
Submitted:
23 August 2024
Posted:
23 August 2024
You are already at the latest version
Abstract
The advancement of computational drug discovery and design necessitates continuous innovation to enhance the accuracy and scope of predictive models for early-stage drug research and development. This article introduces a novel workflow for in silico generation of structural and intermolecular binding affinity data with reasonable accuracy, combining two computational tools: Modeller and Prodigy. By leveraging synthetic structural and biophysical data, this approach addresses the limitations of existing experimental datasets, generating extensive, high-quantity binding affinity data with reasonable accuracy for biomolecular binding pairs, which broadens the horizon of computational biomolecule design and discovery by enabling extensive exploration of the sequence space of biomolecular binding pairs, and narrows the gap between experimental binding affinity data and its unexplored territories. Overall, this article presents a methodological advance to enhance the accuracy and scope of computational biomolecule discovery and design, paves the way for the development of preclinical candidates with improved efficacy and specificity, and holds transformative potential for further advancements in artificial intelligence-enabled biomolecule discovery and design in the future.
Keywords:
Inter-molecular Binding Affinity
; Synthetic Data Augmentation
; Computational Biomolecule Design and Discovery
; Site-specific mutation
; Structural biophysics
1. Background and Motivation
Development of a single novel small molecule drug takes up to 14-15 years and costs over 2.5 billion U.S. dollars from target assessment to regulatory approval [1,2,3]. To date, early-stage drug discovery and design remains an extremely costly and time-consuming process with rather high failure rate, yet it still is essential to ensure safety, quality and profitability of new therapeutic entities entering the market [4]. Historically, drug discovery and design have evolved significantly with the advent of computational methodologies, which have accelerated the identification and optimization of therapeutic candidates [5,6]. Traditional approaches, often involving labor-intensive and time-consuming experimental procedures [7], are increasingly supplemented by computational techniques that predict molecular interactions, binding affinities, and potential off-target effects with high accuracy [8,9,10].
In computational drug discovery and design, a critical task is accurate prediction of drug-target binding affinity [11,12,13], which is crucial for identification and continued optimization of potential drug candidates [14]. However, the availability and quality of experimental binding affinity data [11,15] are often the performance- and speed-limiting factors of these computational models [16,17,18,19,20]. Therefore, this article reports a novel workflow of in silico generation of structural and intermolecular binding affinity data with reasonable accuracy, aiming at improving the accuracy and scope of computational structural biophysics-based biomolecule design, and contributing a little bit to the development of accurate, efficient and cost-effective structural biophysics- and AI-enabled biomolecule discovery and design in future [2].
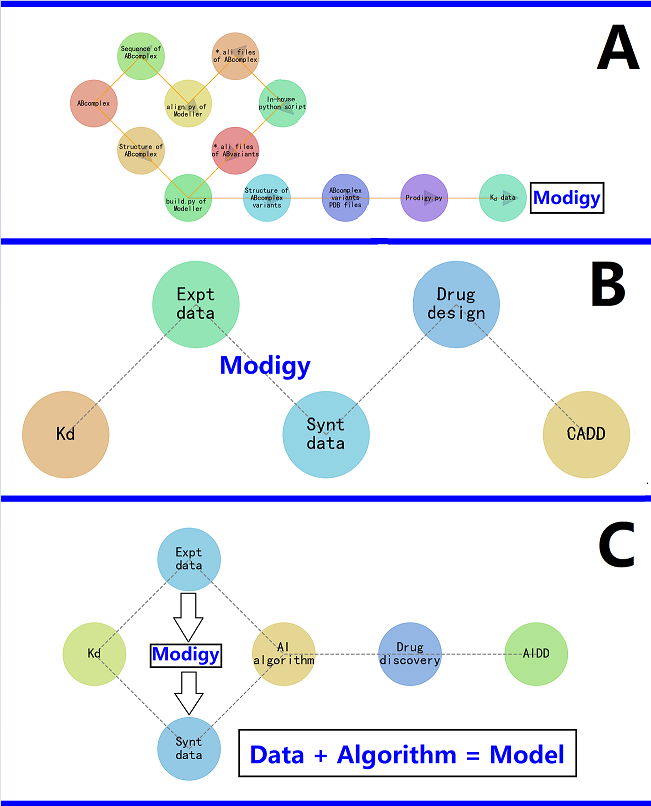
2. Modigy: a Computational Structural Biophysics-Based Workflow
Abbreviated as Modigy (Figure 1), the workflow involves two steps: homology structural modeling using Modeller [21] based on experimental complex structures from Protein Data Bank (PDB) [22] and physics-based calculations of intermolecular Kd using Prodigy [23,24], as illustrated in Figure 1 and described previously in detail [25].
3. How Does the Modigy Workflow Contribute to Biomolecular CADD?
By definition, biomolecule represents any of numerous substances that are produced by cells and living organisms [26,27]. In this article, the term biomolecule specifically refers to recombinant proteins, peptides, monoclonal antibodies, antibody- or peptide-drug conjugates [28,29,30,31,32]. Thus, for computational biomolecule design and discovery, the Modigy workflow (Figure 1) expands its horizons by enabling extensive exploration of the sequence space of biomolecular binding pairs [33,34], and holds transformative potential for computational biomolecule discovery and design (Figure 2).
