
Submitted:
29 August 2024
Posted:
30 August 2024
You are already at the latest version
Abstract
Magnaporthe oryzae is a filamentous heterothallic ascomycete fungus globally distributed in rice-growing regions and serves as the causative agent of rice blast disease. Populations shaped by environmental factors and human intervention play important roles in the formation of genetic structure. In this study, population structures and spatiotemporal dynamics were investigated based on the large-scale whole genomic sequences of rice-infecting M. oryzae around the world. By analyzing the genetic structures, we identified divergent clades that crossed geographic boundaries. While we observed associations between the isolates and their geographic origins, we also found that there were frequent migration events occurring worldwide. The populations in Asia demonstrated the highest genetic diversity due to the continent’s history of rice domestication, followed by separate gene flows to Africa, North America, South America and Europe. Within Asia, China was the migration origin, facilitating gene flows to Japan and South Korea. Additionally, our analysis of the evolutionary history of global M. oryzae populations provided insights into the population expansion that has taken place in recent decades. Overall, our findings indicate that human-mediated gene flows played a pivotal role in shaping the genetic structure of M. oryzae.
Keywords:
Magnaporthe oryzae
; population structure
; genetic diversity
; gene flow
1. Introduction
Rice blast is a devastating disease that occurs in rice-growing areas worldwide, it is caused by the filamentous ascomycete fungus Magnaporthe oryzae and poses a serious threat to global food security [1]. M. oryzae can rapidly overcome the resistance genes in rice and can coexist with resistant varieties within a few years of their initial deployment in rice agrosystems [2,3]. The pathogen infects rice throughout the growth period and also has a wide host range that includes more than 50 cultivated and wild monocot plants, such as rice (Oryza sativa), barley (Hordeum vulgare), wheat (Triticum aestivum), finger millet (Eleusine coracana), goosegrass (Eleusine indica), perennial ryegrass (Lolium perenne) and more [4]. The asexual reproduction is prevalent in most rice fields, resulting in local populations of M. oryzae often exhibiting only one mating type[5,6,7,8,9].
It is necessary to better understand the features of the genetic structure of M. oryzae in order to provide the genetic basis for developing sustainable and effective prevention and control strategies for rice blast disease [10]. Genetic diversity can reflect the survival ability and adaptive potential of natural populations in the face of rapidly changing biotic and abiotic backgrounds [11]. For the plant pathogen M. oryzae, genetic diversity is usually estimated using molecular markers, including microsatellite markers, or DNA fingerprinting via amplified fragment length polymorphism. These methods have been applied in several countries and continents, including China [12,13,14], India [15,16], Thailand [17], the Philippines [18], Africa [19], Europe [20] and America [21], and have revealed regional diversity variations underling local adaptations [22]. The establishment of genetic structures can be driven by geographic isolation and ecological factors. In a previous population genomic analysis, three main genetic clades were identified in M. oryzae, which were associated with the distribution of mating types [23,24]. In a worldwide population structure analysis of M. oryzae, multiple endemic and pandemic lineages were identified, which were distributed in specific rice-growing areas [25]. Based on the amplified fragment length polymorphism, very few genetic differences have been found between the geographically distant populations of M. oryzae in Iran and Uruguay, although evidence of gene flow has been observed [26]. As a broad host pathogen, host specialization has also been reported to be a non-negligible factor in the genetic differentiation of M. oryzae [27,28]. However, the limited genetic markers have hindered population genetic analysis from accurately identifying the comprehensive ensemble population structures of rice-infecting lineages due to insufficient information regarding the incomplete divergent populations. In particular, there remains a scarcity of studies addressing the spatiotemporal dynamics of genetic diversity and population structures of M. oryzae through large-scale genomic analysis.
Gene flow, or migration, implies the movement of genetic material among spatially or temporally separated populations and acts as a major driving force for organisms to establish population structures. It can accelerate novel variations through gene recombination, migration to found new populations and takeovers of other local populations. Island [29] and stepping-stone models [30] have indicated that the migration of one or more subpopulations can decrease the genetic correlation with geographical distance among sexually reproducing species. For plant pathogens, spatial movement occurs in many forms, such as short-distance transfer through rainwater and long-distance global transfer, which counteracts the disadvantage of immobility for large-scale epidemics. Population structures are generally established by genetic variations and natural factors, as well as anthropogenic changes, especially among crop pathogens that are closely linked to human survival. Therefore, human movement significantly influences the migration of plant pathogens [31,32].
In agricultural ecosystems, human movement can aid the extensive spread of plant pathogens by breaking inherent mobility limitations or geographic barriers, such as rivers and mountains, that can restrict population expansion, thereby contributing to the complexity of population structures. Therefore, the globalization of agricultural products, largely driven by human activities, has led plant pathogens to evolve toward metapopulation formation [31]. For example, the transportation of infected seeds is one of that ways that M. oryzae has migrated, demystifying the close genetic relationships that have arisen between geographically separate populations [33]. Through the genome sequence analysis of global M. oryzae populations over different time periods, a sexual recombination signature was detected in the Southeast Asian endemic lineage, suggesting the occurrence of gene flows among geographic population distributions [25]. Based on microsatellite markers, M. oryzae has been found to have weak geographic structures in three islands groups with limited natural migration, which was induced by the transportation of infected seeds around the Philippines [18]. Based on amplified fragment length polymorphism analysis, frequent gene flows have been discovered in East African populations [34], as well as between the different provinces in Korea [35]. This has also occurred with other plant viruses; for example, turnip mosaic potyvirus spread from west to east regions in Eurasia due to historical trade arteries, such as the Silk Road [36].
In this study, we first collected the published whole-genome sequences of 189 rice-infecting M. oryzae isolates from five continents and 22 re-sequenced genomes from China. Then, we analyzed the genetic diversity and population structure characteristics to evaluate the population divergence driven by spatiotemporal changes. We found that a M. oryzae population from Asia exhibited the highest genetic diversity and could be divided into three divergent clades. The diffusion route of M. oryzae followed human activity, suggesting that the Asian population served as the genetic pool for rice-growing regions worldwide, with China being the migration origin within Asia. Our study provides a detailed understanding of the spatiotemporal dynamics of the genetic diversity and population structures, which could be useful for developing cultivars with different resistant genes and could contribute new insights into disease management.
2. Materials and Methods
2.1. Isolate Sampling, DNA Preparation, Genome Sequencing and Published Genome Collection
We collected 22 rice-infecting M. oryzae isolates from rice fields in China. The sampling locations are described in Table S1. Genomic DNA was extracted from fresh mycelium cultures after single-spore isolation, as previous described [37]. An Illumina paired-end DNA sample prep kit was employed to construct the Illumina fragment libraries and an Illumina HiSeq2500 instrument was used for the sequencing.
A total of 189 M. oryzae genomes have been sequenced from rice, which we have downloaded from the NCBI database using the ncbi-genome-download script (https://github.com/kblin/ncbi-genome-download). The genome information is also listed in Table S1.
2.2. Identification of Single-Nucleotide Polymorphism
To obtain high-quality single-nucleotide polymorphism sites from the sequencing reads, the raw data were first trimmed using Trimmomatic software [38]. The eighth version genome of 70-15 was taken as the reference genome and the paired-end reads were aligned against it using the BWA v0.7.17 as the default parameters [39,40]. Then, single-nucleotide polymorphism (SNP) mining was implemented using the Genome Analysis Toolkit (GATK v4.1.4.1), which compared the reference genome to the standard settings [41]. After that, VCFtools was used to filter the biallelic site with minor allele count smaller than 3 and missing data lower than 20% [42].
