
Submitted:
01 September 2024
Posted:
03 September 2024
You are already at the latest version
Abstract
Cyclin-dependent kinase 1 (Cdk1) is a master regulator of the G2-M transition between DNA replication and cell division. This study investigates the regulation of cardiomyocyte (CM) proliferation during the early neonatal period and following ischemic injury in adult mice. We analyzed cell cycle dynamics with the assessment of DNA synthesis, and cytokinesis in murine hearts during the first 15 days after birth. A distinct proliferative block was observed at 1 day, followed by a second wave of DNA synthesis at 4 days, leading to CM binucleation (CMBN) by day 5. Genome-wide mRNA profiling revealed differential expression of cell cycle regulatory genes during this period, with a downregulation of factors involved in cell division and mitosis. The loss of Cdk1 impaired CMBN but extended the neonatal CM proliferation window until day 10 post-birth. In adult hearts, cardiac-specific ablation of Cdk1 triggered CM proliferation post-myocardial infarction (MI) in specific zones, driven by the activation of EGFR1 signaling and suppression of the anti-proliferative p38 and p53 signalling. This was accompanied by restoration of fractional shortening, mitochondrial function, and decreased reactive oxygen species. Additionally, cardiac hypertrophy was mitigated, and survival rates post-MI were increased in Cdk1 knockout mice. These findings reveal a novel role of Cdk1 in regulating cell cycle exit and re-entry in differentiated CM and offer insights into potential strategies for cardiac repair.
Keywords:
cardiac regeneration
; cell cycle
; heart failure
; proliferation
1. Introduction
Heart failure is the leading cause of morbidity and mortality in North America and the second leading cause of extended hospital stays[1]. The most common etiology of heart failure occurs following myocardial infarction (MI), underscoring the heart’s particular vulnerability to ischemic injury[2]. After an ischemic insult, the damaged cardiomyocytes (CM) are replaced with fibrotic tissue[3]. This irreversible loss of CM, coupled with the poor proliferative capacity of the surviving CM, contributes to the development of heart failure[2]. Current treatment strategies primarily address the maladaptive neurohormonal changes but do not necessarily improve cardiac function[4]. Therefore, developing innovative approaches to restore cardiac function will not only improve patients’ quality of life but also reduce associated healthcare costs.
The regenerative capacity of the mammalian heart is lost shortly after birth when CM transition from a proliferative to a quiescent state[5]. During the embryonic stage and up until birth, CM undergo DNA replication, G2/M-phase, and cytokinesis, leading to CM proliferation[6,7,8,9]. Around postnatal day 5, CM complete a final round of partial cell cycle activity, resulting in CMBN[9]. Thereafter, CM exit the cell cycle and growth predominantly occurs through hypertrophy[6,7,8,10,11,12,13,14,15,16,17]. This transition is associated with changes in the levels of cell cycle regulators, where the expression of cell cycle inhibitors increases and cell cycle-activating cyclin/Cdk complexes decrease[18,19,20,21,22,23,24,25,26]. Additionally, CM proliferation is tightly regulated by numerous intricate molecular pathways, including the Hippo/YAP mitogenic signaling pathway[11,27,28,29]. The progression through the cell cycle is positively regulated by Cdks and cyclins, their associated regulatory subunits[30] in a carefully orchestrated manner[31]. However, the factors controlling CM cell cycle progression during the postnatal period remain unknown.
Cdk1, also known as Cdc2, plays a central role in progressing the cell cycle. Cdk1 regulates essential processes during the G2/M-phase transition and mitosis, ensuring accurate chromosome segregation and proliferation[32]. Cdk1 is activated by forming a complex with B-type cyclins in late G2-phase[33]. In contrast, Wee1 kinase can phosphorylate and inhibit Cdk1, thereby delaying entry into mitosis[34]. Active Cdk1-cyclin B governs entry into mitosis by phosphorylating target proteins involved in chromatin condensation, nuclear envelope breakdown, and microtubule formation[33,35]. By controlling these events, Cdk1 ensures proper execution of mitosis, safeguards genome integrity, and maintains cell viability[36].
Based on these findings, we hypothesized that the loss of Cdk1 in murine CM would result in proliferative abnormalities in both neonatal and adult hearts. Here, we report the unexpected finding that Cdk1 is required for normal cell cycle arrest and CMBN during the early postnatal period but is redundant for CM division. Furthermore, cardiac-specific Cdk1 deletion in adult mice promoted cardiac repair through enhanced CM proliferation post-MI, driven by the activation of proliferative pathways such as EGFR1 and the suppression of inhibitory pathways like p38 and p53. This highlights Cdk1’s critical role in regulating the balance between proliferation and differentiation in the heart.
2. Results
2.1. Cell Cycle Regulation of Cardiomyocytes in during the Early Neonatal Period
We began our analysis with the assessment of heart weight/body weight (HBW) ratios in neonatal CM in the first 7d after birth. We found that HBW did not significantly change over this period (Figure 1A). However, with the observation that mRNAs involved in DNA replication and mitosis showed a biphasic periodicity, we investigated DNA synthesis in CM in the first 7d after birth. Neonatal C57BL/6J wild-type (wt) mice were injected subcutaneously with a single dose of the 5-ethynyl-2′-deoxyuridine (EDU), a thymidine analogue[37]. Cardiac specimens were prepared for indirect immunofluorescence microscopy employing an EDU assay, and anti-phospho Histone Pi.H3-Ser28 (H3.Pi-S28) antibodies (Figure 1B)[38] to identify CM in S- and M-phase, respectively, in conjunction with anti-cardiac actinin and 4′,6-diamidino-2-phenylindole (Dapi). We also co-stained neonatal cardiac sections with an antibody to pericentriolar material 1 (Pcm1), a CM-specific peri-nuclear marker[39,40], and EDU to specifically identify CM in S-phase (Figure 1C). Using this approach, the CM labeling index was monitored from embryonic day 21 (0d), neonatal 12h and 1d through 7d (Figure 1C). Exceedingly high labeling indexes were detected at 0d and 12h (Figure 1D). In contrast, levels of EDU/Pcm-1 double-positive CM nuclei dropped dramatically at 1d (Figure 1D,E). This proliferative block was corroborated by immunofluorescence analysis of Pi.H3-Ser28 positive CM in M-phase (Figure 1E), and aurora kinase B- (AurkB) positive CM in cytokinesis (Figure 1F,G)[41]. Re-initiation of DNA synthesis occurred at 4d, with a peak labeling index occurring at 5d (49±4.3%; P<0.001 vs. 2d). The number of EDU-positive CM nuclei decreased again markedly by 6d (5.1±2.1%; P<0.01 vs. 2d) (Figure 1D). there were no sex-dependent differences noted.
To ascertain that this second wave of DNA synthesis represented CMBN, we analyzed this morphological developmental change in 2d- versus 5d-old CM by 3D reconstruction of confocal immunofluorescence micrographs of Dapi-stained cardiac sections[42]. CMBN was essentially absent in 1d old CM (Figure 1H). In contrast, 579.1% of CM were binucleated at the end of 5d (P<0.001 vs. 1d). Combined, all these findings demonstrate that CM proliferation ceases in a sharply defined time window after birth and is followed by a distinct second phase of CMBN.
2.2. Differential mRNA Expression of Cell Cycle Factors in the Early Postnatal Period
To elucidate the molecular mechanisms responsible for cessation of proliferation, DNA binucleation and cell cycle exit, we performed genome-wide mRNA microarray profiling[43]. Unsupervised hierarchical cluster analysis at a high confidence threshold revealed that approximately 1,900 individual transcripts changed significantly in murine hearts at 1d through 15d, relative to 0d (Figure 2A). Three-dimensional principal component analysis was performed to compare gene expression profiles across the postnatal days and revealed a clustering between 1d, 3d, 7d, 10d samples, and 0d and 5d samples, consistent with our microscopic results showing cell cycle re-entry and peaks of DNA synthesis (Figure 2B). The highest ranked ‘Gene Ontology’ (GO) terms comprised transcripts involved in the regulation of nuclear division, negative regulation of cell division and regulation of mitotic cell cycle (Figure 2C,D,E).
Next, we validated the transcriptomic results of well-characterized cell cycle regulatory genes by qRT-qPCR analysis (Figure 2F)[42]. We found that the mRNA levels of a set of factors involved in the transition from S-phase into mitosis (AurkB, Skp2, AurkB), G1-Cdks (Cdk1, Cdk2, Cdk4), G1/G2-cyclins (Cyclins A, B, E, F), and key cell cycle activators (e.g. Cdc25A) and inhibitors (Cdkn2b, Cdnkn3) were significantly downregulated by postnatal day 7-10 coinciding with terminal cell cycle exit (Figure 2F). These analyses revealed that distinct cell cycle factors are differentially regulated in the immediate postnatal period. Our transcriptomic findings were confirmed by Western blotting of CM cell cycle inhibitors, cell cycle regulators, and Mapk signaling pathways, and cardiac-specific genes (Figure 2G)[42]. Among all these factors, Cdk1 was amongst the top ten enriched cell cycle genes that were selectively upregulated at 1d and 5d post-birth (Figure 2F). Thus, we decided to examine the potential role of Cdk1 in the regulation of neonatal CM proliferation in vivo.
2.3. Ablation of Cdk1 Expands the Proliferative Time Window in Neonatal Cardiomyocytes
Factors that control cell cycle exit and binucleation in neonatal CM are still not well characterized [44,45]. To investigate the mitotic regulators during this transition, we generated of cardiac-specific Cdk1 mutant mice (Figure 3A). Transgenic mice homozygous for the loxP-flanked (floxed) allele of Cdk1fl/fl (controls)[46] were bred with αMyh6-Cre+/+ transgenic mice[47] to obtain Cdk1fl/fl;αMyh6 (Cdk1KOc) mice (Figure 3B). These mice had the Cdk1 gene deleted specifically in CM at embryonic day E14 post coitum (p.c.) as analyzed by Western blotting and RT-qPCR (Figure 3C,D).
Cardiac function was not altered in Cdk1KOc mice versus controls as assessed by echocardiographic measurement of fractional shortening (FS; P>0.01) (Figure 3E). Heart weight/tibia length (HTL) ratios in Cdk1KOc mice at 15d and 3 months of age were not significantly different compared to controls (P>0.01) (Figure 3F,G). We also found smaller CM in Cdk1KOc animals compared to controls at 15d (Figure 3H) and at 3 months post-birth (Figure 3H), suggesting that Cdk1 may potentially regulate CM cell division during the early neonatal stage.
2.4. Loss of Cdk1 Prolongs the Proliferative Window of Cardiomyocytes during the Postnatal Stage
Initially, we investigated whether genetic silencing of Cdk1 affects DNA synthesis in Cdk1KOc hearts in the early postnatal period. For analysis by confocal immunofluorescence microscopy, mice were injected subcutaneously with EDU, and hearts were excised, fixed, and sectioned. Subsequently, specimens were co-stained with antibodies to α-actinin, EDU, Pi.H3-Ser28, wheat germ agglutinin (WGA), and Dapi to visualize nuclear DNA. We detected a very high EDU-labeling index in CM from control mice at 1d that dropped dramatically at 2d (P<0.001 vs. 0d) (Figure 4A and Figure S1,S2). This was followed by a re-induction of DNA synthesis that peaked at 5d. A progressive decrease in the number of CM in S-phase was observed over 7d and 10d, until DNA synthesis in this cell type ceased completely by 15d. Intriguingly, the numbers of EDU-positive CM in Cdk1KOc mice remained markedly higher on 1d, 2d and 5d when compared to controls (P<0.001). Subsequently, DNA synthesis in Cdk1-deficient CM progressively decreased over 7d-10d and was undetectable at 15d (Figure 4A).
To analyse the distribution of CM in mitosis, cardiac sections were co-stained with antibodies to H3.Pi-S28, an M-phase-specific nuclear marker, in combination with α-actinin immunostaining to identify CM. Microscopic inspection showed dramatically elevated numbers of H3.Pi-S28-positive CM in control animals at 0d and 5d (Figure 4B and Figure S3). This effect was very much lower in control CM at 7d and undetectable at 10d and 15d (P<0.01 vs 0d). Again, the number of CM in mitosis remained significantly higher in Cdk1KOc mice at between 1d-2d and at 7d post-birth in comparison to control animals (P<0.01). In this strain, H3.Pi-S28-positive CM were never observed at 15d (Figure 4B). Additionally, Cdk1KOc hearts at 4w and 6w of age failed to show any signs of cell cycle activity (Figure S4). Inspection of non-cardiomyocytes (NCM) during the first week after birth revealed no significant differences in the number of cycling NCM in Cdk1KOc compared to control hearts (Figure S5). Importantly, all these cell cycle events in CM are sex-independent processes, since no difference was noted in the number of H3.Pi-S28-positive CM positive CM in female mice in comparison to male mice (Figure S6).
Reportedly, CM undergo a binucleation process at 5d postnatally implying that in these cells M-phase can occur independently of cytokinesis[8]. Therefore, we analyzed whether Cdk1 mutant CM completed cytokinesis by forming two daughter cells. Cytokinesis and polyploidization were distinguished by immunostaining with anti-AurkB antibodies to detect midbody structures between daughter CM in fixed cardiac specimen by confocal microscopy. In the presence of Cdk1, CM cytokinesis was detected only at 0d and 1d post-birth based on the analysis of AurkB positive midbody structures between CM (P<0.01 vs 2d) (Figure 4C). The second phase of DNA synthesis at 5d was associated only with CMBN in control hearts as indicated by the absence of AurkB-positive CM (P<0.01 vs 1d). In contrast, ablation of Cdk1 induced ongoing CM cytokinesis from 1d to 7d (P<0.001 vs 1d-7d controls) (Figure 4C). In particular, there was a significant increase in the percentage of binucleated CM and a decrease in mononucleated CM in control hearts at 7d when compared to hearts from Cdk1KOc mice (P<0.001) (Figure 4D). Thus, the loss of Cdk1 not only extended the normal window of postnatal CM proliferation by 6d but also markedly impaired CMBN. This effect was dependent on the absence of Cdk1 and never observed in controls. Intriguingly, the high numbers of dividing CM in hearts Cdk1KOc mice at 0d are compatible with the concept, that Cdk1 is not absolutely required for prenatal CM proliferation.
