
Submitted:
02 September 2024
Posted:
02 September 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Coronaviruses (CoVs) have recently emerged as significant causes of respiratory disease outbreaks. The novel coronavirus pneumonia of 2019, known as COVID-19, are highly infectious and triggered by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Understanding virus-host interactions and molecular targets in host cell death signalling is crucial for treatment development. Small natural compounds like celastrol and curcumin, acting as proteasome inhibitors, can potentially modify NF-κB signalling for treating SARS-CoV-2 infections. Various natural constituents, including alkaloids, flavonoids, terpenoids, diarylheptanoids, and anthraquinones, inhibit viral infection, progression, and amplification of coronaviruses. Derived from medicinal herbs, these compounds possess anti-inflammatory and antiviral properties, impacting the viral life cycle, including entry, replication, assembly, and release of COVID-19 virions. This review focuses on the development of small molecules of non-covalent inhibitors targeting the Main Protease (Mpro, also called 3CLpro) enzyme of SARS-CoV-2. It highlights the design using molecular dynamics (MD) studies and computational methods for further improvements in Mpro inhibitor design. The in-silico approach, which is pivotal in this process, provides an accelerated virtual avenue for exploring and developing potential inhibitors, representing the latest advancements in drug design.

Keywords:
SARS-CoV-2 virus inhibitors
; main protease (Mpro)
; non-covalent inhibitors
; molecular dynamics simulation
; in silico analysis
1. Introduction
1.1. The main protease (Mpro) enzyme of SARS-CoV-2
The World Health Organisation (WHO) defines the coronavirus that causes severe acute respiratory syndrome (SARS-CoV-2) as COVID-19 [1]. Recently, the virus has become a global threat [2]. With a genome size of about 30 kb, it is a highly contagious positive-strand RNA virus that codes for 29 distinct proteins essential to the virus's survival and life cycle [3]. The beta coronavirus, or SARSCoV-2, is closely linked to the SARS-CoV virus that caused the outbreak of the disease in 2002 and 2004 [4]. The SARS-CoV-2 virion was made up of four structural proteins: spike proteins (S), envelope proteins (E), membrane proteins (M), and nucleocapsid proteins (N) [3]. Like other coronaviruses, these proteins enclose the RNA genome [5]. The Mpro, commonly referred to as 3CLpro, 3-ChymotrypsinLike Protease, or Mpro, and the spike proteins (S) are the primary targets for drug development against SARS-CoV-2 virus [6].
The coronavirus SARS-CoV-2 is a large, enveloped virus with a single-stranded, non-segmented, positive-sense RNA genome broadly distributed in humans and other mammals [7]. As shown in the basic structure of the SARS-CoV-2 virus in Figure 1, SARS-CoV-2 has four primary structural proteins: spike (S) glycoprotein, small envelope (E) glycoprotein, membrane (M) glycoprotein, and nucleocapsid (N) protein, along with several accessory proteins [8].
The spike (S) glycoprotein, a transmembrane protein with a molecular weight of approximately 150 kDa, is located on the virus's outer surface. S protein forms homotrimers that protrude from the viral surface, facilitating the binding of the virus to host cells by interacting with the angiotensinconverting enzyme 2 (ACE2) receptors, which are expressed in the cells of the lower respiratory tract.
This glycoprotein is cleaved by host cell furin-like proteases into two subunits: S1 and S2. The S1 subunit determines the host range and cellular tropism of the virus through its receptor-binding domain, while the S2 subunit mediates the fusion of the virus with host cells, enabling viral transmission [7].
The nucleocapsid, also known as the N protein in Figure 1, is a structural component of coronaviruses (CoV) that localizes in the endoplasmic reticulum-Golgi region. It binds to the viral nucleic acid material, specifically RNA, and plays a crucial role in various processes involving the viral genome, such as the replication cycle and the host cell's response to infection. Due to its binding to RNA, the N protein is integral to these processes. Additionally, the N protein undergoes significant phosphorylation, which is believed to induce structural changes that enhance its affinity for viral RNA. Another key structural element of the virus is the membrane, or M protein, which is the most abundant structural protein and is critical in determining the shape of the viral envelope. The M protein interacts with all other structural proteins and plays a stabilizing role. By binding to the N protein, the M protein helps stabilize the nucleocapsid and supports the final stages of viral assembly by stabilizing the N protein-RNA complex within the virion. The envelope, or E protein, is the smallest structural protein in the SARS-CoV virus, yet it plays a significant role in the production and maturation of the virus [7]. The Mpro enzyme found in the SARS-CoV-2 virus is responsible for the development of COVID19 [2]. This protease is vital for processing virus polyproteins, which are required for viral replication [1]. Mpro is a cysteine protease with three domains: I (residues 8-101), II (102-184), and III (201-306) [9], which works by cleaving viral polypeptides and promoting viral replication [9]. Active Mpro exists as a homodimer made up of two protomers [10], as displayed in Figure 2. Despite differences amongst coronaviruses, Mpro has strong sequence and structural conservation, distinguishing it from human proteases. This property makes Mpro a good choice for drug screening activities aimed at inhibiting the replication and proliferation of SARS-CoV-2 [11].
Drug repurposing has emerged as a highly attractive and practical approach to drug discovery [13]. By exploring new therapeutic applications for drugs that have already been approved for human use, this strategy has proven to be especially valuable in the rapid response to the COVID-19 pandemic [13]. One of the key advantages of drug repurposing, particularly for identifying SARS-CoV-2 inhibitors, is the ability to accelerate the drug development process significantly. Since these drugs have already undergone extensive testing for safety and efficacy, they can quickly move into clinical trials for new indications, bypassing much of the early-stage research and regulatory hurdles typically required for novel compounds. This fast-tracking is crucial in public health emergencies, such as the COVID-19 crisis, where time is critical [13] [14]. In addition to speeding up the development process, drug repurposing reduces the financial risks of drug discovery. Developing entirely new drugs is expensive and uncertain, whereas repurposing existing drugs leverages previous investments in research and development [15, 16]. Another major benefit is the wealth of available data on repurposed drugs. Since these drugs have been previously approved, their safety profiles, potential side effects, and pharmacokinetics are well understood, making it easier to predict their behaviour in new therapeutic contexts [17].
Finally, many of these drugs are already in production and readily available, which is a critical advantage during a pandemic, where immediate access to treatment options can save lives. By utilizing existing drugs, repurposing can provide timely solutions to emerging health crises while minimizing the delays and uncertainties inherent in developing new drugs from scratch [18].
Although several vaccinations are now being developed to target the disease [19], there is still a significant demand for SARS-CoV-2 inhibitors to treat people who become affected [13]. In addition, the recurrence for many people occurs due to the introduction of novel SARS-CoV-2 variants [16]. Some antiviral drugs and prodrugs, such as GS-441524, Molnupiravir, and PF-07321332 (nirmatrelvir), are therapeutically efficacious antiviral drugs, could potentially lead to the development of drug-resistant mutations [17].
The alarming worldwide spread of the virus [20] and the emergence of novel SARS-CoV-2 variants in various regions [18] underscores the urgent need for effective therapeutic strategies. As a result, there is a need to rapidly develop and/or repurpose Mpro inhibitors to combat SARS-CoV-2, which has been a pressing global issue [21]. Recent studies indicate that the effects of SARS-CoV-2 extend beyond the respiratory system (RS) to the central neurological system (CNS) [18]. As a result, therapeutic strategies for combating coronavirus outbreaks are urgently needed [18]. The time-consuming and costly procedure of generating novel inhibitors makes drug repurposing and computer-assisted approaches increasingly attractive [14].
Given the time-consuming and expensive nature of developing new antiviral inhibitors from scratch, drug repurposing and in silico computer-assisted drug design have emerged as an increasingly attractive approach [14]. These strategies can help expedite the identification of effective treatments while mitigating the costs and risks typically associated with traditional drug development pathways.
1.2. The need for novel inhibitors
Existing inhibitors may suffer reduced efficacy, resistance development, or unfavourable adverse side effects [16]. Thus, pursuing novel inhibitor development will overcome these challenges and enhance therapeutic efficacy [16]. Above mentioned antiviral drugs, such as molnupiravir, remdesivir, and PF-07321332 (nirmatrelvir), have demonstrated clinical effectiveness [16], while prolonged use may lead to the emergence of drug-resistant mutations, highlighting the ongoing need for research and development of new inhibitors [17].
Other inhibitors, such as FB2001, SIM0417, vv116, and RAY003, primarily use a peptide-based covalent method. This method provides challenges in terms of target selectivity and pharmacokinetic features [22]. As a result, there is a rising emphasis on developing safer coronavirus antiviral drugs that are neither covalent nor peptidomimetic, such as Shionogi's S-217622 [22]. Compared to other inhibitors such as PF-07321332, S-217622 exhibits improved drug metabolism and pharmacokinetic properties, addressing certain current limitations [22]. While the US Food and Drug Administration (FDA)approved antivirals like boceprevir and telaprevir efficiently suppress Mpro, certain mutants have shown lower binding affinity to these drugs, especially at critical residues likely to undergo positive selection [23]. Novel coronavirus strains can potentially establish endemicity, posing a significant risk of another global pandemic necessitating the development of effective antiviral therapies [24].
Mpro is an essential target of many existing vaccines and antiviral medicines [25]. Despite improvements, challenges remain for advancing Mpro non-covalent inhibitors into clinical trials and eventual approval [26]. Weak inhibitory activity, the necessity for structurally diverse inhibitors, undesirable drug metabolism and pharmacokinetics (DMPK) characteristics, and toxicity concerns are also hurdles [27]. As a result, integrating modern computational approaches with experimental practice, considering numerous factors simultaneously in compound optimization, using efficient screening methods, paying attention to ligand effectiveness, and exploring non-catalytic sites could potentially overcome these challenges [27].
Computer-aided drug design (CADD) has shown considerable promise in identifying lead compounds from traditional medicinal plants, bridging the gap between traditional and modern medicine [25]. Various compounds discovered through virtual screening and drug repurposing using computational approaches have been developed to inhibit the Mpro’s active site [28]. Moreover, combining molecular dynamics (MD) simulations, artificial intelligence (AI), and other computational techniques enhances the prediction of drug efficacy and underscores the importance of in vivo studies to validate findings [28].
Non-covalent inhibitors of Mpro have attracted significant attention in SARS-CoV-2 inhibitors [15]. These inhibitors offer a combination of efficacy, selectivity, safety, and the potential to minimize resistance, making them a promising approach in antiviral drug development, particularly for diseases like COVID-19 caused by SARS-CoV-2 [15]. There has been a shift from earlier peptide-like inhibitors towards non-covalent inhibitors since the SARS-CoV-1 outbreak, which led to the development of promising inhibitors such as ML300 and ML188, as demonstrated in Figure 3 [15]. While covalent inhibitors like GC-376 and PF-00835231 have progressed to clinical trials during the COVID-19 pandemic, exploring non-covalent inhibitors remains essential for their potential therapeutic advantages [15].
This present review provides information through the development of non-covalent therapeutic candidates with potential activity in suppressing SARS-CoV-2's Mpro enzyme, with emphasis on inhibitor optimization and molecular dynamic (MD) simulations in the development of new inhibitors, particularly at the molecular level. This would contribute to understanding the mechanism of action and aid in designing more effective candidates, making this a potential research avenue.
2. Mpro inhibitors for SARS-CoV-2
2.1. Overview of Mpro Inhibitors
The Mpro enzyme of SARS-CoV-2 exhibits a 96% resemblance to SARS-CoV, as illustrated in Figure 4. SARS-CoV-2 and SARS-CoV have 294 amino acids in common among a total of 306 residues, in which Phe140, Leu141, Asn142, His163, and Glu166 amino acid residues create the S1 subsite, whereas His41-Cys145 catalytic dyad, besides Ser144 and Glu143, form the S1′ subsite, S2 comprises His164, Asp187, Arg188, Met49, Tyr54, while S3 is constituted by Leu167, Gln189, Met165, Thr190, and Gln192 [30].
Several studies have identified a range of existing compounds that successfully block SARS-CoV2 Mpro and prevent virus survival [20]. Among them, a potent noncovalent small molecule SARS-CoV Mpro inhibitor, N-(tert-Butyl)-2-(N-arylamido)-2-(pyridin-3-yl) Acetamides (ML188, Figure 5(a)) was discovered and synthesized by Jacob et al. [32] to inhibit SARS, which was later re-used to inhibit SARSCoV-2 [33]. In 2021, Lockbaum et al. produced the cocrystal complex structure of ML188 with SARSCoV2-Mpro, which exhibits structural similarities with the SARS-CoV1-Mpro-ML188 complex [20]. A higher binding affinity of the ML188 with Mpro of SARS-CoV2 was found in comparison to SARS-CoV1. Despite the general similarities, small changes in binding interactions lead to ML188's enhanced effectiveness against SARS-CoV2-Mpro, about twice as much as SARS-CoV1-Mpro [20]. The study revealed that ML188 binds with the catalytic residue His41 and has a specific packing in SARS-CoV2Mpro's subsite S2 [20], suggesting that ML188 can be a suitable non-covalent inhibitor scaffold [20].
