
Submitted:
28 August 2024
Posted:
29 August 2024
You are already at the latest version
Abstract
Applying cold to a bone injury can aid healing, though its mechanisms are complex. This study investigates how cold therapy impacts bone repair to optimize healing. Cold was applied to a rodent bone model, with physiological responses analyzed. Vasoconstriction was mediated by an increase in TRP channels, TRPA1 (p=0.012) and TRPM8 (p<0.001), within cortical defects enhancing sensory response and blood flow regulation. Cold exposure also elevated hypoxia (p < 0.01) and VEGF expression (p < 0.001), promoting angiogenesis vital for bone regeneration. Increased expression of osteogenic proteins PGC-1α (p = 0.039) and RBM3 (p<0.008) suggests stimulated reparative processes. Enhanced osteoblast differentiation and ALP presence at days 5 (three-fold, p=0.014) and 10 (two-fold, p=0.010) were observed, along with increased osteocalcin (OCN) at day 10 (two-fold, p = 0.010) indicating presence of mature osteoblasts capable of mineralization. These findings highlight cold therapy's multifaceted effects on bone repair, offering insights for therapeutic strategies.
Keywords:
Cold
; Bone Healing
; Tissue Engineering
; Hypoxia
; Vasculature
; Osteogenesis
; Shock Proteins
; Osteoblasts
1. Introduction
Novel modalities to approach bone repair may address the burdens patients may face especially within aging populations and complications concerning bone density [1]. One such avenue may be the usage of cold therapy, where intermittent short term cold exposure has been shown to lead to increased bone healing dynamics[2,3]. However, a major bottleneck in ideal application of cold exposure is lack of understanding the mechanistic action. Numerous in vivo studies and observations have shown that prolonged cold exposure impedes bone growth, repair, and morphology[4,5,6], while short-term cold exposure exerts beneficial effects on bone healing and morphology[3,7,8,9]. In vitro investigations have demonstrated that long term cold exposure inhibits specific pathways associated with bone healing, such as osteoblastogenesis and hypoxia signaling[4,5,10,11,12,13]. Conversely, short term cold exposure induces an upregulation of these pathways but have yet to be prominently identified within an in-vivo bone injury model[14,15,16].
Several investigations into short term cold application have elucidated its potential for augmenting bone development and repair, both in vitro and in vivo. In vivo studies have consistently demonstrated enhanced bone formation post-injury- characterized by increased deposition of bone tissue at the injury site, concomitant with elevated levels of osteoblast activity biomarkers such as alkaline phosphatase (ALP)[2,3,17]. Additionally, in vitro experiments have revealed that cold stimulation of mesenchymal stem cells (MSCs) and osteoblast progenitors promotes osteoblast differentiation[14].
Collectively, cold exposure initiates a shift in cellular expression, especially within bone cells, stimulating anabolic bone formation pathways. Established literature has shown that cold exposure induces rapid cellular responses, largely mediated by the activation of cold shock proteins[18,19]. RNA binding motif protein 3 (RBM3) in particular has been established as the most representative protein expressed under cold shock[20]. Similarly, peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) is a key regulator in bone homeostasis affecting primarily osteoblastogenesis as well as mitochondrial biogenesis[21]. Both cold shock proteins hold a prominent role in cellular adaptation necessary for cellular survival [22,23] Recently, these proteins have garnered particular attention for potential impact on bone repair due to their prevalence in osteoblastic cells and pivotal roles in regulating osteoblastogenesis pathways[19]. Additionally, it has been previously established that compromise of the vascular network in and around bone results in hypoxia, leading to the activation of a key mechanism in bone healing to restore the blood flow, i.e., angiogenesis[24]. Angiogenesis is a critical part of the bone healing process in order to restore native blood flow following a bone injury[25]. In-vivo bone injury studies have shown enhancement of vasculature following cold treatment- with angiogenic markers such as vascular endothelial growth factor (VEGF) being upregulated indicative of enhancement of angiogenic pathways. However, there is a lack of findings regarding the mechanism of action at the bone injury site[3,17]. Reports of short cold exposure have been shown to effect bone perfusion[26]; for instance 10 min of immersion in 22°C water causes approximately 40% reduction of whole limb blood flow[7].
The TRP (Transient Receptor Potential) family of receptors are expressed in sensory neurons and vascular endothelial cells. These play a crucial role in sensing various stimuli, including temperature changes[27,28]. Among the TRP family, TRP channels such as transient receptor potential cation channel subfamily M (TRPM8) and transient receptor potential ankyrin 1 (TRPA1) are particularly involved in the perception of cold temperatures and in the conduction of this vasoconstrictive pathway[29]. TRPM8 channels are pivotal in coordinating responses to cold exposure within a temperature range typically from 8°C to 28°C[30]. TRPA1 channels are activated by cold exposure within a range of approximately 8°C to 17°C causing vasoconstriction and changes in blood flow, essential for thermoregulation and tissue viability during cold exposure[31].
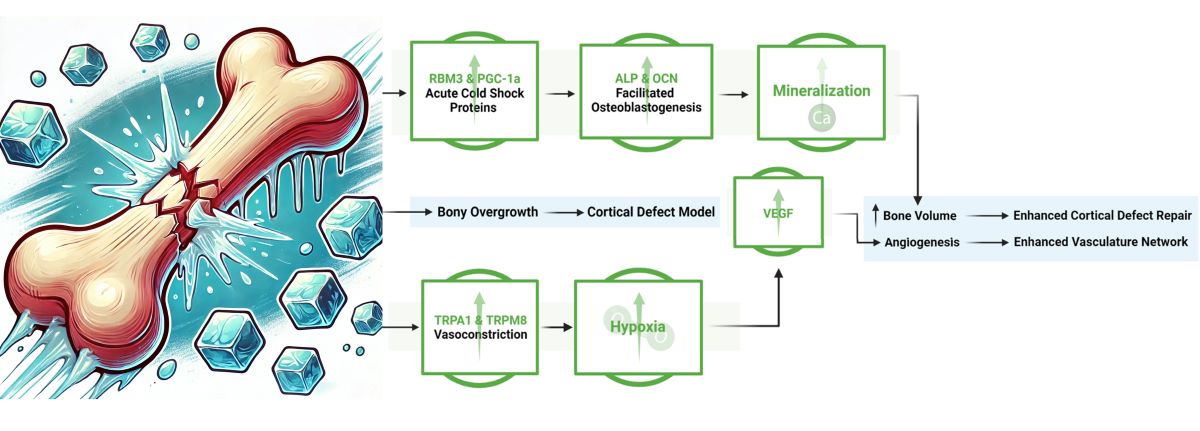
With this in mind, we hypothesized that with the addition of cold there may be an increase in the presence of RBM3 and PGC-1α within a bone injury site, which could facilitate the differentiation of osteoblasts and explain documented cases of increased mineralization of newly formed bone (Figure 1). Additionally, exposure to cold requires a microenvironmental adaptation to maintain heat homeostasis that involves a sensory response to cold temperature leading to the activation of TRPA1 and TRPM8. This in turn may highlight localized vasoconstriction that explains the increase in hypoxia detected and subsequentially the enhanced vasculature network within bone injury sites (Figure 1).
The objective of this study is to delineate the mechanism by which cold therapy affects bone formation in-vivo at the injury site. Building on the potential benefits of acute cold observed in our previous studies, we extended our investigation to the mechanisms through which cold exposure promotes osteogenesis and contributes to the formation of a bony callus. In this work, we implemented a simple and reproducible mouse defect model to demonstrate that localized application of cold temperature leads to elevated induction of hypoxia in the bone injury site indirectly by modifying the vasomotor tone through TRP related pathways in conjunction with increased detection of RBM3 and PGC-1a, responsible for upregulation of osteoblast differentiation (Figure 1).
2. Materials and Methods
2.1. Cortical Defect Animal Model
Following approval from the McGill Facility Animal Care Committee with accordance to the Canada Council on Animal Care and NIH guidelines, a bilateral cortical bone defect model was applied on ten male C3H strain mice aged 2-3 months old (Charles Rivers Laboratories). 30 minutes prior to the surgical intervention, each animal was given subcutaneous injections of analgesic slow release buprenorphine and prophylactic antibiotics baytril for pain management and infection prevention, respectively.Mice were then sedated in a sedation box with an influx of 4% isoflurane at a flow rate of 1.0 LPM of pure oxygen and maintained under anesthesia with an influx of 2.5%-3.0% isoflurane at a flow rate of 0.5 LPM provided through an airflow mask. To ensure thermal regulation, a heating pad was employed, and hydration was maintained with the administration of warm saline solution (0.2- 0.5ml/10 grams, subcutaneously). A lateral 5-mm initial incision was made referencing the trochanter, the muscles were bluntly dissected longitudinally to gain access to the femur. Rectangular cortical defects (1x2.5mm) were made within the ventrolateral aspect of each of the femoral diaphyses using a 1-mm high-speed burr (Stryker, Hamilton, ON, Canada) drill (Figure S1). Following the formation of the defect, the region was flushed with phosphate buffered saline (PBS) to remove any remaining bone fragments.
2.2. Hypoxyprobe Treatments
7 days after operation on the mice, hypoxyprobe treatment commenced. HypoxyprobeTM (Hypoxyprobe Inc.) was diluted in saline to reach a final concentration of 2.45 mg/mL in accordance with manufacture guidelines and 1.5mL of the solution was intraperitoneally injected (Figure S1, S2). Five minutes after the time of injection, the experimental hindlimb of the mouse was exposed to an ice-water bath for fifteen minutes. Only the hindlimb of interest was immersed into the ice-water bath. An internal temperature of 19 degrees Celsius within the mouse hindleg can be obtained through an established methodology[3]. The mice exhibited healthy behaviour throughout the duration of the experiments. The cortical defects formed resulted in no inhibition of animal behavior. No issues arose concerning skin sensitivity during the PO treatment.
2.3. Tissue Harvesting and Staining Analysis
Immediately after the hypoxyprobe treatments, the mice were then euthanized firstly by sedating them with an influx of 5% isoflurane at a flow rate of 4.0 LPM in an induction chamber until unconscious followed by an influx of pure carbon dioxide until there no evidence of heart palpitations the mice are left in the induction chamber for 2 more minutes. Lastly, in accordance with McGill FACC guidelines, chemical asphyxiation is followed by physical asphyxiation via cervical dislocation. Femurs were then harvested and fixated in 4% Paraformaldehyde for 48 hours. Femurs were then decalcified with 10% EDTA, paraffin-embedded, and cut into 5µm sections using a LEICA 2255 microtome (Leica Microsystems, Concord, ON, Canada). Immunohistochemistry was conducted utilizing a secondary antibody approach. Initially, irreversible protein adducts formed between pimonidazole and hypoxic cells (See Supplementary Information) were detected in mice following the hypoxyprobe experiment design, using anti-pimonidazole antibody (Hypoxyprobe Inc). Additionally, cold shock proteins RBM3 and PGC-1a were assessed. Detection utilized a secondary antibody sequence with a fluorescein isothiocyanate (FITC)-conjugated system. Sections were incubated overnight with primary antibodies, followed by goat anti-rabbit conjugated with HRP as a secondary antibody. Antibody binding was detected using the DAKO antigen retrieval system (DAB kit, K3468, USA). Sections were counterstained with Fast Green (Abcam, ab146267), dehydrated, cleared, and mounted. Immunofluorescence staining for CD34 (Santa Cruz, sc-74499 FITC, 1:250) for endothelial vascular cells, vascular endothelial growth factor (VEGF, Santa Cruz, sc-7269 FITC, 1:250) as an angiogenic factor, Transient Receptor Potential Cation Channel Subfamily A Member 1 (TRPA1, Santa Cruz, sc-376495, 1:250), and Transient receptor potential melastatin 8 (TRPM8, Novus Biotech, NB200-145F, 1:250) all involving overnight incubation with primary antibodies and subsequent analysis.