3.1. Design of Semaglutide Analogues with Enhanced Binding Affinity to GLP-1R
Semaglutide, a GLP-1 receptor agonist, is effective in treating type 2 diabetes mellitus by aiding in blood sugar regulation and weight reduction [35,36,37]. In 2021, a Val27-Arg28 substitution was manually designed and introduced into the semaglutide backbone, resulting in an increase in the ligand-receptor Kd [29,38,39]. Afterwards, using the Modigy workflow (Figure 1), 8915 semaglutide analogues were computationally designed with a comprehensive structural biophysics-based strategy [34,40]. Among these, one analogue stood out with a Kd to GLP-1R over 113 times higher than that of native semaglutide, with a Kd of 3.0 × 10-8 M compared to 3.4 × 10-6 M for native semaglutide [34,40]. This study highlights the potential of this Modigy-based approach in designing semaglutide analogues with improved GLP-1R binding affinity and efficacy in diabetes treatment and weight management [41,42,43,44].
3.2. Scalable Antigen-Antibody Binding Affinity Landscape: A Case Study with ENHERTU
Optimizing binding affinities for antibody-drug conjugates (ADCs) is critical for their therapeutic efficacy and specificity. Most ADCs are engineered to achieve equilibrium Kd in the range of 0.1 to 1 nM, but there is a paucity of published data delineating the optimal binding affinity range for improved therapeutic outcomes [45,46]. Trastuzumab deruxtecan (ENHERTU®) is a HER2-directed antibody and DNA topoisomerase I inhibitor conjugate developed for treating HER2-expressing solid tumors [47,48,49]. Trastuzumab, the monoclonal antibody in ENHERTU®, binds to the extracellular domain of the HER2 receptor, inhibiting downstream signaling pathways and mediating antibody-dependent cellular cytotoxicity [32,50].
Using ENHERTU® as an example, a recent computational study [31] reported a scalable antigen-antibody binding affinity landscape using the Modigy workflow [25]. Of particular interest is the HER2-Trastuzumab-Pertuzumab binding affinity landscape as a function of site-specific missense mutations, such as this particular S911F mutation in chain C of Pertuzumab. With the Prodigy server [23,24], the impact of this mutation is reassessed, leading to an antigen-antibody Kd of 2.9 × 10-10 M for the Her2-Pertuzumab interaction, compared to 1.9 × 10-8 M for the native experimental Her2-Trastuzumab-Pertuzumab complex (PDB entry 6OGE [32]). This increase in the antigen-antibody binding affinity due to the S911F substitution underscores the potential of the Modigy workflow [25] for both ADC efficacy optimization [45,46] and monoclonal antibody affinity maturation [51,52].
4. How Does the Modigy Workflow Contribute to Biomolecular AIDD?
The past century, along with the beginning of this one, saw a transformative era in drug discovery and design with the advent of computer-aided drug discovery and design (CADD) and AI-enabled drug discovery and design (AIDD), supported by the development of extensive chemical and biological databases [11,15,22,53,54,55] and machine learning algorithms [56]. For instance, recent advancements in this field have profoundly impacted structural biology (e.g., protein structure prediction by AlphaFold [54]) and drug discovery and design [57,58]. Yet, the predictions made by CADD tools are only as good as the data they are trained on, i.e., if the data are insufficient or of poor quality, the predictions are likely to be inaccurate and thus unreliable.
Figure 3.
A synthetic data and AIDD perspective of the Modigy workflow (Figure 1). In this figure, expt data represents experimentally measured inter-molecular Kd data, while synt data represents drug-target Kd generated by Modigy workflow (Figure 1).
Thus, the Modigy (Figure 1) workflow here addresses the critical bottleneck of limited experimental data by generating synthetic data, which constitutes a paradigm shift (Figure 4) from linear accumulation of experimental Kd data to exponential accumulation of computational Kd data with reasonable accuracy. This paradigm shift facilitates the integration of AI algorithms in biomolecule discovery and design, improving the training of AI models and enhancing their predictive capabilities [59], as combining synthetic data with experimental data [11,15] in hybrid models can enhance the overall accuracy and robustness of predictions by AIDD tools. While the Modigy workflow improves the training and performance of AI models, paves the way for more sophisticated and effective AI-driven drug discovery approaches, it also encourages the development of new AI algorithms tailored to leverage synthetic data, fostering continued innovation and advancement in the field.
Additionally, the Modigy workflow (Figure 1) is inherently scalable and applicable for existing experimental databases like PDB [22], allowing for high-throughput creation of astronomical datasets of synthetic inter-molecular Kd data [25]. Take semaglutide for instance, to introduce three site-specific missense mutations into the semaglutide backbone requires a total of 26,208,000 homology structural models with reasonable accuracy [41] to be built using Modeller [21] and a total of 26,208,000 Prodigy-based [23,24] calculations of the binding affinities between semaglutide analogues and GLP-1R. For Molecule X (a random protein consisting of 100 amino acids), the number soars from 26,208,000 to 1,293,600,000 (Table 1). In practice, an exhaustive exploration of the entire biomolecular space [60,61] is both impossible and unnecessary, which is where AI algorithms come in for continued development of computational biomolecule design and discovery. Moreover, this article proposes an open strategy [62] to making it conceivable to generate astronomical amounts of Kd data, with which to build AIDD models with reasonable accuracy and efficiency [63], as openness in both experimental data acquisition and synthetic data generation, and in AI algorithm development, is essential for promoting transparency, reproducibility, and collaboration within the drug discovery and design community [2,63].
