
Submitted:
01 September 2024
Posted:
02 September 2024
You are already at the latest version
Abstract
The defining characteristic of Alzheimer's disease (AD) is the aberrant deposition of pathogenic proteins, particularly a massive build-up of amyloid-β peptides, specifically in the brain. Amy-loid-β aggregation is neurotoxic and ultimately results in dysfunction of the nervous system. The present investigation aims to predict potential amyloid-β fibrils disaggregating molecules from plant sources through molecular modeling techniques, this appears to be a very promising and appealing treatment philosophy. Here, 500 flavonoids from various plants were considered, ini-tially undergoing ADME studies to screen molecules that could cross the blood-brain barrier. Later, potential Amyloid-β-disaggregating molecules were identified by molecular docking and dynamics (MD) simulation studies. Five molecules, prenylmethoxy flavonol (-7.3 kcal×mol-1), isopentenyl flavonol (-7.3 kcal×mol-1), 7,3'-Dihydroxyflavone (-7.2 kcal×mol-1), 7-Hydroxy-5-methyl-4'-methoxyflavone (-7.2 kcal×mol-1), 8-hydroxy-7-methoxyflavone (-7 kcal×mol-1) showed higher binding affinity score against Alzheimer's Aβ (1-42) fibrils, these are very close to the standard drug (Donepezil) (-7.90 kcal×mol-1). Further, the MD simulation studies established the intermolecular interaction and stability of the five selected ligands-Amyloid-β oligomer protein complexes. These results suggest that the chosen five flavonoid molecules could be used as possible agents to disaggregate amyloid-β fibrils; however, more in vitro and in vivo investigations are required to verify the therapeutic effectiveness.
Keywords:
Alzheimer's disease
; amyloid-β fibrils
; flavonoids
; disaggregation
; blood-brain barrier
1. Introduction
Alzheimer's disease (AD) is a neurodegenerative condition connected with verbal, behavioral, and cognitive problems that eventually interfere with day-to-day functioning [1]. Sadly, there is no known treatment for AD, and each person's experience with the disease is unique [2]. The deposition of abnormal proteins within and around brain cells is thought to be the root cause of AD [3]. Amyloid-beta (Amyloid-β) is a naturally occurring protein that accumulates in the Alzheimer's brain in abnormal proportions to form plaques that damage cell function [4]. Amyloid-β plaques are the main factor behind neuritic plaques in AD, which gradually deteriorates cognitive abilities [5]. Amyloid-β monomers (37–42 residues long) are released when the enzyme Amyloid Precursor Protein (APP) breaks down. These monomers aggregate to create neurotoxic Amyloid-β fibrils [6]. The early formation of Amyloid-β plaques mostly depends on the monomeric Amyloid-β42 peptide [7]. The misfolded Amyloid-β42 peptides undergo self-assembly to generate oligomers, which then combine to form fibrils [8]. The Amyloid-β42 monomer is a crucial indicator of AD and has been extensively utilized in preventing and managing AD.
Two potential strategies exist to prevent the folding error and accumulation of the Amyloid-β42 peptide. The first is limiting the Amyloid-β monomer's conformational change using inhibitors targeting this process. The second approach is to destabilize/disaggregate the native state of the Amyloid-β42 monomer by refolding it from its misfolded configuration [9]. Researchers initiated destabilization/disaggregation as a therapeutic method due to the clinical failure of medicines using an anti-aggregation technique [10]. Amyloid-β fibril destabilization has a double effect: the distorted fibrils stop being neurotoxic in and of themselves and prevent the production of additional higher-order aggregates [11]. Natural chemicals have been the go-to option for breaking up preformed Amyloid-β fibrils because of their minimal toxicity to humans and biocompatibility [12]. Plant-based phytochemicals are the safest option among all-natural compound sources because of their greater potency, fair availability, extractability, and broad-spectrum applicability. Natural polyphenolic molecules, especially flavonoids, are gaining much attention because they inhibit the formation and aggregation of Amyloid-β fibrils [13].
Plants contain a class of secondary metabolites called flavonoids, which have attracted the pharmaceutical and healthcare industries' interest because of their numerous potential therapeutic applications. Flavonoids comprise a benzopyrone ring, including additional phenolic or polyphenolic groups at various locations [14]. These are produced spontaneously via the phenylpropanoid pathway, and their bioactivity relies on their bioavailability and mode of absorption. Flavanoid compounds are present in most plants, fruits, herbs, stems, cereals, nuts, vegetables, flowers, and seeds [15]. To date, over 10,000 distinct flavonoid molecules have been identified and extracted. Most flavonoids are widely acknowledged to be helpful therapeutic agents, including neuroprotection [16]. The development of AD is believed to depend on an overproduction of reactive oxygen species (ROS), which leads to neurofibrillary tangles as tau aggregates and the aggregation of amyloid-β plaques [17]. This can also lead to higher metabolic demand and mitochondrial dysfunction [18]. Some studies have suggested flavonoids may offer protective effects for brain cells [19]. Some early-stage animal studies have shown that flavonoids can block amyloid-β plaque buildup in the brain, a trademark of Alzheimer's [20]. These naturally occurring flavonoids were suggested as possible therapeutic candidates, but their molecular mechanism remains unclear, thus, it would be ideal to increase in silico research.
The technique of using in silico molecular modeling research to discover new drug candidates from plant-derived flavonoids for AD treatment is outstanding and continually changing. There is an increasing agreement that plant-derived flavonoids with aromatic rings are suitable for treating AD. Flavonoids have been found to possess antioxidant and neuroprotective properties, which have been shown to slow down the onset of AD [21]. Molecular dynamics simulation modeling provides atomistic insights into the mechanism of disaggregating Amyloid-β fibrils, which can aid in discovering drugs for treating AD [22]. In this work, five hundred diverse flavonoid compounds derived from plants were randomly chosen from drug libraries. The objective was to anticipate probable molecules that can disaggregate amyloid-β fibrils. The molecules were picked randomly, and their ability to penetrate the blood-brain barrier was determined using in silico ADME analysis. Additionally, these flavonoids were docked with Amyloid-β oligomer to identify potential binders of Amyloid-β oligomer. The screened complexes were further subjected to molecular dynamics (MD) modeling to identify the most promising molecules for the disaggregation of Amyloid-β fibrils.
2. Experimental Section
2.1. Amyloid-β Protein Preparation
The 3-dimensional structure of Alzheimer's amyloid-beta (1-42) fibrils (PDB entry id: 2BEG, Homo sapiens) was downloaded from the RCSB Protein Data Bank (https://www.rcsb.org/structure/2BEG). Before molecular docking studies, the retrieved amyloid-β (1-42) fibrils were pre-processed through Swiss-PDB Viewer v4.1.0, which included the addition of polar hydrogens and Kollmann charges and removing water molecules. The file was then designated as target.pdb and kept for further investigations. A grid box was created based on the outcomes of the ligand binding site prediction tool of PrankWeb (https://prankweb.cz/). The active sites' protein structure and amino acid locations were determined using BIOVIA Discovery Studio Visualiser version 4.0 software (Accelry's Software Inc., San Diego, CA).
2.2. Ligands Selection
Five hundred flavonoid molecules were chosen from different plant sources from IMPPAT (https://cb.imsc.res.in/imppat/) [23] and Dr. Duke's databases (https://phytochem.nal.usda.gov/) [24]. The molecular structures of selected flavonoid molecules and standard drug (Donepezil) were drawn using Chemsketch software and exported as Structure Data Format (SDF). They were then converted using Open Babel to the mol2 chemical representation. Atoms were allocated Gasteiger-type polar hydrogen charges, whereas non-polar hydrogen molecules were combined with carbons. The internal degrees of freedom and torsions were then adjusted to zero. The ligand molecules were subsequently transformed into the dockable PDBQT format using AutoDock Tools.
