
Submitted:
16 August 2024
Posted:
26 August 2024
You are already at the latest version
Abstract
The topic of predictive toxicology has been greatly influenced by recent progress in comprehending drug toxicity processes and enhancing medication development. The integration of omics technologies, such as transcriptomics, proteomics, and metabolomics, with traditional toxicological assessments has yielded extensive knowledge about the biological pathways implicated in drug-induced toxicity. The utilization of a multi-omics method amplifies the ability to identify biomarkers that can detect toxicity at an early stage, hence enhancing the safety profile of novel therapeutic medicines. Machine learning and in silico models, such as QSAR models and multi-task deep learning algorithms, have become essential tools. They have shown great accuracy in predicting toxicity endpoints and have helped in the identification of new biomarkers and therapeutic targets. The introduction of microphysiological systems and PBPK modeling has enhanced the transfer of preclinical discoveries to clinical results, providing more precise forecasts of human reactions to medications. Notwithstanding these progressions, obstacles such as the diversity of data and the complex nature of omics data require sophisticated computational techniques for efficient analysis. Continued cooperation and established procedures are crucial to fully utilize these technologies, guaranteeing the creation of safer and more efficient medicinal agents.
Keywords:
Predictive Toxicology
; Omics Technologies
; Machine Learning
; Structure-Activity Relationship
; Fragment-Based Drug Design
; Microphysiological Systems
; Physiologically Based Pharmacokinetic Modeling
; Virtual Screening
; Biochemical Targets
; Automated De Novo Drug Design
1. Introduction
Gaining insight into the mechanisms underlying medication toxicity is an essential component of the process of creating safer therapeutic medicines. Adverse drug responses (ADRs) provide substantial obstacles in the process of drug development, frequently resulting in the abandonment of drugs and increasing expenses [1]. To understand the complex causes of drug-induced toxicity, it is necessary to use a combination of modern technologies and approaches that can reveal the underlying mechanisms [2]. Advancements in omics technologies, including transcriptomics, proteomics, and metabolomics, have significantly transformed our comprehension of the molecular processes associated with drug toxicity. These technologies allow for a thorough analysis of how medications affect biological systems at a molecular level. This helps in identifying biomarkers that can detect early signs of toxicity and enhance the safety of novel therapeutic drugs [3].
The incorporation of omics data into standard toxicological assessments has fundamentally revolutionized the science of predictive toxicology. Researchers can obtain a comprehensive understanding of the molecular mechanisms behind drug toxicity by combining data from genomics, transcriptomics, proteomics, and metabolomics [3]. The utilization of multi-omics methodology not only improves our comprehension of medication interactions with biological systems, but also facilitates the identification of probable unintended effects and harmful byproducts. Anticipating these harmful consequences at an early stage of drug development might significantly decrease the chances of failures in later stages, therefore improving the safety and effectiveness of new medications [2].
Machine learning and in silico models have become essential tools in enhancing predictive toxicology in recent years. Quantitative structure-activity relationship (QSAR) models and multi-task deep learning algorithms have shown a high level of accuracy in predicting toxicity endpoints [4,5]. Computational models have the ability to analyze large datasets and detect patterns that are not clearly noticeable using traditional experimental methods [4]. By including omics data, these models become even more effective in making predictions, which in turn helps in identifying new biomarkers and therapeutic targets [5].
A recurring obstacle in the field of drug development is the conversion of preclinical discoveries into tangible clinical results. The introduction of microphysiological systems (MPS) and physiologically based pharmacokinetic (PBPK) modeling has successfully tackled this issue by offering more precise forecasts of human reactions to medications [2,6]. MPS, also known as organ-on-a-chip technology, duplicates human organ systems and enables controlled investigations of medication impacts [7]. Pharmacokinetic models (PBPK models) employ mathematical representations of physiological processes to predict how medications are absorbed, distributed, metabolized, and excreted in the human body [8]. When combined with omics data, these technologies provide a strong foundation for assessing the safety and effectiveness of drugs [9].
However, there are still some obstacles that continue to exist in the field of predictive toxicology [10]. The presence of diverse data types, the process of combining them, and the act of understanding their meaning provide significant challenges [11]. Due to the intricate and multi-dimensional nature of omics data, sophisticated computational approaches and tools are required for efficient analysis [3,12]. Furthermore, it is essential to perform biological validation of computational predictions in order to guarantee their significance and precision [10]. It is crucial for academics, doctors, and regulatory authorities to work together in order to provide standardized protocols and standards for using omics technology in drug development [2].
Ultimately, the integration of omics technologies with conventional toxicological assessments has yielded significant understanding of drug toxicity pathways, hence aiding the creation of safer therapeutic agents. The use of machine learning and in silico models has greatly improved the accuracy of predicting toxicity, while the implementation of MPS and PBPK modeling has enhanced the ability to apply preclinical findings to clinical settings. Nevertheless, it is crucial to tackle the difficulties associated with data integration and interpretation in order make further progress in predictive toxicology. To fully exploit the potential of these technologies for safer and more successful medication development, it is crucial to have ongoing collaborative efforts and establish standardized standards [6].
2. Structure-Activity Relationships
Structure-activity relationship (SAR) studies are essential for comprehending the correlation between the chemical structure of a molecule and its biological activity. Recent research has utilized computational algorithms to forecast and enhance the effectiveness of potential medication candidates.
2.1. Computational SAR Models
Novel chemicals’ activity has been predicted using advanced machine learning methods and molecular docking simulations. These techniques have demonstrated potential in discovering powerful inhibitors for several targets, including kinases and G-protein-coupled receptors (GPCRs) [13,14]. Recent research has shown that the combination of machine learning algorithms with atomistic simulations can improve the accuracy of predicting ligand-binding free energies [15]. An example of a hybrid strategy that combines machine learning with molecular dynamics (MD) simulations has been effectively used to discover new inhibitors for SARS-CoV-2 Mpro. This demonstrates the collaborative power of computational tools and experimental validation. Furthermore, structure-based molecular modeling has played a crucial role in the analysis of structure-activity relationships (SAR) and the improvement of lead compounds, by offering valuable information about the specific structural characteristics that contribute to biological activity and aiding in the development of more potent compounds [16,17,18]. By incorporating these computational tools into the drug development process, not only can the identification of potential therapeutic candidates be expedited, but the expenses and time required for experimental testing can also be minimized [13,14] (Figure 1a).
2.2. Fragment-Based Drug Design
Fragment-Based Drug Design (FBDD) is the systematic approach of discovering small chemical fragments with the capacity to bind to a certain target protein [19,20]. Afterwards, these pieces are improved to provide exceptionally potent treatment candidates. The use of high-throughput screening and structural biology methods has been crucial in the recent advancements in fragment-based drug discovery (FBDD) [21,22]. The utilization of biophysical methods, such as Nuclear Magnetic Resonance (NMR) and X-ray crystallography, has significantly enhanced the detection and enhancement of fragment hits [19,23]. Furthermore, computational techniques have been integrated into fragment-based drug discovery (FBDD) in order to augment the efficiency of fragment screening and optimization [22]. The application of Fragment-Based Drug Design (FBDD) has led to the discovery of several drugs that have been authorized for clinical usage, demonstrating its effectiveness in the field of drug development [20]. Vemurafenib, sotorasib, and venetoclax are examples of pharmaceuticals that have demonstrated the effectiveness of FBDD. The potential of the method in oncology was demonstrated by Vemurafenib, the first FBDD-derived drug to be approved by the FDA for the treatment of BRAF-mutant melanoma [24,25]. Sotorasib, a KRAS G12C inhibitor, emphasizes the capacity of FBDD to target previously impassable proteins, thereby providing novel treatment options for cancer. Venetoclax, a BCL-2 inhibitor, is another success story of FBDD. It has been approved for chronic lymphocytic leukemia, demonstrating the method’s capacity to develop highly selective and potent inhibitors [24]. The efficacy of FBDD is further illustrated by its application in central nervous system disorders, where it has facilitated the development of medications with enhanced pharmacokinetic properties [26]. In general, FBDD is gaining momentum as a mainstream drug discovery strategy, with the potential to improve the efficiency and success rate of the development of novel therapeutic agents [27]. The combination of experimental and computational methods in fragment-based drug discovery (FBDD) is continuously progressing, offering new opportunities for the advancement of novel therapies [21,22] (Figure 1b).
