in a colorimetric assay to determine the transport activity of the MgCBT2 permease, as already described for the maltose permeases of S. cerevisiae [66]. After growth on cellobiose the B7-CBT2 strain was capable to transport 7.4 ± 1.6 nmol of pNPβG mg-1 min-1. The B7-CBT2 cells were also capable to transport pNPβX (3.3 ± 0.1 nmol mg-1 min-1), indicating that the MgCBT2 permease probably also transports xylobiose. Figure 3 shows the intracellular β-glucosidase activity (determined with pNPβG), as well as the pNPβG transport activity (both activities were determined with 5 mM of substrate), during the stuck cellobiose fermentation by strain B7-CBT2.
It is evident in Figure 3B that the intracellular β–glucosidase activity remained high along all the fermentation period, while the transport activity was higher only in the first 36 h of the fermentation (at the time in which cellobiose was rapidly consumed and fermented, Figure 3A), and from 48 h on the transport activity dropped, and was accompanied by a significant drop in cellobiose consumption by the cells. Thus, our results indicate that the stuck cellobiose consumption is a consequence of a drop in the transport activity, and not in the intracellular hydrolysis of the disaccharide. Since a possible explanation for the drop in the transport activity is the well-known post-transcriptional mechanism of down-regulation of yeast sugar permeases, involving ubiquitinylation, endocytosis and vacuolar degradation [71], we next identified lysine residues (the site of ubiquitinylation) in the N- and C-terminal domains of the MgCBT2 transporter, in order to develop strategies to avoid this drop in transport activity causing cellobiose stuck fermentations.
3.3. Identification of Possible Lysine Residues Involved in Ubiquitinylation and Down-Regulation of the MgCBT2 Cellobiose Transporter Expressed in S. cerevisiae
Our data indicated that the best cellobiose transporter from yeasts that we cloned was MgCBT2 from M. guilliermondii, but it still could not allow the use and fermentation of all cellobiose (stuck fermentation) present in the medium. The analysis of the presence of lysine residues (K) with ubiquitinylation potential at the cytoplasmic C-terminal domain of the MgCBT2 transporter revealed 3 lysine residues (K534, K536 and K544) with high ubiquitinylation potential present in the last 12 amino acids of the transporter (Figure 4), while at the cytoplasmic N-terminal domain only 2 lysine residues (K10 and K20) were found with medium ubiquitinylation potential (Figure 4).
Considering that lysine residues with ubiquitinylation potential are involved in removing the transporters from the plasma membrane through endocytosis [71,72], we decided to remove these terminal lysine residues by simply truncating the MgCBT2 permease. We removed the last 12 amino acid residues of the protein (with 3 lysine residues with high ubiquitinylation potential) by introducing a premature stop codon during cloning as described in Materials and Methods, and this modified transporter was denominated MgCBT2ΔC (Figure 4). Another truncated version of MgCBT2 was produced were we not only removed the last 12 amino acid residues, but also the first 19 amino acid residues of the protein (after the initial methionine), producing the MgCBT2ΔNΔC transporter lacking all lysine residues with ubiquitinylation potential from both the N- and C-terminal domains (Figure 4).
These cellobiose transporters (MgCBT2, MgCBT2ΔC, and MgCBT2ΔNΔC), as well as the intracellular SpBGL7 β-glucosidase, were introduced into the genome of an industrial S. cerevisiae strain (MP-C5H1) capable of fermenting xylose, using CRISPR-Cas9 as described in Materials and Methods. As can be seen in Figure 5, while the strain harboring the MgCBT2ΔC transporter continued to produce stuck cellobiose fermentations as the strain with the wild-type MgCBT2 permease, the strain with the MgCBT2ΔNΔC permease was able to consume and ferment all the cellobiose present in the medium, producing higher levels of ethanol. However, is important to note that although this yeast strain with MgCBT2ΔNΔC consumes all the sugar, the ethanol yield (Yp/s = 0.38 ± 0.01 g of ethanol/g of consumed cellobiose) is very similar to the one obtained with the strain harboring the truncated MgCBT2ΔC transporter (Yp/s = 0.39 ± 0.01 g of ethanol/g of consumed cellobiose), and lower when compared with the wild-type MgCBT2 permease (Yp/s = 0.47 ± 0.02 g of ethanol/g of consumed cellobiose).
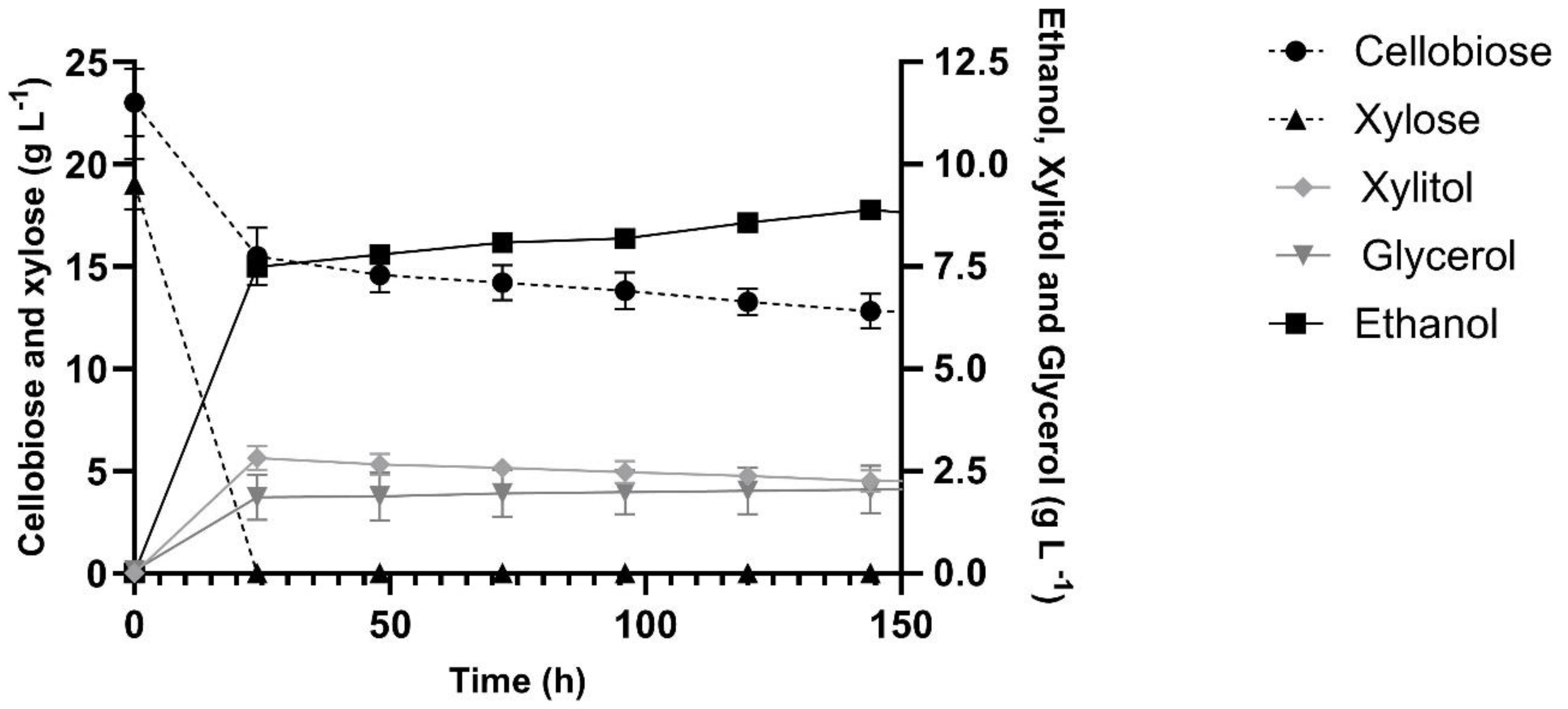
Finally, since direct cellobiose transport and intracellular hydrolysis by recombinant S. cerevisiae is interesting in the context of cellobiose-xylose co-fermentations [29,30,31], we performed such fermentations with the industrial xylose-fermenting strain MP-C5H1 harboring the MgCBT2ΔNΔC transporter and SpBGL7 β-glucosidase (strain MP-B7-CBT2ΔNΔC, Table 1). As can be seen in Figure 6, xylose is indeed totally consumed and fermented in the first 24 h, and although cellobiose is also consumed during that period, it is consumed very slowly after that time point (thus a stuck cellobiose fermentation), even considering that the strain harbors the MgCBT2ΔNΔC transporter lacking lysine residues in both the N- and C-terminal domains. While cellobiose is consumed slowly, also the xylitol produced from xylose fermentation also drops, thus both are probably contributing to the slight increase in ethanol produced (from 7.5 to 8.9 g/L) by the cells during the slow fermentation.
Figure 5.
Cellobiose consumption (A) and ethanol production (B) during fermentation of 20 g/L cellobiose in rich YP medium by the indicated industrial yeast strains harboring the intracellular SpBGL7 β-glucosidase and the MgCBT2, MgCBT2ΔC, and MgCBT2ΔNΔC cellobiose transporters.
Figure 5.
Cellobiose consumption (A) and ethanol production (B) during fermentation of 20 g/L cellobiose in rich YP medium by the indicated industrial yeast strains harboring the intracellular SpBGL7 β-glucosidase and the MgCBT2, MgCBT2ΔC, and MgCBT2ΔNΔC cellobiose transporters.