2.3. In Silico ADME Screening
The blood-brain barrier, or BBB, keeps larger substances and undesirable cells out of the brain while selectively allowing ions, nutrition, and small molecules (less than 400 Da) to enter [25]. Consequently, the SwissADME web tool (https://www.swissadme.ch) was used to anticipate possible BBB crossing compounds from the five hundred compounds that were previously selected. In this regard, SMILES (Simplified Molecular Input Line Entry System) was translated into their 3D structure using an online SMILES translator and structure file generator (https://cactus.nci.nih.gov/translate/).
2.4. Active site-Targeted Molecular Docking of Flavonoids
The previously stated Swiss-ADME study screened the flavonoid molecules selected for molecular docking studies. These investigations aim to identify compounds with a solid capacity to break down amyloid-β fibrils. Using the AutoDock Vina tool in PyRx 0.8 software, the chosen compounds and the conventional medication donepezil were docked against amyloid-β fibrils (PDB id: 2BEG), emphasizing the active amino acid residues. The molecules and the standard medication, donepezil, were imported using the Open Babel tool, and then the energy was minimized. Then, PyRx 0.8 was loaded with the optimised compounds. Conjugate gradient descent was the energy optimization algorithm, while the Universal Force Field (UFF) was the energy minimization parameter. The amyloid-β fibrils (2BEG) active region was used for the docking studies. It had a grid box size of 30.34 × 25.0 × 18.93 Å and was centered at coordinates (x, y, z) of (-5.73, 0.024, 0.247) Å. The remaining parameters were all kept at their original configurations. The Discovery Studio Visualiser version 16 was used to observe the intermolecular interactions.
2.5. Molecular Dynamics (MD) Simulation Studies
The stability of binding, confirmation, and intermolecular interactions between selected highly ranked bioactive molecules (ligands) and the target protein, amyloid-β fibrils (2BEG), were investigated using MD simulation studies. A 100-nanosecond time-dependent evolution of the complexes was estimated on a Linux system using the Desmond dynamic package 2017 in Schrodinger (academic version). Under physiological settings, the MD simulation (Schrodinger, LLC, Schrodinger Release: QikProp) highlights the likely outcomes of protein-ligand complexes (PLCs) at target binding sites. The system developers' panel: Initially, the panel lets us build a cubic (10 × 10 × 10) container that can house physiological parameters like pH and molecules of water. Na+ or Cl- ions can be added if the pH is low or needs to be raised to satisfy the particular demands of the research methodology. The orthorhombic point-charge (SPC) water model was used to solve the docked protein-ligand complexes straightforwardly. Counter ions were added to the solvated system to make it electrically neutral while keeping the physiological salt concentration at 0.15 M [26]. The OPLS AA (Optimal Potentials for Liquid Simulation - All Atom) force field was applied to the PLC system [27]. In about 100 picoseconds, the system builder panel will moderately reduce the prepared PLC. Two picoseconds were used as the relaxation length in the MD simulation. The Nose-Hoover chain thermostat, the Martyna-Tobias-Klein barostat, and the Reversible Reference System Propagator Algorithms (RESPA) integrator were utilised [28]. The MD simulation's last iteration was produced with the system in equilibrium. The NPT ensemble was used for the MD simulation, which keeps the temperature, pressure, and particle count constant [29]. Using the default relaxation parameters, the simulation ran for 100 nanoseconds at a temperature of 310.15 Kelvin and a pressure of 1.0 bar. After the experiment, the results were assessed using a simulated interaction diagram.
2.6. Density Functionality Theory (DFT)
One well-known and valid "ab initio" technique for examining the structure of quantum many-body systems, such as atoms, molecules, and solids, is DFT [30]. The DFT analysis of the highest-scoring compounds was carried out using the Gaussian 03W package and the Gauss View molecular visualization program. They used a DFT/Becke-3–Lee–Yang–Parr (B3LYP)/6-311G (d, p) technique to ascertain the chosen bioactive chemicals' ideal molecular structure and vibrational frequencies. Furthermore, the optimal configurations and border molecular orbital energies of specific bioactive compounds were calculated. The energies are comprised of the energy difference (Eg) between the highest occupied orbital (EHOMO) and the lowest unoccupied orbital (ELUMO) of a macromolecule. The molecular orbital energy diagrams of the chosen bioactive compounds were examined using the Gauss View molecular visualization software.
3. Result and Discussion
3.1. Binding Site Identification
The binding site can be found by evaluating the physicochemical and shape characteristics of the protein region. To create novel drugs based on the target protein's structure, molecular docking requires discovering new chemical entities, which is vital. In this case, the Alzheimer's amyloid-β (1-42) fibrils' three-dimensional structure contains ten possible binding sites that the PrankWeb tool found (PDB entry id: 2BEG). The binding of amino acid residues to amyloid-β fibrils is shown in Figure 1. The 10 binding pockets were identified by a spectrum of colours, including red, light-yellow, dark-yellow, light green, dark green, light blue, tan, grey, pink, light orange, and dark bluish-green. The same binding pockets of the amyloid-β fibrils were utilized in the molecular docking process to screen the selected active compounds. The ligand posture grading accuracy was enhanced by molecular docking grid generation. We developed a receptor grid tailored to the chosen amyloid-β fibrils to improve our ligand posture score accuracy. The binding site residues discovered in earlier studies created this grid. With the box dimensions of X = 67.1909, Y = 91.6671, and Z = 88.4240 in angstrom (Å), a receptor grid was made to achieve this.
3.2. Active Molecules and Their Structures
The conventional medication, donepezil, and the three-dimensional structures of 500 active compounds from different plants were obtained and optimized from the IMPPAT and Dr. Dukes databases. The results of in silico ADME and molecular docking analyses utilizing the optimized structures against the Alzheimer's amyloid-β (1-42) fibrils are shown in Supplementary Table 1.
3.3. In Silico ADME Screening
The blood-brain barrier (BBB) impedes the entry of most medications into the brain. The existence of the BBB hinders the discovery of drugs for Alzheimer's disease (AD). The BBB prevents over 98% of small-molecule medications and almost 100% of large-molecule pharmaceuticals from entering the brain. While most drug candidates for Alzheimer's disease (AD) are unable to pass through the blood-brain barrier (BBB), the current focus of AD drug development is heavily skewed towards discovering drugs that target the central nervous system (CNS), with less than 1% of the effort dedicated to developing drugs that can effectively reach the CNS [25]. Through ADME analysis, this study screened 125 blood-brain barrier crossing ability of flavonoids from 500 molecules. Supplementary Table 2 displays the chosen molecules for subsequent molecular docking.
3.4. Molecular Docking
Molecular docking analyses were conducted to find possible candidates for Alzheimer's amyloid-β (1-42) fibril disaggregation. Using AutoDock Vina, the binding affinity of 125 active compounds and the conventional medication donepezil to the disaggregation of amyloid-β fibrils was assessed to establish their potential for protein interaction with the target. The study revealed that five active compounds had the lowest binding energy (-7.3 kcal×mol-1) against Alzheimer's amyloid-β (1-42) fibrils, the target protein. Furthermore, it was observed that the active compounds displayed a spectrum of binding energies, which varied from -7.7 to -5 kcal×mol-1, as illustrated in Supplementary Table 2. Five molecules, namely prenylmethoxy flavonol (-7.3 kcal×mol-1), isopentenyl flavonol (-7.3 kcal×mol-1), 7,3'-Dihydroxyflavone (-7.2 kcal×mol-1), 7-Hydroxy-5-methyl-4'-methoxyflavone (-7.2 kcal×mol-1), and 8-hydroxy-7-methoxyflavone (-7 kcal×mol-1), were selected for further investigation due to their potentially binding with the amino acid residues in the active site of amyloid-β (1-42) fibrils. The outcomes are also consistent with the publication [31], in which it was discovered that docking a flavonoid derivative (63), along with other flavones, destabilized the Aβ oligomer [31]. Compared to previous results, the least binding energy value of ΔG indicates that it would make an excellent binder to Aβ fibril [32]. The benchmark binding energy for the medication donepezil was -7.9 kcal×mol-1. Because donepezil effectively breaks down the build-up of amyloid-β (1-42) fibril protein in the human brain, it was selected as the standard medication.