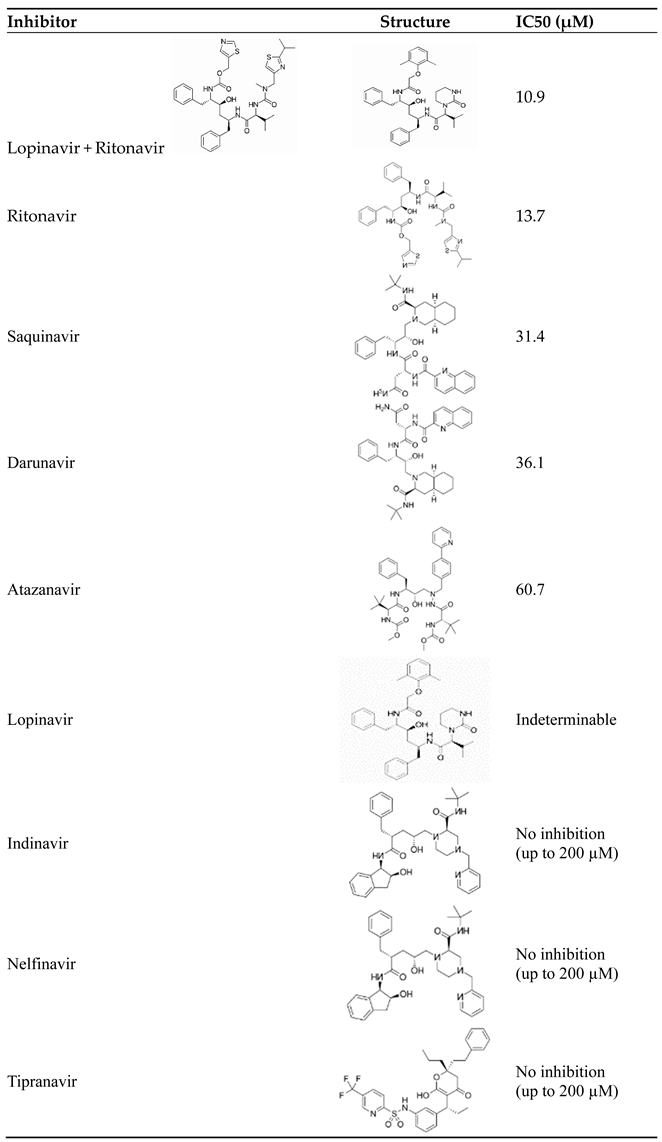
Table 1 collects existing inhibitors of the Mpro enzyme of SARS-CoV-2 [34]. Among these inhibitors, lopinavir and ritonavir combination had the lowest IC50 value (more potent) in cell culture but negatively affected cell survival, whereas other inhibitors, such as darunavir and atazanavir required substantially higher concentrations for inhibition, with no resulting cytotoxicity even at 200 µM [34]. Given these observations, the clinical use of HIV PIs to combat COVID-19 may be limited, and any beneficial effects may be attributed to their interactions with other molecular targets; thus, in vivo trials are needed to understand their actual clinical efficacy [34].
Sayed et al. explored the prospect of natural products, particularly structurally diverse microbialderived metabolites, as promising sources for potential drug candidates with a strong likelihood of modulating or inhibiting Mpro [35]. Figure 6 displays the preferred compounds (1–6) identified through virtual screening on the Mpro active site [35].
Thakur et al. applied structure-activity relationships (SAR) to investigate the active site subpockets (S1, S1′, S2, and S3) inside the SARS-CoV-2 Mpro (refer to Figure 7) to reveal key interactions for developing potent inhibitors [36], and presented four guidelines for developing highly potent inhibitors of the SARS-CoV-2 Mpro [36]. Rule 1: The S1 sub pocket requires a ring of five or six members with a proton donor or acceptor (-NH or ═O group) to make crucial hydrogen bonds with particular residues [36]. Rule 2: In the S1' sub pocket, which is known for hydration and design issues, it is critical to form a water bridge between the scaffold and Thr26, as well as hydrogen/covalent bonding with the 'Asn142-Gly143-Ser144-Cys145 (NGSC) motif' [36].
According to Thakur et al. (2022), the S2 sub-pocket favours cyclic moiety interactions with His41 or hydrogen bonding with His41 and Gln189, which increases efficacy. Rule 4: The S3 binding site must have a significant aromatic or aliphatic component [36]. It is found that while the S1 and S2 sub-pockets have significance for noncovalently bound inhibitors, the S1′ and S3 sub-pockets may affect inhibitor efficacy [36].
A group of newly designed thioesters intended for Mpro enzyme inhibition in SARS-CoV-2 [38] was developed by Pillaiyar et al. using virtual screening from The Tübingen Kinase Inhibitor Collection (TüKIC) database. These thioesters had inhibitory solid activity against SARS-CoV-2 Mpro, and their co-crystal structures provided insights into the target's molecular interactions [38]. Figure 8 reports the synthesis of thioesters 3w-z [38]. Particularly, compounds 3w and 3x substantially inhibit SARS-CoV-2 Mpro [38].
On the other hand, Verma et al. evaluated up to 73 withanolides and reference compounds using Lipinski's rule of five to determine their binding affinity in the active site of SARS-CoV-2 Mpro [40]. Nine molecules exhibit higher docking scores than the reference compounds [40]. The ADME tests were carried out to evaluate drug-likeness and bioavailability, and the screened compounds with favourable ADME characteristics may serve as potential leads for further therapeutic development [40].
2.2. Computer-aided drug design
Computer-aided drug design (CADD) based on existing drugs or inhibitors involves several key steps [25]. These steps help optimize the chemical structure to enhance the efficacy, selectivity, and pharmacokinetic properties of drug candidates [27]. The process begins with identifying a lead compound, such as ML188, from existing drugs or inhibitors with known biological activity and a specific biological target, such as Mpro [15]. The initial data is organized in databases using drug-like criteria to screen and identify potential candidates [28]. Techniques such as X-ray crystallography, NMR, or cryo-EM are employed to determine the 3D structure of the target [34]. The compounds are then subjected to an ensemble docking approach, which uses conformers generated during molecular dynamics simulations (MDS) [35] to calculate the binding free energy. This step predicts the strength and stability of the drug-target interaction [34]. Following this, the process involves synthesis, validation, refinement, and preclinical trials [34]. This strategy is crucial for identifying ligands with strong binding capabilities, especially considering the dynamic nature of the active site, and for confirming the findings [35].
Examination of the latest trends and advancements in drug design has made the use of computer screens grow in significance [16]. The successful discovery of antivirals that target illnesses such as HIV and HCV by attacking Mpro, has prompted the search for small compounds that can inhibit Mpro activity in treating SARS-CoV-2 [41]. For example, Ibrahim et al. employed a structure-based computational approach to identify potential Mpro inhibitors by focusing on the interactions between ligands and receptors [42]. Virtual screening was used to assess the binding affinity of terpene metabolites from a Red Sea coral reef ecosystem with SARS-CoV-2 Mpro [42]. Natural products, notably flavonoids, alkaloids, and terpenoids, play a significant role in the quest for efficient COVID19 therapies. Figure 9 shows the 2D structure of Lopinavir, Depresosterol, 178, and Erylosides B [42].
In a different study, up to eighty-one bioactive compounds were isolated from medicinal plants and tested for efficacy against SARS-CoV-2 Mpro using several in silico approaches, including quantum mechanics, molecular docking, and molecular dynamics simulations [43]. As shown in Figure 10, the average binding affinities of hesperidin, rutin, diosmin, and apiin exceeded the positive control compound, Nelfinavir's (−7.4 ± 0.6 kcal/mol) [43]. In a plan for future experimental examinations of these drugs/inhibitors obtained for COVID-19 therapy [43], in silico screening tools are on the list before further confirmation, such as in vitro protease activity assays and in vivo studies are conducted [43].
Computers can also help understand the binding of inhibitors. Hicks et al. found that the IC50 values of inhibitors are ranging from 6.8 μM to 138.0 μM [45]. Hicks et al. developed that aloesin and aloeresin D derived from the aloe plant as new scaffolds for Mpro inhibitors. Through simulation of the binding modes of the inhibitors, as shown in Figure 11, more details of the inhibitors are revealed.
The findings contribute to the development of novel molecular scaffolds for designing new inhibitors [45].
While traditional drug discovery approaches tend to be time-consuming, in silico-docking analysis offers a quicker, more reliable, and cost-effective solution for large-scale screening [47]. Due to the apparent structural similarity between SARS-CoV-2 and SARS-CoV and the recognition of Mpro as a fundamental CoV enzyme, molecular docking and molecular dynamics (MD) simulations were utilised to identify potential inhibitors for SARS-CoV-2 Mpro, with a focus on inhibitors previously reported for SARS-Mpro [48]. Motiwale et al. conducted docking investigations using existing SARS-CoV main protease inhibitors that showed high binding potency with SARS-CoV-2 Mpro. However, further experimental validation is essential to assess their suitability for clinical trials [48].
A molecular docking approach was adopted to screen a naturally small pool of phenolic compounds, resulting in a prediction of the primary binding modes and the identification of HIT-1 (corilagin) and HIT-2 (oxyresveratrol) as promising candidates, which is apparent in Figure 12 [49]. Extended molecular dynamics simulations boosted the drug-ligand complex's resilience, confirming HIT-1's potential and the strength of HIT-1 and HIT-2 for future drug development exploration [49]. Aleissa et al. demonstrated the effectiveness of computational tools in the early phases of drug development, providing cost-effective alternatives to traditional methods [49].
Hicks et al. identified aloesin and aloeresin D as unique inhibitors of SARS-CoV-2 Mpro after reviewing an extensive protease inhibitor library [45]. To gain a better understanding of how these compounds work, Hicks et al. conducted a thorough molecular docking analysis using the AutoDock Vina algorithm, which revealed these compounds as valuable candidates for developing Mpro inhibitors due to their safety profiles and adjustable functional groups [45]. For example, the estimated aloesin and aleoresin D affinities in Figure 11 are given by 7.5 and 6.8 kcal/mol, respectively [45].
2.3. Molecular dynamics (MD) in drug development
Computer-aided drug design includes a variety of in silico tools, such as MD simulations [50]. Beyond early drug discovery, MD simulation is one of the most important in silico tools in pharmaceutical development and formulation research, as it provides molecular-level insights to improve properties such as solubility and stability [50]. This section will concentrate on the role of MD simulations in developing Mpro inhibitors. By simulating the dynamic behavior of the protein-inhibitor complex, MD simulations help identify key interactions, such as hydrogen bonds, hydrophobic interactions, and van der Waals forces, that contribute to the inhibitor's efficacy [51]. These insights are crucial for optimizing inhibitor design, as they allow researchers to predict how modifications to the inhibitor might improve its binding affinity, specificity, or overall therapeutic potential [50]. With continued advances in machine learning (ML), drug development has an increased dependence on MD simulations to produce novel pharmacological therapeutics [50].
The stability of top potent lead compounds from Withania sps. in the active site of Mpro of SARSCoV-2 are studied using MD simulation. Although the lead compounds need further in-vitro analysis, the 50 ns MD simulation confirmed the stability of an inhibitor - withacoagulin H (W30) - with a binding energy of -63.463 KJ/mol in Figure 13 [40]. This inhibitor becomes the compound of choice among the screened compounds as W30 possesses an excellent binding affinity compared to other compounds with the Mpro of SARS-CoV-2 [40].
Inhibitors of SARS-CoV-2 Mpro are also screened from marine sources using MD simulations and binding energy calculations [42]. A computer screen for 227 terpenes revealed that six of them, notably erylosides B, had a substantial inhibitory potential against Mpro, with a binding affinity of as large as -51.9 kcal/mol with respect to -33.6 kcal/mol for Lopinavir [42] from a 100 ns MD simulation, as shown in Figure 14 which verified the stability of the complex generated by erylosides B and Mpro [42].
Chemical structures of newly synthesized sulfonamide SARS-CoV-2 Mpro inhibitors are given in Figure 15, together with the structures of N3 and Remdesivir as references [51]. The study revealed favourable binding energy values and significant ligand effectiveness in inhibiting Mpro, superior to conventional inhibitors N3 and remdesivir [51]. The binding energy of remdesivir against the SARSCoV-2 Mpro (PDB ID: 6Y84) is -9.85 kcal/mol [51]. improved binding capabilities in comparison to remdesivir's potency (-9.85 kcal/mol) against the SARS-CoV-2 main protease (PDB ID: 6Y84) [51].
Other studies have prescreened compounds capable of inhibiting the activity of Mpro of SARSCoV-2 [53]. For example, Rutin in Figure 16, a flavonoid derivative with a high binding affinity to the virus, can be a good candidate [53]. MD simulations and docking investigations reveal that the interaction between rutin and Mpro is confirmed by the alignment with the 6GLU7 binding site [53] 6GLU7 binding site [53].
MD simulation has also been employed to estimate the ligand properties by calculating the parameters such as ligand root mean square deviation (RMSD), molecular surface area (MolSA), the radius of gyration (rGyr), polar surface area (PSA) and solvent-accessible surface area (SASA) [55]. The RMSD values fluctuated until 5 and 10 ns for Comp.1-6LU7 complex before gradually reaching an equilibrium as given in Figure 17 [55].
2.4. Leveraging quantum mechanics for ADME-optimized drugs
In addition to pre-screening of inhibitors for particular proteins using molecular docking and MD simulations as well as other in silico tools, these methods can also provide valuable insights into druglikeness, pharmacokinetics, toxicity profiles and other properties at the molecular level, which are essential for optimizing new drug candidates [57]. Many inhibitors can be small flexible compounds, such as some epidermal growth factor receptor (EGFR) inhibitors [58-63], noncovalent small molecule Mpro inhibitors, including ML188 and its potent derivative 23R [64]. As a result, a detailed quantum mechanical study of the inhibitors and their conformations becomes necessary.
Quantum mechanics (QM) density functional theory (DFT) study of inhibitors and drugs has been widely applied to obtain quantitative structure-property relationship (QSRP) of inhibitors in drug design [61]. DFT allows for the accurate prediction of electronic properties, enabling detailed insights into the conformation, reactivity, spectroscopy and interaction of inhibitors with target proteins at an atomic level [61]. These properties are associated with ADMET properties as inhibitors and drugs [61]. Many inhibitors' drug potency and binding are closely related to their flexibility and conformational space. However, conformational sampling still has major hurdles for effective drug design methods [61].
Most of the algorithms in drug design are based on geometry in three-dimensional (3D) space, for the binding sites are always located on the concave surface of the proteins [65]. Estimating the range of 3D conformations of a molecule is a key component of computer-aided drug design [66]. As summarized by Habgood et al.[66] that three major challenges in drug design once the scaffold has been identified, are (1) the development of a good method to generate ensembles of a molecule's bioactive conformation; (2) rational analysis and modification of a pre-established bioactive conformation; and (3) approximation of real solution phase conformational ensembles in tandem with spectroscopic data such as IR, NMR and UV–vis. Although crystal structures usually provide single 3D geometries, they do not cover the full conformational space of the preferred conformers of a flexible inhibitor with multiple rotatable single bonds or unsaturated ring systems [66].
