
Submitted:
28 August 2024
Posted:
29 August 2024
You are already at the latest version
Abstract
Nanopore raw read accuracy has improved to over 99%, making it a potential tool for metabarcoding. For broad adoption, guidelines on quality filtering are needed to ensure reliable taxonomic unit recovery. This study aims to provide those guidelines for a fungal metabarcoding context and to apply them to a case-study of ectomycorrhizae in decaying bark of Fagus sylvatica.We introduce the eNano pipeline to test two standard metabarcoding approaches: (1) reference-based mapping leveraging the UNITE’s species hypothesis system (SH-approach) and (2) constructing 98% OTUs (OTU-approach). We evaluate these approaches using a mock and natural community. Our results demonstrate that both approaches are effective with Nanopore data. When using a reference database, we recommend strict mapping criteria rather than Phred-based filtering. For 98% OTUs, filtering reads at ≥Q25 is recommended. Our case-study reveals that the decay gradient is a primary determinant of community composition, and that specific mycorrhizal fungi colonize decaying bark. Complementing our metabarcoding results with root tip morphotypification, we identify Laccaria amethystina and Tomentella sublilacina as key ectomycorrhizae of saplings on decaying logs. These findings demonstrate that Nanopore sequencing can provide valuable ecological insights and support its broader use in fungal metabarcoding as read quality continues to improve.
Keywords:
Nanopore
; deadwood
; ectomycorrhiza
; Fagus sylvatica
; quality filtering
; Phred score
; eNano pipeline
; natural regeneration
; metabarcoding
1. Introduction
1.1. Fungal Metabarcoding
A significant hurdle in fungal ecology is that most species elude visual detection. This affects especially community ecological studies, where the aim is to include all community members. While second generation sequencing propelled fungal ecology by enabling large-scale metabarcoding, the taxonomic resolution using the dominant Illumina platform is constrained by short amplicon lengths at maximum 2×300 bp. This hampers the exploitation of the fungal barcode region, the Internal Transcribed Spacer (ITS), which ranges between 250-1500 base-pairs (bp) [1–3]. Third-generation sequencing technologies, such as Nanopore (Oxford Nanopore Technologies Inc.) and PacBio SMRT (Pacific BioSciences Inc.), have overcome read length limitations, enabling the targeting of the full-length ITS region. While Nanopore initially lagged in quality, advancements in chemistry (V14) and algorithms (transformer model-based architecture, dorado ≥v0.7) are closing this gap. Raw read accuracy now exceeds 99% correct base call probability [4]. This accuracy surpasses the interspecific distances typically observed for the ITS region [5], potentially making Nanopore a viable tool in fungal metabarcoding.
Despite these advancements, studies on the application of Nanopore sequencing in fungal metabarcoding remain limited, with conclusions varying from unsuitable to feasible for simple communities, depending on the version of the technology used [6–11]. Broadly speaking, two approaches have thus far been used for processing Nanopore metabarcoding data. The first, standard metabarcoding, approach aligns raw reads against a reference database, retaining reads that can be confidently mapped [7,9,10]. Most often some minimum Phred score is set. This approach is dependent on the comprehensiveness and accuracy of the reference database and shifts the quality issue to defining what constitutes a confidently mapped sequence. The second approach is more sophisticated and improves raw sequence quality by drafting consensus sequences from clusters [6,9,11], as implemented in tools like Decona, ONTrack2, and CONCOMPRA [12–14]. Notably, the most widely adopted approach in metabarcoding—generating operational taxonomic units (OTUs) de novo from raw data— has been considered unfeasible with Nanopore data as high error rates significantly impact clustering. This standard approach involves setting a minimum Phred score —often Q28 as implemented in FASTQC [15]— and clustering the filtered reads at a predefined similarity threshold. At the time of writing, enforcing a Q28 threshold results in a near-total reduction of usable data. Like consensus sequence generation, OTU clustering bypasses the need for a reference database, offering an advantage for taxonomic units absent from databases. As outlined above, setting a minimum Phred score is standard practice in all approaches. Therefore, establishing a robust minimal quality threshold for each approach is crucial for the reliable recovery of taxonomic units, which underpins all subsequent analyses. However, guidelines for determining an appropriate threshold remain sparse, complicating the analysis of Nanopore data and raising the question of what read quality suffices in a metabarcoding context.
In this paper, we aim to provide guidelines for quality filtering of Nanopore metabarcoding data using the standard metabarcoding reference-based and OTU-clustering approaches. We detail our experiences with analyzing Nanopore data from a mock community and share our bioinformatics pipeline eNano. Building on these insights, we apply our methods to an often-overlooked microsite—the community within decaying bark of European beech (Fagus sylvatica) and validate our results through morphotypification of EcM mantles.
1.2. Case-Study: Deadwood Bark Ectomycorrhizae
A specific aspect of habitat creation by deadwood is the case where large logs in intermediate to advanced decay stages act as a tree regeneration site (Figure 1). In mycorrhizal research, these logs are of particular interest because the excavation of root systems can confirm the establishment of ectomycorrhizal fungi (EcM) through morphotypification.
While EcM are abundant in soil and readily mycorrhize roots after germination [16], they are initially absent or scarce in deadwood. Their prevalence increases as the wood decays and becomes more penetrable [17–21]. Eventually, the fungal community in deadwood transitions to resemble that of soil, completing a full ecological transformation [20]. In contrast to fruitbody based inventories, metabarcoding techniques have detected EcM surprisingly early in the decay process. For example, Rajala et al. (2011) found metabolically active EcM in slightly decayed Picea abies logs, suggesting a continuum of tree regeneration in old natural forests. Several reasons for colonization have been put forward, including nutrient mining [22–24], elevating fruiting positions, and associating with sapling roots in decaying logs [21,25].
with EcM is believed to be crucial for survival, especially in environments where nutrients are hard to access such as recent deadwood [26]. For example, in North-America Tsuga heterophylla regenerating on deadwood is known to associate with a wide range of EcM species [16,27,28]. In Europe, Tedersoo et al. [25,29] studied sapling EcM in mixed Estonian forests and found the most frequent species on roots in deadwood also to be common in soil, consistent with metabarcoding results.
Over the past decade, we have occasionally observed seedlings of European beech growing on relatively fresh deadwood. These seedlings primarily root in the decaying inner bark—comprising the secondary phloem and nutrient-rich cambium—rather than in the denser wood underneath (Figure 1). This layer, often overlooked except in studies on bark beetles (e.g., [30]), offers a potentially favorable environment for EcM colonization due to its higher moisture content, higher nutrient availability, less compact nature and faster decay rate [31–34]. Such conditions could facilitate the penetration of sapling roots and EcM, either originating from soil or from propagules. However, there is limited literature available to contextualize our observations, making it an intriguing case-study for this underexplored EcM niche.
2. Materials and Methods
2.1. Study Site and Log Selection
Sampling was conducted in the Joseph Zwaenepoel forest reserve (Brussels, Belgium).This forest reserve is dominated by European beech (Fagus sylvatica) and has been left unmanaged since 1983. Further information on the study site can be found in Vandekerkhove et al. [35] and De Keersmaeker et al. [36]. For this study, we specifically selected large beech logs of intermediate decay stage (Log decay stage 2—3 according to Renvall et al. [37]) bearing newly established saplings. This means that the wood is still mostly hard, and the bark is still present, but starts to loosen. We selected 29 suitable logs, an overview of the sampled logs is given in Table S1.
2.2. Root Tip Morphotypification and Sanger Barcoding
From the selected logs, a set of beech saplings were collected in September – November 2022. Sapling age was approximated by counting the number of leaf bud scars zones along the dominant axis. To extract saplings from the logs, a knife was used to pry away the outer bark (periderm). Then, the roots were carefully pulled out of the decaying bark (secondary phloem). Saplings were transported separately, with their roots wrapped in aluminium foil to prevent contamination and desiccation of EcM mantles. In addition, five saplings growing on soil in close proximity of the log were collected to serve as a baseline. All plants were stored at 4°C and processed within three days from collection.
We morphotypified the EcM mantles, using the work of Agerer (1991, 2001). Mantle coverage was estimated by sectioning the fine root tips (⌀ < 1 mm) into fragments of approximately 5mm. Each fragment was inspected for mantle presence to estimate total coverage, expressed as the ratio of colonized to total root tips. Mantle micromorphology was assessed using brightfield microscopy. For each morphotype, three root tips were separately preserved for Sanger sequencing.
Kruskal-Wallis rank sum , and Dunn’s tests (stats 4.2.3, FSA 0.9.4) were used to test for differences in EcM species richness and colonization rates between bark– and soil-growing saplings.
Genomic DNA was isolated from the mycorrhized root tips of the collected saplings following a modified cetyltrimethylammonium bromide (CTAB) method (Doyle & Doyle, 1987). Homogenized mantles were suspended in CTAB extraction buffer and incubated on a heat block at 55°C for at least two hours. The lysate was purified with two rounds of chloroform:isoamyl alcohol cleanups and precipitated using isopropanol (0.54x)|ammonium acetate (0.08x). The ITS region was amplified using Dream Taq Mastermix (Thermo Fisher) and primer pair ITS1F/ITS4 (10 µM) (Gardes & Bruns, 1993; White et al., 1990; Schoch et al., 2012). Amplification conditions were: initial denaturation at 94°C for 5 min; 30 cycles of 94°C for 30 sec, 55°C for 30 sec, and 72°C for 45 sec; and a final extension at 70°C for 7 min. Amplicons were purified using EXOFastAP and sequenced by Macrogen™.
2.3. Nanopore Metabarcoding
2.3.1. Mock Community
We constructed a mock community comprising 16 taxa that represent major ectomycorrhizal lineages and are phylogenetically diverse. Moreover, we included some closely related species to assess the sensitivity of our run at lower taxonomic levels. DNA extracts were sourced from previous collections (45–48) and opportunistically pooled without prior quality control. Included taxa are detailed in Table S2.
2.3.2. Bark Sampling
We gathered decaying inner bark of 16 beech logs with Beech regeneration, with two of these logs sampled at three distinct locations on the log to assess community turnover at the log level. Three logs without regeneration were selected as a control group.
We assessed inner bark decay stage (bark DS) on a 3 level scale from early to late as described in Table 1.
Each substrate sample constitutes a pool of 4 subsamples. Each subsample was taken at a 30° angle from the top of the log, half a meter apart, repeated in two places and on both sides of the log. A sterile knife was used to cut out a 25 cm x 25 cm square in the outer bark layer (periderm) that was carefully peeled back. In each subsample area, three tablespoons of degraded inner bark were added to a sterile Ziplock bag and pooled with other subsamples. The spoon and knife were disinfected between samples by wiping and burning with EtOH. To assess background soil diversity, three soil samples were collected according to the protocol of Tedersoo et al. [45]. All samples were transferred to the lab in a cooling box. Samples were airdried at 40°C and homogenised by vigorously rubbing in a Ziplock bag. Two grams of sample material were used for DNA extraction.
2.3.3. Wet Lab
Samples were ground under liquid nitrogen. gDNA was isolated as previously described and cleaned using 0.8X SPRI beads.
15 µl PCR reactions were run in duplicate. Each reaction mix consisted of: 200µM dNTP’s, 0,5µM of each primer, 1x Q5 reaction buffer, 0.02U/µl Q5 HIFI polymerase (New England Biolabs, Massachusetts, US) and 1 µl sample DNA. Cycling conditions were as follows: (1) Initial denaturation for 30 sec at 98°C, (2) 30 cycles of denaturation for 30 sec at 98°C, annealing for 20 sec at 60°C, extension for 20 sec at 72°C, (3) final extension for 2 minutes at 72°C. Amplicons were purified using SPRI beads and quantified on a Qubit Fluorometer (Invitrogen, California, USA).
Amplicons were normalized to 200 fmol, assuming an average amplicon size of 700 bp, and barcoded using the Native Barcoding kit 96 V14 (SQK-NBD114.96). 25 fmol was loaded on an R10.4.1 MinION flowcell. Sequencing ran for 72 hours at 260 bp/s.
2.4. Bioinformatics
2.4.1. Reference Database
We used the SINTAX-formatted version of UNITEv10 (50). Fasta header taxonomy annotation was manipulated to include UNITE species hypotheses (SH), by replacing or adding species-level annotations with the RefSeq’s associated SH code (e.g., >UDB05341741;tax=d:Fungi,p:Rozellomycota,s:SH0910702.10FU;)
Nine reference sequences were added as dummy SH codes (e.g., SH0000001.10FU) for easier downstream manipulation (Table 2). Three previously generated sanger sequences that did not match any SH. Two references for taxa in the Russula nigrifacta species complex: Russula ustulata as it is part of our mock community, and R. ambusta to serve as a control for false positives (GenBank accession number MW172301). In addition, because some of our root tip Sanger sequences could also not be confidently matched to any SH, references for Tomentella sublilacina and Laccaria amethystina were added. Finally, two large Laccaria zOTUs identified in the SH-approach were also added (see 3.2.2).