3.5. Interpretation of Ligands-Amyloid-β Fibrils Interactions
The protein-ligand interaction profiler web tool (https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index) was used to visualize the developed residue interactions between the chosen ligands with amyloid-β fibrils. One of the top-scored molecules, prenylmethoxy flavonol, binds with the target amyloid-β fibrils with the binding energy -7.3 kcal×mol-1. The active molecule, prenylmethoxy flavonol, binds with amyloid-β fibrils protein to create four hydrogen bonds (27B ASN (2.95Å), 27C ASN (2.07Å), 27B ASN (2.79Å), and 29C GLY (2.04Å)), 2D and 3D interactions presented in Figure 2(a) and 2(b). The molecule, isopentenyl flavonol, binds with the target amyloid-β fibrils with the binding energy of -7.3 kcal×mol-1. The isopentenyl flavonol binds with amyloid-β fibrils protein to create two hydrophobic interactions (30A ALA (3.84 Å) and 30B ALA (3.76Å)) and one hydrogen bonding (27C ASN (1.99 Å)), 2D and 3D interactions presented in Figure 2(c) and 2(d). The 7,3'-Dihydroxyflavone molecule is also docked with amyloid-β fibrils protein, with a binding energy of -7.2 kcal×mol-1. 7,3'-Dihydroxyflavone formed one hydrophobic interaction (30C ALA (3.88Å)) and four hydrogen bonding (27B ASN (2.82 Å), 27C ASN (1.77 Å), 27D ASN (2.32 Å), and 30D ALA (2.61 Å)) with the target protein amyloid-β fibrils, 2D and 3D interactions presented in Figure 2(e) and 2(f.) The molecule 7-Hydroxy-5-methyl-4'-methoxyflavone binds with the target amyloid-β fibrils with the binding energy of -7.2 -7.2 kcal×mol-1. The 7-Hydroxy-5-methyl-4'-methoxyflavone bind with amyloid-β fibrils protein to create two hydrophobic interactions (30C ALA (3.96 Å) and 30D ALA (3.72 Å)) and three hydrogen bonding (27B ASN (3.15 Å), 27D ASN (2.14 Å), and 30D ALA (2.58 Å)), 2D and 3D interactions presented in Figure 2(g) and 2(h). The 8-hydroxy-7-methoxyflavone molecule binds with the target amyloid-β fibrils with the binding energy of -7 -7.2 kcal×mol-1. The 8-hydroxy-7-methoxyflavone bind with amyloid-β fibrils protein to create two hydrophobic interactions (30B ALA (3.90 Å) and 30C ALA (3.65 Å)) and five hydrogen bonding (27B ASN (3.03 Å), 27B ASN (2.30 Å), 27C ASN (2.24 Å), 30C ALA (3.11 Å) and 30D ALA (2.13 Å)), 2D and 3D interactions presented in Figure 2(i) and 2(j). The standard drug, Donepezil, binds with the target amyloid-β fibrils with a binding energy of -7.9 -7.2 kcal×mol-1. The Donepezil binding with amyloid-β fibrils protein created two hydrophobic interactions (30B ALA (3.75 Å) and 30D ALA (3.60 Å)) and two hydrogen bonding (27D ASN (3.11 Å), and 30B ALA (2.61 Å)), 2D and 3D interactions presented in Figure 2(k) and 2(l).
3.6. MD Simulation Studies
Molecular dynamics simulation studies were used to investigate the stability of protein-ligand interactions. The protein-ligand complexes of amyloid-β fibrils-prenyl methoxy flavonol, amyloid-β fibrils-isopentenyl flavonol, amyloid-β fibrils-7,3'-Dihydroxyflavone, amyloid-β fibrils-7-Hydroxy-5-methyl-4'-methoxyflavone, and amyloid-β fibrils-8-hydroxy-7-methoxyflavone, as well as amyloid-β fibrils-donepezil complexes, were studied using MD simulations. The RMSD graph and ligand-protein interactions (2D interaction diagram) were examined to grasp the simulated results thoroughly. The RMSD graphs show the evolution of the protein (left y-axis) and its ligand (right y-axis). The amyloid-β fibrils-prenylmethoxy flavonol complex was stable, with protein root-mean-square deviation (RMSD) ranging from 4.8 to 7.1 Å and ligand RMSD ranging from 12.5 to 18.2 Å (Figure 3a). The MD trajectory events of the amyloid-β fibrils-isopentenyl flavonol complex showed that the protein RMSD was between 4.2 and 4.8 Å. In comparison, the ligand RMSD varied from 24 to 32 Å, indicating the stable complex (see Figure 3b). The MD trajectory events of the amyloid-β fibrils-7,3'-Dihydroxyflavone complex showed that the protein RMSD was between 8 and 20 Å, while the ligand RMSD ranged from 36 to 42 Å, showing the complex is non-stable (Figure 3c). The MD trajectory events of the amyloid-β fibrils-7-Hydroxy-5-methyl-4'-methoxyflavone complex showed that the protein RMSD was between 3.8 and 4.6 Å, while the ligand RMSD varied from 4 to 6 Å, indicating the stable complex (Figure 3d). The MD trajectory events of the amyloid-β fibrils-8-hydroxy-7-methoxyflavone complex showed that the protein RMSD was between 4 and 4.5 Å, while the ligand RMSD ranged from 10 to 14 Å, indicating a stable complex (Figure 3e). The MD trajectory events of the amyloid-β fibrils-donepezil combination showed that the protein RMSD was between 31 and 32 Å. In contrast, the ligand RMSD varied from 20 to 21 Å, indicating a highly stable complex (Figure 6(f)). The RMSF analysis of stable six protein-ligand complexes showed no significant changes when ligands were bound to the key functional groups of amyloid-β fibrils (Figure 3g–l). The amyloid-β fibrils- prenylmethoxy flavonol complex MD trajectory investigated the interactions of two specific amino acids (ALA30 and GLY29) with the protein. Based on the residue index, it was noted that these amino acids remained constant during the MD simulation (Figure 4a,b). The MD trajectory of the amyloid-β fibrils-isopentenyl flavonol complex revealed a stable link between two specific amino acids (ALA30 and GLY29), as demonstrated by their residue index and lack of volatility during simulation. The protein-ligand interactions of the amyloid-β fibrils-isopentenyl flavonol complex revealed that amino acid residues ALA30 and GLY29 had the most significant interaction with isopentenyl flavonol (Figure 4d,e). The protein-ligand interactions of the amyloid-β fibril-7,3'-Dihydroxyflavone complex revealed that amino acid residues 27B ASN, 27C ASN, 27D ASN, and 30D ALA contributed the most significant contact with 7,3'-Dihydroxyflavone (Figure 4g,h). The protein-ligand interactions of the amyloid-β fibrils-7-Hydroxy-5-methyl-4'-methoxyflavone complex revealed that amino acid residues 27D ASN and 30D ALA contributed the most significant interactions with 7-Hydroxy-5-methyl-4'-methoxyflavone (Figure 4j,k). The protein-ligand interactions of the amyloid-β fibril-8-hydroxy-7-methoxyflavone complex revealed that amino acid residues 27B ASN, 27C ASN, and 30C ALA had the most significant contact with 8-hydroxy-7-methoxyflavone (Figure 4m,n). The protein-ligand interactions of the amyloid-β fibrils-donepezil complex revealed that amino acid residues 30B ALA, 30D ALA, and 27D ASN had the most significant interaction with Donepezil (Figure 4p,q). Figure 4c,f,I,l,o,r) depict hydrogen bond interactions and a timeline of all amino acid residues that form H-bonds, hydrophobic, ionic, or water bridges. Darker lines imply a steady interaction with the object. These connections kept the protein-ligand combination stable throughout the molecular docking simulations. Based on our findings, in vitro studies with prenylmethoxy flavonol, isopentenyl flavonol, 7,3'-Dihydroxyflavone, 7-Hydroxy-5-methyl-4'-methoxyflavone, and 8-hydroxy-7-methoxyflavone should be done to determine their capacity to disaggregate amyloid-β fibrils. These compounds could be used as the cornerstone for future lead optimization.