2.5. Loss of Cdk1 Triggers Cell Cycle Re-Entry and Proliferation of Adult Cardiomyocytes Post-MI
To investigate the potential role of Cdk1 loss in CM proliferation and cardiac remodeling in a clinically relevant model of ischemic stress, 10-12 weeks old male and female Cdk1KOc and control mice were subjected to MI by permanent ligation of the Left Anterior Descending Artery. Next, RNA-seq analysis on total left ventricular (LV) tissues from Cdk1KOc and control mice was performed at 4 days post-MI for genome-wide mRNA profiling. Our previous study identified the 4-day post-MI time point as a critical ‘window’ that drives the proliferative cardiac phenotype in vivo [42]. Three-dimensional principal component analysis (3D-PCA) revealed distinct clustering of Cdk1KOc and control samples, both with and without MI, as analyzed using Strand-NGS software (Figure 5A). Unsupervised hierarchical clustering at a high-confidence threshold identified 1,433 transcripts that significantly differed between Cdk1KOc, and control mice post-MI compared to sham-operated controls (Figure 5B,C). This finding was further supported by Volcano plot analysis, which showed that Cdk1 ablation led to the destabilization of mRNAs in control versus Cdk1KOc mice (Figure 5D).
To explore gene clusters with similar biological functions in mutant and wild type transcriptomes, we performed Gene Set Enrichment Analysis (GSEA) at 4 days post-MI treatment. We found that transcripts related to ventricular remodeling, mitochondrial function, cardiac contraction, and mitosis were among the most enriched GSEA terms (Figure 5E). Notably, transcripts for cell cycle-promoting factors, such as Mcm2, Mcm3, and Tert, were selectively enriched at 4 days post-MI (Figure 5F). At 4d post-MI, the area at risk (AR) in Cdk1KOc and control groups was of equal size and involved greater than 80% of the cross-sectional area of the LV, measured at the level of the papillary muscles (Figure S7). We also noted a decreased sarcomeric α-actinin expression and lower extracellular matrix content, as evaluated by wheat germ agglutinin (WGA) staining. The border zone (BZ) showed α-actinin expression comparable to that of the area at risk with a markedly higher level of WGA reactivity. This is in stark contrast to the remote area (RA) where both α-actinin and WGA are observed at levels comparable to sham Cdk1KOc and sham controls (Figure S7, S8).
To further define the role of Cdk1 loss in cardiac remodeling after MI, DNA synthesis was assessed using EdU labeling in adult mice, followed by confocal immunofluorescence microscopy. Intriguingly, we observed the following numbers of S-phase CM in Cdk1KOc mice at 4d post-MI: (1) AR 61±10.9 nuclei/mm2, (2) BZ 33±7.6 CM nuclei/mm2, (3) RA 1.2±3.4 CM nuclei/mm2, and (4) total number (TN) 98±11.3 CM nuclei/mm2 (P<0.01) (Figure 5G,H and Figure S7,S8). These results clearly demonstrate that induction of DNA synthesis in CM lacking Cdk1 requires an ischemic stress.
Assessment of numbers of CM in Cdk1KOc mice in M-phase by immunofluorescence microscopy employing anti-phospho-histone 3 phosphorylated at H3.Pi-S28 showed: (1) AR 19.8±7.6 CM nuclei/mm2, (2) BZ 9.7±3.2 CM nuclei/mm2, (3) RA zero CM nuclei/mm2, and (4) TN 30±8.6 CM nuclei/mm2 (P<0.01) (Figure 5I,J,K and Figure S9). In all these experiments, EDU-positive and H3.Pi-S28-positive CM were absent from control mice. In addition, the induction of CM in M-phase post-MI was restricted to CM residing in the BZ and was absent from the right ventricle.
Next, we analyzed whether CM from Cdk1KOc mice completed cytokinesis by dividing into 2 daughter cells by detecting midbody structures in the final phase of daughter cell separation employing anti-AurkB antibodies. MI in Cdk1KOc animals induced cytokinesis in 15.2±4.6 CM/mm2 in the AR and minimally in the BZ (P<0.001) (Figure 5L,M,N and Figure S10) that was not observed in controls. This supports the notion, that Cdk1 deletion restricts proliferation to a specific subpopulation located in an ischemic milieu. Of note, CM proliferation in hearts from sham Cdk1KOc and sham control mice was never observed.
Infarct ‘wound healing’ is characterized by initiation of non-CM (NCM) proliferation that mainly comprises endothelial cells and fibroblasts[48]. We noticed a significant increase in the numbers of NCM in S- and M-phase in Cdk1KOc and control mice at 4d following MI (P<0.001 vs. sham Cdk1KOc and sham controls) (Figure 5G,J and Figure S7). Numbers of proliferating EDU-positive NCM was markedly lower in hearts of Cdk1KOc mice compared to controls (1506.1 vs 3519.6 NCM nuclei/mm2). All these findings support the view that Cdk1 ablation induces adult CM proliferation after ischemic injury. We and others have previously shown that cell cycle re-entry in adult CM is tightly regulated by Cdk2[42]. At 4d post MI, sham control hearts lacked an induction of Cdk2 protein relative to Cdk1KOc post-MI (Figure 5O). Elevated Cdk2 protein levels were accompanied by sustained induction of proliferative Cyclins A, B, D2, and E (Figure 5O). Intriguingly, ablation of Cdk1 also led to extremely low protein levels of Cdk2-inhibitory p21 and p19, a cdk4/6 inhibitor.
We have established that adult Cdk1KOc mice have developed a hypertrophic heart (Figure 3I) that mainly consists of mononucleated CM (78±7.6%; P<0.001 vs. control) (Figure 4D). Thus, we examined whether Cdk1-deficiency can further influence the ploidy of CM post-MI. We observed a small but significant increase (11%) of mononucleated CM, and a decrease (−75.4%) in binucleated CM in the AR/BZ areas in Cdk1KOc post-MI (P<0.001) (Figure 5P). This data provide additional evidence that genetic Cdk1 ablation leads to CM proliferation rather than polyploidy.
2.6. Activation of EGFR1 Signaling Promotes Cardiomyocyte Proliferation in Cdk1KOc Mice Post-MI
RNA-seq analysis at 4d post-MI identified differential pathway activation between Cdk1KOc and control mice, as analyzed employing the pathway analysis module of Strand-NGS (Figure 5Q). The EGFR1 signaling pathway was significantly upregulated in the Cdk1KOc MI group, as indicated by the enrichment of 88 out of 159 genes within this pathway (P< 0.001). This activation is associated with enhanced CM proliferation in the Cdk1KOc mice post-MI. In contrast, the p53- and p38-MAPK signaling pathways were predominantly activated in the control group. The activation of these pathways is known to inhibit CM division, suggesting that control mice respond to MI with signals that block cell cycle re-entry[49,50]. In contrast, the absence of Cdk1 allows for the activation of proliferative pathways, such as EGFR1, potentially facilitating CM proliferation (Figure 5Q)[51]. The heatmap in Figure 5R further corroborates the pathway analysis by displaying the expression profiles of key genes involved in the EGFR1, p38, and p53 signaling pathways across the four different experimental groups. Notably, key genes involved in the EGFR1 pathway, such as ErbB4, Erk1, and Sos2, show marked upregulation in the Cdk1KOc MI group when compared to controls, aligning with the observed pathway activation. Conversely, key genes associated with the p38-MAPK and p53 pathways, including p38α, Cdkn1, and Gadd45g, are upregulated in the control group post-MI, which is consistent with the inhibition of CM proliferation in this cohort (Figure 5R). Western blot analysis confirms the activation of these pathways at the protein level (Figure 5S). In Cdk1KOc mice post-MI, there is an increase in phosphorylated ERK1/2 (Pi-ERK1/2), a key downstream effector of EGFR1 signaling, supporting the role of this pathway in promoting CM proliferation. In contrast, control mice show increased levels of p53 and phosphorylated p38α (Pi-38α), further validating the RNA-seq findings that these pathways are involved in blocking CM division following MI. The presence of these protein markers in the Cdk1KOc and control groups provides strong evidence for the differential activation of proliferative vs. inhibitory pathways depending on the genetic background of the experimental strains.
2.7. Cdk1-Deficiency Mitigates Hypertrophy, Boosts Survival and Improves Heart Function Post-MI
Next, we determined the physiological consequences of cardiac-specific Cdk1 knockout after ischemic injury. Cdk1 ablation significantly improve cardiac function in Cdk1KOc mice post-MI versus sham controls as assessed by echocardiographic measurement of FS at 21d post-MI (Figure 6A). Measurements of LVEDD and LVESD dimensions revealed that Cdk1KOc animals were protected from developing significant LV dilation post-MI that was not seen in hearts of sham control mice (Figure 6A).
Western blot analysis in Figure 6B shows several key cardiac proteins, including Serca2a and RyR2, along with their phosphorylated activated forms. The Cdk1KOc cohort demonstrates altered protein expression post-MI that correlates with improved calcium handling and contractility when compared to controls. Figure 6C shows levels of sarcomeric genes, which are crucial for cardiac muscle contraction, across different experimental groups. Notably, key genes such as Ttn, Myh7b and Tnnt1 are significantly upregulated in the Cdk1KOc MI group relative to controls. This elevated expression of sarcomeric genes in Cdk1KOc mice post-MI suggests an enhanced contractile machinery, contributing to improved cardiac function. The heatmap in Figure 6D shows the differential expression of genes involved in the cardiac cycle. Key genes such as Kcnj3, Scn4b, and Prkca, which are involved in cardiac rhythm and contraction regulation, are specifically upregulated in Cdk1KOc mice post-MI. This upregulation implies that the Cdk1 knockout promotes a more efficient cardiac cycle post-MI, likely contributing to the observed preservation of heart function in these mice.
Figure 6E illustrates the significant upregulation of transcript levels of various cardiac transcription factors, highlighting Gata4, Srf, and Nkx2-5 in Cdk1KOc mice post-MI compared to controls. These transcription factors are known to regulate genes critical for cardiac function and remodeling, and their increased expression in the knockout mice may play a pivotal role in the observed enhancement of cardiac function post-MI. Collectively, these findings indicate that the upregulation of sarcomeric and cardiac cycle genes, along with key transcription factors, contributes to the superior cardiac function observed in Cdk1KOc mice post-MI.
Increases in HBW ratios in Cdk1KOc mice were significantly less post-MI versus MI/controls (Figure 6F). Moreover, lung weight/body weight (LBW) ratios, an index of pulmonary congestion, was also less elevated in Cdk1KOc mice as opposed to controls post-MI (Figure 6G). Importantly, Mantel-Cox test showed significantly improved survival in Cdk1KOc at 21d post-MI relative to controls (P<0.001) (Figure 6H). RT-qPCR analysis of canonical hypertrophic marker genes revealed decreased transcript levels ANP, BNP, and -MHC in Cdk1KOc mice post-MI against controls (P<0.01) at 3d and 21d(Figure 6I,J)). Moreover, CM cross-sectional area (Figure 6K) and infarct size (Figure 6L,M) as measured at the peri-infarct zone, were significantly lower in Cdk1KOc mice in contrast to controls post-MI (P<0.01).
Inflammation typically precedes angiogenesis in the sequence of biological responses following MI[3,4,52]. The heatmap in Figure 6N shows the expression levels of inflammation-related genes across different experimental groups. In Cdk1KOc mice post-MI, several key pro-inflammatory genes (Stat1, Il6ra, Nfkb1, Tgfbr1) are significantly upregulated against the control cohort. This indicates a heightened inflammatory response in Cdk1KOc mice following MI. Notably, genes involved in promoting angiogenesis (Vegfa, Flt1, Pecam1, Angpt1) are upregulated in the Cdk1KOc mice relative to controls post-MI (Figure 6O). This suggests that Cdk1KOc mice may have enhanced angiogenic responses post-MI, potentially contributing to better tissue repair and remodeling. Additionally, immunofluorescence images of fixed Cdk1KOc-derived cardiac tissue samples exhibit significantly higher vWF expression post-MI (P < 0.01) versus controls, indicating increased endothelial cell activity and angiogenesis (Figure 6P). All these findings indicate a higher degree of coronary angiogenesis in Cdk1KOc mice post-MI as opposed to their wt counterparts.
Our heatmap analysis reveals that key apoptotic genes (Bax, Casp3, Fas, and Bcl2) are upregulated in the controls in comparison to Cdk1KOc animals post-MI (Figure 6Q). Quantification of apoptotic CM through TUNEL staining and fluorescence microscopic analysis, demonstrates a significantly higher number of apoptotic CMs in controls versus. Cdk1KOc mice post-MI (P<0.01) (Figure 6R) suggesting that Cdk1 deletion protects against CM apoptosis post-MI.
2.8. Cdk1 Loss Preserves Mitochondrial Energetics by Protecting Against Ischemic Oxidative Stress
We have previously shown that MI-induced oxidative stress decreases in ATP levels negatively impact the contractile function and survival of CM[42]. Thus, we analyzed gene expression of mt-DNA replication and mt-dynamics in Cdk1KOc mice post-MI. The heatmap in Figure 7A shows that transcripts levels of key genes involved in mt-DNA replication such as Tfam, PrimPol, and Tfb2m are upregulated in Cdk1KOc mice post-MI. Notably, the expression of genes related to mt-fission and fusion like Fis1, Mfn2, and Mff are also significantly higher in the Cdk1KOc mice post-MI (Figure 7B). Next, mt-DNA copy number was quantified by qPCR using mt-Cytb that were normalized to the nuclear-encoded gene B2m. In Cdk1KOc mice post-MI mice post-MI, we found a significant restoration of mt DNA copy number (P<0.01) (Figure 7C) in contrast to controls. This restoration was accompanied by an increase in mitochondrial respiratory capacity (P<0.01) (Figure 7D) and notably higher ATP/ADP ratios in the myocardium of Cdk1KOc mice vs. controls (P<0.01) (Figure 7E,F,G).
Moreover, we also observed significantly decreased levels of cytotoxic 4-HAE, an indicator of ROS-dependent lipid peroxidation[27] (Figure 7H,I) with upregulated glutathione/oxidized glutathione (GSH/GSSG) ratios in Cdk1KOc mice post-MI (Figure 7J,K,L). The heatmap in Figure 7M illustrates transcript levels of genes involved in the pentose phosphate pathway (PPP), which is critical for generating glutathione, a key molecule for producing NADPH to combat oxidative stress[53]. In Cdk1KOc mice post-MI, there is an upregulation of genes such as Tkt and G6pdx, indicating increased PPP activity compared to controls. This upregulation correlates with higher NADPH/NADP+ ratios in Cdk1KOc hearts subjected to MI (Figure 7N) and implies an enhanced capacity to neutralize oxidative stress (P< 0.01). This view agrees with our Western blot analysis showing elevated levels of antioxidative enzymes such as Sod2, Nqo1, and Pink1 in Cdk1KOc mice post-MI, underscoring the improved oxidative stress response associated with Cdk1 knockout (Figure 7O).