Microscopic images of immunohistochemical stained samples were captured using either a 1) Zeiss Axioskop 40 microscope (Carl Zeiss, Toronto, ON, Canada) or 2) Aperio digital pathology slide scanners from Leica Biosystems. Immunofluorescent stained samples were imaged using an EVOS M5000 Imaging system (Life Technologies Corporation, Bothell, WA, USA). Histomorphometric calculation of stained cell percentages in defined regions of interest was performed using ImageJ v.1.6.0 software (NIH, Bethesda, MD, USA). SPSS version 20 (IBM) compared means of parameters in control and experimental groups using a Paired Student T-test, with statistical significance set at p<0.05.
2.4. Femoral Fracture Animal Model
The next segment of the study was carried out following approval from the McGill Facility Animal Care Committee and in adherence to guidelines from the Canada Council on Animal Care and National Institutes of Health. Bilateral femoral fractures were induced in 21 male C3H strain mice, selected for their distinct bone characteristics. 30 minutes prior to the surgical intervention, each animal was given subcutaneous injections of analgesic slow release buprenorphine and prophylactic antibiotics baytril for pain management and infection prevention, respectively.Mice were then sedated in a sedation box with an influx of 4% isoflurane at a flow rate of 1.0 LPM of pure oxygen and maintained under anesthesia with an influx of 2.5%-3.0% isoflurane at a flow rate of 0.5 LPM provided through an airflow mask. To ensure thermal regulation, a heating pad was employed, and hydration was maintained with the administration of warm saline solution (0.2- 0.5ml/10 grams, subcutaneously).
2.5. Treatments and Cell Study
The same cold treatment was applied daily for five days in 9 mice, 10 days in 9 mice, and 14 days in 3 mice. A summary of the study segments has been provided in supplementary figure S3. The mice were then euthanized firstly by sedating them with an influx of 5% isoflurane at a flow rate of 4.0 LPM in an induction chamber until unconscious followed by an influx of pure carbon dioxide until there no evidence of heart palpitations the mice are left in the induction chamber for 2 more minutes. Lastly, in accordance with McGill FACC guidelines, chemical asphyxiation is followed by physical asphyxiation via cervical dislocation. To prevent contamination, the whole animals were transported to a sterile cell culture hood in 70% ethanol solution. The femurs were then isolated, and a puncture needle used to enter the medullary cavity of the bone to aspirate the bone marrow. The femurs were then washed 3 times with PBS. To maximize the amount of osteoblast extraction, each group consisted of 3 femurs paired together and crushed coarsely inside a MEM (Gibco 12561056, Thermofisher Scientific) enriched with amino acids (Gibco 50X MEM Amino Acids, 11130051 Thermofisher Scientific), fetal bovine serum (12483020, Thermofisher Scientific), and penicillin-streptomycin (15140122, Thermofisher Scientific). A digestion sequence followed in order to specifically isolate osteoblast cells using a modification of pre-existing procedures from the literature[32]. Bone fragments were digested sequentially in 10 ml of each of the following enzymes: trypL (1 mg/ml; 12605010 Thermofisher Scientific), dispase II (2 mg/ml; 17105041 Thermofisher Scientific), and twice in collagenase II (3 mg/ml; 17101015 Thermofisher Scientific). The supernatants from the trypsin and dispase digestions were discarded whereas those from the collagenase digestions were retained and combined. The cells were then assessed for morphology to confirm them as osteoblasts. The OBs were then cultured to reach confluency. Once confluency was attained, alamar blue cell viability testing was done to normalize cell counts.
The secretion levels of biomarkers related to osteoblast differentiation was measured after adding TrypL for dissolution of OBs. The 2mL cell suspensions were then transferred into Eppendorf tubes and underwent lysing. The supernatant was carefully collected and underwent first assessment of osteocalcin (OCN) via ELISA (ab285236, Abcam) followed by alkaline phosphatase (ALP) via colorimetric assay (ab83369, Abcam). Statistical analyses of the assays were conducted by taking absorbance measurements using Varioskan LUX (Thermofisher Scientific, N16044) with 406nm absorbance for ALP readings and 450nm absorbance for OCN readings. Statistical analysis was done using SPSS version 20 (IBM) via unpaired student T-test, with significance set at p<0.05.
Another group of 3 mice using the femoral fracture model were used to assess for mineralization following daily cold exposure. The bilateral femoral fracture model mentioned was used followed by daily usage of the same cold treatment regime for 14 days. Following osteoblast isolation after day 14, the cells were cultured in a 6 well plate and stained using alizarin red to assess for mineralization nodules found within osteoblasts. Histomorphometric calculation of stained cell percentages in defined regions of interest was performed using ImageJ v.1.6.0 software (NIH, Bethesda, MD, USA). SPSS version 20 (IBM) compared means of parameters in control and experimental groups using a Paired Student T-test, with statistical significance set at p<0.05.
2.6. Statistical Design and Analysis
The purpose of this study is to assess the mechanisms upon which cold therapy affects positively the bone healing by comparison of 2 means representing the presence of specific biomarkers affiliated with the potential repair pathways. Statistical power analysis was performed for sample size estimation (clinicalc.com/stats/samplesize), and an estimated sample size was calculated that was requisite for a significance level of 0.05 and power of 0.9. We used our previous pilot study for anticipated means and a standard deviation of +/- 5.5 [3]. All assessments were performed in a blinded manner regarding the source of the tissue. To prevent asymmetric expression in osteoblast differentiations/mineralization, right or left limbs were used as experimental group. Student paired T-tests were used to assess quantitative data for paired femora from the same mouse to account for varying baseline fluctuation between mice while unpaired t-tests were used to account for two groups of means unaffected by baseline fluctuation between mice. Statistical analysis was done using SPSS version 20 (IBM). Differences considered significant at P<0.05.
3. Results
The assessment of vasoconstriction receptors revealed a significant increase (Figure 2C) in TRPA1 presence following cold treatment (p = 0.012), with levels rising to 9.03% ± 2.78 in the cortical defect region, compared to 5.38 ± 2.32 in untreated controls (Figure 2A vs. Figure 2B). Similarly, TRPM8 levels were significantly elevated (p < 0.001) after cold exposure, showing an increase to 8.75 ± 1.68 in treated femurs versus 3.86 ± 1.72 in untreated ones (Figure 2D vs. Figure 2E). Elevation of receptors responsible for triggering a vasoconstrictive response confirms the detection of cold exposure within the microenvironment of the bone injury site.
There was a 5.6% increase in the number of hypoxic cells within and around the cortical bone defect of experimental (Figure 3A) hindlegs (23.5% ± 5.8) compared to untreated (Figure 3B) controls (17.9% ± 3.8) following exposure to an acute cold stimulus (p-value<0.01) reflecting an increase in local hypoxia following the application of cold exposure (Figure 3C). Investigating cold-induced cortical defect repair revealed significant angiogenic changes, particularly for Vascular Endothelial Growth Factor (VEGF) expression. VEGF expression in the cold-treated group (Figure 3D) notably surpassed the control group (Figure 3E), with levels at 9.69% ± 1.75 and 4.05% ± 1.83, respectively (Figure 3F; p<0.001), suggesting accelerated angiogenesis. Endothelial cells within the cortical defect were identified using the endothelial-specific marker CD34. There was no statistically significant change (Figure 3I) in the presence of these cells in the cold-treated group (8.00% ± 2.76; Figure 3G) compared to the control group (6.92% ± 2.77; Figure 3H).
Simultaneously, increased detection of PGC-1a and RBM3 were prevalent in newly forming bone cells and the surrounding matrix within cortical defects following cold exposure. RBM3 significantly (p-value<0.008) increased (Figure 4C) within cold (Figure 4A) treated femora (11.0% ± 4.26%) compared to nontreated (Figure 4B) femora (4.47% ± 2.84%)). Synonymously, PGC-1a was significantly (p-value = 0.039) elevated (Figure 4F) in cold (Figure 4D) treated cortical defects (5.85% ± 1.01%) in comparison to nontreated (Figure 4E) defects (2.57% ± 0.36%). Increase in presence of RBM3 and PGC-1a indicates that cells within the bone injury site are affected by cold exposure resulting in the stimulation of proteins responsible for cellular adaptation required for cell survival [19,33,34]. Furthermore, the percentage of PGC-1a and RBM3 positive cells marked by DAB staining (brown) within and around the cortical bone defect in the hindlimbs of mice exposed to a cold stimulus is higher in comparison to the control hindlimbs is of particular interest in part due to the prominent roles RBM3 and PGC-1a have in osteoblastogenesis pathways.
At the junction of old bone and new bone formation, there was a distinct observational contrast in the prevalence of PGC-1a within the new bone formation region within cold treated femora (Figure 5A) and non-treated femora (Figure 5E). Within both cold and control treated femora, 70.56% ± 7.08 of total PGC-1a was detected at the junction of new and old bone (Figure 5B-D) corresponding with PGC-1a’s role as a cold shock protein and deterring necessary cellular changes as well as PGC-1a’s prominence in the bone repair process as this finding was consistently found within non-treated legs (Figure 5F-H)
Assessment of the differentiation of osteoblasts following cold therapy shows that there was a statistically significant facilitative impact on the osteogenic pathway. ALP, an early marker of immature osteoblasts, was shown to be significantly elevated (p-value = 0.021) following cold treatment at day 5 (Figure 6A) compared to non-treated femurs (2.72 U/L ± 0.85 vs. 0.74 U/L (µmol/min/L) ± 0.35). This similar trend was prevalent at day 10 (Figure 6B) as well where ALP was significantly elevated (p-value < 0.001) following cold exposure (2.25 U/L ± 0.04 vs. 1.03 U/L ± 0.74). OCN, a marker for mature osteoblasts capable of mineralization, was not significant at day 5 (p-value = 0.29; Figure 6C).
However, OCN was significantly (p-value = 0.019) more prominent within cold-treated femurs (0.84 mg/mL ± 0.17 vs. 0.35mg/mL ± 0.10) at day 10 (Figure 6D) indicating the presence of mature osteoblasts capable of mineralization. ARS further confirmed the detection on calcium deposits with a statistically significant increase (p-value = 0.030) in the amount of mineralization from isolated osteoblasts at day 14 (Figure 6G) following cold exposure (Figure 6E) in comparison to non-treated (Figure 6F) samples (1.04% ± 0.40 vs. 0.27% ± 0.025).
4. Discussion
The biological mechanism that promotes reparative processes and osteogenic differentiation upon cold exposure is multifaceted as different pathways related to bone repair are upregulated. Short-term cold exposure elicits a complex physiological response involving vasoconstriction, impacting bone healing through modulation of angiogenesis and vascular responses[7,27]. The hypoxic microenvironment ensuing from diminished blood flow elicits the production of angiogenic factors such as vascular endothelial growth factor (VEGF)[35]. Adaptations to cold exposure lead to activation of cell hemostasis regulators within bone cells that corroborate the stimulation of osteogenic pathways and bone formation.
The findings of this study show that an upregulation of TRPA1 (3.65% increase, Figure 2) and TRPM8 (4.89% increase, Figure 2) followed by a hypoxic environment (5.0% increase, Figure 3) upon cold application. Given their role in sensory perception, TRPA1 and TRPM8 cold stimulated upregulation within cortical defects signifies an amplified sensory response to lower temperature[27]. The vasoconstriction, a pivotal process for blood flow regulation, is induced by the sympathetic nervous system through activation of TRPA1 and TRPM8 that release norepinephrine, activating α-adrenergic receptors on vascular smooth muscle cells (Figure 7A)[31,36]. Then, an intracellular signaling cascade occurs with the release of intracellular calcium ions causes the contraction of vascular smooth muscle cells (Figure 7B), ultimately resulting in vasoconstriction and reduced blood flow to peripheral tissue leading to hypoxia (Figure 7C)[27]. To measure the acute hypoxic microenvironment, we developed a novel approach based on pimonidazole based staining [37] to measure hypoxic cells within a bone injury site (Supplementary Figure 1A; Supplementary Figure 4) as the short half-life of the Hypoxia Inducible Factor 1-α protein is impractical to directly analyze the level of hypoxia[10].