3.7. DFT Studies
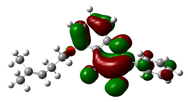
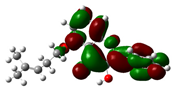
Frontier molecular orbitals (FMOs) are used to predict the chemical systems' most reactive regions and describe different reactions. In the sphere of material research, the FMO features—which include the HOMO and LUMO orbitals—are invaluable for characterizing materials. They are used to measure the molecules' chemical reactivity [33]. The energies of the highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), and the HOMO-LUMO energy gap were calculated at the B3LYP level using the 6-311 G (d, p) basis set. Table 1 showed the HOMO-LUMO images of the five highest-binding compounds in addition to the reference medication donepezil.
4. Conclusion
Traditionally, plant extracts and their secondary metabolites have exhibited numerous chemical constituents that aid in preventing various diseases. These molecules possess high levels of security, exhibit low toxicity levels, and demonstrate economic efficiency, thus enhancing the overall quality of human existence. The aggregation of amyloid-β fibrils in the brain is a characteristic feature of Alzheimer's disease. This work has found five potential molecules, namely prenylmethoxy flavonol, isopentenyl flavonol, 7,3'-Dihydroxyflavone, 7-Hydroxy-5-methyl-4'-methoxyflavone, and 8-hydroxy-7-methoxyflavone, that can disaggregate amyloid-β fibrils. Molecular docking experiments identified these molecules. MD simulation assessed the ligand-receptor complex's stability, revealing that all five Aβ oligomer protein complexes remained stable throughout the simulation. The results obtained from density functional theory also indicated that all five compounds exhibit high stability. In conclusion, the five discovered molecules have the potential to serve as lead compounds for further testing as potential neuroprotective agents that can act on Aβ fibril disaggregation. These compounds can be evaluated utilizing both in vitro and in vivo experiments.
Funding
This research received no funding.
Acknowledgments
The authors are grateful to the Management of Kalasalingam Academy of Research and Education for the research fellowships and utilization of the research facilities.
Contributions
S.K developed the concept and designed the experiments. U.L.R., G.A.K., T.P., P.P (4th Author)., S.V., P.P (6th Author)., S.K.B. and S.K performed the experiments. U.L.R., G.A.K., T.P., P.P (4th Author), SV, PP (6th Author)., S.K.B. and S.K analyzed and interpreted the data. All authors drafted and revised the manuscript.
Conflicts of Interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- Atri A. The Alzheimer’s disease clinical spectrum: diagnosis and management. Medical Clinics 2019; 103: 263-293. [CrossRef]
- Harris PB. The person with Alzheimer's disease: Pathways to understanding the experience. JHU Press; 2002.
- Rajeshkumar RR, Kumar BK, Parasuraman P, Sundar K, Ammunje DN,. Pandian SRK, Murugesan S, Kabilan SJ, Kunjiappan S. Graph theoretical network analysis, in silico exploration, and validation of bioactive compounds from Cynodon dactylon as potential neuroprotective agents against α-synuclein. BioImpacts: BI 2022; 12: 487. [CrossRef]
- Rajmohan R, Reddy PH. Amyloid-beta and phosphorylated tau accumulations cause abnormalities at synapses of Alzheimer’s disease neurons. J Alzheimer's Dis 2017; 57: 975-999. [CrossRef]
- van der Kant R, Goldstein LS, Ossenkoppele R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci 2020; 21: 21-35. [CrossRef]
- Bignante EA, Heredia F, Morfini G, Lorenzo A. Amyloid β precursor protein as a molecular target for amyloid β–induced neuronal degeneration in Alzheimer's disease. Neurobiol Aging 2013; 34: 2525-2537. [CrossRef]
- Butterfield DA, Swomley AM, Sultana R. Amyloid β-peptide (1–42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxidants Redox Signal 2013; 19: 823-835. [CrossRef]
- Michaels TC, Šarić A, Curk S, Bernfur K, Arosio P, Meisl G, Dear AJ, Cohen SI, Dobson CM, Vendruscolo M. Dynamics of oligomer populations formed during the aggregation of Alzheimer’s Aβ42 peptide. Nat Chem 2020; 12: 445-451. [CrossRef]
- Barucker C, Bittner HJ, Chang PK-Y, Cameron S, Hancock MA, Liebsch F, Hossain S, Harmeier A, Shaw H, Charron FM. Aβ42-oligomer Interacting Peptide (AIP) neutralizes toxic amyloid-β42 species and protects synaptic structure and function. Sci Rep 2015; 5: 15410. [CrossRef]
- Salahuddin P, Khan RH, Furkan M, Uversky VN, Islam Z, Fatima MT. Mechanisms of amyloid proteins aggregation and their inhibition by antibodies, small molecule inhibitors, nano-particles and nano-bodies. Int J Biol Macromol 2021; 186: 580-590. [CrossRef]
- Jakhria TC. Amyloid fibrils are nanoparticles that target lysosomes. University of Leeds; 2014. eThesis id: uk.bl.ethos.638882.