3. Biochemical and Pharmacological Targets
Gaining knowledge about the biochemical and pharmacological targets of therapeutic action is essential for the creation of successful treatments. Current research has prioritized the identification of novel targets and the clarification of the processes by which existing medicines work.
3.1. Target Identification
Advanced techniques in molecular biology, including as proteomics and CRISPR-Cas9 screening, have been instrumental in identifying novel therapeutic targets [28,29]. Proteomics-based techniques have identified new targets for cancer therapy, leading to the development of inhibitors that are both more precise and potent [29,30,31]. The application of CRISPR-Cas9 screening has enabled the precise identification and validation of medicinal targets. This method allows scientists to deactivate, activate, or modify the expression of particular genes, thus uncovering their roles in disease pathways [28,32]. The integration of these high-throughput screening approaches with advanced computational tools has also enhanced the efficiency and accuracy of target identification [29,30]. Recent study has demonstrated the advantageous use of combining CRISPR-based techniques with proteomics to find novel targets for therapy and acquire a deeper understanding of the mechanisms of action of small molecules [28,30,31]. These advancements have significantly accelerated the drug discovery process, providing a solid basis for the development of targeted medications [29,32] (Figure 2a).
3.2. Mechanism of Action
Understanding the mechanism of action of medications requires comprehending how they interact with their targets on a molecular level [33]. Recent studies have utilized advanced methods such as cryo-electron microscopy (cryo-EM) and X-ray crystallography to see and understand the interactions between drugs and their targets. These techniques have allowed researchers to gain insights into how drugs bind to their targets and the structural changes that occur as a result of drug binding [34,35]. Cryo-EM offers structural insights at resolutions finer than 3 Å and can even achieve ultra-high resolutions up to 1.2 Å. By freezing samples at extremely low temperatures, Cryo-EM allows for the observation of structures in a state close to their natural form, facilitating the identification of dynamic processes. On the other hand, X-ray analysis of protein structures is limited to the crystallized state, making it challenging to directly observe dynamic changes in living organisms. However, X-ray analysis provides high resolutions ranging from 1.5 to 2.5 Å, enabling precise structural analysis. Cryo-EM has had a significant impact in the study of complex biological macromolecules, providing almost atomic-level resolution and uncovering dynamic processes that were previously impossible to observe [36,37]. X-ray crystallography is a fundamental technique in the field of structural biology, allowing for the precise visualization of protein-ligand complexes and providing guidance for the development of drugs based on their structure [38]. By combining structural approaches with computer modeling, we have gained a deeper knowledge of how drugs work. This has helped us build more efficient therapies [39]. The progress made in these areas has greatly expedited the process of finding new drugs, offering a strong structure for creating specific treatments and enhancing the effectiveness and safety of medications [40,41] (Figure 2b).
4. Drug Design and Synthetic Chemistry
The design and synthesis of novel pharmaceutical candidates are crucial stages in the drug discovery process. The progress in synthetic chemistry and computational approaches has expedited the creation of novel pharmaceuticals.
4.1. Automated De Novo Drug Design
Recent advancements in automated de novo drug design have facilitated the swift creation of new chemical compounds that possess specific desirable characteristics. Evolutionary algorithms and deep generative models have been used to investigate the extensive chemical space and discover potential therapeutic candidates [42]. Recent research has shown that deep generative models, such as variational autoencoders (VAEs) and generative adversarial networks (GANs), are effective in creating drug-like compounds that are both highly unique and easy to synthesize [43]. The incorporation of these models with structure-based techniques, such as molecular docking and molecular dynamics simulations, has additionally amplified their ability to predict and their effectiveness [42]. Furthermore, the utilization of reinforcement learning algorithms has demonstrated potential in enhancing the pharmacokinetic and pharmacodynamic characteristics of synthesized drugs [44]. The progress made in these areas has greatly expedited the process of finding new drugs, offering a strong structure for the creation of medicines that specifically target certain conditions [42] (Figure 3a).
4.2. Combinatorial Synthetic Chemistry
Combinatorial chemistry is the process of quickly creating extensive collections of chemicals, which may then be tested for their biological effects [45]. Current advancements in this domain have prioritized enhancing the effectiveness and variety of compound libraries, resulting in the identification of novel lead compounds for multiple therapeutic domains. The progress in high-throughput screening methods has greatly improved the capacity to efficiently and precisely assess a huge quantity of chemicals [46]. The incorporation of computer methods, such as virtual screening and quantitative structure-activity relationship (QSAR) modeling, has enhanced the refinement of identifying potential candidates [47]. In addition, the utilization of solid-phase synthesis techniques has simplified the creation of various chemical libraries, resulting in a more efficient and scalable procedure [48]. These advancements have not only expedited the process of discovering new drugs, but also decreased the related expenses and time required [47]. Advancements in combinatorial chemistry are expected to continue evolving and contribute to the creation of new therapies. This progress will help meet medical demands that have not been satisfied in different disease areas [45] (Figure 3b).
5. Virtual Screening
Virtual screening is a computational method employed to discover new drug candidates from extensive collections of chemicals. Recent progress in virtual screening has enhanced its precision and effectiveness.
5.1. Library Size and Diversity
The magnitude and variety of chemical libraries are essential factors in determining the effectiveness of virtual screening. Recent research indicates that the presence of larger and more diversified libraries enhances the likelihood of detecting active chemicals [49]. Nevertheless, the library’s quality is crucial, as it must encompass molecules that possess drug-like characteristics. Computational techniques such as docking have played a crucial role in expanding virtual screening libraries from millions to billions of molecules. These approaches help prioritize genuine ligands from a broad chemical space [49]. The expansion has resulted in the identification of molecules that fit better, as seen by the improvement in docking scores, which increase logarithmically with the size of the library. However, when the library size increases, there is also an increased probability of artifacts that take advantage of vulnerabilities in docking scoring and sampling. Therefore, it is necessary to implement techniques to reduce the negative effects of these artifacts. Moreover, the inclination towards bio-like compounds diminishes considerably in larger libraries, hence augmenting the investigation of novel chemical domains [50]. Hence, it is crucial to prioritize the preservation of high-quality, drug-like compounds and the effective management of artifacts in order to achieve successful virtual screening. Additionally, the inclusion of bigger and more diverse libraries can also be advantageous in this process [49,51,52].
5.2. Scoring Functions and Docking Algorithms
The accuracy of virtual screening has been greatly improved by advancements in scoring systems and docking algorithms. These technological breakthroughs have made it possible to identify high-affinity binders for a range of targets, including difficult ones such as protein-protein interactions. Recent research has demonstrated that combining machine learning with conventional scoring methods has enhanced performance in many targets, resulting in more accurate predictions of binding affinities [53,54]. In addition, the advancement of empirical scoring systems and the integration of knowledge-based methodologies have enhanced the accuracy of predicting binding modes and affinities [54]. Advanced sampling approaches, including shape matching, systematic search, and stochastic methods, have enhanced the precision of docking simulations [53]. The methodological improvements have increased the usefulness of virtual screening for a wider variety of targets, including those that were previously deemed challenging, such as protein-protein interactions. This is achieved by accurately simulating the intricate energy landscapes involved in these interactions [55]. As a result, the use of enhanced scoring functions and advanced docking algorithms has increased the effectiveness of virtual screening in drug discovery. This has made it easier to identify new therapeutic agents with great accuracy [56].
6. Drug Safety and Toxicology
Verifying the safety and effectiveness of novel medications is a crucial component of the drug development process. Current studies have prioritized enhancing the accuracy of medication safety prediction and comprehending the mechanisms that cause drug toxicity.