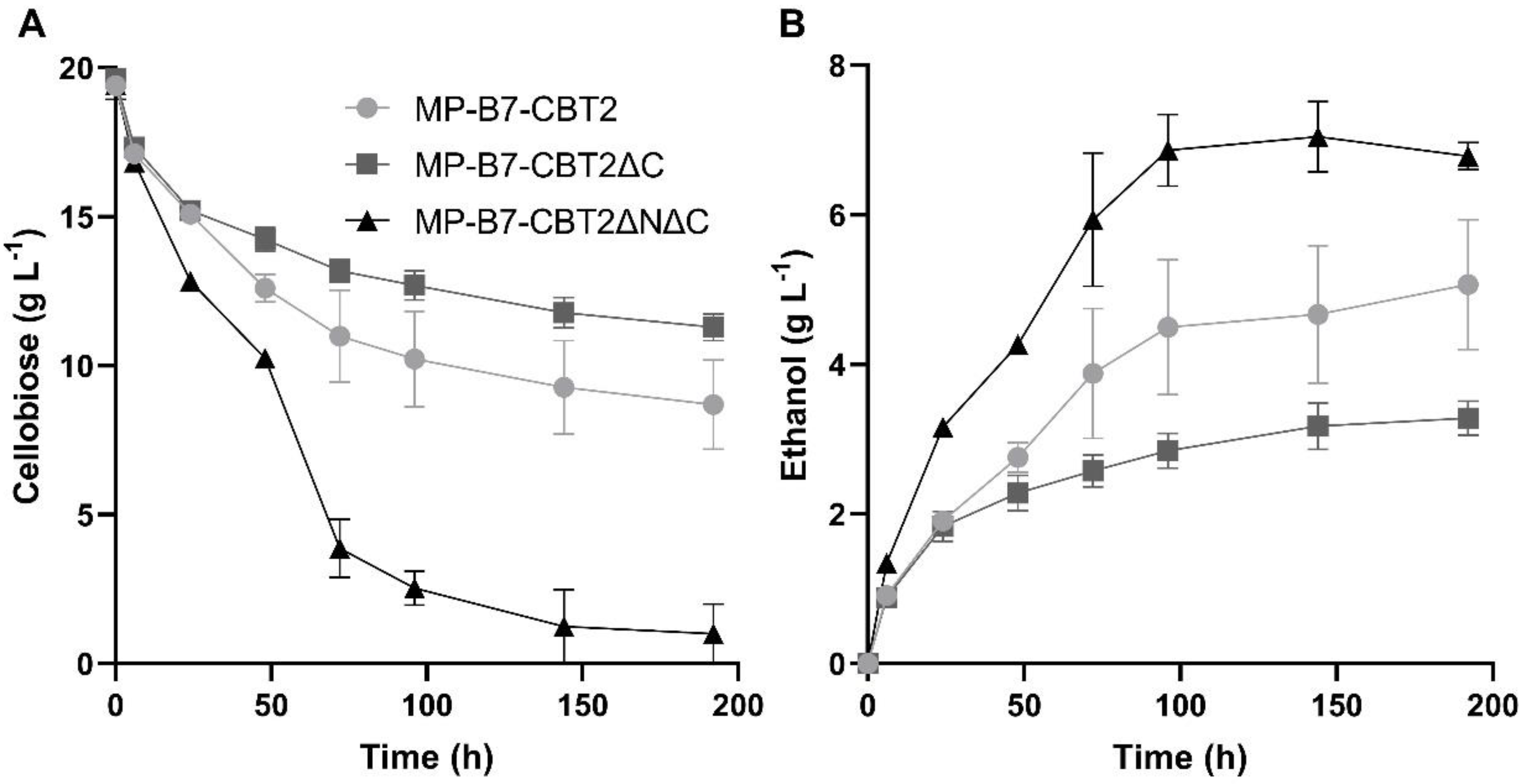
Figure 6.
Co-fermentation of 20 g/L cellobiose plus 20 g/L xylose in rich YP medium by the recombinant industrial yeast strain MP-B7-CBT2ΔNΔC harboring the intracellular SpBGL7 β-glucosidase and the MgCBT2ΔNΔC cellobiose transporter.
Figure 6.
Co-fermentation of 20 g/L cellobiose plus 20 g/L xylose in rich YP medium by the recombinant industrial yeast strain MP-B7-CBT2ΔNΔC harboring the intracellular SpBGL7 β-glucosidase and the MgCBT2ΔNΔC cellobiose transporter.
4. Discussion
We have cloned an intracellular β-glucosidase gene (SpBGL7) from the cellobiose fermenting yeast Sp. passalidarum [46], and expressed it in S. cerevisiae. While the β-glucosidase gh1-1 from N. crassa belongs to the GH1 family of glycosyl hydrolases, the cloned enzyme belongs to the GH3 family, where are found not only β-glucosidases, but also β-xylosidases and other enzymes. The SpBGL7 enzyme had higher affinity with pNPβG and pNPβX, and lower affinity with cellobiose, as already reported for other β-glucosidases characterized biochemically from yeasts [73,74]. Using the S. cerevisiae strain expressing the SpBGL7 enzyme allowed the identification and cloning of 3 yeast cellobiose transporters that permitted growth of the strain in the presence of cellobiose. Figure 7 shows that the 3 transporters (we included the HXT2.4 permease from Sc. stipitis [44], although the transporter we cloned was from Sc. illinoinensis) it is close to the known yeast cellobiose transporters, including the one cloned from L. starkeyi [45] and a cellobiose transporter CEL1 from Kluyveromyces dobzhanskii (that allows growth of S. cerevisiae on cellobiose, but no fermentation data is available) [75]. Note that in general Kluyveromyces yeasts can utilize cellobiose through the lactose permease encoded by LAC12. The cloned transporters are also closer to CDT-2 from N. crassa, and other transporters known to transport cellodextrins and xylobiose.
Although the 3 transporters allowed growth on cellobiose by the recombinant S. cerevisiae strain, only the MgCBT2 permease from M. guilliermondii allowed cellobiose fermentation, but the consumption of the sugar stopped after 50-75 h, leading to a stuck (incomplete) fermentation. Our analysis revealed that the reduced transport activity over time was responsible for this stuck fermentation, and that prompted us to look for lysine residues in the N- or C-terminal cytoplasmic domains of the MgCBT2 transporter that could be ubiquitinylated, a signal that triggers the endocytosis and vacuolar degradation of plasma membrane transporters. Indeed, some years ago Sen and co-workers [76] showed that S. cerevisiae cells expressing the CDT-2 cellobiose transporter also produced stuck cellobiose fermentations, and that cellobiose transport triggers the internalization of the permease, and they were able to identify that four α-arrestins (ROD1, ROG3, ALY1, and ALY2) are primarily responsible for this ubiquitinylation and internalization of the CDT-2 transporter. We have also shown that other heterologous sugar transporters expressed in S. cerevisiae are removed from the plasma membrane through action of the ROD1 and ROG3 α-arrestins [77]. In another approach, mutated versions of the CDT-2 permease were engineered changing cytoplasmic lysine residues into arginine, and revealed that lysine residues in the C-terminal domain were responsible for the α–arrestin mediated internalization of the transporter. A truncated C-terminal CDT-2 transporter (losing the last lysine, K522) was also shown to remain stable at the plasma membrane, and either the mutant CDT-2 permease lacking lysine residues at the C-terminal domain, or the same transporter truncated at this C-terminal domain, showed improved cellobiose consumption and fermentation [76]. Truncation at the C-terminal domain of other sugar transporters is an interesting strategy that allows stable expression of the permeases at the plasma membrane, allowing better sugar consumption and fermentation [77,78,79]. However, for other permeases (e.g. HXT1, GAL2) the lysine residues involved in ubiquitinylation and endocytosis can be present at the N-terminal domain [80,81]. For example, the HXT1 hexose transporter from S. cerevisiae lacking practically all the N-terminal domain is stable and functional at the plasma membrane, allowing efficient xylose fermentation by recombinant S. cerevisiae cells [56,81,82,83].
In the case of the MgCBT2 transporter, we found 3 lysine residues at the C-terminal domain with high ubiquitinylation potential, and two others (with medium ubiquitinylation potential) at the N-terminal domain. Truncation of the MgCBT2 permease at the C-terminal domain did not improved cellobiose consumption, when the transporter was expressed in an industrial S. cerevisiae strain (Figure 5), but when both cytoplasmic terminal domains were truncated (removing the first 19 amino acids, and the last 12 amino acids), the MgCBT2ΔNΔC transporter allowed total cellobiose consumption and fermentation by the recombinant yeast cells. However, during co-fermentation of cellobiose with xylose, the cells expressing the MgCBT2ΔNΔC transporter could not consume all the cellobiose present in the medium after the total consumption and fermentation of xylose, indicating that probably other factors are impairing the activity of the MgCBT2ΔNΔC permease, or the intracellular metabolism of cellobiose. One such factor could be the ethanol produced during fermentation, as many nutrient transporters are sensitive to high ethanol concentrations [84,85]. Indeed, when we performed a co-fermentation of 20 g/L sucrose plus 20 g/L cellobiose by the industrial yeast strain harboring the MgCBT2ΔNΔC transporter, sucrose was efficiently fermented in 24 h, producing 10 g/L ethanol, and although some cellobiose was consumed initially, from that time point on, cellobiose was consumed very slowly, producing a stuck fermentation.
In general cellobiose fermentation by recombinant S. cerevisiae yeasts is much more slower than the fermentation of other sugar (e.g. glucose), with also lower ethanol yields [26,27]. Thus, several other strategies have been employed to increase cellobiose consumption and fermentation, including mutagenesis, direct evolution/selection (also called evolutionary engineering) [44,86,87,88,89,90], as well as increasing the copy number of transporter or β–glucosidase genes [91,92]. Our results show that the identification of cytoplasmatic lysine residues in the N- or C-terminal domain capable of ubiquitinylation, and removing such residues by truncating the transporter, is another interesting strategy for enhancing cellobiose consumption and fermentation. Another issue that needs to be considered is the fact that cellobiose is not recognized by S. cerevisiae as a sugar to be fermented [93], and thus it is also required to introduce/engineer transcriptional factors to increase cellobiose fermentation by this industrial yeast [94,95]. Another strategy recently described to increase cellobiose fermentation is the deletion of the mitochondrial adenylate kinase encoded by the ADK2 gene, or the deletion of the MRX1 gene involved in the response to oxidative stress that was shown to increase cellobiose (and xylose) fermentation by recombinant S. cerevisiae [96,97]. It would be interesting to verify if some of these strategies would enhance cellobiose fermentation by an industrial S. cerevisiae expressing the intracellular β-glucosidase encoded by the SpBGL7 gene and the MgCBT2ΔNΔC cellobiose transporter.