For the genomes downloaded from the NCBI database, genome-to-genome SNP identification was performed using the -maxmatch and -c 100 options in MUMMer [43] against the reference genome.
2.3. Characterization of Genetic Diversity and Population Genetic Structures
To estimate the genetic diversity of M. oryzae populations that were divided by spatiotemporal boundaries, the nucleotide diversity of the whole-genome SNPs was calculated using the “PopGenome” v2.1.6 package with a 20 kb sliding window and 2 kb step size [44]. The statistical significance between each pair of geographical populations was estimated using Student’s t-test (two-tailed). A SNP-based neighbor-joining phylogenetic tree was reconstructed according to the bitwise distance with 1000 bootstrap replicates using the “poppr” packages in R [45]. A principal component analysis (PCA) of the genetic structures of global populations from Asia, Europe, North America and South America was implemented on the matrix of binary allele sites using the “adegenet” package [46].
To establish the population structure compositions, the SNP dataset was pruned based on linkage disequilibrium (LD) with a value of r2 = 0.5 using Plink v.1.9 (https://www.cog-genomics.org/plink/). To evaluate the individual ancestry components and admixture proportions, a population admixture analysis was performed using the ADMIXTURE v1.3.0 program [47]. In ADMIXTURE, 5-fold and 10-fold cross-validation procedures with K values between 2 and 10 were implemented to identify the optimal number of population clusters (K) with the lowest cross-validation error values. The admixture proportions of each sample from the geographic populations were visualized using PopHelper [48].
2.4. Phylogeographic Analysis of M. oryzae
To detect the reticulation events of the populations, such as hybridization, horizontal gene transfer and recombination, the phylogenetic networks were reconstructed using the SplitsTree algorithm [49]. In order to assess the robustness of the tree topology, 1000 bootstrap replicates were performed using the “Neighbor-Net” and “Uncorrected p-distance” parameters.
To ascertain the phylogeographical dispersal routes (including migration routes and directionality) of M. oryzae across the world and in Asia (four regions), we employed the asymmetric discrete trait substitution model with the Bayesian stochastic search variable selection approach [50] using BEAST 1.10.4 [51]. Based on the Bayesian information criterion, the best nucleotide substitution model was estimated using jModeltest [52] after converting the SNP dataset into the Phylip format using vcf2phylip v2.0 [53]. The posterior distributions of the parameters were estimated using two independent Markov chain Monte Carlo (MCMC) methods for 100 million generations, with sampling every 20,000 generations. Tracer v1.7 was used to check model convergence (effective sample size >200) after excluding 10% of the initial samples as burn-in [54]. Using the SpreaD3 v0.9.6 software, a Bayes factor analysis (BF >3) was performed to identify diffusion routes that were strongly supported by the data [55].
3. Results
3.1. Spatiotemporal Dynamics of Genetic Diversity in M. oryzae Populations
After the SNP mining and filtering, a final SNP dataset of 605,369 whole-genome SNPs was obtained from the samples, which was applied for the evaluation of the genetic diversity of each geographical population of M. oryzae. A nucleotide diversity statistical analysis revealed the variations among the populations in Asia, Europe, Africa, North America and South America, indicating an average nucleotide diversity index (Pi) of 553.57–774.30 for each analysis window (Figure 1). Additionally, according to the t-test statistic between each of the pairwise populations, the M. oryzae samples from Asia had the highest nucleotide diversity (774.30). By contrast, the populations in Europe had the lowest nucleotide diversity (553.57), while those in Africa, South America and North America had moderate diversity.
3.2. Genetic Structures of Worldwide Geographic Populations
A neighbor-joining (NJ) tree was constructed for the global populations of M. oryzae based on the whole-genome SNPs, which showed that the worldwide samples formed three distinct sub-clades (Figure 2A). Out of these clades, the samples from Asia were distributed into three sub-clades. However, for the populations from Europe, North America, South America and Africa, although there were a few isolates that were localized in other clades, the majority of the individual isolates were assigned to a single clade. Therefore, there was no obvious segregation of individuals based on geographical location. The results of the PCA corresponded to the phylogenetic tree topology, indicating that individuals were grouped to three clusters with incomplete separation among the geographic populations (Figure 2B).
Furthermore, population structure composition analysis using ADMIXTURE revealed the individual ancestry and admixture proportions within each population. The optimum cluster number was K = 3 (Table S2), as it had the lowest cross-validation error and was thus selected as the number of potential ancestors for the worldwide M. oryzae populations (Figure 2C). Among them, the genetic compositions of the populations from Asia, Africa, Europe and North America contained three ancestral lineages, although the individuals were dominated by a single ancestral lineage. However, some samples contained two ancestral lineages. Notably, the European populations consisted of three independent ancestral lineages, without any mixed multilineage isolates. In addition, the South American populations were descended from two independent lineages.
3.3. Migration Patterns of M. oryzae
The genetic differentiation between populations (FST) ranged from 0.088 to 0.237 (Figure 3A, Table S3) with the 20 Kb window length and 2 Kb steps, indicating moderate genetic differentiation. This result implies the likelihood of M. oryzae migration occurring among the worldwide geographic populations. At the same time, the results from the phylogenetic network reconstruction exhibit the reticulate relationships within the three clades, suggesting the occurrence of spatial migration (Figure 3B). In addition, the migration pathways of this pathogen that were inferred from the phylogeographic analysis revealed that migration events could be detected from the spatial diffusion of M. oryzae across the world and Asia. The results also show that five major migration events occurred across five continents. Of these, Asia was the major source gene pool, which then flowed to other continents (Figure 4A). Interestingly, Europe served as a pivotal transfer station, facilitating gene flow to North America. In Asia, five major migration events were detected among China (including Taiwan and the mainland), Japan and South Korea (Figure 4B). Mainland China acted as the primary source of gene migration, initially spreading to Japan and South Korea and subsequently extending to Taiwan via Japan. Additonally, bidirectional gene flows were observed between South Korea and Japan.