6.1. Predictive Toxicology
Novel chemicals’ toxicity can be predicted using computational models and in vitro testing. These techniques have been employed to detect possible unintended consequences and harmful byproducts, hence minimizing the likelihood of negative outcomes in clinical studies. Machine learning algorithms have been combined with classical in vitro assays in recent developments in predictive toxicology, resulting in improved accuracy of toxicity predictions [57,58]. In silico models, such as quantitative structure-activity relationship (QSAR) models, offer reliable and efficient alternatives to experimental procedures. However, it is still essential to have expert review of these predictions [57]. In addition, the advancement of multi-task deep learning models has made it possible to model in vitro, in vivo, and clinical toxicity data simultaneously, leading to a significant enhancement in the prediction of clinical toxicity endpoints Furthermore, the incorporation of micro-physiological systems (MPS) and physiologically based pharmacokinetic (PBPK) modeling into predictive toxicology frameworks has significantly improved the capacity to forecast human toxicity without the need for animal experimentation [59]. These developments collectively lead to a more efficient and ethical strategy in drug development, reducing the probability of late-stage failures caused by unanticipated toxicity [5] (Figure 5a).
6.2. Drug Toxicity Mechanisms
Comprehending the mechanisms behind drug toxicity is crucial in the development of safer pharmaceuticals [1]. Recent research has utilized omics technologies, such as transcriptomics and metabolomics, to examine the pathways implicated in drug-induced toxicity. These observations have resulted in the discovery of biomarkers that can be used to detect toxicity at an early stage For example, transcriptomics has been utilized to detect changes in gene expression that are linked to toxic reactions, whereas metabolomics has uncovered modifications in metabolic pathways that are connected to toxicity. In addition, the incorporation of these omics data with conventional toxicological evaluations has yielded a more thorough comprehension of the molecular pathways that underlie drug toxicity [1,60]. This comprehensive approach not only facilitates the early detection of potential harmful effects but also contributes to the creation of safer medical treatments [2,11] (Figure 5b).
7. Conclusions
Overall, the recent progress in Structure-Activity Relationship (SAR) studies, specifically in computational SAR models and Fragment-Based Drug Design (FBDD), has greatly improved our capacity to forecast and enhance the effectiveness of potential drugs. This highlights the strong collaboration between computational and experimental methods. Advancements in tools like CRISPR-Cas9 and cryo-electron microscopy have enhanced our ability to identify and comprehend biochemical and pharmacological targets. This has led to a greater understanding of drug mechanisms, enabling the development of more accurate and efficient therapeutic treatments. In addition, the combination of automated de novo drug design and combinatorial chemistry has sped up the creation and improvement of novel compounds, broadening the range of chemicals that could be used for therapeutic purposes. Virtual screening methodologies have experienced advancements in library size, scoring functions, and docking algorithms, resulting in enhanced accuracy and efficiency in the identification of potential drug candidates. Furthermore, the integration of predictive toxicology and mechanistic investigations of drug toxicity has improved the safety profiles of novel medications, guaranteeing a more efficient and morally sound drug development procedure. Collectively, these enhancements underscore the substantial impact of integrating computational and experimental methodologies in drug discovery, resulting in the development of more sophisticated therapies that are both safer and more effective.
Author Contributions
Conceptualization, investigation, writing, and original draft preparation, A.S. (Ahrum Son); H.K. (Hyunsoo Kim) – Visualization, and proofreading, J.P. (Jongham Park); W.K. (Woojin Kim); Y.Y. (Yoonki Yoon); S.L. (Sangwoon Lee) – Supervision, Project Administration, Funding Acquisition, Review and Editing, H.K. (Hyunsoo Kim). All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. RS-2023-00209456). This work was supported by the Korea Basic Science Institute (National research Facilities and Equipment Center) grant funded by the Korea government (MSIT) (No. RS-2024-00402298). This work was supported by Institute of Information & communications Technology Planning & Evaluation (IITP) grant funded by the Korea government (MSIT) (No. RS-2022-00155857, Artificial Intelligence Convergence Innovation Human Resources Development (Chungnam National University).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created.
Acknowledgments
This manuscript was proofread and edited with the assistance of ChatGPT-4, a language model by OpenAI.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Guengerich, F.P. Mechanisms of drug toxicity and relevance to pharmaceutical development. Drug Metab Pharmacokinet 2011, 26, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Pognan, F.; Beilmann, M.; Boonen, H.C.M.; Czich, A.; Dear, G.; Hewitt, P.; Mow, T.; Oinonen, T.; Roth, A.; Steger-Hartmann, T.; et al. The evolving role of investigative toxicology in the pharmaceutical industry. Nat Rev Drug Discov 2023, 22, 317–335. [Google Scholar] [CrossRef]
- Nguyen, N.; Jennen, D.; Kleinjans, J. Omics technologies to understand drug toxicity mechanisms. Drug Discov Today 2022, 27, 103348. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Scardino, V. Machine Learning Toxicity Prediction: Latest Advances by Toxicity End Point. ACS Omega 2022, 7, 47536–47546. [Google Scholar] [CrossRef]
- Tran, T.T.V.; Surya Wibowo, A.; Tayara, H.; Chong, K.T. Artificial Intelligence in Drug Toxicity Prediction: Recent Advances, Challenges, and Future Perspectives. J Chem Inf Model 2023, 63, 2628–2643. [Google Scholar] [CrossRef]
- Ingber, D.E. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat Rev Genet 2022, 23, 467–491. [Google Scholar] [CrossRef] [PubMed]
- Baker, T.K.; Van Vleet, T.R.; Mahalingaiah, P.K.; Grandhi, T.S.P.; Evers, R.; Ekert, J.; Gosset, J.R.; Chacko, S.A.; Kopec, A.K. The Current Status and Use of Microphysiological Systems by the Pharmaceutical Industry: The International Consortium for Innovation and Quality Microphysiological Systems Affiliate Survey and Commentary. Drug Metab Dispos 2024, 52, 198–209. [Google Scholar] [CrossRef]
- Rowland Yeo, K.; Gil Bergland, E.; Chen, Y. Dose Optimization Informed by PBPK Modeling: State-of-the Art and Future. Clin Pharmacol Ther 2024. [Google Scholar] [CrossRef]