3. Discussion
Cdk1 has a distinct role in embryonic mammalian proliferation that is not fulfilled by Cdk2/4/6[54]. Yet, there’s a paucity of experimental evidence regarding the role of Cdk1 in regulating CM proliferation during the neonatal phase and post-MI. Our data demonstrates that Cdk1 activity is crucial for CM cell cycle arrest on 1d and CMBN at 5d after birth. Notably, the ablation of Cdk1 causes CM to remain in a proliferative state until 10d. Additionally, the genetic ablation of Cdk1 promotes CM proliferation and aids in cardiac repair after MI in the murine adult heart. This study is the first to reveal Cdk1’s dual roles in CMM.
It was surprising to discover that Cdk1 intrinsically inhibits the proliferation of both neonatal and adult CMs post-MI. This finding contradicts the prevailing belief that Cdk1 is necessary for cell proliferation. While germline Cdk1KO mice do not progress beyond the two-cell embryonic stage[54], it is interesting that Cdk1 can offset the lack of individual Cdks in Cdk2/4/6 triple-KO embryos, allowing them to develop normally[55,56]. The triple KO embryos maintain Cdk1 expression, supporting regular cell proliferation until mid-gestation. Interestingly, the introduction of Cdk2 into the Cdk1 gene locus in mice didn’t prevent early embryonic death. In our study, Cdk1KOc mice are born at expected Mendelian ratios and exhibit hypertrophic ventricular walls but maintain normal cardiac function.
The rate of CM proliferation in mouse hearts varies between embryonic and post-natal stages. The four-chambered mammalian mouse heart is completely developed by embryonic day E12.5[57]. In mice, CM proliferation peaks around E10-12 and then gradually diminishes until birth. This pattern might shed light on our observation that Cdk1 is not essential for CM proliferation after E14d, which is after the initiation of α-MHC Cre-dependent homologous recombination, and during the early post-natal phase. Rather, we hypothesize that Cdk1 is primarily required during the initial phases of heart formation in embryogenesis. Our data further corroborates the notion that Cdk2/4/6, either individually or in conjunction, can offset the absence of Cdk1 activity during the cell cycle progression of fetal and neonatal CMs. Comparable to Cdk1KOc mice, transgenic mice with cardiac-specific overexpression of Cdk2 in adults displayed an increase in the number of smaller, mononucleated CMs, suggesting enhanced CM division[58]. Hearts with Cdk2 overexpression exhibited normal FS, even with elevated levels of the canonical hypertrophic marker genes ANF and β-MHC. In contrast, the triple-KO mice lacking cyclin D1/2/3 present hypoplastic ventricular walls accompanied by severe anemia. These animals typically die around mid to late gestation around embryonic day E16.5[59].
Our study revealed that the loss of Cdk1 post-MI leads to the activation of the EGFR1 signaling pathway, which promotes CM proliferation[51] and the suppression of the p38[50] and p53 pathways[49], associated with blocking CM division, thereby allowing for an extended proliferative window in Cdk1KOc mice. This shift in signaling pathways is crucial for enhancing cardiac repair and preserving cardiac function post-MI. Furthermore, Cdk1 ablation was found to significantly enhance mitochondrial function and reduce the production of ROS, as evidenced by the upregulation of mitochondrial biogenesis and antioxidative enzymes in the Cdk1KOc mice. This improvement in mitochondrial energetics likely contributes to the preserved cardiac function observed in these mice.
We also demonstrate that Cdk1 is important for the regulation of the CM division cycle early after birth. Unexpectedly, the genetic deletion of Cdk1 in neonatal mouse CM effectively disabled the intrinsic proliferative block at 1d post-birth, extending the postnatal proliferative window to 10 days. However, there are still gaps in our understanding of the mechanisms that govern CMBN [60,61]. In inbred C57BL/6 mice, commonly employed for cardiological studies, 8-10% of CM are mononucleated[60]. Notably, non-epigenetic mechanisms such as certain polymorphisms located in X-linked and autosomal genes can influence the outcome of mitosis and karyokinesis in a CM-nonautonomous fashion[60]. Our findings found no evidence to support this view during adolescent murine growth, as HTW of Cdk1KOc mice at 15d and 3m did not significantly alter from controls (Figure 3F,G). In addition, a proliferative burst at 15d post-birth has been suggested to give rise to binucleated CM of mouse CM[62]. This event is mediated by the thyroid hormone-IGF-1-Akt signaling pathway. However, several independent studies, including our present work, were unable to confirm this preadolescent event[63,64].
4. Conclusions
In this study, we investigated the regulation of the cell division cycle in CM during the early neonatal period and its impact on heart regeneration following MI. Our results revealed a tightly controlled sequence of events: CM proliferation ceases shortly after birth, followed by a distinct phase of CMBN. Cdk1 plays a pivotal role in regulating both CMBN and proliferation during this period. The loss of Cdk1 extended the neonatal proliferative window, increasing the number of dividing CMs. In adult mice, Cdk1 ablation post-MI activated the EGFR1 signaling pathway, promoting CM proliferation, while downregulating the p38 and p53 pathways, which are typically antiproliferative. This was associated with restoration of FS, enhanced mt function and reduction in oxidative stress post-MI.
Cdk1 emerges as a potential key regulator of heart regeneration post-MI. Targeting Cdk1-related pathways holds significant therapeutic potential for enhancing heart regeneration after MI. Future research should explore the interplay between Cdk1 and other cell cycle regulators to gain a more comprehensive understanding of the balance between CMBN and CM proliferation. In conclusion, loss of Cdk1 is a potential key regulator of heart regeneration post-infarction. Targeting Cdk1-related pathways could hold therapeutic potential for improving heart regeneration after MI that reduce HF risks and improve quality of life.
5. Materials and Methods
5.1. Generation of Cardiac-Specific Cdk1 Knockout Mice
All animal usage were performed in accordance with the institutional animal care guidelines of University Health Network (AUP 1379; Canadian Council in Animal Care). As outlined in the ensemble website (http://useast.ensembl.org/index.html), the murine Cdk1-202 mRNA (strain reference CL57BL6) is located on chromosome 10 on the reverse strand. This transcript has 9 exons, of which 7 are coding exons. The length of the Cdk1-202 transcript is 1,253 bp that codes for 297 residues and has a molecular weight of 34.1 kDa. To examine the potential role of Cdk1 in mice, a constitutive cardiac-specific Cdk1 knockout (Cdk1KOc) was generated. This was accomplished by crossing Cdk1f/f mice (Jackson laboratory, 129S(B6N)-Cdk1 tm1Eddy/J)[46] with transgenic mice carrying the a-MHC Cre recombinase construct (Jackson laboratory, B6.FVB-Tg(Myh6-cre)2182Mds/J)[47]. During generation of the Cdk1f/f strain, mice were crossed to mice expressing FLP1 recombinase to excise the neomycin selection cassette. The cardiac muscle specific α-myosin heavy chain 6 promoter drives the expression of Cre recombinase at birth and results in a recombination efficiency of more than 90%[47]. Cre recombinase fuses together the flanking loxP site of the engineered Cdk1f/f gene at either side of exon 3, thereby deleting the translational initiation site. Cdk1KOc had exon 3 from Cdk1 deleted in CM fromE14 which leads to a reading frameshift mutation that induces a premature translational stop of the Cdk1 mRNA. These mice did not express functional forms of Cdk1.
5.2. Isolation of DNA and Genotyping
DNA isolated from fresh tail snips (<5 mm) was used for genotyping. Samples in were incubated in 300 l of 50 mM NaOH for 2h, 80°C while rocking, and neutralized with 25 l 1.0 M HCl in 700 l H2O. Sample were vigorously vortexed, centrifuged for 13,000 rpm at 10 minutes and stored at 4°C. 0.5 l of DNA sample was used per PCR reaction with the following primers: Cdk1 forward 5’-CCAGGGTGA CCTTGTGCT-3’; Cdk1 reverse 5’-AGCCTGCCTCCACTTCCA-3’; Cdk1 Post-Cre forward 5’-GCACTCGGCCTCTAAGCTC-3’; Cdk1 Post-Cre reverse 5’-TCCACTTGGGAAAGGTGTTC-3’; Myh6-Cre forward 5’-ATGACAGACAGATCCCTCCT ATCTCC-3’; Myh6-Cre reverse 5’-CTCATCA CTCGTGCATCATCGAC-3’. PCR analysis was done with the Applied Biosystems ProFlex PCR system by Life Technologies (Thermo Fisher Scientific, 4484073); Cdk1 wild-type allele, 299 bp; Cdk1 floxed allele, 320 bp; Cdk1 Post-Cre, 838 bp (Figure 3B). Myh6-Cre transgene, 300 bp; Myh6-Cre internal positive control, 200 bp.
5.3. Coronary Artery Ligation
To create a MI, permanent ligation of the left descending coronary artery was performed in in 10-12 weeks old male and female Cdk1KOc and control mice. At the time of surgery and at 24h post MI, a single subcutaneous dose of buprenorphine (30 mg/g body weight) (B9275; Sigma-Aldrich) was given as described previously[42].
5.4. Echocardiography
Echocardiography was performed in anesthetized mice pre-MI and at 21d post-MI.
5.5. DNA Synthesis, TUNEL, ROS Assays and 3-Dimensional Immunofluorescence Microscopy
For EDU-labeling experiments, EDU (160 mg/kg body weight cumulative dosage) intraperitoneally injected twice at 4d post-MI. Hearts were sampled 2 hours after the second injection. For detection of incorporated EDU, the Click-iT™ EdU Cell Proliferation Kit for Imaging (Alexa Fluor™ 488 dye; C10337; Thermo Fisher) was employed according to the manufacturer’s specifications.
Detection of fragmented genomic DNA (apoptosis assay) was performed by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling according to the manufacturer’s instructions (no. 1684795910; Roche)[53]. For immunofluorescence measurements of cell cycle distribution, and CMBN, 3 independent fields of approximately of 0.25 mm2 per left ventricular (LV) sample were counted on 3 consecutive sections. 3D-reconstruction of digital microphotographs were performed using Imaris Imaging software version 10.0.
5.6. Primary Mouse Neonatal Ventricular Cardiomyocyte Isolation
5.7. Preparation of Protein Extracts from Left Ventricular Heart Tissue Samples
Only left ventricular (LV) specimens were used from neonatal hearts derived from Cdk1KOc mice and controls and they were snap frozen and stored at -80°C. Preparation of total protein extracts were performed as described previously[42].
5.8. Western Blotting
Western blotting was done as described previously[42]. The antibodies employed in this study are summarized in the Supplementary Table S1.
5.9. Total RNA Isolation, Reverse Transcription, and Quantitative Real Time PCR Assays
Total RNA isolation, reverse transcription and quantitative real time PCR assays were performed as described previously[42]. The following oligonucleotide primers were used:
α-smooth muscle actin (Acta2) forward 5’-CTGACAGAGGCACCACTGAA-3’, reverse 5’-CAT CTCCAGAGTCCAGCACA-3’. β-Actin forward 5’-GGCTGTATTCCCCTCCATCG-3’, reverse 5’-CCAGTTGGTAACAATGCCATGT-3’. ANP forward 5’-GCTTCCAGGCCATATTGGAG-3’, reverse 5’-GGGGGCATGACCTCATCTT-3. BNP forward 5’- GAGGTCACTCCTATCC TCTGG-3’, reverse 5’-GCCATTTCCTCCGACTTTTCTC-3’. α-MHC forward 5’-GCCCAGTA CCTCCGAAAGTC-3’, reverse 5’-GCCTTAACATACTCCTCCTT GTC -3’. β-MHC forward 5’-ACTGTCAACACTAAG AGGGTCA-3’, reverse 5’-TTGGATGATTTGATCTTCCAGGG-3’.
5.10. Cell Cycle RT-qPCR Array
RNA was purified from hearts of neonatal wild-type mice as described previously[28]. Equal amounts of RNA (400ng/sample) were converted into cDNA using the First Strand cDNA Synthesis kit (Qiagen). The Cell cycle PCR Array was obtained from Qiagen (330171) and profiled the expression of 87 cell cycle genes, 5 housekeeping genes, controls for genomic DNA contamination, and efficiency of both the reverse transcriptase and PCR reaction as described previously[42].
5.11. Gene Expression Analysis and Bioinformatics
Affymetrix Mouse Gene 2.0 ST expression arrays were processed at the Centre for Applied Genomics (Toronto, Canada). Processing of probe level data and subsequent data analyses were performed using GeneSpring (Version 13.2; Agilent Technologies Inc.) as described previously [53]. Genome wide data from the gene expression microarrays were normalized, filtered on expression in the range of -5.2 to 3.5) in the normalized data, filtered on Error-CV < 50.0 percent, filtered for genes with significant differences in expression levels (log2 fold change ±1.3; P<0.05), followed by statistical significance according to t test with Benjamini-Hochberg correction (log2 fold change ±1.3; P<0.01) in wild-type neonatal mice (0d through 15d) in comparison to 0d employing GeneSpring. n=6 to20 per time point. GSEA and GO term analysis of differentially expressed genes were done with GeneSpring. Microarray data were submitted to the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress). Accession number: E-MTAB-9256. Name: “Analysis of cardiac miRNA expression in the early neonatal period”.
RNA-seq samples were processed at the Princess Margaret Genomics Centre, 101 College St., 9-601, Toronto, ON, M5G 1L7, Canada. Briefly, whole exome RNA-seq was performed using paired-end 150 nucleotide sequencing with 40 million reads. Processing of probe level data and subsequent data analyses were performed using Strand-NGS software (Version; Agilent Technologies Inc.). Genome wide data were normalized, filtered for genes with significant differences in expression levels (log2 fold change ±2.0; P<0.01), followed by statistical significance according to t test with Benjamini-Hochberg correction (log2 fold change ±2.0; P<0.01) employing Strand-NGS. 3d-PCA analysis, Volcano plot, Heatmaps, GSEA, GO, and Pathway analysis of differentially expressed genes were done with Strand-NGS. RNA-seq data were submitted to the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress). Accession number: pending.