3.4. Demographic History of M. oryzae Populations
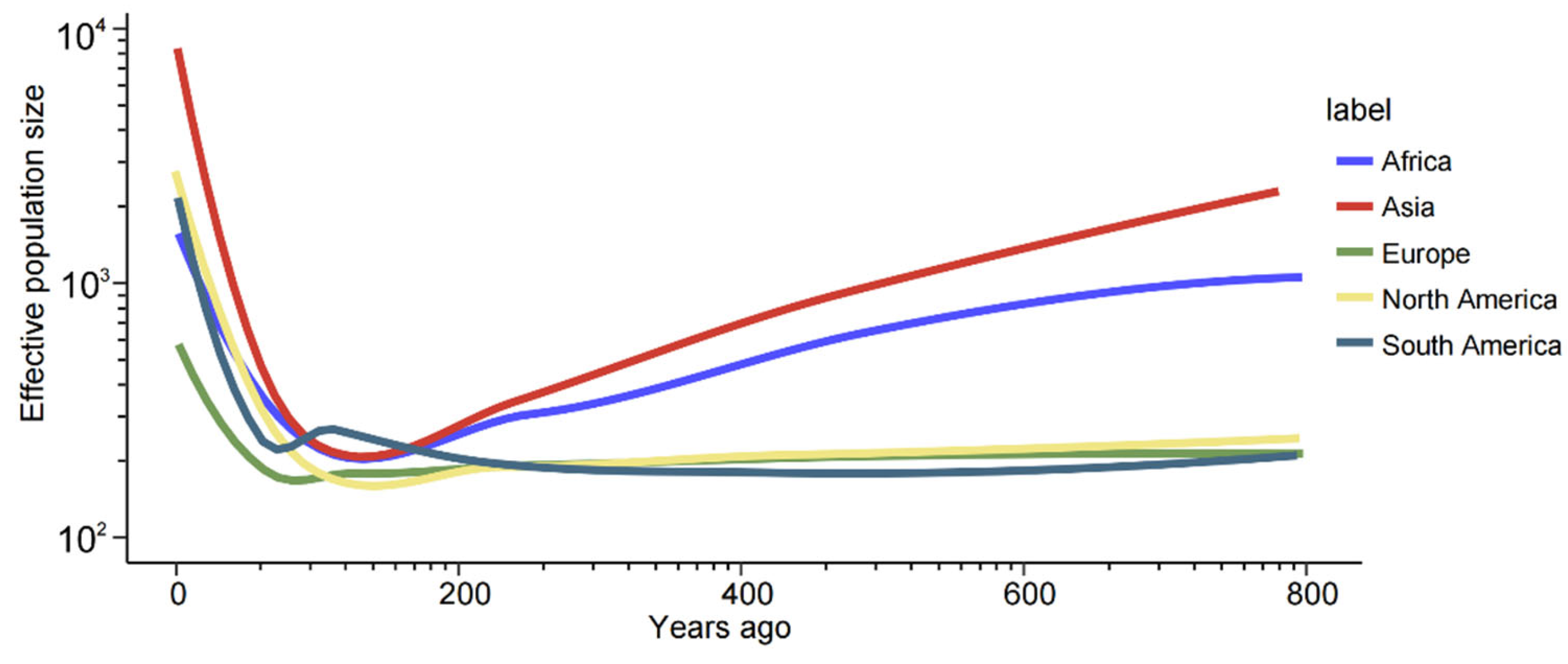
To investigate the demographic history of M. oryzae, effective population sizes were calculated using a pairwise sequentially Markovian coalescent (PSMC) model. As previously reported, rice-infecting isolates of M. oryzae evolved from grass-infecting isolates around 1000 years ago [23]. Therefore, the effective population sizes were predicted for the last 800 years (Figure 5). Our finding indicated that M. oryzae underwent a striking population expansion across five continents approximately 100 years ago.
4. Discussion
For fungal pathogens of plants in agroecosystems, genetic diversity can reflect their capacity to adapt and survive under biotic and abiotic stresses [56,57]. This diversity is closely linked to their population origin and evolution, influencing their formation and maintenance [58,59]. In this study, Asian populations of M. oryzae exhibited the highest nucleotide diversity based on whole-genome polymorphism, in contrast to populations from Africa, Europe, North America and South America. This suggests that Asian populations could serve as potential reservoirs of M. oryzae variants containing more novel genotypes. Furthermore, among the three Asian countries studied, including Taiwan island and mainland of China, Japan and South Korea, the highest nucleotide diversity was observed in Chinese populations. This suggests that China serves as a center of genetic diversity of M. oryzae in Asia, as indicated by the microsatellite markers [24]. Based on its history of rice domestication, China is also deemed to be the center of origin for rice [60,61], meaning that M. oryzae would have accumulated more abundant genetic resources for adaptation and expansion across its host range.
For M. oryzae populations, the temporal dynamics of genetic diversity are often closely related to rice cultivation. According to the US Department of Agriculture (https://www.ers.usda.gov/data-products/rice-yearbook/), worldwide rice cultivation areas have expanded rapidly, increasing by 11% from 1960 to 2019. The main rice-planting acreage and production regions are concentrated in Asia, where M. oryzae populations have maintained a high level of genetic diversity. Factors contributing to this diversity include genomic diversification, genetic instability and high variation rates, such as the presence of abundant variable number tandem repeats (VNTRs) and transposable elements (TEs) [62,63,64]. The rapidly decreasing genetic diversity observed in the 2020s could potentially be attributed to insufficient sampling from this period. In addition, the frequent host-shifting and host range expansion of this pathogen in crops and weeds have historically contributed to its sustained high genetic diversity [37,65,66], along with the gene flow events between cereal- and grass-specific lineages [28]. However, the continually growing genetic diversity of M. oryzae poses a significant threat to rice production, as these pathogens with diversified genetic resources could more rapidly overcome resistant genes in rice. While this genetic diversity could provide important new insights into the development and deployment of resistant rice varieties in practice. Under these scenarios, our findings underscore the need for greater attention to the effective monitoring and control of rice blast disease.
However, past hybridization events have imprinted on individual genome samples. In this study, the phylogenetic reticulate network and population differentiation indices indicated that incompatible events occurred within worldwide geographic populations, reflecting their harbored admixture status. In addition, three main clades were identified in worldwide populations from the genetic structure analysis, with each clade containing individuals from different continents, suggesting that geographical location could not sufficiently account for the population genetic differentiation of M. oryzae. Although geographical isolation plays an important role in shaping genetic divergence and leading to the geographic differentiation of many plant pathogen populations [67,68], the genetic differentiation of M. oryzae appears to be more vulnerable to its mating types [23]. While both MAT1-1 and MAT1-2 mating types have been identified in China [69], the individuals from China are distributed across three divergent clades. The population differentiation indices, as represented by FST values, demonstrate that the African populations are genetically distant from other populations, aligning with previous findings that African populations maintain relatively distant genetic relationships with Asian populations and are predominantly distributed in MAT1-2 [19,70].
In terms of the population structures of M. oryzae, populations with only one mating type occupied the principal position, indicating that clonal lineages dominated by asexual reproduction were prevalent in natural populations. The species can be geographically separated into a series of genetically less closely related subpopulations, according to population communication decreases [71] or gradual subpopulation increases following extinction and recolonization events. However, migration mediated by human activity and the transportation of infected seed [33] has made plant pathogen populations deviate from their evolutionary routes through recombination. In this study, an incomplete separation signature was also detected in the SplitsTree analysis for the reticular structures in the clades, which contained samples from Asia, Africa, Europe and North America. Additionally, five migration events among five continents were identified, consistent with findings reported by Tharreau et al. [33], indicating a global distribution of virulent genotypes. In this study, Asia was considered as a gene pool, which agreed with the result that Southeast Asia was recognized as a center of origin and diversity based on microsatellite markers [72]. This result is further corroborated by previous genetic diversity studies, which consistently revealed a higher genetic diversity within Asian populations [22,72]. Additionally, the Asian populations exhibited the highest nucleotide diversity in this study, underscoring Asia’s role as a center of genetic diversity and a source of migration. This finding aligns with the putative origins of rice in southern China and northeast India [60,61,73,74]. As a seed-borne pathogen, M. oryzae can infect two major subspecies: Oryzae sativa subsp. Japonica and Indica. The Japonica subspecies originated from rice domestication and then diverged into temperate and tropical Japonica subspecies, which progressively led to the formation of the Indica subspecies. This implies the possibility of pathogen migration following the domestication and introduction of rice crops [73,75,76]. Within Asia, migration events were observed in South Korea, Japan and China. Given that China is considered as the global origin of M. oryzae, it likely served as a foundational source for migration within Asia. Our results suggest that migration pathways originated from mainland China, initially extending to Japan and subsequently to Taiwan. These migration pathways have mainly been attributed to rice cultivars, as Japonica rice is preferred in both Japan and Taiwan. Furthermore, the limited trade and agricultural exchange between Taiwan and mainland China in recent decades could also have contributed to the limited gene flow. Similarly, a reciprocal gene flow was observed between populations in Japan and South Korea, potentially influenced by frequent interstate trade between these two countries. A similar pattern of results has been observed in Fusarium head blight pathogens, where agricultural practices and human migration have been critical driving forces in shaping the population genetic structure of Fusarium asiaticum [77].