- Gupta S, Dasmahapatra AK. Lycopene destabilizes preformed Aβ fibrils: Mechanistic insights from all-atom molecular dynamics simulation. Comput Biol Chem 2023; 105: 107903. [CrossRef]
- Stefani M, Rigacci S. Protein folding and aggregation into amyloid: the interference by natural phenolic compounds. Int J Mol Sci 2013; 14: 12411-12457. [CrossRef]
- Kunjiappan S, Pandian SRK, Panneerselvam T, Pavadai P, Kabilan SJ, Sankaranarayanan M. Exploring the Role of Plant Secondary Metabolites for Aphrodisiacs. In: Plant Specialized Metabolites: Phytochemistry, Ecology and Biotechnology. Springer; 2023: 1-19. [CrossRef]
- Sawicka B, Ziarati P, Messaoudi M, Agarpanah J, Skiba D, Bienia B, Barbaś P, Rebiai A, Krochmal-Marczak B, Yeganehpoor F. Role of Herbal Bioactive Compounds as a Potential Bioavailability Enhancer for Active Pharmaceutical Ingredients. In: Handbook of Research on Advanced Phytochemicals and Plant-Based Drug Discovery. IGI Global; 2022: 450-495. [CrossRef]
- Ullah A, Munir S, Badshah SL, Khan N, Ghani L, Poulson BG, Emwas AH, Jaremko M. Important flavonoids and their role as a therapeutic agent. Molecules 2020; 25: 5243. [CrossRef]
- Sun X, Chen W-D, Wang Y-D. β-Amyloid: the key peptide in the pathogenesis of Alzheimer’s disease. Front Pharmacol 2015; 6: 221. [CrossRef]
- Wang X, Su B, Perry G. Smith MA, Zhu X. Insights into amyloid-β-induced mitochondrial dysfunction in Alzheimer disease. Free Radic Biol Med 2007; 43: 1569-1573. [CrossRef]
- Dajas F, Rivera-Megret F, Blasina F, Arredondo F, Abin-Carriquiry J, Costa G, Echeverry C, Lafon L, Heizen H, Ferreira M. Neuroprotection by flavonoids. Braz J Med Biol Res 2003; 36: 1613-1620. [CrossRef]
- Pritam P, Deka R, Bhardwaj A, Srivastava R, Kumar D, Jha AK, Jha NK, Villa C, Jha SK. Antioxidants in Alzheimer’s disease: Current therapeutic significance and future prospects. Biology 2022; 11: 212. [CrossRef]
- Calderaro A, Patanè GT, Tellone E, Barreca D, Ficarra S, Misiti F, Laganà G. The neuroprotective potentiality of flavonoids on Alzheimer’s disease. Int J Mol Sci 2022; 23: 14835. [CrossRef]
- Palanichamy C, Pavadai P, Panneerselvam T, Arunachalam S, Babkiewicz E, Ram Kumar Pandian S, Shanmugampillai Jeyarajaguru K, Nayak Ammunje D, Kannan S,. Chandrasekaran J, Kunjiappan S. Aphrodisiac performance of bioactive compounds from Mimosa pudica Linn.: In silico molecular docking and dynamics simulation approach. Molecules 2022; 27: 3799. [CrossRef]
- Mohanraj K, Karthikeyan BS, Vivek-Ananth R, Chand RB, Aparna S, Mangalapandi P, Samal A. IMPPAT: A curated database of Indian Medicinal Plants, Phytochemistry And Therapeutics. Sci Rep 2018; 8: 4329. [CrossRef]
- Duke JA. Dr. Duke's phytochemical and ethnobotanical databases. 1994.
- Wu D, Chen Q, Chen X, Chen, Han F, Chen Z, Wang Y. The blood–brain barrier: structure, regulation, and drug delivery. Signal Transduct Target Ther 2023; 8: 217. [CrossRef]
- Kuntz CP. Molecular Mechanisms of Ligand Recognition by the Human Serotonin Transporter: A Molecular Modeling Approach. Purdue University; 2015.
- Espinosa JR, Wand CR, Vega C, Sanz E, Frenkel D. Calculation of the water-octanol partition coefficient of cholesterol for SPC, TIP3P, and TIP4P water. J Chem Phys 2018; 149. [CrossRef]
- Liu Y. Novel theoretical analysis methods and algorithms for classical and ab initio molecular dynamics. New York University; 2002. [CrossRef]
- Kim M, Kim E, Lee S, Kim JS, Lee S. New method for constant-NPT molecular dynamics. J Chem Phys Chem A 2019; 123: 1689-1699. [CrossRef]
- Ghosal A, Roy AK. DFT calculations of atoms and molecules in Cartesian grids. 2016. [CrossRef]
- Kumar A, Srivastava S, Tripathi S, Singh SK, Srikrishna S, Sharma A. Molecular insight into amyloid oligomer destabilizing mechanism of flavonoid derivative 2-(4′ benzyloxyphenyl)-3-hydroxy-chromen-4-one through docking and molecular dynamics simulations. J Biomol Struct Dyn 2016; 34: 1252-1263. [CrossRef]
- Gupta S, Dasmahapatra AK. Destabilization potential of phenolics on Aβ fibrils: Mechanistic insights from molecular dynamics simulation. Phys Chem Chem Phys 2020; 22: 19643-19658. [CrossRef]
- Fahim AM, Farag AM. Synthesis, antimicrobial evaluation, molecular docking and theoretical calculations of novel pyrazolo [1, 5-a] pyrimidine derivatives. J Mol Struct 2020; 1199: 127025. [CrossRef]
Figure 1.
The PrankWeb tool is used to predict the binding pockets and correspondence binding sites of the Alzheimer's amyloid-β (1-42) fibrils. The 10 binding pockets were distinguished by various colours: red, light-yellow, dark-yellow, light green, dark green, light blue, tan, grey, pink, light orange, and dark bluish-green.
Figure 1.
The PrankWeb tool is used to predict the binding pockets and correspondence binding sites of the Alzheimer's amyloid-β (1-42) fibrils. The 10 binding pockets were distinguished by various colours: red, light-yellow, dark-yellow, light green, dark green, light blue, tan, grey, pink, light orange, and dark bluish-green.
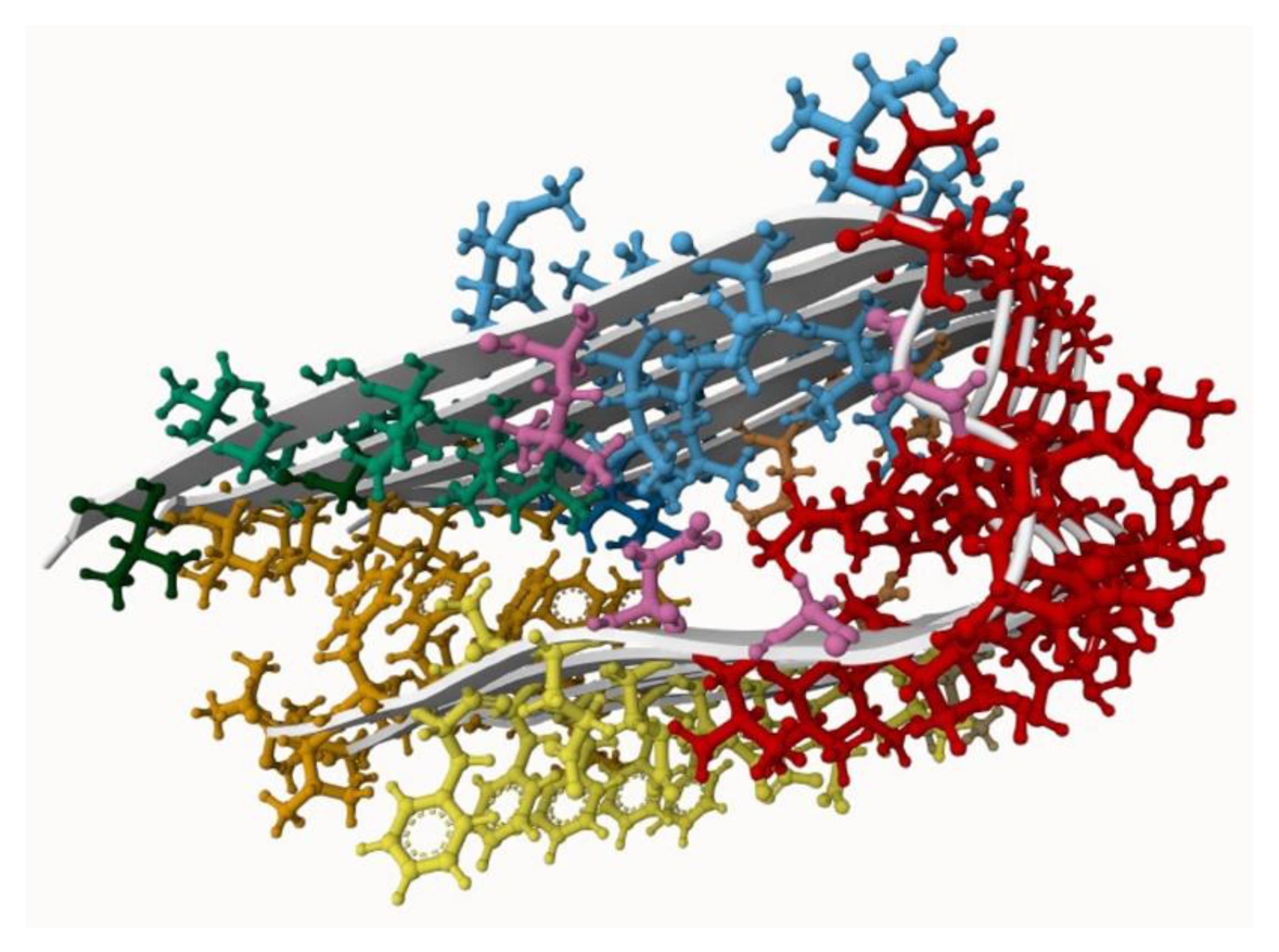
Figure 2.