2.4.2. eNano Pipeline
Demultiplexed fastq files as outputted by MinKow (v. 22.10.7) were run through our eNano pipeline that is opensource and available on the GitHub page of the Research Group Mycology (https://github.com/MycoMatics/eNano). The pipeline takes a directory with subfolders (each representing sample barcodes) containing fastq files and a SINTAX formatted database as input. It outputs a (LULU-curated) OTU table with assigned taxonomy. Processing steps are detailed in the README file on GitHub. In short, Porechop (51) trims adapters and barcodes, Cutadapt (52) reorients sequences and cuts primers, Chopper (53) quality filters on average Phred score, Vsearch (54) appends sample information and performs clustering, chimera detection, creates a LULU match list and assigns taxonomy with the SINTAX algorithm [46]. It is also possible to run R (49) within the pipeline to perform LULU curation [47] and aggregate OTUs at the species level.
We explored two standard metabarcoding approaches for processing read data, here explained as the SH-approach (reference-based) and the OTU-approach (de novo clustering).
2.4.3. SH-Approach
Reads were deduplicated into zero-radius Operational Taxonomic Units (zOTUs) and classified into Species Hypotheses (SH). Given that our mock community comprises databased taxa — which are theoretically classifiable — we established the threshold for the SINTAX classification algorithm by plotting the distribution of SINTAX confidence scores across Phred scores in Figure S1. Using high-quality reads (≥Q28), the SINTAX threshold was determined based on the density of the right tail, which shows an increase starting at 0.80 and a slight dip between 0.90 and 0.95. We therefore set the threshold for singletons at 0.95 and relaxed the criterion for multiton zOTUs to 0.8, considering such reads are less likely to contain sequencing errors. zOTUs meeting this criterium were retained, and their conspecific abundances were aggregated per SH. This method prioritizes read classifiability over read quality, hereafter referred to as the ‘SH-approach’.
2.4.4. OTU-Approach
Given that not all taxa are represented in databases and thus not all reads are classifiable, we also evaluated whether our datasets could support a traditional approach of quality filtering and clustering into 98% OTUs. This method allows the incorporation of sequence clusters that have no obvious match in databases but might be large and influential in driving ecological patterns and is hereafter referred to as the ‘OTU-approach’.
2.4.5. Minimum Quality Evaluation
To determine the minimum acceptable quality threshold, we divided the reads from our mock and bark communities based on their average Phred scores. We subsampled Phred-specific datasets to read count at Q28 (4245 mock and 42560 bark reads per Phred score) and ran eNano on each set. The minimum Phred score for quality filtering was determined as the point where the recovery of taxonomic units (either 98% OTUs or SHs) stabilized. Due to the probabilistic nature of several steps including chimera removal, clustering, and classification, we ran each dataset one hundred times. Additionally, because singleton exclusion is a common practice in quality filtering, but problematic in Nanopore data due to high error rates resulting in many singletons, we also evaluated the effect of singleton taxonomic unit exclusion for both approaches. Finally, to assess species turnover between Phred-specific datasets, Beta-diversity (Bray-Curtis distance) is visualized in Figure S2.
2.4.6. Bark Metabarcoding
Sequence data was processed using insights from both the SH- and OTU-approaches. For the SH-approach, eNano was run with clustering at 100% identity (--id 1), without a quality threshold (--q 0), using our modified version of the UNITEv10 database. zOTUs were filtered based on SINTAX threshold (0.95 for singletons, 0.8 multitons), and conspecific abundances were aggregated into a SH-table used in all subsequent analyses.
For the OTU-approach, eNano was run with a clustering identity of 98% (--id 0.98), a quality threshold of Q25 (--q 25), and a SINTAX threshold of 0.80 (--sintax 0.8) with LULU-curation (--skip-lulu 0). Singleton SHs and OTUs were excluded from analysis.
OTU-table handling was performed using phyloseq 1.42.0 [48], metagMisc 0.5.0 [49] and vegan [50]. Alpha diversity indices—species richness, Shannon-Wiener, and Simpson—were analysed using a generalized linear model (GLM) with sequencing depth as a covariate to assess variations across bark decay stages. Read abundances were converted to Aitchison distances for Non-metric Multidimensional Scaling (NMDS) and Principal Coordinates Analysis (PCoA). Variable selection for PERMANOVA was conducted through forward and backward selection using AICcPermanova 0.0.2 [51], considering factors such as decay stages of the log and bark (DS Log, DS Bark) and presence of regeneration. Indicator species analysis was carried out with the indicspecies package 1.7.13 [52], applying Pearson’s phi coefficient for association (fidelity) and the standard Invdal.g function. Ecological guild assignments were made using FUNGuildR version 0.2.0.9000 [53]. Data processing was performed on the HPC of Ghent University, using eNano and R 4.2.3 (49)
3. Results
3.1. Morphotyping
Saplings growing on logs were primarily found in the intermediate (n=6) and late (n=17) stages of bark decay. Although seedlings were occasionally observed in early bark decay stages, their delicate root systems were not successfully retrieved.
For this study, 23 beech saplings growing on 22 distinct logs were selected for root tip morphotyping analysis. Additionally, five saplings growing in soil were included as a reference group (Table S3). We identified ten morphotypes, eight of which were identified by sanger sequencing (Table S4). Two others failed sequencing and were identified based on morphology, the jet-black Cenococcum geophilum and Elaphomyces cf. muricatus, which is recognizable by the spindle shape of the emanating hyphae.
Among the log-based saplings, over half (n=13) were colonized by EcM (Table S3). Specifically, two EcM species, Laccaria amethystina and Tomentella sublilacina were identified, with the former being dominant. In contrast, all soil-grown saplings were colonized by EcM. Also, species diversity is higher with in total nine species identified (Laccaria amethystina, Lactarius subdulcis, Xerocomellus pruinatus, Cenococcum geophilum, Russula ochroleuca, Melanogaster cf. intermedius, Scleroderma citrinum, Inocybe napipes and Elaphomyces cf. muricatus). EcM coverage (proportion of colonized root tips) and EcM species richness is significantly higher for those saplings growing in soil (Dunn test, resp. p=0.02 and p=0.001.). Eight out of ten of the uncolonized saplings on the log were estimated to be only two years old. The two older saplings (8 and 9 years old) did not show accumulation of EcM.
3.2. Metabarcoding
SH and OTU tables for mock and bark datasets can be found in File S1. The negative control was sequenced conducted to account for potential contaminants. Two OTUs mapped to Malassezia spp., these were removed in further analysis.
Average Phred score was 15.3, but is best re-evaluated after trimming the low-quality, non-informative adapter region. With the first 20bp trimmed, average Phred score increased to Q17.3. Median amplicon size was 709 bp.
3.2.1. Minimum Quality Evaluation
Both approaches demonstrated stabilization in taxonomic unit recovery, with consistent patterns observed for mock and bark datasets. Recovery in the bark community was consistently slower with increasing quality, highlighting the complexity of the dataset (Figure 2b,c).
For the 98% OTU-approach, the stabilization in taxonomic unit recovery began at Q24, but never fully plateaued with 26 and 2605 OTUs recovered at Q28 in the mock and bark communities respectively (Figure 2b). This trend persisted when excluding singleton OTUs, yielding 15 and 1770 OTUs at Q28.
In the SH-approach, the intermediate zOTUs step did not show any signs of plateauing before Q28, indicating that the sequence error is too impactful to reach a stable number of taxonomic (Figure 2a). When singleton zOTUs were excluded, zOTU recovery slowed at Q25 but never stabilized. At Q28, singleton zOTUs accounted for 28,6% and 37,2% of reads in mock and bark respectively. The SH-approach exhibited stable recovery of SHs across all Phred scores for both datasets, with a slight increase when using higher quality reads in the bark dataset (Figure 2c). At Q28, excluding singleton SH resulted in fewer SHs in the bark community, reflecting the presence of rare SHs. In the mock community, the average difference when excluding singleton SH was one SH, Curvibasidium cygneicollum (14 vs 15, Figure 2c and Figure 3).
Merging 98% OTUs into SHs was excluded a priori because clustering at 98% blurs taxonomic resolution well beyond the minimum difference of 0.5% between SHs and the 1.5% clustering threshold of the dynamic UNITE release (Figure 2d).
3.2.2. Mock Community
The OTU-approach resulted in 188 OTUs, of which 107 were identified to SH, corresponding to 18 unique SH codes. 14 taxa from our input mock were recovered as well as four contaminant OTUs.
The SH-approach generated 127075 zOTUs, of which 46% could be mapped to SH. 44 SH were recovered, 17 of which are singletons. 75% of total reads were classified into SHs, unmatched reads (not detected as fungal) occurred only at Q5-7 (0.27%). Classification rates show a rapid increase up until Q16, after which increase slows down and finally stabilized at Q27 (88 ± 0.2% of reads/Phred classified). This pattern was partly repeated for the more complex bark community, whose classification rates stabilized at Q16 as seen in Figure S3.
Taxon recovery at each Phred score is depicted in Figure 3. Our mock community showed high recovery, with all taxa (excl. Lactifluus russulisporus) recovered at Phred scores up to Q27. L. russulisporus is detected at Q16 (3 reads) and Q20 (2 reads) showing that it was picked up in low abundances. Next to our 16 mock taxa, 28 contaminant SH were picked up, most of which are singletons.
3.2.2. Bark Substrate Community
The OTU-approach produced 2431 OTUs out of 332874 reads (9,4% of total reads), while the SH-approach used 758755 reads (24,4%) to recover 901 species hypotheses, including two contaminants and 62 singletons SH.
At the phylum level, recovery of both methods was similar, with most taxonomic units belonging to Ascomycota, then Basidiomycota and Rozellomycota (Figure 4). The OTU-approach produced more units that belong to Mortierellomycota and more units that could not be classified. The SH-approach recovered relatively more Basidiomycota and Rozellomycota. Because the main target of our study was to identify ectomycorrhizal taxa – most of which we assume to be databased in our study area – metabarcoding results and discussion focus mostly on our SH-approach.
The inner bark samples were composed of 778 SH. Subulicystidium longisporum (SH0924096.10FU) is the most common SH, with high abundance in both early and intermediate bark decay stages (Figure 4). 19 SH represent the most common taxa, most of which have no associated species name, for example two Rozellomycota SHs (SH0131567.10FU and SH0146816.10FU) (Figure 4).
Species richness (SR) increased with bark decay from 106 ± 21 SH in early decay to 125 ± 27 and 135 ± 28 SH in intermediate and late decay stages respectively. Observed and Shannon diversity differed significantly between early and late bark decay stages (Tukey’s HSD, p=0.02 and 0.05 respectively) but not between other pairs. Simpson diversity was similar for all bark decay stages. While richness patterns were consistent in both approaches, significance was more pronounced in the OTU-approach.
Both NMDS and PCoA ordination reveal clustering of samples according to bark decay stage (Figure 5), with most overlap between early and intermediate bark decay stages. A notable exception is the NMDS on OTU-approach, that shows little data structure with decay (Figure 5b). Two outlier samples can be identified (ZF321_1 and ZF327) in the PCoA of OTU-approach (Figure 5d). ZF321_1 is dominated by a single OTU, mapping to a species of Saccharomycetales (66% sample reads). ZF327 contains an OTU that doesn’t occur in any other sample, identified as Pluteus podospileus (SH0855453.10FU). This SH is present across multiple samples in the SH-approach, thereby not producing an outlier by common absence. As explained before, for two logs, three separate composite samples were taken, at several meters distance on the same log. These samples from the same log exhibit greater dissimilarity in ordination space than those from different logs, as illustrated in (Figure 5). Stepwise model selection indicated that bark decay stage is the best predictor of community composition. PERMANOVA analysis showed that bark decay stage explains 15.60% of the variance (p = 0.001). The effect of log decay stage was less pronounced, explaining an additional 7.41% (p = 0.005). The presence regeneration (p = 0.4) was a non-significant predictor.
In the OTU-approach, 50.18% of reads could not be assigned at the species level, including many large clusters. The two largest of these OTUs account for 7% of reads and both belong to Saccharomycetales. Another 4.5% of reads can be assigned to three OTUs in the Sordariales. Other large OTUs belong to Trichoderma (1.6% reads), Mortierella (1.4% reads) and Helotiales (1.3% reads).
Fidelity analysis identified 27 and 65 taxa in the SH- and OTU-approach respectively. Results at the 0.01 significance level are presented in Table 2. Of the OTUs identified at the SH level, 18 were shared with those identified by the SH-approach. Additionally, 14 SH-level high-fidelity OTUs were exclusive to the OTU-approach, with only one (Rhinocladiella, SH0970950.10FU) surpassing the 0.01 significance threshold. In addition, indicator analysis (Indval.g function) corroborated these high-fidelity taxa as indicators at the 0.01 level and additionally identified Pleurothecium recurvatum (SH0926211.10FU) as an indicator for early bark decay across both approaches.
3.2.2. Mycorrhizae in Decaying Beech Inner Bark
Our SH-dataset was filtered for reads of mycorrhizal taxa, which were low in abundance. Their relative read abundances in each sample are visualized in Figure 6. Ectomycorrhizal SHs accounted for slightly less reads than endomycorrhizal SHs (1.18% and 1.4% respectively). In soil 3.6% of reads are considered mycorrhizal. Four Glomeromycotan and six ectomycorrhizal taxa are identified: Lactarius subdulcis and Russula nigricans (Russulaceae), Inocybe maculata (Inocybaceae), Paxillus involutus (Paxillaceae), Tomentella sublilacina (Thelephoraceae) and three Laccaria species (Hydnangiaceae). Two of these were detected as large Laccaria zOTUs, containing 36 and 12 sequences respectively in our SH-approach, but failed to align with our included reference sequence for Laccaria amethystina. At the same time, they BLASTn match with more than 99 % identity to different Laccaria taxa. Pairwise distance between these three sequences ranges from 1.4-2.2%. Consequently, we incorporated these zOTUs into our reference database (SH0000009-10.10FU) and conducted a re-analysis with the updated database. These two additional references are only present in a single sample (ZF321_1) while Laccaria amethystina occurs in six other samples.