5. Limitations of the Modigy Workflow in Drug Design and Discovery
While the proposed Modigy workflow (Figure 1) holds potential for biomolecular CADD and AIDD, it is essential to recognize its limitations and identify areas for future research and development. The Modigy approach (Figure 1) does not work in the absence of accurate structural information and is tailored specifically for biomolecules, making it unsuitable for small molecule discovery and design. Although Prodigy [23,24] does account for temperature in Kd calculation, the Modigy approach (Figure 1) does not consider other parameters such as pH [64,65], site-specific protonation states (e.g., protein side chain pKa) [66,67], post-translational modifications [68,69], post-expression modifications [29,30] and buffer conditions [70]. Furthermore, the Modigy approach (Figure 1) is not applicable for the inclusion of unnatural amino acids into biomolecules [56,71] and is limited to biomolecules, excluding other molecular types and drug modalities [60,61].
6. Conclusion
The Modigy workflow (Figure 1) transforms the landscape of computational biomolecule design and discovery by generating high-quality synthetic structural and intermolecular Kd data. By addressing the limitations of existing experimental datasets [11,15] and integrating advanced computational techniques with AI algorithms, this Modigy workflow (Figure 1) provides a technically feasible approach for accelerating the development of therapeutic biomolecular candidates with improved efficacy and specificity, as it is able to not only improves the accuracy and efficiency of computational models, but also contributes to future advancements in biomolecule discovery and design enabled by both AI algorithms and structural biophysics [2,7].
7. Ethical Statement
No ethical approval is required.
8. Declaration of Generative AI and AI-Assisted Technologies in the Writing Process
During the preparation of this work, the author used OpenAI’s ChatGPT in order to improve the readability of the manuscript, and to make it as concise and short as possible. After using this tool, the author reviewed and edited the content as needed and takes full responsibility for the content of the publication.
Author Contributions
Conceptualization, W.L.; methodology, W.L.; software, W.L.; validation, W.L.; formal analysis, W.L.; investigation, W.L.; resources, W.L.; data duration, W.L.; writing–original draft preparation, W.L.; writing–review and editing, W.L.; visualization, W.L.; supervision, W.L.; project administration, W.L.; funding acquisition, not applicable.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Schlander, M.; Hernandez-Villafuerte, K.; Cheng, C.Y.; Mestre-Ferrandiz, J.; Baumann, M. How Much Does It Cost to Research and Develop a New Drug? A Systematic Review and Assessment. PharmacoEconomics 2021, 39, 1243–1269. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2018, 20, 273–286. [Google Scholar] [CrossRef]
- Mikulic, M. Average R&D cost to develop a pharmaceutical compound from discovery to launch from 2010 to 2020, 2022. Accessed: (Aug 5, 2024).
- Niazi, S.K.; Mariam, Z. Computer-Aided Drug Design and Drug Discovery: A Prospective Analysis. Pharmaceuticals 2023, 17, 22. [Google Scholar] [CrossRef] [PubMed]
- Tropsha, A.; Bajorath, J. Computational Methods for Drug Discovery and Design. Journal of Medicinal Chemistry 2015, 59, 1–1. [Google Scholar] [CrossRef]
- Brogi, S. Computational Approaches for Drug Discovery. Molecules 2019, 24, 3061. [Google Scholar] [CrossRef] [PubMed]
- Steven, A.C.; Baumeister, W. The future is hybrid. Journal of Structural Biology 2008, 163, 186–195. [Google Scholar] [CrossRef]
- Nayarisseri, A. Experimental and Computational Approaches to Improve Binding Affinity in Chemical Biology and Drug Discovery. Current Topics in Medicinal Chemistry 2020, 20, 1651–1660. [Google Scholar] [CrossRef]
- Tinberg, C.E.; Khare, S.D.; Dou, J.; Doyle, L.; Nelson, J.W.; Schena, A.; Jankowski, W.; Kalodimos, C.G.; Johnsson, K.; Stoddard, B.L.; Baker, D. Computational design of ligand-binding proteins with high affinity and selectivity. Nature 2013, 501, 212–216. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Nguyen, N.T.; Malagon-Lopez, J.; Topkar, V.V.; Aryee, M.J.; Joung, J.K. CIRCLE-seq: A highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nature Methods 2017, 14, 607–614. [Google Scholar] [CrossRef]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Research 2007, 35, D198–D201. [Google Scholar] [CrossRef]
- Ballester, P.J.; Mitchell, J.B.O. A machine learning approach to predicting protein-ligand binding affinity with applications to molecular docking. Bioinformatics 2010, 26, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Li, J.; He, X. Fragment-based quantum mechanical calculation of protein-protein binding affinities. Journal of Computational Chemistry 2018, 39, 1617–1628. [Google Scholar] [CrossRef]
- Mason, D.M.; Friedensohn, S.; Weber, C.R.; Jordi, C.; Wagner, B.; Meng, S.M.; Ehling, R.A.; Bonati, L.; Dahinden, J.; Gainza, P.; Correia, B.E.; Reddy, S.T. Optimization of therapeutic antibodies by predicting antigen specificity from antibody sequence via deep learning. Nature Biomedical Engineering 2021, 5, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Fang, X.; Lu, Y.; Yang, C.Y.; Wang, S. The PDBbind Database: Methodologies and Updates. Journal of Medicinal Chemistry 2005, 48, 4111–4119. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, S.; Prema, K.; Balaji, S. Machine learning models for drug-target interactions: Current knowledge and future directions. Drug Discovery Today 2020, 25, 748–756. [Google Scholar] [CrossRef]
- Emami, N.; Ferdousi, R. AptaNet as a deep learning approach for aptamer-protein interaction prediction. Scientific Reports 2021, 11. [Google Scholar] [CrossRef]
- Kwon, Y.; Shin, W.H.; Ko, J.; Lee, J. AK-Score: Accurate Protein-Ligand Binding Affinity Prediction Using an Ensemble of 3D-Convolutional Neural Networks. International Journal of Molecular Sciences 2020, 21, 8424. [Google Scholar] [CrossRef]
- de Ávila, M.B.; Xavier, M.M.; Pintro, V.O.; de Azevedo, W.F. Supervised machine learning techniques to predict binding affinity. A study for cyclin-dependent kinase 2. Biochemical and Biophysical Research Communications 2017, 494, 305–310. [Google Scholar] [CrossRef]
- Fuji, H.; Qi, F.; Qu, L.; Takaesu, Y.; Hoshino, T. Prediction of Ligand Binding Affinity to Target Proteins by Molecular Mechanics Theoretical Calculation. Chemical and Pharmaceutical Bulletin 2017, 65, 461–468. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. In Methods in Molecular Biology; Springer US, 2020; pp. 239–255.