Interaction between the molecule Prenylmethoxy flavonol and Amyloid-β fibrils. Left side representing 3D (a) and the right side representing 2D complex protein–ligand interaction (b); the interaction between the molecule Isopentenyl flavonol and Amyloid-β fibrils. Left side representing 3D (c) and the right side representing 2D complex protein–ligand interaction (d); and the interaction between the molecule 7,3'-Dihydroxyflavone and Amyloid-β fibrils. Left side representing 3D (e) and the right side representing 2D complex protein–ligand interaction (f); and the interaction between the molecule 7-Hydroxy-5-methyl-4'-methoxyflavone and Amyloid-β fibrils. Left side representing 3D (g) and the right side representing 2D complex protein–ligand interaction (h); and the interaction between the molecule 8-hydroxy-7-methoxyflavone and Amyloid-β fibrils. Left side representing 3D (i) and the right side representing 2D complex protein–ligand interaction (j); and the interaction between the molecule Donepezil and Amyloid-β fibrils. Left side representing 3D (k) and the right side representing 2D complex protein–ligand interaction (l).
Figure 2.
Interaction between the molecule Prenylmethoxy flavonol and Amyloid-β fibrils. Left side representing 3D (a) and the right side representing 2D complex protein–ligand interaction (b); the interaction between the molecule Isopentenyl flavonol and Amyloid-β fibrils. Left side representing 3D (c) and the right side representing 2D complex protein–ligand interaction (d); and the interaction between the molecule 7,3'-Dihydroxyflavone and Amyloid-β fibrils. Left side representing 3D (e) and the right side representing 2D complex protein–ligand interaction (f); and the interaction between the molecule 7-Hydroxy-5-methyl-4'-methoxyflavone and Amyloid-β fibrils. Left side representing 3D (g) and the right side representing 2D complex protein–ligand interaction (h); and the interaction between the molecule 8-hydroxy-7-methoxyflavone and Amyloid-β fibrils. Left side representing 3D (i) and the right side representing 2D complex protein–ligand interaction (j); and the interaction between the molecule Donepezil and Amyloid-β fibrils. Left side representing 3D (k) and the right side representing 2D complex protein–ligand interaction (l).
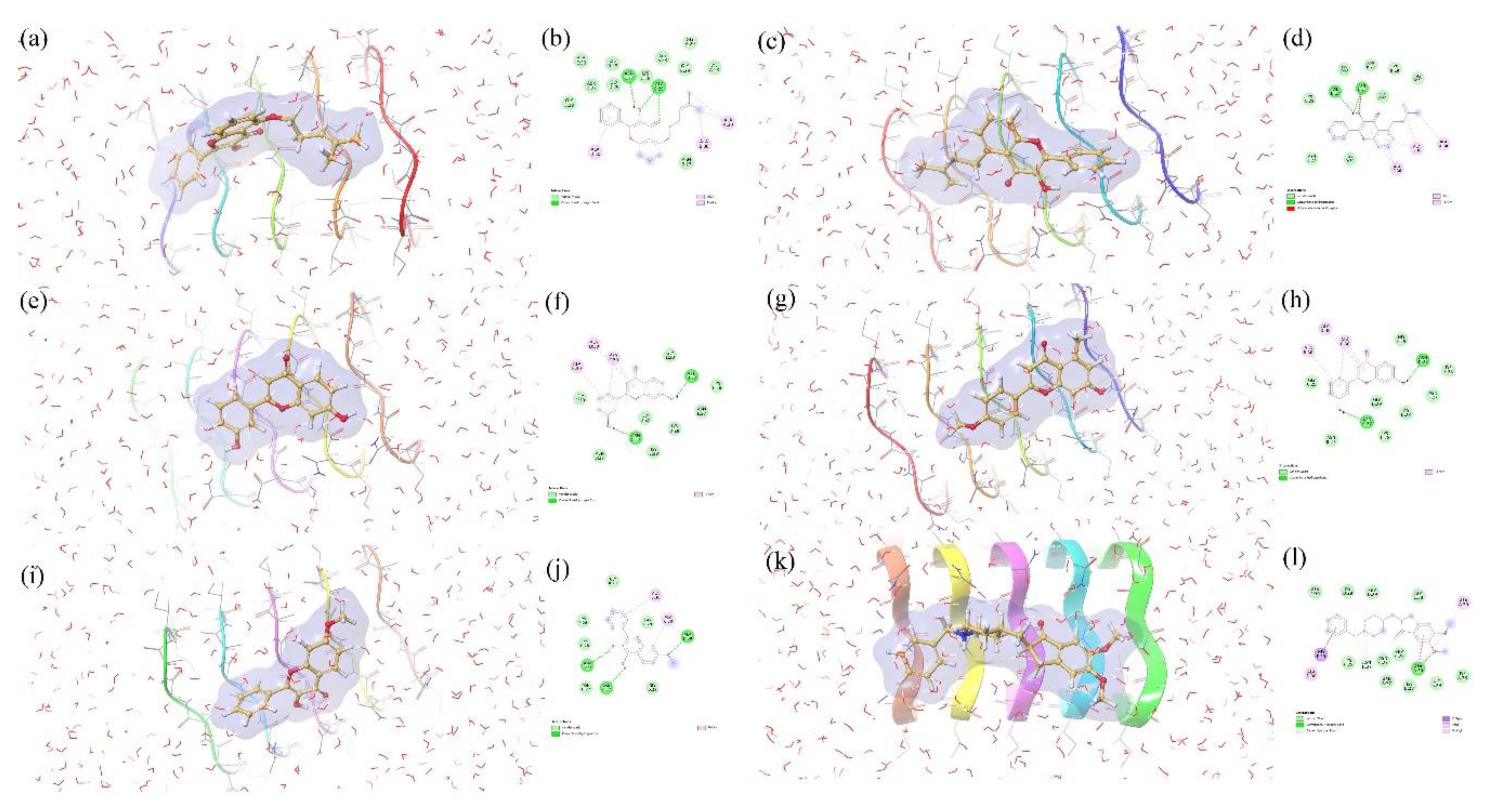
Figure 3.