In summary, gene flows at different geographical scales have exerted a significant influence on the establishment of the genetic structure of M. oryzae. Understanding the spatiotemporal dynamics of population compositions following the success of introduced fungal pathogens is essential for combatting plant disease pandemics [78,79].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Genome information for M. oryzae strains. Table S2: Cross-validation errors for different models. Table S3: Genetic differentiation among populations (Fst).
Author Contributions
Conceptualization, Z.W. and M.C.; methodology, G.D.; software, J.X.; validation, Y.L. and C.Z.; formal analysis, K.Y.; investigation, H.Z. and B.W.; resources, J.B. and W.T.; writing—original draft preparation, G.D.; writing—review and editing, Z.W. and M.C.; funding acquisition, Z.W., M.C. and H.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the National Natural Science Foundations of China (32001976, 32272513, 32172365).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Sequencing raw reads and assembled contigs are available at NCBI under BioProject ID: PRJNA1138824, BioSample: SAMN42726964 to SAMN42726985.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Talbot, N.J. On the trail of a cereal killer: Exploring the biology of Magnaporthe grisea. Annu Rev Microbiol 2003, 57, 177–202. [Google Scholar] [CrossRef] [PubMed]
- Khang, C.H.; Park, S.Y.; Lee, Y.H.; Valent, B.; Kang, S. Genome organization and evolution of the AVR-Pita avirulence gene family in the Magnaporthe grisea species complex. Mol Plant Microbe Interact 2008, 21, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Zeigler, R.S. Recombination in Magnaporthe grisea. Annu Rev Phytopathol 1998, 36, 249. [Google Scholar] [CrossRef]
- Valent, B. The Impact of Blast Disease: Past, Present, and Future. Methods Mol Biol 2021, 2356, 1–18. [Google Scholar]
- Notteghem, J.L.; Silué, D. Distribution of the mating type alleles in Magnaporthe grisea populations pathogenic on rice. Phytopathology 1992, 82, 421–424. [Google Scholar] [CrossRef]
- Dayakar, B.V.; Narayanan, N.N.; Gnanamanickam, S.S. Cross-Compatibility and Distribution of Mating Type Alleles of the Rice Blast Fungus Magnaporthe grisea in India. Plant Dis 2000, 84, 700–704. [Google Scholar] [CrossRef]
- Maciel, J.L.; Ceresini, P.C.; Castroagudin, V.L.; Zala, M.; Kema, G.H.; McDonald, B.A. Population structure and pathotype diversity of the wheat blast pathogen Magnaporthe oryzae 25 years after its emergence in Brazil. Phytopathology 2014, 104, 95–107. [Google Scholar] [CrossRef]
- Tredway, L.P.; Stevenson, K.L.; Burpee, L.L. Mating type distribution and fertility status in Magnaporthe grisea populations from turfgrasses in georgia. Plant Dis 2003, 87, 435–441. [Google Scholar] [CrossRef]
- Consolo, V.F.; Cordo, C.A.; Salerno, G.L. Mating-type distribution and fertility status in Magnaporthe grisea populations from Argentina. Mycopathologia 2005, 160, 285–290. [Google Scholar] [CrossRef]
- Zhan, J.; Thrall, P.H.; Burdon, J.J. Achieving sustainable plant disease management through evolutionary principles. Trends Plant Sci 2014, 19, 570–575. [Google Scholar] [CrossRef]
- Markert JA, Champlin DM, Gutjahr-Gobell R, Grear JS, Kuhn A, McGreevy TJ Jr; et al. Population genetic diversity and fitness in multiple environments. BMC Evol Biol 2010, 10, 205.
- Xu X, Yang W, Tian K, Zheng J, Liu X, Li K; et al. Genetic diversity and pathogenicity dynamics of Magnaporthe oryzae in the wuling mountain area of china. Eur J Plant Pathol 2019, 153, 731–742. [CrossRef]
- Xu X, Tang X, Han H, Yang W, Liu X, Li K; et al. Pathogenicity, mating type distribution and avirulence gene mutation of Magnaporthe oryzae populations in the wuling mountain region of china. Physiol Mol Plant Pathol 2021, 116, 101716. [CrossRef]
- Li, Y.; Liu, E.M.; Dai, L.Y.; Li, C.Y.; Liu, L. Genetic diversity among populations as related to pathotypes for Magnaporthe grisea in hunan province. Chin J Rice Sci 2007, 21, 304. [Google Scholar]
- Yadav MK, Aravindan S, Raghu S, Prabhukarthikeyan SR, Keerthana U, Ngangkham U; et al. Assessment of genetic diversity and population structure of Magnaporthe oryzae causing rice blast disease using ssr markers. Physiol Mol Plant P 2019, 106, 157–165. [CrossRef]
- Jagadeesh, D.; Kumar, M.K.P.; Amruthavalli, C.; Devaki, N.S. Genetic diversity of Magnaporthe oryzae, the blast pathogen of rice in different districts of karnataka, india determined by simple sequence repeat (ssr) markers. Indian Phytopathology 2020, 73, 713–723. [Google Scholar] [CrossRef]
- Longya, A.; Talumphai, S.; Jantasuriyarat, C. Morphological characterization and genetic diversity of rice blast fungus, Pyricularia oryzae, from thailand using issr and srap markers. J Fungi (Basel) 2020, 6, 38. [Google Scholar] [CrossRef]
- Lopez, A.L.C.; Cumagun, C.J.R. Genetic structure of Magnaporthe oryzae populations in three island groups in the philippines. Eur J Plant Pathol 2019, 153, 101–118. [Google Scholar] [CrossRef]
- Odjo T, Diagne D, Adreit H, Milazzo J, Raveloson H, Andriantsimialona D; et al. Structure of african populations of Pyricularia oryzae from rice. Phytopathology 2021, 111, 1428–1437. [CrossRef]
- Roumen, E.; Levy, M.; Notteghem, J.L. Characterisation of the european pathogen population of Magnaporthe grisea by DNA fingerprinting and pathotype analysis. Eur J Plant Pathol 1997, 103, 363–371. [Google Scholar] [CrossRef]
- Pagliaccia D, Urak RZ, Wong F, Douhan LI, Greer CA, Vidalakis G; et al. Genetic structure of the rice blast pathogen (Magnaporthe oryzae) over a decade in North Central California rice fields. Microb. Ecol 2018, 75, 310–317. [CrossRef] [PubMed]
- Onaga G, Suktrakul W, Wanjiku M, Quibod IL, Entfellner JBD, Bigirimana J; et al. Magnaporthe oryzae populations in sub-saharan africa are diverse and show signs of local adaptation. BioRxiv, 2020. [CrossRef]
- Zhong Z, Chen M, Lin L, Han Y, Bao J, Tang W; et al. Population genomic analysis of the rice blast fungus reveals specific events associated with expansion of three main clades. ISME J 2018, 12, 1867–1878. [CrossRef]
- Saleh, D.; Milazzo, J.; Adreit, H.; Fournier, E.; Tharreau, D. South-East Asia is the center of origin, diversity and dispersion of the rice blast fungus, Magnaporthe oryzae. New Phytol 2014, 201, 1440–1456. [Google Scholar] [CrossRef] [PubMed]
- Gladieux P, Ravel S, Rieux A, Cros-Arteil S, Adreit H, Milazzo J; et al. Coexistence of multiple endemic and pandemic lineages of the rice blast pathogen. mBio 2018, 9, e01806–17.