We detected no correlation between the presence of regeneration on logs and the number of mycorrhizal reads (r=-0.16). PERMANOVA on mycorrhizal taxa confirmed that neither the presence of regeneration (R²=0.08, p=0.47), the decay stage of bark (R²=0.11, p=0.75), nor decay stage of the log (R²=0.03, p=0.93) had significant effects on mycorrhizal community composition.
4. Discussion
4.1. Minimum Quality Evaluation
We evaluated Nanopore sequencing for fungal metabarcoding using the full-length ITS region. We defined acceptable read quality as the point where taxonomic unit recovery stabilized, indicated by consistent recovery rates of 98% for OTUs (OTU-approach) or zOTUs aggregated into SH (SH-approach). While we acknowledge that annotation accuracy might improve with even fewer sequence errors, we believe such changes would be minimal and unlikely to impact biodiversity patterns in community ecology studies.
Beta-diversity comparisons of OTU tables from Q24 and higher quality reads showed minimal differences, supporting the robustness of this threshold (Figure S2). Furthermore, the difference in OTU numbers with and without singleton exclusion remained stable within this range, indicating consistent clustering. As illustrated in Figure 2b, the number of recovered 98% OTUs declines rapidly when sequencing accuracy exceeds the clustering threshold (Q17 for 98% OTUs) and stabilizes at Q24-25, where errors minimally impact clustering.
The SH-approach bypasses Phred-based filtering by prioritizing read classifiability over average base call quality. Any zOTU that could be confidently mapped to an SH was deemed of sufficient quality, irrespective of its Phred score. Surprisingly, the proportion of classified reads is only partly correlated to Phred score, with strong increase at the low end of quality, up to Q16, especially in more complex datasets like our bark community (Figure S3). For higher Phred scores, classifiability is independent on average Phred score. Therefore, beyond Q16, average Phred score does not reliably predict classifiability.
Our experimental results demonstrate that both the SH- and OTU-approach are viable and complementary due to their distinct quality filtering methods The SH-approach avoids signal loss from Phred score filtering and does not blur biological signal by pre-defining clustering thresholds, using mapping confidence as the quality filter instead. Because SH recovery remains stable regardless of Phred score, also when singleton zOTUs are included, the SH-approach eliminates the need for Phred score filtering and singleton zOTU exclusion
Our results show that raw Nanopore data, when appropriately filtered, can support a traditional metabarcoding approach without needing to generate consensus sequences or use complex protocols like Unique Molecular Identifiers [54,55] or Rolling Circle Amplification [56]. We recommend filtering at Q25 for 98% OTUs and to re-evaluate for other clustering thresholds or marker genes. While this currently still loses much data, it effectively identifies larger OTUs, likely key drivers of ecological patterns. Moreover, this threshold can be maintained with expected improvements in Nanopore data quality [57].
While the presented methods of quality filtering are effective, there remains an unoccupied space for more sophisticated techniques. Projecting the zOTU trend (Figure 2a) suggests that Phred scores of >Q30 are necessary for zOTUs to serve as a reliable analysis unit. If such a quality threshold is reached, it opens up the possibility to use Amplicon Sequence Variants (ASVs) with DADA2-like error correction techniques, which assumes the data contains a non-trivial fraction of error-free reads [58].
4.2. Mock Community
Both approaches on the mock community yielded similar results in terms of identified taxa. The OTU-approach’ shortcomings of both low data retention and a priori clustering are obvious, with Lactifluus russulisporus filtered out due to low abundance (no presence at ≥Q25) and Russula nigrifacta and R. ustulata being merged to a single taxon (R. ustulata) as they differ less than the clustering threshold.
In contrast, the SH-approach, demonstrated a higher resolution with 46% of zOTUs being confidently mapped to SH, accounting for 75% of total reads (Figure S3). Classification rates increased rapidly up to a Phred score of Q16, confirming that even modest-quality reads can be reliably classified.
The taxon recovery graph (Figure 3) reveals that nearly all taxa, except for Lactifluus russulisporus) from our mock community were effectively recovered at Phred scores up to Q27. We attribute the low recovery of L. russulisporus to poor DNA extract quality and PCR bias [59]. Despite expecting some false positives, our analysis found no misidentifications, demonstrating the robustness of our classification thresholds.
Our study demonstrated the effectiveness of the SH-approach, rather than the OTU-approach, in accurately identifying closely related taxa. This is shown by the consistent detection of all species from the included included Lactifluus and Russula groups across various Phred scores. Notably, even at lower read qualities (as low as Q14), the closely related species R. ustulata and R. nigrifacta were reliably identified (using our reference sequence for R. ustulata). Although it is impossible to discern whether each read was correctly assigned to either R. ustulata or R. nigrifacta, we did not pick up any false positive. For example no zOTUs matched the reference sequence for the closely related R. ambusta. Similar precision was observed for other challenging taxa, such as Cortinarius alboadustus. In addition to the mock taxa, we detected a small number of contaminants (0.07% of reads in the mock), likely originating from co-extraction with the voucher material.
We recommend the routine use of mock communities as positive controls. Their use offers a low-effort strategy to evaluate both qualitative and quantitative aspects of sequencing runs. A more sophisticated mock should include taxa that are not present in the sample’s locality to ensure that index-switching and cross-contamination can be attributed to the control [1]. An interesting extension of this concept are non-biological synthetic spike-in controls, like SynMock, which mimic biological diversity without the risk of overlapping with natural taxa [60].
4.3. Inner Bark Community
Both approaches classified less than a quarter of the bark dataset reads. While the SH-approach still manages to use more than double the amount of reads of the OTU-approach, it is obvious that in complex and natural communities, many reads cannot be matched to a database confidently. Even with high-quality reads (Q28), only 36% of reads (31% of zOTUs, Figure S3) were identifiable at the SH level.
At the phylum level, both approaches produced broadly similar outcomes (Figure 4). However, the OTU-approach detected a notably higher proportion of Mortierellomycota, largely due to two large OTUs. One of these OTUs is annotated as Linnemannia amoeboidea (SH0777285.10FU), with 4099 reads, although it also appears in the SH-approach, albeit in lower abundance (1586 reads). The other notable OTU is identified as Mortierella sp., represented by 3058 reads. In contrast, the SH-approach yielded a greater recovery of Basidiomycota and Rozellomycota. The subtle nature of these differences suggest both approaches adequately capture diversity, with the main distinction being the higher number of unclassified phylum-level units in the OTU-approach, reflecting its lack of a priori filtering for database taxa. Unless stated otherwise, the species-level results discussed hereafter pertain to the SH-approach.
In our bark samples, across decay stages, we found a co-dominating mix of typical soil (Aspergillus fumigatus, Leuconeurospora, Sporothrix) and wood inhabiting fungi (Subulicystidium longisporum, Pleurothecium recurvatum, Pluteus podospileus, Helicogloea insularis, Postia tephroleuca and Basidiodendron caesiocinereum). This mix reflects our characterization of the substrate as ‘soil-like wood’. A significant number of reads were assignable to taxa capable of growing as yeasts (Saccharomycetales) and known mycoparasites (Tremella), alongside some less clearly defined taxa such as the enigmatic Rozellomycetous ‘GS05’ and a Hyaloriaceae SH. Interestingly, the latter is also present in the OTU-approach, and a manual BLASTn search suggests these sequences likely originate from Myxarium podlachicum, a common jelly fungus in the reserve. Interestingly, members of our lab have described a mycoparasite inhabiting this species, Slooffia micra [61], which, despite targeted efforts, has not been found in the sampled area. S. micra was incorporated in the latest version of UNITE [62]) as SH0867261.10FU and was recovered in relatively high abundance (213 and 64 reads in SH- and OTU-approach respectively). This surprising result hints that this species has part of its life cycle outside of its host basidiomes (fruitbodies were excluded from samples). Indeed, Slooffia spp. have been isolated as yeasts from soils, litter and insect faeces [61]. Nevertheless, at the time of writing it had not been observed in eDNA samples on UNITE. The abundant recovery of Helicogloea insularis was also striking, as this taxon is only known from two localities in Estonia and Norway where it was collected from strongly rotten elm logs [63]. If we expected to pick up any Helicogloea species at all, it was H. sebaceae, as we know it to be present in the sampling area. A false positive seems unlikely as H. sebaceae was properly retrieved in our mock community. Possible explanations include significant intraspecific ITS variation or incomplete lineage sorting where H. sebaceae might retain an ITS sequence similar to H. insularis. Evidently, it is also possible that the species is truly occurring as it concerns an understudied and often overlooked group. Additionally, several species typically associated with beech deadwood such as Simocybe centunculus, Coprinellus micaceus and eight species of Pluteus were found. Strangely, Mycena species, expected to be diverse and abundant, were conspicuously absent from our bark dataset. This is particularly striking given that Mycena galericulata and M. haematopus are among the most prevalent fungi on deadwood in the reserve [64,65]. The findings outlined in this paragraph highlight the added value of solid field knowledge when interpreting eDNA data.
We observed significant changes in species richness and composition corresponding to bark decay. Species richness (SR) and Shannon diversity metrics varied between early and late decay stages, but never with intermediate decay. This pattern suggests that early and late decay stages represent opposite ends of a decay continuum characterized by a gradual species gain and turnover. In the early decay stage, which exhibited the lowest species richness (106 ± 21 SH), we anticipated a high number of reads from pyrenomycetous endophytes, such as Jackrogersiella cohaerens and Eutypa spinosa. However, these were found only in relatively low abundance and showed no correlation with the bark decay stage. We hypothesize that their signals have been diluted over time, as even our samples from the earliest decay stage were collected from logs already in the second phase of decay. As the bark decays, it becomes more soil-like, as demonstrated by in Figure 5. However, it does not fully converge to resemble the soil community. We attribute this to the presence of colonization barriers and greater environmental fluctuations, such as variations in temperature.
Bark decay stage best explains variation in beta diversity, accounting for 15.6% of the variation, while a model incorporating both bark and log decay stage explains approximately 23% of the variation. This suggests that the decay gradient is a primary determinant of community composition and aligns with patterns in coarse woody debris, where fungal communities vary significantly between decay stages due to shifting abiotic conditions attracting different specialized species [21,65,66]. We observed high community turnover, with intra-log communities exhibiting greater beta-diversity than those across different logs (Figure 5). This suggests that our sampling was insufficient to capture bark community at the log level, and we hypothesize that bark volume is an important factor for community richness provided an adequate surface is sampled. Given that several strategies to maintain species richness in wood and soil have been proposed and demonstrated [67], we expect priority effects, along with stochastic and dispersal processes, to play crucial roles in the assembly of these communities
Both fidelity and indicator analyses underscore the utility of the OTU- and SH-approaches in pinpointing critical taxa, with all but one SH-level taxon being shared at a significance level of p≤0.01. Simultaneously, these analyses also emphasize the importance of incorporating OTUs at all taxonomic levels, as OTU-only indicators were detected at each stage of bark decay (early: Tulasnella; Mid: Hydnodontaceae, Rhinocladiella, and Helotiales; Late: Rozellomycota, Trichoderma, and Hypocreales).
The identification of decay stage-specific indicators, further underpins the successional nature of bark fungal communities. In early bark decay, only wood saprotrophs are recovered, all but Tulasnella are Sordariomycetes.
In intermediate bark decay most taxa - excl. Ganoderma adspersum and a Hydnodontaceae OTU that BLASTn matches Subulicystidium spp., are difficult to interpret. Rhinocladiella (Herpotrichiellaceae, ‘black yeasts’) and Mortierella are common in organic soils [68–71]. Serendipita species (e.g., S. vermifera) can occur free-living on wood [72], but are best known as root endophytes and mycoparasites. Chaetosphaeria decastyla is a common saprobe on decaying wood of many hosts, incl. Beech [73].
In late bark DS, most annotations are above the species level, complicating interpretation. Only SH1450888.10FU is annotated at the species level (Ilyonectria mors-panacis), and while this SH seems to be geographically widespread, its exact taxonomic annotation should be cautiously interpreted as evidenced by the ten Ilyonectria species names associated with sequences incorporated in the SH. Nevertheless, we know from surveys that many Nectria(-like) species occur in the sampling area. Likewise, the identified Trichoderma OTU (Hypocreales) BLASTn matches to T. viride, for which we have often observed both ana- and teleomorph stages on Beech deadwood. However, caution is warranted as the ITS region shows little variation in Trichoderma, complicating identification beyond the genus level [74].