In the context of bone healing, angiogenesis is essential for supplying oxygen, nutrients, and growth factors to the healing site, facilitating the formation of new bone tissue[11]. Here, we have shown that the application of localized non-invasive cold therapy for a short duration has demonstrated an elevation of baseline hypoxia within a bone injury site (23.5%, Figure 3) and upregulation of VEGF (9.69%, Figure 3) in cortical defect following cold therapy. Under normal conditions, Hypoxia Inducible Factor 1-alpha (HIF-1α) undergoes degradation because oxygen presence induces its prolyl hydroxylation, marking it for targeted destruction but, in a localized acute hypoxic microenvironment, HIF-1α is stabilized and forms a dimer with HIF-1β to activate the expression of VEGF through the Hypoxia-Response Element (HRE) pathway (Figure 8). The resulting VEGF is released to trigger 1) endothelial cell activation through its receptor vascular endothelial growth factor receptor 2 (VEGFR2), initiating signaling pathways such as PI3K-Akt and MAPK, which are crucial for endothelial cell proliferation and migration (Figure 8)[38,39] and 2) TRPA1 activation which directs the development of the direction of vessel elongation[40]. Furthermore, elevated VEGF following short term cold exposure has been shown to be consistent with enhanced maturation of vasculature within bone injury models [2,3,17] especially within calluses where heightened presence of VEGF overlaps in the same regions of elevated presence of CD34, a biomarker of mature endothelial cells essential for vasculature [2]. Within cortical defects specifically, previous results have shown that bone vessel volume over tissue volume (BVV/TV) was found to be higher in the cold group compared with control (7.8% vs. 6.7% p<0.001) [3].
Nonetheless, TRPM8’s role in angiogenesis is less defined but appears to be implicated in influencing endothelial function, suggesting its potential contribution to the angiogenic process[40,41]. Consequently, observed upregulation of VEGF suggests an augmented angiogenic response, essential for fostering bone regeneration within cortical defects[3,17]. Moreover, the collaborative action of VEGF, TRPA1, and TRPM8 indirectly supports osteogenic processes by securing an adequate blood supply to regenerating tissue, facilitating osteoblast proliferation, and thereby promoting bone formation and maturation within cortical defects.
While vasomotor tone and hypoxia plays an important role in regulating angiogenesis in bone repair following cold exposure, the expression of essential cell hemostasis regulators such as RBM3 and PGC-1α are elevated[42,43]. Initially, cold denaturation of proteins results in proliferation of heat shock proteins (HSPs) for stabilization and proper folding. Dissociation of the HSP/heat shock factor-1 (HSF1) dimer releases free HSF1, which becomes phosphorylated and nuclear localized, leading to HSP expression (Figure 9A). Heat shock protein 70 (HSP70) activates Toll-like receptor 4 (TLR4), which triggers the MyD88/NF-kB signaling pathway. The phosphorylated and nuclear-localized NF-kB dimer is crucial for RBM3 expression (Figure 9B) [44,45], RBM3 activates the MAPK/ERK pathway, leading to the expression of runx2 and osteocalcin (OCN), essential for osteoblast differentiation and consistent with preceding findings showing that RBM3 has previously shown to stimulate the osteoblast differentiation via the ERK signalling pathway and activates two osteogenic genes, Runx2 and OCN (Figure 9C)[14,42]. Runx2 has been shown to be a critical gene in inducing osteoblast differentiation[46], whereas OCN has an important role in calcifying new bone tissue to reform the mechanical integrity of native bone at an injury site[47]. Several in-vitro studies have shown elevated expression of Runx2 following cold exposure stimulation indicating Runx2 as a crucial gene important in the osteogenic pathway impacted by cold exposure [14,48]. Within fractures, this is important in transitioning from a fibrogenesis callus to a bony callus[49]. The increased expression of RBM3 confirms the activation of osteoblast differentiation associated with elevation of ALP levels at days 5 and 10 (2.72U/L, 2.25U/L, Figure S4) and calcium-binding pathways, which are aligned with enhanced bone formation following acute cold therapy. This is consistent with in-vitro studies that have shown elevated levels of ALP and OCN following short term cold exposure indicating activation of osteoblastogenesis pathways essential for osteoblast maturation [14]. ‘Under the same cold treatment conditions, ALP levels have been shown to increase in the callus of femoral fractures, correlating with enhanced mineralized bone formation. These results are consistent with similar observations in cortical defects, further supporting the role of cold exposure in stimulating bone repair pathways [2,3,17].
Studies have already demonstrated that cold exposure leads to activation of PGC-1α via the PKA/CaMKII pathway where cold activation of adrenergic receptors and transient receptor proteins (TRPs) stimulates cAMP and calcium influx, leading to cAMP-response element binding protein (CREB) phosphorylation via the PKA/Ca2+/CaMK pathway (Figure 9D)[50,51]. PGC-1α has synergistic properties with ERRα which is highly expressed in osteoblasts and osteoclasts, and interacts cooperatively with PGC-1α to amplify the levels of the key proteins in bone formation, including OCN, Runx2, and osteopontin (OPN) necessary for mineralization (Figure 9E)[52]. For example, OPN facilitates blood vessel maturation following angiogenesis and is essential for early callus formation.[11] Furthermore, in-vitro studies have demonstrated upregulation of OPN essential for osteoblast differentiation following cold exposure resulting from Runx2 stimulation [48]. Distinctive contrast of PGC-1α detected at the junction between old bone and new bone formed further reinforces its importance in being responsible for establishing the baseline for new bone formed by osteoblasts to interconnect with old bone [53] (Figure 5). These osteoblasts then secrete further OPN and create a positive feedback cycle for bone regeneration [53]. Additionally, PGC-1alpha activation stimulates the transcription of mitochondrial genes by forming a complex with nuclear respiratory factor 1 & 2 (NRF-1 and NRF-2), activating Mitochondrial transcription factor A (TFAM) and NCMP expression [50], that are crucial for mitochondrial biogenesis and intracellular calcium storage, enhancing oxidative phosphorylation, and energy production (Figure 9F)[19]. This also induces genes involved in calcium homeostasis, leading to increased calcium storage in mitochondrial granules, which is crucial for osteoblasts and bone formation[54]. Adequate calcium levels are essential for the mineralization process in osteoblasts. This is aligned with elevated alizarin red staining in osteoblasts from cold-treated bone injury samples at day 14 (1.04%, Figure S5) indicating an enhanced extracellular matrix mineralization, reflecting improved osteoblast functionality and bone formation. This may be due to cold exposure influencing cellular metabolism and energy homeostasis via RBM3 and PGC-1α, supporting osteoblast differentiation and matrix mineralization (Figure S4). By boosting mitochondrial function and calcium storage, PGC-1alpha activation via cold therapy supports osteoblastic activity and bone formation. This dual action improves mitochondrial efficiency and ensures sufficient calcium availability, facilitating proper osteoblast function and differentiation [54]. Future studies can further explore the influence of mitochondrial biogenesis on bone healing and matrix mineralization.
This work shows the dual effect of cold exposure on 1) bone and vasculature morphology and 2) on pathways involved in bone repair (Figure 10). Upregulation of TRPA1 and TRPM8 in the injury region induces vasoconstriction resulting in a heightened acute hypoxic microenvironment, leading to elevated VEGF levels and activation of angiogenic pathways, promoting a robust vasculature network in cold-treated bone injuries. Elevated RBM3 and PGC-1α levels at the injury site support osteoblast differentiation necessary for new bone mineralization, thereby enhancing bone production following cold exposure.
5. Conclusion
In conclusion, the accelerated osteoblast differentiation with increased ALP activity and enhanced OCN-mediated bone matrix synthesis and mineral deposition underscore the critical role of cold exposure in promoting advanced osteoblastic maturation and improving bone healing. The heightened presence of TRPA1 and TRPM8 within cortical bone defects highlights their pivotal involvement in cold-induced skeletal repair mechanisms. These transient receptor potential channels initiate vasoconstriction and trigger reparative responses crucial for bone regeneration, intensified by the observed increase in hypoxia following cold therapy. Elevated VEGF levels further support this process by stimulating angiogenesis, essential for fostering bone growth within cold-treated defects. The upregulation of RBM3 and PGC-1α underscores the adaptation of bone cells to cold stimulation, enhancing osteogenic processes and facilitating osteoblast differentiation crucial for bone repair and regeneration. This comprehensive interplay between sensory perception, vascular regulation, angiogenesis, and osteogenesis reveals the intricate dynamics behind cold-based therapeutic interventions for bone injuries, promising avenues for future research and clinical applications. With the rise of interest in exosomes and further discoveries regarding the Jumonji C domain-containing (JMJD) protein family within the field of epigenetics regarding cold exposure and bone repair pathways [55,56,57,58], exosomes could be utilized to prime a bone injury microenvironment to respond favorably to an external stimuli such as cold by optimizing bone repair pathways through miRNA and LncRNA based epigenetic modification locally. Pioneering research in biomaterials has begun to advance the development of 4D based materials that respond to external stimuli such as temperature which formulates an active scaffold that can dynamically change over time allowing for an optimizable approach to bone healing [59,60,61,62,63].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Hypoxyprobe is a chemical compound designed to detect and measure tissue hypoxia, or low oxygen levels, in biological samples. Pimonidazole hydrochloride, a 2-nitroimidazole derivative, serves as the active component due to its ability to undergo reduction in hypoxic conditions. Upon administration, typically through injection, pimonidazole diffuses throughout tissues. In normoxic conditions, it remains unaltered; however, under hypoxic conditions, it undergoes reduction reactions facilitated by cellular reductases. This reduced form becomes chemically reactive, binding to thiol groups on proteins, peptides, and amino acids within hypoxic cells, forming stable irreversible adducts in regions where oxygen levels are critically low [37]. These pimonidazole-protein adducts can then be detected using specific antibodies through immunohistochemical techniques, such as fluorescent or chromogenic staining, allowing for the visualization of hypoxic regions in tissue samples. The extent of hypoxia is quantified by measuring the intensity and distribution of the staining, which correlates with the level of pimonidazole binding and the severity and distribution of hypoxia within the tissue [37]. This pimonidazole-based staining approach is particularly effective in quantifying acute hypoxic induction due to the compound’s properties. Acute hypoxia is associated with fluctuating partial oxygen pressure (pO2) resulting from blood flow instabilities [37]. Cells experiencing fluctuating hypoxia are typically located near blood vessels and at a relatively high, weakly basic pH. In these conditions, weakly basic 2-nitroimidazole hypoxic markers like pimonidazole are concentrated within these cells, leading to higher levels of binding compared to hypoxia markers lacking weakly basic moieties. As such, pimonidazole is highly effective for detecting acute hypoxia [64].
Author Contributions
Matthew Zakaria: Experimental design, implementation, performance, data collection, analysis, preparation of the manuscript. Justin Matta: Data collections, experimental design, implementation, and performance. Yazan Honjol: Experimental Design, Implementation, and performance. Drew Schupbach: Experimental Design, Implementation, and performance. Michael Grant: Data analysis, preparation of the manuscript. Fackson Mwale: revised of the manuscript. Edward Harvey and Geraldine Merle: conceived the ideas, Experimental design, provided funding, and supervised the project.
Funding
This research was funded by Merle, NSERC discovery, and FRQS chercheur boursier J1.
Institutional Review Board Statement
The study was conducted in adherence to guidelines from the Canada Council on Animal Care and National Institutes of Health and was carried out following approval from the McGill Facility Animal Care Committee (protocol code AUP-7780; approved 2020).
Informed Consent Statement
Not applicable.
Data Availability Statement
Dataset available on request from the authors.