- Taheri, P.; Irannejad, A. Genetic structure of various Magnaporthe oryzae populations in iran and uruguay. Aus Plant Pathol 2014, 43, 287–297. [Google Scholar] [CrossRef]
- Duan G, Bao J, Chen X, Xie J, Liu Y, Chen H; et al. Large-scale genome scanning within exonic regions revealed the contributions of selective sweep prone genes to host divergence and adaptation in Magnaporthe oryzae species complex. Microorganisms 2021, 9, 562. [CrossRef]
- Gladieux P, Condon B, Ravel S, Soanes D, Maciel JLN, Nhani A Jr; et al. Gene Flow between Divergent Cereal- and Grass-Specific Lineages of the Rice Blast Fungus Magnaporthe oryzae. mBio 2018, 9, e01219–17.
- Lande, R. Neutral theory of quantitative genetic variance in an island model with local extinction and colonization. Evolution 1992, 46, 381–389. [Google Scholar] [CrossRef]
- Kimura, M.; Weiss, G.H. The stepping stone model of population structure and the decrease of genetic correlation with distance. Genetics 1964, 49, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Meng JW, He DC, Zhu W, Yang LN, Wu EJ, Xie JH; et al. Human-mediated gene flow contributes to metapopulation genetic structure of the pathogenic fungus Alternaria alternata from potato. Front Plant Sci 2018, 9, 00198. [CrossRef]
- Gao, F.; Chen, C.; Li, B.; Weng, Q.; Chen, Q. The gene flow direction of geographically distinct Phytophthora infestans populations in China corresponds with the route of seed potato exchange. Front Microbiol 2020, 11, 1077. [Google Scholar] [CrossRef]
- Tharreau D, Fudal I, Andriantsimialona D, Utami D, Fournier E, Lebrun MH; et al. World population structure and migration of the rice blast fungus, Magnaporthe oryzae. Advances in genetics, genomics and control of rice blast disease, Springer 2009; pp:209-15.
- Onaga, G.; Wydra, K.; Koopmann, B.; Séré, Y.; von Tiedemann, A. Population structure, pathogenicity, and mating type distribution of Magnaporthe oryzae isolates from east Africa. Phytopathology 2015, 105, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Milgroom, M.; Han, S.; Kang, S.; Lee, Y.H. Genetic differentiation of Magnaporthe oryzae populations from scouting plots and commercial rice fields in Korea. Phytopathology 2008, 98, 436–442. [Google Scholar] [CrossRef]
- Kawakubo S, Gao F, Li S, Tan Z, Huang YK, Adkar-Purushothama CR; et al. Genomic analysis of the brassica pathogen turnip mosaic potyvirus reveals its spread along the former trade routes of the Silk Road. Proc Natl Acad Sci U S A 2021, 118, e2021221118. [CrossRef] [PubMed]
- Zhong Z, Norvienyeku J, Chen M, Bao J, Lin L, Chen L; et al. Directional selection from host plants is a major force driving host specificity in Magnaporthe species. Sci Rep 2016, 6, 25591. [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Dean RA, Talbot NJ, Ebbole DJ, Farman ML, Mitchell TK, Orbach MJ; et al. The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 2005, 434, 980–986. [CrossRef]
- Li, F.; Fast, R. Accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A; et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010, 20, 1297–1303. [CrossRef]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158.
- Delcher, A.L.; Phillippy, A.; Carlton, J.; Salzberg, S.L. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res 2002, 30, 2478–2483. [Google Scholar] [CrossRef]
- Pfeifer, B.; Wittelsbürger, U.; Ramos-Onsins, S.E.; Lercher, M.J. PopGenome: An efficient Swiss army knife for population genomic analyses in R. Mol Biol Evol 2014, 31, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal or partially clonal reproduction. PeerJ 2013, 2, e281. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Francis, RM. pophelper: An R package and web app to analyse and visualize population structure. Mol Ecol Resour 2017, 17, 27–32. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput Biol 2009, 5, e1000520. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol 2018, 4, vey016. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. Modelfinder: Fast model selection for accurate phylogenetic estimates. Nat Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Ortiz EJUhdoz. Vcf2phylip v2. 0: Convert a vcf matrix into several matrix formats for phylogenetic analysis. URL https://doi org/105281/zenodo 2019;2540861.
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in bayesian phylogenetics using tracer 1.7. Syst Biol 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive visualization of spatiotemporal history and trait evolutionary processes. Mol Biol Evol 2016, 33, 2167–2169. [Google Scholar] [CrossRef]
- Li, Q.; Xu, K.; Yu, R.J. Genetic variation in chinese hatchery populations of the japanese scallop (patinopecten yessoensis) inferred from microsatellite data. Aquaculture 2007, 269, 211–219. [Google Scholar] [CrossRef]
- Edwards, A.W. The genetical theory of natural selection. Genetics 2000, 154, 1419–1426. [Google Scholar] [CrossRef]
- Zhan, J. Population genetics of plant pathogens. In B. McDonald (eds) eLS. John Wiley & Sons, Ltd 2016. [CrossRef]
- McDonald, B.A.; McDermott, J.M. Population genetics of plant pathogenic fungi. Bioscience 1993, 43, 311–319. [Google Scholar] [CrossRef]
- Gutaker RM, Groen SC, Bellis ES, Choi JY, Pires IS, Bocinsky RK; et al. Genomic history and ecology of the geographic spread of rice. Nat Plants 2020, 6, 492–502. [CrossRef]
- Gross, B.L.; Zhao, Z. Archaeological and genetic insights into the origins of domesticated rice. Proc Natl Acad Sci U S A 2014, 111, 6190–6197. [Google Scholar] [CrossRef]
- Thon MR, Pan H, Diener S, Papalas J, Taro A, Mitchell TK; et al. The role of transposable element clusters in genome evolution and loss of synteny in the rice blast fungus Magnaporthe oryzae. Genome Biol 2006, 7, R16.
- Couch, B.C.; Kohn, L.M. A multilocus gene genealogy concordant with host preference indicates segregation of a new species, Magnaporthe oryzae, from M. grisea. Mycologia 2002, 94, 683–693. [Google Scholar] [CrossRef]
- Yoshida K, Saunders DG, Mitsuoka C, Natsume S, Kosugi S, Saitoh H; et al. Host specialization of the blast fungus Magnaporthe oryzae is associated with dynamic gain and loss of genes linked to transposable elements. BMC Genomics 2016, 17, 370.