Ectomycorrhizal Colonization of Beech Deadwood
In the morphotyping analysis, around half of the investigated saplings growing on logs were colonized by EcM fungi. EcM coverage, species diversity and proportion of colonized saplings were significantly lower compared to saplings growing in soil. Only two species could be identified forming associations with saplings growing on deadwood: Laccaria amethystina and Tomentella sublilacina. Tomentella is multistage, highly competitive ECM genus in mature forests [75]. T. sublilacina is a corticoid species forming fruiting bodies on wood and was also identified by Tedersoo et al. [25,76] on saplings growing on decaying logs of Betula pendula, Picea abies and Nothofagus cunninghamii. Baldrian et al. [17] metabarcoded course woody debris in a Beech dominated forest and found the species to be the second most common EcM at 0,45% of reads. Interestingly, only one sapling was colonized with both L. amethystina and T. sublilacina, which hints at competitive exclusion. Indeed T. sublilacina has been found to be capable of competitive exclusion of e.g., Tylospora fibrillosa [25] and Lactarius spp. [26].
Laccaria amethystina is considered a late successional species In Europe [77], and can form basidiomes on decaying wood of Beech [65], Norway spruce [78] and Silver fir [79]. This species produces small, short-lived genets dedicated to the production of meiospores [80]. Moreover, it prefers to associate with beech saplings within our study area [81]. Supporting this, Grebenc et al. [82] found a positive association between the occurrence of L. amethystina and the number of Beech seedlings and Hortal et al. [83] demonstrated that beech roots can be simultaneously colonized by multiple L. amethystina genets. These characteristics underscore its classification as an R-strategist, adept at spore production and dispersal, which explains its propensity to rapidly colonize new microsites within its late successional habitat.
Despite the relatively low abundance of mycorrhizal reads, several mycorrhizal SH were detected. Ectomycorrhizal (EcM) species accounted for 1.18% of the total reads, while endomycorrhizal species made up a slightly higher proportion at 1.4%, as illustrated in Figure 6. We visually confirmed the presence of arbuscular mycorrhizal fungi by staining Acer saplings growing on certain Beech logs (results not shown, staining method according to Vierheilig et al. [84]). Supporting our observations, Baldrian et al. [17] found EcM fungi in similarly low abundances across early and intermediate log decay stages of Beech, only becoming more abundant in late log DS. Our analysis revealed no significant correlation between the presence of logs with and without regeneration. Suggesting that, while specific mycorrhizal taxa are effectively colonizing decaying bark, their distribution is independent of sapling presence. The absence of correlation indicates that other environmental or biological factors have a more pronounced impact on shaping these mycorrhizal communities. Our metabarcoding results offered limited insights into the presence of ectomycorrhizal (EcM) fungi in deadwood. In contrast, morphotyping is per definition more detailed as it also demonstrates successful establishment. Like the results of our morphotyping analysis, EcM read abundance and diversity are higher in soil than in bark, with EcM diversity in bark being a subset of that in soil. Thus, both methods indicate that soil is the more beneficial habitat for mycorrhization.
Across our analyses, L. amethystina and T. sublilacina are identified as the key EcM in bark. In terms of EcM establishment, two critical factors can be identified: the source of the inoculum (via propagules or mycelium emerging from the soil) and the timing of arrival (before or after seedlings arrive). Mycelia in the soil may respond to the presence of deadwood by initiating growth into it. Initially, wood is resistant to penetration, but inner bark, with its softer consistency and quicker degradation, may serve as an early colonization pathway for soil-based EcM. Another possibility is that mycelium grows out of the soil regardless, as some fungi, such as Tomentella spp., are known to use wood as a substrate for fruiting. Alternatively, EcM can establish within deadwood bark through propagules, deposited either from the air or vectored by arthropods. The latter route benefits from direct deposition into the decaying substrate, a process well-supported by studies demonstrating the effectiveness of arthropods as fungal vectors [85–87].
While EcM are able to colonize deadwood, their viability in the absence of seedlings remains uncertain. Without seedlings, deadwood could act as a refuge from intense competition in the soil among well-established mycelia linked within the EcM network, especially among mature beeches. Such EcM may utilize delignified carbohydrates as a carbon source while awaiting their symbiotic partners [88,89], although such capabilities are now questioned [23]. This process, involving the formation of common mycelial networks prior to the arrival of saplings, could explain the enhanced sapling establishment sometimes observed in these specific forest microhabitats [25]. Alternatively, successful EcM establishment might require germination in the presence of roots, raising questions about whether the EcM reads detected in logs without seedlings are from spores, mycelia doomed to perish, or necromass. Our data present mixed signals. First, there is no evidence that EcM accumulation correlates with bark decay stage (DS), suggesting either that establishment is difficult after early bark DS, or more likely, that EcM do not accumulate in time. Second, the absence of a signal of Beech regeneration on mycorrhizal reads indicates that they occur independently of their partners. This dual pattern supports our belief that colonization can happen in the absence of seedlings, but forming symbiosis is crucial for the continued survival of mycorrhizae in decaying bark. Mechanistic experiments (e.g., Fukasawa and Kitabatake [90]), or sampling forests without natural regeneration would enhance our understanding.
Database
By prioritizing the classifiability of our data within the SH framework, our study demonstrates that sequences, even those with moderate average quality, can be accurately classified. In some instances, our data aligned more closely with UNITE’s reference sequences for a given SH than with our own Sanger-generated references (e.g., for Cortinarius alboadustus). User-defined references can also be included if existing SHs are inadequate, as demonstrated in our analysis of mock and bark samples. For example, we added references for mock and ectomycorrhizal taxa that could not be confidently assigned to a SH. Unfortunately, incorporating such references also hampers cross-study communication of taxa. In that regards, the nascent SH-matching tool [91] deserves further attention as it make possible the public assignment of new SH, but we did not extensively test it.
We assumed EcM in our sampling area to be well databased, and indeed many reference sequences are available for e.g., Laccaria amethystina. Nevertheless, we encountered difficulties in mapping zOTUs of Laccaria and Tomentella to SH, as well as issues with the interpretation of some taxon names, such as Illyonectria mors-panis. These problems may stem from the proliferation of sequence data and inconsistent sequence annotations, which complicate the precise definition and annotation of SH. Addressing these challenges will require manual curation, a daunting task at the scale of UNITE.
We recommend leveraging reference-based approaches, such as the SH-approach, in well-characterized communities, i.e., in regions or substrates that have been extensively sampled. A significant benefit of this approach is superior data retention and aggregation at the SH level, facilitating communication and comparisons of taxonomic units across analyses and studies and enhancing reproducibility.
Nanopore Metabarcoding
We found Nanopore to be a suitable platform for metabarcoding, although currently only a fraction of the generated data meets the quality required for broad-scale adoption. Despite these limitations, the platform offers unique advantages beyond the elimination of traditional read length constraints, making it particularly advantageous in the field of mycology, which heavily involves citizen scientists [92,93]. The low initial capital investment and the presence of an active user community contribute to its accessibility, which makes it a practical option for both amateurs and smaller research groups. Moreover, the ability to conduct in-house sequencing eliminates the need for outsourcing, thereby providing autonomy over workflows and data, empowering especially citizen scientists.
The Nanopore metabarcoding field is experiencing rapid growth, with a variety of tools and analysis strategies being developed. We built eNano to be a simple to understand tool, with straightforward integration of standard metabarcoding steps. Therefore, it is not designed to outperform emerging tools but rather to serve as a valuable baseline for comparison. The simple installation process (a single binary file) and the clarity of the procedures make eNano also suitable for educational purposes.
Considering the dynamic nature of this field, we recommend that researchers also explore other strategies like implementing Unique Molecular Identifiers [54,55] or tools such as EMU [94] and PIMENTA [95]. Noteworthy alternatives such as Decona [12] and CONCOMPRA [14] do not require a reference databases to generate taxonomic units, and have demonstrated to be viable alternatives [11,14,96]. Regardless of the approach, benchmarking tools should ideally include a comparison to a properly quality-filtered OTU table as a baseline.
Moreover, targeting longer marker regions such as the full rDNA operon shows promise for improved identification and phylogenetic placement [97,98], though it requires optimization of long-range PCR and handling increased chimera formation. In our dataset, 9.2% of reads were chimeric, consistent with other reports of elevated chimera rates compared to short-read sequencing [14,99–102]. While our Vsearch-based chimera detection is partially effective, some chimeras likely remain undetected. Even in our SH-approach we found several per chance assigned to an SH, which we flagged as chimeric in UNITE. Artificial Intelligence could be particularly useful in the detection of chimeric sequences, which, although readily identifiable by humans in multiple sequence alignments, often elude conventional bioinformatic detection methods.
As the field continues to evolve, engaging with a variety of tools will be crucial for maximizing the effectiveness and accuracy of Nanopore metabarcoding analyses.
5. Conclusions
This study evaluates the effectiveness of Nanopore sequencing for fungal metabarcoding using the full-length ITS region using both a mock and natural community. We demonstrate that both the reference-based SH-approach (mapping reads to a database) and the OTU-approach (de novo clustering) are viable options. The SH-approach ensures stable taxonomic unit recovery, and is only minimally impacted by low Phred scores (<Q16) when strict mapping criteria are applied. The OTU-approach is feasible but requires stricter quality filtering, which currently results in significant data loss. However, it is capable of identifying taxonomic units that are not represented in existing databases. We recommend using reads with a quality score of ≥ Q25 when constructing 98% OTUs.
Our analysis of fungal communities in decaying bark emphasizes the decay gradient as a primary determinant of community composition. Ectomycorrhizal fungi are found to be less diverse and abundant in bark compared to soil, indicating that soil provides a more favorable environment for mycorrhization. Despite this, Laccaria amethystina and Tomentella sublilacina are identified as key species colonizing decaying bark and capable of successful establishment. These findings suggest that decaying bark can serve as a microhabitat for specific mycorrhizal species, providing a potential explanation for the presence of mycorrhizae observed in early decay stages in other metabarcoding studies on deadwood.
We introduce the eNano tool, which offers a simple, baseline approach to Nanopore metabarcoding, suitable for broad adoption. Overall, Nanopore offers a promising platform for fungal metabarcoding, with the potential to yield valuable ecological insights and engage a wider audience of researchers and citizen scientists. As read quality continues to improve, the use of Nanopore data in a more classical metabarcoding fashion, whether through reference databases or by constructing OTUs, is likely to take hold.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1. Density histogram of SINTAX confidence values at SH-level; Figure S2. Heatmaps of Beta diversity in Bray-Curtis distance between subsampled Phred-specific datasets; Table S1: Overview of sampled logs; Table S2: Mock community composition; Table S2. Mycorrhization and morphotyping results; Table S4. Sanger sequencing results of root tip morphotypes, File S1: OpenDocument Spreadsheet with OTU-tables.
Author Contributions
Conceptualization, Glen Dierickx, Kris Vandekerkhove and Annemieke Verbeken; Formal analysis, Glen Dierickx and Lowie Tondeleir; Funding acquisition, Glen Dierickx, Kris Vandekerkhove and Annemieke Verbeken; Investigation, Glen Dierickx and Lowie Tondeleir; Methodology, Glen Dierickx, Lowie Tondeleir and Pieter Asselman; Project administration, Glen Dierickx, Kris Vandekerkhove and Annemieke Verbeken; Resources, Kris Vandekerkhove and Annemieke Verbeken; Software, Glen Dierickx and Pieter Asselman; Supervision, Kris Vandekerkhove and Annemieke Verbeken; Visualization, Glen Dierickx; Writing – original draft, Glen Dierickx and Lowie Tondeleir; Writing – review & editing, Glen Dierickx, Lowie Tondeleir, Pieter Asselman, Kris Vandekerkhove and Annemieke Verbeken. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Research Foundation Flanders (Belgium), grant numbers 1126221N and 1126223N, the Research Institute for Nature and Forest (INBO) and Research Group Mycology at Ghent University.
Data Availability Statement
The original contributions presented in the study are included in the article and supplementary materials. Sequence data can be accessed as fastq files at the NCBI Sequence Read Archive under BioProject ID PRJNA1151640.
Acknowledgments
We thank the forest ecology and management team at INBO for providing data on deadwood.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Tedersoo, L.; Bahram, M.; Zinger, L.; Nilsson, R.H.; Kennedy, P.G.; Yang, T.; Anslan, S.; Mikryukov, V. Best Practices in Metabarcoding of Fungi: From Experimental Design to Results. Molecular Ecology 2022, 31, 2769–2795. [CrossRef]
- Nilsson, R.H.; Tedersoo, L.; Ryberg, M.; Kristiansson, E.; Hartmann, M.; Unterseher, M.; Porter, T.M.; Bengtsson-Palme, J.; Walker, D.M.; De Sousa, F.; et al. A Comprehensive, Automatically Updated Fungal ITS Sequence Dataset for Reference-Based Chimera Control in Environmental Sequencing Efforts. Microbes and environments 2015, 30, 145–150. [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Bolchacova, E.; Voigt, K.; Crous, P.W.; et al. Nuclear Ribosomal Internal Transcribed Spacer (ITS) Region as a Universal DNA Barcode Marker for Fungi. Proceedings of the National Academy of Sciences 2012, 109, 6241–6246. [CrossRef]
- Oxford Nanopore Technologies Available online: https://nanoporetech.com/platform/accuracy/simplex (accessed on 4 January 2024).