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nature Structural & Molecular Biology 2003, 10, 980–980. [Google Scholar]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein–protein complexes. Bioinformatics 2016, btw514. [Google Scholar] [CrossRef]
- Vangone, A.; Bonvin, A.M. Contacts-based prediction of binding affinity in protein–protein complexes. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Li, W. In Silico Generation of Structural and Intermolecular Binding Affinity Data with Reasonable Accuracy: Expanding Horizons in Drug Discovery and Design 2024. [CrossRef]
- Capecchi, A.; Probst, D.; Reymond, J.L. One molecular fingerprint to rule them all: Drugs, biomolecules, and the metabolome. Journal of Cheminformatics 2020, 12. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nature structural biology 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Sørensen, H.P.; Mortensen, K.K. Advanced genetic strategies for recombinant protein expression in Escherichia coli. J. Biotechnol. 2005, 115, 113–128. [Google Scholar] [CrossRef]
- Li, W. Strengthening Semaglutide-GLP-1R Binding Affinity via a Val27-Arg28 Exchange in the Peptide Backbone of Semaglutide: A Computational Structural Approach. Journal of Computational Biophysics and Chemistry 2021, 20, 495–499. [Google Scholar] [CrossRef]
- Weiss, M. Design of ultra-stable insulin analogues for the developing world. Journal of Health Specialties 2013, 1, 59. [Google Scholar] [CrossRef]
- Li, W. Scalable Antigen-Antibody Binding Affinity Landscape: A Case Study with ENHERTU 2024. [CrossRef]
- Hao, Y.; Yu, X.; Bai, Y.; McBride, H.J.; Huang, X. Cryo-EM Structure of HER2-trastuzumab-pertuzumab complex. PLOS ONE 2019, 14, e0216095. [Google Scholar] [CrossRef]
- Reymond, J.L.; van Deursen, R.; Blum, L.C.; Ruddigkeit, L. Chemical space as a source for new drugs. MedChemComm 2010, 1, 30. [Google Scholar] [CrossRef]
- Li, W. An Exhaustive Exploration of the Semaglutide-GLP-1R Sequence Space towards the Design of Semaglutide Analogues with Elevated Binding Affinity to GLP-1R 2024. [CrossRef]
- Granhall, C.; Donsmark, M.; Blicher, T.M.; Golor, G.; Sondergaard, F.L.; Thomsen, M.; Bakdal, T.A. Safety and Pharmacokinetics of Single and Multiple Ascending Doses of the Novel Oral Human GLP-1 Analogue, Oral Semaglutide, in Healthy Subjects and Subjects with Type 2 Diabetes. Clinical Pharmacokinetics 2018, 58, 781–791. [Google Scholar] [CrossRef]
- Garg, S.K.; Kaur, G.; Haider, Z.; Rodriquez, E.; Beatson, C.; Snell-Bergeon, J. Efficacy of Semaglutide in Overweight and Obese Patients with Type 1 Diabetes. Diabetes Technology & Therapeutics 2024, 26, 184–189. [Google Scholar] [CrossRef]
- Europe, T.L.R.H. Semaglutide and beyond a turning point in obesity pharmacotherapy. The Lancet Regional Health Europe 2024, 37, 100860. [Google Scholar] [CrossRef]
- Ahrén, B.; Atkin, S.L.; Charpentier, G.; Warren, M.L.; Wilding, J.P.H.; Birch, S.; Holst, A.G.; Leiter, L.A. Semaglutide induces weight loss in subjects with type 2 diabetes regardless of baseline BMI or gastrointestinal adverse events in the SUSTAIN 1 to 5 trials. Diabetes, Obesity and Metabolism 2018, 20, 2210–2219. [Google Scholar] [CrossRef]
- Aroda, V.R.; Rosenstock, J.; Terauchi, Y.; Altuntas, Y.; Lalic, N.M.; Villegas, E.C.M.; Jeppesen, O.K.; Christiansen, E.; Hertz, C.L.; Haluzík, M. PIONEER 1: Randomized Clinical Trial of the Efficacy and Safety of Oral Semaglutide Monotherapy in Comparison With Placebo in Patients With Type 2 Diabetes. Diabetes Care 2019, 42, 1724–1732. [Google Scholar] [CrossRef] [PubMed]