5.12. Isolation of Mitochondria, Detection of Oxidative Damage, Antioxidants, and ATP Levels
Isolation of mitochondria, detection of oxidative damage, antioxidants and ATP levels was done as described previously[65].
5.13. Statistical Analyses
Statistical analyses were done using GraphPad InStat (version 3.1) and GraphPad Prism (GraphPad Software, version 10.0.2; La Jolla, CA 92037 USA). Data are reported as meanss.e.m. Two-tailed P values of < 0.05 were considered as significant. When groups passed the normality test, we performed data evaluation between 2 groups by an unpaired Student t-test. The statistical significance of 4 groups was calculated using one-way analysis of variance (ANOVA) and Tukey-Kramer multiple comparison post-test.
We calculated experimental power with GraphPad Statmate (GraphPad Software, version 1 La Jolla, CA 92037 USA). For example, for the FS data presented in Figure 6L we used the value of 1 for the standard deviation (S.D.) for Cdk1KOc mice versus controls post-MI, together with a significance level alpha = 0.01 (two-tailed), and 80% power. Therefore, at least n=4 per group was required in each experiment, that was increased to n=6 per group.
Survival analysis was performed using a conservative log-rank test (Mantel-Cox). For calculation of experimental power, the expected a proportion of event-free or surviving was 20% for the Pkm2KOi versus controls. Thus, a sample size of n=20 for Cdk1KOc mice and 24 for controls post-MI in each group provided 80% power to detect an increase in survival proportion of 0.115 with a significance level alpha of 0.01 (two-tailed). We used for the proliferation data (Figure 5) the S.D. value of 3.5 and n=6 per group in each experiment for Cdk1KOc mice versus controls post-MI. This results in an 80% power to detect a difference between means of 5.78 with a significance level (alpha) of 0.01 (two-tailed).
Author Contributions
F.B., L.H. and D.M. designed research; D.M., B.P. and L.H. performed research; F.B., D.M. and L.H. analyzed data; F.B. and D.M. wrote the manuscript.
Funding
This work was supported by grants awarded by the Canadian Institute of Health Research to F. Billia.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data and reagents in this publication can be shared upon request to the PI.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors disclose no other conflicts of interest, financial or otherwise.
References
- Farmakis D, Stafylas P, Giamouzis G, Maniadakis N, Parissis J. The medical and socioeconomic burden of heart failure: A comparative delineation with cancer. Int J Cardiol. 2016;203:279-281. [CrossRef]
- Minicucci MF, Azevedo PS, Polegato BF, Paiva SA, Zornoff LA. Heart failure after myocardial infarction: clinical implications and treatment. Clin Cardiol. 2011;34:410-414. [CrossRef]
- Anderson JL, Morrow DA. Acute Myocardial Infarction. N Engl J Med. 2017;376:2053-2064. [CrossRef]
- Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000;101:2981-2988. [CrossRef]
- Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am J Physiol. 1997;272:H220-226. [CrossRef]
- Soonpaa MH, Field LJ. Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res. 1998;83:15-26. [CrossRef]
- Lopaschuk GD, Jaswal JS. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J Cardiovasc Pharmacol. 2010;56:130-140. [CrossRef]
- Breckenridge RA. Molecular control of cardiac fetal/neonatal remodeling. J Cardiovascular Dev Disease. 2014;1:29-36.
- Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol. 1996;271:H2183-2189. [CrossRef]
- Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. 2013;14:38-48. [CrossRef]
- Foglia MJ, Poss KD. Building and re-building the heart by cardiomyocyte proliferation. Development. 2016;143:729-740. [CrossRef]
- Zebrowski DC, Engel FB. The cardiomyocyte cell cycle in hypertrophy, tissue homeostasis, and regeneration. Rev Physiol Biochem Pharmacol. 2013;165:67-96. [CrossRef]
- Agata Y, Hiraishi S, Oguchi K, Misawa H, Horiguchi Y, Fujino N, Yashiro K, Shimada N. Changes in left ventricular output from fetal to early neonatal life. J Pediatr. 1991;119:441-445. [CrossRef]
- Lopaschuk GD, Spafford MA, Marsh DR. Glycolysis is predominant source of myocardial ATP production immediately after birth. Am J Physiol. 1991;261:H1698-1705. [CrossRef]
- Lavrentyev EN, He D, Cook GA. Expression of genes participating in regulation of fatty acid and glucose utilization and energy metabolism in developing rat hearts. Am J Physiol Heart Circ Physiol. 2004;287:H2035-2042. [CrossRef]
- Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, Medeiros DM, Kovacs A, Kelly DP. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008;22:1948-1961. [CrossRef]
- RA B. Molecular control of cardiac fetal/neonatal remodeling. J Cardiovascular Dev Disease. 2014;1:29-36.
- von Harsdorf R, Hauck L, Mehrhof F, Wegenka U, Cardoso MC, Dietz R. E2F-1 overexpression in cardiomyocytes induces downregulation of p21CIP1 and p27KIP1 and release of active cyclin-dependent kinases in the presence of insulin-like growth factor I. Circ Res. 1999;85:128-136. [CrossRef]
- Tane S, Okayama H, Ikenishi A, Amemiya Y, Nakayama KI, Takeuchi T. Two inhibitory systems and CKIs regulate cell cycle exit of mammalian cardiomyocytes after birth. Biochem Biophys Res Commun. 2015;466:147-154. [CrossRef]
- Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, Ding H, Wylie JN, Pico AR, Capra JA, et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206-220. [CrossRef]
- Sim CB, Ziemann M, Kaspi A, Harikrishnan KN, Ooi J, Khurana I, Chang L, Hudson JE, El-Osta A, Porrello ER. Dynamic changes in the cardiac methylome during postnatal development. FASEB J. 2015;29:1329-1343. [CrossRef]
- Brodsky WY, Arefyeva AM, Uryvaeva IV. Mitotic polyploidization of mouse heart myocytes during the first postnatal week. Cell Tissue Res. 1980;210:133-144. [CrossRef]
- Brodsky WY, Tsirekidze NN, Arefyeva AM. Mitotic-cyclic and cycle-independent growth of cardiomyocytes. J Mol Cell Cardiol. 1985;17:445-455. [CrossRef]
- O’Meara CC, Wamstad JA, Gladstone RA, Fomovsky GM, Butty VL, Shrikumar A, Gannon JB, Boyer LA, Lee RT. Transcriptional reversion of cardiac myocyte fate during mammalian cardiac regeneration. Circ Res. 2015;116:804-815. [CrossRef]
- Mathiyalagan P, Keating ST, Du XJ, El-Osta A. Chromatin modifications remodel cardiac gene expression. Cardiovasc Res. 2014;103:7-16. [CrossRef]
- He A, Kong SW, Ma Q, Pu WT. Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc Natl Acad Sci U S A. 2011;108:5632-5637. [CrossRef]
- von Gise A, Lin Z, Schlegelmilch K, Honor LB, Pan GM, Buck JN, Ma Q, Ishiwata T, Zhou B, Camargo FD, et al. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc Natl Acad Sci U S A. 2012;109:2394-2399. [CrossRef]
- Lin Z, Zhou P, von Gise A, Gu F, Ma Q, Chen J, Guo H, van Gorp PR, Wang DZ, Pu WT. Pi3kcb links Hippo-YAP and PI3K-AKT signaling pathways to promote cardiomyocyte proliferation and survival. Circ Res. 2015;116:35-45. [CrossRef]
- Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, Martin JF. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332:458-461. [CrossRef]
- Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140:3079-3093. [CrossRef]
- Rhind N, Russell P. Signaling pathways that regulate cell division. Cold Spring Harb Perspect Biol. 2012;4. [CrossRef]
- Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2:21-32. [CrossRef]
- Riabowol K, Draetta G, Brizuela L, Vandre D, Beach D. The cdc2 kinase is a nuclear protein that is essential for mitosis in mammalian cells. Cell. 1989;57:393-401. [CrossRef]
- Morgan DO. Principles of CDK regulation. Nature. 1995;374:131-134. [CrossRef]
- Parker LL, Piwnica-Worms H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science. 1992;257:1955-1957. [CrossRef]
- Szmyd R, Niska-Blakie J, Diril MK, Renck Nunes P, Tzelepis K, Lacroix A, van Hul N, Deng LW, Matos J, Dreesen O, et al. Premature activation of Cdk1 leads to mitotic events in S phase and embryonic lethality. Oncogene. 2019;38:998-1018. [CrossRef]
- Huynh N, Dickson C, Zencak D, Hilko DH, Mackay-Sim A, Poulsen SA. Labeling of Cellular DNA with a Cyclosal Phosphotriester Pronucleotide Analog of 5-ethynyl-2’-deoxyuridine. Chem Biol Drug Des. 2015;86:400-409. [CrossRef]
- Crosio C, Fimia GM, Loury R, Kimura M, Okano Y, Zhou H, Sen S, Allis CD, Sassone-Corsi P. Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol Cell Biol. 2002;22:874-885. [CrossRef]
- Gilsbach R, Preissl S, Gruning BA, Schnick T, Burger L, Benes V, Wurch A, Bonisch U, Gunther S, Backofen R, et al. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat Commun. 2014;5:5288. [CrossRef]
- Gilsbach R, Schwaderer M, Preissl S, Gruning BA, Kranzhofer D, Schneider P, Nuhrenberg TG, Mulero-Navarro S, Weichenhan D, Braun C, et al. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat Commun. 2018;9:391. [CrossRef]
- Yasui Y, Urano T, Kawajiri A, Nagata K, Tatsuka M, Saya H, Furukawa K, Takahashi T, Izawa I, Inagaki M. Autophosphorylation of a newly identified site of Aurora-B is indispensable for cytokinesis. J Biol Chem. 2004;279:12997-13003. [CrossRef]
- Hauck L, Dadson K, Chauhan S, Grothe D, Billia F. Inhibiting the Pkm2/b-catenin axis drives in vivo replication of adult cardiomyocytes following experimental MI. Cell Death Differ. 2021;28:1398-1417. [CrossRef]
- Mak TW, Hauck L, Grothe D, Billia F. p53 regulates the cardiac transcriptome. Proc Natl Acad Sci U S A. 2017;114:2331-2336. [CrossRef]
- Gonzalez-Rosa JM, Sharpe M, Field D, Soonpaa MH, Field LJ, Burns CE, Burns CG. Myocardial Polyploidization Creates a Barrier to Heart Regeneration in Zebrafish. Dev Cell. 2018;44:433-446 e437. [CrossRef]
- Kirillova A, Han L, Liu H, Kuhn B. Polyploid cardiomyocytes: implications for heart regeneration. Development. 2021;148. [CrossRef]
- Chaffee BR, Shang F, Chang ML, Clement TM, Eddy EM, Wagner BD, Nakahara M, Nagata S, Robinson ML, Taylor A. Nuclear removal during terminal lens fiber cell differentiation requires CDK1 activity: appropriating mitosis-related nuclear disassembly. Development. 2014;141:3388-3398. [CrossRef]
- Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100:169-179. [CrossRef]
- Virag JI, Murry CE. Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair. Am J Pathol. 2003;163:2433-2440. [CrossRef]
- Chen J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb Perspect Med. 2016;6:a026104. [CrossRef]
- Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, Jiang H, Wang Y, Keating MT. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005;19:1175-1187. [CrossRef]
- Xie Y, Su N, Yang J, Tan Q, Huang S, Jin M, Ni Z, Zhang B, Zhang D, Luo F, et al. FGF/FGFR signaling in health and disease. Signal Transduct Target Ther. 2020;5:181. [CrossRef]
- Bhatt AS, Ambrosy AP, Velazquez EJ. Adverse Remodeling and Reverse Remodeling After Myocardial Infarction. Curr Cardiol Rep. 2017;19:71. [CrossRef]
- Hauck L, Stanley-Hasnain S, Fung A, Grothe D, Rao V, Mak TW, Billia F. Cardiac-specific ablation of the E3 ubiquitin ligase Mdm2 leads to oxidative stress, broad mitochondrial deficiency and early death. PLoS One. 2017;12:e0189861. [CrossRef]
- Santamaria D, Barriere C, Cerqueira A, Hunt S, Tardy C, Newton K, Caceres JF, Dubus P, Malumbres M, Barbacid M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448:811-815. [CrossRef]
- Berthet C, Klarmann KD, Hilton MB, Suh HC, Keller JR, Kiyokawa H, Kaldis P. Combined loss of Cdk2 and Cdk4 results in embryonic lethality and Rb hypophosphorylation. Dev Cell. 2006;10:563-573. [CrossRef]
- Satyanarayana A, Berthet C, Lopez-Molina J, Coppola V, Tessarollo L, Kaldis P. Genetic substitution of Cdk1 by Cdk2 leads to embryonic lethality and loss of meiotic function of Cdk2. Development. 2008;135:3389-3400. [CrossRef]
- Yuan X, Braun T. Multimodal Regulation of Cardiac Myocyte Proliferation. Circ Res. 2017;121:293-309. [CrossRef]
- Liao HS, Kang PM, Nagashima H, Yamasaki N, Usheva A, Ding B, Lorell BH, Izumo S. Cardiac-specific overexpression of cyclin-dependent kinase 2 increases smaller mononuclear cardiomyocytes. Circ Res. 2001;88:443-450. [CrossRef]
- Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, Geng Y, Yu Q, Bhattacharya S, Bronson RT, et al. Mouse development and cell proliferation in the absence of D-cyclins. Cell. 2004;118:477-491. [CrossRef]
- Gan P, Patterson M, Watanabe H, Wang K, Edmonds RA, Reinholdt LG, Sucov HM. Allelic variants between mouse substrains BALB/cJ and BALB/cByJ influence mononuclear cardiomyocyte composition and cardiomyocyte nuclear ploidy. Sci Rep. 2020;10:7605. [CrossRef]
- Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev. 2007;87:521-544. [CrossRef]
- Naqvi N, Li M, Calvert JW, Tejada T, Lambert JP, Wu J, Kesteven SH, Holman SR, Matsuda T, Lovelock JD, et al. A proliferative burst during preadolescence establishes the final cardiomyocyte number. Cell. 2014;157:795-807. [CrossRef]
- Alkass K, Panula J, Westman M, Wu TD, Guerquin-Kern JL, Bergmann O. No Evidence for Cardiomyocyte Number Expansion in Preadolescent Mice. Cell. 2015;163:1026-1036. [CrossRef]
- Soonpaa MH, Zebrowski DC, Platt C, Rosenzweig A, Engel FB, Field LJ. Cardiomyocyte Cell-Cycle Activity during Preadolescence. Cell. 2015;163:781-782. [CrossRef]
- Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci U S A. 2011;108:9572-9577. [CrossRef]
Figure 1.