The solution is to develop a conformational ensemble. For this reason, Wang and Vasilyev [61] developed a robust QM conformational sampling method/program (PrefQMCon), which is able for QM determination of a large number of target drug conformer clusters and their properties given in Figure 18(a) [61]. These conformers of the AG-1478 EGFR inhibitor can be superpositioned in Figure 18(b).
Numerous studies using DFT calculations are able to predict various spectroscopic properties, such as IR, NMR, and UV-Vis spectra and their possible link to the potency of the inhibitors. Figure 19 presents the correlation between optical spectral shift and the potency of the EGFR tyrosine kinase inhibitors (TKIs) with respect to halogen substitutions [62]. In the figure, the larger the shifts in their optical spectra (the orange colour in the figure) between the global minimum structures and their corresponding crystal structures from the EGFR database, the more potent the inhibitors, that is (negative) larger the ln(IC50) function [62]. The more potent TKIs, TKI-Cl and TKI-Br, behave very differently from the less potent inhibitors. These predictions are valuable for characterising inhibitors and validating their structure experimentally.
Non-covalent small molecule inhibitors of SARS-CoV-2 Mpro, such as ML188 (see Figure 5(a)), are flexible compounds. ML188 contains several single bonds, contributing to a wide range of possible conformers. Rigorous benchmarking studies [62] have shown that DFT methods, with appropriately chosen exchange-correlation functionals and basis sets, can accurately predict the molecular properties of such inhibitors. These properties are crucial for understanding their drug-like behaviour. This approach can also be extended to study other inhibitors of SARS-CoV-2 Mpro, including ML188, to enhance drug development efforts.
3. Summary and outlook
This article updates recent efforts in developing inhibitory compounds targeting the main protease (Mpro) of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus responsible for the COVID-19 pandemic. The study focuses on various approaches for the identification and development of potential drug candidates, particularly in silico studies such as molecular dynamics (MD) simulations and quantum mechanical (QM) density functional theory (DFT) calculations in recent years.
Potential Mpro inhibitors can be derived from a range of sources, including natural products, peptides, and synthetic molecules. In this discovery process, in silico techniques have become indispensable computational experiments in addition to physical experiments. MD simulations provide a dynamic perspective on biomolecular systems, offering insights into molecules' movement and conformational changes over time. This understanding is crucial for assessing the flexibility of drug targets and ligands.
MD simulations reveal details at atomic-level interactions, such as hydrogen bonds, hydrophobic contacts, and ionic interactions, which are essential for identifying key residues involved in binding. MD simulations also provide valuable information about various ligand properties. When combined with QM-based DFT calculations, the information offers deeper insights into inhibitors, aiding in the further development of drugs.
However, existing studies of effective inhibitors targeting SARS-CoV-2 Mpro provided limited information on inhibitors as scaffolds for new inhibitor development. When combined with significant computation demand and human work [66] in the process, it presents a bottleneck challenge in response to the request of the urgent need for inhibitors targeting the SARS-CoV-2 virus. One of the urgent needs is to develop a robust process to develop new inhibitors or to rediscover existing drugs that can inhibit the SARS-CoV-2 virus efficiently.
Author Contributions
The review was written with contributions from all authors who have approved the final version.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflicts of interest in this research.
Acknowledgment
IA acknowledges Swinburne University of Technology Tuition Fee Scholarship (TFS).
References
- Kumar, D., G. Chauhan, S. Kalra, B. Kumar, and M.S. Gill, A perspective on potential target proteins of COVID-19: Comparison with SARS-CoV for designing new small molecules. Bioorganic chemistry, 2020. 104: p. 104326. [CrossRef]
- Liu, Y., C. Liang, L. Xin, X. Ren, L. Tian, X. Ju, H. Li, Y. Wang, Q. Zhao, and H. Liu, The development of Coronavirus 3C-Like protease (3CLpro) inhibitors from 2010 to 2020. European journal of medicinal chemistry, 2020. 206: p. 112711. [CrossRef]
- Sharma, P., V. Vijayan, P. Pant, M. Sharma, N. Vikram, P. Kaur, T. Singh, and S. Sharma, Identification of potential drug candidates to combat COVID-19: a structural study using the main protease (mpro) of SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 2021. 39(17): p. 6649-6659. [CrossRef]
- Krammer, F., SARS-CoV-2 vaccines in development. Nature, 2020. 586(7830): p. 516-527. [CrossRef]
- Sharun, K., R. Tiwari, and K. Dhama, Protease inhibitor GC376 for COVID-19: Lessons learned from feline infectious peritonitis. Annals of medicine and surgery, 2021. 61: p. 122-125. [CrossRef]
- Badavath, V.N., A. Kumar, P.K. Samanta, S. Maji, A. Das, G. Blum, A. Jha, and A. Sen, Determination of potential inhibitors based on isatin derivatives against SARS-CoV-2 main protease (mpro): a molecular docking, molecular dynamics and structure-activity relationship studies. Journal of Biomolecular Structure and Dynamics, 2022. 40(7): p. 3110-3128. [CrossRef]
- Astuti, I., Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes & Metabolic Syndrome: Clinical Research & Reviews, 2020. 14(4): p. 407-412. [CrossRef]
- Mondol, W.C., Exploring the potential of organic molecules in the treatment of covid-19. 2021, Brac University.
- Xu, Y.-S., J.-Z. Chigan, J.-Q. Li, H.-H. Ding, L.-Y. Sun, L. Liu, Z. Hu, and K.-W. Yang, Hydroxamate and thiosemicarbazone: Two highly promising scaffolds for the development of SARS-CoV-2 antivirals. Bioorganic Chemistry, 2022. 124: p. 105799. [CrossRef]
- Dai, W., B. Zhang, X.-M. Jiang, H. Su, J. Li, Y. Zhao, X. Xie, Z. Jin, J. Peng, and F. Liu, Structurebased design, synthesis and biological evaluation of peptidomimetic aldehydes as a novel series of antiviral drug candidates targeting the SARS-CoV-2 main protease. BioRxiv, 2020. [CrossRef]
- Li, X., M. Geng, Y. Peng, L. Meng, and S. Lu, Molecular immune pathogenesis and diagnosis of COVID-19. Journal of pharmaceutical analysis, 2020. 10(2): p. 102-108. [CrossRef]
- Mengist, H.M., T. Dilnessa, and T. Jin, Structural basis of potential inhibitors targeting SARSCoV-2 main protease. Frontiers in Chemistry, 2021. 9: p. 622898. [CrossRef]
- Adelusi, T.I., A.-Q.K. Oyedele, O.E. Monday, I.D. Boyenle, M.O. Idris, A.T. Ogunlana, A.M. Ayoola, J.O. Fatoki, O.E. Kolawole, and K.B. David, Dietary polyphenols mitigate SARS-CoV-2 main protease (Mpro)–Molecular dynamics, molecular mechanics, and density functional theory investigations. Journal of molecular structure, 2022. 1250: p. 131879. [CrossRef]
- Muratov, E.N., R. Amaro, C.H. Andrade, N. Brown, S. Ekins, D. Fourches, O. Isayev, D. Kozakov, J.L. Medina-Franco, and K.M. Merz, A critical overview of computational approaches employed for COVID-19 drug discovery. Chemical Society Reviews, 2021. 50(16): p. 9121-9151. [CrossRef]
- Han, S.H., C.M. Goins, T. Arya, W.-J. Shin, J. Maw, A. Hooper, D.P. Sonawane, M.R. Porter, B.E. Bannister, R.D. Crouch, A.A. Lindsey, G. Lakatos, S.R. Martinez, J. Alvarado, W.S. Akers, N.S.
- Wang, J.U. Jung, J.D. Macdonald, and S.R. Stauffer, Structure-Based Optimization of ML300-Derived, Noncovalent Inhibitors Targeting the Severe Acute Respiratory Syndrome Coronavirus 3CL Protease (SARS-CoV-2 3CLpro). Journal of Medicinal Chemistry, 2022. 65(4): p. 2880-2904. [CrossRef]
- Fernandes, Q., V.P. Inchakalody, M. Merhi, S. Mestiri, N. Taib, D. Moustafa Abo El-Ella, T. Bedhiafi, A. Raza, L. Al-Zaidan, M.O. Mohsen, M.A. Yousuf Al-Nesf, A.A. Hssain, H.M. Yassine, M.F. Bachmann, S. Uddin, and S. Dermime, Emerging COVID-19 variants and their impact on SARS-CoV-2 diagnosis, therapeutics and vaccines. Annals of Medicine, 2022. 54(1): p. 524-540. [CrossRef]
- Cooper, M.S., L. Zhang, M. Ibrahim, K. Zhang, X. Sun, J. Röske, M. Göhl, M. Brönstrup, J.K. Cowell, and L. Sauerhering, Diastereomeric Resolution Yields Highly Potent Inhibitor of SARSCoV-2 Main Protease. Journal of Medicinal Chemistry, 2022. 65(19): p. 13328-13342. [CrossRef]
- Alexandris, N., G. Lagoumintzis, C.T. Chasapis, D.D. Leonidas, G.E. Papadopoulos, S.J. Tzartos, A. Tsatsakis, E. Eliopoulos, K. Poulas, and K. Farsalinos, Nicotinic cholinergic system and COVID19: In silico evaluation of nicotinic acetylcholine receptor agonists as potential therapeutic interventions. Toxicology reports, 2021. 8: p. 73-83. [CrossRef]
- Yang, F., T.N.-A. Tran, E. Howerton, M.F. Boni, and J.L. Servadio, Benefits of near-universal vaccination and treatment access to manage COVID-19 burden in the United States. BMC Medicine, 2023. 21(1): p. 321. [CrossRef]
- Lockbaum, G.J., A.C. Reyes, J.M. Lee, R. Tilvawala, E.A. Nalivaika, A. Ali, N. Kurt Yilmaz, P.R. Thompson, and C.A. Schiffer, Crystal structure of SARS-CoV-2 main protease in complex with the non-covalent inhibitor ML188. Viruses, 2021. 13(2): p. 174. [CrossRef]
- Li, Q. and C. Kang, Progress in developing inhibitors of SARS-CoV-2 3C-like protease. Microorganisms, 2020. 8(8): p. 1250. [CrossRef]
- Wu, Q., S. Yan, Y. Wang, M. Li, Y. Xiao, and Y. Li, Discovery of 4′-O-methylscutellarein as a potent SARS-CoV-2 main protease inhibitor. Biochemical and biophysical research communications, 2022. 604: p. 76-82.