5. Conclusions
In the present work we have cloned an intracellular β-glucosidase encoded by the SpBGL7 gene from the xylose-fermenting yeasts Sp. passalidarum, and three cellobiose transporters from yeasts (HXT2.4 from Sc. illinoinensis, CBT1 from C. tropicalis and CBT2 from M. guilliermondii) that allowed growth of recombinant S. cerevisiae cells on this carbon source. While the CBT2 transporter allowed incomplete (stuck) fermentation of cellobiose, the truncation of the N- and C-terminal domains, removing lysine residues with ubiquitinylation potential, allowed the complete consumption and fermentation of cellobiose by an industrial yeast strain. This work therefore highlights the importance of post-translational modifications in the correct expression of novel sugar transporters in recombinant S. cerevisiae strains.
Author Contributions
Conceptualization, L.G.K., M.M.K., and B.U.S.; investigation, methodology, and formal analysis, L.G.K., M.M.K., L.C.S., I.C.C.F., and M.J.L.; resources, C.F., M.J.L., K.J.V. and B.U.S.; writing—review and editing, C.F., M.J.L. and B.U.S.; project administration and funding acquisition, C.F., K.J.V., and B.U.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by grants and fellowships from the Brazilian agencies CAPES (process no 7207-15-8 and 88887.575075/2020-00), CNPq (process no 307290/2012-3, 478841/2013-2, 30862/2015-6, 308389/2019-0 and 309047/2023-4), and FINEP (process no 01.09.0566.00/1421-08), and also by the Fundação para a Ciência e a Tecnologia (FCT), Portugal (CAPES/FCT process no 359/14 and postdoctoral fellowship SFRH/BPD/102803/2014 to M.J.L.) and I.P., Project MOSTMICRO-ITQB. K.J.V. acknowledges funding from VIB, VLAIO, KU Leuven, FWO and project FWO SBO S004624N LAPLACE. This work is also part of the project “INCT Yeasts: Biodiversity, preservation and biotechnological innovation”, supported by grants and fellowships from CNPq (process 406564/2022-1).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data used in this study are included in the manuscript, and further inquiries can be directed to the corresponding author.
Acknowledgments
We would like to thank Dr. Carlos A. Rosa (Federal University of Minas Gerais, Brazil) for providing some of the yeast strains used in the present work.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Jaiswal, D.; de Souza, A.; Larsen, S.; Lebauer, D.S.; Miguez, F.E.; Sparovek, G.; Bollero, G.; Buckeridge, M.S.; Long, S.P. Brazilian sugarcane ethanol as an expandable green alternative to crude oil use. Nat. Clim. Chang. 2017, 7, 788–792. [Google Scholar] [CrossRef]
- Liu, Y.; Cruz-Morales, P.; Zargar, A.; Belcher, M.S.; Pang, B.; Englund, E.; Dan, Q.; Yin, K.; Keasling, J.D. Biofuels for a sustainable future. Cell 2021, 184, 1636–1647. [Google Scholar] [CrossRef] [PubMed]
- Jacobus, A.P.; Gross, J.; Evans, J.H.; Ceccato-Antonini, S.R.; Gombert, A.K. Saccharomyces cerevisiae strains used industrially for bioethanol production. Essays Biochem. 2021, 65, 147–161. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, L.V.; de Barros Grassi, M.C.; Gallardo, J.C.M.; Pirolla, R.A.S.; Calderón, L.L.; de Carvalho-Netto, O.V.; et al. Second-generation ethanol: The need is becoming a reality. Ind. Biotechnol. 2016, 12, 40–57. [Google Scholar] [CrossRef]
- Igwebuike, C.M.; Awad, S.; Andrès, Y. Renewable energy potential: Second-generation biomass as feedstock for bioethanol production. Molecules 2024, 29, 1619. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Ploessl, D.; Shao, Z. Enhancing the co-utilization of biomass-derived mixed sugars by yeasts. Front. Microbiol. 2019, 9, 3264. [Google Scholar] [CrossRef]
- Broda, M.; Yelle, D.J.; Serwańska, K. Bioethanol production from lignocellulosic biomass-Challenges and solutions. Molecules 2022, 27, 8717. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Sharma, V.; Tsai, M.L.; Chen, C.W.; Sun, P.P.; Nargotra, P.; Wang, J.X.; Dong, C.D. Development of lignocellulosic biorefineries for the sustainable production of biofuels: Towards circular bioeconomy. Bioresour. Technol. 2023, 381, 129145. [Google Scholar] [CrossRef]
- da Silva, A.S.; Espinheira, R.P.; Teixeira, R.S.S.; de Souza, M.F.; Ferreira-Leitão, V.; Bon, E.P.S. Constraints and advances in high-solids enzymatic hydrolysis of lignocellulosic biomass: a critical review. Biotechnol. Biofuels. 2020, 13, 58. [Google Scholar] [CrossRef]
- Østby, H.; Hansen, L.D.; Horn, S.J.; Eijsink, V.G.H.; Várnai, A. Enzymatic processing of lignocellulosic biomass: principles, recent advances and perspectives. J. Ind. Microbiol. Biotechnol. 2020, 47, 623–657. [Google Scholar] [CrossRef]
- Sørensen, A.; Lübeck, M.; Lübeck, P.S.; Ahring, B.K. Fungal β-glucosidases: a bottleneck in industrial use of lignocellulosic materials. Biomolecules 2013, 3, 612–631. [Google Scholar] [CrossRef]
- Singhania, R.R.; Patel, A.K.; Sukumaran, R.K.; Larroche, C.; Pandey, A. Role and significance of β-glucosidases in the hydrolysis of cellulose for bioethanol production. Bioresour. Technol. 2013, 127, 500–507. [Google Scholar] [CrossRef]
- Pryor, S.W.; Nahar, N. β-glucosidase supplementation during biomass hydrolysis: How low can we go? Biomass Bioenergy 2015, 80, 298–302. [Google Scholar] [CrossRef]
- Ferreira, R.G.; Azzoni, A.R.; Freitas, S. On the production cost of lignocellulose-degrading enzymes. Biofuels Bioprod. Bioref. 2021, 15, 85–99. [Google Scholar] [CrossRef]
- Barbosa, A.M.; Giese, E.C.; Dekker, R.F.; Borsato, D.; Briones Pérez, A.I.; Ubeda Iranzo, J.F. Extracellular β-glucosidase production by the yeast Debaryomyces pseudopolymorphus UCLM-NS7A: optimization using response surface methodology. N. Biotechnol. 2010, 27, 374–381. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Z.L.; Weber, S.A.; Zhang, X. Two new native β-glucosidases from Clavispora NRRL Y-50464 confer its dual function as cellobiose fermenting ethanologenic yeast. PLoS One 2016, 11, e0151293. [Google Scholar] [CrossRef]
- Vaz, J.E.; Rabelo, L.; Zaiter, M.A.; Pereira, W.E.S.; Metzker, G.; Boscolo, M.; da Silva, R.; Gomes, E.; da Silva, R.R. Functional properties and potential application of ethanol tolerant β-glucosidases from Pichia ofunaensis and Trichosporon multisporum yeasts. 3 Biotech. 2021, 11, 467. [Google Scholar] [CrossRef]
- Albarello, M.L.; Giehl, A.; Tadioto, V.; dos Santos, A.A.; Milani, L.M.; Bristot, J.C.; Tramontin, M.A.; Treichel, H.; Bernardi, O.; et al. Analysis of the holocellulolytic and fermentative potentials of yeasts isolated from the gut of Spodoptera frugiperda larvae. Bioener. Res. 2023, 16, 2046–2057. [Google Scholar] [CrossRef]
- Jeffries, T.W.; Van Vleet, J.R. Pichia stipitis genomics, transcriptomics, and gene clusters. FEMS Yeast Res. 2009, 9, 793–807. [Google Scholar] [CrossRef]
- Santos, R.O.; Cadete, R.M.; Badotti, F.; Mouro, A.; Wallheim, D.O.; Gomes, F.C.; Stambuk, B.U.; Lachance, M.A.; Rosa, C.A. Candida queiroziae sp. nov., a cellobiose-fermenting yeast species isolated from rotting wood in Atlantic Rain Forest. Antonie Van Leeuwenhoek 2011, 99, 635–642. [Google Scholar] [CrossRef]