- Chen, C.; Wang, J.; Pan, D.; Wang, X.; Xu, Y.; Yan, J.; Wang, L.; Yang, X.; Yang, M.; Liu, G.P. Applications of multi-omics analysis in human diseases. MedComm (2020) 2023, 4, e315. [Google Scholar] [CrossRef]
- Marques, L.; Costa, B.; Pereira, M.; Silva, A.; Santos, J.; Saldanha, L.; Silva, I.; Magalhaes, P.; Schmidt, S.; Vale, N. Advancing Precision Medicine: A Review of Innovative In Silico Approaches for Drug Development, Clinical Pharmacology and Personalized Healthcare. Pharmaceutics 2024, 16. [Google Scholar] [CrossRef]
- Ivanisevic, T.; Sewduth, R.N. Multi-Omics Integration for the Design of Novel Therapies and the Identification of Novel Biomarkers. Proteomes 2023, 11. [Google Scholar] [CrossRef]
- Chicco, D.; Cumbo, F.; Angione, C. Ten quick tips for avoiding pitfalls in multi-omics data integration analyses. PLoS Comput Biol 2023, 19, e1011224. [Google Scholar] [CrossRef]
- Guha, R. On exploring structure-activity relationships. Methods Mol Biol 2013, 993, 81–94. [Google Scholar] [CrossRef]
- Béquignon, O.J.M.; Gómez-Tamayo, J.C.; Lenselink, E.B.; Wink, S.; Hiemstra, S.; Lam, C.C.; Gadaleta, D.; Roncaglioni, A.; Norinder, U.; Water, B.v.d.; et al. Collaborative SAR Modeling and Prospective In Vitro Validation of Oxidative Stress Activation in Human HepG2 Cells. Journal of Chemical Information and Modeling 2023, 63, 5433–5445. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.; Thai, Q.M.; Pham, M.Q.; Minh, P.T.H.; Phung, H.T.T. Machine learning combines atomistic simulations to predict SARS-CoV-2 Mpro inhibitors from natural compounds. Molecular Diversity 2023, 28, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Batool, M.; Ahmad, B.; Choi, S. A Structure-Based Drug Discovery Paradigm. International Journal of Molecular Sciences 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W.; Barker, E.L. Computational Methods in Drug Discovery. Pharmacological Reviews 2013, 66, 334–395. [Google Scholar] [CrossRef]
- Naithani, U.; Guleria, V. Integrative computational approaches for discovery and evaluation of lead compound for drug design. Frontiers in Drug Discovery 2024, 4. [Google Scholar] [CrossRef]
- Kirsch, P.; Hartman, A.M.; Hirsch, A.K.H.; Empting, M. Concepts and Core Principles of Fragment-Based Drug Design. Molecules 2019, 24. [Google Scholar] [CrossRef]
- Li, Q. Application of Fragment-Based Drug Discovery to Versatile Targets. Frontiers in Molecular Biosciences 2020, 7. [Google Scholar] [CrossRef]
- Chan, B.W.G.L.; Lynch, N.B.; Tran, W.; Joyce, J.M.; Savage, G.P.; Meutermans, W.; Montgomery, A.P.; Kassiou, M. Fragment-based drug discovery for disorders of the central nervous system: designing better drugs piece by piece. Frontiers in Chemistry 2024, 12. [Google Scholar] [CrossRef]
- Bon, M.; Bilsland, A.; Bower, J.; McAulay, K. Fragment-based drug discovery—the importance of high-quality molecule libraries. Molecular Oncology 2022, 16, 3761–3777. [Google Scholar] [CrossRef] [PubMed]
- Mallakuntla, M.K.; Togre, N.S.; Santos, D.B.; Tiwari, S. Implications of Fragment-Based Drug Discovery in Tuberculosis and HIV. Pharmaceuticals 2022, 15. [Google Scholar] [CrossRef]
- Bon, M.; Bilsland, A.; Bower, J.; McAulay, K. Fragment-based drug discovery-the importance of high-quality molecule libraries. Mol Oncol 2022, 16, 3761–3777. [Google Scholar] [CrossRef]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: the impact of fragments on drug discovery. Nat Rev Drug Discov 2016, 15, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.; Lynch, N.B.; Tran, W.; Joyce, J.M.; Savage, G.P.; Meutermans, W.; Montgomery, A.P.; Kassiou, M. Fragment-based drug discovery for disorders of the central nervous system: designing better drugs piece by piece. Front Chem 2024, 12, 1379518. [Google Scholar] [CrossRef]
- Villemagne, B.; Faion, L.; Tangara, S.; Willand, N. Recent advances in Fragment-based strategies against tuberculosis. Eur J Med Chem 2023, 258, 115569. [Google Scholar] [CrossRef]
- Jost, M.; Weissman, J.S. CRISPR Approaches to Small Molecule Target Identification. ACS Chem Biol 2018, 13, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Schenone, M.; Dancik, V.; Wagner, B.K.; Clemons, P.A. Target identification and mechanism of action in chemical biology and drug discovery. Nat Chem Biol 2013, 9, 232–240. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Y.; Ma, N.; Tian, J.; Shao, Y.; Zhu, B.; Wong, Y.K.; Liang, Z.; Zou, C.; Wang, J. Target identification of natural medicine with chemical proteomics approach: probe synthesis, target fishing and protein identification. Signal Transduct Target Ther 2020, 5, 72. [Google Scholar] [CrossRef]
- Hedl, T.J.; San Gil, R.; Cheng, F.; Rayner, S.L.; Davidson, J.M.; De Luca, A.; Villalva, M.D.; Ecroyd, H.; Walker, A.K.; Lee, A. Proteomics Approaches for Biomarker and Drug Target Discovery in ALS and FTD. Front Neurosci 2019, 13, 548. [Google Scholar] [CrossRef]
- Tabana, Y.; Babu, D.; Fahlman, R.; Siraki, A.G.; Barakat, K. Target identification of small molecules: an overview of the current applications in drug discovery. BMC Biotechnol 2023, 23, 44. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.H.; Shimoni, Y.; Yang, W.S.; Subramaniam, P.; Iyer, A.; Nicoletti, P.; Rodriguez Martinez, M.; Lopez, G.; Mattioli, M.; Realubit, R.; et al. Elucidating Compound Mechanism of Action by Network Perturbation Analysis. Cell 2015, 162, 441–451. [Google Scholar] [CrossRef]
- Maveyraud, L.; Mourey, L. Protein X-ray Crystallography and Drug Discovery. Molecules 2020, 25. [Google Scholar] [CrossRef]
- Kausar, S.; Said Khan, F.; Ishaq Mujeeb Ur Rehman, M.; Akram, M.; Riaz, M.; Rasool, G.; Hamid Khan, A.; Saleem, I.; Shamim, S.; Malik, A. A review: Mechanism of action of antiviral drugs. Int J Immunopathol Pharmacol 2021, 35, 20587384211002621. [Google Scholar] [CrossRef]
- Lees, J.A.; Dias, J.M.; Han, S. Applications of Cryo-EM in small molecule and biologics drug design. Biochem Soc Trans 2021, 49, 2627–2638. [Google Scholar] [CrossRef] [PubMed]
- Van Drie, J.H.; Tong, L. Cryo-EM as a powerful tool for drug discovery. Bioorg Med Chem Lett 2020, 30, 127524. [Google Scholar] [CrossRef]
- Zheng, H.; Handing, K.B.; Zimmerman, M.D.; Shabalin, I.G.; Almo, S.C.; Minor, W. X-ray crystallography over the past decade for novel drug discovery - where are we heading next? Expert Opin Drug Discov 2015, 10, 975–989. [Google Scholar] [CrossRef] [PubMed]
- Cebi, E.; Lee, J.; Subramani, V.K.; Bak, N.; Oh, C.; Kim, K.K. Cryo-electron microscopy-based drug design. Front Mol Biosci 2024, 11, 1342179. [Google Scholar] [CrossRef]
- Banerjee, S.; Banerjee, D.; Singh, A.; Kumar, S.; Pooja, D.; Ram, V.; Kulhari, H.; Saharan, V.A. A Clinical Insight on New Discovered Molecules and Repurposed Drugs for the Treatment of COVID-19. Vaccines (Basel) 2023, 11. [Google Scholar] [CrossRef]