Acknowledgments
We would like to thank Dr. Chan Gao and Dr. Aileen Li for their assistance in helping with CD34 staining. We would also like to thank Parsa Azizi for his assistance in ImageJ analysis. Lastly, we’d like to thank the RI-MUHC histopathology core at the Glen site for their help with the histology process.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Morgan R, Kalbarczyk A, Mohan D, Jacobs C, Mishra M, Tyagi P, Cox-Roman C, Williamson C. Counting older women: Measuring the health and wellbeing of older women in LMICs. Cell Rep Med. 2024 Jun 18;5(6):101607. [CrossRef]
- Zakaria M, Allard J, Garcia J, Matta J, Honjol Y, Schupbach D, Grant M, Mwale F, Harvey E, Merle G. Enhancing Bone Healing Through Localized Cold Therapy in a Murine Femoral Fracture Model. Tissue Eng Part A. 2024 Aug 7; [CrossRef]
- Castano D, Comeau-Gauthier M, Ramirez-GarciaLuna JL, Drager J, Harvey E, Merle G. Noninvasive Localized Cold Therapy: A New Mode of Bone Repair Enhancement. Tissue Eng Part A. 2019 Apr;25(7–8):554–62. [CrossRef]
- Du J, He Z, Cui J, Li H, Xu M, Zhang S, Zhang S, Yan M, Qu X, Yu Z. Osteocyte Apoptosis Contributes to Cold Exposure-induced Bone Loss. Front Bioeng Biotechnol. 2021;9:733582. [CrossRef]
- Tanaka T, Wakamatsu T, Daijo H, Oda S, Kai S, Adachi T, Kizaka-Kondoh S, Fukuda K, Hirota K. Persisting mild hypothermia suppresses hypoxia-inducible factor-1alpha protein synthesis and hypoxia-inducible factor-1-mediated gene expression. Am J Physiol Regul Integr Comp Physiol. 2010 Mar;298(3):R661-671. [CrossRef]
- Janský L, Pospíšilová D, Honzová S, Uličný B, Šrámek P, Zeman V, Kamínková J. Immune system of cold-exposed and cold-adapted humans. European Journal of Applied Physiology and Occupational Physiology. 1996 Mar 1;72(5):445–50.
- Gregson W, Black MA, Jones H, Milson J, Morton J, Dawson B, Atkinson G, Green DJ. Influence of cold water immersion on limb and cutaneous blood flow at rest. Am J Sports Med. 2011 Jun;39(6):1316–23. [CrossRef]
- FOWLER EP JR, OSMUN PM. NEW BONE GROWTH DUE TO COLD WATER IN THE EARS. Archives of Otolaryngology. 1942 Oct 1;36(4):455–66.
- White GE, Wells GD. Cold-water immersion and other forms of cryotherapy: physiological changes potentially affecting recovery from high-intensity exercise. Extrem Physiol Med. 2013 Sep 1;2(1):26. [CrossRef]
- Iommarini L, Porcelli AM, Gasparre G, Kurelac I. Non-Canonical Mechanisms Regulating Hypoxia-Inducible Factor 1 Alpha in Cancer. Frontiers in Oncology. 2017;7:286. [CrossRef]
- Duvall CL, Taylor WR, Weiss D, Wojtowicz AM, Guldberg RE. Impaired angiogenesis, early callus formation, and late stage remodeling in fracture healing of osteopontin-deficient mice. J Bone Miner Res. 2007 Feb;22(2):286–97. [CrossRef]
- Coassin M, Duncan KG, Bailey KR, Singh A, Schwartz DM. Hypothermia reduces secretion of vascular endothelial growth factor by cultured retinal pigment epithelial cells. Br J Ophthalmol. 2010 Dec;94(12):1678–83. [CrossRef]
- Leegwater NC, Bakker AD, Hogervorst JMA, Nolte PA, Klein-Nulend J. Hypothermia reduces VEGF-165 expression, but not osteogenic differentiation of human adipose stem cells under hypoxia. PLOS ONE. 2017 Feb 6;12(2):e0171492. [CrossRef]
- Mohd Din A, Nor-Ashikin MNK, Ab. Rahim S, Nawawi H, Kapitonova M, Froemming G. Short-term moderate hypothermia stimulates alkaline phosphatase activity and osteocalcin expression in osteoblasts by upregulating Runx2 and osterix in vitro. Experimental Cell Research. 2014 Aug 1;326.
- Kim JC, Yi HK, Hwang PH, Yoon JS, Kim HJ, Kawano F, Ohira Y, Kim CK. Effects of cold-water immersion on VEGF mRNA and protein expression in heart and skeletal muscles of rats. Acta Physiol Scand. 2005 Apr;183(4):389–97. [CrossRef]
- Xue Y, Petrovic N, Cao R, Larsson O, Lim S, Chen S, Feldmann HM, Liang Z, Zhu Z, Nedergaard J, Cannon B, Cao Y. Hypoxia-Independent Angiogenesis in Adipose Tissues during Cold Acclimation. Cell Metabolism. 2009 Jan 7;9(1):99–109. [CrossRef]
- Shakurov AV, Lukina YuS, Skriabin AS, Bionyshev-Abramov LL, Serejnikova NB, Smolencev DV. Enhanced bone healing using local cryostimulation: In vivo rat study. Journal of Thermal Biology. 2023 Apr 1;113:103501. [CrossRef]
- Ihsan M, Abbiss CR, Allan R. Adaptations to Post-exercise Cold Water Immersion: Friend, Foe, or Futile? Front Sports Act Living. 2021;3:714148. [CrossRef]
- Lindquist JA, Mertens PR. Cold shock proteins: from cellular mechanisms to pathophysiology and disease. Cell Communication and Signaling. 2018 Sep 26;16(1):63. [CrossRef]
- Muhlig Nielsen M, Overgaard J, Sørensen JG, Holmstrup M, Justesen J, Loeschcke V. Role of HSF activation for resistance to heat, cold and high-temperature knock-down. Journal of Insect Physiology. 2005 Dec 1;51(12):1320–9. [CrossRef]
- Chen H, Fan W, He H, Huang F. PGC-1: a key regulator in bone homeostasis. J Bone Miner Metab. 2022 Jan;40(1):1–8. [CrossRef]
- Hu Y, Liu Y, Quan X, Fan W, Xu B, Li S. RBM3 is an outstanding cold shock protein with multiple physiological functions beyond hypothermia. J Cell Physiol. 2022 Oct;237(10):3788–802. [CrossRef]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A Cold-Inducible Coactivator of Nuclear Receptors Linked to Adaptive Thermogenesis. Cell. 1998 Mar 20;92(6):829–39. [CrossRef]
- Oryan A, Monazzah S, Bigham-Sadegh A. Bone Injury and Fracture Healing Biology. Biomedical and Environmental Sciences. 2015 Jan 1;28(1):57–71. [CrossRef]
- Grzegorz Szczęsny ED1 - Ozgur Karcioglu ED2 - Hakan Topacoglu. Fracture Repair: Its Pathomechanism and Disturbances. In: Trauma Surgery [Internet]. Rijeka: IntechOpen; 2018 [cited 2021 Feb 12]. p. Ch. 1. Available from: . [CrossRef]
- Schoutens A, Bergmann P, Verhas M. Bone blood flow measured by 85 Sr microspheres and bone seeker clearances in the rat. Am J Physiol. 1979 Jan;236(1):H1-6. [CrossRef]
- Pan Y, Thapa D, Baldissera L, Argunhan F, Aubdool AA, Brain SD. Relevance of TRPA1 and TRPM8 channels as vascular sensors of cold in the cutaneous microvasculature. Pflügers Archiv - European Journal of Physiology. 2018 May 1;470(5):779–86. [CrossRef]
- Lieben L, Carmeliet G. The Involvement of TRP Channels in Bone Homeostasis. Frontiers in Endocrinology. 2012;3:99. [CrossRef]
- Earley S, Brayden JE. Transient receptor potential channels in the vasculature. Physiol Rev. 2015 Apr;95(2):645–90. [CrossRef]
- McKemy DD. The molecular and cellular basis of cold sensation. ACS Chem Neurosci. 2013 Feb 20;4(2):238–47. [CrossRef]
- Aubdool AA, Graepel R, Kodji X, Alawi KM, Bodkin JV, Srivastava S, Gentry C, Heads R, Grant AD, Fernandes ES, Bevan S, Brain SD. TRPA1 is essential for the vascular response to environmental cold exposure. Nature Communications. 2014 Dec 11;5(1):5732. [CrossRef]
- Ali E, Birch M, Hopper N, Rushton N, McCaskie AW, Brooks RA. Human osteoblasts obtained from distinct periarticular sites demonstrate differences in biological function in vitro. Bone Joint Res. 2021 Sep;10(9):611–8. [CrossRef]
- Dey P, Rajalaxmi S, Saha P, Thakur PS, Hashmi MA, Lal H, Saini N, Singh N, Ramanathan A. Cold-shock proteome of myoblasts reveals role of RBM3 in promotion of mitochondrial metabolism and myoblast differentiation. Communications Biology. 2024 Apr 30;7(1):515. [CrossRef]
- Xu L, Ma X, Bagattin A, Mueller E. The transcriptional coactivator PGC1α protects against hyperthermic stress via cooperation with the heat shock factor HSF1. Cell Death & Disease. 2016 Feb 1;7(2):e2102–e2102. [CrossRef]
- Krock BL, Skuli N, Simon MC. Hypoxia-induced angiogenesis: good and evil. Genes Cancer. 2011 Dec;2(12):1117–33. [CrossRef]
- Thapa D, Valente J de S, Barrett B, Smith MJ, Argunhan F, Lee SY, Nikitochkina S, Kodji X, Brain SD. Dysfunctional TRPM8 signalling in the vascular response to environmental cold in ageing. Mangoni AA, Barton M, Garami A, editors. eLife. 2021 Nov 2;10:e70153. [CrossRef]
- Arteel GE, Thurman RG, Raleigh JA. Reductive metabolism of the hypoxia marker pimonidazole is regulated by oxygen tension independent of the pyridine nucleotide redox state. Eur J Biochem. 1998 May 1;253(3):743–50. [CrossRef]
- Abhinand CS, Raju R, Soumya SJ, Arya PS, Sudhakaran PR. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J Cell Commun Signal. 2016 Dec;10(4):347–54.
- Wang X, Bove AM, Simone G, Ma B. Molecular Bases of VEGFR-2-Mediated Physiological Function and Pathological Role. Frontiers in Cell and Developmental Biology [Internet]. 2020;8. Available from: https://www.frontiersin.org/articles/10.3389/fcell.2020.599281. [CrossRef]
- Negri S, Faris P, Berra-Romani R, Guerra G, Moccia F. Endothelial Transient Receptor Potential Channels and Vascular Remodeling: Extracellular Ca2 + Entry for Angiogenesis, Arteriogenesis and Vasculogenesis. Frontiers in Physiology [Internet]. 2020;10. Available from: https://www.frontiersin.org/journals/physiology/articles/10.3389/fphys.2019.01618. [CrossRef]
- Vrenken KS, Jalink K, Leeuwen FN van, Middelbeek J. Beyond ion-conduction: Channel-dependent and -independent roles of TRP channels during development and tissue homeostasis. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2016;1863(6, Part B):1436–46. [CrossRef]
- Kim DY, Kim KM, Kim EJ, Jang WG. Hypothermia-induced RNA-binding motif protein 3 (RBM3) stimulates osteoblast differentiation via the ERK signaling pathway. Biochemical and Biophysical Research Communications. 2018 Apr 6;498(3):459–65. [CrossRef]
- Buccoliero C, Dicarlo M, Pignataro P, Gaccione F, Colucci S, Colaianni G, Grano M. The Novel Role of PGC1α in Bone Metabolism. Int J Mol Sci. 2021 Apr 28;22(9). [CrossRef]
- Shi H, Yao R, Lian S, Liu P, Liu Y, Yang YY, Yang H, Li S. Regulating glycolysis, the TLR4 signal pathway and expression of RBM3 in mouse liver in response to acute cold exposure. Stress. 2019 May;22(3):366–76. [CrossRef]
- Hang K, Ye C, Chen E, Zhang W, Xue D, Pan Z. Role of the heat shock protein family in bone metabolism. Cell Stress Chaperones. 2018 Nov;23(6):1153–64. [CrossRef]
- McGee-Lawrence ME, Carpio LR, Bradley EW, Dudakovic A, Lian JB, van Wijnen AJ, Kakar S, Hsu W, Westendorf JJ. Runx2 is required for early stages of endochondral bone formation but delays final stages of bone repair in Axin2-deficient mice. Bone. 2014 Sep;66:277–86. [CrossRef]
- Einhorn TA, Gundberg CM, Devlin VJ, Warman J. Fracture healing and osteocalcin metabolism in vitamin K deficiency. Clin Orthop Relat Res. 1988 Dec;(237):219–25. [CrossRef]
- Nie Y, Yan Z, Yan W, Xia Q, Zhang Y. Cold exposure stimulates lipid metabolism, induces inflammatory response in the adipose tissue of mice and promotes the osteogenic differentiation of BMMSCs via the p38 MAPK pathway in vitro. Int J Clin Exp Pathol. 2015;8(9):10875–86.