- Lopes, M.R.; Lara, C.A.; Moura, M.E.F.; Uetanabaro, A.P.T.; Morais, P.B.; Vital, M.J.S.; Rosa, C.A. Characterization of the diversity and physiology of cellobiose-fermenting yeasts isolated from rotting wood in Brazilian ecosystems. Fungal Biol. 2018, 122, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Barrilli, É.T.; Tadioto, V.; Milani, L.M.; Deoti. J.R.; Fogolari, O.; Müller, C.; Barros, K.O.; Rosa, C.A.; dos Santos, A.A.; et al. Biochemical analysis of cellobiose catabolism in Candida pseudointermedia strains isolated from rotten wood. Arch. Microbiol. 2020, 202, 1729–1739. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Choi, H.J.; Lee, Y.R.; Kim, S.J.; Lee, S.; Lee, W.H. Comprehensive characterization of mutant Pichia stipitis co-fermenting cellobiose and xylose through genomic and transcriptomic analyses. J. Microbiol. Biotechnol. 2022, 32, 1485–1495. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.; Hipp, J.; Trinh, C.T. Activating and elucidating metabolism of complex sugars in Yarrowia lipolytica. Appl. Environ. Microbiol. 2015, 82, 1334–1345. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Duquesne, S.; Bozonnet, S.; Cioci, G.; Nicaud, J,M. ; Marty, A.; O'Donohue, M.J. Development of cellobiose-degrading ability in Yarrowia lipolytica strain by overexpression of endogenous genes. Biotechnol. Biofuels 2015, 8, 109. [Google Scholar] [CrossRef]
- Galazka, J.M.; Tian, C.; Beeson, W.T.; Martinez, B.; Glass, N.L.; Cate, J.H. Cellodextrin transport in yeast for improved biofuel production. Science 2010, 330, 84–86. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.H.; Kang, K.H.; Jin, Y.S.; Seo, J.H. Molecular cloning and expression of fungal cellobiose transporters and β-glucosidases conferring efficient cellobiose fermentation in Saccharomyces cerevisiae. J. Biotechnol. 2014, 169, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, D.L.; Matsushika, A.; de Sales, B.B.; Goshima, T.; Bon, E.P.S.; Stambuk, B.U. Xylose and xylose/glucose co-fermentation by recombinant Saccharomyces cerevisiae strains expressing individual hexose transporters. Enzyme Microb. Technol. 2014, 63, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Du, J.; Sun, J.; Galazka, J.M.; Glass, N.L.; Cate, J.H.; Yang, X.; Zhao, H. Overcoming glucose repression in mixed sugar fermentation by co-expressing a cellobiose transporter and a β-glucosidase in Saccharomyces cerevisiae. Mol. Biosyst. 2010, 6, 2129–2132. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.J.; Galazka, J.M.; Kim, S.R.; Choi, J.H.; Yang, X.; Seo, J.H.; Glass, N.L.; Cate, J.H.; Jin, Y.S. Engineered Saccharomyces cerevisiae capable of simultaneous cellobiose and xylose fermentation. Proc. Natl. Acad. Sci. USA 2011, 108, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.J.; Kim, S.R.; Kim, H.; Du, J.; Cate, J.H.; Jin, Y.S. Continuous co-fermentation of cellobiose and xylose by engineered Saccharomyces cerevisiae. Bioresour, Technol. 2013, 149, 525–531. [Google Scholar] [CrossRef]
- Parisutham, V.; Chandran, S.P.; Mukhopadhyay, A.; Lee, S.K.; Keasling, J.D. Intracellular cellobiose metabolism and its applications in lignocellulose-based biorefineries. Bioresour. Technol. 2017, 239, 496–506. [Google Scholar] [CrossRef]
- Oh, E.J.; Jin, Y.S. Engineering of Saccharomyces cerevisiae for efficient fermentation of cellulose. FEMS Yeast Res. 2020, 20, foz089. [Google Scholar] [CrossRef]
- Cai, P.; Gu, R.; Wang, B.; Li, J.; Wan, L.; Tian, C.; Ma, Y. Evidence of a critical role for cellodextrin transporter 2 (CDT-2) in both cellulose and hemicellulose degradation and utilization in Neurospora crassa. PLoS One 2014, 9, e89330. [Google Scholar] [CrossRef]
- Znameroski, E.A.; Li, X.; Tsai, J.C.; Galazka, J.M.; Glass, N.L.; Cate, J.H. Evidence for transceptor function of cellodextrin transporters in Neurospora crassa. J. Biol. Chem. 2014, 289, 2610–2619. [Google Scholar] [CrossRef]
- Li, X.; Yu, V.Y.; Lin, Y.; Chomvong, K.; Estrela, R.; Park, A.; Liang, J.M.; Znameroski, E.A.; Feehan, J.; Kim, S.R.; Jin, Y.S.; Glass, N.L.; Cate, J.H. Expanding xylose metabolism in yeast for plant cell wall conversion to biofuels. Elife 2015, 4, e05896. [Google Scholar] [CrossRef]
- Kim, H.; Lee, W.H.; Galazka, J.M.; Cate, J.H.; Jin, Y.S. Analysis of cellodextrin transporters from Neurospora crassa in Saccharomyces cerevisiae for cellobiose fermentation. Appl. Microbiol. Biotechnol. 2014, 98, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Kou, Y.; Xu, J.; Cao, Y.; Zhao, G.; Shao, J.; Wang, H.; Wang, Z.; Bao, X.; Chen, G.; Liu, W. Two major facilitator superfamily sugar transporters from Trichoderma reesei and their roles in induction of cellulase biosynthesis. J. Biol. Chem. 2013, 288, 32861–72. [Google Scholar] [CrossRef]
- dos Reis, T.F.; de Lima, P.B.; Parachin, N.S.; Mingossi, F.B.; de Castro Oliveira, J.V.; Ries, L.N.; Goldman, G.H. Identification and characterization of putative xylose and cellobiose transporters in Aspergillus nidulans. Biotechnol. Biofuels 2016, 9, 204. [Google Scholar] [CrossRef]
- Zhang, C.; Acosta-Sampson, L.; Yu, V.Y.; Cate, J.H.D. Screening of transporters to improve xylodextrin utilization in the yeast Saccharomyces cerevisiae. PLoS One 2017, 12, e0184730. [Google Scholar] [CrossRef]
- Nogueira, K.M.V.; de Paula, R.G.; Antoniêto, A.C.C.; dos Reis, T.F.; Carraro, C.B.; Silva, A.C.; Almeida, F.; Rechia, C.G.V.; et al. Characterization of a novel sugar transporter involved in sugarcane bagasse degradation in Trichoderma reesei. Biotechnol. Biofuels 2018, 11, 84. [Google Scholar] [CrossRef]
- Havukainen, S.; Valkonen, M.; Koivuranta, K.; Landowski, C.P. Studies on sugar transporter CRT1 reveal new characteristics that are critical for cellulase induction in Trichoderma reesei. Biotechnol. Biofuels 2020, 13, 158. [Google Scholar] [CrossRef]
- Lin, H.; Zhao, J.; Zhang, Q.; Cui, S.; Fan, Z.; Chen, H.; Tian, C. Identification and characterization of a cellodextrin transporter in Aspergillus niger. Front. Microbiol. 2020, 11, 145. [Google Scholar] [CrossRef]
- Ha, S.J.; Kim, H.; Lin, Y.; Jang, M.U.; Galazka, J.M.; Kim, T.J.; Cate, J.H.; Jin, Y.S. Single amino acid substitutions in HXT2.4 from Scheffersomyces stipitis lead to improved cellobiose fermentation by engineered Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2013, 79, 1500–1507. [Google Scholar] [CrossRef] [PubMed]
- de Ruijter, J.C.; Igarashi, K.; Penttilä, M. The Lipomyces starkeyi gene Ls120451 encodes a cellobiose transporter that enables cellobiose fermentation in Saccharomyces cerevisiae. FEMS Yeast Res. 2020, 20, foaa019. [Google Scholar] [CrossRef] [PubMed]
- Long, T.M.; Su, Y.K.; Headman, J.; Higbee, A.; Willis, L.B.; Jeffries, T.W. Cofermentation of glucose, xylose, and cellobiose by the beetle-associated yeast Spathaspora passalidarum. Appl. Environ. Microbiol. 2012, 78, 5492–500. [Google Scholar] [CrossRef] [PubMed]
- Lauwers, E.; Erpapazoglou, Z.; Haguenauer-Tsapis, R.; André, B. The ubiquitin code of yeast permease trafficking. Trends Cell Biol. 2010, 20, 196–204. [Google Scholar] [CrossRef]