- Naidu, S.A.G., Y.B. Tripathi, P. Shree, R.A. Clemens, and A.S. Naidu, Phytonutrient Inhibitors of SARS-CoV-2/NSP5-Encoded Main Protease (Mpro) Autocleavage Enzyme Critical for COVID-19 Pathogenesis. Journal of dietary supplements, 2023. 20(2): p. 284-311. [CrossRef]
- Kouam, A.F., F.D. Mabou, L. Fu, R.R. Koagne, Y. Li, B.A. Owona, E.M. Zeuko'o, A.G.K. Fepa, B.R.T. Galani, F. Reyes, F.N. Njayou, P.F. Moundipa, and G.F. Gao, Insights into the inhibition mechanisms of MERS-CoV and SARS-CoV2 papain-like proteases by inhibitors from Crinum distichum: In vitro and in silico analysis. South African Journal of Botany, 2024. 165: p. 290306. [CrossRef]
- Ebenezer, O. and M. Shapi, Promising inhibitors against main protease of SARS CoV-2 from medicinal plants: In silico identification. Acta Pharmaceutica, 2022. 72(2): p. 159-169. [CrossRef]
- Zhu, J., H. Zhang, Q. Lin, J. Lyu, L. Lu, H. Chen, X. Zhang, Y. Zhang, and K. Chen, Progress on SARS-CoV-2 3CLpro Inhibitors: Inspiration from SARS-CoV 3CLpro Peptidomimetics and SmallMolecule Anti-Inflammatory Compounds. Drug Design, Development and Therapy, 2023. 16(null): p. 1067-1082. [CrossRef]
- Song, L., S. Gao, B. Ye, M. Yang, Y. Cheng, D. Kang, F. Yi, J.-P. Sun, L. Menéndez-Arias, and J. Neyts, Medicinal chemistry strategies towards the development of non-covalent SARS-CoV-2 Mpro inhibitors. Acta Pharmaceutica Sinica B, 2023. [CrossRef]
- Singh, E., R.J. Khan, R.K. Jha, G.M. Amera, M. Jain, R.P. Singh, J. Muthukumaran, and A.K. Singh, A comprehensive review on promising anti-viral therapeutic candidates identified against main protease from SARS-CoV-2 through various computational methods. Journal of Genetic Engineering and Biotechnology, 2020. 18(1): p. 69. [CrossRef]
- Reprinted (adapted) with permission from Structure-Based Optimization of ML300-Derived, Noncovalent Inhibitors Targeting the Severe Acute Respiratory Syndrome Coronavirus 3CL Protease (SARS-CoV-2 3CLpro). Sang Hoon Han, Christopher M. Goins, Tarun Arya, Woo-Jin Shin, Joshua Maw, Alice Hooper, Dhiraj P. Sonawane, Matthew R. Porter, Breyanne E. Bannister, Rachel D. Crouch, A. Abigail Lindsey, Gabriella Lakatos, Steven R. Martinez, Joseph Alvarado, Wendell S. Akers, Nancy S. Wang, Jae U. Jung, Jonathan D. Macdonald, and Shaun Journal of Medicinal Chemistry 2022 65 (4), 2880-2904. Copyright 2022 American Chemical Society. [CrossRef]
- Tumskiy, R.S. and A.V. Tumskaia, Multistep rational molecular design and combined docking for discovery of novel classes of inhibitors of SARS-CoV-2 main protease 3CLpro. Chemical physics letters, 2021. 780: p. 138894. [CrossRef]
- Fischer, A., M. Sellner, S. Neranjan, M. Smieško, and M.A. Lill, Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds. International Journal of Molecular Sciences, 2020. 21(10): p. 3626. [CrossRef]
- Jacobs, J., V. Grum-Tokars, Y. Zhou, M. Turlington, S.A. Saldanha, P. Chase, A. Eggler, E.S. Dawson, Y.M. Baez-Santos, and S. Tomar, Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. Journal of medicinal chemistry, 2013. 56(2): p. 534-546. [CrossRef]
- Berry, M., B. Fielding, and J. Gamieldien, Human coronavirus OC43 3CL protease and the potential of ML188 as a broad-spectrum lead compound: Homology modelling and molecular dynamic studies. BMC structural biology, 2015. 15: p. 1-10. [CrossRef]
- Mahdi, M., J.A. Mótyán, Z.I. Szojka, M. Golda, M. Miczi, and J. Tőzsér, Analysis of the efficacy of HIV protease inhibitors against SARS-CoV-2′s main protease. Virology Journal, 2020. 17(1): p. 190. [CrossRef]
- Sayed, A.M., H.A. Alhadrami, A.O. El-Gendy, Y.I. Shamikh, L. Belbahri, H.M. Hassan, U.R. Abdelmohsen, and M.E. Rateb, Microbial natural products as potential inhibitors of SARS-CoV2 main protease (Mpro). Microorganisms, 2020. 8(7): p. 970. [CrossRef]
- Thakur, A., G. Sharma, V.N. Badavath, V. Jayaprakash, K.M. Merz, Jr., G. Blum, and O. Acevedo, Primer for Designing Main Protease (Mpro) Inhibitors of SARS-CoV-2. The Journal of Physical Chemistry Letters, 2022. 13(25): p. 5776-5786. [CrossRef]
- Reprinted (adapted) with permission from Primer for Designing Main Protease (Mpro) Inhibitors of SARS-CoV-2. Abhishek Thakur, Gaurav Sharma, Vishnu Nayak Badavath, Venkatesan Jayaprakash, Kenneth M. Merz Jr., Galia Blum, and Orlando Acevedo. The Journal of Physical Chemistry Letters 2022 13 (25), 5776-5786. Copyright 2022 American Chemical Society. [CrossRef]
- Pillaiyar, T., P. Flury, N. Krüger, H. Su, L. Schäkel, E. Barbosa Da Silva, O. Eppler, T. Kronenberger, T. Nie, S. Luedtke, C. Rocha, K. Sylvester, M.R.I. Petry, J.H. McKerrow, A. Poso, Pöhlmann, M. Gütschow, A.J. O’Donoghue, Y. Xu, C.E. Müller, and S.A. Laufer, SmallMolecule Thioesters as SARS-CoV-2 Main Protease Inhibitors: Enzyme Inhibition, Structure– Activity Relationships, Antiviral Activity, and X-ray Structure Determination. Journal of Medicinal Chemistry, 2022. 65(13): p. 9376-9395. [CrossRef]
- Reprinted (adapted) with permission from Small-Molecule Thioesters as SARS-CoV-2 Main Protease Inhibitors: Enzyme Inhibition, Structure–Activity Relationships, Antiviral Activity, and X-ray Structure Determination Thanigaimalai Pillaiyar, Philipp Flury, Nadine Krüger, Haixia Su, Laura Schäkel, Elany Barbosa Da Silva, Olga Eppler, Thales Kronenberger, Tianqing Nie, Stephanie Luedtke, Cheila Rocha, Katharina Sylvester, Marvin R.I. Petry, James H. McKerrow, Antti Poso, Stefan Pöhlmann, Michael Gütschow, Anthony J. O’Donoghue, Yechun Xu, Christa E. Müller, and Stefan A. Laufer. Journal of Medicinal Chemistry 2022 65 (13), 9376-9395. Copyright 2022 American Chemical Society. [CrossRef]
- Verma, S., C.N. Patel, and M. Chandra, Identification of novel inhibitors of SARS-CoV-2 main protease (Mpro) from Withania sp. by molecular docking and molecular dynamics simulation. Journal of computational chemistry, 2021. 42(26): p. 1861-1872. [CrossRef]
- Amin, S.A., S. Banerjee, S. Singh, I.A. Qureshi, S. Gayen, and T. Jha, First structure–activity relationship analysis of SARS-CoV-2 virus main protease (Mpro) inhibitors: an endeavor on COVID-19 drug discovery. Molecular diversity, 2021. 25(3): p. 1827-1838. [CrossRef]
- Ibrahim, M.A.A., A.H.M. Abdelrahman, T.A. Mohamed, M.A.M. Atia, M.A.M. Al-Hammady, K.A.A. Abdeljawaad, E.M. Elkady, M.F. Moustafa, F. Alrumaihi, K.S. Allemailem, H.R. El-Seedi, P.W. Paré, T. Efferth, and M.-E.F. Hegazy, In Silico Mining of Terpenes from Red-Sea Invertebrates for SARS-CoV-2 Main Protease (Mpro) Inhibitors. Molecules, 2021. 26(7): p. 2082. [CrossRef]
- Adem, Ş., V. Eyupoglu, I.M. Ibrahim, I. Sarfraz, A. Rasul, M. Ali, and A.A. Elfiky, Multidimensional in silico strategy for identification of natural polyphenols-based SARS-CoV-2 main protease (Mpro) inhibitors to unveil a hope against COVID-19. Computers in biology and medicine, 2022. 145: p. 105452-105452. [CrossRef]
- Reprinted from Computers in Biology and Medicine, Vol 145, Adem, Şevki; Eyupoglu, Volkan; Ibrahim, Ibrahim M.; Sarfraz, Iqra; Rasul, Azhar; Ali, Muhammad; Elfiky, Abdo A., Multidimensional in silico strategy for identification of natural polyphenols-based SARS-CoV2 main protease (Mpro) inhibitors to unveil a hope against COVID-19, 105452-105452, Copyright 2022, with permission from Elsevier. [CrossRef]
- Hicks, E.G., S.E. Kandel, and J.N. Lampe, Identification of Aloe-derived natural products as prospective lead scaffolds for SARS-CoV-2 main protease (Mpro) inhibitors. Bioorganic & Medicinal Chemistry Letters, 2022. 66: p. 128732.
- Reprinted from Bioorganic & Medicinal Chemistry Letters, Vol 66, Hicks,Emily G. Kandel,Sylvie E. Lampe,Jed N., Identification of Aloe-derived natural products as prospective lead scaffolds for SARS-CoV-2 main protease (Mpro) inhibitors, 128732, Copyright 2022, with permission from Elsevier. [CrossRef]
- Vincent, S., S. Arokiyaraj, M. Saravanan, and M. Dhanraj, Molecular docking studies on the anti-viral effects of compounds from Kabasura Kudineer on SARS-CoV-2 3CLpro. Frontiers in molecular biosciences, 2020. 7: p. 613401. [CrossRef]
- Motiwale, M., N.S. Yadav, S. Kumar, T. Kushwaha, G. Choudhir, S. Sharma, and P.K. Singour, Finding potent inhibitors for COVID-19 main protease (Mpro): an in silico approach using SARSCoV-3CL protease inhibitors for combating CORONA. Journal of Biomolecular Structure and Dynamics, 2022. 40(4): p. 1534-1545. [CrossRef]
- Aleissa, M.S., M. Al-Zharani, M.S. Hasnain, and S. Alkahtani, Screening, molecular simulation & in silico kinetics of virtually designed covid-19 main protease inhibitors. Journal of King Saud University - Science, 2022. 34(8): p. 102283. [CrossRef]
- Salo-Ahen, O.M.H., I. Alanko, R. Bhadane, A.M.J.J. Bonvin, R.V. Honorato, S. Hossain, A.H. Juffer, A. Kabedev, M. Lahtela-Kakkonen, A.S. Larsen, E. Lescrinier, P. Marimuthu, M.U. Mirza, G. Mustafa, A. Nunes-Alves, T. Pantsar, A. Saadabadi, K. Singaravelu, and M. Vanmeert, Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development. Processes, 2021. 9(1): p. 71. [CrossRef]
- Sarfraz, M., A. Rauf, P. Keller, and A.M. Qureshi, N,N'-dialkyl-2-thiobarbituric acid based sulfonamides as potential SARS-CoV-2 main protease inhibitors. Canadian Journal of Chemistry, 2021. 99: p. 330+. [CrossRef]
- Reprinted (adapted) with permission from Verma, Surjeet. Patel, Chirag N. Chandra, Muktesh., Identification of novel inhibitors of SARS-CoV-2 main protease (Mpro) from Withania sp. by molecular docking and molecular dynamics simulation, Journal of Computational Chemistry, copyright © 2021 Wiley Periodicals LLC. All rights preserved.
- Agrawal, P.K., C. Agrawal, and G. Blunden, Rutin: A Potential Antiviral for Repurposing as a SARS-CoV-2 Main Protease (Mpro) Inhibitor. Natural product communications, 2021. 16(4): p. 1934578. [CrossRef]
- Adapted from Rutin: A Potential Antiviral for Repurposing as a SARS-CoV-2 Main Protease (Mpro) Inhibitor by Agrawal, Pawan K. Agrawal, Chandan. Blunden, Gerald, used under Creative Commons Attribution 4.0 International License. No changes were made.
- Mohaptra, R.K., K. Dhama, A.A. El-Arabey, A.K. Sarangi, R. Tiwari, T. Bin Emran, M. Azam, M.K. Raval, V. Seidel, and M. Abdalla, Repurposing benzimidazole and benzothiazole derivatives as potential inhibitors of SARS-CoV-2: DFT, QSAR, molecular docking, molecular dynamics simulation, and in-silico pharmacokinetic and toxicity studies. Journal of King Saud UniversityScience, 2021. 33(8). [CrossRef]
- Adapted from Repurposing benzimidazole and benzothiazole derivatives as potential inhibitors of SARS-CoV-2: DFT, QSAR, molecular docking, molecular dynamics simulation, and in-silico pharmacokinetic and toxicity studies by R. K. Mohaptra, K. Dhama, A. A. El-Arabey, A. K. Sarangi, R. Tiwari, T. Bin Emran, et al. Journal of King Saud University-Science, 2021. 33(8), used under Creative Commons Attribution 4.0 International License. No changes were made.
- Elgohary, A.M., A.A. Elfiky, F. Pereira, T.M. Abd El-Aziz, M. Sobeh, R.K. Arafa, and A. El-Demerdash, Investigating the structure-activity relationship of marine polycyclic batzelladine alkaloids as promising inhibitors for SARS-CoV2 main protease (Mpro). Computers in biology and medicine, 2022. 147: p. 105738-105738. [CrossRef]
- Khattab, M., C. Chatterjee, A. Clayton, and F. Wang, Tyrosine kinase inhibitor (AG-1478): two conformers of a tyrosine kinase inhibitor (AG-1478) disclosed using simulated UV-vis absorption spectroscopy. New J. Chem, 2016. 40: p. 8296.
- Khattab, M., F. Wang, and A.H. Clayton, UV–Vis spectroscopy and solvatochromism of the tyrosine kinase inhibitor AG-1478. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 2016. 164: p. 128-132. [CrossRef]
- Khattab, M., F. Wang, and A.H. Clayton, A pH-induced conformational switch in a tyrosine kinase inhibitor identified by electronic spectroscopy and quantum chemical calculations. Scientific Reports, 2017. 7(1): p. 16271. [CrossRef]
- Wang, F. and V. Vasilyev, Accelerating optical reporting for conformation of tyrosine kinase inhibitors in solutions. International Journal of Quantum Chemistry, 2021. 121(20): p. e26765. [CrossRef]
- Alagawani, S., V. Vasilyev, and F. Wang, Optical spectra of EGFR inhibitor AG-1478 for benchmarking DFT functionals. Electronic Structure, 2023. 5(2): p. 024011. [CrossRef]
- Alagawani, S., V. Vasilyev, A.H. Clayton, and F. Wang, Insights into Halogen-Induced Changes in 4-Anilinoquinazoline EGFR Inhibitors: A Computational Spectroscopic Study. Molecules, 2024. 29(12): p. 2800. [CrossRef]
- Ashraf-Uz-Zaman, M., T.K. Chua, X. Li, Y. Yao, B.K. Moku, C.B. Mishra, V. Avadhanula, P.A. Piedra, and Y. Song, Design, Synthesis, X-ray Crystallography, and Biological Activities of Covalent, Non-Peptidic Inhibitors of SARS-CoV-2 Main Protease. ACS Infectious Diseases, 2024. 10(2): p. 715-731. [CrossRef]
- Wang, K., J. Gao, S. Shen, J.A. Tuszynski, J. Ruan, and G. Hu, An Accurate Method for Prediction of Protein-Ligand Binding Site on Protein Surface Using SVM and Statistical Depth Function. BioMed Research International, 2013. 2013(1): p. 409658. [CrossRef]
- Habgood, M., T. James, and A. Heifetz, Conformational searching with quantum mechanics. Quantum Mechanics in Drug Discovery, 2020: p. 207-229. [CrossRef]
- Reprinted (adapted) with permission from Wang, Vasilyev., Accelerating optical reporting for conformation of tyrosine kinase inhibitors in solutions, International Journal of Quantum Chemistry, copyright © 2021 Wiley Periodicals LLC. All rights are preserved.
Figure 1.
Basic structure of the SARS-CoV-2 virus responsible for the COVID-19 pandemic [7]. Licensed under CCBY license.
Figure 1.
Basic structure of the SARS-CoV-2 virus responsible for the COVID-19 pandemic [7]. Licensed under CCBY license.

Figure 2.
The crystal structure of unbound SARS-CoV-2 Mpro (PDB entry: 6Y2E) and the pocket dedicated to substrate interaction [12] Licensed under CC-BY license.
Figure 2.
The crystal structure of unbound SARS-CoV-2 Mpro (PDB entry: 6Y2E) and the pocket dedicated to substrate interaction [12] Licensed under CC-BY license.