Cell cycle distribution and nucleation status of murine CM during the first week after birth. (A) Heart weight, body weight and heart/body weight ratios of neonatal mouse CM during the first week after birth. Data are mean±s.e.m. n=6. *P<0.01 vs. 4d. (B) Wide field immunofluorescence micrographs of neonatal mouse hearts. Tissue sections of whole hearts were fixed, permeabilized and stained for EDU (green) to label S-phase nuclei, or anti-Pi.H3-Ser28 (green) antibodies to detect mitotic nuclei and were co-stained with anti-sarcomeric alpha-actinin (red), a cytosolic CM-specific marker, and Dapi (blue) to visualize genomic DNA. RA, right atrium. LA, left atrium. RV, right ventricle. LV, left ventricle. S, interventricular septum. One representative result of 3 independent experiments is shown. (C) Wide field immunofluorescence micrographs of CM in S-phase in neonatal wt-mice. Fixed specimens were co-stained for indirect immunofluorescence microscopy with S-phase marker EdU and with anti-Pcm1 antibodies, recognizing a CM-specific perinuclear marker. Arrow heads denote representative CM nuclei in S-phase. One representative result of 3 independent experiments is shown. (D) Quantification of neonatal CM in S-phase. #P<0.001 vs. 2d. *P<0.01 vs. 2d. Data are mean±s.e.m. n=6. (E) Quantification (left panel) and confocal immunofluorescence microscopic analysis (right panel) of neonatal CM in mitosis at 12h and 2d employing antibodies to Histone H3Pi-Ser28 (green) and cardiac actinin (red). Data are mean±s.e.m. n=6. (F) Analysis of neonatal CM undergoing cytokinesis at 12h and 2d. Three-dimensional (3D) reconstitution of typical confocal micrographs visualizes AurkB positive midbody structure (white arrowheads) between 2 dividing daughter CM. Fixed LV tissue sections were stained for indirect immunofluorescence microscopy analysis with antibodies to actinin (red), and AurkB (green). Specimen were co-stained with Dapi to detect genomic DNA. (G) CM cell division and exit from the cell cycle occurs in a sex-independent manner. Quantitative analysis of AurkB-positive neonatal CM at 12 h and 2d. n.s., not significant. Data are mean±s.e.m. n=6. (H) Quantification (left) and 3D reconstitution of typical confocal immunofluorescence microscopical analysis (right panel) of mono- vs. binucleated CM in ventricular tissue sections. Cardiac specimens were stained with antibodies to AurkB (green) and actinin (red). Nuclear DNA was visualized employing Dapi. Data are mean±s.e.m. n=6.
Figure 1.
Cell cycle distribution and nucleation status of murine CM during the first week after birth. (A) Heart weight, body weight and heart/body weight ratios of neonatal mouse CM during the first week after birth. Data are mean±s.e.m. n=6. *P<0.01 vs. 4d. (B) Wide field immunofluorescence micrographs of neonatal mouse hearts. Tissue sections of whole hearts were fixed, permeabilized and stained for EDU (green) to label S-phase nuclei, or anti-Pi.H3-Ser28 (green) antibodies to detect mitotic nuclei and were co-stained with anti-sarcomeric alpha-actinin (red), a cytosolic CM-specific marker, and Dapi (blue) to visualize genomic DNA. RA, right atrium. LA, left atrium. RV, right ventricle. LV, left ventricle. S, interventricular septum. One representative result of 3 independent experiments is shown. (C) Wide field immunofluorescence micrographs of CM in S-phase in neonatal wt-mice. Fixed specimens were co-stained for indirect immunofluorescence microscopy with S-phase marker EdU and with anti-Pcm1 antibodies, recognizing a CM-specific perinuclear marker. Arrow heads denote representative CM nuclei in S-phase. One representative result of 3 independent experiments is shown. (D) Quantification of neonatal CM in S-phase. #P<0.001 vs. 2d. *P<0.01 vs. 2d. Data are mean±s.e.m. n=6. (E) Quantification (left panel) and confocal immunofluorescence microscopic analysis (right panel) of neonatal CM in mitosis at 12h and 2d employing antibodies to Histone H3Pi-Ser28 (green) and cardiac actinin (red). Data are mean±s.e.m. n=6. (F) Analysis of neonatal CM undergoing cytokinesis at 12h and 2d. Three-dimensional (3D) reconstitution of typical confocal micrographs visualizes AurkB positive midbody structure (white arrowheads) between 2 dividing daughter CM. Fixed LV tissue sections were stained for indirect immunofluorescence microscopy analysis with antibodies to actinin (red), and AurkB (green). Specimen were co-stained with Dapi to detect genomic DNA. (G) CM cell division and exit from the cell cycle occurs in a sex-independent manner. Quantitative analysis of AurkB-positive neonatal CM at 12 h and 2d. n.s., not significant. Data are mean±s.e.m. n=6. (H) Quantification (left) and 3D reconstitution of typical confocal immunofluorescence microscopical analysis (right panel) of mono- vs. binucleated CM in ventricular tissue sections. Cardiac specimens were stained with antibodies to AurkB (green) and actinin (red). Nuclear DNA was visualized employing Dapi. Data are mean±s.e.m. n=6.
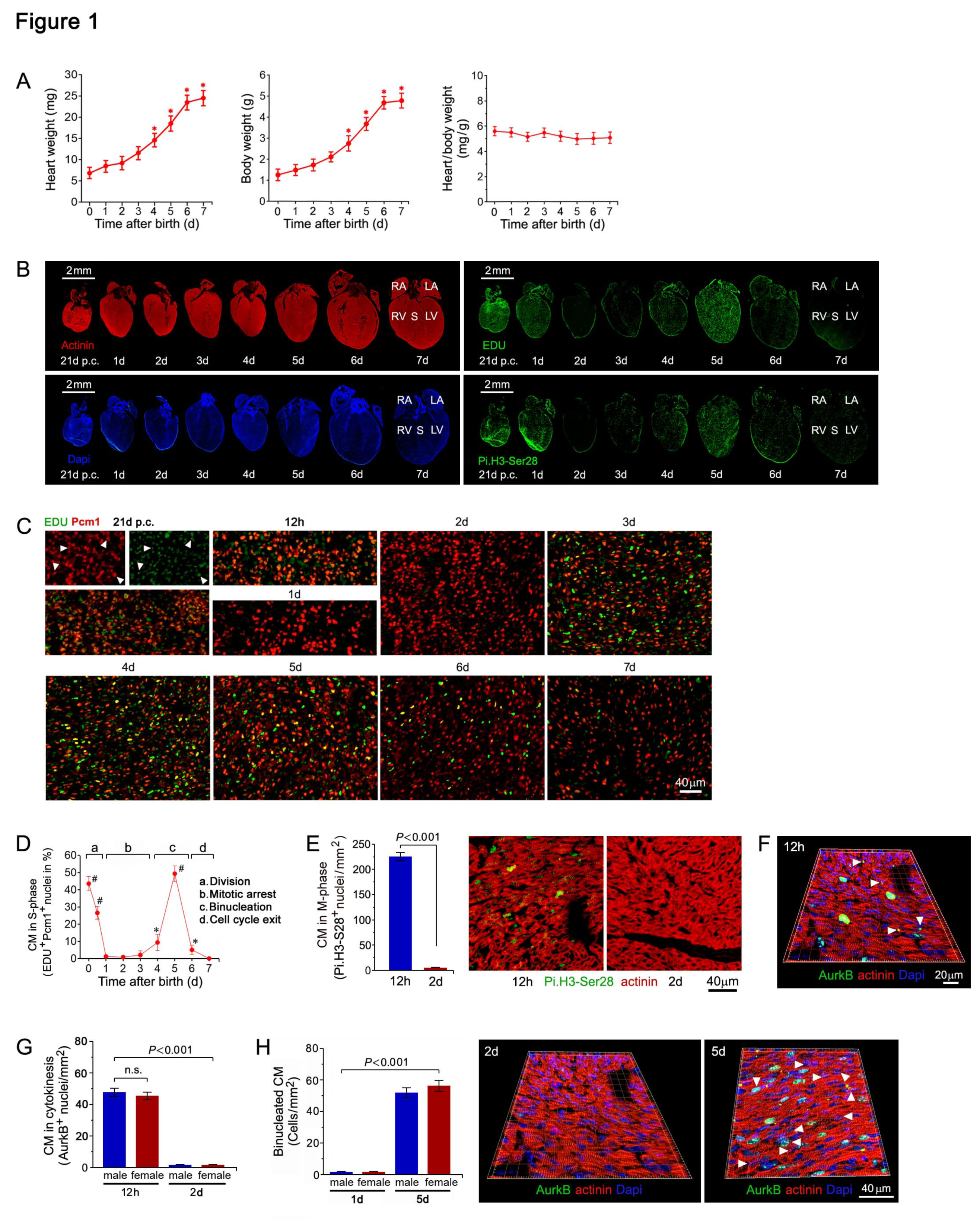
Figure 2.
Genome wide transcriptional changes in cell cycle factors in the early postnatal period. (A) Genome wide transcriptional changes in the myocardium of neonatal mice. Heat map examining unsupervised hierarchical cluster analysis identified approximately 1,900 of 43,000 individual genes (rows) that were enriched in the hearts of mice at 1 day to 15 days (columns), relative to controls at 0 days. Values (log2 expression) are shown by color and intensity of shading. Blue, repressed. Red, induced. n=3 biological samples. Each biological sample consists of 12 hearts (0d) to 3 hearts (15d) serving as technical replicates. P<0.01. Fold change cut-off 1.3. (B) Comparison of neonatal mouse transcriptomics by three-dimensional principal component analysis (3D-PCA). The transcriptome profiles are projected onto PC space. The 3D-PCA analysis demonstrates the presence of distinct developmental stages in neonatal mouse hearts during the early postnatal period. (C,D) Gene Set Enrichment Analysis (GSEA) of biological processes among all differentially expressed transcripts, assessed by over-representation of GSEA terms for the biological function of each transcript in neonatal mouse hearts at 3d (C) and 5d (D) vs. 0d. NES, normalized enrichment scores. (E) GO term enrichment representing upregulated (red) and downregulated (blue) biological processes involved in the regulation of cellular proliferation in ventricular samples (vs. 0d) from neonatal mice in the early postnatal period. (F) Significantly changed mRNA contents of cell cycle factors in murine hearts during the first 2 weeks after birth. RT-qPCR analysis to profile cell cycle related genes in neonatal mouse hearts. Data are mean±s.e.m. *P<0.01. n=4. (G) Significantly changed protein levels of cell cycle regulators in murine neonatal hearts. Western blot analysis was done employing specific antibodies as shown on the left of each panel. Immunoblots were repeated at least once yielding similar results.
Figure 2.
Genome wide transcriptional changes in cell cycle factors in the early postnatal period. (A) Genome wide transcriptional changes in the myocardium of neonatal mice. Heat map examining unsupervised hierarchical cluster analysis identified approximately 1,900 of 43,000 individual genes (rows) that were enriched in the hearts of mice at 1 day to 15 days (columns), relative to controls at 0 days. Values (log2 expression) are shown by color and intensity of shading. Blue, repressed. Red, induced. n=3 biological samples. Each biological sample consists of 12 hearts (0d) to 3 hearts (15d) serving as technical replicates. P<0.01. Fold change cut-off 1.3. (B) Comparison of neonatal mouse transcriptomics by three-dimensional principal component analysis (3D-PCA). The transcriptome profiles are projected onto PC space. The 3D-PCA analysis demonstrates the presence of distinct developmental stages in neonatal mouse hearts during the early postnatal period. (C,D) Gene Set Enrichment Analysis (GSEA) of biological processes among all differentially expressed transcripts, assessed by over-representation of GSEA terms for the biological function of each transcript in neonatal mouse hearts at 3d (C) and 5d (D) vs. 0d. NES, normalized enrichment scores. (E) GO term enrichment representing upregulated (red) and downregulated (blue) biological processes involved in the regulation of cellular proliferation in ventricular samples (vs. 0d) from neonatal mice in the early postnatal period. (F) Significantly changed mRNA contents of cell cycle factors in murine hearts during the first 2 weeks after birth. RT-qPCR analysis to profile cell cycle related genes in neonatal mouse hearts. Data are mean±s.e.m. *P<0.01. n=4. (G) Significantly changed protein levels of cell cycle regulators in murine neonatal hearts. Western blot analysis was done employing specific antibodies as shown on the left of each panel. Immunoblots were repeated at least once yielding similar results.