- Barata-Antunes, C.; Alves, R.; Talaia, G.; Casal, M.; Gerós, H.; Mans, R.; Paiva, S. Endocytosis of nutrient transporters in fungi: The ART of connecting signaling and trafficking. Comput. Struct. Biotechnol. J. 2021, 19, 1713–1737. [Google Scholar] [CrossRef] [PubMed]
- Meselson, M.; Yuan, R. DNA restriction enzyme from E. coli. Nature 1968, 217, 1110–1114. [Google Scholar] [CrossRef]
- Lara, C.A.; Santos, R.O.; Cadete, R.M.; Ferreira, C.; Marques, S.; Gírio, F.; Oliveira, E.S.; Rosa, C.A.; Fonseca, C. Identification and characterisation of xylanolytic yeasts isolated from decaying wood and sugarcane bagasse in Brazil. Antonie Van Leeuwenhoek. 2014, 105, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Cadete, R.M.; Melo-Cheab, M.A.; Dussán, K.J.; Rodrigues, R.C.L.B.; da Silva, S.S.; Gomes, F.C.O.; Rosa, C.A. Production of bioethanol in sugarcane bagasse hemicellulosic hydrolysate by Scheffersomyces parashehatae, Scheffersomyces illinoinensis and Spathaspora arborariae isolated from Brazilian ecosystems. J. Appl. Microbiol. 2017, 123, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Mouro, A.; dos Santos, A.A.; Agnolo, D.D.; Gubert, G.F.; Bon, E.P.S.; Rosa, C.A.; Fonseca, C.; Stambuk, B.U. Combining xylose reductase from Spathaspora arborariae with xylitol dehydrogenase from Spathaspora passalidarum to promote xylose consumption and fermentation into xylitol by Saccharomyces cerevisiae. Fermentation 2020, 6, 72. [Google Scholar] [CrossRef]
- Entian, K.; Kotter, P. Yeast genetic strain and plasmid collections. Methods Microbiol. 2007, 36, 629–666. [Google Scholar] [CrossRef]
- Babrzadeh, F.; Jalili, R.; Wang, C.; Shokralla, S.; Pierce, S.; Robinson-Mosher, A.; Nyren, P.; Shafer, R.W.; Basso, L.C.; et al. . Whole-genome sequencing of the efficient industrial fuel-ethanol fermentative Saccharomyces cerevisiae strain CAT-1. Mol. Genet. Genomics 2012, 287, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Muller, G.; de Godoy, V.R.; Dário, M.G.; Duval, E.H.; Alves-Jr, S.L.; Bücker, A.; Rosa, C.A.; Dunn, B.; Sherlock, G.; Stambuk, B.U. Improved sugarcane-based fermentation processes by an industrial fuel-ethanol yeast strain. J. Fungi 2023, 9, 803. [Google Scholar] [CrossRef] [PubMed]
- Pereira, I.O.; dos Santos, Â.A.; Gonçalves, D.L.; Purificação, M.; Guimarães, N.C.; Tramontina, R.; Coutouné, N.; Zanella, E.; et al. Comparison of Spathaspora passalidarum and recombinant Saccharomyces cerevisiae for integration of first- and second-generation ethanol production. FEMS Yeast Res. 2021, 21, foab048. [Google Scholar] [CrossRef]
- Mumberg, D.; Müller, R.; Funk, M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 1995, 156, 119–122. [Google Scholar] [CrossRef]
- Vyas, V.K.; Bushkin, G.G.; Bernstein, D.A.; Getz, M.A.; Sewastianik, M.; Barrasa, M.I.; Bartel, D.P.; Fink, G.R. New CRISPR mutagenesis strategies reveal variation in repair mechanisms among fungi. mSphere 2018, 3, e00154–18. [Google Scholar] [CrossRef]
- Gietz, D.; St Jean, A.; Woods, R.A.; Schiestl, R.H. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992, 20, 1425. [Google Scholar] [CrossRef]
- Ausubel, F.M.; Brent, R.; Kingston, R.E.; Moore, D.D.; Seidman, J.G.; Smith, J.A.; Struhl, K. Short Protocols in Molecular Biology, 3rd ed.; John Wiley & Sons: New York, USA, 1995. [Google Scholar]
- Butler, G.; Rasmussen, M.D.; Lin, M.F.; Santos, M.A.; Sakthikumar, S.; Munro, C.A.; Rheinbay, E.; Grabherr, M.; Forche, A.; et al. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 2009, 459, 657–662. [Google Scholar] [CrossRef]
- Jeffries, T.W.; Grigoriev, I.V.; Grimwood, J.; Laplaza, J.M.; Aerts, A.; Salamov, A.; Schmutz, J.; Lindquist, E.; Dehal, P.; Shapiro, H.; et al. Genome sequence of the lignocellulose-bioconverting and xylose-fermenting yeast Pichia stipitis. Nat. Biotechnol. 2007, 25, 319–326. [Google Scholar] [CrossRef]
- Wohlbach, D.J.; Kuo, A.; Sato, T.K.; Potts, K.M.; Salamov, A.A.; Labutti, K.M.; Sun, H.; Clum, A.; Pangilinan, J.L.; Lindquist, et al. Comparative genomics of xylose-fermenting fungi for enhanced biofuel production. Proc. Natl. Acad. Sci. USA 2011, 108, 13212–13217. [Google Scholar] [CrossRef]
- Reider Apel, A.; d'Espaux, L.; Wehrs, M.; Sachs, D.; Li, R.A.; Tong, G.J.; Garber, M.; Nnadi, O.; Zhuang, W.; Hillson, N.J.; Keasling, J.D.; Mukhopadhyay, A. A Cas9-based toolkit to program gene expression in Saccharomyces cerevisiae. Nucleic Acids Res. 2017, 45, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Stambuk, B.U. A simple experiment illustrating metabolic regulation: Induction versus repression of yeast α-glucosidase. Biochem. Educ. 1999, 27, 177–180. [Google Scholar] [CrossRef]
- Hollatz, C.; Stambuk, B.U. Colorimetric determination of active α-glucoside transport in Saccharomyces cerevisiae. J. Microbiol. Methods. 2001, 46, 253–259. [Google Scholar] [CrossRef]
- Pirovano W, Feenstra KA, Heringa J. PRALINETM: a strategy for improved multiple alignment of transmembrane proteins. Bioinformatics 2008, 24, 492–497. [Google Scholar] [CrossRef]
- Li, A.; Gao, X.; Ren, J.; Jin, C.; Xue, Y. BDM-PUB: Computational prediction of protein ubiquitination sites with a bayesian discriminant method, 2009. Available online: http://bdmpub.biocuckoo.org/prediction.php (accessed on 30 November 2021).
- Radivojac, P.; Vacic; V. ; Haynes, C.; Cocklin, R.R.; Mohan, A.; Heyen, J.W.; Goebl, M.G.; Iakoucheva, L.M. Identification, analysis, and prediction of protein ubiquitination sites. Proteins 2010, 78, 365–380. [Google Scholar] [CrossRef]
- Urbina, H.; Blackwel,l M. Multilocus phylogenetic study of the Scheffersomyces yeast clade and characterization of the N-terminal region of xylose reductase gene. PLoS One 2012, 7, e39128. [Google Scholar] [CrossRef]
- Stambuk, B.U. Transcriptional and posttranslational regulation of a membrane nutrient transporter. Biochem. Mol. Biol. Educ. 2002, 30, 388–393. [Google Scholar] [CrossRef]
- Horák, J. The role of ubiquitin in down-regulation and intracellular sorting of membrane proteins: insights from yeast. Biochim. Biophys. Acta 2003, 1614, 139–155. [Google Scholar] [CrossRef]
- Skory, C.D.; Freer, S.N.; Bothast, R.J. Properties of an intracellular β-glucosidase purified from the cellobiose-fermenting yeast Candida wickerhamii. Appl. Microbiol. Biotechnol. 1996, 46, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Gueguen, Y.; Chemardin, P.; Arnaud, A. Purification and characterization of an intracellular β-glucosidase from a Candida sake strain isolated from fruit juices. Appl. Biochem. Biotechnol. 2001, 95, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Varela, J.A.; Puricelli, M.; Ortiz-Merino, R.A.; Giacomobono, R.; Braun-Galleani, S.; Wolfe, K.H.; Morrissey, J.P. Origin of lactose fermentation in Kluyveromyces lactis by interspecies transfer of a neo-functionalized gene cluster during domestication. Curr. Biol. 2019, 29, 4284–4290e2. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Acosta-Sampson, L.; Alvaro, C.G.; Ahn, J.S.; Cate, J.H.; Thorner, J. Internalization of heterologous sugar transporters by endogenous α-arrestins in the yeast Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2016, 82, 7074–7085. [Google Scholar] [CrossRef] [PubMed]