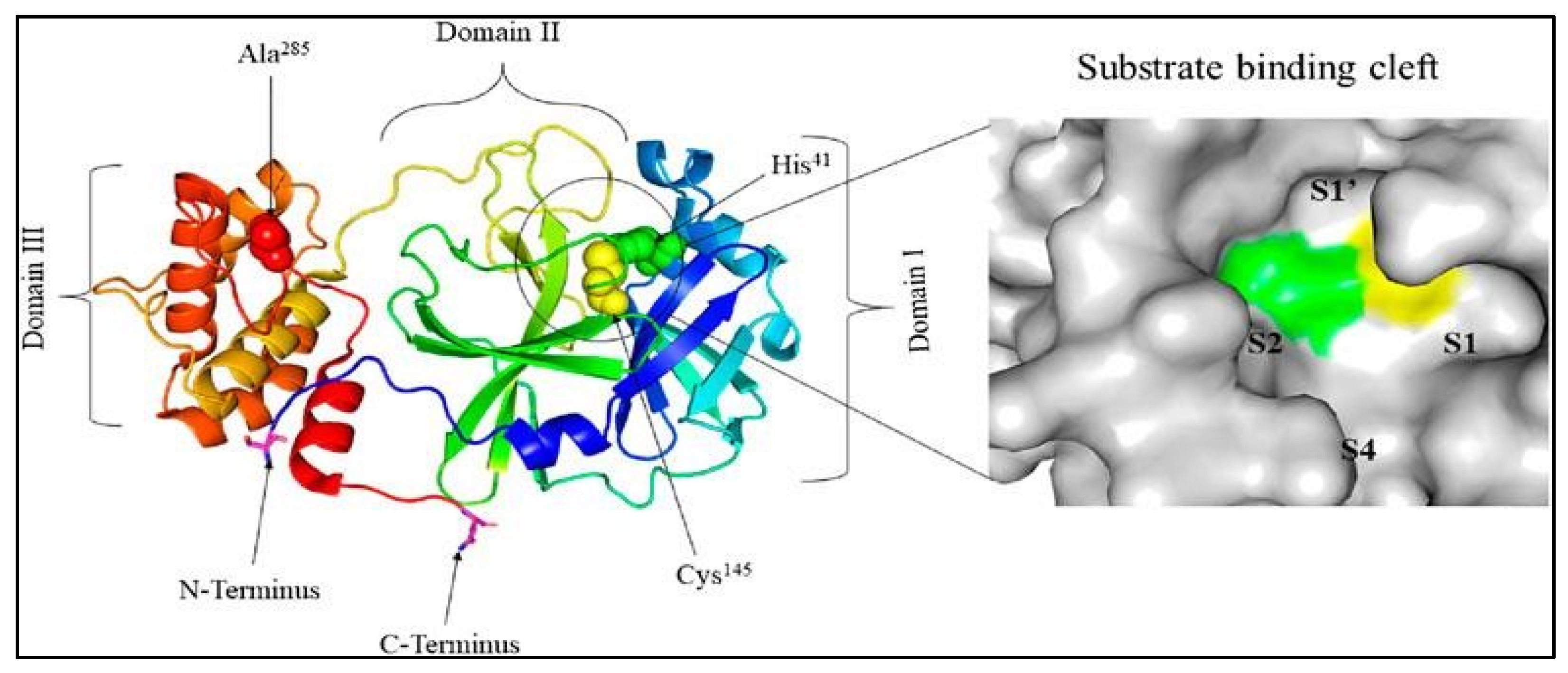
Figure 3.
Representative structures of identified 3CLpro inhibitors for SARS-CoV-2 using either a noncovalent (top) or covalent (bottom) mechanism of action [29].
Figure 3.
Representative structures of identified 3CLpro inhibitors for SARS-CoV-2 using either a noncovalent (top) or covalent (bottom) mechanism of action [29].
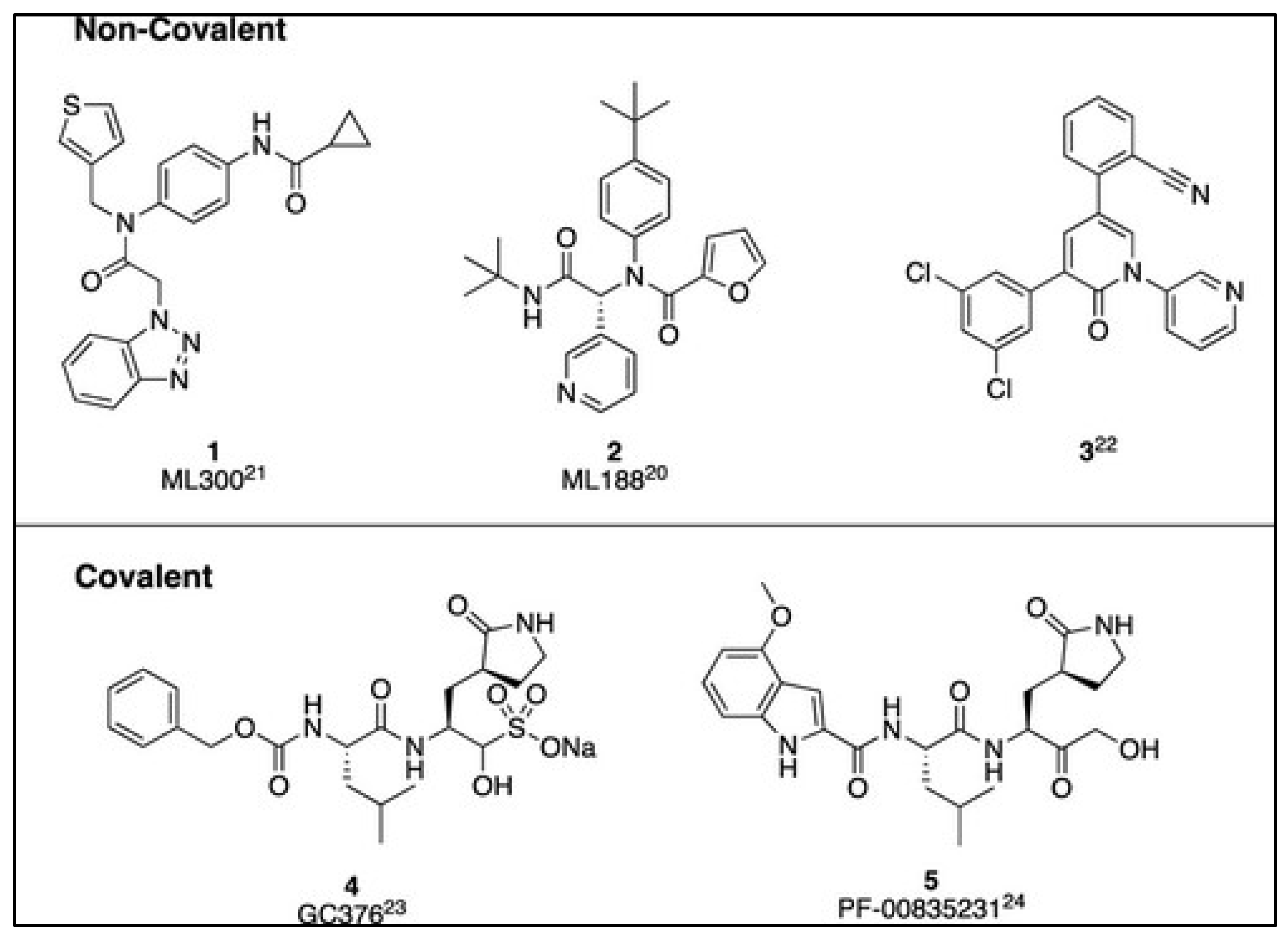
Figure 4.
The architecture of the Mpro enzyme of both viruses (SARS-CoV2 and SARS-CoV): (a) Mpro domains are presented (I, II, and III, coloured green, grey, and red, respectively). The active site pockets of the SARS-CoV2 Mpro (b) and SARS-CoV Mpro (c) PDB ID 6LU7, respectively [31] Licensed under CC-BY License.
Figure 4.
The architecture of the Mpro enzyme of both viruses (SARS-CoV2 and SARS-CoV): (a) Mpro domains are presented (I, II, and III, coloured green, grey, and red, respectively). The active site pockets of the SARS-CoV2 Mpro (b) and SARS-CoV Mpro (c) PDB ID 6LU7, respectively [31] Licensed under CC-BY License.
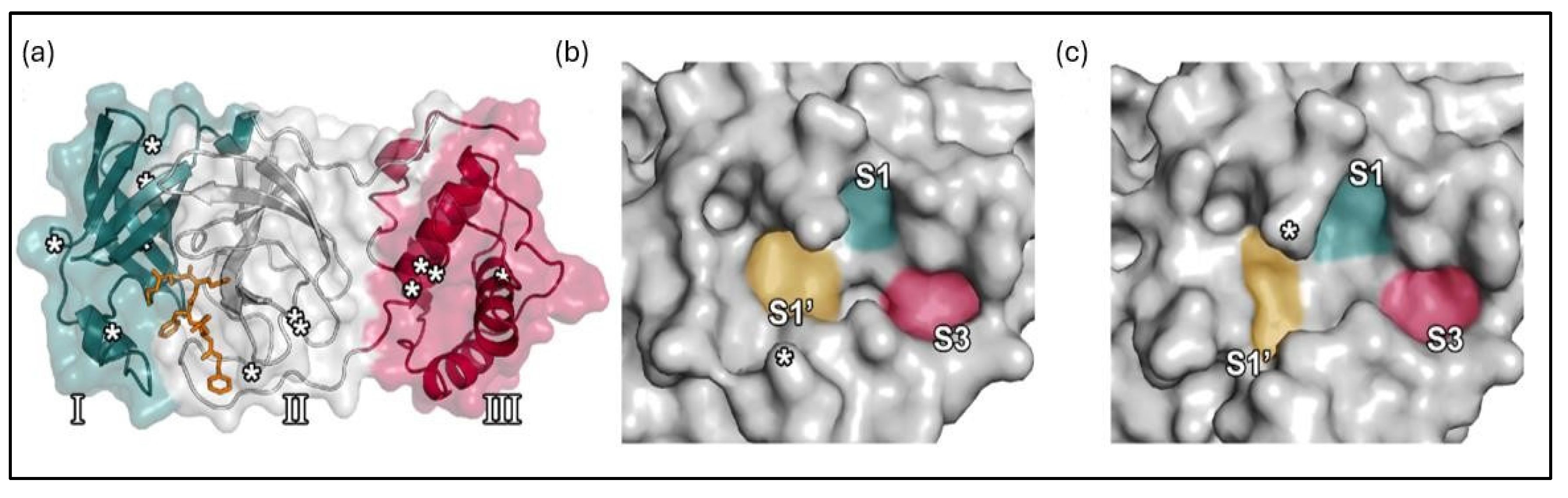
Figure 5.
(a) The 2D structure of ML188; (b) The IC50 values of ML188 against Mpro of SARS-CoV-2 and SARSCoV-1 [20] Licensed under CC-BY License.
Figure 5.
(a) The 2D structure of ML188; (b) The IC50 values of ML188 against Mpro of SARS-CoV-2 and SARSCoV-1 [20] Licensed under CC-BY License.
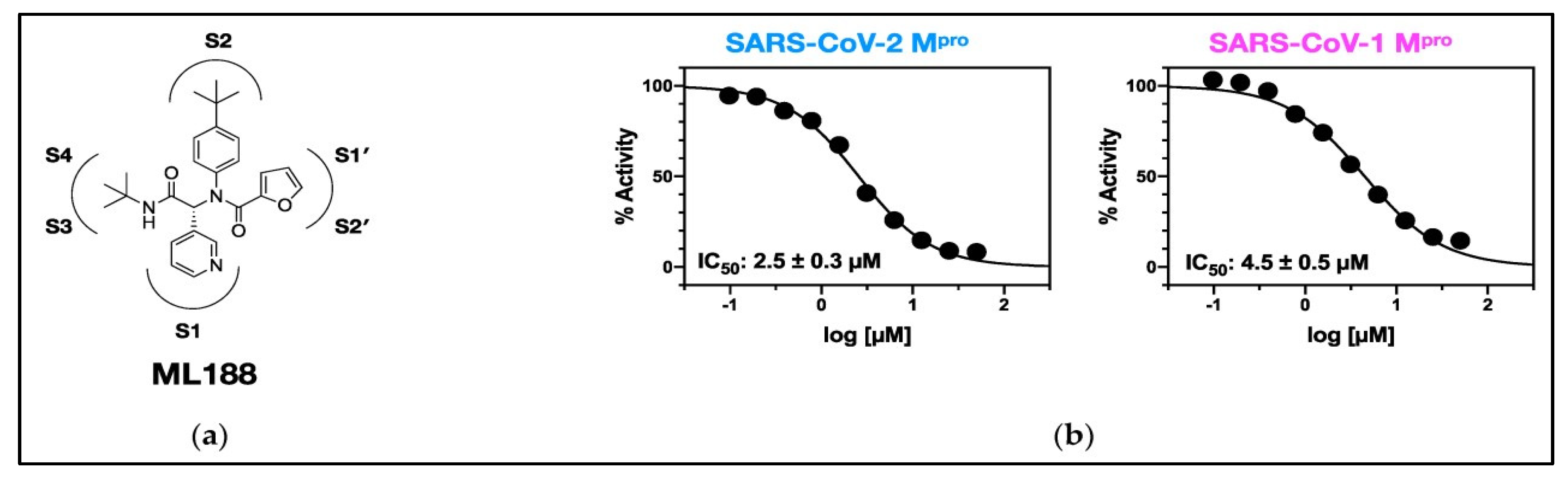
Figure 6.
Highest-ranking compounds (1–6) identified through virtual screening on the Mpro active site [35] Licensed under CC-BY License.
Figure 6.
Highest-ranking compounds (1–6) identified through virtual screening on the Mpro active site [35] Licensed under CC-BY License.