- Chung H, Goh J, Han SS, Roh JH, Kim Y, Heu S; et al. Comparative pathogenicity and host ranges of Magnaporthe oryzae and related species. Plant Pathol J 2020, 36, 305–313. [CrossRef]
- Woolhouse, M.E.; Haydon, D.T.; Antia, R. Emerging pathogens: The epidemiology and evolution of species jumps. Trends Ecol Evol 2005, 20, 238–244. [Google Scholar] [CrossRef]
- Parada-Rojas, C.H.; Quesada-Ocampo, L.M. Phytophthora capsici populations are structured by host, geography, and fluopicolide sensitivity. Phytopathology 2022, 112, 1559–1567. [Google Scholar] [CrossRef]
- Diao Y, Zhang C, Xu J, Lin D, Liu L, Mtung'e OG; et al. Genetic differentiation and recombination among geographic populations of the fungal pathogen Colletotrichum truncatum from chili peppers in China. Evol Appl 2015, 8, 108–118. [CrossRef] [PubMed]
- Li, J.; Lu, L.; Jia, Y.; Wang, Q.; Fukuta, Y.; Li, C. Characterization of field isolates of Magnaporthe oryzae with mating type, DNA fingerprinting, and pathogenicity assays. Plant Dis 2016, 100, 298–303. [Google Scholar] [CrossRef]
- Onaga, G.; Wydra, K.; Koopmann, B.; Séré, Y.; von Tiedemann, A. Population structure, pathogenicity, and mating type distribution of Magnaporthe oryzae isolates from east Africa. Phytopathology 2015, 105, 1137–1145. [Google Scholar] [CrossRef]
- Teixeira, M.M.; Barker, B.M. Use of population genetics to assess the ecology, evolution, and population structure of coccidioides. Emerg Infect Dis 2016, 22, 1022–1030. [Google Scholar] [CrossRef]
- Saleh, D.; Milazzo, J.; Adreit, H.; Fournier, E.; Tharreau, D. South-East Asia is the center of origin, diversity and dispersion of the rice blast fungus, Magnaporthe oryzae. New Phytol 2014, 201, 1440–1456. [Google Scholar] [CrossRef]
- Huang X, Wei X, Wang ZX, Wang A, Zhao Q; et al. A map of rice genome variation reveals the origin of cultivated rice. Nature 2012, 490, 497–501. [CrossRef] [PubMed]
- Londo, J.P.; Chiang, Y.C.; Hung, K.H.; Chiang, T.Y.; Schaal, B.A. Phylogeography of asian wild rice, Oryza rufipogon, reveals multiple independent domestications of cultivated rice, Oryza sativa. Proc Natl Acad Sci U S A 2006, 103, 9578–9583. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Han, B. Rice domestication occurred through single origin and multiple introgressions. Nat Plants 2015, 2, 15207. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Platts, A.E.; Fuller, D.Q.; Hsing, Y.I.; Wing, R.A.; Purugganan, M.D. The rice paradox: Multiple origins but single domestication in asian rice. Mol Biol Evol 2017, 34, 969–979. [Google Scholar] [CrossRef]
- Yang M, Smit S, de Ridder D, Feng J, Liu T, Xu J, et al. Adaptation of Fusarium Head Blight Pathogens to Changes in Agricultural Practices and Human Migration. Adv Sci (Weinh) 2024; 5:e2401899.
- Thrall PH, Oakeshott JG, Fitt G, Southerton S, Burdon JJ, Sheppard A; et al. Evolution in agriculture: The application of evolutionary approaches to the management of biotic interactions in agro-ecosystems. Evol Appl 2011, 4, 200–215. [CrossRef]
- Williams, P.D. Darwinian interventions: Taming pathogens through evolutionary ecology. Trends Parasitol 2010, 26, 83–92. [Google Scholar] [CrossRef]
Figure 1.
The nucleotide diversity indices for rice-infecting M. oryzae populations by continent. The spatiotemporal dynamics of the nucleotide diversity were based on the whole-genome SNPs with 20 kb sliding windows and 2 kb steps. The nucleotide diversity distribution in different continents include AS (Asia), AF (Africa), EU (Europe), SA(South America), NA (North America) and ALL (all continents) shown in X-axis. Boxes show the first quartile, median and third quartile, and whiskers extend 1.5 times the interquartile range. Values for each of the window are shown as points scattered in boxplots. The dotted line is connecting the median values at each time period.
Figure 1.
The nucleotide diversity indices for rice-infecting M. oryzae populations by continent. The spatiotemporal dynamics of the nucleotide diversity were based on the whole-genome SNPs with 20 kb sliding windows and 2 kb steps. The nucleotide diversity distribution in different continents include AS (Asia), AF (Africa), EU (Europe), SA(South America), NA (North America) and ALL (all continents) shown in X-axis. Boxes show the first quartile, median and third quartile, and whiskers extend 1.5 times the interquartile range. Values for each of the window are shown as points scattered in boxplots. The dotted line is connecting the median values at each time period.
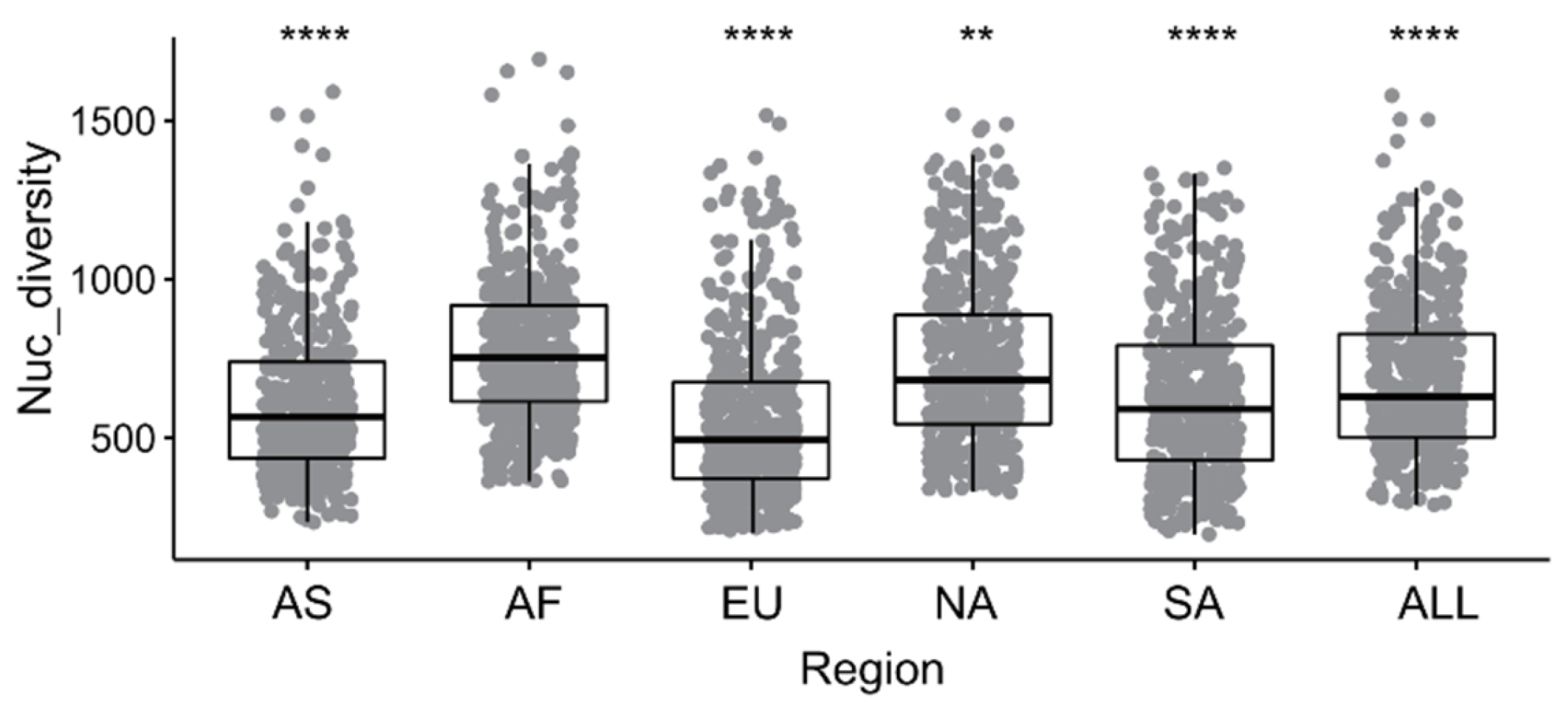
Figure 2.