- Zhu, K.F.; Yuan, C.; Du, Y.M.; Sun, K.L.; Zhang, X.K.; Vogel, H.; Jia, X.D.; Gao, Y.Z.; Zhang, Q.F.; Wang, D.P.; et al. Applications and prospects of cryo-EM in drug discovery. Mil Med Res 2023, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Moretti, R.; Meiler, J. Recent Advances in Automated Structure-Based De Novo Drug Design. J Chem Inf Model 2024, 64, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Atz, K.; Cotos, L.; Isert, C.; Hakansson, M.; Focht, D.; Hilleke, M.; Nippa, D.F.; Iff, M.; Ledergerber, J.; Schiebroek, C.C.G.; et al. Prospective de novo drug design with deep interactome learning. Nat Commun 2024, 15, 3408. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Dai, H.; Knight, E.; Wu, F.; Li, Y.; Li, T.; Gerstein, M. A survey of generative AI for de novo drug design: new frontiers in molecule and protein generation. Brief Bioinform 2024, 25. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, X.; Lam, K.S. Combinatorial chemistry in drug discovery. Curr Opin Chem Biol 2017, 38, 117–126. [Google Scholar] [CrossRef]
- Galloway, W.R.; Isidro-Llobet, A.; Spring, D.R. Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat Commun 2010, 1, 80. [Google Scholar] [CrossRef]
- Sadybekov, A.V.; Katritch, V. Computational approaches streamlining drug discovery. Nature 2023, 616, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, K.K.; Ha, H.H.; Kang, N.Y.; Chandran, Y.; Chang, Y.T. Solid phase combinatorial synthesis of a xanthone library using click chemistry and its application to an embryonic stem cell probe. Chem Commun (Camb) 2011, 47, 7488–7490. [Google Scholar] [CrossRef]
- Lyu, J.; Irwin, J.J.; Shoichet, B.K. Modeling the expansion of virtual screening libraries. Nat Chem Biol 2023, 19, 712–718. [Google Scholar] [CrossRef]
- Kuan, J.; Radaeva, M.; Avenido, A.; Cherkasov, A.; Gentile, F. Keeping pace with the explosive growth of chemical libraries with structure-based virtual screening. WIREs Computational Molecular Science 2023, 13. [Google Scholar] [CrossRef]
- Brenk, R.; Schipani, A.; James, D.; Krasowski, A.; Gilbert, I.H.; Frearson, J.; Wyatt, P.G. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem 2008, 3, 435–444. [Google Scholar] [CrossRef]
- Grotsch, K.; Sadybekov, A.V.; Hiller, S.; Zaidi, S.; Eremin, D.; Le, A.; Liu, Y.; Smith, E.C.; Illiopoulis-Tsoutsouvas, C.; Thomas, J.; et al. Virtual Screening of a Chemically Diverse “Superscaffold” Library Enables Ligand Discovery for a Key GPCR Target. ACS Chem Biol 2024, 19, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Shamsian, S.; Sokouti, B.; Dastmalchi, S. Benchmarking different docking protocols for predicting the binding poses of ligands complexed with cyclooxygenase enzymes and screening chemical libraries. Bioimpacts 2024, 14, 29955. [Google Scholar] [CrossRef] [PubMed]
- Guedes, I.A.; Pereira, F.S.S.; Dardenne, L.E. Empirical Scoring Functions for Structure-Based Virtual Screening: Applications, Critical Aspects, and Challenges. Front Pharmacol 2018, 9, 1089. [Google Scholar] [CrossRef]
- Vakser, I.A. Protein-protein docking: from interaction to interactome. Biophys J 2014, 107, 1785–1793. [Google Scholar] [CrossRef]
- Leung, C.H.; Ma, D.L. Recent advances in virtual screening for drug discovery. Methods 2015, 71, 1–3. [Google Scholar] [CrossRef]
- Giorgini, M.; Taroncher, M.; Ruiz, M.J.; Rodriguez-Carrasco, Y.; Tolosa, J. In Vitro and Predictive Computational Toxicology Methods for the Neurotoxic Pesticide Amitraz and Its Metabolites. Brain Sci 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Chenthamarakshan, V.; Dhurandhar, A.; Pereira, S.; Hendler, J.A.; Dordick, J.S.; Das, P. Accurate clinical toxicity prediction using multi-task deep neural nets and contrastive molecular explanations. Sci Rep 2023, 13, 4908. [Google Scholar] [CrossRef] [PubMed]
- Najjar, A.; Kramer, N.; Gardner, I.; Hartung, T.; Steger-Hartmann, T. Editorial: Advances in and applications of predictive toxicology: 2022. Front Pharmacol 2023, 14, 1257423. [Google Scholar] [CrossRef]
- Barnes, D.A.; Firman, J.W.; Belfield, S.J.; Cronin, M.T.D.; Vinken, M.; Janssen, M.J.; Masereeuw, R. Development of an adverse outcome pathway network for nephrotoxicity. Arch Toxicol 2024, 98, 929–942. [Google Scholar] [CrossRef]
Figure 1.
An overview of advanced computational approaches in drug discovery, demonstrating the integration of machine learning with atomic-level simulations and the optimization of drug candidates through Fragment-Based Drug Discovery (FBDD). (a) The identification of potential drug candidates is considerably improved while time and cost are reduced by the synergy between atomic-level in silico docking simulations and machine learning techniques. More precise and realistic simulations are facilitated by detailed atomic-level docking data, which includes the rotation and coordinates of individual atoms. This hybrid approach, when combined with sophisticated machine learning algorithms, expedites the overall drug discovery process by enhancing both efficiency and accuracy. (b) A schematic illustration of the drug optimization process that employs Fragment-Based Drug Discovery (FBDD). The efficiency of FBDD has been significantly enhanced by recent advancements in high-throughput screening and structural biology, resulting in a reduction in the time necessary for drug development. Potential matches are identified through the screening of small molecular fragments to evaluate their interactions with target proteins in FBDD. The broader drug discovery pipeline has been substantially impacted by the combination of these technological and methodological advancements.
Figure 1.
An overview of advanced computational approaches in drug discovery, demonstrating the integration of machine learning with atomic-level simulations and the optimization of drug candidates through Fragment-Based Drug Discovery (FBDD). (a) The identification of potential drug candidates is considerably improved while time and cost are reduced by the synergy between atomic-level in silico docking simulations and machine learning techniques. More precise and realistic simulations are facilitated by detailed atomic-level docking data, which includes the rotation and coordinates of individual atoms. This hybrid approach, when combined with sophisticated machine learning algorithms, expedites the overall drug discovery process by enhancing both efficiency and accuracy. (b) A schematic illustration of the drug optimization process that employs Fragment-Based Drug Discovery (FBDD). The efficiency of FBDD has been significantly enhanced by recent advancements in high-throughput screening and structural biology, resulting in a reduction in the time necessary for drug development. Potential matches are identified through the screening of small molecular fragments to evaluate their interactions with target proteins in FBDD. The broader drug discovery pipeline has been substantially impacted by the combination of these technological and methodological advancements.
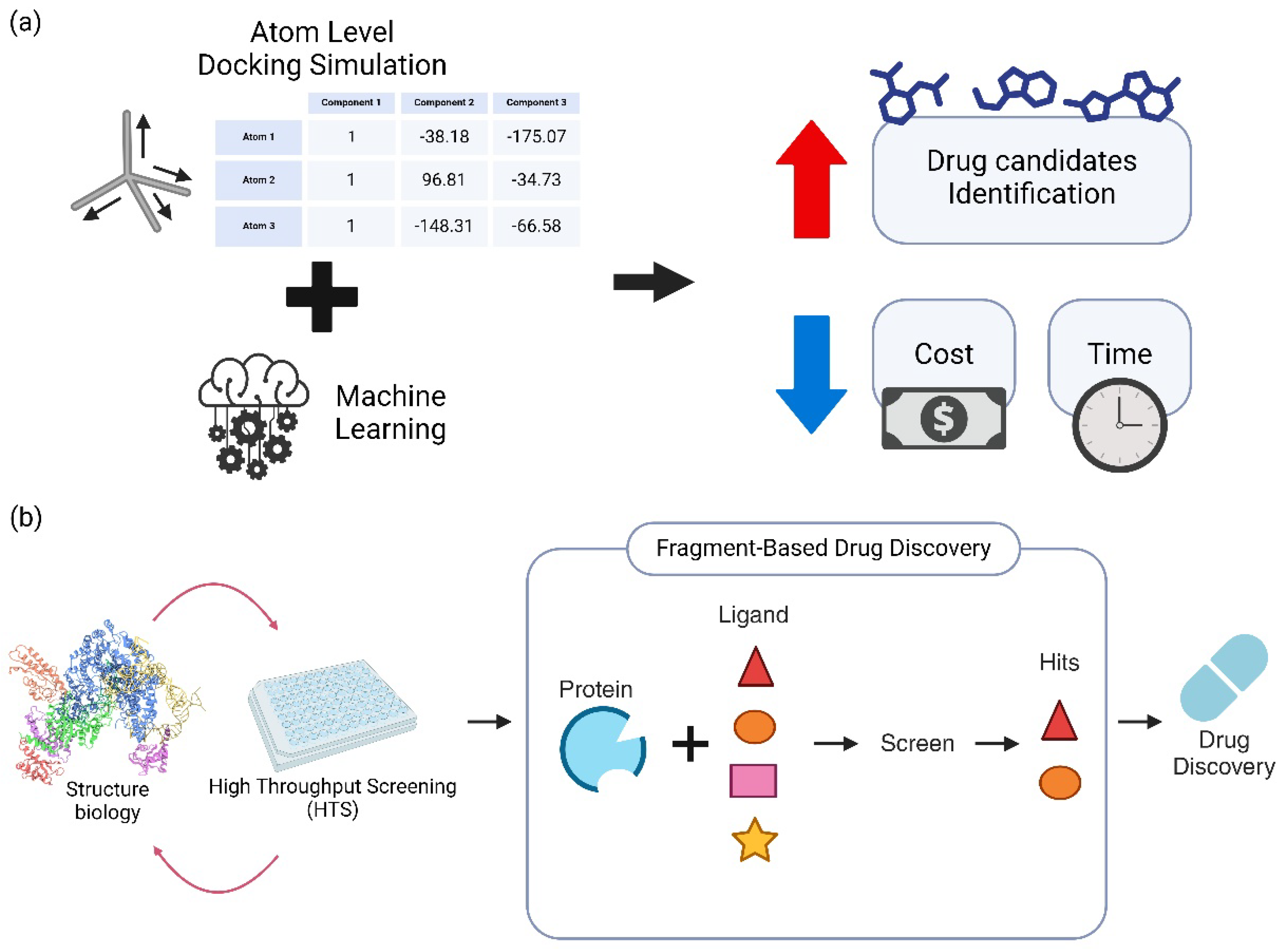
Figure 2.