RMSD study plot for 100 ns MD simulation of prenylmethoxy flavonol-Amyloid-β fibrils docked complex (a), isopentenyl flavonol-Amyloid-β fibrils docked complex (b), 7,3'-Dihydroxyflavone-Amyloid-β fibrils docked complex (c), 7-Hydroxy-5-methyl-4'-methoxyflavone-Amyloid-β fibrils docked complex (d), 8-hydroxy-7-methoxyflavone-Amyloid-β fibrils docked complex (e), and Donepezil-Amyloid-β fibrils docked complex (f). Root mean square fluctuation (RMSF) of the Prenylmethoxy flavonol for characterizing changes in the ligand atom positions (a), root mean square fluctuation of the isopentenyl flavonol for characterizing changes in the ligand atom positions (b), root mean square fluctuation of the 7,3'-Dihydroxyflavone for characterizing changes in the ligand atom positions (c), root mean square fluctuation of the 7-Hydroxy-5-methyl-4'-methoxyflavone for characterizing changes in the ligand atom positions (d), root mean square fluctuation of the 8-hydroxy-7-methoxyflavone for characterizing changes in the ligand atom positions (e), and root mean square fluctuation of the Donepezil for characterizing changes in the ligand atom positions (f).
Figure 3.
RMSD study plot for 100 ns MD simulation of prenylmethoxy flavonol-Amyloid-β fibrils docked complex (a), isopentenyl flavonol-Amyloid-β fibrils docked complex (b), 7,3'-Dihydroxyflavone-Amyloid-β fibrils docked complex (c), 7-Hydroxy-5-methyl-4'-methoxyflavone-Amyloid-β fibrils docked complex (d), 8-hydroxy-7-methoxyflavone-Amyloid-β fibrils docked complex (e), and Donepezil-Amyloid-β fibrils docked complex (f). Root mean square fluctuation (RMSF) of the Prenylmethoxy flavonol for characterizing changes in the ligand atom positions (a), root mean square fluctuation of the isopentenyl flavonol for characterizing changes in the ligand atom positions (b), root mean square fluctuation of the 7,3'-Dihydroxyflavone for characterizing changes in the ligand atom positions (c), root mean square fluctuation of the 7-Hydroxy-5-methyl-4'-methoxyflavone for characterizing changes in the ligand atom positions (d), root mean square fluctuation of the 8-hydroxy-7-methoxyflavone for characterizing changes in the ligand atom positions (e), and root mean square fluctuation of the Donepezil for characterizing changes in the ligand atom positions (f).
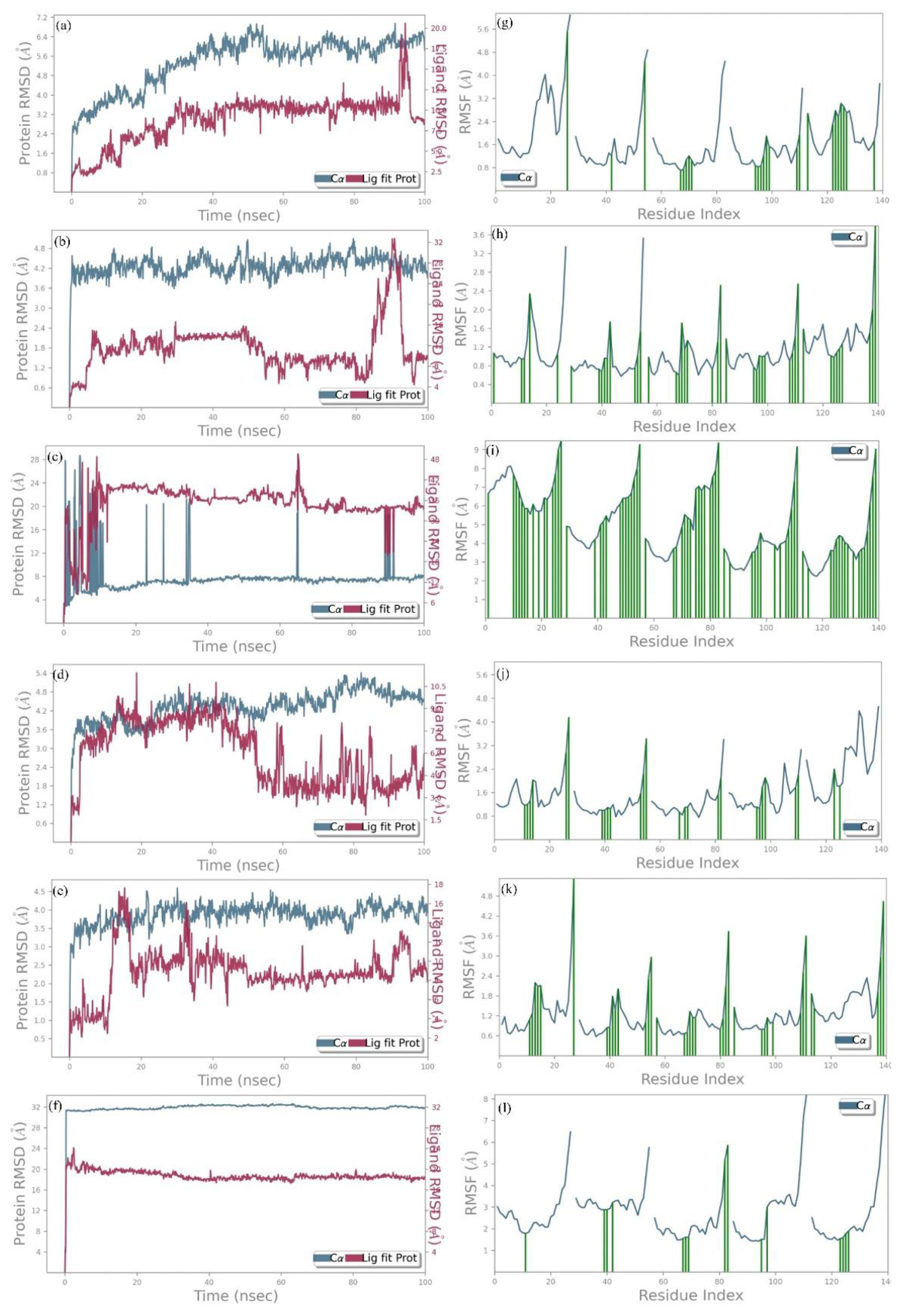
Figure 4.
Prenylmethoxy flavonol-Amyloid-β fibrils docked complex timeline representation of the Prenylmethoxy flavonol (right side) (a), contacts with respect to the amino acids in the target (centre) (a). Percentage of amino acid and water-mediated interactions in MD simulations with Prenylmethoxy flavonol (left side) (c); Isopentenyl flavonol -Amyloid-β fibrils docked complex timeline representation of the isopentenyl flavonol (right side) (d), contacts with respect to the amino acids in the target (centre) (e). Percentage of amino acid and water-mediated interactions in MD simulations with isopentenyl flavonol (left side) (f); 7,3'-Dihydroxyflavone-Amyloid-β fibrils docked complex timeline representation of the 7,3'-Dihydroxyflavone (right side) (g), contacts with respect to the amino acids in the target (centre) (h). Percentage of amino acid and water-mediated interactions in MD simulations with 7,3'-Dihydroxyflavone (left side) (i); 7-Hydroxy-5-methyl-4'-methoxyflavone-Amyloid-β fibrils docked complex timeline representation of the 7-Hydroxy-5-methyl-4'-methoxyflavone (right side) (j), contacts with respect to the amino acids in the target (centre) (k). Percentage of amino acid and water-mediated interactions in MD simulations with 7-Hydroxy-5-methyl-4'-methoxyflavone (left side) (l); 8-hydroxy-7-methoxyflavone-Amyloid-β fibrils docked complex timeline representation of the 8-hydroxy-7-methoxyflavone (right side) (m), contacts with respect to the amino acids in the target (centre) (n). Percentage of amino acid and water-mediated interactions in MD simulations with 8-hydroxy-7-methoxyflavone (left side) (o); Donepezil-Amyloid-β fibrils docked complex timeline representation of the Donepezil (right side) (p), contacts with respect to the amino acids in the target (centre) (q). Percentage of amino acid and water-mediated interactions in MD simulations with Donepezil (left side) (r).