- Wilson, A.W.; Eberhardt, U.; Nguyen, N.; Noffsinger, C.R.; Swenie, R.A.; Loucks, J.L.; Perry, B.A.; Herrera, M.; Osmundson, T.W.; DeLong-Duhon, S.; et al. Does One Size Fit All? Variations in the DNA Barcode Gaps of Macrofungal Genera. Journal of Fungi 2023, 9, 788. [CrossRef]
- Davidov, K.; Iankelevich-Kounio, E.; Yakovenko, I.; Koucherov, Y.; Rubin-Blum, M.; Oren, M. Identification of Plastic-Associated Species in the Mediterranean Sea Using DNA Metabarcoding with Nanopore MinION. Sci Rep 2020, 10, 17533. [CrossRef]
- Loit Kaire; Adamson Kalev; Bahram Mohammad; Puusepp Rasmus; Anslan Sten; Kiiker Riinu; Drenkhan Rein; Tedersoo Leho Relative Performance of MinION (Oxford Nanopore Technologies) versus Sequel (Pacific Biosciences) Third-Generation Sequencing Instruments in Identification of Agricultural and Forest Fungal Pathogens. Applied and Environmental Microbiology 2019, 85, e01368-19. [CrossRef]
- Theologidis, I.; Karamitros, T.; Vichou, A.-E.; Kizis, D. Nanopore-Sequencing Metabarcoding for Identification of Phytopathogenic and Endophytic Fungi in Olive (Olea Europaea) Twigs. Journal of Fungi 2023, 9, 1119. [CrossRef]
- Langsiri, N.; Worasilchai, N.; Irinyi, L.; Jenjaroenpun, P.; Wongsurawat, T.; Luangsa-ard, J.J.; Meyer, W.; Chindamporn, A. Targeted Sequencing Analysis Pipeline for Species Identification of Human Pathogenic Fungi Using Long-Read Nanopore Sequencing. IMA Fungus 2023, 14, 18. [CrossRef]
- Groben, G.; Clarke, B.B.; Kerkhof, L.J.; Bonos, S.A.; Meyer, W.A.; Qu, Y.; Luo, J.; Walsh, E.; Zhang, N. Mycobiome Analysis of Tall Fescue Grass Under Drought Stress Using the Illumina MiSeq and Oxford Nanopore Technology MinION. Phytobiomes Journal 2023, 7, 413–423. [CrossRef]
- Lysenko, L.; Griem, E.; Wagener, P.; Langer, E.J. Fungi Associated with Fine Roots of Fraxinus Excelsior Affected by Ash Dieback Detected by Next-Generation Sequencing. J Plant Dis Prot 2024. [CrossRef]
- Oosterbroek, S.; Doorenspleet, K.; Nijland, R.; Jansen, L. Decona: From Demultiplexing to Consensus for Nanopore Amplicon Data. ACA 2021, 4, e65029. [CrossRef]
- Maestri, S.; Cosentino, E.; Paterno, M.; Freitag, H.; Garces, J.M.; Marcolungo, L.; Alfano, M.; Njunjić, I.; Schilthuizen, M.; Slik, F.; et al. A Rapid and Accurate MinION-Based Workflow for Tracking Species Biodiversity in the Field. Genes 2019, 10, 468. [CrossRef]
- Stock, W.; Rousseau, C.; Dierickx, G.; D’hondt, S.; Martínez, L.A.; Dittami, S.M.; Loos, L. van der; Clerck, O.D. Breaking Free from References: A Consensus-Based Approach for Community Profiling with Long Amplicon Nanopore Data 2024, 2024.07.04.602031. [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010.
- Christy, E.J.; Sollins, P.; Trappe, J.M. First-Year Survival of Tsuga Heterophylla without Mycorrhizae and Subsequent Ectomycorrhizal Development on Decaying Logs and Mineral Soil. Can. J. Bot. 1982, 60, 1601–1605. [CrossRef]
- Baldrian, P.; Zrůstová, P.; Tláskal, V.; Davidová, A.; Merhautová, V.; Vrška, T. Fungi Associated with Decomposing Deadwood in a Natural Beech-Dominated Forest. Fungal Ecology 2016, 23, 109–122. [CrossRef]
- Kubartová, A.; Ottosson, E.; Dahlberg, A.; Stenlid, J. Patterns of Fungal Communities among and within Decaying Logs, Revealed by 454 Sequencing. Molecular Ecology 2012, 21, 4514–4532. [CrossRef]
- Rajala, T.; Peltoniemi, M.; Hantula, J.; Mäkipää, R.; Pennanen, T. RNA Reveals a Succession of Active Fungi during the Decay of Norway Spruce Logs. Fungal Ecology 2011, 4, 437–448. [CrossRef]
- Mäkipää, R.; Rajala, T.; Schigel, D.; Rinne, K.T.; Pennanen, T.; Abrego, N.; Ovaskainen, O. Interactions between Soil- and Dead Wood-Inhabiting Fungal Communities during the Decay of Norway Spruce Logs. ISME J 2017, 11, 1964–1974. [CrossRef]
- Rajala, T.; Tuomivirta, T.; Pennanen, T.; Mäkipää, R. Habitat Models of Wood-Inhabiting Fungi along a Decay Gradient of Norway Spruce Logs. Fungal Ecology 2015, 18, 48–55. [CrossRef]
- Bödeker, I.T.M.; Nygren, C.M.R.; Taylor, A.F.S.; Olson, A.; Lindahl, B.D. ClassII Peroxidase-Encoding Genes Are Present in a Phylogenetically Wide Range of Ectomycorrhizal Fungi. ISME J 2009, 3, 1387–1395. [CrossRef]
- Lindahl, B.D.; Tunlid, A. Ectomycorrhizal Fungi–Potential Organic Matter Decomposers, yet Not Saprotrophs. New Phytologist 2015, 205, 1443–1447. [CrossRef]
- Lindahl, B.; Stenlid, J.; Olsson, S.; Finlay, R. Translocation of 32P between Interacting Mycelia of a Wood-Decomposing Fungus and Ectomycorrhizal Fungi in Microcosm Systems. The New Phytologist 1999, 144, 183–193. [CrossRef]
- Tedersoo, L.; Suvi, T.; Jairus, T.; Kõljalg, U. Forest Microsite Effects on Community Composition of Ectomycorrhizal Fungi on Seedlings of Picea Abies and Betula Pendula. Environmental Microbiology 2008. [CrossRef]
- Poznanovic, S.K.; Lilleskov, E.A.; Webster, C.R. Sharing Rotting Wood in the Shade: Ectomycorrhizal Communities of Co-Occurringbirch and Hemlock Seedlings. Mycorrhiza 2015, 25, 153–164. [CrossRef]
- Kropp, B.R. Fungi from Decayed Wood as Ectomycorrhizal Symbionts of Western Hemlock. Can. J. For. Res. 1982, 12, 36–39. [CrossRef]
- Orrego, G. Western Hemlock Regeneration on Coarse Woody Debris Is Facilitated by Linkage into a Mycorrhizal Network in an Old-Growth Forest. master thesis, THE UNIVERSITY OF BRITISH COLUMBIA: Vancouver, 2018.
- Tedersoo, L.; Kõljalg, U.; Hallenberg, N.; Larsson, K.-H. Fine Scale Distribution of Ectomycorrhizal Fungi and Roots across Substrate Layers Including Coarse Woody Debris in a Mixed Forest. New Phytologist 2003, 159, 153–165. [CrossRef]
- Kluting, K.; Strid, Y.; Six, D.; Rosling, A. Forest Fire Influence on Tomicus Piniperda-Associated Fungal Communities and Phloem Nutrient Availability of Colonized Pinus Sylvestris. Microb Ecol 2023, 86, 224–239. [CrossRef]
- Rumpf, S.; Schönfelder, E.; Ahrends, B. 3 Biometrische Schätzmodelle für Nährelementgehalte in Baumkompartimenten. 2018.
- Mussche, S.; Bussche, B.; De Schrijver, A.; Neirynck, J.; Nachtergale, L.; Lust, N. Nutrient Uptake of a Mixed Oak/Beech Forest in Flanders (Belgium). SG 1998, 63. [CrossRef]
- André, F.; Jonard, M.; Ponette, Q. Biomass and Nutrient Content of Sessile Oak (Quercus Petraea (Matt.) Liebl.) and Beech (Fagus Sylvatica L.) Stem and Branches in a Mixed Stand in Southern Belgium. Science of The Total Environment 2010, 408, 2285–2294. [CrossRef]
- Ahrends, B.; Von Wilpert, K.; Weis, W.; Vonderach, C.; Kändler, G.; Zirlewagen, D.; Sucker, C.; Puhlmann, H. Merits and Limitations of Element Balances as a Forest Planning Tool for Harvest Intensities and Sustainable Nutrient Management—A Case Study from Germany. Soil Systems 2022, 6, 41. [CrossRef]
- Vandekerkhove, K.; Deforce, K.; Bastiaens, J. Historic-ecological position of beech in the area of the Sonian Forest and an overview of beech-forest- related biodiversity present in the forest; Instituut voor Natuur- en Bosonderzoek, 2018; [CrossRef]
- De Keersmaeker, L.; Esprit, M.; Goessens, S.; Anja, L.; Thomaes, A.; Van de Kerckhove, P.; Vandekerkhove, K. Monitoring Programme on Strict Forest Reserves in Flanders (Belgium) - Site Level Stand Structure, Regeneration and Vegetation Data 2023. [CrossRef]
- Renvall, P. Community Structure and Dynamics of Wood-Rotting Basidiomycetes on Decomposing Conifer Trunks in Northern Finland. Karstenia 1995, 35, 1–51. [CrossRef]
- Agerer, R. Studies on Ectomycorrhizae II. Introducing Remarks on Characterisation and Identification. Mycotaxon 1986, 26, 473–492.
- Agerer, R. Studies on Ectomycorrhizae III. Mycorrhizae Formed by Four Fungi in the Genera Lactarius and Russula on Spruce. Mycotaxon 1986, 27, 1–59.
- Agerer, R. (ed. ) Colour Atlas of Ectomycorrhizae.; 1st–11th ed.; Einhorn-Verlag: Schwäbisch Gmünd, 1987;
- Agerer, R. Characterisation of Ectomycorrhiza. In Methods in Microbiology; Norris, J.R., D.J., R., Varma, A.K., Eds.; Academic Press Limited, 1991; Vol. 23, pp. 25–73. [CrossRef]
- Agerer, R. Anatomical Characteristics of Identified Ectomycorrhizas: An Attempt towards a Natural Classification.; Mycorrhiza: structure, function, molecular biology and biotechnology.; Springer-Verlag: Berlin Heidelberg, 1995; [CrossRef]
- Agerer, R.; R.M., D.; S., E.; Ingleby K., L. Descriptions of Ectomycorrhizae.; Einhorn-Verlag: Schwäbisch Gmünd, 1996;
- Nuytinck, J.; Verbeken, A. Lactarius Sanguifluus versus Lactarius Vinosus — Molecular and Morphological Analyses. Mycol Progress 2003, 2, 227–234. [CrossRef]
- Tedersoo, L.; Bahram, M.; Ryberg, M.; Otsing, E.; Koljalg, U.; Abarenkov, K. Global Biogeography of the Ectomycorrhizal /Sebacina Lineage (Fungi, Sebacinales) as Revealed from Comparative Phylogenetic Analyses. Molecular ecology 2014, 23, 4168–4183. [CrossRef]
- Edgar, R.C. SINTAX: A Simple Non-Bayesian Taxonomy Classifier for 16S and ITS Sequences 2016, 074161. [CrossRef]
- Frøslev, T.G.; Kjøller, R.; Bruun, H.H.; Ejrnæs, R.; Brunbjerg, A.K.; Pietroni, C.; Hansen, A.J. Algorithm for Post-Clustering Curation of DNA Amplicon Data Yields Reliable Biodiversity Estimates. Nat Commun 2017, 8, 1188. [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PloS one 2013, 8, e61217. [CrossRef]
- Mikryukov, V. metagMisc: Miscellaneous Functions for Metagenomic Analysis. View Article 2019.
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Package ‘Vegan.’ Community ecology package, version 2013, 2, 1–295.
- Corcoran, D.; Corcoran, M.D. Package ‘AICcPermanova.’ 2023.
- De Caceres, M.; Jansen, F.; De Caceres, M.M. Package ‘Indicspecies.’ indicators 2016, 8.
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An Open Annotation Tool for Parsing Fungal Community Datasets by Ecological Guild. Fungal ecology 2016, 20, 241–248. [CrossRef]
- Karst, S.M.; Ziels, R.M.; Kirkegaard, R.H.; Sørensen, E.A.; McDonald, D.; Zhu, Q.; Knight, R.; Albertsen, M. Enabling High-Accuracy Long-Read Amplicon Sequences Using Unique Molecular Identifiers with Nanopore or PacBio Sequencing 2019. [CrossRef]
- Lin, X.; Waring, K.; Tyson, J.; Ziels, R.M. High-Accuracy Meets High-Throughput for Microbiome Profiling with near Full-Length 16S rRNA Amplicon Sequencing on the Nanopore Platform 2023, 2023.06.19.544637. [CrossRef]
- Baloğlu, B.; Chen, Z.; Elbrecht, V.; Braukmann, T.; MacDonald, S.; Steinke, D. A Workflow for Accurate Metabarcoding Using Nanopore MinION Sequencing. Methods in Ecology and Evolution 2021, 12, 794–804. [CrossRef]
- Oxford Nanopore Announces Breakthrough Technology Performance to Deliver Complete Human Genome Assemblies and Richer Multiomic Data in London Calling Available online: https://nanoporetech.com/news/oxford-nanopore-announces-breakthrough-technology-performance-to-deliver-complete-human-genomes-and-richer-multiomic-data-in-london-calling-tech-update (accessed on 26 August 2024).