Technical methods for target identification and the understanding of mechanisms of action for the detection of biochemical and pharmacological targets. (a) The schematic illustrates the benefits of integrating sophisticated computational tools, such as machine learning and AI, with proteomics and Crispr-Cas 9 screening for Target Identification. It has the advantage of facilitating the efficient discovery of novel therapeutic targets and the accurate identification and validation of drug targets, thereby increasing the potential for the development of targeted therapies. (b) The schematic demonstrates the potential to expedite the drug discovery process and enhance the efficacy and safety of drugs by visualizing biomacromolecules and protein-ligand complexes using cryo-electron microscopy (cryo-EM) and X-ray crystallography.
Figure 2.
Technical methods for target identification and the understanding of mechanisms of action for the detection of biochemical and pharmacological targets. (a) The schematic illustrates the benefits of integrating sophisticated computational tools, such as machine learning and AI, with proteomics and Crispr-Cas 9 screening for Target Identification. It has the advantage of facilitating the efficient discovery of novel therapeutic targets and the accurate identification and validation of drug targets, thereby increasing the potential for the development of targeted therapies. (b) The schematic demonstrates the potential to expedite the drug discovery process and enhance the efficacy and safety of drugs by visualizing biomacromolecules and protein-ligand complexes using cryo-electron microscopy (cryo-EM) and X-ray crystallography.
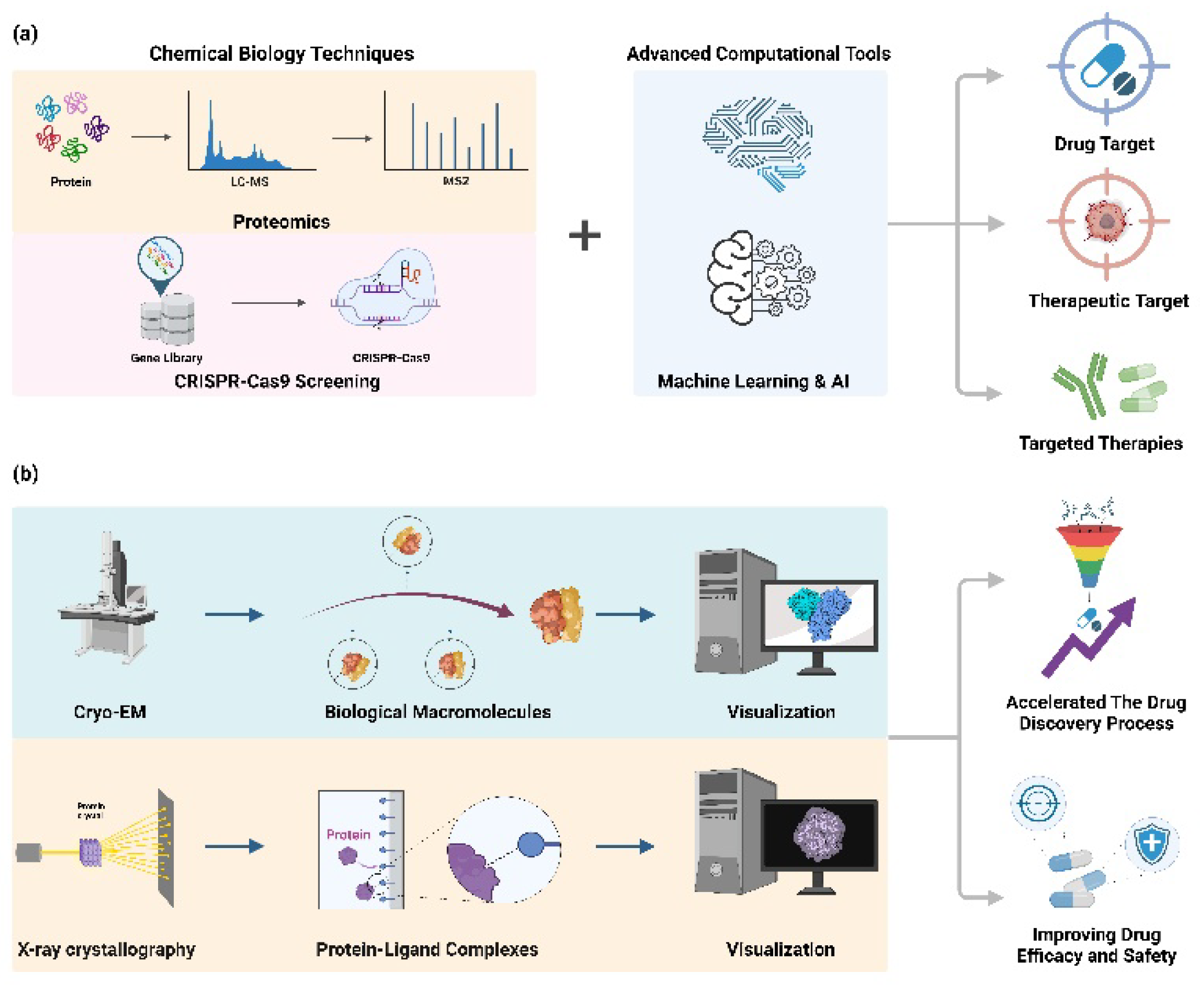
Figure 3.
Optimization of the synthesis process and advancement in new drug candidate design and synthesis through an AI-based integrated approach. (a) Deep generative models, including variational autoencoders (VAEs) and generative adversarial networks (GANs), and evolutionary algorithms are potent instruments for the identification of potential drug candidates. In particular, it generates novel candidate medications by employing a generative AI model. In conjunction with structure-based simulation analysis tools, such as molecular docking or molecular dynamics simulations, these potent AI-based new drug candidate derivation algorithms can be employed to identify more dependable candidate substances. (b) The efficacy and diversity of large compound libraries have been significantly enhanced by advancements in high-throughput screening technologies, such as combinatorial chemistry. The diversity of large-scale compounds can be secured, evaluation can be conducted more swiftly, and the accuracy has been significantly enhanced through methods such as quantitative structure-activity relationship (QSAR) modeling. Furthermore, the production of a wide range of compound libraries has been simplified by advancements in solid-phase synthesis technology. High-throughput screening and synthesis technologies have the potential to significantly address a variety of unmet medical requirements.
Figure 3.