- Li, W. Optimizing GLP-1R Agonist: A Computational Semaglutide Analogue with 112-fold Enhanced Binding Affinity to GLP-1R 2024. [CrossRef]
- Lau, J.; Bloch, P.; Schäffer, L.; Pettersson, I.; Spetzler, J.; Kofoed, J.; Madsen, K.; Knudsen, L.B.; McGuire, J.; Steensgaard, D.B.; Strauss, H.M.; Gram, D.X.; Knudsen, S.M.; Nielsen, F.S.; Thygesen, P.; Reedtz-Runge, S.; Kruse, T. Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide. Journal of Medicinal Chemistry 2015, 58, 7370–7380. [Google Scholar] [CrossRef]
- Marijic, J.; Neelankavil, J.P. Semaglutide: A New Medical Swiss Army Knife? Journal of Cardiothoracic and Vascular Anesthesia 2024, 38, 871–873. [Google Scholar] [CrossRef] [PubMed]
- Cowart, K. Oral Semaglutide: First-in-Class Oral GLP-1 Receptor Agonist for the Treatment of Type 2 Diabetes Mellitus. Annals of Pharmacotherapy 2019, 54, 478–485. [Google Scholar] [CrossRef]
- Knudsen, L.B.; Lau, J. The Discovery and Development of Liraglutide and Semaglutide. Frontiers in Endocrinology 2019, 10, 1–32. [Google Scholar] [CrossRef]
- Jin, Y.; Schladetsch, M.A.; Huang, X.; Balunas, M.J.; Wiemer, A.J. Stepping forward in antibody-drug conjugate development. Pharmacology & Therapeutics 2022, 229, 107917. [Google Scholar]
- Kang, J.C.; Sun, W.; Khare, P.; Karimi, M.; Wang, X.; Shen, Y.; Ober, R.J.; Ward, E.S. Engineering a HER2-specific antibody–drug conjugate to increase lysosomal delivery and therapeutic efficacy. Nature Biotechnology 2019, 37, 523–526. [Google Scholar] [CrossRef]
- Keam, S.J. Trastuzumab Deruxtecan: First Approval. Drugs 2020, 80, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, Y.H. Trastuzumab Deruxtecan for HER2+ Advanced Breast Cancer. Future Oncology 2021, 18, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Oitate, M.; Hagihara, K.; Shiozawa, H.; Furuta, Y.; Ogitani, Y.; Kuga, H. Pharmacokinetics of trastuzumab deruxtecan (T-DXd), a novel anti-HER2 antibody-drug conjugate, in HER2-positive tumour-bearing mice. Xenobiotica 2020, 50, 1242–1250. [Google Scholar] [CrossRef]
- Ruedas, R.; Vuillemot, R.; Tubiana, T.; Winter, J.M.; Pieri, L.; Arteni, A.A.; Samson, C.; Jonic, S.; Mathieu, M.; Bressanelli, S. Structure and conformational variability of the HER2-trastuzumab-pertuzumab complex. Journal of Structural Biology 2024, 216, 108095. [Google Scholar] [CrossRef]
- Rathore, A.S.; Sarker, A.; Gupta, R.D. Recent Developments Toward Antibody Engineering and Affinity Maturation. Protein & Peptide Letters 2018, 25, 886–896. [Google Scholar] [CrossRef]
- Comeau, S.R.; Thorsteinson, N.; Kumar, S. Structural Considerations in Affinity Maturation of Antibody-Based Biotherapeutic Candidates. In Methods in Molecular Biology; Springer US, 2022; p. 309–321. [CrossRef]
- Roy, R.; Al-Hashimi, H.M. AlphaFold3 takes a step toward decoding molecular behavior and biological computation. Nature Structural & Molecular Biology 2024, 31, 997–1000. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; Bridgland, A.; Meyer, C.; Kohl, S.A.A.; Ballard, A.J.; Cowie, A.; Romera-Paredes, B.; Nikolov, S.; Jain, R.; Adler, J.; Back, T.; Petersen, S.; Reiman, D.; Clancy, E.; Zielinski, M.; Steinegger, M.; Pacholska, M.; Berghammer, T.; Bodenstein, S.; Silver, D.; Vinyals, O.; Senior, A.W.; Kavukcuoglu, K.; Kohli, P.; Hassabis, D. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Greenidge, P.A.; Kramer, C.; Mozziconacci, J.C.; Wolf, R.M. MM/GBSA Binding Energy Prediction on the PDBbind Data Set: Successes, Failures, and Directions for Further Improvement. Journal of Chemical Information and Modeling 2012, 53, 201–209. [Google Scholar] [CrossRef]