Figure 4.
Prenylmethoxy flavonol-Amyloid-β fibrils docked complex timeline representation of the Prenylmethoxy flavonol (right side) (a), contacts with respect to the amino acids in the target (centre) (a). Percentage of amino acid and water-mediated interactions in MD simulations with Prenylmethoxy flavonol (left side) (c); Isopentenyl flavonol -Amyloid-β fibrils docked complex timeline representation of the isopentenyl flavonol (right side) (d), contacts with respect to the amino acids in the target (centre) (e). Percentage of amino acid and water-mediated interactions in MD simulations with isopentenyl flavonol (left side) (f); 7,3'-Dihydroxyflavone-Amyloid-β fibrils docked complex timeline representation of the 7,3'-Dihydroxyflavone (right side) (g), contacts with respect to the amino acids in the target (centre) (h). Percentage of amino acid and water-mediated interactions in MD simulations with 7,3'-Dihydroxyflavone (left side) (i); 7-Hydroxy-5-methyl-4'-methoxyflavone-Amyloid-β fibrils docked complex timeline representation of the 7-Hydroxy-5-methyl-4'-methoxyflavone (right side) (j), contacts with respect to the amino acids in the target (centre) (k). Percentage of amino acid and water-mediated interactions in MD simulations with 7-Hydroxy-5-methyl-4'-methoxyflavone (left side) (l); 8-hydroxy-7-methoxyflavone-Amyloid-β fibrils docked complex timeline representation of the 8-hydroxy-7-methoxyflavone (right side) (m), contacts with respect to the amino acids in the target (centre) (n). Percentage of amino acid and water-mediated interactions in MD simulations with 8-hydroxy-7-methoxyflavone (left side) (o); Donepezil-Amyloid-β fibrils docked complex timeline representation of the Donepezil (right side) (p), contacts with respect to the amino acids in the target (centre) (q). Percentage of amino acid and water-mediated interactions in MD simulations with Donepezil (left side) (r).
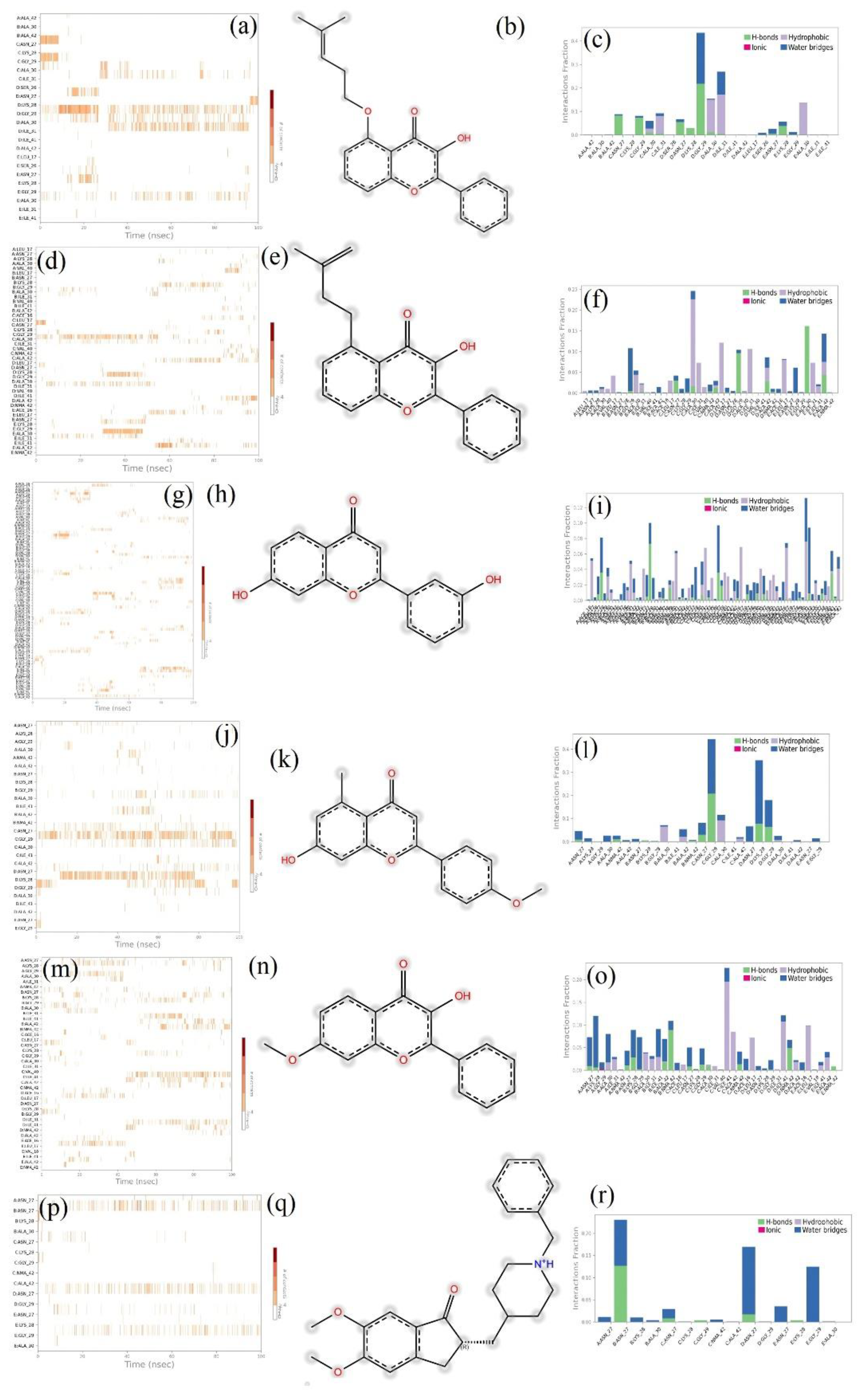

Table 1.
EHOMO and ELUMO and ∆ev values of selected top binding scored compounds and standard drug Donepezil.
Table 1.
EHOMO and ELUMO and ∆ev values of selected top binding scored compounds and standard drug Donepezil.
| Compound Name | HOMO | EHOMO (ev) | LUMO | ELUMO (ev) | Energy gap (Δev) |
|---|---|---|---|---|---|
| Prenylmethoxy flavonol |
 |
-8.578 |
 |
-5.828 | 2.750 |
| Isopentenyl flavonol | 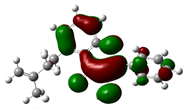 |
-8.604 | 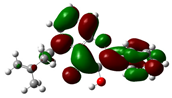 |
-5.917 | 2.687 |
| 7,3'-Dihydroxyflavone | 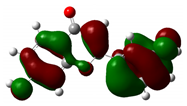 |
-9.285 | 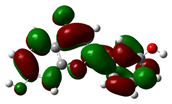 |
-5.880 | 3.404 |
| 7-Hydroxy-5-methyl-4'-methoxyflavone |
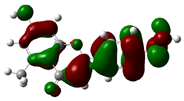 |
-8.771 | 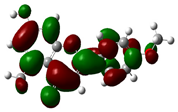 |
-5.654 | 3.117 |
| 8-hydroxy-7-methoxyflavone |
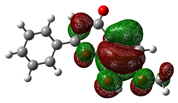 |
-8.515 | 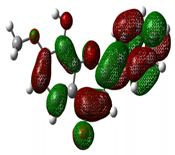 |
-5.850 | 2.665 |
| Donepezil | 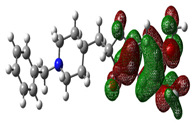 |
-8.794 | 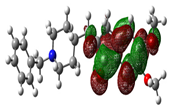 |
-5.667 | 3.127 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