Optimization of the synthesis process and advancement in new drug candidate design and synthesis through an AI-based integrated approach. (a) Deep generative models, including variational autoencoders (VAEs) and generative adversarial networks (GANs), and evolutionary algorithms are potent instruments for the identification of potential drug candidates. In particular, it generates novel candidate medications by employing a generative AI model. In conjunction with structure-based simulation analysis tools, such as molecular docking or molecular dynamics simulations, these potent AI-based new drug candidate derivation algorithms can be employed to identify more dependable candidate substances. (b) The efficacy and diversity of large compound libraries have been significantly enhanced by advancements in high-throughput screening technologies, such as combinatorial chemistry. The diversity of large-scale compounds can be secured, evaluation can be conducted more swiftly, and the accuracy has been significantly enhanced through methods such as quantitative structure-activity relationship (QSAR) modeling. Furthermore, the production of a wide range of compound libraries has been simplified by advancements in solid-phase synthesis technology. High-throughput screening and synthesis technologies have the potential to significantly address a variety of unmet medical requirements.
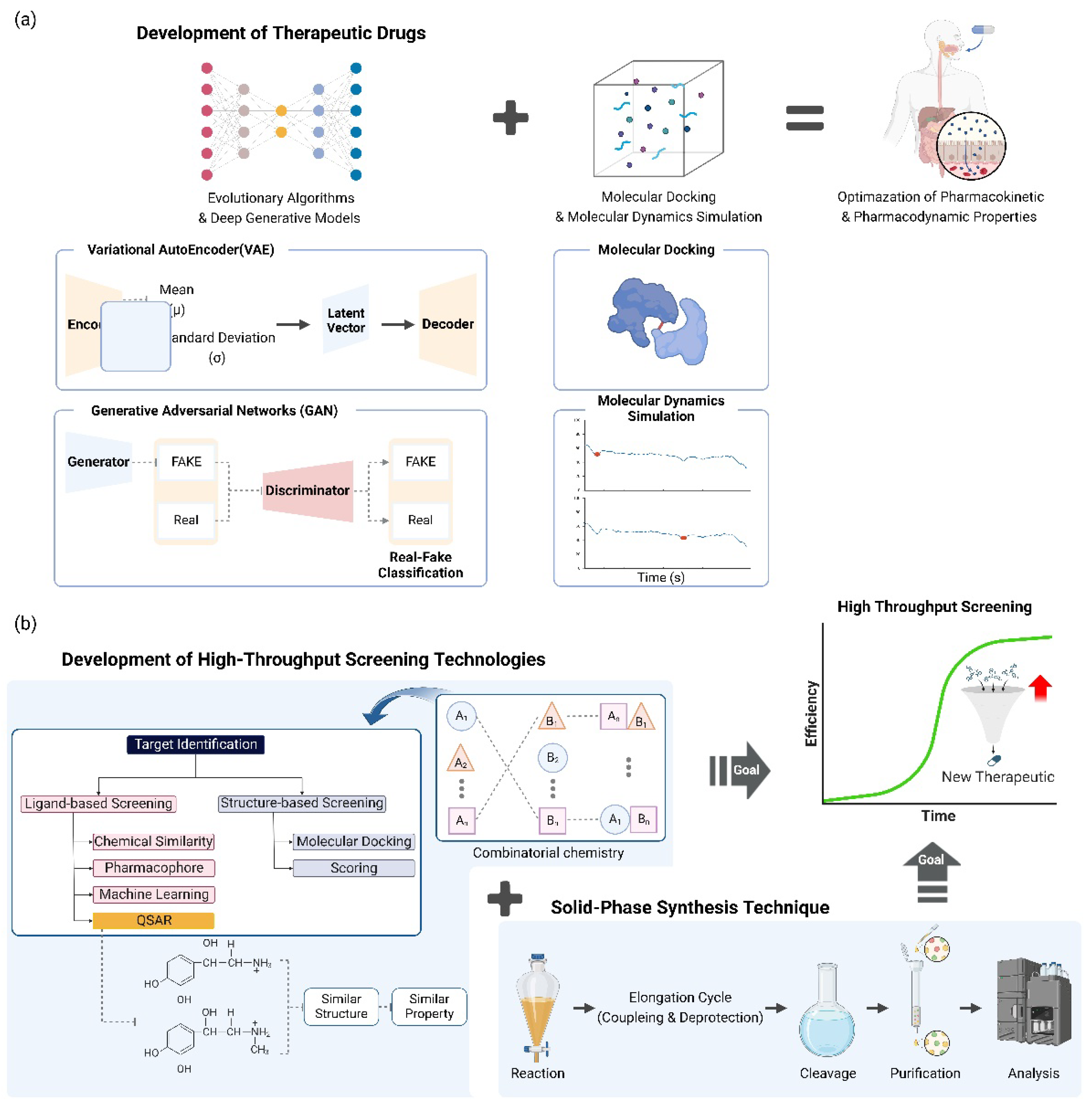
Figure 4.
Illustrates the primary factors that should be taken into account when conducting virtual screening in order to optimize efficiency and accuracy. (a) Benefits of library diversity and size. The accuracy of Docking Score calculations and the likelihood of identifying promising candidates are among the numerous advantages that the size and diversity of a chemical library provide. Nevertheless, the library may encounter a variety of structural challenges as its volume increases. Effective quality management can alleviate these obstacles, suggesting that the preservation of high quality is equally critical as the library’s size and diversity. (b) A schematic that illustrates the role of enhanced docking algorithms and scoring functions in improving the efficiency of the virtual screening process, thereby furthering drug discovery. The overall efficacy of the drug discovery pipeline has been significantly enhanced by the integration of docking algorithms and scoring functions in virtual screening, which has resulted in significant improvements in performance and binding affinity prediction.
Figure 4.
Illustrates the primary factors that should be taken into account when conducting virtual screening in order to optimize efficiency and accuracy. (a) Benefits of library diversity and size. The accuracy of Docking Score calculations and the likelihood of identifying promising candidates are among the numerous advantages that the size and diversity of a chemical library provide. Nevertheless, the library may encounter a variety of structural challenges as its volume increases. Effective quality management can alleviate these obstacles, suggesting that the preservation of high quality is equally critical as the library’s size and diversity. (b) A schematic that illustrates the role of enhanced docking algorithms and scoring functions in improving the efficiency of the virtual screening process, thereby furthering drug discovery. The overall efficacy of the drug discovery pipeline has been significantly enhanced by the integration of docking algorithms and scoring functions in virtual screening, which has resulted in significant improvements in performance and binding affinity prediction.