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat Methods 2016, 13, 581–583. [CrossRef]
- Dierickx, G.; Froyen, M.; Halling, R.; Wisitrassameewong, K.; Delgat, L.; De Crop, E.; Verbeken, M. Updated Taxonomy of Lactifluus Section Luteoli: L. Russulisporus from Australia and L. Caliendrifer from Thailand. Mycokeys 2019, 56, 13–32. [CrossRef]
- Palmer, J.M.; Jusino, M.A.; Banik, M.T.; Lindner, D.L. Non-Biological Synthetic Spike-in Controls and the AMPtk Software Pipeline Improve Mycobiome Data. PeerJ 2018, 6, e4925. [CrossRef]
- Schoutteten, N.; Yurkov, A.; Leroux, O.; Haelewaters, D.; Van Der Straeten, D.; Miettinen, O.; Boekhout, T.; Begerow, D.; Verbeken, A. Diversity of Colacosome-Interacting Mycoparasites Expands the Understanding of the Evolution and Ecology of Microbotryomycetes Available online: https://www.ingentaconnect.com/content/wfbi/sim/pre-prints/content-a2_sim_vol106_art2;jsessionid=1tl834f1c8ks.x-ic-live-03# (accessed on 3 August 2023).
- Abarenkov, K.; Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; May, T.W.; Frøslev, T.G.; Pawlowska, J.; Lindahl, B.; Põldmaa, K.; Truong, C.; et al. The UNITE Database for Molecular Identification and Taxonomic Communication of Fungi and Other Eukaryotes: Sequences, Taxa and Classifications Reconsidered. Nucleic Acids Research 2024, 52, D791–D797. [CrossRef]
- Malysheva, V.; Spirin, V.; Schoutteten, N.; De Lange, R.; Pennanen, J.; Larsson, K.-H. New and Noteworthy Species of Helicogloea (Atractiellomycetes, Basidiomycota) from Europe. Annales Botanici Fennici 2020, 57, 1. [CrossRef]
- Walleyn, R.; Vandekerkhove, K. Diversiteit, ecologie en indicatorwaarde van paddestoelen op groot dood beukenhout in het bosreservaat kersselaerspleyn (zoniënwoud). 2002.
- Ódor, P.; Heilmann-Clausen, J.; Christensen, M.; Aude, E.; van Dort, K.W.; Piltaver, A.; Siller, I.; Veerkamp, M.T.; Walleyn, R.; Standovár, T.; et al. Diversity of Dead Wood Inhabiting Fungi and Bryophytes in Semi-Natural Beech Forests in Europe. Biological Conservation 2006, 131, 58–71. [CrossRef]
- Heilmann-Clausen, J.; Christensen, M. Fungal Diversity on Decaying Beech Logs - Implications for Sustainable Forestry. Biodiversity and Conservation - BIODIVERS CONSERV 2003, 12, 953–973. [CrossRef]
- Abrego, N. Wood-Inhabiting Fungal Communities: Opportunities for Integration of Empirical and Theoretical Community Ecology. Fungal Ecology 2022, 59, 101112. [CrossRef]
- Ozimek, E.; Hanaka, A. Mortierella Species as the Plant Growth-Promoting Fungi Present in the Agricultural Soils. Agriculture 2021, 11, 7. [CrossRef]
- Beatrice Schol-Schwarz, M. Rhinocladiella, Its Synonym Fonsecaea and Its Relation to Phialophora. Antonie van Leeuwenhoek 1968, 34, 119–152. [CrossRef]
- Vicente, V.A.; Attili-Angelis, D.; Pie, M.R.; Queiroz-Telles, F.; Cruz, L.M.; Najafzadeh, M.J.; de Hoog, G.S.; Zhao, J.; Pizzirani-Kleiner, A. Environmental Isolation of Black Yeast-like Fungi Involved in Human Infection. Studies in Mycology 2008, 61, 137–144. [CrossRef]
- Marčiulynas, A.; Marčiulynienė, D.; Mishcherikova, V.; Franić, I.; Lynikienė, J.; Gedminas, A.; Menkis, A. High Variability of Fungal Communities Associated with the Functional Tissues and Rhizosphere Soil of Picea Abies in the Southern Baltics. Forests 2022, 13, 1103. [CrossRef]
- Roberts, P. Exidiopsis Species from Devon, Including the New Segregate Genera Ceratosebacina, Endoperplexa, Microsebacina, and Serendipita. Mycological Research 1993, 97, 467–478. [CrossRef]
- Réblová, M.; Nekvindová, J. New Genera and Species with Chloridium-like Morphotype in the Chaetosphaeriales and Vermiculariopsiellales. Studies in Mycology 2023, 106, 199–258. [CrossRef]
- Cai, F.; Druzhinina, I.S. In Honor of John Bissett: Authoritative Guidelines on Molecular Identification of Trichoderma. Fungal Diversity 2021, 107, 1–69. [CrossRef]
- Burke, D.J.; López-Gutiérrez, J.C.; Smemo, K.A.; Chan, C.R. Vegetation and Soil Environment Influence the Spatial Distribution of Root-Associated Fungi in a Mature Beech-Maple Forest. Applied and Environmental Microbiology 2009, 75, 7639–7648. [CrossRef]
- Tedersoo, L.; Gates, G.; Dunk, C.W.; Lebel, T.; May, T.W.; Kõljalg, U.; Jairus, T. Establishment of Ectomycorrhizal Fungal Community on Isolated Nothofagus Cunninghamii Seedlings Regenerating on Dead Wood in Australian Wet Temperate Forests: Does Fruit-Body Type Matter? Mycorrhiza 2009, 19, 403–416. [CrossRef]
- Vincenot, L.; Nara, K.; Sthultz, C.; Labbé, J.; Dubois, M.; Tedersoo, L.; Martin, F.; Selosse, M. Extensive Gene Flow over Europe and Possible Speciation over Eurasia in the Ectomycorrhizal Basidiomycete Laccaria Amethystina Complex. Molecular Ecology 2012, 21, 281–299. [CrossRef]
- Holec, J.; Kučera, T.; Běťák, J.; Hort, L. Macrofungi on Large Decaying Spruce Trunks in a Central European Old-Growth Forest: What Factors Affect Their Species Richness and Composition? Mycol Progress 2020, 19, 53–66. [CrossRef]
- Holec, J.; Kučera, T. Richness and Composition of Macrofungi on Large Decaying Trees in a Central European Old-Growth Forest: A Case Study on Silver Fir (Abies Alba). Mycol Progress 2020, 19, 1429–1443. [CrossRef]
- Fiore-Donno, A.-M.; Martin, F. Populations of Ectomycorrhizal Laccaria Amethystina and Xerocomus Spp. Show Contrasting Colonization Patterns in a Mixed Forest. New Phytologist 2001, 152, 533–542. [CrossRef]
- Boeraeve, M.; Everts, T.; Vandekerkhove, K.; De Keersmaeker, L.; Van de Kerckhove, P.; Jacquemyn, H. Partner Turnover and Changes in Ectomycorrhizal Fungal Communities during the Early Life Stages of European Beech (Fagus Sylvatica L.). Mycorrhiza 2021, 31, 43–53. [CrossRef]
- Grebenc, T.; Christensen, M.; Vilhar, U.; Cater, M.; Martin, M.P.; Simoncic, P.; Kraigher, H. Response of Ectomycorrhizal Community Structure to Gap Opening in Natural and Managed Temperate Beech-Dominated Forests. Can. J. For. Res. 2009, 39, 1375–1386. [CrossRef]
- Hortal, S.; Trocha, L.K.; Murat, C.; Chybicki, I.J.; Buée, M.; Trojankiewicz, M.; Burczyk, J.; Martin, F. Beech Roots Are Simultaneously Colonized by Multiple Genets of the Ectomycorrhizal Fungus Laccaria Amethystina Clustered in Two Genetic Groups. Molecular Ecology 2012, 21, 2116–2129. [CrossRef]
- Vierheilig, H.; Coughlan, A.P.; Wyss, U.; Piche, Y. Ink and Vinegar, a Simple Staining Technique for Arbuscular-Mycorrhizal Fungi. Appl. Environ. Microbiol. 1998, 64, 5004–5007. [CrossRef]
- Lilleskov, E.A.; Bruns, T.D. Spore Dispersal of a Resupinate Ectomycorrhizal Fungus, Tomentella Sublilacina, via Soil Food Webs. Mycologia 2005, 97, 762–769. [CrossRef]
- Persson, Y.; Ihrmark, K.; Stenlid, J. Do Bark Beetles Facilitate the Establishment of Rot Fungi in Norway Spruce? Fungal Ecology 2011, 4, 262–269. [CrossRef]
- Seibold, S.; Müller, J.; Baldrian, P.; Cadotte, M.W.; Štursová, M.; Biedermann, P.H.W.; Krah, F.-S.; Bässler, C. Fungi Associated with Beetles Dispersing from Dead Wood – Let’s Take the Beetle Bus! Fungal Ecology 2019, 39, 100–108. [CrossRef]
- Frank, A. Die Bedeutung Der Mykorrhiza-Pilze Für Die Gemeine Kiefer. Forstwissenschaftliches Centralblatt 1894, 185–190. [CrossRef]
- Kuyper, T.W. De rol van ectomycorrhiza-schimmels in de nutriëntenkringloop. Coolia 1990, 33, 32–37.
- Fukasawa, Y.; Kitabatake, H. Which Is the Best Substrate to Regenerate? A Comparative Pot Experiment for Tree Seedling Growth on Decayed Wood and in Soil. Forests 2022, 13, 1036. [CrossRef]
- Abarenkov, K.; Kõljalg, U.; Nilsson, R.H. UNITE Species Hypotheses Matching Analysis. Biodiversity Information Science and Standards 2022, 6, e93856. [CrossRef]
- Haelewaters, D.; Quandt, C.A.; Bartrop, L.; Cazabonne, J.; Crockatt, M.E.; Cunha, S.P.; De Lange, R.; Dominici, L.; Douglas, B.; Drechsler-Santos, E.R.; et al. The Power of Citizen Science to Advance Fungal Conservation. Conservation Letters 2024, 17, e13013. [CrossRef]
- Heilmann-Clausen, J.; Bruun, H.H.; Ejrnæs, R.; Frøslev, T.G.; Læssøe, T.; Petersen, J.H. How Citizen Science Boosted Primary Knowledge on Fungal Biodiversity in Denmark. Biological Conservation 2019, 237, 366–372. [CrossRef]
- Curry, K.D.; Wang, Q.; Nute, M.G.; Tyshaieva, A.; Reeves, E.; Soriano, S.; Wu, Q.; Graeber, E.; Finzer, P.; Mendling, W.; et al. Emu: Species-Level Microbial Community Profiling of Full-Length 16S rRNA Oxford Nanopore Sequencing Data. Nat Methods 2022, 19, 845–853. [CrossRef]
- Vorst, V. van der; Thijssen, M.; Fronen, B.J.; Groot, A. de; Maathuis, M.A.M.; Nijhuis, E.; Polling, M.; Stassen, J.; Voorhuijzen-Harink, M.M.; Jak, R. PIMENTA: PIpeline for MEtabarcoding through Nanopore Technology Used for Authentication 2024, 2024.02.14.580249. [CrossRef]
- Doorenspleet, K.; Jansen, L.; Oosterbroek, S.; Kamermans, P.; Bos, O.; Wurz, E.; Murk, A.; Nijland, R. The Long and the Short of It: Nanopore Based eDNA Metabarcoding of Marine Vertebrates Works; Sensitivity and Specificity Depend on Amplicon Lengths 2023, 2021.11.26.470087.
- Tedersoo, L.; Tooming-Klunderud, A.; Anslan, S. PacBio Metabarcoding of Fungi and Other Eukaryotes: Errors, Biases and Perspectives. New Phytologist 2018, 217, 1370–1385. [CrossRef]
- Wurzbacher, C.; Larsson, E.; Bengtsson-Palme, J.; Van den Wyngaert, S.; Svantesson, S.; Kristiansson, E.; Kagami, M.; Nilsson, R.H. Introducing Ribosomal Tandem Repeat Barcoding for Fungi. Molecular ecology resources 2019, 19, 118–127. [CrossRef]
- Laver, T.W.; Caswell, R.C.; Moore, K.A.; Poschmann, J.; Johnson, M.B.; Owens, M.M.; Ellard, S.; Paszkiewicz, K.H.; Weedon, M.N. Pitfalls of Haplotype Phasing from Amplicon-Based Long-Read Sequencing. Sci Rep 2016, 6, 21746. [CrossRef]
- White, R.; Pellefigues, C.; Ronchese, F.; Lamiable, O.; Eccles, D. Investigation of Chimeric Reads Using the MinION. F1000Research 2017, 6. [CrossRef]
- Xu, Y.; Lewandowski, K.; Lumley, S.; Pullan, S.; Vipond, R.; Carroll, M.; Foster, D.; Matthews, P.C.; Peto, T.; Crook, D. Detection of Viral Pathogens With Multiplex Nanopore MinION Sequencing: Be Careful With Cross-Talk. Front. Microbiol. 2018, 9. [CrossRef]
- Makhoul, M.; Chawla, H.S.; Wittkop, B.; Stahl, A.; Voss-Fels, K.P.; Zetzsche, H.; Snowdon, R.J.; Obermeier, C. Long-Amplicon Single-Molecule Sequencing Reveals Novel, Trait-Associated Variants of VERNALIZATION1 Homoeologs in Hexaploid Wheat. Front. Plant Sci. 2022, 13. [CrossRef]
Figure 1.