- Knychala, M.M.; dos Santos, A.A.; Kretzer, L.G.; Gelsleichter, F.; Leandro, M.J.; Fonseca, C.; Stambuk, B.U. Strategies for efficient expression of heterologous monosaccharide transporters in Saccharomyces cerevisiae. J. Fungi 2022, 8, 84. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Schmitz, O.; Alper, H.S. Enabling glucose/xylose co-transport in yeast through the directed evolution of a sugar transporter. Appl. Microbiol. Biotechnol. 2016, 100, 10215–10223. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Tao, X.; Ran, G.; Deng, Y.; Wang, H.; Tan, L.; Pang, Z. Molecular modification enhances xylose uptake by the sugar transporter KM_SUT5 of Kluyveromyces marxianus. Int. J. Mol. Sci. 2024, 25, 8322. [Google Scholar] [CrossRef] [PubMed]
- Tamayo Rojas, S.A.; Schmidl, S.; Boles, E.; Oreb, M. Glucose-induced internalization of the S. cerevisiae galactose permease Gal2 is dependent on phosphorylation and ubiquitination of its aminoterminal cytoplasmic tail. FEMS Yeast Res. 2021, 21, foab019. [Google Scholar] [CrossRef]
- Roy, A.; Kim, Y.B.; Cho, K.H.; Kim, J.H. Glucose starvation-induced turnover of the yeast glucose transporter Hxt1. Biochim. Biophys. Acta 2014, 1840, 2878–2885. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Jimenez, F.D.; Pereira, I.O.; Ribeiro, M.P.A.; Sargo, C.R.; dos Santos, A.A.; Zanella, E.; Stambuk, B.U.; et al. Integration of first- and second-generation ethanol production: Evaluation of a mathematical model to describe sucrose and xylose co-fermentation by recombinant Saccharomyces cerevisiae. Renew. Energy 2022, 192, 326–339. [Google Scholar] [CrossRef]
- de Oliveira Pereira, I.; dos Santos, Â.A.; Guimarães, N.C.; Lima, C.S.; Zanella, E.; Matsushika, A.; Rabelo, S.C.; Stambuk, B.U.; Ienczak, J.L. First- and second-generation integrated process for bioethanol production: Fermentation of molasses diluted with hemicellulose hydrolysate by recombinant Saccharomyces cerevisiae. Biotechnol. Bioeng. 2024, 121, 1314–1324. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.; Sousa, M.J.; Cardoso, H.; Inácio, J.; Silva, S.; Spencer-Martins, I.; Leão, C. Ethanol tolerance of sugar transport, and the rectification of stuck wine fermentations. Microbiology 2008, 154, 422–430. [Google Scholar] [CrossRef]
- Swinnen, S.; Goovaerts, A.; Schaerlaekens, K.; Dumortier, F.; Verdyck, P.; Souvereyns, K.; Van Zeebroeck, G.; Foulquié-Moreno, M.R.; Thevelein, J.M. Auxotrophic mutations reduce tolerance of Saccharomyces cerevisiae to very high levels of ethanol stress. Eukaryot. Cell 2015, 14, 884–897. [Google Scholar] [CrossRef]
- Eriksen, D.T.; Hsieh, P.C.; Lynn, P.; Zhao, H. Directed evolution of a cellobiose utilization pathway in Saccharomyces cerevisiae by simultaneously engineering multiple proteins. Microb. Cell Fact. 2013, 12, 61. [Google Scholar] [CrossRef]
- Lian, J.; Li, Y.; HamediRad, M.; Zhao, H. Directed evolution of a cellodextrin transporter for improved biofuel production under anaerobic conditions in Saccharomyces cerevisiae. Biotechnol. Bioeng. 2014, 111, 1521–31. [Google Scholar] [CrossRef]
- Hu, M.L.; Zha, J.; He, L.W.; Lv, Y.J.; Shen, M.H.; Zhong, C.; Li, B.Z.; Yuan, Y.J. Enhanced bioconversion of cellobiose by industrial Saccharomyces cerevisiae used for cellulose utilization. Front. Microbiol. 2016, 7, 241. [Google Scholar] [CrossRef]
- Oh, E.J.; Kwak, S.; Kim, H.; Jin, Y.S. Transporter engineering for cellobiose fermentation under lower pH conditions by engineered Saccharomyces cerevisiae. Bioresour. Technol. 2017, 245, 1469–1475. [Google Scholar] [CrossRef]
- Kim, H.; Oh, E.J.; Lane, S.T.; Lee, W.H.; Cate, J.H.D.; Jin, Y.S. Enhanced cellobiose fermentation by engineered Saccharomyces cerevisiae expressing a mutant cellodextrin facilitator and cellobiose phosphorylase. J. Biotechnol. 2018, 275, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.J.; Skerker, J.M.; Kim, S.R.; Wei, N.; Turner, T.L.; Maurer, M.J.; Arkin, A.P.; Jin, Y.S. Gene amplification on demand accelerates cellobiose utilization in engineered Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2016, 82, 3631–3639. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, WH.; Turner, T.L.; Kwak, S.; Jin, Y.S. An extra copy of the β-glucosidase gene improved the cellobiose fermentation capability of an engineered Saccharomyces cerevisiae strain. 3 Biotech, 2019, 9, 367. [Google Scholar] [CrossRef]
- Chomvong, K.; Benjamin, D.I.; Nomura, D.K.; Cate, J.H.D. Cellobiose consumption uncouples extracellular glucose sensing and glucose metabolism in Saccharomyces cerevisiae. mBio. 2017, 8, e00855–17. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Chomvong, K.; Acosta-Sampson, L.; Estrela, R.; Galazka, J.M.; Kim, S.R.; Jin, Y.S.; Cate, J.H. Leveraging transcription factors to speed cellobiose fermentation by Saccharomyces cerevisiae. Biotechnol. Biofuels 2014, 7, 126. [Google Scholar] [CrossRef]
- Sullivan, S.F.; Shetty, A.; Bharadwaj, T.; Krishna, N.; Trivedi, V.D.; Endalur Gopinarayanan, V.; Chappell, T.C.; Sellers, D.M.; et al. Towards universal synthetic heterotrophy using a metabolic coordinator. Metab. Eng. 2023, 79, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, Z.; Lu, M.; Ding, B.; Chen, S.; Wen, Z.; Yu, Y.; Zhou, L.; Jin, M. Rapid evolution and mechanism elucidation for efficient cellobiose-utilizing Saccharomyces cerevisiae through synthetic chromosome rearrangement and modification by LoxPsym-mediated evolution. Bioresour. Technol. 2022, 356, 127268. [Google Scholar] [CrossRef]
- Blount, B.A.; Gowers, G.F.; Ho, J.C.H.; Ledesma-Amaro, R.; Jovicevic, D.; McKiernan, R.M.; Xie, Z.X.; Li, B.Z.; Yuan, Y.J.; Ellis, T. Rapid host strain improvement by in vivo rearrangement of a synthetic yeast chromosome. Nat. Commun. 2018, 9, 1932. [Google Scholar] [CrossRef]
Figure 1.
Cell growth (A) and cellobiose consumption (B) in YNB medium containing 20 g/L cellobiose by the indicated yeast strains harboring the intracellular SpBGL7 β-glucosidase (strain B7), or this strain also transformed with the pGPD-426 plasmids containing the genes (CtCBT1, MgCBT2 and SiHXT2.4) encoding sugar transporters.
Figure 1.
Cell growth (A) and cellobiose consumption (B) in YNB medium containing 20 g/L cellobiose by the indicated yeast strains harboring the intracellular SpBGL7 β-glucosidase (strain B7), or this strain also transformed with the pGPD-426 plasmids containing the genes (CtCBT1, MgCBT2 and SiHXT2.4) encoding sugar transporters.
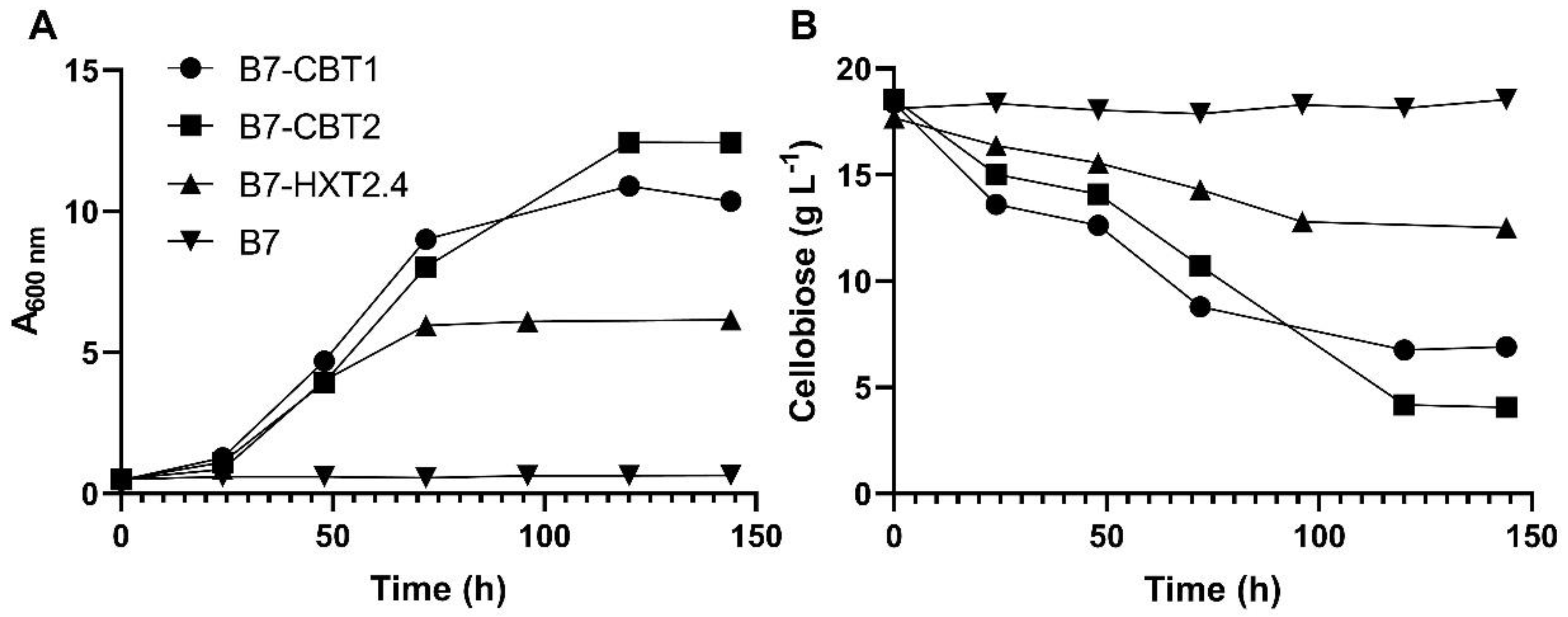
Figure 2.
Cellobiose consumption (A) and ethanol production (B) during fermentation of 20 g/L cellobiose in rich YP medium by 10 g dry cell weight/L of the indicated yeast strains harboring the SpBGL7 β-glucosidase and the genes CtCBT1, MgCBT2 and SiHXT2.4 encoding sugar transporters.
Figure 2.
Cellobiose consumption (A) and ethanol production (B) during fermentation of 20 g/L cellobiose in rich YP medium by 10 g dry cell weight/L of the indicated yeast strains harboring the SpBGL7 β-glucosidase and the genes CtCBT1, MgCBT2 and SiHXT2.4 encoding sugar transporters.