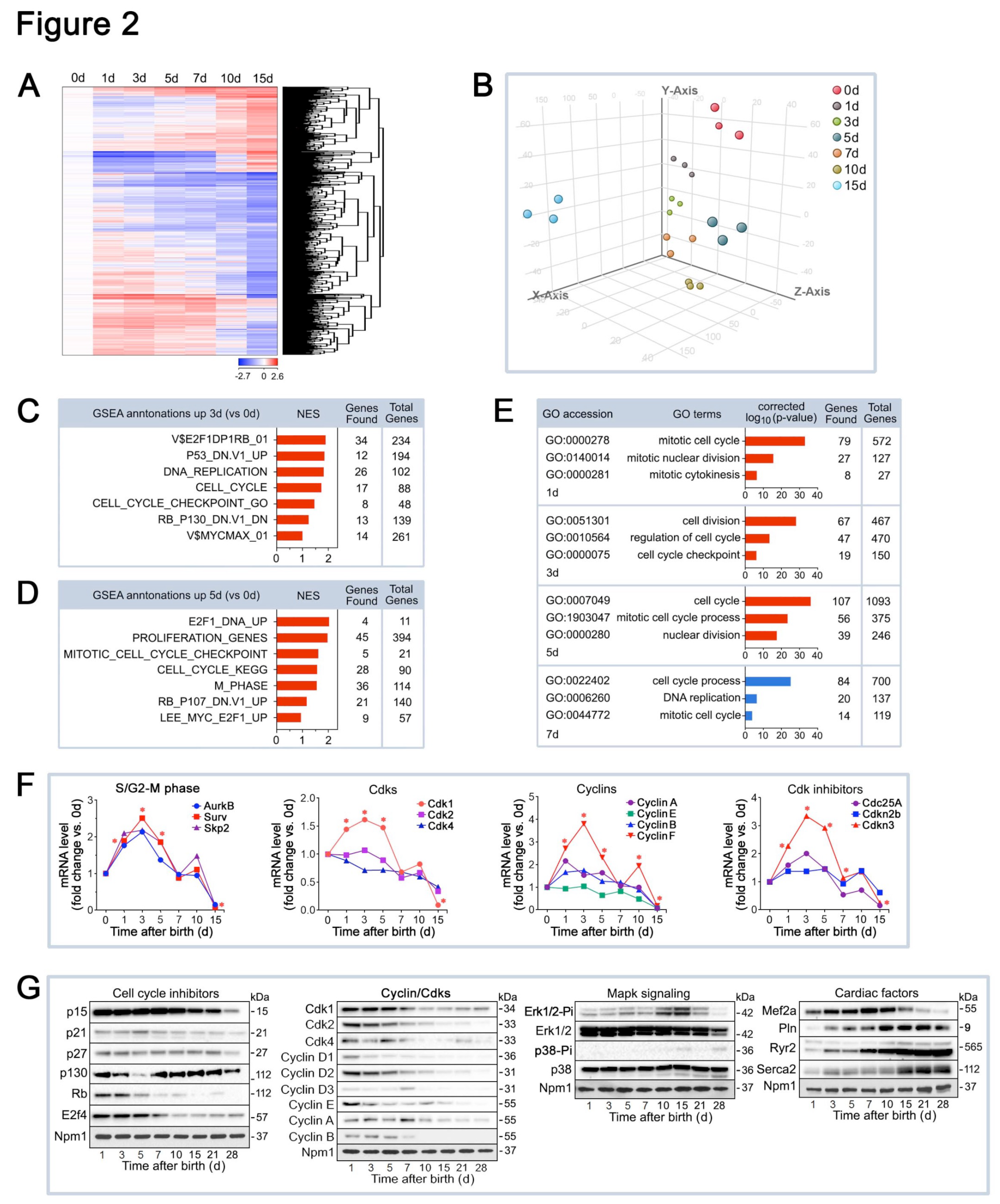
Figure 3.
Normal heart function and tissue architecture in cardiac-specific Cdk1KOc mice. (A) Targeted Cdk1 gene locus showing wild type (top), floxed (middle), and deleted alleles (bottom). (B) CM specific knockout of Cdk1. PCR genotyping of genomic tail DNA from controls (left panel). PCR genotyping of DNA from Cdk1KOc hearts (right panel). (C) Western blot analysis of Cdk1 expression in LV extracts in neonatal CM isolated from 1d old Cdk1KOc mice employing anti-Cdk1 antibodies as indicated on the left. To confirm equal loading, membranes were re-probed with anti-nucleophosmin (Npm1). Western blots were repeated twice with similar results. (D) Quantification of Cdk1 mRNA levels in isolated neonatal CM and in LV extracts of Cdk1KOc mice as analyzed by RT-qPCR. Data are meanss.e.m. n4. *P<0.001 vs. controls. (E,G) Heart weight corrected for tibia length at 15d after birth (E), and 3 months of age (G). Data are meanss.e.m. n6. (H,I) Quantification (top panel) of cross-sectional area of CM at 15d (H) and 3 months of age (I), as analyzed by confocal immunofluorescence microscopy (bottom panel). Data are meanss.e.m. n6. .
Figure 3.
Normal heart function and tissue architecture in cardiac-specific Cdk1KOc mice. (A) Targeted Cdk1 gene locus showing wild type (top), floxed (middle), and deleted alleles (bottom). (B) CM specific knockout of Cdk1. PCR genotyping of genomic tail DNA from controls (left panel). PCR genotyping of DNA from Cdk1KOc hearts (right panel). (C) Western blot analysis of Cdk1 expression in LV extracts in neonatal CM isolated from 1d old Cdk1KOc mice employing anti-Cdk1 antibodies as indicated on the left. To confirm equal loading, membranes were re-probed with anti-nucleophosmin (Npm1). Western blots were repeated twice with similar results. (D) Quantification of Cdk1 mRNA levels in isolated neonatal CM and in LV extracts of Cdk1KOc mice as analyzed by RT-qPCR. Data are meanss.e.m. n4. *P<0.001 vs. controls. (E,G) Heart weight corrected for tibia length at 15d after birth (E), and 3 months of age (G). Data are meanss.e.m. n6. (H,I) Quantification (top panel) of cross-sectional area of CM at 15d (H) and 3 months of age (I), as analyzed by confocal immunofluorescence microscopy (bottom panel). Data are meanss.e.m. n6. .
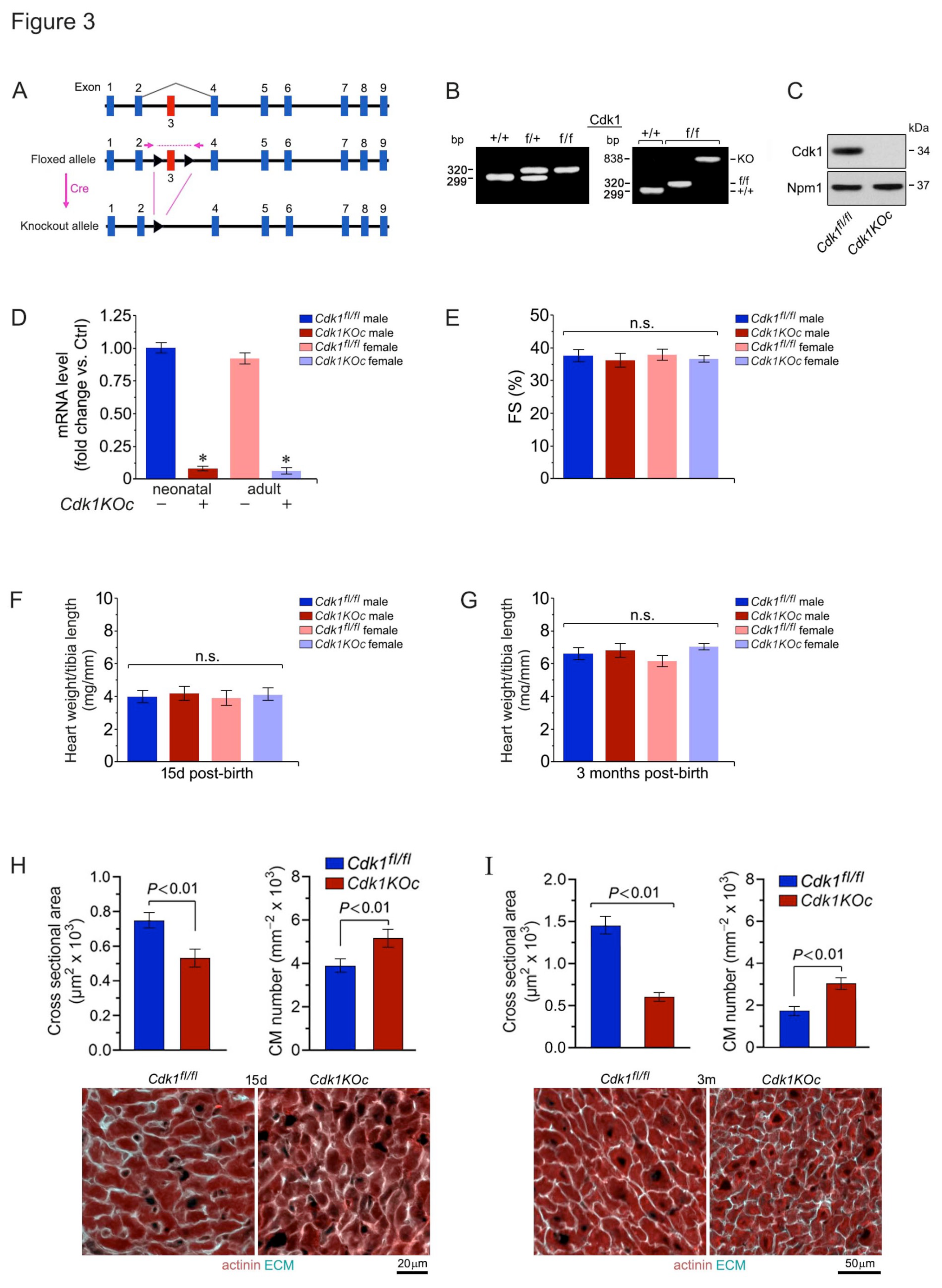
Figure 4.
Cdk1 ablation increases neonatal cardiomyocyte proliferative in vivo. (A) Analysis of CM in S-phase in response to Cdk1 ablation. Heart samples were collected from Cdk1KOc mice and controls. CMs were analyzed by confocal immunofluorescence microscopic employing belled the S-phase marker EDU (green) in conjunction with antibodies to CM-specific cytoplasmic marker α-actinin (red). Data are mean ± S.D. n=4 biological replicates. *P<0.01 vs. controls/0d. #P<0.01 vs. Cdk1KOc/0d. δP <0.001. φP<0.01. n.s, not significant. (B) Analysis of CM in M-phase in Cdk1KOc mice. Fixed tissue sections were stained with antibodies to nuclear H3.Pi-S28 (green), an M-phase marker, in conjunction with α-actinin (red). Data are mean ± S.D. n=4 biological replicates. *P<0.01 vs. controls/0d. #P<0.01 vs. Cdk1KOc/0d. δP <0.001. φP<0.01. (C) Analysis of CM in cytokinesis in the absence of Cdk1. CM were quantified by confocal immunofluorescence microscopic and 3D-reconstruction of serial confocal microphotographs by IMARIS software. Fixed tissue sections were co-immuno-stained for AurkB, a mid-body-specific cytokinesis marker, and α-actinin (red), in conjunction with DAPI to stain genomic DNA (blue). Data are mean ± S.D. n=4 biological replicates. *P<0.01 vs. controls/0d. #P<0.01 vs. Cdk1KOc/0d. δP <0.001. φP<0.01. n.s, not significant. (D) CM-specific ablation of Cdk1 decreases the ploidy of CM. Quantification of CM nucleation in neonatal Cdk1KOc mice at 7d post-birth (left panel) by confocal immunofluorescence microscopy and 3D reconstruction to distinguish mononucleated CM from binucleated CM (right panel). Fifty CM located in 3 different regions were counted per sample. Data are meanss.e.m. n6.
Figure 4.
Cdk1 ablation increases neonatal cardiomyocyte proliferative in vivo. (A) Analysis of CM in S-phase in response to Cdk1 ablation. Heart samples were collected from Cdk1KOc mice and controls. CMs were analyzed by confocal immunofluorescence microscopic employing belled the S-phase marker EDU (green) in conjunction with antibodies to CM-specific cytoplasmic marker α-actinin (red). Data are mean ± S.D. n=4 biological replicates. *P<0.01 vs. controls/0d. #P<0.01 vs. Cdk1KOc/0d. δP <0.001. φP<0.01. n.s, not significant. (B) Analysis of CM in M-phase in Cdk1KOc mice. Fixed tissue sections were stained with antibodies to nuclear H3.Pi-S28 (green), an M-phase marker, in conjunction with α-actinin (red). Data are mean ± S.D. n=4 biological replicates. *P<0.01 vs. controls/0d. #P<0.01 vs. Cdk1KOc/0d. δP <0.001. φP<0.01. (C) Analysis of CM in cytokinesis in the absence of Cdk1. CM were quantified by confocal immunofluorescence microscopic and 3D-reconstruction of serial confocal microphotographs by IMARIS software. Fixed tissue sections were co-immuno-stained for AurkB, a mid-body-specific cytokinesis marker, and α-actinin (red), in conjunction with DAPI to stain genomic DNA (blue). Data are mean ± S.D. n=4 biological replicates. *P<0.01 vs. controls/0d. #P<0.01 vs. Cdk1KOc/0d. δP <0.001. φP<0.01. n.s, not significant. (D) CM-specific ablation of Cdk1 decreases the ploidy of CM. Quantification of CM nucleation in neonatal Cdk1KOc mice at 7d post-birth (left panel) by confocal immunofluorescence microscopy and 3D reconstruction to distinguish mononucleated CM from binucleated CM (right panel). Fifty CM located in 3 different regions were counted per sample. Data are meanss.e.m. n6.
Figure 5.