Schematic representation of Fagus sylvatica deadwood microsite for regeneration. Seedlings root in the soft decaying outer layer, the inner bark. Ectomycorrhizal fungi may also proliferate in this substrate, helping seedlings grow.
Figure 1.
Schematic representation of Fagus sylvatica deadwood microsite for regeneration. Seedlings root in the soft decaying outer layer, the inner bark. Ectomycorrhizal fungi may also proliferate in this substrate, helping seedlings grow.
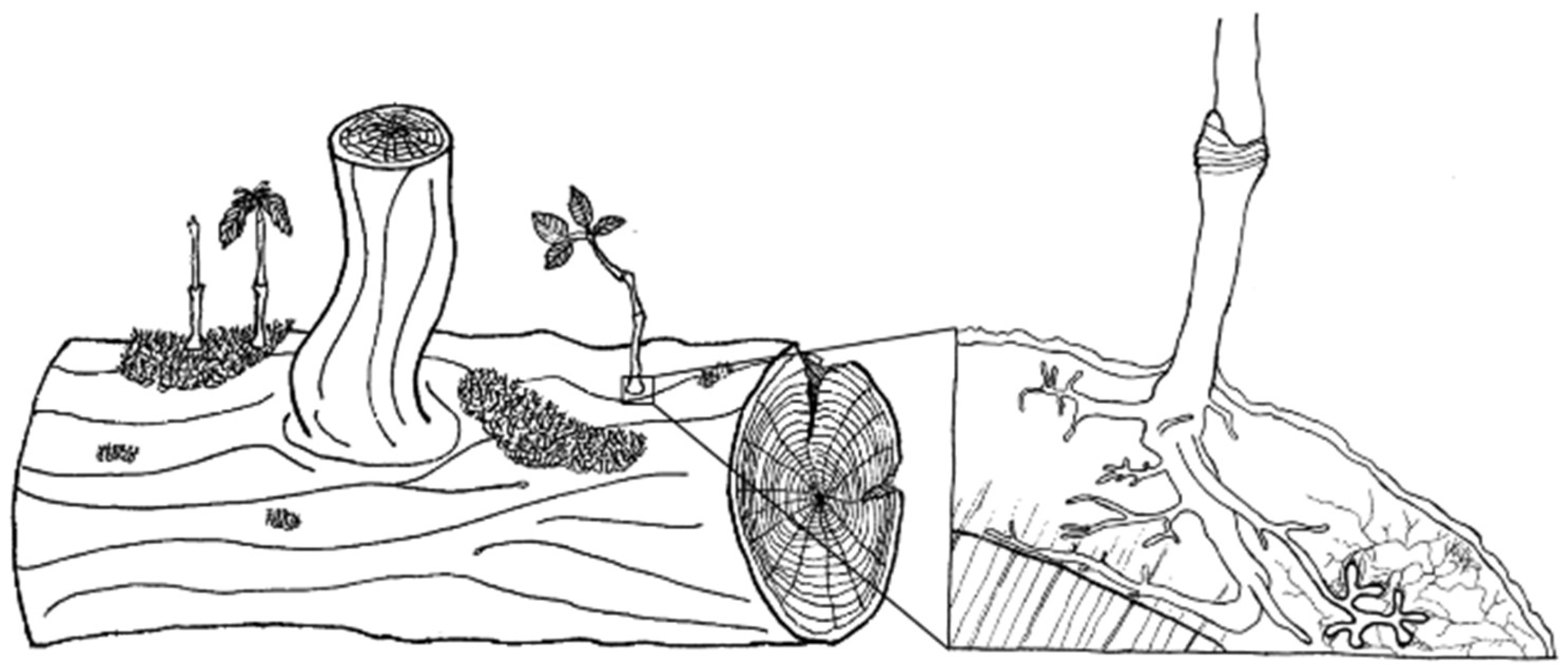
Figure 2.
Taxonomic unit recovery using all reads and excluding singleton units (zOTUS, 98% OTUs or SHs), Phred-specific datasets subsampled to amount of reads at Q28. Each datapoint represent 4245 reads for the mock dataset and 42560 for the bark dataset. Lines represent a loess smoother to visualize trends. (a) intermediate zOTU step in SH-approach; (b) OTU-approach (c) SH-approach (d) 98% OTUs merged into SH was excluded a priori (gray-shaded), shown for completeness. Graphs standardized by maximum recovered units per analysis per dataset. Nmax-zOTU-bark = 39382; Nmax-zOTU-mock=3818; Nmax-98%OTU-bark=39348; Nmax-98%OTU-bark=3722; Nmax-SH-bark=546; Nmax-SH-mock=20.
Figure 2.
Taxonomic unit recovery using all reads and excluding singleton units (zOTUS, 98% OTUs or SHs), Phred-specific datasets subsampled to amount of reads at Q28. Each datapoint represent 4245 reads for the mock dataset and 42560 for the bark dataset. Lines represent a loess smoother to visualize trends. (a) intermediate zOTU step in SH-approach; (b) OTU-approach (c) SH-approach (d) 98% OTUs merged into SH was excluded a priori (gray-shaded), shown for completeness. Graphs standardized by maximum recovered units per analysis per dataset. Nmax-zOTU-bark = 39382; Nmax-zOTU-mock=3818; Nmax-98%OTU-bark=39348; Nmax-98%OTU-bark=3722; Nmax-SH-bark=546; Nmax-SH-mock=20.
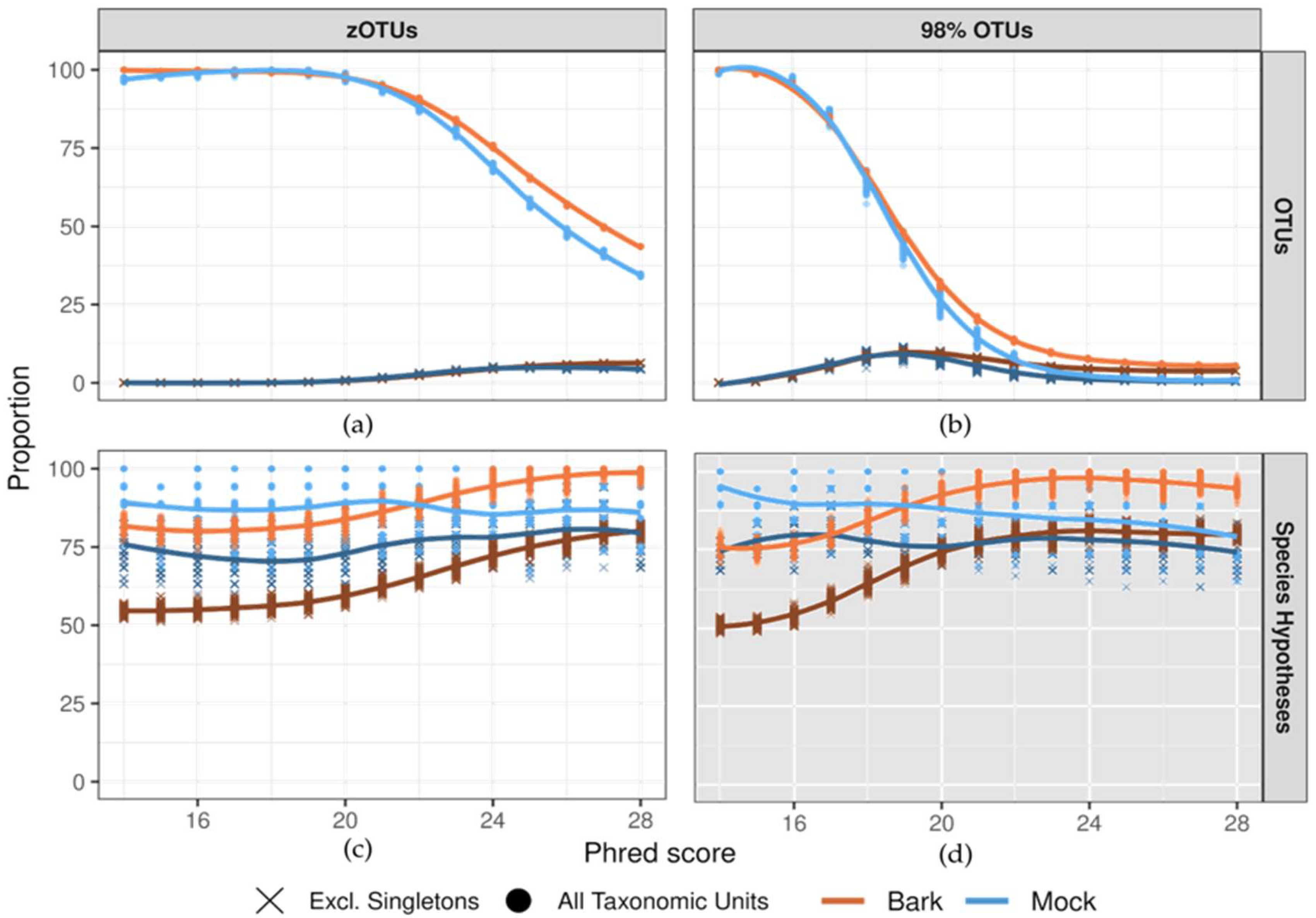
Figure 3.
Mock community recovered at different Phred scores using SH-approach. *Reference sequences added to database.
Figure 3.
Mock community recovered at different Phred scores using SH-approach. *Reference sequences added to database.
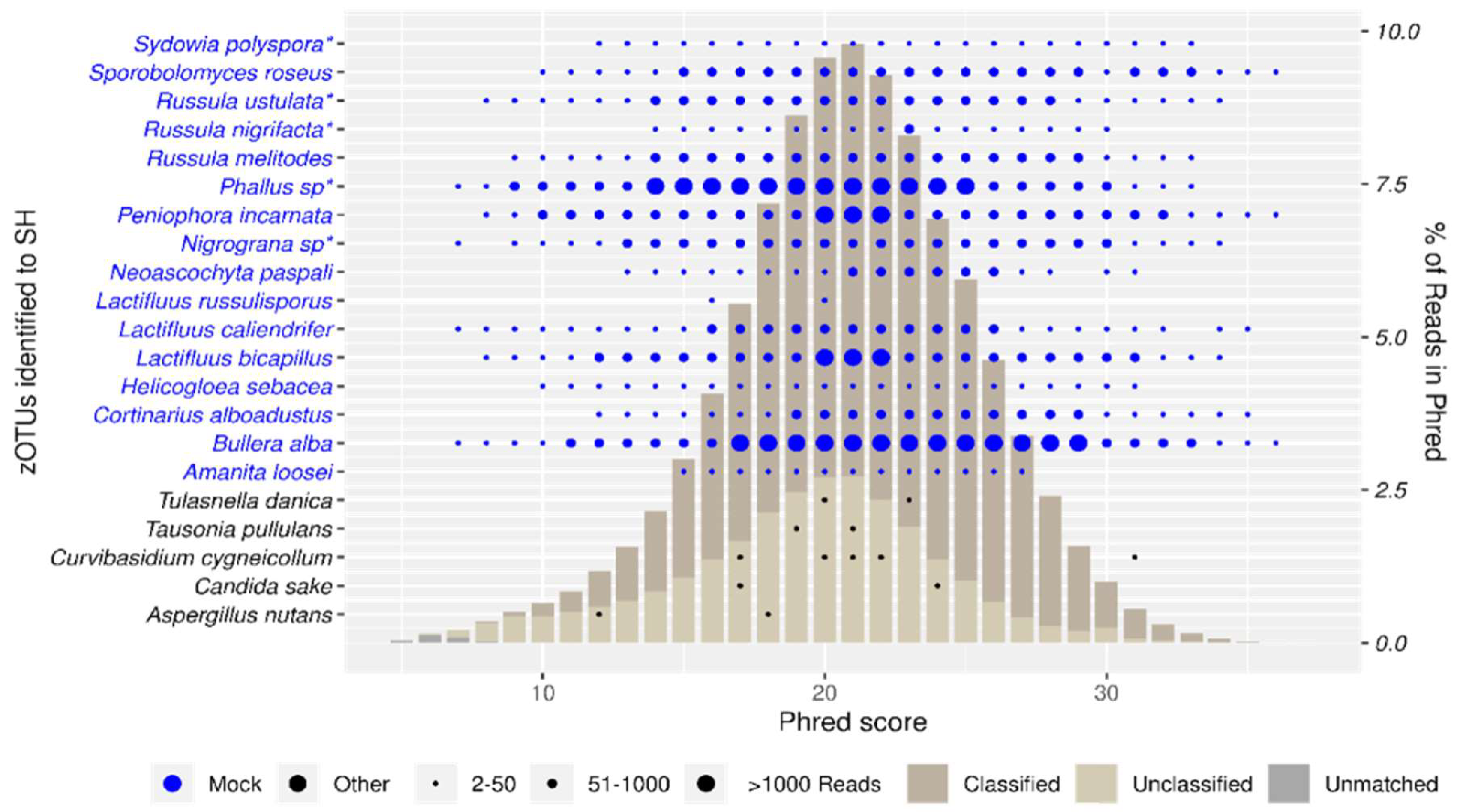
Figure 4.