Population genetic structure analysis of M. oryzae from five continents. (A) phylogenetic tree based on the whole-genome single-nucleotide genetic variant data for worldwide samples using the neighbor-joining (NJ) method with 1000 bootstrap replicates (the different colors denote the various geographic populations); (B) principal component analysis (PCA) for populations from five continents (each dot represents one individual and the colors correspond to the sample collection locations); (C) ADMIXTURE bar plot of the ancestral assignments of individual isolates using the optimal K = 3 (the length of each colored segment corresponds to the proportion of the individual’s genome ancestry).
Figure 2.
Population genetic structure analysis of M. oryzae from five continents. (A) phylogenetic tree based on the whole-genome single-nucleotide genetic variant data for worldwide samples using the neighbor-joining (NJ) method with 1000 bootstrap replicates (the different colors denote the various geographic populations); (B) principal component analysis (PCA) for populations from five continents (each dot represents one individual and the colors correspond to the sample collection locations); (C) ADMIXTURE bar plot of the ancestral assignments of individual isolates using the optimal K = 3 (the length of each colored segment corresponds to the proportion of the individual’s genome ancestry).
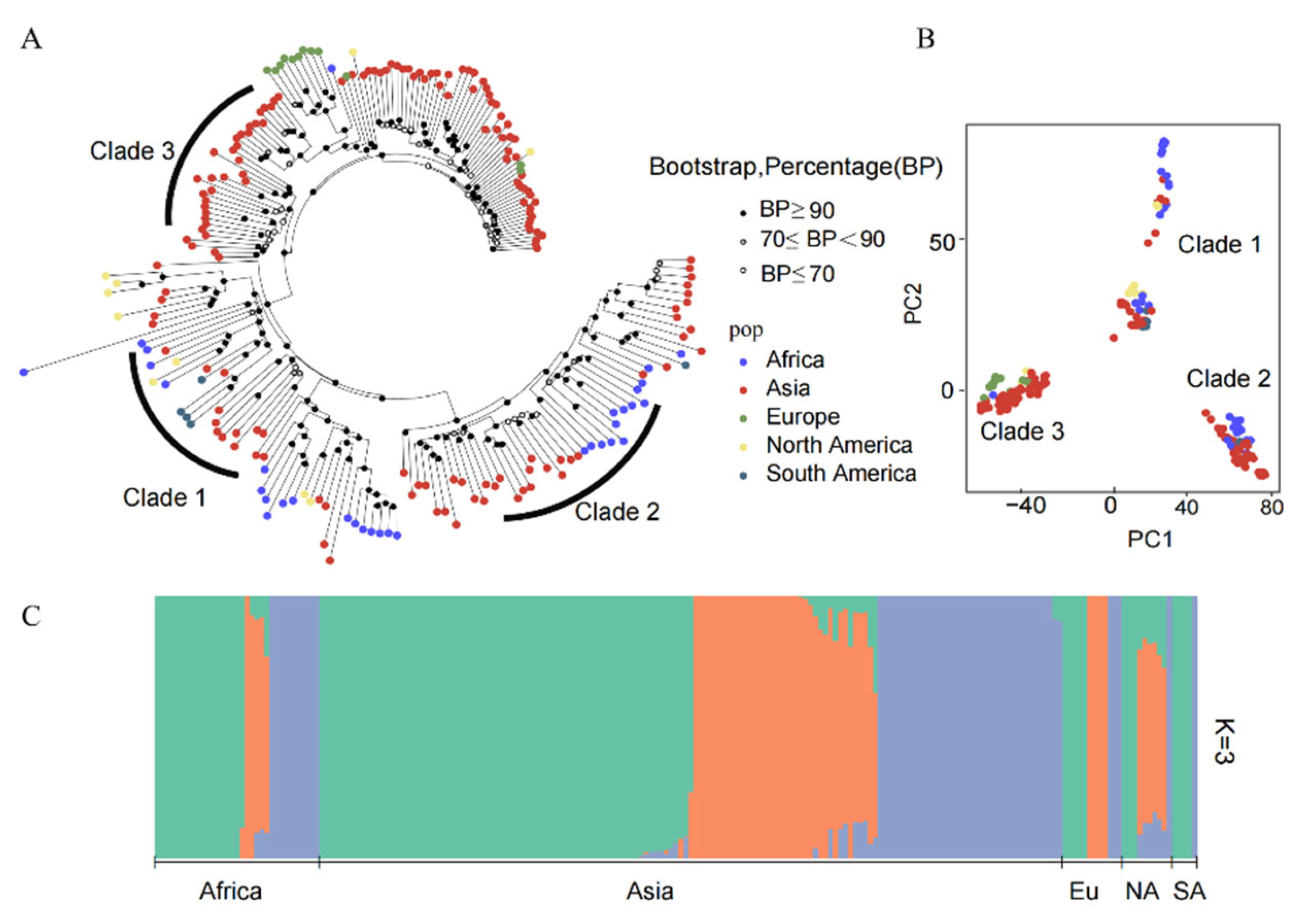
Figure 3.
Phylogenetic network of worldwide M. oryzae populations, according to a Neighbor-Net analysis. (A) The Heatmap of genetic differentiation (FST) between populations. (B) The analysis was implemented using SplitsTree software with the “Neighbor-Net” and “Uncorrected p-distance” parameters. The reticulate dendrogram in the tree represents the incompatible signals, implying incompatible or ambiguous relationships between the samples.
Figure 3.
Phylogenetic network of worldwide M. oryzae populations, according to a Neighbor-Net analysis. (A) The Heatmap of genetic differentiation (FST) between populations. (B) The analysis was implemented using SplitsTree software with the “Neighbor-Net” and “Uncorrected p-distance” parameters. The reticulate dendrogram in the tree represents the incompatible signals, implying incompatible or ambiguous relationships between the samples.