- Hofmann, D.W.M.; Kuleshova, L.N. A general force field by machine learning on experimental crystal structures. Calculations of intermolecular Gibbs energy with iFlexCryst. Acta Crystallographica Section A Foundations and Advances 2023, 79, 132–144. [Google Scholar] [CrossRef]
- Cramer, P. AlphaFold2 and the future of structural biology. Nature Structural & Molecular Biology 2021, 28, 704–705. [Google Scholar]
- Read, R.J.; Baker, E.N.; Bond, C.S.; Garman, E.F.; van Raaij, M.J. AlphaFold and the future of structural biology. Acta Crystallographica Section D Structural Biology 2023, 79, 556–558. [Google Scholar] [CrossRef]
- Xu, S.; Shen, L.; Zhang, M.; Jiang, C.; Zhang, X.; Xu, Y.; Liu, J.; Liu, X. Surface-based multimodal protein–ligand binding affinity prediction. Bioinformatics 2024, 40. [Google Scholar] [CrossRef]
- Wang, T.; He, X.; Li, M.; Shao, B.; Liu, T.Y. AIMD-Chig: Exploring the conformational space of a 166-atom protein Chignolin with ab initio molecular dynamics. Scientific Data 2023, 10. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.E.; Hansen, F.K.; Gütschow, M.; Lindsley, C.W.; Liotta, D. New Drug Modalities in Medicinal Chemistry, Pharmacology, and Translational Science. ACS Pharmacology & Translational Science 2021, 4, 1712–1713. [Google Scholar]
- Hanson, B.; Stall, S.; Cutcher-Gershenfeld, J.; Vrouwenvelder, K.; Wirz, C.; Rao, Y.; Peng, G. Garbage in, garbage out: Mitigating risks and maximizing benefits of AI in research. Nature 2023, 623, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Vottevor, G. Towards a Truly General Intermolecular Binding Affinity Calculator for Drug Discovery & Design 2023. [CrossRef]
- Yang, A.S.; Honig, B. On the pH Dependence of Protein Stability. Journal of Molecular Biology 1993, 231, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.K.; Turner, G.J. Structural Basis of Perturbed pKa Values of Catalytic Groups in Enzyme Active Sites. IUBMB Life (International Union of Biochemistry and Molecular Biology: Life) 2002, 53, 85–98. [Google Scholar] [CrossRef]
- Li, W. Gravity-driven pH adjustment for site-specific protein pKa measurement by solution-state NMR. Measurement Science and Technology 2017, 28, 127002. [Google Scholar] [CrossRef]
- Hansen, A.L.; Kay, L.E. Measurement of histidine pKa values and tautomer populations in invisible protein states. Proceedings of the National Academy of Sciences 2014, 111, E1705–E1712. [Google Scholar] [CrossRef]
- Herget, S.; Ranzinger, R.; Maass, K.; Lieth, C.W. GlycoCT—a unifying sequence format for carbohydrates. Carbohydrate Research 2008, 343, 2162–2171. [Google Scholar] [CrossRef]
- Foster, J.M.; Moreno, P.; Fabregat, A.; Hermjakob, H.; Steinbeck, C.; Apweiler, R.; Wakelam, M.J.O.; Vizcaíno, J.A. LipidHome: A Database of Theoretical Lipids Optimized for High Throughput Mass Spectrometry Lipidomics. PLoS ONE 2013, 8, e61951. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. Journal of Chemical Theory and Computation 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Ju, T.; Niu, W.; Guo, J. Fine-tuning Interaction between Aminoacyl-tRNA Synthetase and tRNA for Efficient Synthesis of Proteins Containing Unnatural Amino Acids. ACS Synthetic Biology 2014, 4, 207–212. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A detailed flowchart of automated in silico generation of synthetic structural and Kd data. In this figure, Modigy represents an abbreviated name of the workflow consisting of Modeller [21] and Prodigy [23,24].
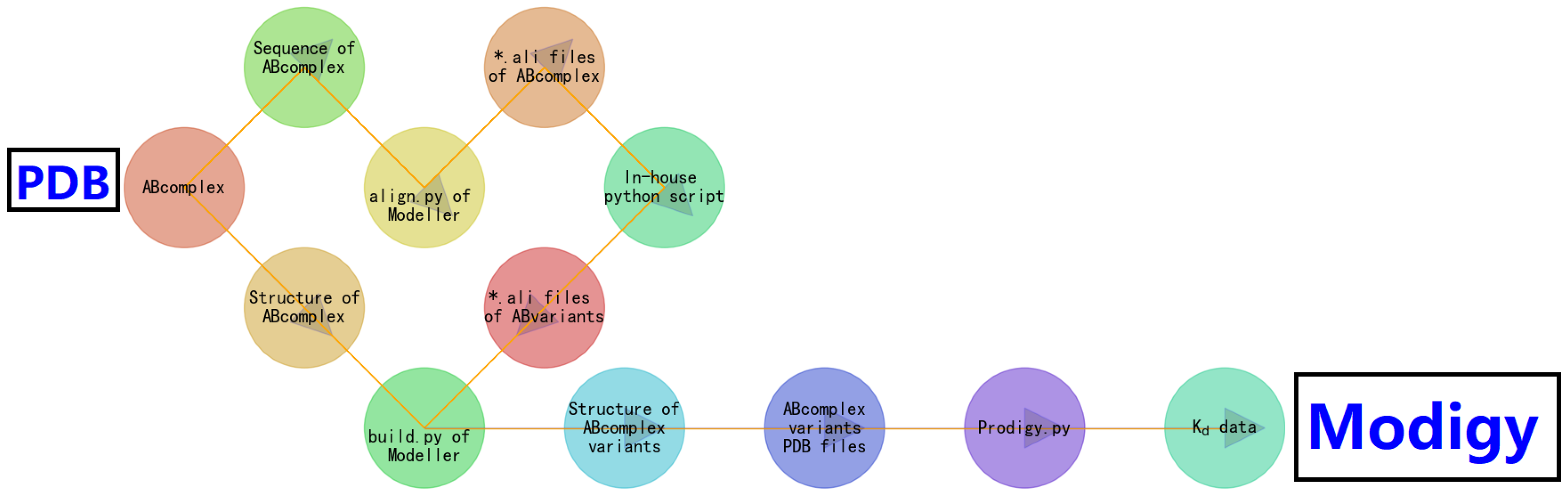
Figure 2.