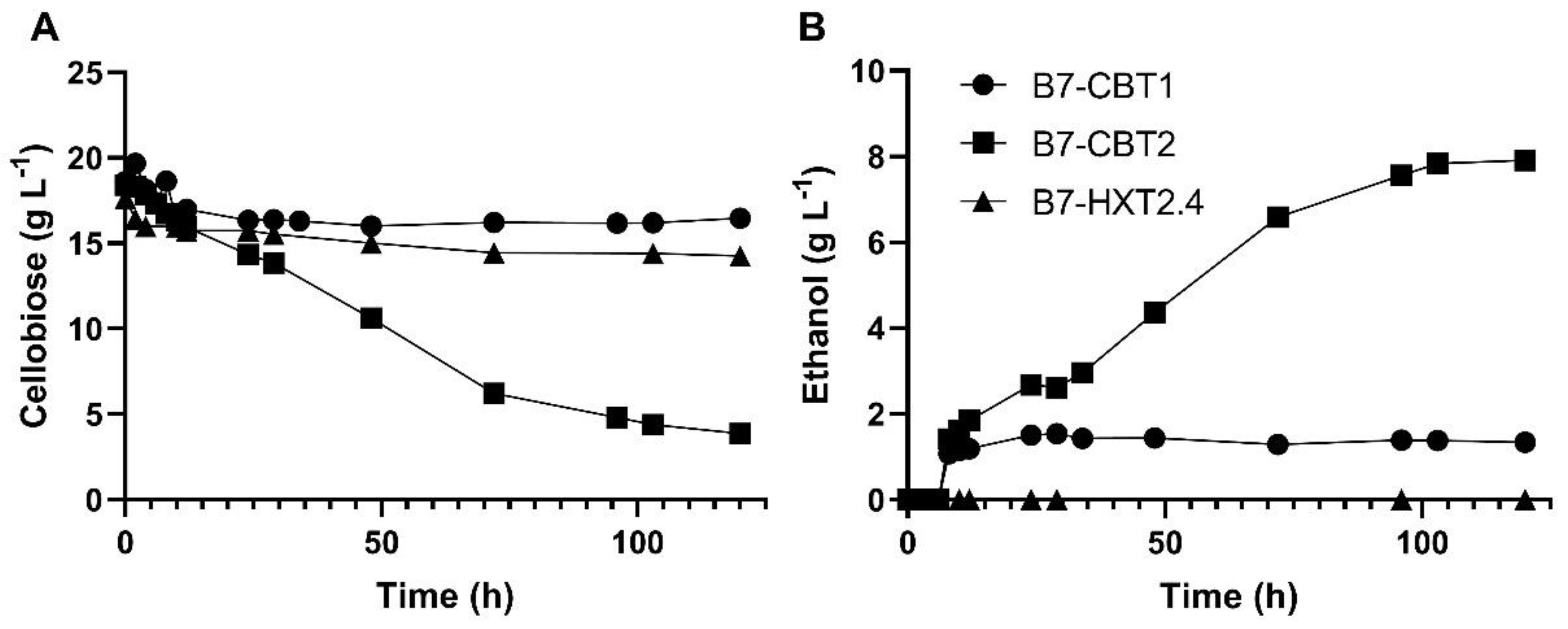
Figure 3.
Cellobiose consumption and ethanol production (A), and pNPβG intracellular hydrolysis and transport activities (B) by strain B7-CBT2 during fermentation of 20 g/L cellobiose in rich YP medium.
Figure 3.
Cellobiose consumption and ethanol production (A), and pNPβG intracellular hydrolysis and transport activities (B) by strain B7-CBT2 during fermentation of 20 g/L cellobiose in rich YP medium.
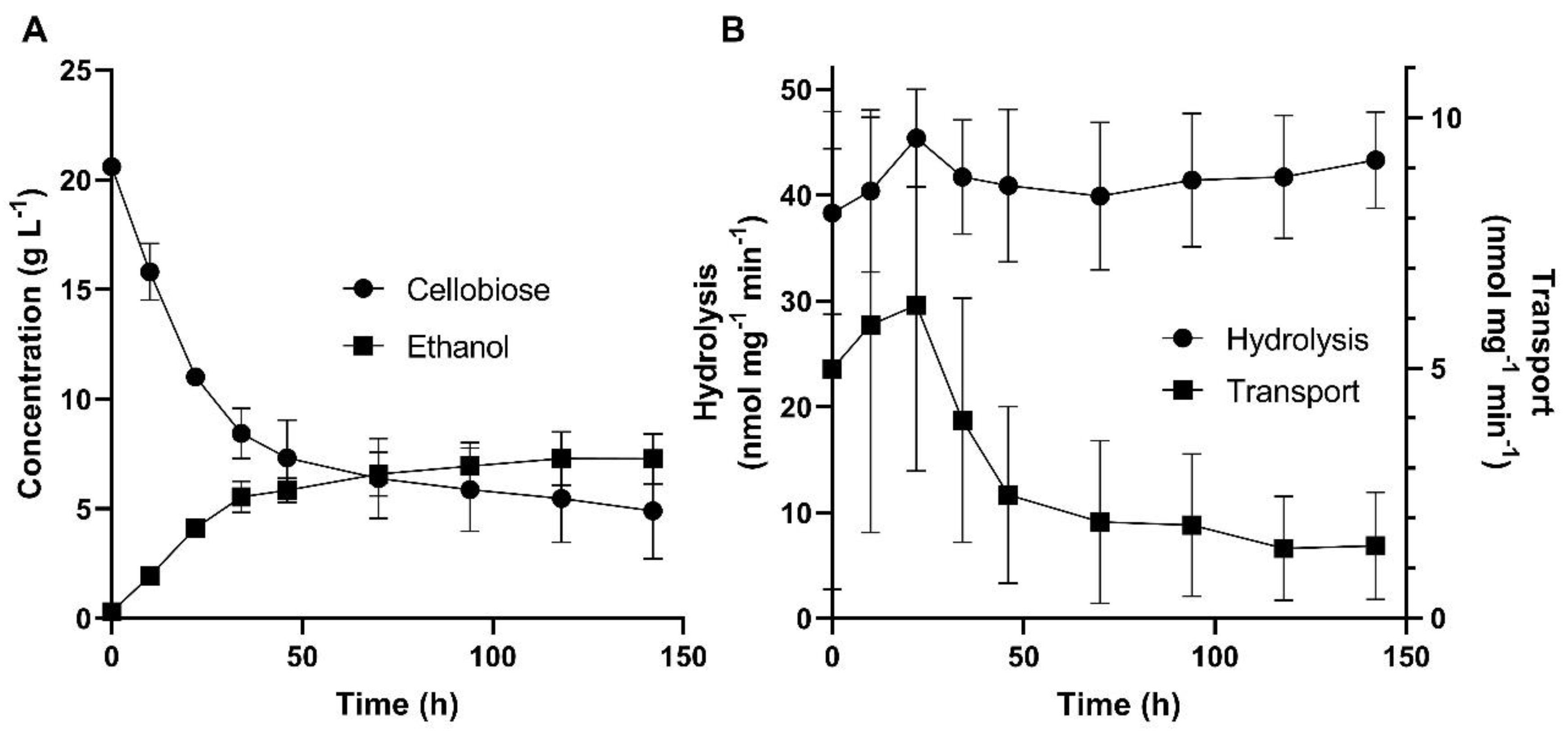
Figure 4.
Sequence alignment of the cytoplasmic N-terminal first 43 amino acids (A) and the cytoplasmic C-terminal last 43 amino acids of the protein, after the last transmembrane domain (B), deduced from the MgCBT2 gene, and the truncated versions of MgCBT2 at the C-terminal domain (MgCBT2ΔC), or at both the N- and C-terminal domains (MgCBT2ΔNΔC). The lysine residues with medium (blue) or high (red) ubiquitinylation potential were determined with the BDM-PUB [68] and UbPred programs [69].
Figure 4.
Sequence alignment of the cytoplasmic N-terminal first 43 amino acids (A) and the cytoplasmic C-terminal last 43 amino acids of the protein, after the last transmembrane domain (B), deduced from the MgCBT2 gene, and the truncated versions of MgCBT2 at the C-terminal domain (MgCBT2ΔC), or at both the N- and C-terminal domains (MgCBT2ΔNΔC). The lysine residues with medium (blue) or high (red) ubiquitinylation potential were determined with the BDM-PUB [68] and UbPred programs [69].

Figure 7.
Phylogenetic classification of cellobiose transporters from various yeast and fungal hosts. The phylogenetic tree contains 24 transporter sequences, and the numbers at the nodes represent percentage bootstrap values based on 1500 samplings. The abbreviation of each species is added after the name of the transporter genes: As = Aspergillus nidulans, Tt = Thielavia terrestris, Af = Aspergillus flavus, Pp = Postia placenta, Nc = Neurospora crassa, Fg = Fusarium graminearum, Ls = Lipomyces starkeyi, Kd = Kluyveromyces dobzhanskii, Ss = Scheffersomyces stipitis, Ct = Candida tropicalis, Mg = Meyerozyma guilliermondii, Km = Kluyveromyces marxianus, Kw = Kluyveromyces wickerhamii, Ka = Kluyveromyces aestuarii, Kl = Kluyveromyces lactis, Po = Penicillium oxalicum, An = Aspergillus niger, Tr = Trichoderma reesei, and Pc = Penicillium chrysogenum.
Figure 7.
Phylogenetic classification of cellobiose transporters from various yeast and fungal hosts. The phylogenetic tree contains 24 transporter sequences, and the numbers at the nodes represent percentage bootstrap values based on 1500 samplings. The abbreviation of each species is added after the name of the transporter genes: As = Aspergillus nidulans, Tt = Thielavia terrestris, Af = Aspergillus flavus, Pp = Postia placenta, Nc = Neurospora crassa, Fg = Fusarium graminearum, Ls = Lipomyces starkeyi, Kd = Kluyveromyces dobzhanskii, Ss = Scheffersomyces stipitis, Ct = Candida tropicalis, Mg = Meyerozyma guilliermondii, Km = Kluyveromyces marxianus, Kw = Kluyveromyces wickerhamii, Ka = Kluyveromyces aestuarii, Kl = Kluyveromyces lactis, Po = Penicillium oxalicum, An = Aspergillus niger, Tr = Trichoderma reesei, and Pc = Penicillium chrysogenum.