Cdk1 deletion induces proliferation of adult ventricular cardiomyocytes after myocardial infarction. (A) 3D-Principal component analysis of ventricular transcriptomes as analyzed by RNA-seq at 4d post-MI. (B) Heatmap of genome-wide differentially enriched ventricular transcriptomes in Cdk1KOc mice and controls at 4d post-MI vs. sham. n=3. Values (log2 expression) are shown by color and intensity of shading. Blue, repressed; red, induced. n=3 biological replicates. P< 0.01. Fold change ± 2.0. (C) Venn diagram of differentially regulated genes in Cdk1KOc mice vs. controls at 4d days post-MI. n=3 biological replicates. P< 0.01. Fold change ± 2.0. (D) Volcano plot of RNA-seq results of transcripts selectively upregulated in Cdk1KOc mice post-MI vs. controls. (E) GSEA of different biological processes assessed by overrepresentation of GSEA terms for the biological function of each transcript in Cdk1KOc mice at 4d post-MI. NES, normalized enrichment scores. (F) Heatmap of selectively enriched genes within the GO category of DNA replication, examining the impact of genomic modifications in Cdk1-deficient hearts (columns) on mRNA levels (rows). Arrows indicate key functional genes within this group. (G) Quantitative analysis of CM and non-cardiomyocytes (NCM) in S-phase in Cdk1KOc mice at 4d post-MI. Data are means±s.e.m. n=6. *P<0.001 vs. sham/controls. #P<0.001 vs. sham/ Cdk1KOc. (H) Confocal immunofluorescence microscopic analysis of S-phase CM was done employing co-immunostaining of EDU (green) and anti-α-actinin (red). White asterisks, CM. (I) Heatmap of selectively enriched genes in the GO category G2/M phase. (J) Quantitative analysis of CM and non-cardiomyocytes (NCM) in M-phase in Cdk1KOc mice at 4d post-MI. Data are means±s.e.m. n=6. *P<0.001 vs. sham/controls. #P<0.001 vs. sham/ Cdk1KOc. AR, area at risk. BZ, border zone. RA, remote area. NCM, non-cardiomyocytes. (K) Confocal immunofluorescence microscopic analysis of M-phase CM was performed by confocal immunofluorescence microscopic analysis employing antibodies to Histone H3Pi-Ser28 (green) and α-actinin (red). White asterisks, CM. Data are means±s.e.m. n=6. *P<0.001 vs. sham/controls. #P<0.001 vs. sham/Cdk1KOc. (L) Heatmap of selectively enriched genes in the GO category cytokinesis. (M) Quantitative analysis of CM in cytokinesis in Cdk1KOc mice at 4d post-MI as analyzed by 3D reconstitution of immunofluorescence micrographs. Data are means±s.e.m. n=6. #P<0.001 vs. sham/ Cdk1KOc.(N) 3D reconstitution of immunofluorescence micrographs employing antibodies recognizing AurkB positive midbody structures between 2 dividing daughter cells during cytokinesis. (O) Western blot analysis of cell cycle factors total left-ventricular extracts using antibodies as indicated on the left. Membranes were re-probed with anti-α-Tubulin for control of equal loading. Western blots were done twice employing two independent biological replicates yielding similar results. (P) Analysis of mono- and bi-nucleated CM in the AR at 21d post-MI. Quantification of CM ploidy in adult Cdk1KOc mice (left) by confocal immunofluorescence microscopy and 3D reconstruction analysis (right). Serial confocal microphotographs were reconstructed into 3D employing IMARIS software to unambiguously distinguish mononucleated CM from binucleated CM. Fifty CM located in 3 different regions in the AR/BZ zone were counted per specimen. Data are means±s.e.m. n=6. (Q) Pathway analysis based on differentially enriched mRNAs in Cdk1KOc mice relative to controls at 4d post-MI.(R) Heatmap of selectively enriched transcripts within the top signaling pathways associated with CM proliferation, comparing Cdk1KOc and controls post-MI. (S) Immunoblot analysis of key genes in the EGFR1, p53 and p38α pathways.
Figure 5.
Cdk1 deletion induces proliferation of adult ventricular cardiomyocytes after myocardial infarction. (A) 3D-Principal component analysis of ventricular transcriptomes as analyzed by RNA-seq at 4d post-MI. (B) Heatmap of genome-wide differentially enriched ventricular transcriptomes in Cdk1KOc mice and controls at 4d post-MI vs. sham. n=3. Values (log2 expression) are shown by color and intensity of shading. Blue, repressed; red, induced. n=3 biological replicates. P< 0.01. Fold change ± 2.0. (C) Venn diagram of differentially regulated genes in Cdk1KOc mice vs. controls at 4d days post-MI. n=3 biological replicates. P< 0.01. Fold change ± 2.0. (D) Volcano plot of RNA-seq results of transcripts selectively upregulated in Cdk1KOc mice post-MI vs. controls. (E) GSEA of different biological processes assessed by overrepresentation of GSEA terms for the biological function of each transcript in Cdk1KOc mice at 4d post-MI. NES, normalized enrichment scores. (F) Heatmap of selectively enriched genes within the GO category of DNA replication, examining the impact of genomic modifications in Cdk1-deficient hearts (columns) on mRNA levels (rows). Arrows indicate key functional genes within this group. (G) Quantitative analysis of CM and non-cardiomyocytes (NCM) in S-phase in Cdk1KOc mice at 4d post-MI. Data are means±s.e.m. n=6. *P<0.001 vs. sham/controls. #P<0.001 vs. sham/ Cdk1KOc. (H) Confocal immunofluorescence microscopic analysis of S-phase CM was done employing co-immunostaining of EDU (green) and anti-α-actinin (red). White asterisks, CM. (I) Heatmap of selectively enriched genes in the GO category G2/M phase. (J) Quantitative analysis of CM and non-cardiomyocytes (NCM) in M-phase in Cdk1KOc mice at 4d post-MI. Data are means±s.e.m. n=6. *P<0.001 vs. sham/controls. #P<0.001 vs. sham/ Cdk1KOc. AR, area at risk. BZ, border zone. RA, remote area. NCM, non-cardiomyocytes. (K) Confocal immunofluorescence microscopic analysis of M-phase CM was performed by confocal immunofluorescence microscopic analysis employing antibodies to Histone H3Pi-Ser28 (green) and α-actinin (red). White asterisks, CM. Data are means±s.e.m. n=6. *P<0.001 vs. sham/controls. #P<0.001 vs. sham/Cdk1KOc. (L) Heatmap of selectively enriched genes in the GO category cytokinesis. (M) Quantitative analysis of CM in cytokinesis in Cdk1KOc mice at 4d post-MI as analyzed by 3D reconstitution of immunofluorescence micrographs. Data are means±s.e.m. n=6. #P<0.001 vs. sham/ Cdk1KOc.(N) 3D reconstitution of immunofluorescence micrographs employing antibodies recognizing AurkB positive midbody structures between 2 dividing daughter cells during cytokinesis. (O) Western blot analysis of cell cycle factors total left-ventricular extracts using antibodies as indicated on the left. Membranes were re-probed with anti-α-Tubulin for control of equal loading. Western blots were done twice employing two independent biological replicates yielding similar results. (P) Analysis of mono- and bi-nucleated CM in the AR at 21d post-MI. Quantification of CM ploidy in adult Cdk1KOc mice (left) by confocal immunofluorescence microscopy and 3D reconstruction analysis (right). Serial confocal microphotographs were reconstructed into 3D employing IMARIS software to unambiguously distinguish mononucleated CM from binucleated CM. Fifty CM located in 3 different regions in the AR/BZ zone were counted per specimen. Data are means±s.e.m. n=6. (Q) Pathway analysis based on differentially enriched mRNAs in Cdk1KOc mice relative to controls at 4d post-MI.(R) Heatmap of selectively enriched transcripts within the top signaling pathways associated with CM proliferation, comparing Cdk1KOc and controls post-MI. (S) Immunoblot analysis of key genes in the EGFR1, p53 and p38α pathways.
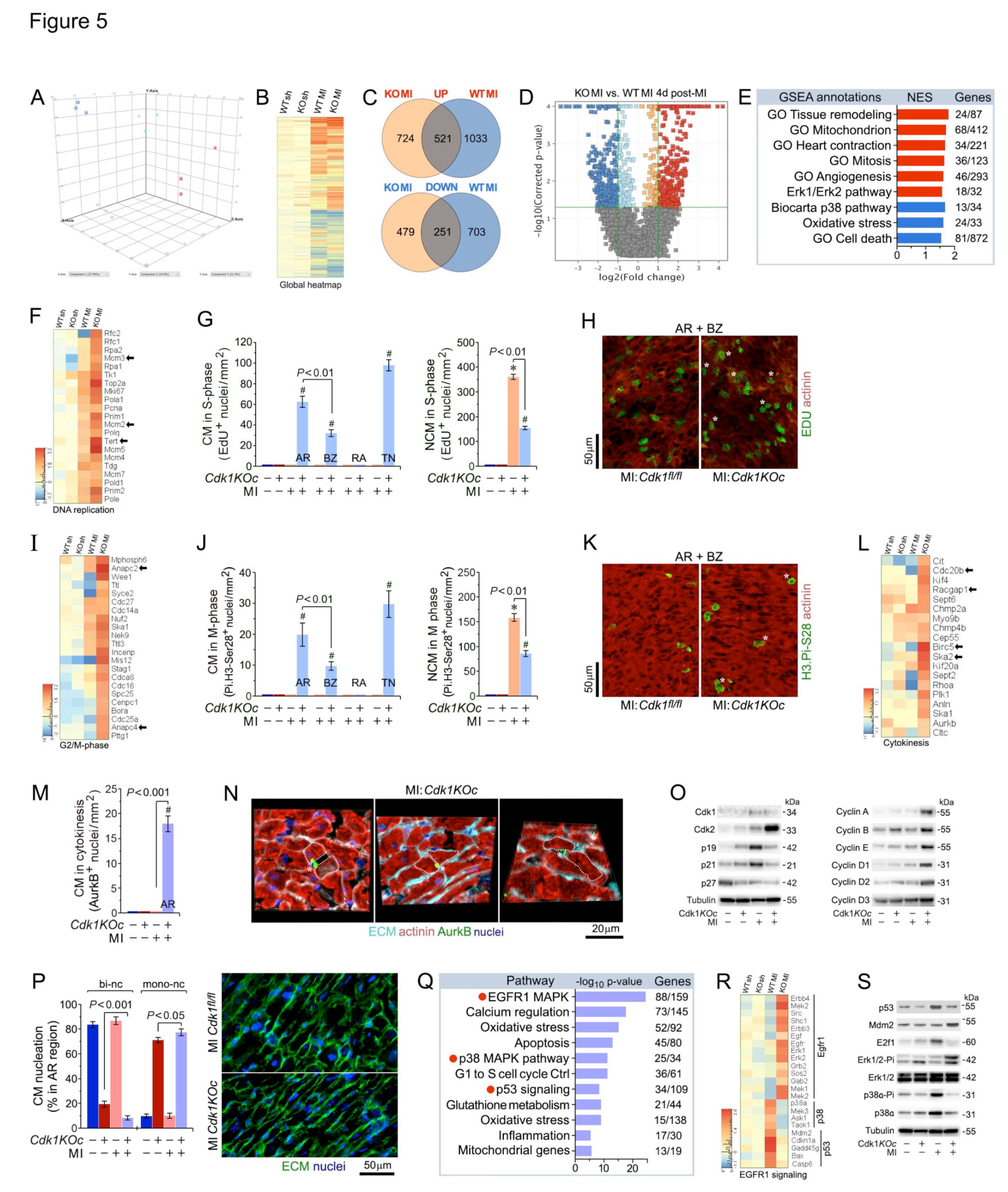
Figure 6.
Cdk1-ablation normalizes contractility with improved survival post-MI. .(A) Echocardiographic assessment of cardiac function. FS, LVEDD , and LVESD in Cdk1KOc mice at 21d post-MI. Data are mean±s.e.m. n=6.(B) Immunoblot analysis of key sarcomeric and calcium-handling proteins in Cdk1KOc and controls at 21d post-MI employing specific antibodies as shown on the left. Membranes were re-probed with anti-Tubulin for control of equal loading. Western blots were done twice employing two independent biological replicates yielding similar results. (C-E) Heatmap analysis of transcript levels in key cardiac Gene Ontology terms: Sarcomeric structure (C), cardiac cycle (D), and cardiac transcription factors (Cardiac TransFacs) (E) at 4d post-MI.(F) Heart weight corrected for body weight at 21d post-MI. Data are means±s.e.m. n=6.(G) Lung weight corrected for body weight at 21d post-MI. Data are means±s.e.m. n=6. *P<0.01 vs. sham controls. *P<0.01 vs. sham Cdk1KOc.(H) Mantel-Cox test shows improved survival in Cdk1KOc mice compared to controls at 21d post-MI (P<0.001). MI Cdk1KO n=20; MI controls n=24; sham Cdk1KO n=6; sham controls n=6.(I) Heat map examining the impact of genomic modifications in Cdk1-deficient hearts (columns) on transcript levels of the fetal gene program (rows) at 4d post-MI.(J) RNA-seq quantification of mRNA levels for hypertrophic marker genes at 21 days post-MI. Data are presented as mean ± s.e.m., n=3. *P<0.01 vs. sham controls. #P<0.01 vs. sham Cdk1KOc.(K) Quantification of cross-sectional area of CM in the border zone at 21d post-MI (left) as analyzed by confocal immunofluorescence microscopy (right). Data are means±s.e.m. n=6. *P<0.01 vs. sham controls. #P<0.01 vs. sham Cdk1KOc.(L) Heat maps examining protein coding transcripts important for the regulation of collagen synthesis at 4d post-MI.(M) Quantification of infarct sizes at 21d post-MI (left panel). Representative confocal immunofluorescence micrographs of WGA-stained cardiac cross-sections recognizing fibrotic ECM (right). Data are means±s.e.m. n=6. (N,O) Heatmap analysis of gene expression in key cardiac Gene Ontology terms: Inflammation (N), and angiogenesis (O) at 4d post-MI.(P) Quantification of capillary density at 4d post-MI (left), as determined in myocardial LV cross-sections employing anti-Von Willebrand factor staining (right; green). Data are means±s.e.m. n=6. *P<0.01 vs. sham controls. #P<0.01 vs. sham Cdk1KOc. (Q) Heat map examining the impact of Cdk1-ablation on validated pro-apoptotic transcript levels.(R) Quantification of CM apoptosis (left) in LV-sections (right) at 2d post-MI. White stars, CM. Data are means±s.e.m. n=6. *P<0.01 vs. sham controls. #P<0.01 vs. sham Cdk1KOc.
Figure 6.