- Cheng Z, Li A, Tu CL, Maria CS, Szeto N, Herberger A, Chen TH, Song F, Wang J, Liu X, Shoback DM, Chang W. Calcium-Sensing Receptors in Chondrocytes and Osteoblasts Are Required for Callus Maturation and Fracture Healing in Mice. J Bone Miner Res. 2020 Jan;35(1):143–54. [CrossRef]
- Larson C, Opichka M, McGlynn ML, Collins CW, Slivka D. Exercise- and Cold-Induced Human PGC-1α mRNA Isoform Specific Responses. Int J Environ Res Public Health. 2020 Aug 8;17(16). [CrossRef]
- Zhang Z, Yang D, Xiang J, Zhou J, Cao H, Che Q, Bai Y, Guo J, Su Z. Non-shivering Thermogenesis Signalling Regulation and Potential Therapeutic Applications of Brown Adipose Tissue. Int J Biol Sci. 2021;17(11):2853–70. [CrossRef]
- Wang H, Wang J. Estrogen-related receptor alpha interacts cooperatively with peroxisome proliferator-activated receptor-gamma coactivator-1alpha to regulate osteocalcin gene expression. Cell Biol Int. 2013 Nov;37(11):1259–65. [CrossRef]
- McKee MD, Pedraza CE, Kaartinen MT. Osteopontin and wound healing in bone. Cells Tissues Organs. 2011;194(2–4):313–9. [CrossRef]
- Boonrungsiman S, Gentleman E, Carzaniga R, Evans ND, McComb DW, Porter AE, Stevens MM. The role of intracellular calcium phosphate in osteoblast-mediated bone apatite formation. Proceedings of the National Academy of Sciences. 2012 Aug 28;109(35):14170–5. [CrossRef]
- Abe Y, Fujiwara Y, Takahashi H, Matsumura Y, Sawada T, Jiang S, Nakaki R, Uchida A, Nagao N, Naito M, Kajimura S, Kimura H, Osborne TF, Aburatani H, Kodama T, Inagaki T, Sakai J. Histone demethylase JMJD1A coordinates acute and chronic adaptation to cold stress via thermogenic phospho-switch. Nature Communications. 2018 Apr 19;9(1):1566. [CrossRef]
- Casali C, Galgano L, Zannino L, Siciliani S, Cavallo M, Mazzini G, Biggiogera M. Impact of heat and cold shock on epigenetics and chromatin structure. European Journal of Cell Biology. 2024 Mar 1;103(1):151373. [CrossRef]
- Li JY, Wang TT, Ma L, Zhang Y, Zhu D. Silencing of Jumonji domain-containing 1C inhibits the osteogenic differentiation of bone marrow mesenchymal stem cells via nuclear factor-κB signaling. World J Stem Cells. 2024 Feb 26;16(2):151–62.
- Zeng ZL, Xie H. Mesenchymal stem cell-derived extracellular vesicles: a possible therapeutic strategy for orthopaedic diseases: a narrative review. Biomater Transl. 2022;3(3):175–87. [CrossRef]
- Wang Q. Biomaterials Translational - The New Vehicle for Translational Medicine. Biomater Transl. 2020;1(1):1–2.
- Zhang C, Cai D, Liao P, Su JW, Deng H, Vardhanabhuti B, Ulery BD, Chen SY, Lin J. 4D Printing of shape-memory polymeric scaffolds for adaptive biomedical implantation. Acta Biomaterialia. 2021 Mar 1;122:101–10. [CrossRef]
- El-Husseiny HM, Mady EA, Hamabe L, Abugomaa A, Shimada K, Yoshida T, Tanaka T, Yokoi A, Elbadawy M, Tanaka R. Smart/stimuli-responsive hydrogels: Cutting-edge platforms for tissue engineering and other biomedical applications. Materials Today Bio. 2022 Jan 1;13:100186. [CrossRef]
- Cheng Y, Yu Y, Zhang Y, Zhao G, Zhao Y. Cold-Responsive Nanocapsules Enable the Sole-Cryoprotectant-Trehalose Cryopreservation of β Cell-Laden Hydrogels for Diabetes Treatment. Small. 2019 Dec;15(50):e1904290.
- Molkenova A, Choi HE, Lee G, Baek H, Kwon M, Lee SB, Park JM, Kim JH, Han DW, Park J, Hahn SK, Kim KS. Cold-Responsive Hyaluronated Upconversion Nanoplatform for Transdermal Cryo-Photodynamic Cancer Therapy. Advanced Science. 2024 May 1;11(19):2306684. [CrossRef]
- Kleiter MM, Thrall DE, Malarkey DE, Ji X, Lee DYW, Chou SC, Raleigh JA. A comparison of oral and intravenous pimonidazole in canine tumors using intravenous CCI-103F as a control hypoxia marker. Int J Radiat Oncol Biol Phys. 2006 Feb 1;64(2):592–602. [CrossRef]
Figure 1.
Potential mechanisms activated in response to cold exposure.
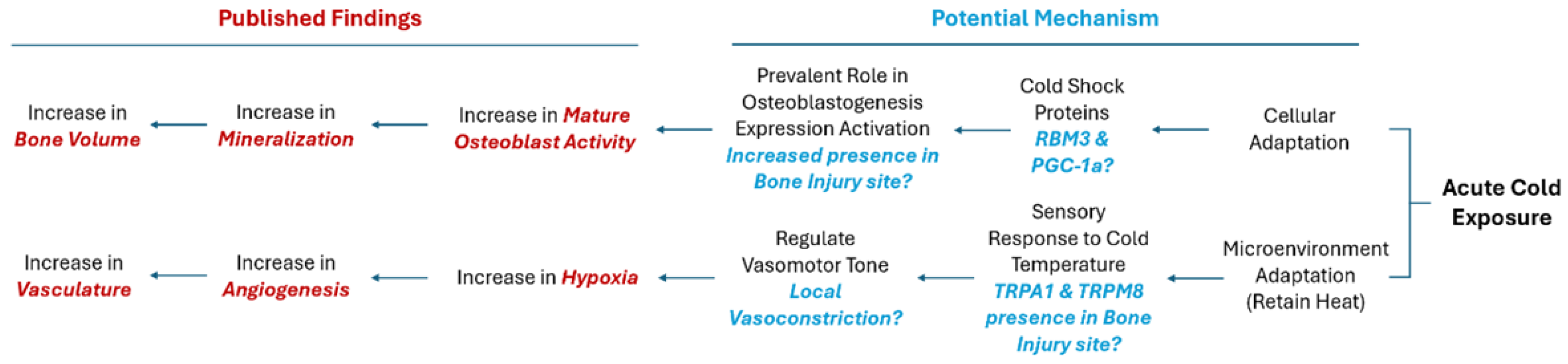
Figure 2.
Identification of receptor proteins involved in vasoconstriction within a cortical defect following cold exposure. TRPM8 and TRPA1 detection (bright green) within the cortical defect region (4x magnification). Scale bar represents 500 µm. A, D) Non-treated femurs serving as baseline. B, E) Cold treated femurs. C) TRPA1 Staining Analysis Expression (n=8) of positive staining for TRPA1 in the control group was 5.38 ± 2.32 while in the experimental group it was 9.03% ± 2.78 (*p-value = 0.012). F) TRPM8 Staining Analysis Expression (n=8) of positive staining for TRPM8 in the control group was 3.86% ± 1.72 while in the experimental group it was 8.75% ± 1.68 (**p-value < 0.001).
Figure 2.
Identification of receptor proteins involved in vasoconstriction within a cortical defect following cold exposure. TRPM8 and TRPA1 detection (bright green) within the cortical defect region (4x magnification). Scale bar represents 500 µm. A, D) Non-treated femurs serving as baseline. B, E) Cold treated femurs. C) TRPA1 Staining Analysis Expression (n=8) of positive staining for TRPA1 in the control group was 5.38 ± 2.32 while in the experimental group it was 9.03% ± 2.78 (*p-value = 0.012). F) TRPM8 Staining Analysis Expression (n=8) of positive staining for TRPM8 in the control group was 3.86% ± 1.72 while in the experimental group it was 8.75% ± 1.68 (**p-value < 0.001).
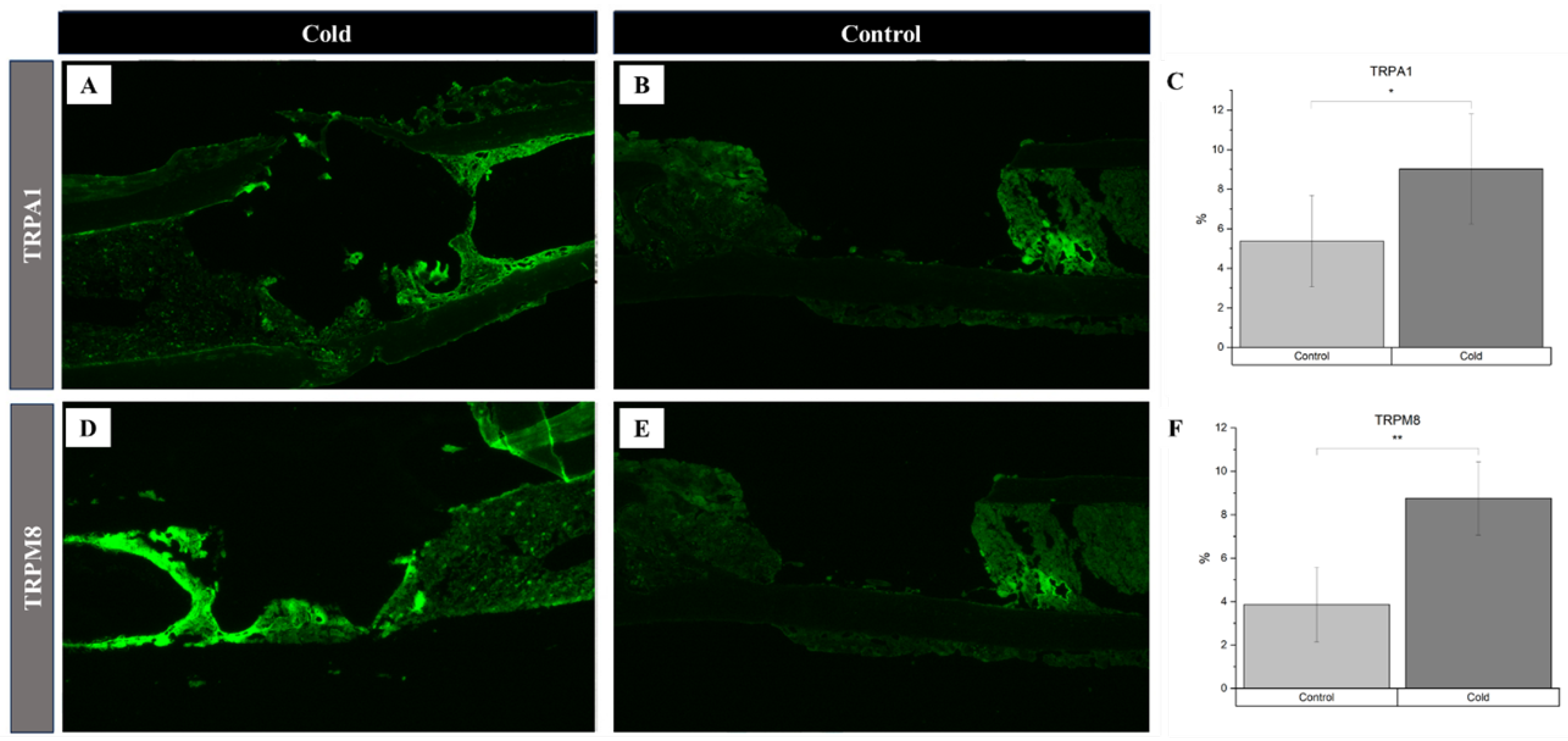
Figure 3.