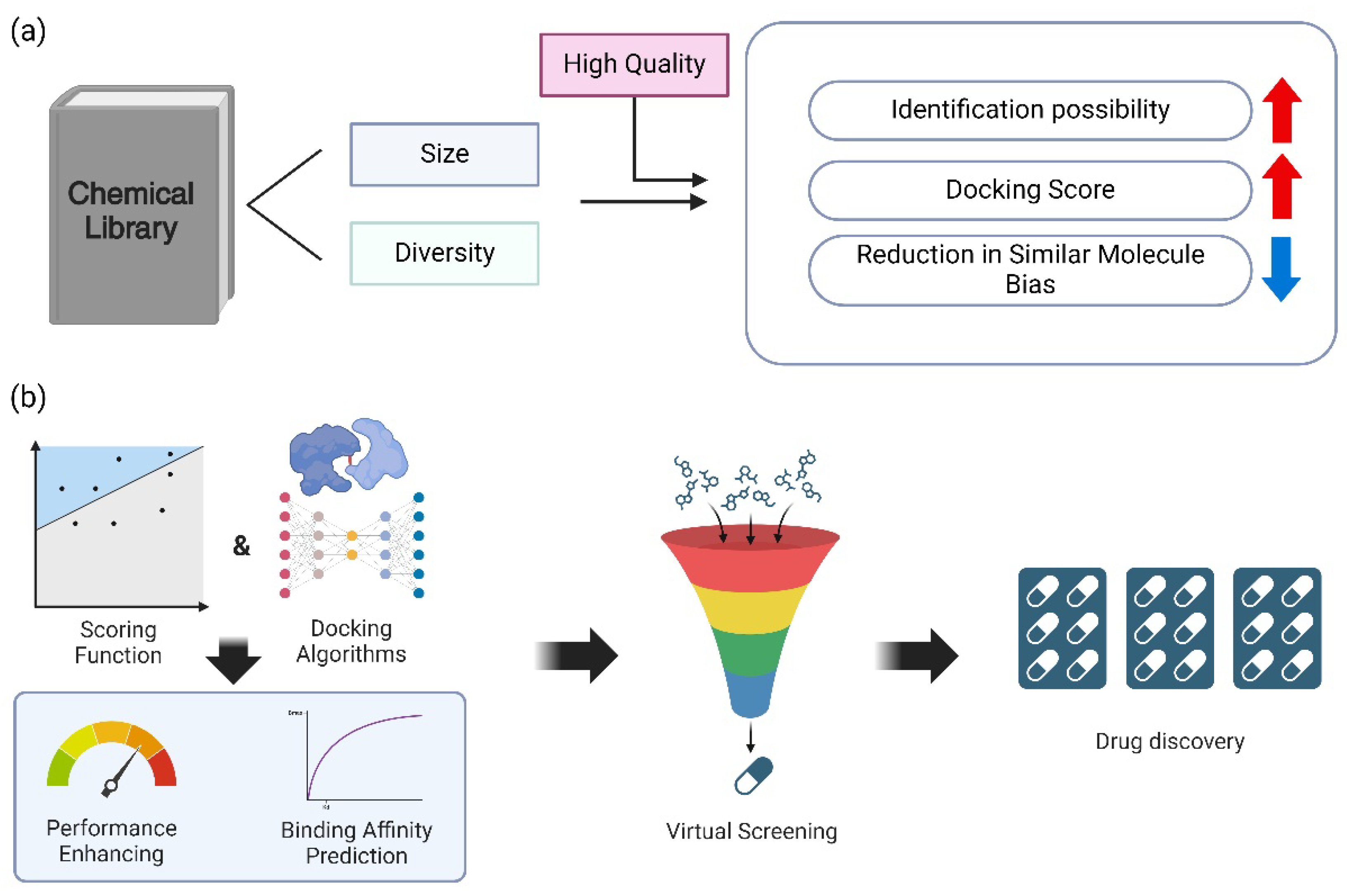
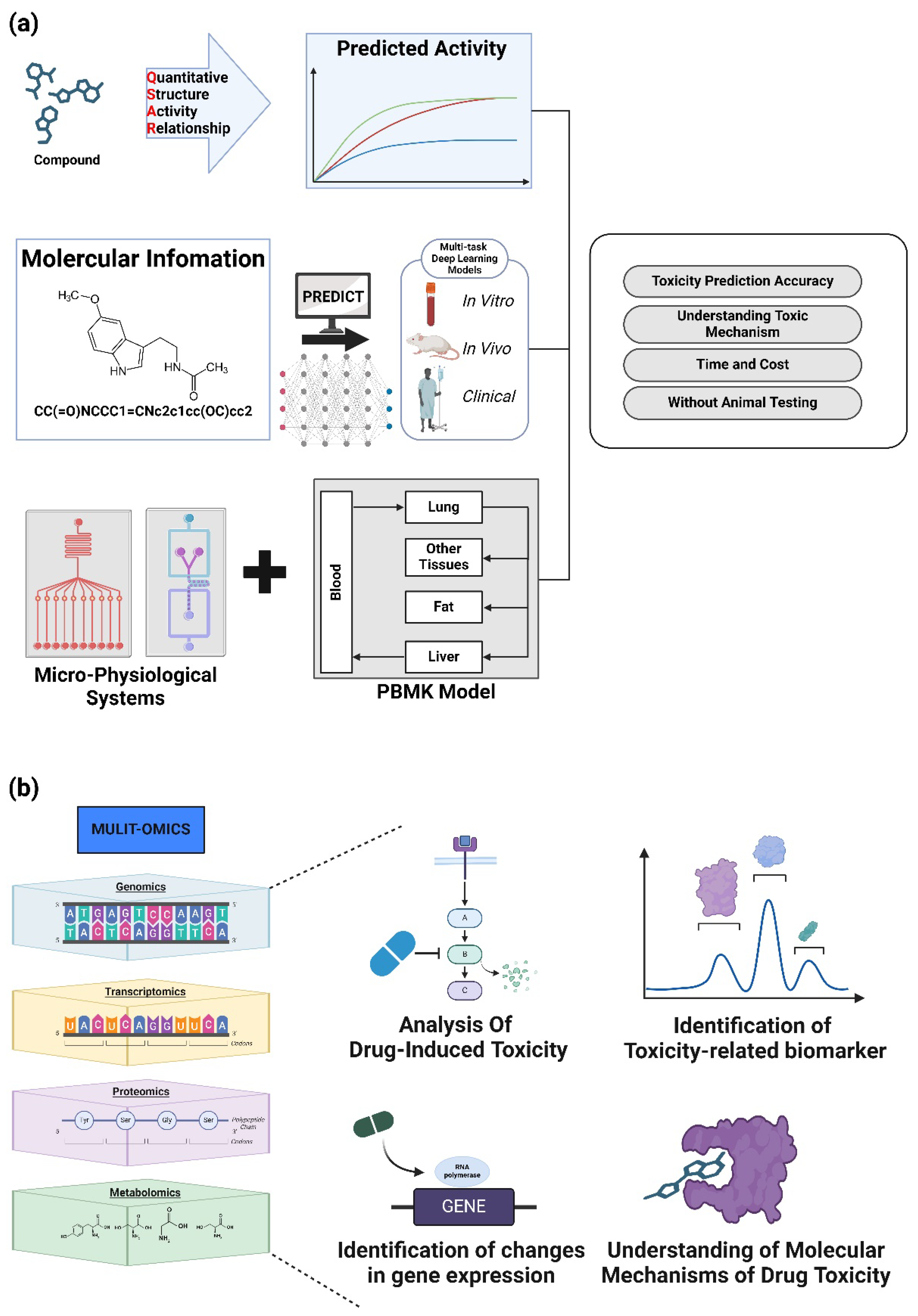
Figure 5.
Toxicology and Drug Safety. (a) Recent advancements in in silico research have improved the efficacy and accuracy of drug development in predictive toxicology. Quantitative Structure-Activity Relationship (QSAR) models provide a swift and dependable alternative to traditional experimental methods by predicting the activity of specific compounds. The prediction of clinical toxicity outcomes is enhanced by the simultaneous modeling of in vitro, in vivo, and clinical toxicity data, which is facilitated by multi-task deep learning models. The drug development process is rendered more efficient and ethical through the integration of physiologically based pharmacokinetic (PBPK) modeling and micro-physiological systems (MPS). This further enhances the capacity to predict human toxicity without relying on animal testing. (b) To identify alterations in metabolic pathways associated with toxicity and to detect gene expression changes linked to toxic responses, multi-omics approaches are employed. The molecular mechanisms that drive drug toxicity are more comprehensively understood when these omics data are integrated with traditional toxicological assessments. This integrative approach not only facilitates the early detection of potential toxicities but also contributes to the development of safer therapeutic agents.
Figure 5.
Toxicology and Drug Safety. (a) Recent advancements in in silico research have improved the efficacy and accuracy of drug development in predictive toxicology. Quantitative Structure-Activity Relationship (QSAR) models provide a swift and dependable alternative to traditional experimental methods by predicting the activity of specific compounds. The prediction of clinical toxicity outcomes is enhanced by the simultaneous modeling of in vitro, in vivo, and clinical toxicity data, which is facilitated by multi-task deep learning models. The drug development process is rendered more efficient and ethical through the integration of physiologically based pharmacokinetic (PBPK) modeling and micro-physiological systems (MPS). This further enhances the capacity to predict human toxicity without relying on animal testing. (b) To identify alterations in metabolic pathways associated with toxicity and to detect gene expression changes linked to toxic responses, multi-omics approaches are employed. The molecular mechanisms that drive drug toxicity are more comprehensively understood when these omics data are integrated with traditional toxicological assessments. This integrative approach not only facilitates the early detection of potential toxicities but also contributes to the development of safer therapeutic agents.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