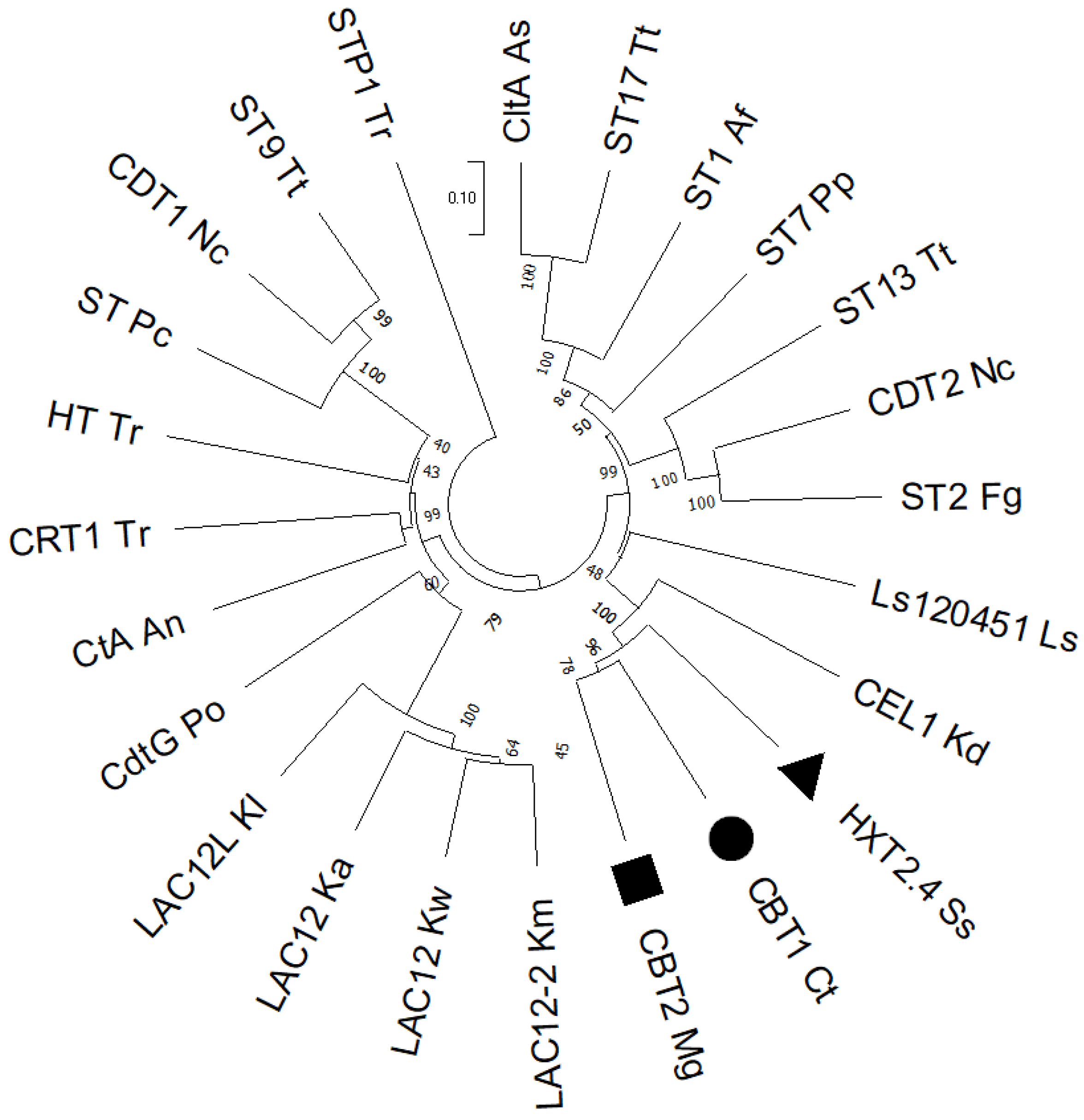

Table 1.
Yeast strains and plasmids used in this study.
| Strains and plasmids | Relevant features or genotype | Source |
|---|---|---|
| Yeast strains: | ||
| C. tropicalis UFMG-HB-93a | Isolated from decaying sugarcane bagasse in São Paulo, Brazil | [50] |
| M. guilliermondii NRRL Y-27844 | Clinical isolate | USDA-ARC Culture Collection |
| Sc. illinoinensis UFMG-CM-Y512 | Isolated from rotting wood in Rio de Janeiro, Brazil | [51] |
| Sp. passalidarum UFMG-CM-Y474 | Isolated from rotting wood in Roraima, Brazil | [52] |
| S. cerevisiae CEN.PK2-1C | MATa leu2-3,112 ura3-52 trp1-289 his3-Δ1 MAL2-8c SUC2 | [53] |
| S. cerevisiae B7 | CEN.PK2-1C + pGPD-424-SpBGL7 | This work |
| S. cerevisiae B7-HXT2.4 | CEN.PK2-1C + pGPD-424-SpBGL7 + pGPD-426-SsHXT2.4 | This work |
| S. cerevisiae B7-CBT1 | CEN.PK2-1C + pGPD-424-SpBGL7 + pGPD-426-CtCBT1 | This work |
| S. cerevisiae B7-CBT2 | CEN.PK2-1C + pGPD-424-SpBGL7 + pGPD-426-MgCBT2 | This work |
| S. cerevisiae CAT-1 | Industrial strain isolated from Usina VO Catanduva, São Paulo, Brazil | [54,55] |
| S. cerevisiae MP-C5H1 | Isogenic to CAT-1, but AUR1::pAUR-XKXDHXR loxP-KanMX-loxP-PADH1::[4- 59∆]HXT1 | [56] |
| S. cerevisiae MP-B7 | Isogenic to MP-C5H1, but ARS208::PTEF1-SpBGL7-TPGK1 | This work |
| S. cerevisiae MP-B7-CBT2 | Isogenic to MP-B7, but ARS1309::PTDH3-MgCBT2-TCYC1 | This work |
| S. cerevisiae MP-B7-CBT2ΔC | Isogenic to MP-B7, but ARS1309::PTDH3-MgCBT2ΔC-TCYC1 | This work |
| S. cerevisiae MP-B7-CBT2ΔNΔC | Isogenic to MP-B7, but ARS1309::PTDH3-MgCBT2ΔNΔC-TCYC1 | This work |
| Plasmids: | ||
| pGPD-424 | AmpR ori 2µ TRP1 PTDH3-TCYC1 | ATCC® 87357TM [57] |
| pGPD-426 | AmpR ori 2µ URA3 PTDH3-TCYC1 | ATCC® 87361TM [57] |
| pGPD-424-SpBGL7 | AmpR ori 2µ TRP1 PTDH3-SpBGL7-TCYC1 | This work |
| pGPD-426-SsHXT2.4 | AmpR ori 2µ URA3 PTDH3-SsHXT2.4-TCYC1 | This work |
| pGPD-426-CtCBT1 | AmpR ori 2µ URA3 PTDH3-CtCBT1-TCYC1 | This work |
| pGPD-426-MgCBT2 | AmpR ori 2µ URA3 PTDH3-MgCBT2-TCYC1 | This work |
| pGPD-426-MgCBT2ΔC | AmpR ori 2µ URA3 PTDH3-MgCBT2ΔC-TCYC1 | This work |
| pGPD-426-MgCBT2ΔNΔC | AmpR ori 2µ URA3 PTDH3-MgCBT2ΔNΔC-TCYC1 | This work |
| pV1382 | AmpR ori CEN ARS URA3 NATR PTEF1-CaCas9-TCYC1 PSNR52-sgRNA-TSNR52 | [58] |
| pV1382-ARS1309 | pV1382 PSNR52-sgRNA(ARS1309)-TSNR52 | This work |
| pV1382-ARS208 | pV1382 PSNR52-sgRNA(ARS208)-TSNR52 | This work |
| pMV | AmpR ori (pBR322 derivative) | BGI Group |
| pMV-SpBGL7 | AmpR ori [5’ARS208-PTEF1-SpBGL2-TPGK1-3’ARS208] | This work |
Table 2.
Primers used in this study.
| Primer 1 | Sequence 2 |
|---|---|
| Cloning: | |
| SpBGL7-F | GAATTCATGACCGTGTCTGATTTCGATGTTG |
| SpBGL7-R | CTCGAGCTAATTACCTTTCCAGAAGAAACTTTGATC |
| HXT2.4-F | GGCGGATCCAAAATGTCTGACAAACTTCACAACATCAAG |
| HXT2.4-R | GGCCTCGAGGTCGACATAATCAGGTATAATTTATTGACTAAAGCTTAG |
| CtCBT1-F | GGCGAATTCAAAATGTCATCCAAAGATAATATCATCATCACTGAAG |
| CtCBT1-R | GGCCTCGAGGTCGACCTAGGCCAATTTTTCAACGTGATCAACC |
| MgCBT2-F | GGCGGATCCATGGTTTCCAATTCGTCTTCATACTGG |
| MgCBT2-R | GGCAAGCTTTCATACTTTTTCAGCATGTTCAAGCG |
| MgCBT2ΔC-F | CATGGATCCATGGGTTTCCATTCGTCTTC |
| MgCBT2ΔC-R | TGAAAGCTTTCACGGAGTGGCAAGAATATGGA |
| MgCBT2ΔNΔC-F | CATGGATCCATGCACCAGGATATCGCTACTCA |
| CRISPR-Cas9: | |
| sgRNA.ARS1309-F | 5P-GATCGCCTGTGGTGACTACGTATCCG |
| sgRNA.ARS1309-R | 5P-AAAACGGATACGTAGTCACCACAGGC |
| sgRNA.ARS208-F | 5P-GATCGGTCCGCTAAACAAAAGATCTG |
| sgRNA.ARS208-R | 5P-AAAACAGATCTTTTGTTTAGCGGACC |
| ARS208-F | CCGCAGTGTCTTGCGTCTCTGATCTTACCTGGTGAATTGG |
| ARS208-R | TTGGCAGTGACTCCGTCTCTAGTAGGTGCCAGTTGAATAG |
| Sequencing: | |
| seq.p1382.sgRNA-F | GCTGTAGAAGTGAAAGTTGG |
| seq.p1382.sgRNA-R | CAAGTTGATAACGGACTAGC |
1 All primers were synthesised by Integrated DNA Technologies (IDT, Coralville, IA, USA). 2 Bold sequences indicate restriction enzyme sites (BamHI, EcoRI, HindIII, SalI or XhoI) used for cloning.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