Identification of angiogenic related factors and endothelial cells within a cortical defect following cold exposure. VEGF and CD34 (bright green) detection within the cortical defect region (4x magnification). Scale bar represents 500 µm. A,D,G) Cold treated femurs. B,E,H) Non-treated femurs serving as baseline C) Hypoxia Staining Analysis Expression (n=10) of positive staining for hypoxia in the control group was 17.9% ± 3.8 while in the experimental group it was 23.5% ± 05.8 (*p-value <0.01). F) VEGF Staining Analysis Expression (n=8) of positive staining for VEGF in the control group was 4.05% ± 1.83 while in the experimental group it was 9.69% ± 1.75 (*p-value<0.001). I) CD34 Staining Analysis Expression (n=8) of positive staining for CD34 in the control group was 6.92% ± 2.77 while in the experimental group it was 8.00% ± 2.76 (p-value = 0.42).
Figure 3.
Identification of angiogenic related factors and endothelial cells within a cortical defect following cold exposure. VEGF and CD34 (bright green) detection within the cortical defect region (4x magnification). Scale bar represents 500 µm. A,D,G) Cold treated femurs. B,E,H) Non-treated femurs serving as baseline C) Hypoxia Staining Analysis Expression (n=10) of positive staining for hypoxia in the control group was 17.9% ± 3.8 while in the experimental group it was 23.5% ± 05.8 (*p-value <0.01). F) VEGF Staining Analysis Expression (n=8) of positive staining for VEGF in the control group was 4.05% ± 1.83 while in the experimental group it was 9.69% ± 1.75 (*p-value<0.001). I) CD34 Staining Analysis Expression (n=8) of positive staining for CD34 in the control group was 6.92% ± 2.77 while in the experimental group it was 8.00% ± 2.76 (p-value = 0.42).
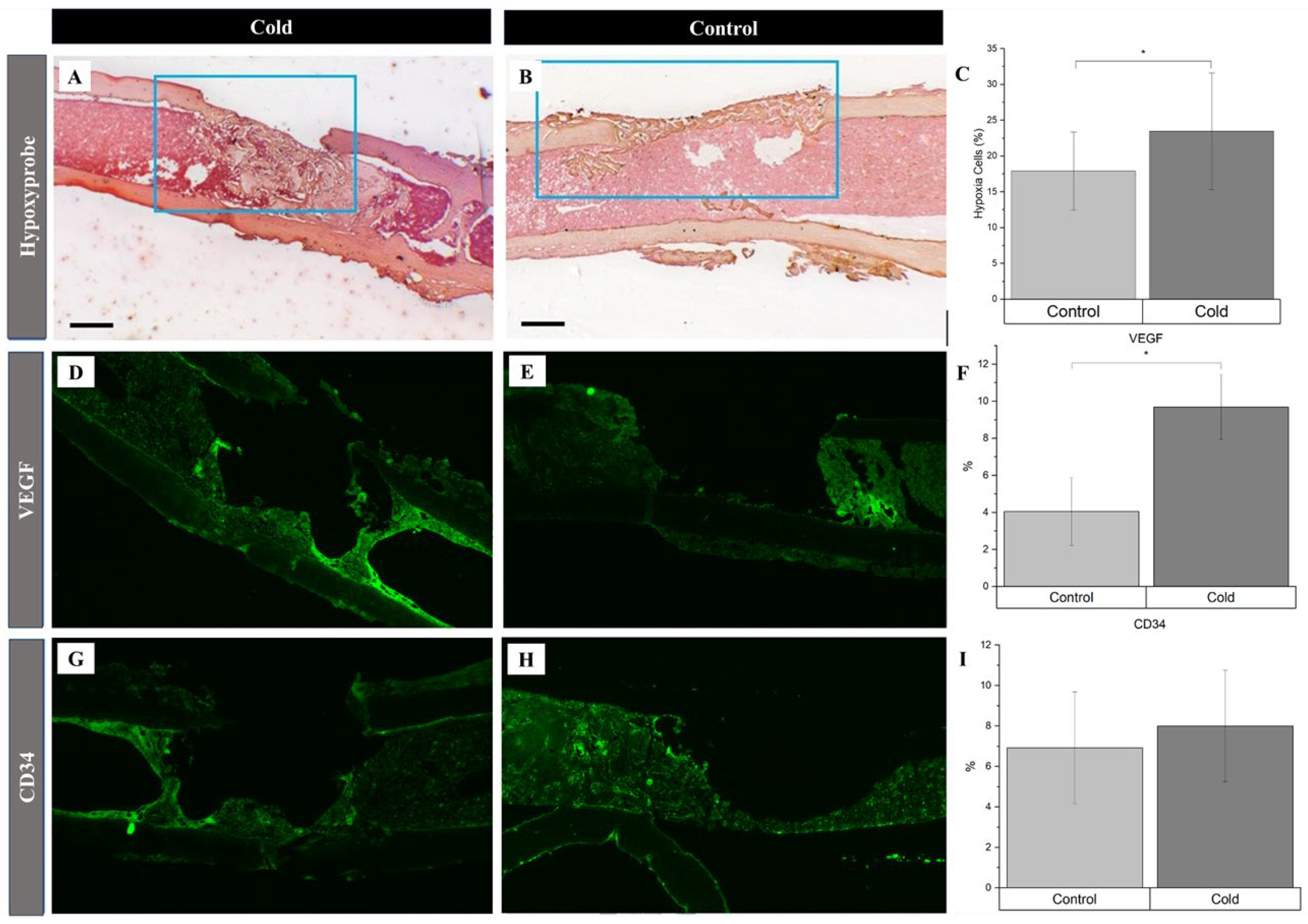
Figure 4.
Identification of cold shock proteins and hypoxia in regenerating bone following cold exposure. PGC-1a (brown), RBM3 (brown), and hypoxia (brown) detection within the cortical defect region (4x magnification). Scale bar represents 500 µm. (A, D) Cold treated femurs. B, E) Non-treated femurs serving as baseline C) RBM3 Staining Analysis Expression (n=8) of positive staining for RBM3 in the control group was 4.47% ± 2.84 while in the experimental group it was 11.0% ± 4.26 (**p-value<0.008). F) PGC-1a Staining Analysis Expression (n=8) of positive staining for PGC-1a in the control group was 2.57% ± 0.36 while in the experimental group it was 5.85% ± 1.01 (*p-value = 0.039).
Figure 4.
Identification of cold shock proteins and hypoxia in regenerating bone following cold exposure. PGC-1a (brown), RBM3 (brown), and hypoxia (brown) detection within the cortical defect region (4x magnification). Scale bar represents 500 µm. (A, D) Cold treated femurs. B, E) Non-treated femurs serving as baseline C) RBM3 Staining Analysis Expression (n=8) of positive staining for RBM3 in the control group was 4.47% ± 2.84 while in the experimental group it was 11.0% ± 4.26 (**p-value<0.008). F) PGC-1a Staining Analysis Expression (n=8) of positive staining for PGC-1a in the control group was 2.57% ± 0.36 while in the experimental group it was 5.85% ± 1.01 (*p-value = 0.039).
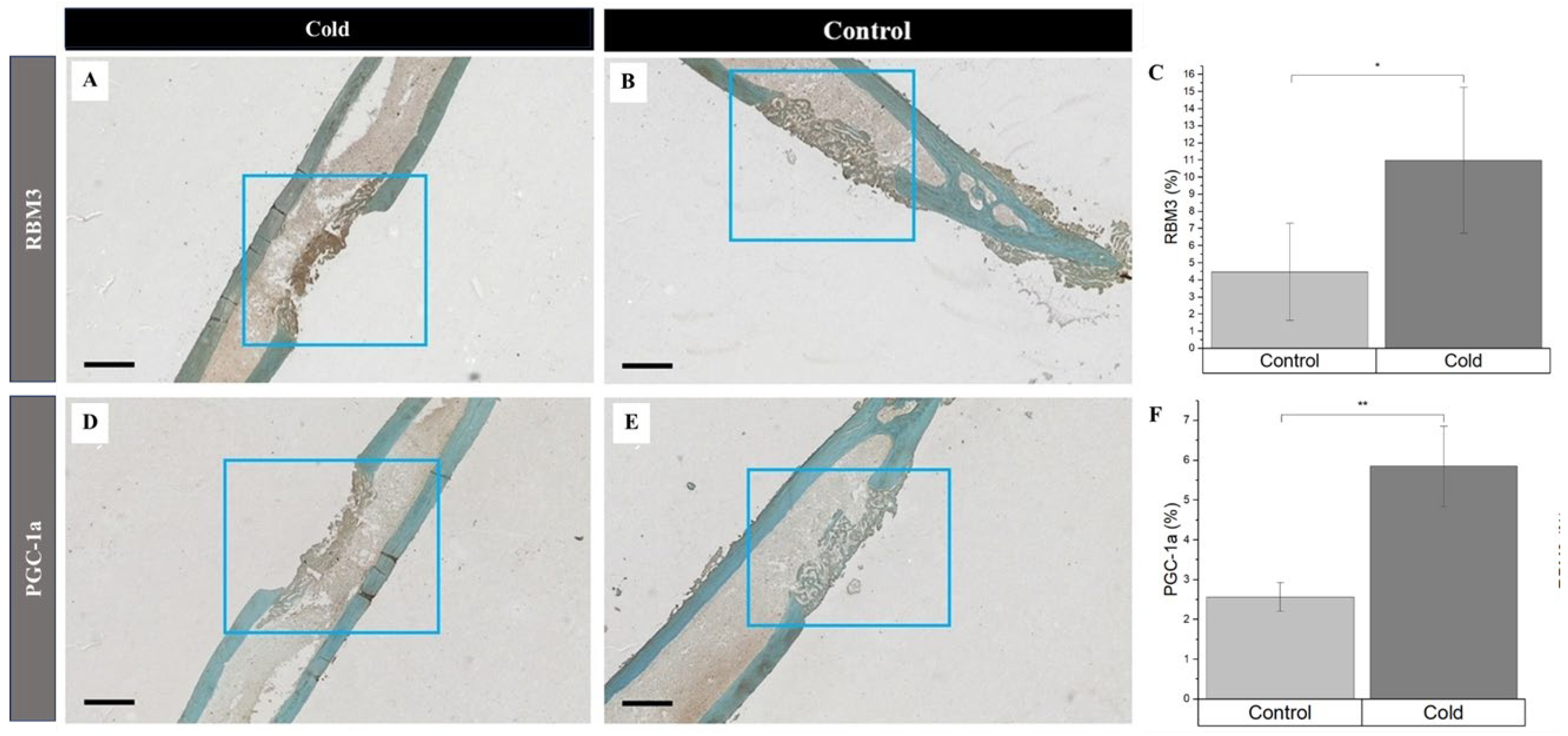
Figure 5.
Prevalence of PGC-1a at the junction of old and new bone formation following cold therapy treatment. A, E) 2.5x Magnification. Scale bar represents 500 µm. B-D, F-H) 20x Magnification. Scale Bar represents 50 µm. PGC-1a has been shown to be upregulated (I) at the junction of new and old bone (F-H) Furthermore, PGC-1a expression within these junctions appears to be elevated following cold exposure (B-D) indicating PGC-1a upregulation. I) Detected location of PGC-1α Staining Analysis Expression (n=8) of PGC-1α within cells at the junction of newly formed bone and old bone was 70.56% ± 7.08 while in non-junctional regions it was 29.44% ± 7.08 (*p-value <0.00001).
Figure 5.
Prevalence of PGC-1a at the junction of old and new bone formation following cold therapy treatment. A, E) 2.5x Magnification. Scale bar represents 500 µm. B-D, F-H) 20x Magnification. Scale Bar represents 50 µm. PGC-1a has been shown to be upregulated (I) at the junction of new and old bone (F-H) Furthermore, PGC-1a expression within these junctions appears to be elevated following cold exposure (B-D) indicating PGC-1a upregulation. I) Detected location of PGC-1α Staining Analysis Expression (n=8) of PGC-1α within cells at the junction of newly formed bone and old bone was 70.56% ± 7.08 while in non-junctional regions it was 29.44% ± 7.08 (*p-value <0.00001).