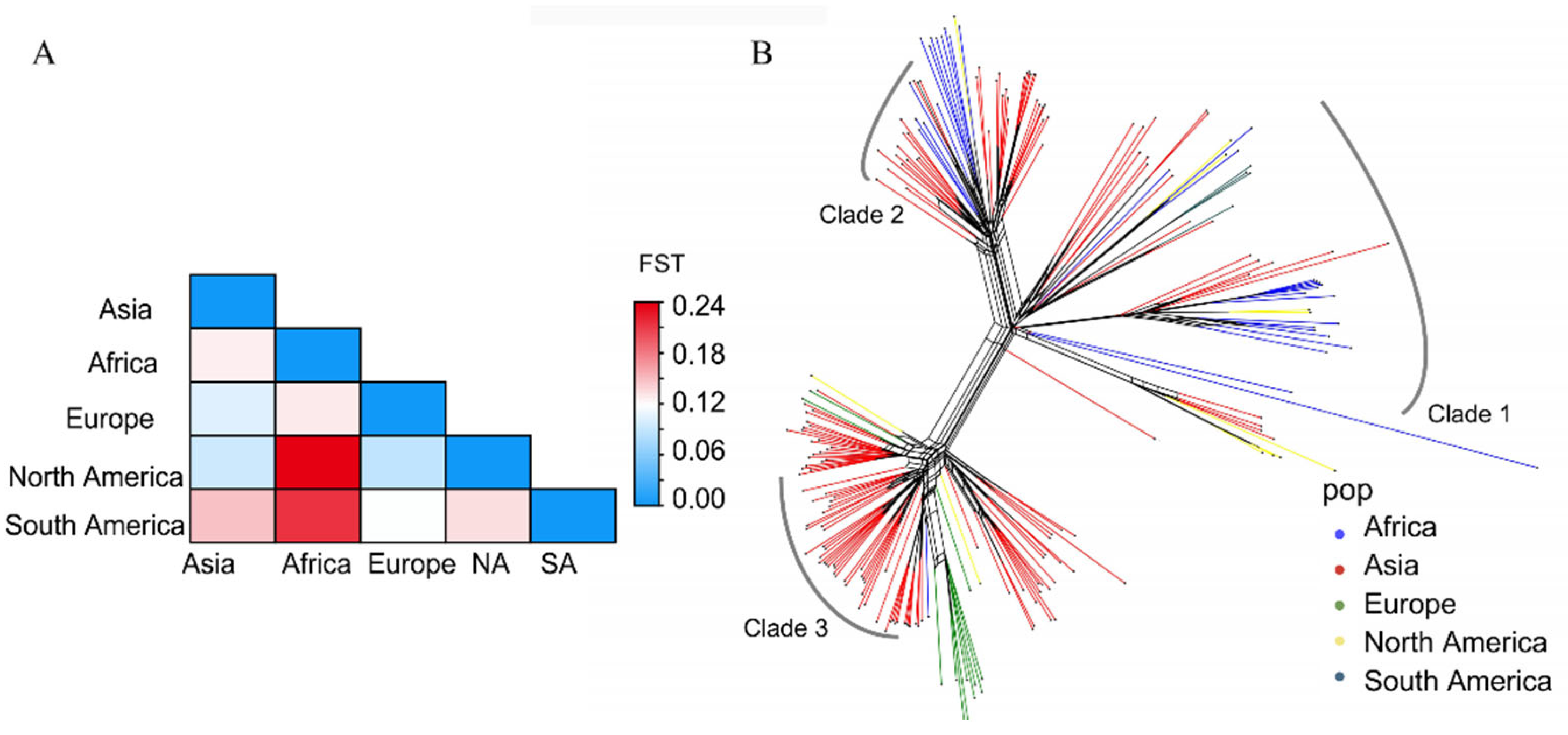
Figure 4.
Schematic presentation of migration pathways of M. oryzae. (A) The migration pathways across five continents. (B) The migration pathways In Asian regions. The arrows show the directions of immigration and emigration between the different locations.
Figure 4.
Schematic presentation of migration pathways of M. oryzae. (A) The migration pathways across five continents. (B) The migration pathways In Asian regions. The arrows show the directions of immigration and emigration between the different locations.
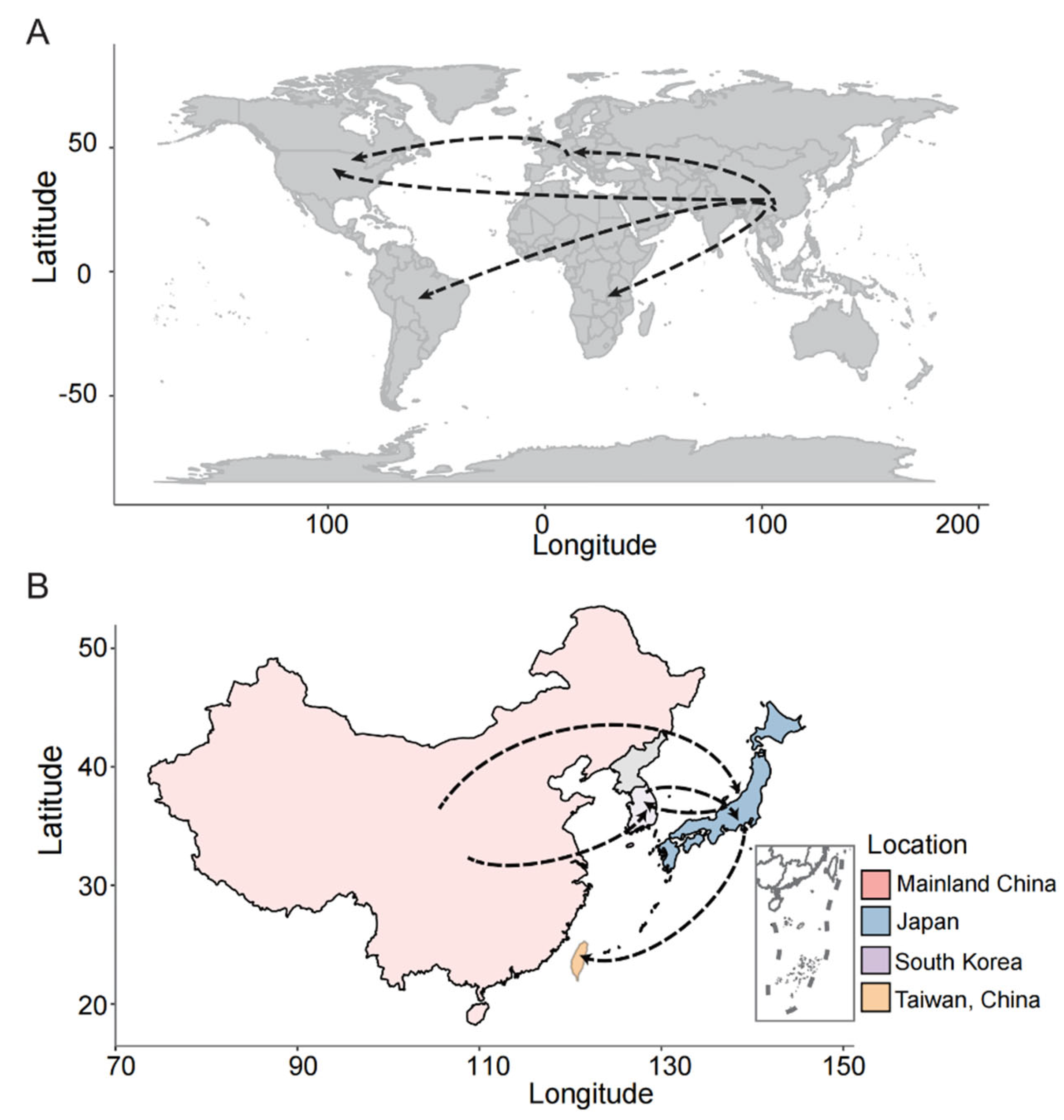
Figure 5.
The demographic history of M. oryzae, illustrating the effective population sizes from the different geographical groups. The coordinates were logarithmically scaled. The colored lines represent the different geographical populations.
Figure 5.
The demographic history of M. oryzae, illustrating the effective population sizes from the different geographical groups. The coordinates were logarithmically scaled. The colored lines represent the different geographical populations.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