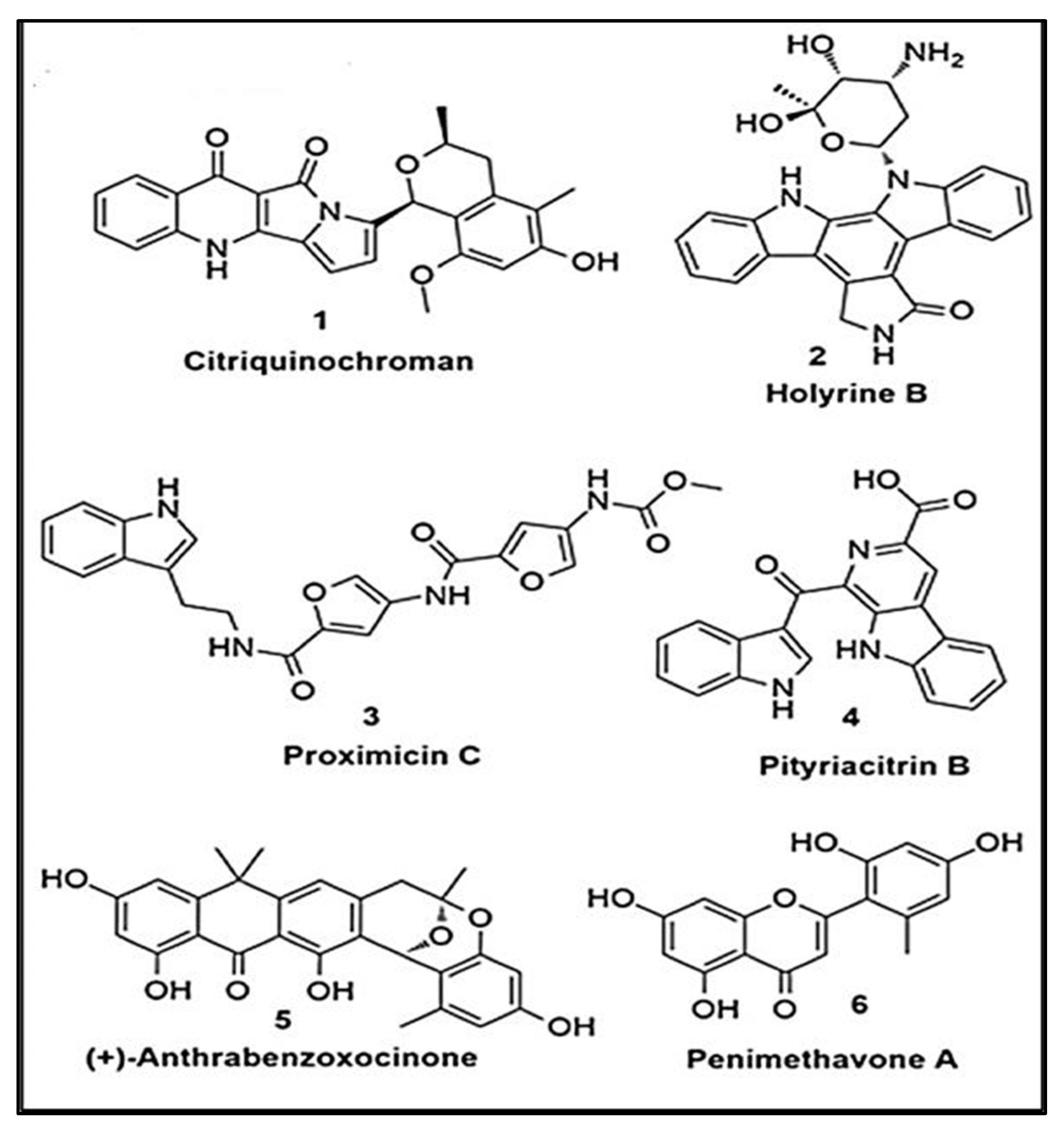
Figure 7.
The structures of cathepsin inhibitor GB111-NH2 and Mpro inhibitor N3 [37].
Figure 7.
The structures of cathepsin inhibitor GB111-NH2 and Mpro inhibitor N3 [37].
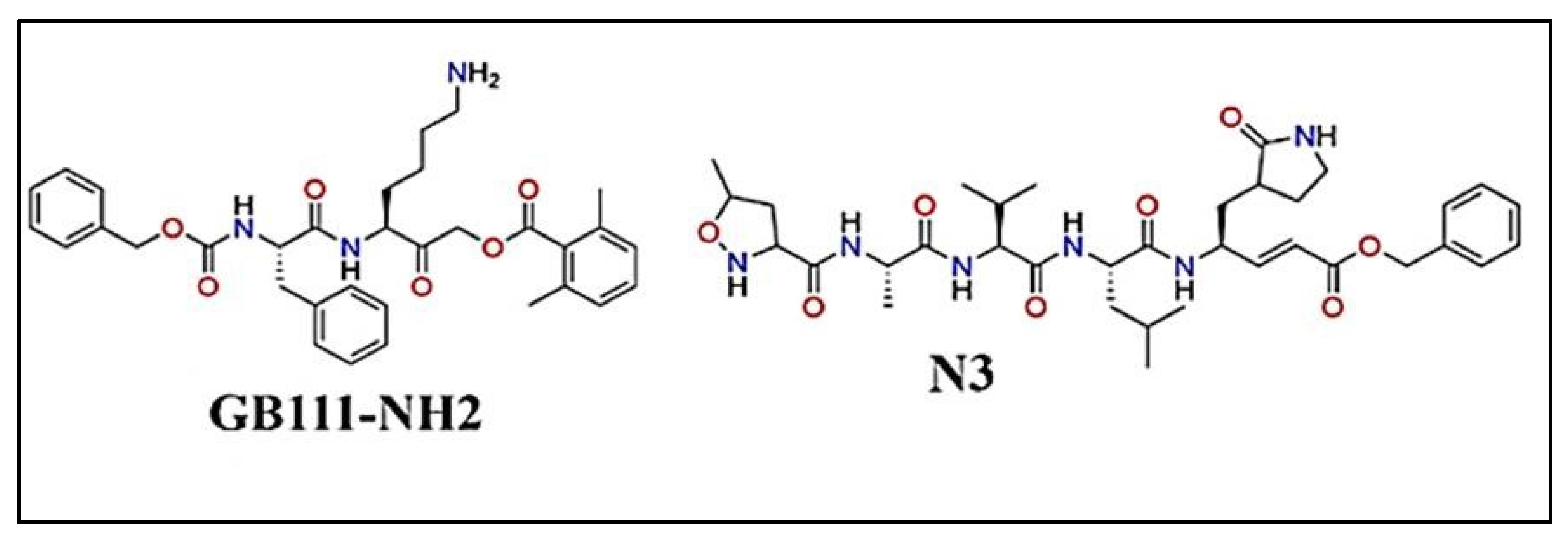
Figure 8.
Synthesis of Thioesters 3w+z [39].
Figure 8.
Synthesis of Thioesters 3w+z [39].
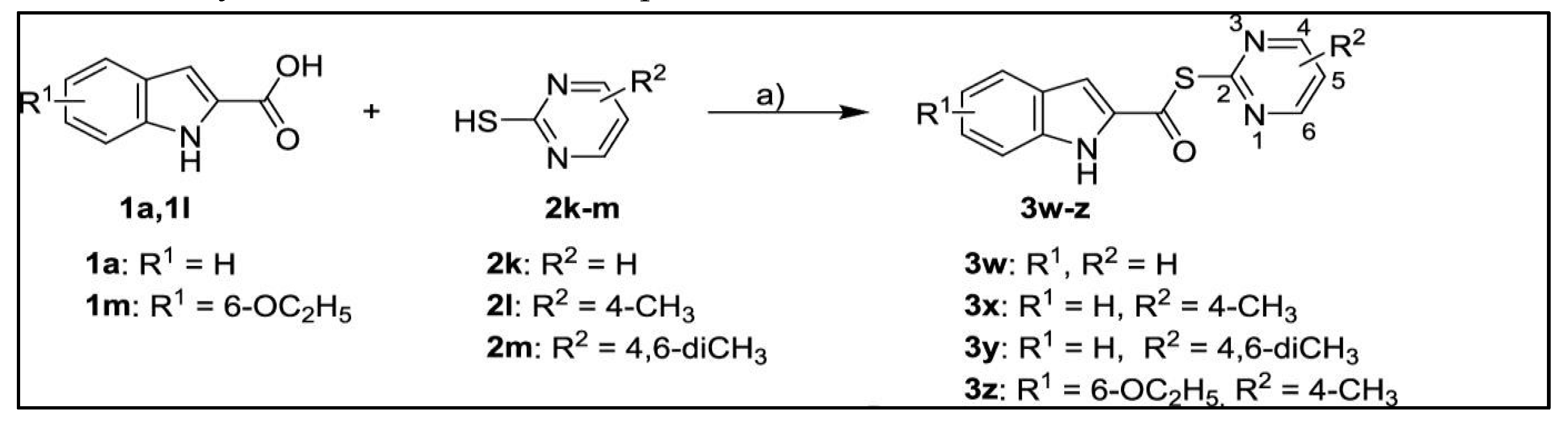
Figure 9.
2D structures of the promising marine natural products as Mpro inhibitors [42] Licensed under CC-BY License.
Figure 9.
2D structures of the promising marine natural products as Mpro inhibitors [42] Licensed under CC-BY License.
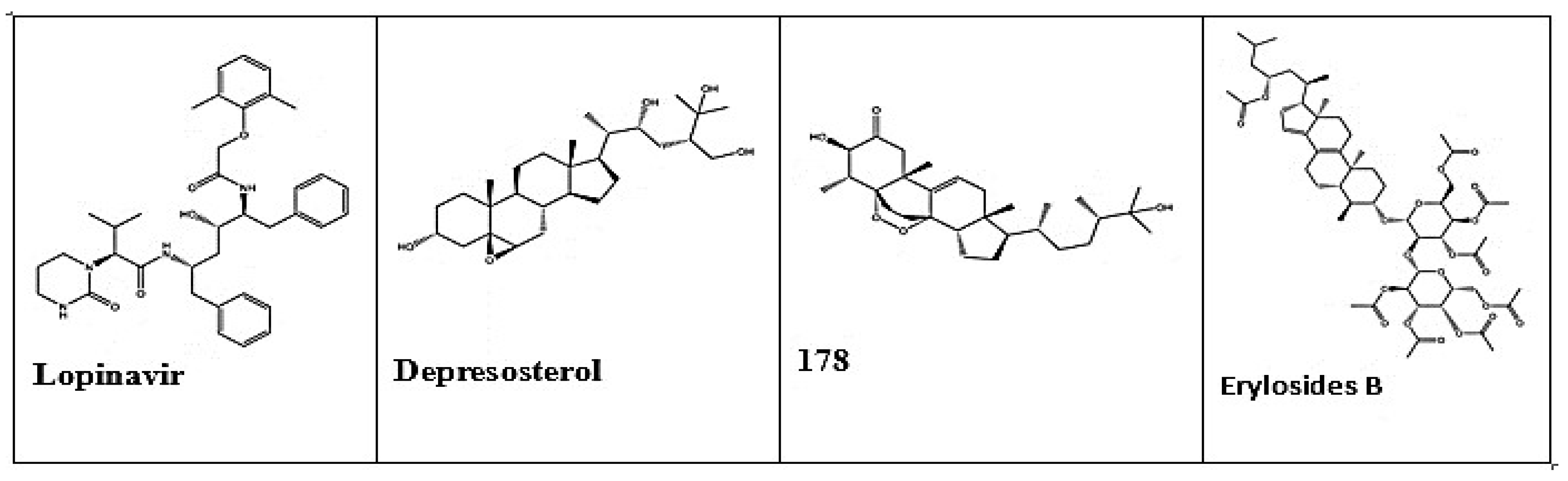
Figure 10.
The polyphenolic compounds with their docking scores, Hesperidin, Rutin, Diosmin, and Apiin, respectively [44].
Figure 10.
The polyphenolic compounds with their docking scores, Hesperidin, Rutin, Diosmin, and Apiin, respectively [44].
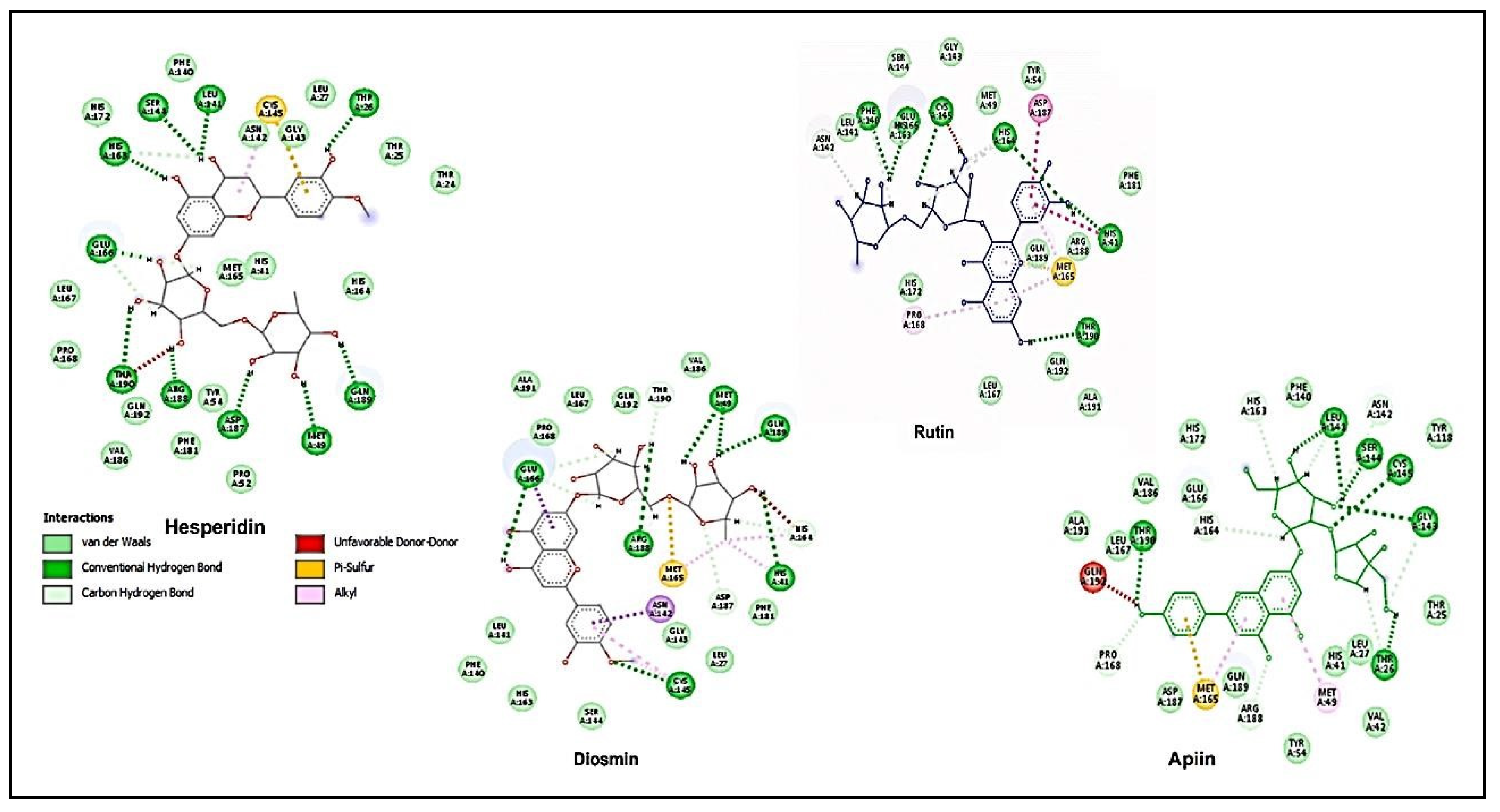
Figure 11.