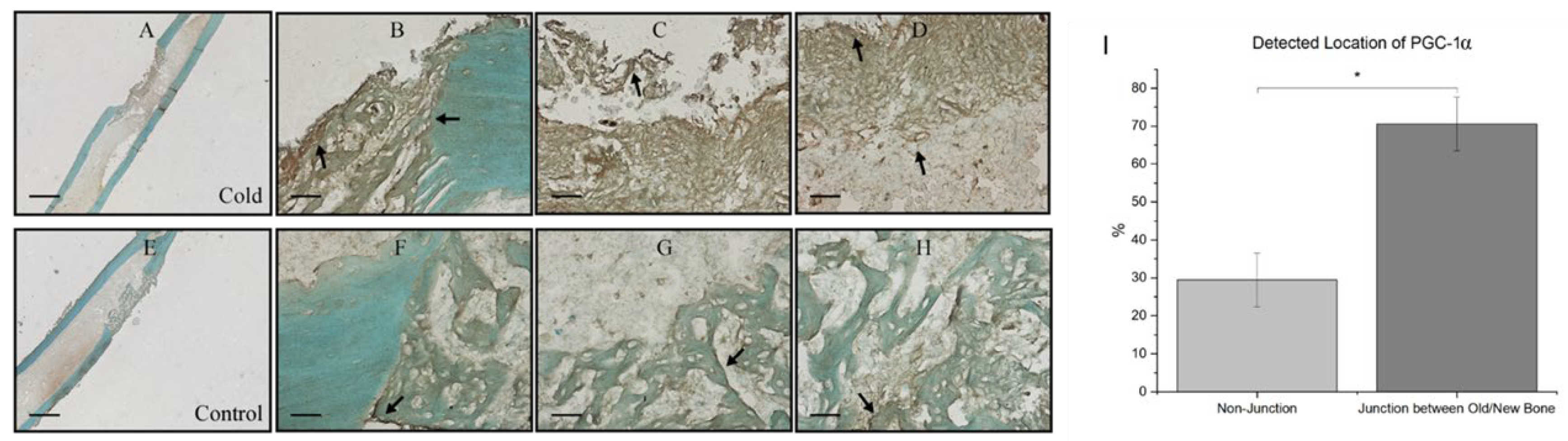
Figure 6.
Detection of ALP and OCN at Day 5 and Day 10 within isolate osteoblasts following cold exposure and mineralization within isolate osteoblasts following cold exposure for 14 days. A) ALP Activity after 5 days of cold exposure was 2.72 U/L (µmol/min/L) ± 0.85 while in the non-treated group it was 0.74 U/L ± 0.35 (*p-value = 0.021). B) ALP Activity after 10 days of cold exposure was 2.25 U/L ± 0.04 while in the non-treated group it was 1.0301 U/L ± 0.24 (**p-value < 0.001) C) OCN levels after 5 days of cold exposure was 0.34 mg/mL ± 0.10 while in the non-treated group it was 0.43 mg/mL ± 0.10 (p-value = 0.29) D) OCN levels after 10 days of cold exposure was 0.84 mg/mL ± 0.17 while in the non-treated group it was 0.35 mg/mL ± 0.10 (***p-value = 0.019) ARS (red) detection within isolate osteoblasts (10x magnification). Scale bar represents 250 µm. E) Osteoblasts isolated from non-treated fractured femurs. F) Osteoblasts isolated from daily cold-treated fractured femurs. G) ARS Staining Analysis: Detection of ARS after 14 days of cold exposure was 1.04% ± 0.40 while in the non-treated group it was 0.27% ± 0.025 (***p-value = 0.030).
Figure 6.
Detection of ALP and OCN at Day 5 and Day 10 within isolate osteoblasts following cold exposure and mineralization within isolate osteoblasts following cold exposure for 14 days. A) ALP Activity after 5 days of cold exposure was 2.72 U/L (µmol/min/L) ± 0.85 while in the non-treated group it was 0.74 U/L ± 0.35 (*p-value = 0.021). B) ALP Activity after 10 days of cold exposure was 2.25 U/L ± 0.04 while in the non-treated group it was 1.0301 U/L ± 0.24 (**p-value < 0.001) C) OCN levels after 5 days of cold exposure was 0.34 mg/mL ± 0.10 while in the non-treated group it was 0.43 mg/mL ± 0.10 (p-value = 0.29) D) OCN levels after 10 days of cold exposure was 0.84 mg/mL ± 0.17 while in the non-treated group it was 0.35 mg/mL ± 0.10 (***p-value = 0.019) ARS (red) detection within isolate osteoblasts (10x magnification). Scale bar represents 250 µm. E) Osteoblasts isolated from non-treated fractured femurs. F) Osteoblasts isolated from daily cold-treated fractured femurs. G) ARS Staining Analysis: Detection of ARS after 14 days of cold exposure was 1.04% ± 0.40 while in the non-treated group it was 0.27% ± 0.025 (***p-value = 0.030).
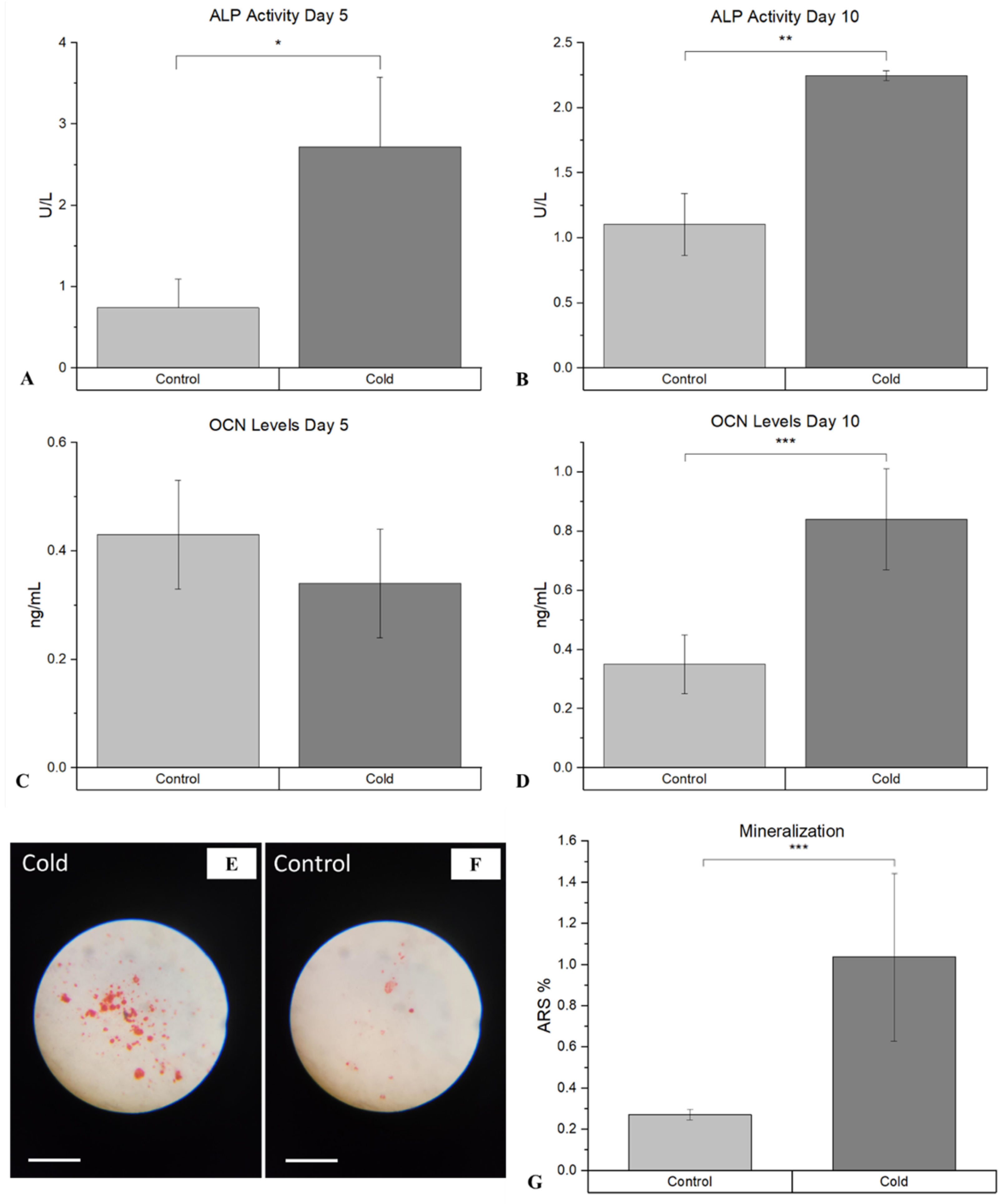
Figure 7.
Cold based activation of vasoconstrictive pathways and hypoxic impact. A & B) TRPA1 and TRPM8 Mechanism Cold exposure leads to activation of TRPM8 (8 - 28°C) and TRPA1 (8 - 17°C) initiating a contractive response leading to vasoconstriction. C) Hypoxic Impacts. A reduced influx of blood supply coincides with a decrease in oxygen availability leading to the formation of an acute hypoxic microenvironment with respect to nearby cells. NA: Norepinephrine.
Figure 7.
Cold based activation of vasoconstrictive pathways and hypoxic impact. A & B) TRPA1 and TRPM8 Mechanism Cold exposure leads to activation of TRPM8 (8 - 28°C) and TRPA1 (8 - 17°C) initiating a contractive response leading to vasoconstriction. C) Hypoxic Impacts. A reduced influx of blood supply coincides with a decrease in oxygen availability leading to the formation of an acute hypoxic microenvironment with respect to nearby cells. NA: Norepinephrine.
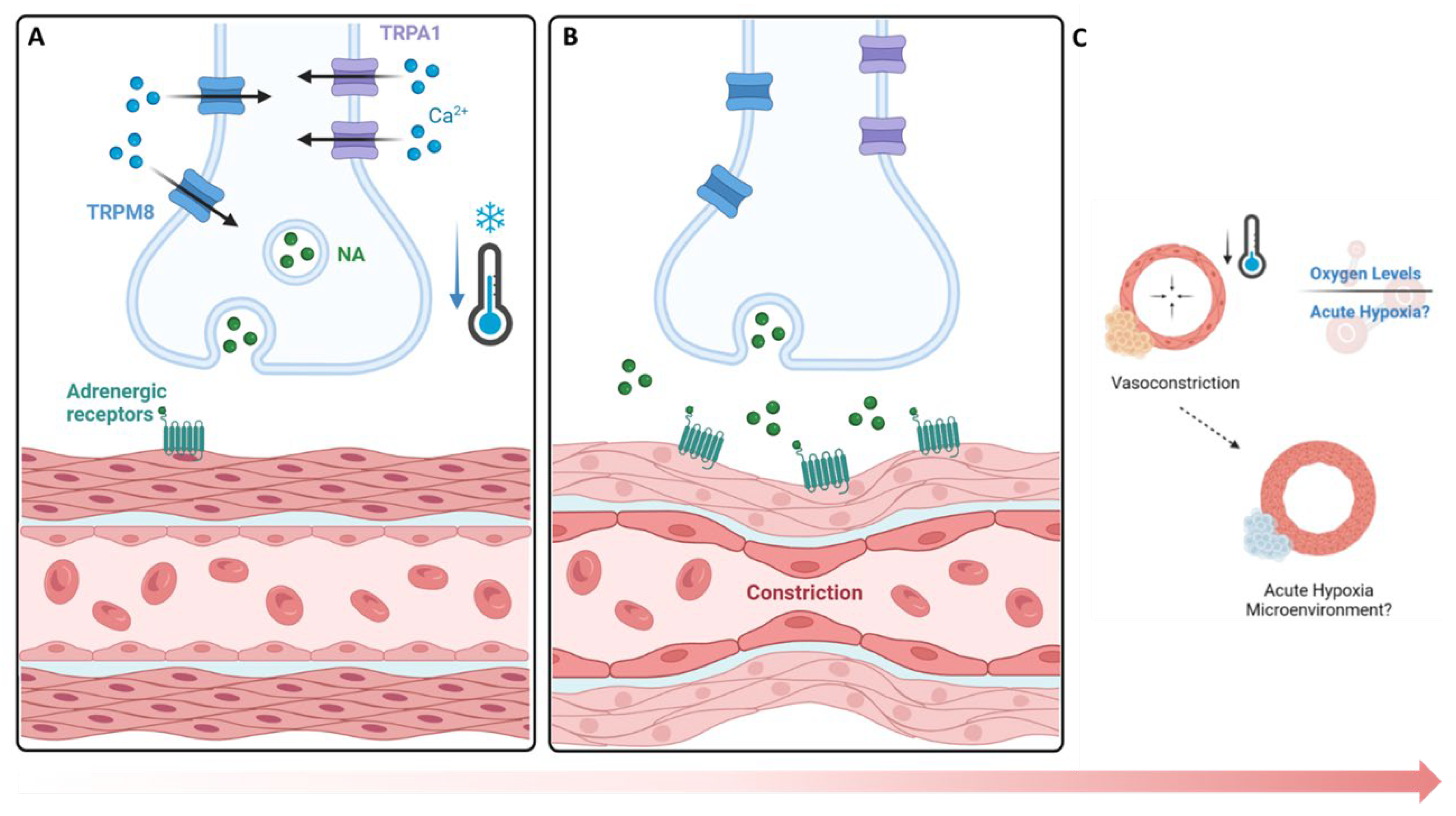
Figure 8.