Taxonomic composition across approaches. Right: Composition per Bark decay stage for the SH-approach.
Figure 4.
Taxonomic composition across approaches. Right: Composition per Bark decay stage for the SH-approach.
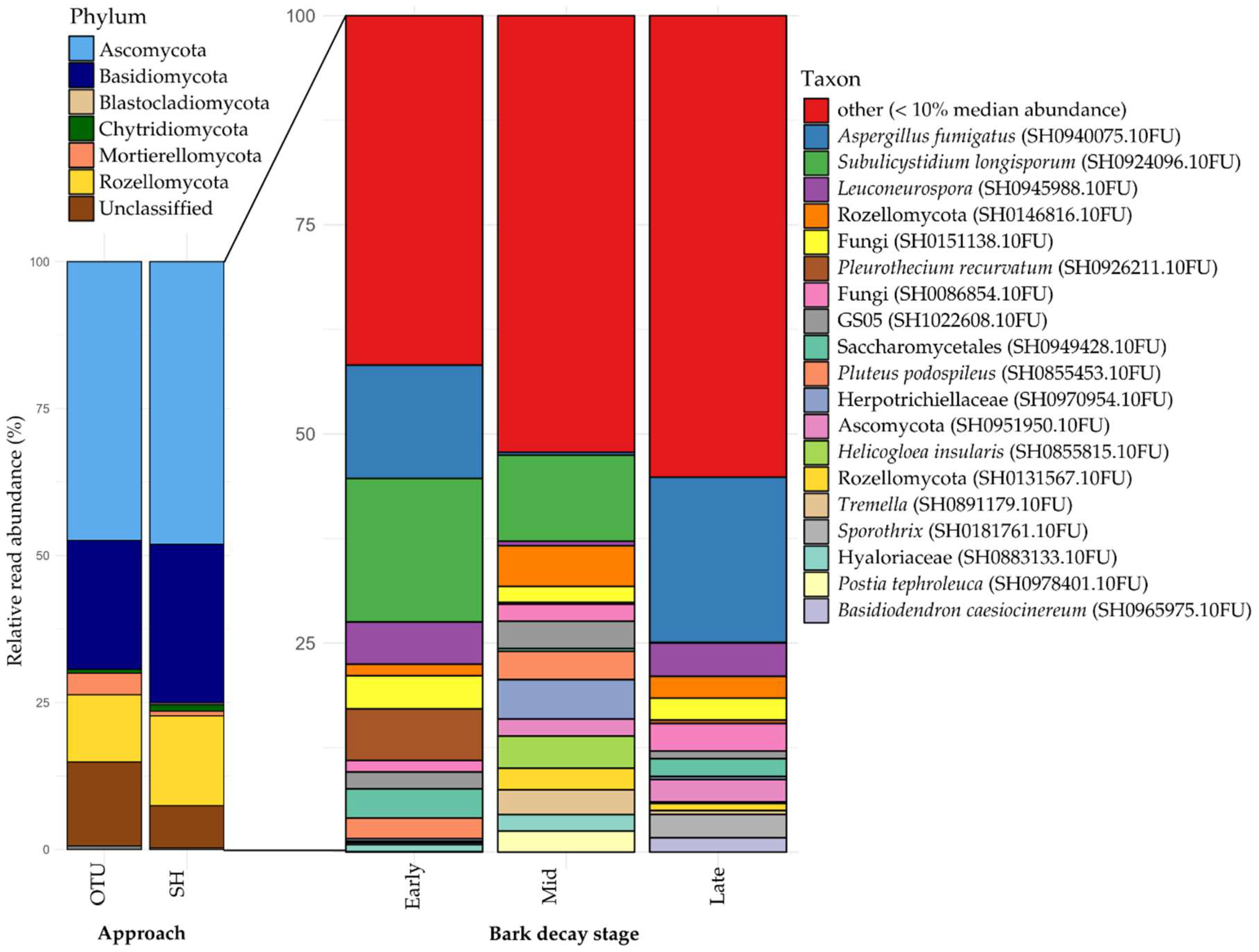
Figure 5.
Ordination of both approaches on Aitchison distances. Samples originating from the same logs are connected by the same line types. (a) NMDS of SH-approach; (b) NMDS of OTU-approach; (c) PCoA of SH-approach; (d) PCoA of OTU-approach.
Figure 5.
Ordination of both approaches on Aitchison distances. Samples originating from the same logs are connected by the same line types. (a) NMDS of SH-approach; (b) NMDS of OTU-approach; (c) PCoA of SH-approach; (d) PCoA of OTU-approach.
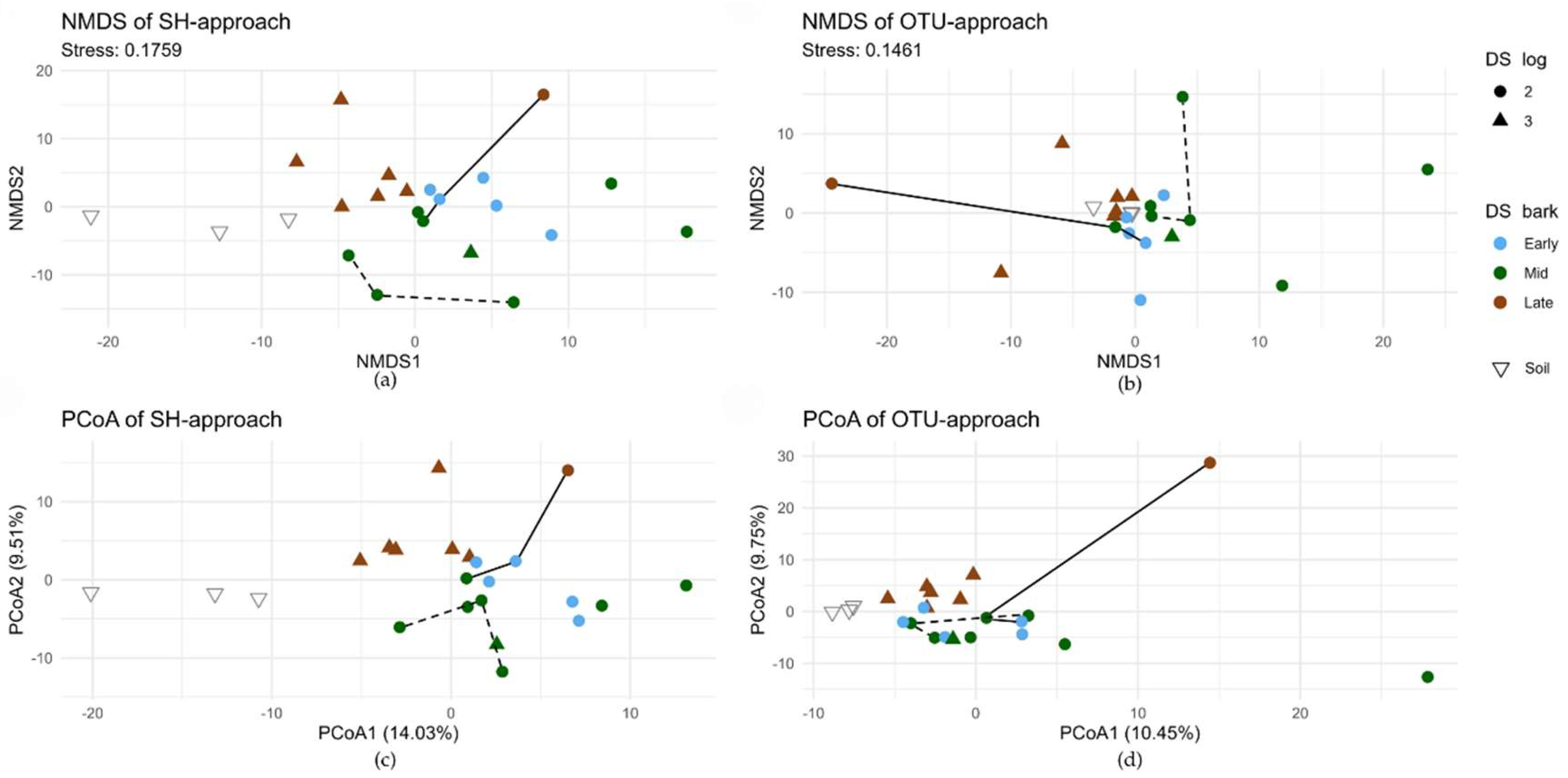
Figure 6.
Relative read abundance of EcM species in bark according to presence of regeneration. Bars indicate samples from the same log. Circles indicate logs where EcM where found by morphotyping. Large asterisks under bars indicate samples not used in morphotyping. Sample names from left to right: ZF031, ZF302, ZF311, ZF319_1, ZF319_2, ZF319_3, ZF320, ZF321_1, ZF321_2, ZF321_3, ZF322, ZF323, ZF324, ZF325, ZF326, ZF327, ZF’681’, ZF317, ZF328, ZF401. *dummy SH codes.
Figure 6.
Relative read abundance of EcM species in bark according to presence of regeneration. Bars indicate samples from the same log. Circles indicate logs where EcM where found by morphotyping. Large asterisks under bars indicate samples not used in morphotyping. Sample names from left to right: ZF031, ZF302, ZF311, ZF319_1, ZF319_2, ZF319_3, ZF320, ZF321_1, ZF321_2, ZF321_3, ZF322, ZF323, ZF324, ZF325, ZF326, ZF327, ZF’681’, ZF317, ZF328, ZF401. *dummy SH codes.


Table 1.
Decay stages of inner bark layer in European beech (Fagus sylvatica) deadwood. Note that Bark decay stage (DS) may differ within the same log.
Table 1.
Decay stages of inner bark layer in European beech (Fagus sylvatica) deadwood. Note that Bark decay stage (DS) may differ within the same log.
| Bark decay stage | Description |
|---|---|
| Early | Layer (incl. inner bark) difficult to penetrate with a metal spoon (force needed), almost all fibers intact, dark yellow in color. Corresponds to log decay stage (DS) 1 or early 2. |
| Intermediate | Layer crumbles and can be scooped up with a metal spoon. Crumbles with minimal force between fingers, some fibers intact, light brown in color. Corresponds to a late log DS 2 or early 3. |
| Late | Layer is almost fully decayed, penetrable without force, it easily disintegrates between fingertips – resembles soil, no fibers intact, (dark) brown in color. Corresponds to a late log DS 3 or 4. |
Table 2.
High-fidelity taxa for the OTU- and SH-approaches that met a significance threshold of p<0.01. Fidelity scores are those from SH-approach if taxon is present in both approaches.
Table 2.
High-fidelity taxa for the OTU- and SH-approaches that met a significance threshold of p<0.01. Fidelity scores are those from SH-approach if taxon is present in both approaches.
| DS Bark | lowest taxon name | fidelity | p | SH | OTU | |
|---|---|---|---|---|---|
| Early | Chaetosphaeriaceae (SH0980871.10FU) | 0.798 | 0,001 | ● | ● |
| Tulasnella (OTU 1097) | 0.728 | 0,003 | ● | ||
| Sordariales (SH0840221.10FU) | 0.779 | 0,008 | ● | ● | |
| Pleurothecium recurvatum (SH0926211.10FU)* | 0.950 | 0.007 | ● | ● | |
| Mid | Ganoderma adspersum (SH0762773.10FU) | 0.839 | 0,001 | ● | |
| Hydnodontaceae (OTU 448) | 0.729 | 0,004 | ● | ||
| Herpotrichiellaceae (SH0970954.10FU) | 0.751 | 0,005 | ● | ● | |
| Rhinocladiella (OTU 75 | SH0970950.10FU) | 0.697 | 0,005 | ● | ||
| Helotiales (OTU 474) | 0.729 | 0,006 | ● | ||
| Mortierella (SH0960682.10FU) | 0.745 | 0,008 | ● | ● | |
| Serendipita (SH0743656.10FU) | 0.730 | 0,008 | ● | ● | |
| Chaetosphaeria decastyla (SH0980872.10FU) | 0.655 | 0,010 | ● | ||
| Late | Hyaloscyphaceae (SH0973189.10FU) | 0.707 | 0,006 | ● | ● |
| Rozellomycota (SH0910702.10FU) | 0.707 | 0,006 | ● | ||
| Ilyonectria mors-panacis (SH1450888.10FU) | 0.707 | 0,007 | ● | ||
| Rozellomycota (OTU 1883) | 0.711 | 0,007 | ● | ||
| Pezizomycotina (SH0755218.10FU) | 0.808 | 0,008 | ● | ● | |
| Trichoderma (OTU 4045) | 0.707 | 0,008 | ● | ||
| Hypocreales (OTU 2917) | 0.707 | 0,009 | ● | ||
| Leotiomycetes (SH0948543.10FU) | 0.707 | 0,010 | ● | ● | |
* Additional taxon identified using Indval.g function in multiipatt, fidelity value is the indicator statistic.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