A synthetic data and CADD perspective of the Modigy (Figure 1) workflow. In this figure, expt data represents experimentally measured drug-target Kd, while synt data represents drug-target Kd generated by Modigy workflow (Figure 1).
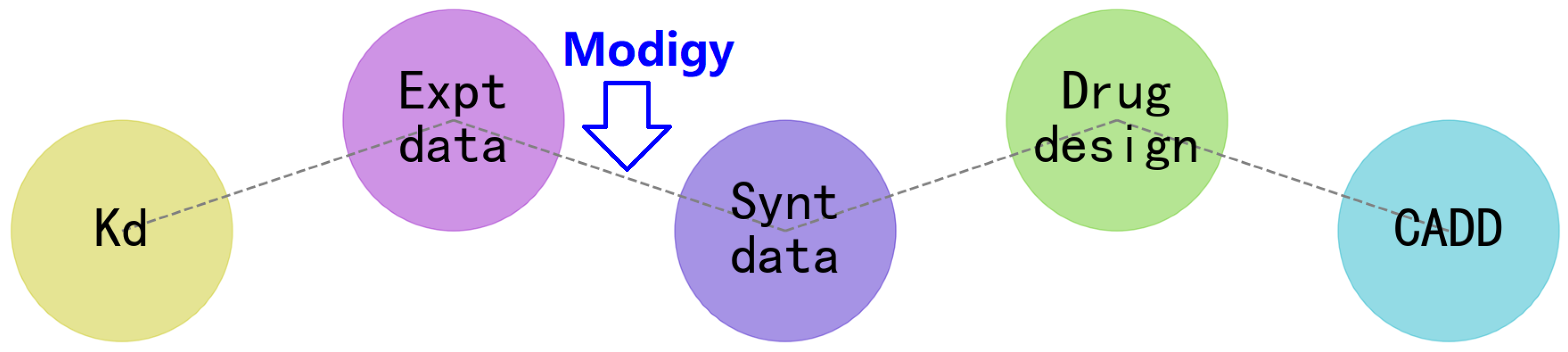
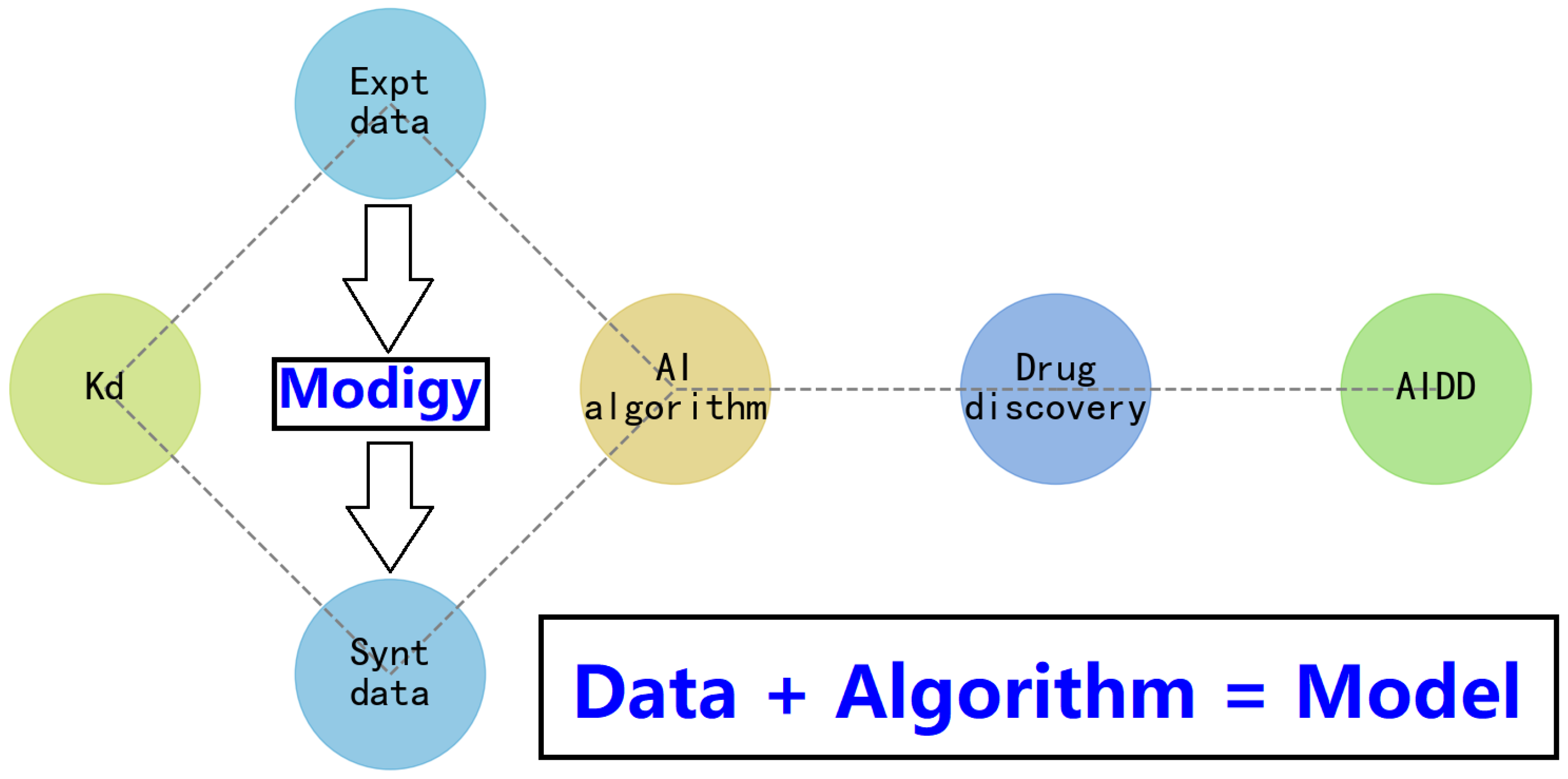
Figure 4.