Hypoxic upregulation following vasoconstriction via local cold exposure and impact on angiogenic pathways. Under normal oxygen conditions, HIF-1a is degraded via von Hippel–Lindau (VHL) after prolyl hydroxylation, but in hypoxia, HIF-1a forms a dimer with HIF-1B, nucleolocalizes with aryl hydrocarbon receptor nuclear translocator (ARNT), leading to VEGF expression through the HRE pathway, which then activates VEGFR2 on endothelial cells, upregulating cellular proliferation and angiogenesis via the Ras/Raf signaling pathway.
Figure 8.
Hypoxic upregulation following vasoconstriction via local cold exposure and impact on angiogenic pathways. Under normal oxygen conditions, HIF-1a is degraded via von Hippel–Lindau (VHL) after prolyl hydroxylation, but in hypoxia, HIF-1a forms a dimer with HIF-1B, nucleolocalizes with aryl hydrocarbon receptor nuclear translocator (ARNT), leading to VEGF expression through the HRE pathway, which then activates VEGFR2 on endothelial cells, upregulating cellular proliferation and angiogenesis via the Ras/Raf signaling pathway.
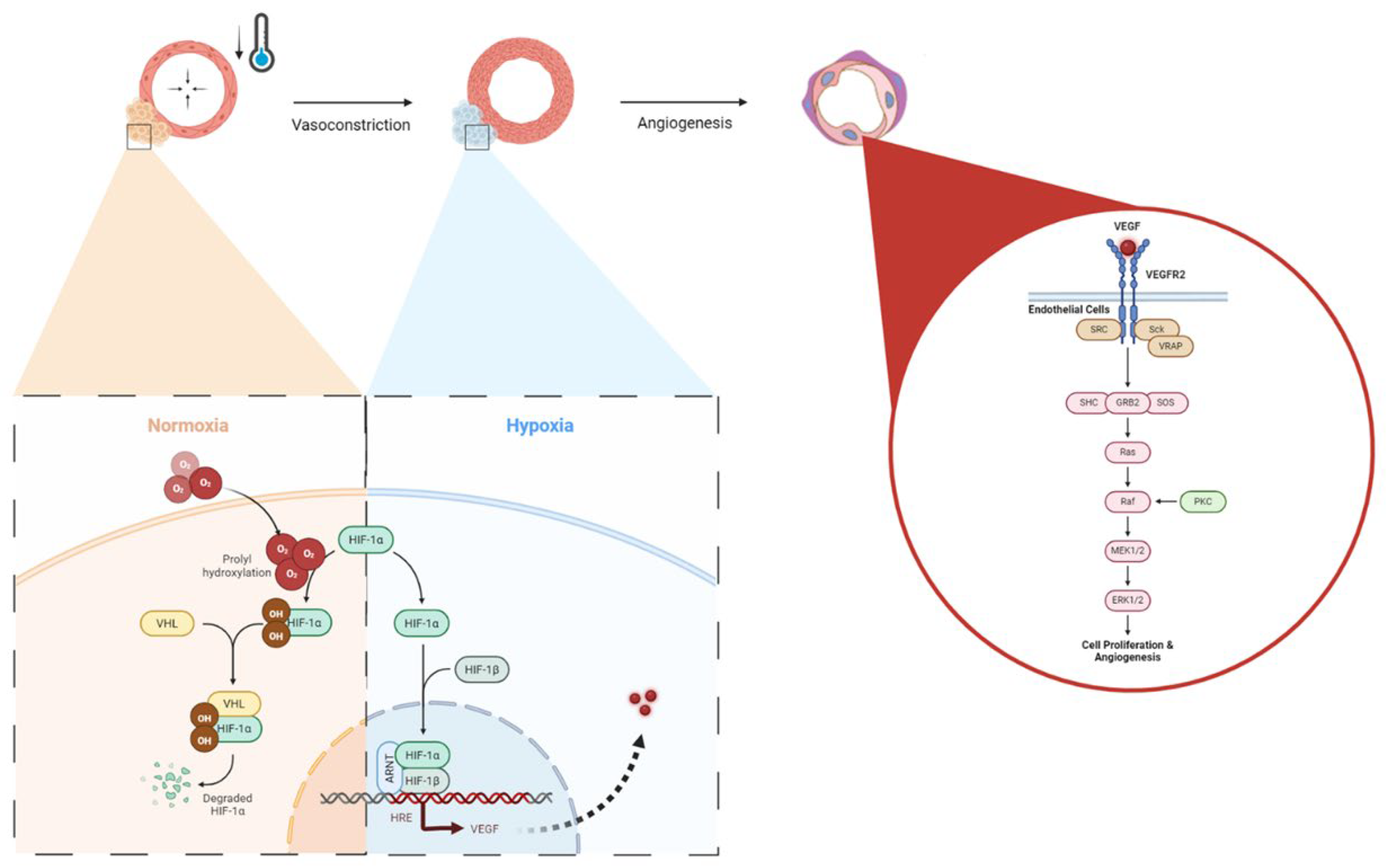
Figure 9.
Cellular Adaptive Signaling Pathways activated by Cold Exposure.RBM3 activation and downstream osteogenic potential following short term cold exposure. A) Cold exposure triggers a cellular cold shock response, activating RBM3 via HSPs released by phosphorylated and nuclear-localized HSF1. B) HSP70 activates TLR4, initiating the MyD88/NF-kB pathway, with phosphorylated NF-kB essential for RBM3 expression. C) RBM3 activates the MAPK/ERK pathway, leading to runx2 and osteocalcin (OCN) expression, crucial for osteoblast differentiation.PGC-1a activation and downstream osteogenic potential following short term cold exposure. PGC-1α Activation and Osteogenic Potential Following Short Term Cold Exposure D) Cold activation of adrenergic receptors and TRPs stimulates cAMP and calcium influx, leading to CREB phosphorylation via the PKA/Ca2+/CaMK pathway. E) PGC-1α forms a complex with ERRα in osteoblastic cells, promoting differentiation via the RUNX2/OCN pathway for mineralization. F) PGC-1α and NRF-1/NRF-2 activate TFAM and NCMP, crucial for mitochondrial biogenesis and intracellular calcium storage.
Figure 9.
Cellular Adaptive Signaling Pathways activated by Cold Exposure.RBM3 activation and downstream osteogenic potential following short term cold exposure. A) Cold exposure triggers a cellular cold shock response, activating RBM3 via HSPs released by phosphorylated and nuclear-localized HSF1. B) HSP70 activates TLR4, initiating the MyD88/NF-kB pathway, with phosphorylated NF-kB essential for RBM3 expression. C) RBM3 activates the MAPK/ERK pathway, leading to runx2 and osteocalcin (OCN) expression, crucial for osteoblast differentiation.PGC-1a activation and downstream osteogenic potential following short term cold exposure. PGC-1α Activation and Osteogenic Potential Following Short Term Cold Exposure D) Cold activation of adrenergic receptors and TRPs stimulates cAMP and calcium influx, leading to CREB phosphorylation via the PKA/Ca2+/CaMK pathway. E) PGC-1α forms a complex with ERRα in osteoblastic cells, promoting differentiation via the RUNX2/OCN pathway for mineralization. F) PGC-1α and NRF-1/NRF-2 activate TFAM and NCMP, crucial for mitochondrial biogenesis and intracellular calcium storage.
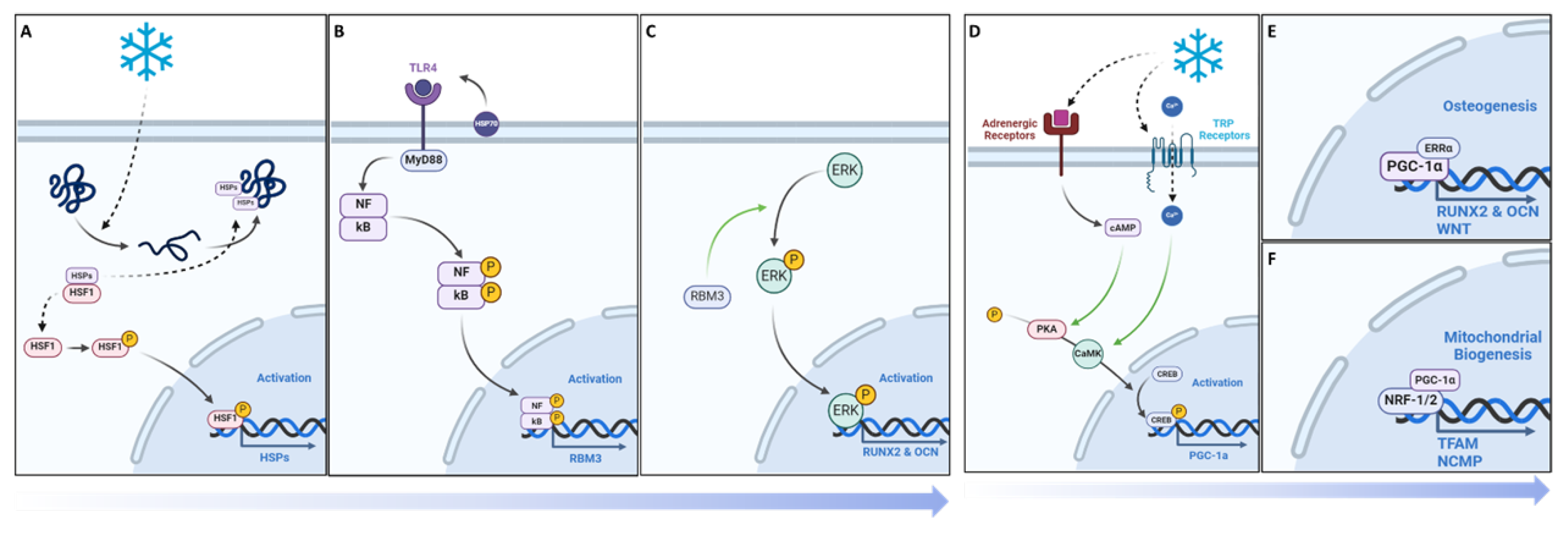
Figure 10.
Overview of the mechanistic propensities of short duration cold exposure and potential impacts in a bone injury site. Blue: Observed impacts of cold exposure on bone and vasculature morphology Green: Observed impacts of cold exposure on pathways involved in bone repair.
Figure 10.
Overview of the mechanistic propensities of short duration cold exposure and potential impacts in a bone injury site. Blue: Observed impacts of cold exposure on bone and vasculature morphology Green: Observed impacts of cold exposure on pathways involved in bone repair.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