Aloesin and Aloesin D binding modes [46].
Figure 11.
Aloesin and Aloesin D binding modes [46].

Figure 12.
2D structure of HIT1 and HIT2 [49] Licensed under CC-BY 4.0 License.
Figure 12.
2D structure of HIT1 and HIT2 [49] Licensed under CC-BY 4.0 License.
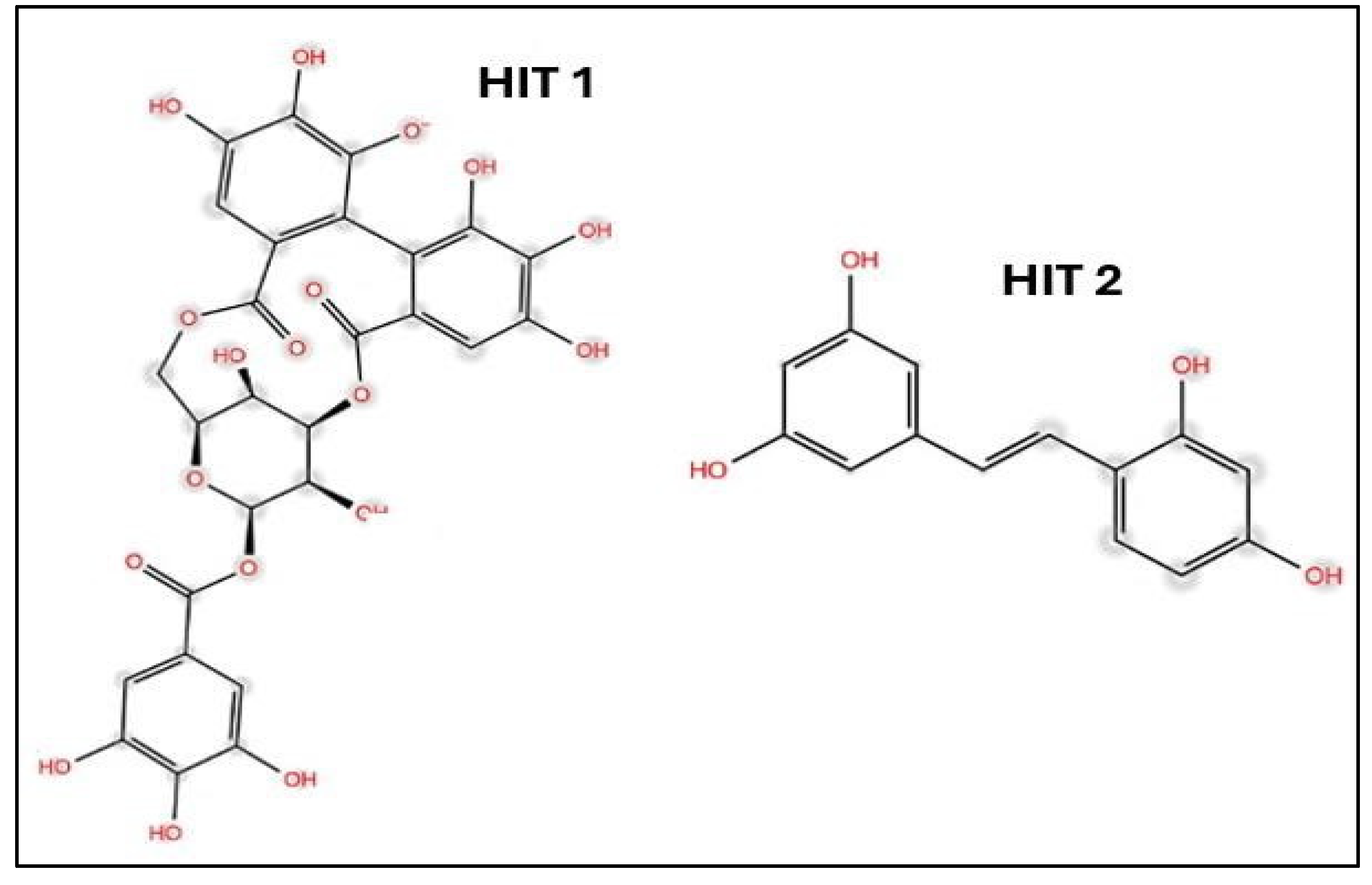
Figure 13.
(a) The binding mode of the most stable compound with the lowest binding energy compound with a coagulin H (W30); (b) Root mean square deviation (RMSD) plot of withacoagulin H (W30) as a function of time (top) and root mean square fluctuations (RMSF) plot of withacoagulin H (W30) [52].
Figure 13.
(a) The binding mode of the most stable compound with the lowest binding energy compound with a coagulin H (W30); (b) Root mean square deviation (RMSD) plot of withacoagulin H (W30) as a function of time (top) and root mean square fluctuations (RMSF) plot of withacoagulin H (W30) [52].

Figure 14.
Calculated MM/GBSA binding energy per frame for erylosides B (black) and lopinavir (red) with Mpro over 100 ns MD simulations [42] Licensed under CC-BY 4.0 License.
Figure 14.
Calculated MM/GBSA binding energy per frame for erylosides B (black) and lopinavir (red) with Mpro over 100 ns MD simulations [42] Licensed under CC-BY 4.0 License.
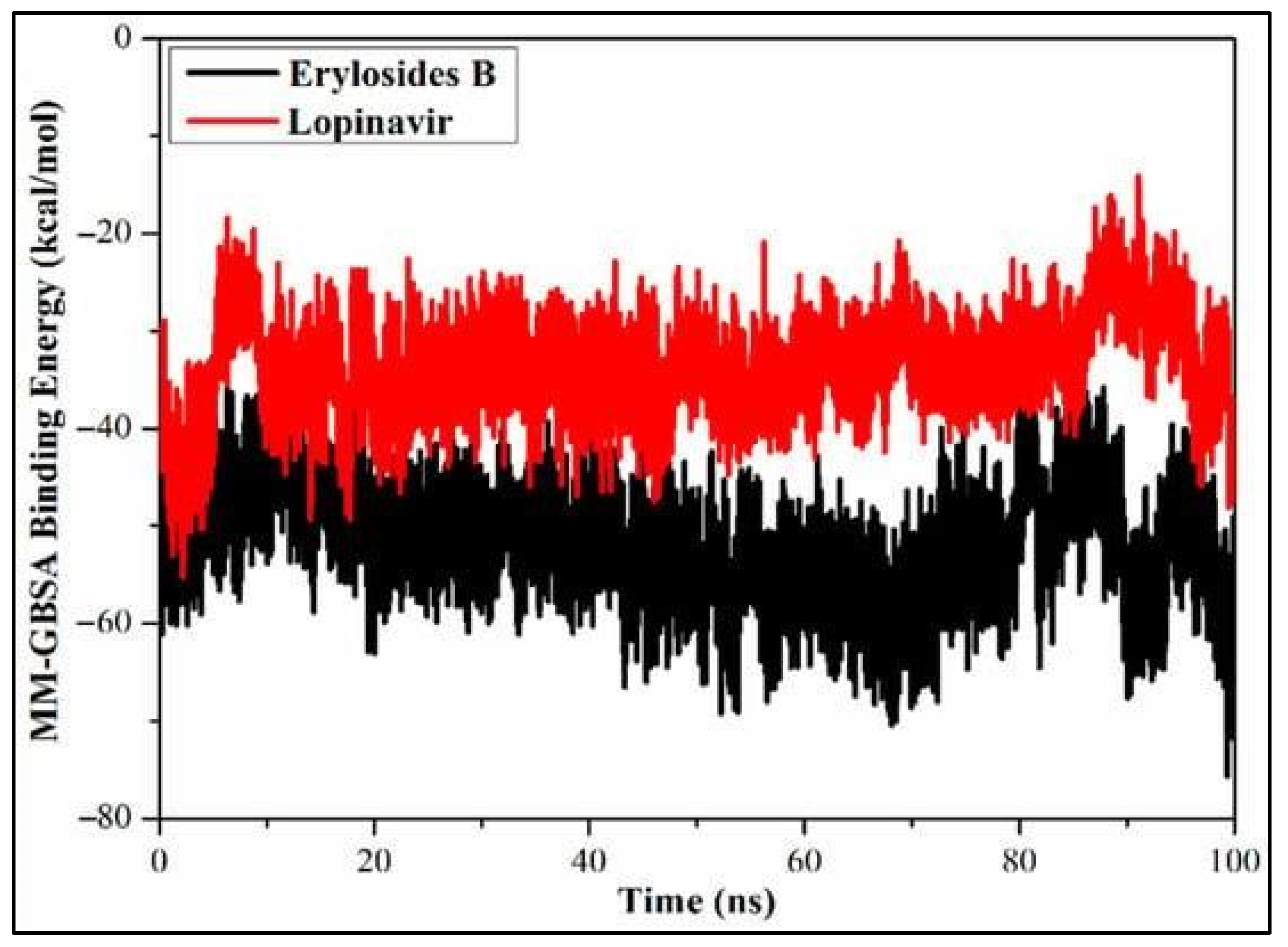
Figure 15.
The chemical structures of compounds S1–S4 besides remdesivir and N3 Mpro inhibitor as reference compounds [51] Licensed under CC-BY 4.0 License.
Figure 15.
The chemical structures of compounds S1–S4 besides remdesivir and N3 Mpro inhibitor as reference compounds [51] Licensed under CC-BY 4.0 License.
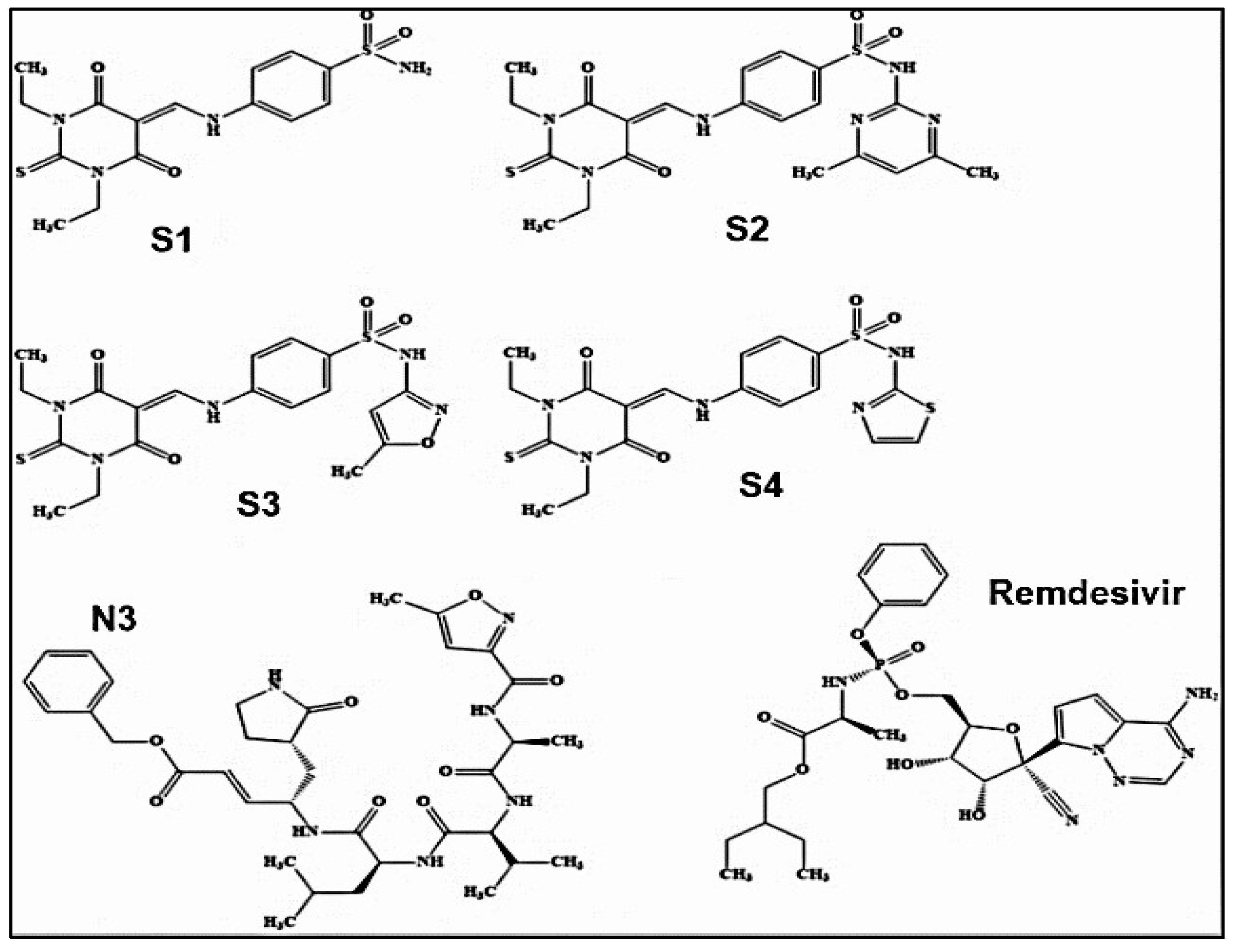
Figure 16.