Inter-molecular binding affinity synthetic data augmentation expands the horizon of computational biomolecule design and discovery. In this three-tier sketch of the sequence space of biomolecular binding pairs, the Modigy workflow (Figure 1) bridges the gap between experimentally measured Kd data and its uncharted territory.
Figure 4.
Inter-molecular binding affinity synthetic data augmentation expands the horizon of computational biomolecule design and discovery. In this three-tier sketch of the sequence space of biomolecular binding pairs, the Modigy workflow (Figure 1) bridges the gap between experimentally measured Kd data and its uncharted territory.
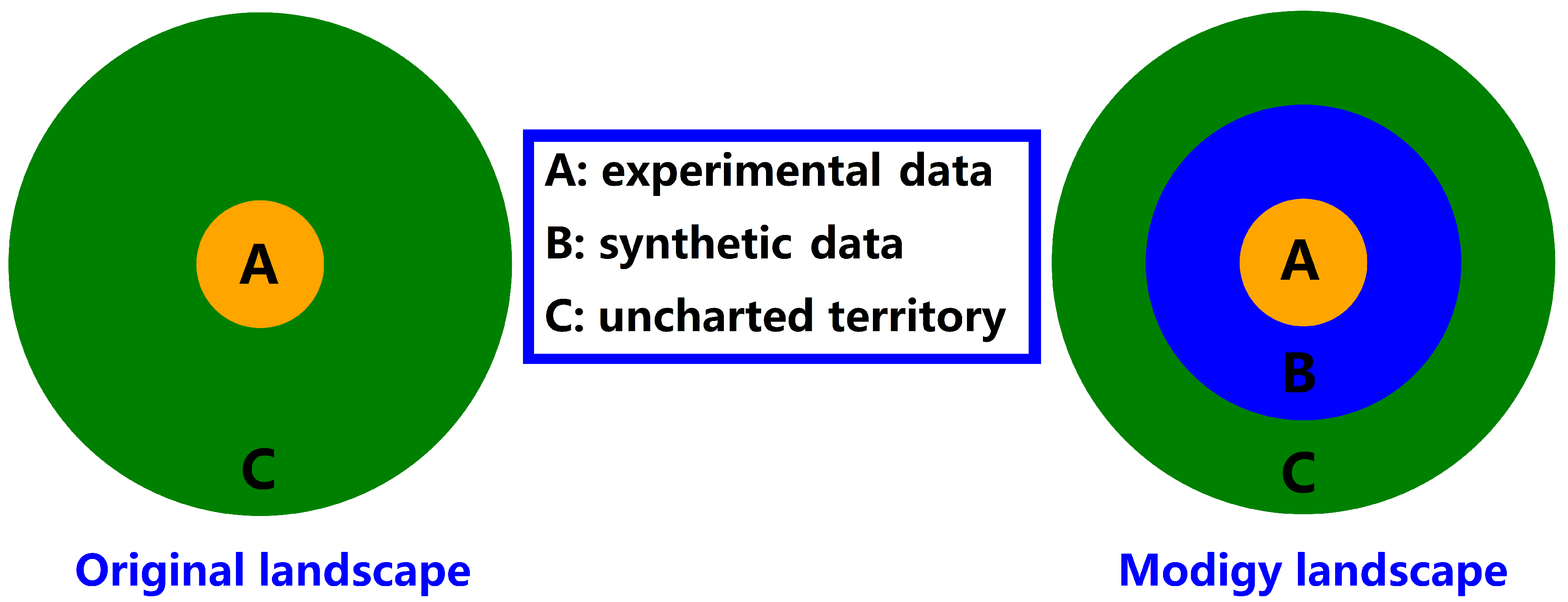

Table 1.
The size () [25] of the synthetic structural data set based on the semaglutide-GLP-1R complex structure. Here, k represents the length of the semaglutide backbone, and n represents the number of missense mutations introduced into the semaglutide backbone, with the value of being key to ensuring the overall reasonable accuracy of the synthetic data of inter-molecular binding affinities.
Table 1.
The size () [25] of the synthetic structural data set based on the semaglutide-GLP-1R complex structure. Here, k represents the length of the semaglutide backbone, and n represents the number of missense mutations introduced into the semaglutide backbone, with the value of being key to ensuring the overall reasonable accuracy of the synthetic data of inter-molecular binding affinities.
| 6|cSize (s) of the synthetic structural and biophysical data set | |||||
|---|---|---|---|---|---|
| Semaglutide backbone (28 Aa) | Molecule X (100 Aa) | ||||
| g(28,1) | 560 | g(100,1) | 2000 | ||
| g(28,2) | 151200 | g(100,2) | 1980000 | ||
| g(28,3) | 26208000 | g(100,3) | 1293600000 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