Cdk1-ablation normalizes contractility with improved survival post-MI. .(A) Echocardiographic assessment of cardiac function. FS, LVEDD , and LVESD in Cdk1KOc mice at 21d post-MI. Data are mean±s.e.m. n=6.(B) Immunoblot analysis of key sarcomeric and calcium-handling proteins in Cdk1KOc and controls at 21d post-MI employing specific antibodies as shown on the left. Membranes were re-probed with anti-Tubulin for control of equal loading. Western blots were done twice employing two independent biological replicates yielding similar results. (C-E) Heatmap analysis of transcript levels in key cardiac Gene Ontology terms: Sarcomeric structure (C), cardiac cycle (D), and cardiac transcription factors (Cardiac TransFacs) (E) at 4d post-MI.(F) Heart weight corrected for body weight at 21d post-MI. Data are means±s.e.m. n=6.(G) Lung weight corrected for body weight at 21d post-MI. Data are means±s.e.m. n=6. *P<0.01 vs. sham controls. *P<0.01 vs. sham Cdk1KOc.(H) Mantel-Cox test shows improved survival in Cdk1KOc mice compared to controls at 21d post-MI (P<0.001). MI Cdk1KO n=20; MI controls n=24; sham Cdk1KO n=6; sham controls n=6.(I) Heat map examining the impact of genomic modifications in Cdk1-deficient hearts (columns) on transcript levels of the fetal gene program (rows) at 4d post-MI.(J) RNA-seq quantification of mRNA levels for hypertrophic marker genes at 21 days post-MI. Data are presented as mean ± s.e.m., n=3. *P<0.01 vs. sham controls. #P<0.01 vs. sham Cdk1KOc.(K) Quantification of cross-sectional area of CM in the border zone at 21d post-MI (left) as analyzed by confocal immunofluorescence microscopy (right). Data are means±s.e.m. n=6. *P<0.01 vs. sham controls. #P<0.01 vs. sham Cdk1KOc.(L) Heat maps examining protein coding transcripts important for the regulation of collagen synthesis at 4d post-MI.(M) Quantification of infarct sizes at 21d post-MI (left panel). Representative confocal immunofluorescence micrographs of WGA-stained cardiac cross-sections recognizing fibrotic ECM (right). Data are means±s.e.m. n=6. (N,O) Heatmap analysis of gene expression in key cardiac Gene Ontology terms: Inflammation (N), and angiogenesis (O) at 4d post-MI.(P) Quantification of capillary density at 4d post-MI (left), as determined in myocardial LV cross-sections employing anti-Von Willebrand factor staining (right; green). Data are means±s.e.m. n=6. *P<0.01 vs. sham controls. #P<0.01 vs. sham Cdk1KOc. (Q) Heat map examining the impact of Cdk1-ablation on validated pro-apoptotic transcript levels.(R) Quantification of CM apoptosis (left) in LV-sections (right) at 2d post-MI. White stars, CM. Data are means±s.e.m. n=6. *P<0.01 vs. sham controls. #P<0.01 vs. sham Cdk1KOc.
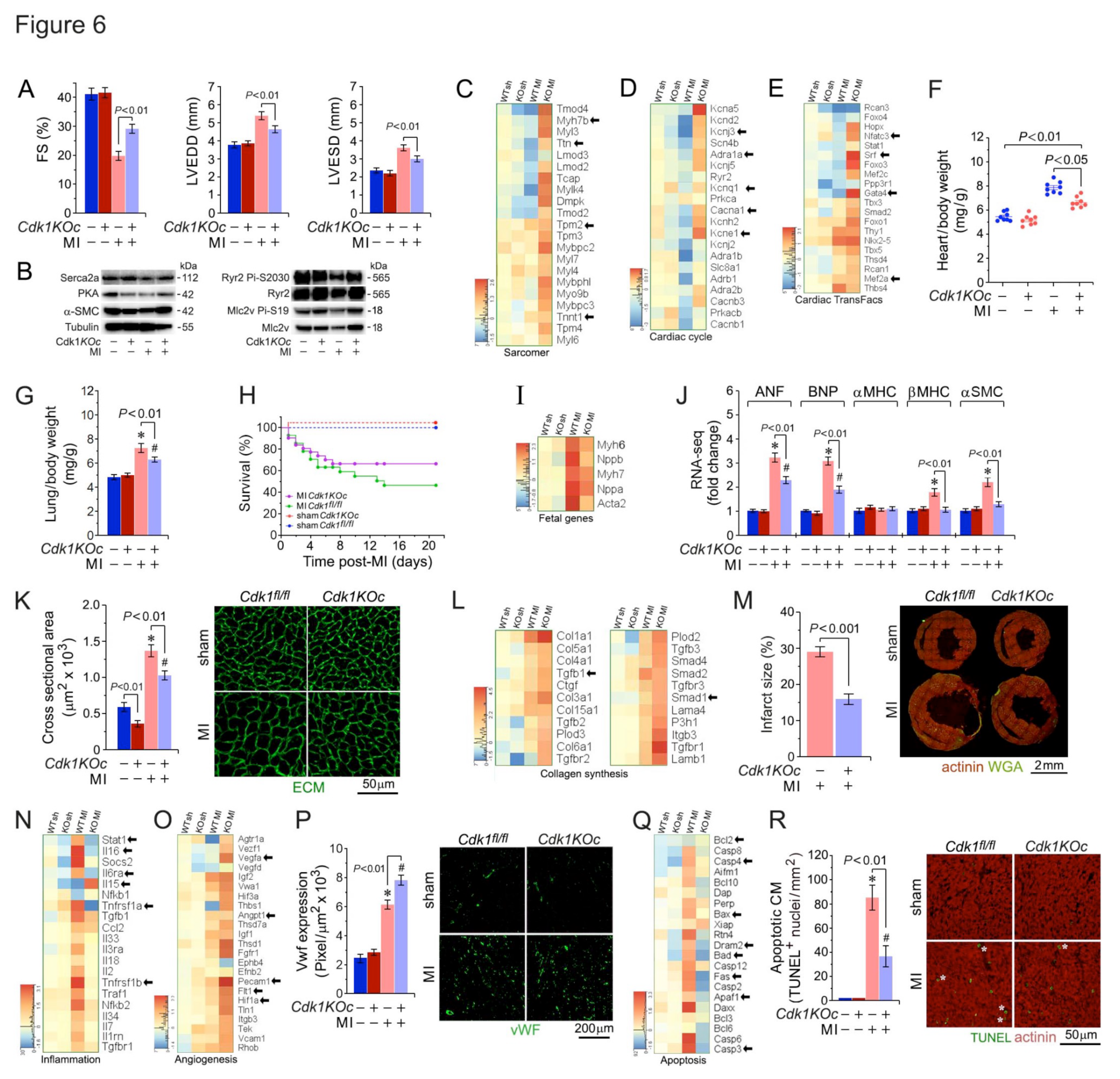
Figure 7.
Cdk1-deficiency inhibits cardiac oxidative stress and improves mitochondrial ATP production. (A,B) Heat map examining the impact of Cdk1-loss on mRNA levels of mt-DNA replication (A) and mt-fission/fusion (B) at 4d post-MI. (C,D) Mt biogenesis (C), defined as relative DNA copy number of mt encoded Cytb gene normalized to the copy number of the nuclear gene B2m, was determined by qPCR. mice. Mt capacity, defined as relative mRNA (D) levels of the nuclear gene cytochrome b (Cytb), a constituent of OxPhos complex III, normalized to B2m transcript expression, was determined by RT-qPCR at 4d post-MI. Data are means±s.e.m. n=4.(E) Heat maps examining the impact of Cdk1-deficiency on components of mt oxidative phosphorylation.(F) Quantification of ATP/ADP ratios in Cdk1KOc mice vs. controls at 4d post-MI. Data are means±s.e.m. n=4.(G) Immunoblot analysis of proteins in Cdk1KOc important for the mt electron transport chain (ATP5a, complex V; Sdh1, complex II) and key factors necessary for mt Fusion/fission (Drp1, Mfn2, Opa1) at 4d post-MI employing specific antibodies as shown on the left. Membranes were re-probed with anti-hexokinase 2 (Hk2) for control of equal loading. Western blots were done twice employing two independent biological replicates yielding similar results. (H) Heat maps investigating the consequences of Cdk1-ablation on hypoxia-related gene expression.(I) Levels of 4-HAE, a biomarker for oxidative lipid damage, in hearts of Cdkn1KOc mice at 4d post-MI. Data are means±s.e.m. n=4.(J) Heat maps showing enrichment of induced (red) and repressed (blue) protein coding gene transcripts involved in the regulation of the GO term “reactive oxygen species (ROS) defence”. (K,L) Levels of GSSG (K) and GSH (L), indicators of cardiac oxidative stress, in Cdk1KOc mice in vs. controls at 4d post-MI. Data are means±s.e.m. n=4. *P<0.001 vs. sham control. (M) Heat maps showing enrichment of induced (red) and repressed (blue) protein coding gene transcripts involved in the regulation of the pentose phosphate pathway (PPP). (N) Levels NAPDH, an important metabolite in GSH synthesis, in Cdk1KOc mice in vs. controls at 4d post-MI. Data are means±s.e.m. n=4. (O) Immunoblot analysis of important anti-oxidative factors Cdk1KOc mice and controls at 4d post-MI employing specific antibodies as shown on the left. Membranes were re-probed with Hk2 antibodies for loading control. Western blots were done twice employing two independent biological replicates giving similar results.
Figure 7.
Cdk1-deficiency inhibits cardiac oxidative stress and improves mitochondrial ATP production. (A,B) Heat map examining the impact of Cdk1-loss on mRNA levels of mt-DNA replication (A) and mt-fission/fusion (B) at 4d post-MI. (C,D) Mt biogenesis (C), defined as relative DNA copy number of mt encoded Cytb gene normalized to the copy number of the nuclear gene B2m, was determined by qPCR. mice. Mt capacity, defined as relative mRNA (D) levels of the nuclear gene cytochrome b (Cytb), a constituent of OxPhos complex III, normalized to B2m transcript expression, was determined by RT-qPCR at 4d post-MI. Data are means±s.e.m. n=4.(E) Heat maps examining the impact of Cdk1-deficiency on components of mt oxidative phosphorylation.(F) Quantification of ATP/ADP ratios in Cdk1KOc mice vs. controls at 4d post-MI. Data are means±s.e.m. n=4.(G) Immunoblot analysis of proteins in Cdk1KOc important for the mt electron transport chain (ATP5a, complex V; Sdh1, complex II) and key factors necessary for mt Fusion/fission (Drp1, Mfn2, Opa1) at 4d post-MI employing specific antibodies as shown on the left. Membranes were re-probed with anti-hexokinase 2 (Hk2) for control of equal loading. Western blots were done twice employing two independent biological replicates yielding similar results. (H) Heat maps investigating the consequences of Cdk1-ablation on hypoxia-related gene expression.(I) Levels of 4-HAE, a biomarker for oxidative lipid damage, in hearts of Cdkn1KOc mice at 4d post-MI. Data are means±s.e.m. n=4.(J) Heat maps showing enrichment of induced (red) and repressed (blue) protein coding gene transcripts involved in the regulation of the GO term “reactive oxygen species (ROS) defence”. (K,L) Levels of GSSG (K) and GSH (L), indicators of cardiac oxidative stress, in Cdk1KOc mice in vs. controls at 4d post-MI. Data are means±s.e.m. n=4. *P<0.001 vs. sham control. (M) Heat maps showing enrichment of induced (red) and repressed (blue) protein coding gene transcripts involved in the regulation of the pentose phosphate pathway (PPP). (N) Levels NAPDH, an important metabolite in GSH synthesis, in Cdk1KOc mice in vs. controls at 4d post-MI. Data are means±s.e.m. n=4. (O) Immunoblot analysis of important anti-oxidative factors Cdk1KOc mice and controls at 4d post-MI employing specific antibodies as shown on the left. Membranes were re-probed with Hk2 antibodies for loading control. Western blots were done twice employing two independent biological replicates giving similar results.
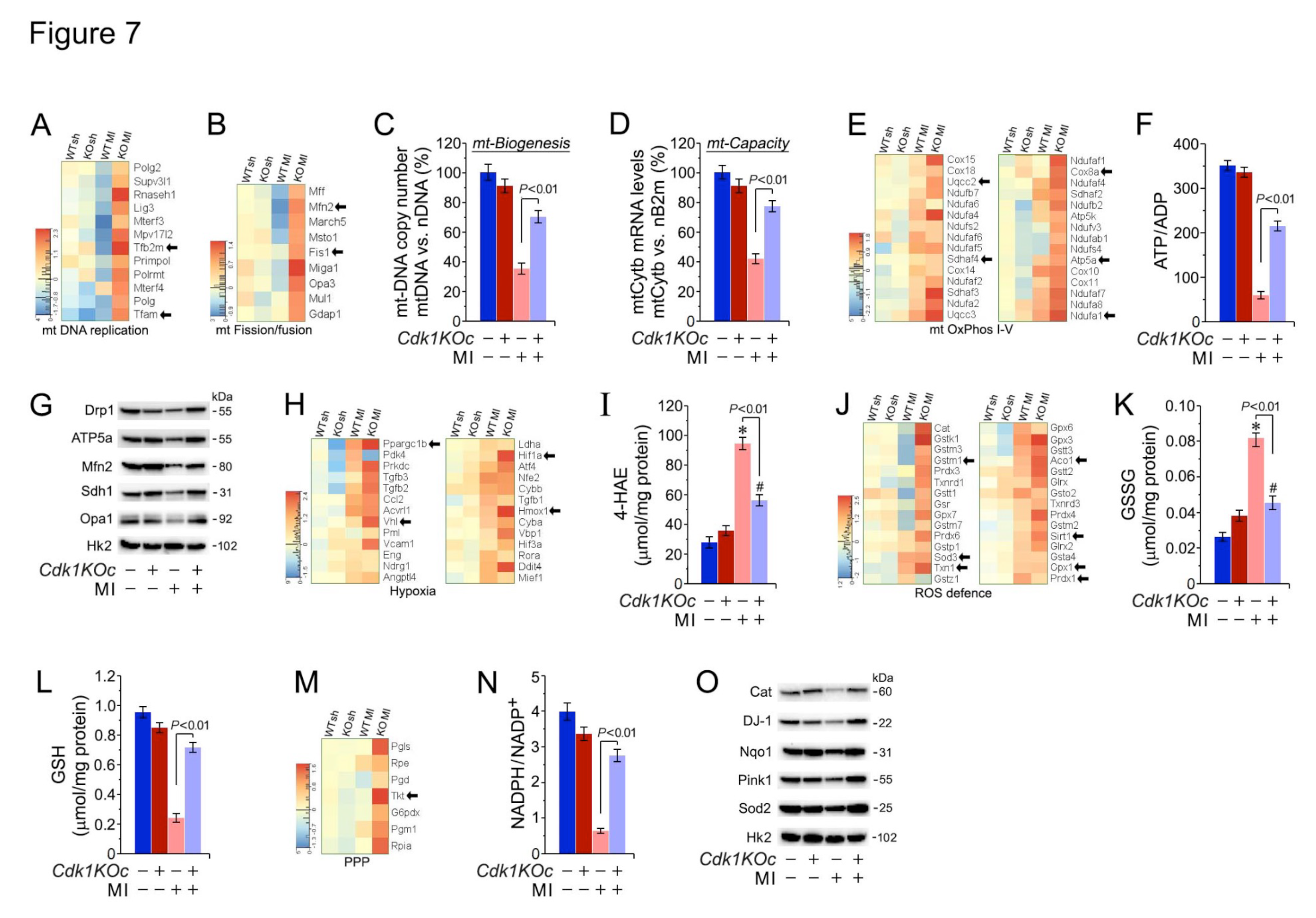
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