Structure of Rutin, an inhibitor of Mpro [54].
Figure 16.
Structure of Rutin, an inhibitor of Mpro [54].
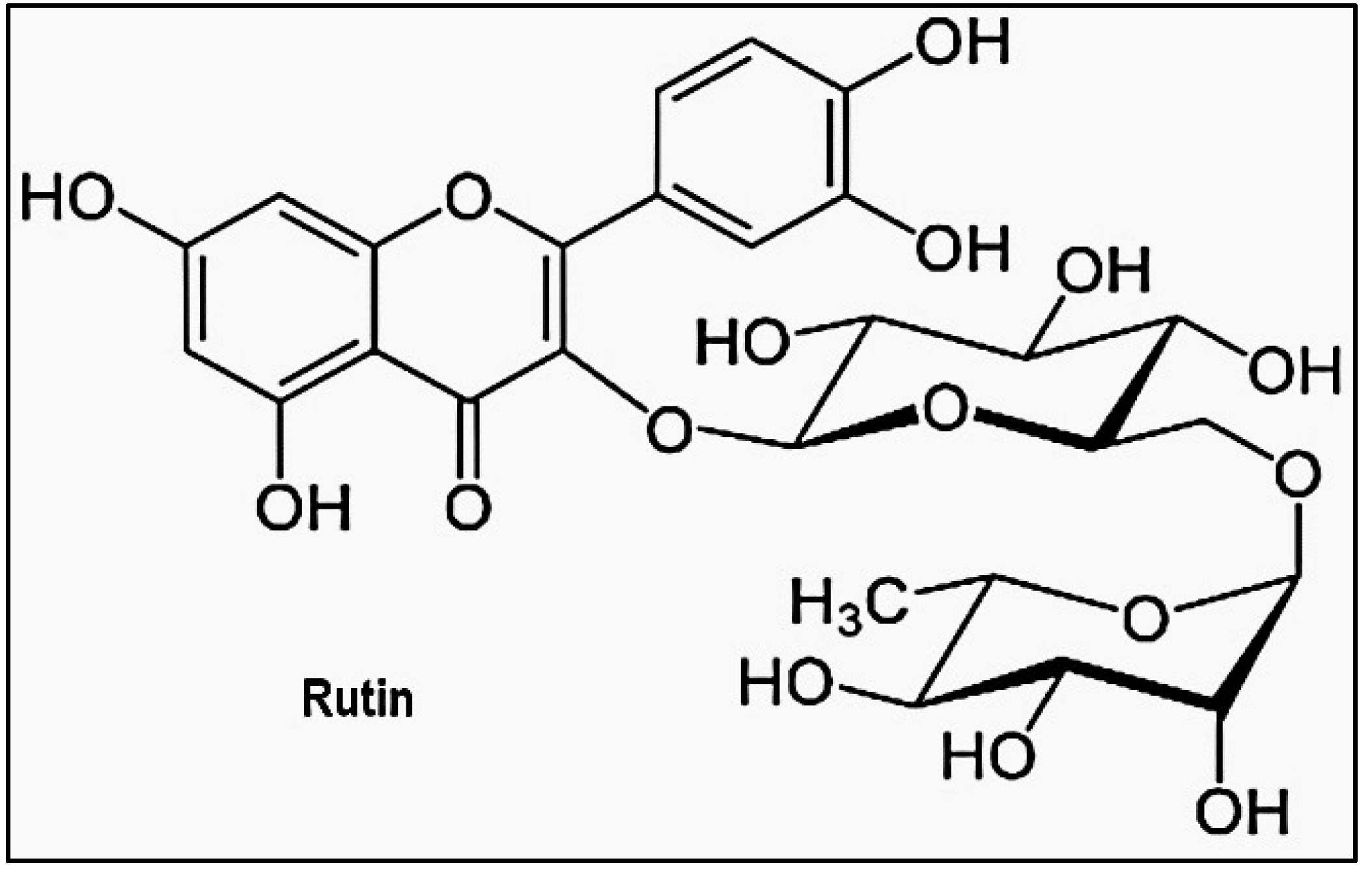
Figure 17.
Ligand property trajectories for Comp.1-6LU7 complex [56].
Figure 17.
Ligand property trajectories for Comp.1-6LU7 complex [56].

Figure 18.
(a) Comparison of the top twelve AG-1478 preferred conformers above the strain energy cutoff (10.5 kcal/mol), dipole moment (D), cluster size and probability distribution (in %), together with the optimized structures in the DMSO; (b) Superposition of all preferred conformers of AG-1478 in DMSO [67].
Figure 18.
(a) Comparison of the top twelve AG-1478 preferred conformers above the strain energy cutoff (10.5 kcal/mol), dipole moment (D), cluster size and probability distribution (in %), together with the optimized structures in the DMSO; (b) Superposition of all preferred conformers of AG-1478 in DMSO [67].
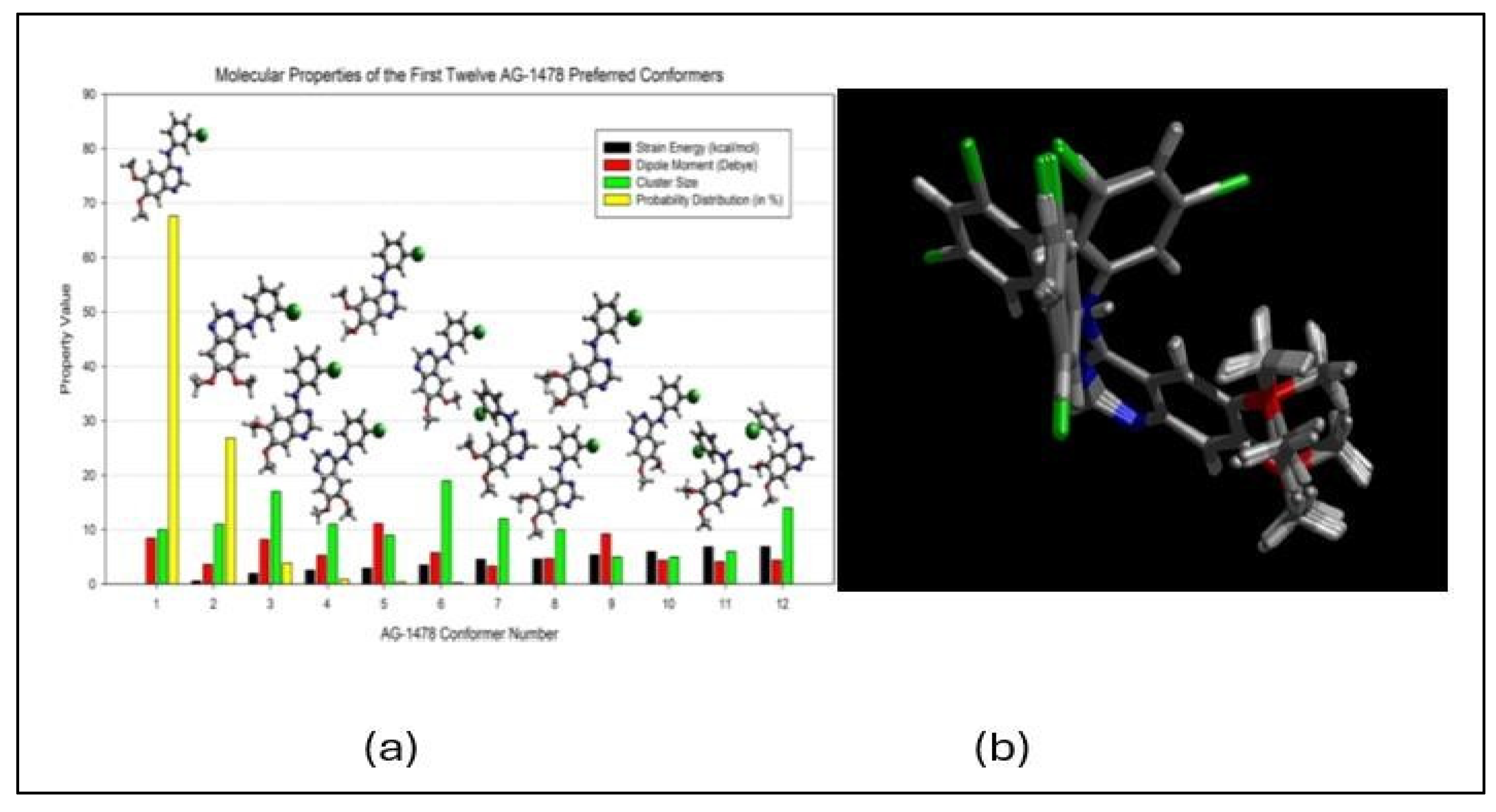
Figure 19.
Correlation between optical shift and the potency of the EGFR TKIs [62] Licensed under CC-BY 4.0 License.
Figure 19.
Correlation between optical shift and the potency of the EGFR TKIs [62] Licensed under CC-BY 4.0 License.
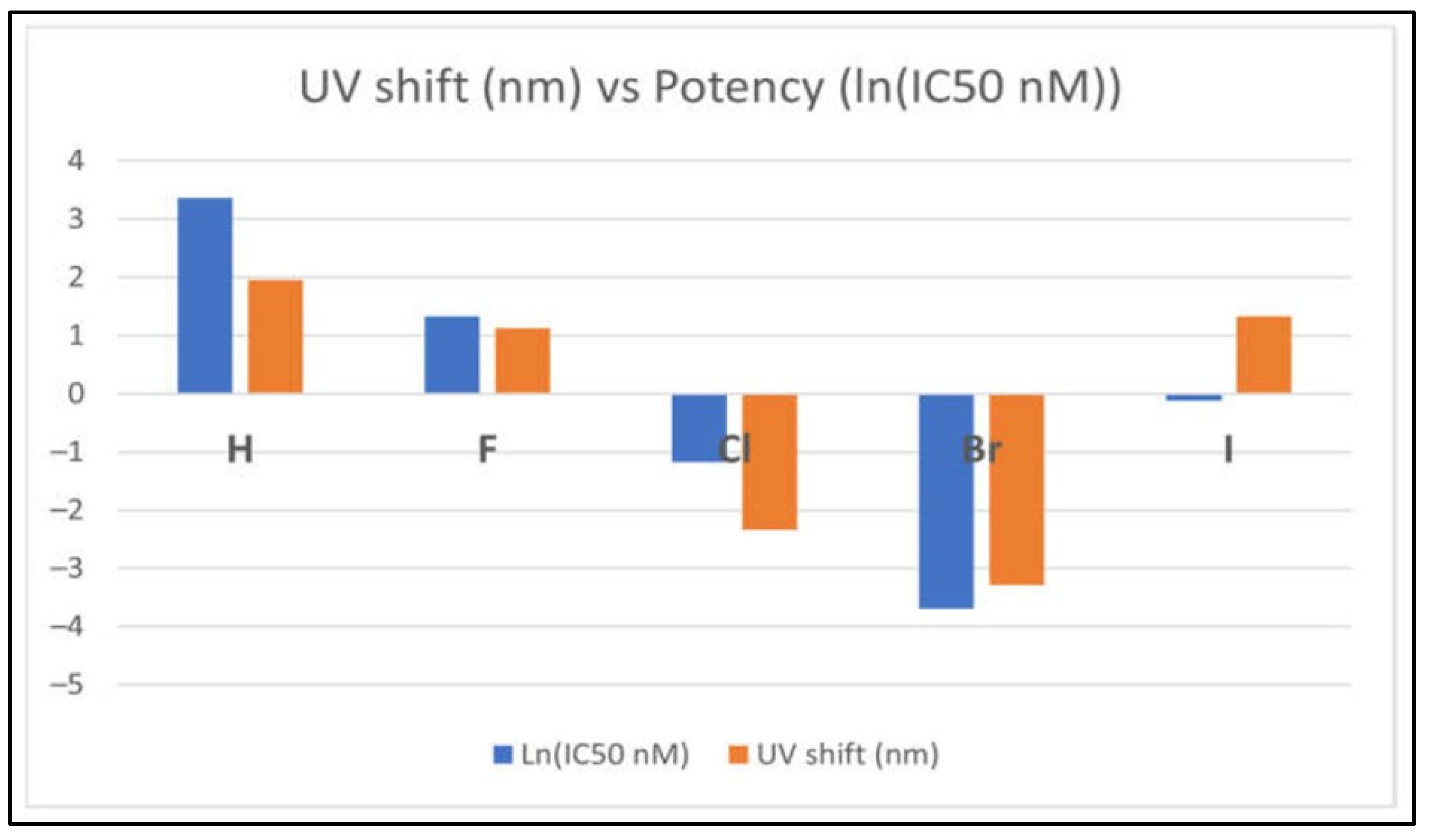

Table 1.
Inhibition results in cell culture of HIV protease inhibitors targeting SARS-CoV-2 Mpro. Licensed under CC-BY License.
Table 1.
Inhibition results in cell culture of HIV protease inhibitors targeting SARS-CoV-2 Mpro. Licensed under CC-BY License.
 |
*Ref [34].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



