
Submitted:
02 September 2024
Posted:
03 September 2024
You are already at the latest version
Abstract
Conduction of vasodilator signals plays an important role in the coordination of blood flow distribution. It is thought that endothelium-dependent vasodilation’s are conducted by electrotonic spread through gap junctions of the endothelial cell hyperpolarization generated at the stimulation site. However, nitric oxide (NO) was found to reduce the propagation of vasodilator responses, suggesting the participation of this signaling molecule in the mechanism of conduction. Mouse cremasteric arterioles were stimulated through micro-application of ACh or the NO donor, S-nitroso-N-acetylpenicillamine (SNAP) and the responses were evaluated at the stimulation site (local) and at sites located 500, 1000 and 2000 µm upstream from the vessel segment stimulated with ACh. The response to ACh spread along the entire vessel showing a slight decay and, in contrast, the dilation evoked by SNAP was restricted to the stimulation site, independently of the magnitude of the response. Blockade of NO production with 100 µM NG-nitro-L-arginine methyl ester (L-NAME) or 100 µM NG-nitro-L-arginine (L-NA) reduced the resting diameter by 10-12% (n=4), but the combination of two NO blockers enhanced the basal vasoconstrictor tone by ~38% (n=5) and inhibited the local (~45%) and conducted (~20-35%) responses to ACh. Interestingly, the conduction of ACh-induced vasodilation increased along the vessel length in the presence of L-NAME and L-NA. In addition, endothelial cell hyperpolarization blockade exclusively at the stimulation site through microsuperfusion of tetraethylammonium inhibited the local vasodilation, but not the conduction of the response. These results indicate that ACh activates a NO-sensitive regenerative vasodilator mechanism that is coupled to the activation of NO production and endothelium-dependent hyperpolarization along the vessel length.
Keywords:
Conducted vasodilation
; Endothelial cells
; Nitric Oxide
; resting arteriolar diameter
; Mouse Cremaster Arterioles
Introduction
Most of the resistance to blood flow resides on feed arteries and arterioles (i.e. resistance arteries) in the microcirculation, and then, changes in diameter of these vessels play a central role in the control of systemic arterial blood pressure and blood flow distribution(1–3). Blood vessels are complex, multicellular structures that must work as a unit to rapidly adjust the distribution of blood flow according to the changing metabolic demand of cells of the surrounding tissue (1,4). In addition to the response directly activated in the site of stimulation, changes in diameter are also conducted along the length of resistance arteries and conduction of vasomotor signals has emerged as an important physiological mechanism to coordinate vascular resistance within the microvascular bed, connecting function of distal and proximal segments of the vasculature (1,4–6). Cells of the vessel wall are functionally connected via gap junctions and conducted vasomotor responses are associated with the propagation of an electrical signal (7,8). Then, it is thought that conduction of the changes in vessel diameter is the result of the electrotonic spread via gap junctions of the variations in membrane potential observed at the stimulation site. In this context, depolarization is associated with the conduction of vasoconstriction and hyperpolarization with the spread of vasodilation (7,9,10).
Although the magnitude of vessel diameter is determined by the degree of smooth muscle contraction (i.e. vasomotor tone), Ca2+-dependent production of vasodilator signals by endothelial cells plays a critical role in the fine control of vascular resistance to blood flow along the time (4,11,12). Nitric oxide (NO) has widely been recognized as the primary endothelium-dependent vasodilator signal in large, conduit vessels (13). However, in small resistance arteries and arterioles, the relevance of a complementary vasodilator component associated with endothelial cell-mediated smooth muscle hyperpolarization was also identified (14). This additional vasodilator signal was first thought to be a factor released by endothelial cells, but it is currently recognized the importance of Ca2+-activated K+ channels (KCa) of small (SKCa) and intermediate (IKCa) conductance to trigger this vasodilator signal by generating a hyperpolarization that is transmitted from endothelial cells to smooth muscle cells through the gap junctions connecting these two cell types (i.e., myoendothelial gap junctions), which led to call this vasodilator signaling as endothelium-derived hyperpolarization (EDH) (15). It must be noted that, in blood vessels, SKCa and IKCa are only expressed in endothelial cells (16–18).
NO is generated by the enzyme NO synthase (NOS) and, of the three isoforms of NOS, the endothelial isoform (eNOS) is expressed in the endothelium (13). As the vasodilation activated by NO production is mainly mediated by a reduction in the Ca2+ sensitivity of smooth muscle contractile machinery (19,20), the conduction of endothelium-dependent vasodilator responses is thought to rely exclusively on the spread of the EDH signaling along the vessel length, whereas NO only contributes to the local vasodilation observed at the site of stimulation (21,22). However, NO may also be involved in the functional coordination of vasomotor tone among the arterioles in the microcirculation, since the conducted vasodilation observed in response to acetylcholine (ACh) was shown to be inhibited by histamine in a NO-dependent manner (23). Furthermore, the response was enhanced in PECAM-1 knockout mice (i.e., CD31) after blocking the NO synthesizing enzyme, suggesting that NO may be involved in the regulation of the mechanism of conduction of the vasodilator signal (23).
Based on the above-described findings, we hypothesized that NO participates in the regulation of the mechanism involved in the coordination of the changes in diameter of resistance arteries in the microcirculation. We evaluated the effect of the blockade of NO production on the conduction of vasodilator signals activated by ACh. Our findings indicate that stimulation with ACh triggers the initiation of a regenerative mechanism that mediates the propagation of a vasodilator signal coupled to the activation of NO production and EDH signaling along the arteriolar length. In addition, the ACh-evoked regenerative vasodilator mechanism is sensitive to NO, which, consequently, works as negative feedback signaling on the conducted vasodilation.
Materials and Methods
Male C57 Bl/6 (wild type) mice between 22 and 28 g were used. Mice were bred and maintained in the Research Animal Facility of the Pontificia Universidad Católica de Chile and all studies were approved by the Institutional Bioethics Committee (protocol ID 210422002). Experiments were conducted according to the Helsinki Declaration and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8523, revised 2011) were followed. All efforts were made to minimize the suffering and number of animals used.
Mouse Cremaster Preparation
Mice were anaesthetized with pentobarbital sodium (40 mg/Kg, i.p., diluted in isotonic saline to 5 mg/mL), placed on a Plexiglas board and the cremaster muscle microcirculation was prepared as described previously (5). The right cremaster muscle was exposed, opened by a longitudinal incision on its ventral surface and the testis and epididymis were excised after ligating the supply vessels. The cremaster was pinned out on a silicone rubber pedestal, and thus, the mouse was placed on a Gibraltar Platform coupled to an Olympus microscope (BX 50 WI). Body temperature was maintained at 35-36°C with a heating pad and the cremaster muscle was continuously superfused at 3 ml/min with a bicarbonate-buffered saline solution (mM: 131.9 NaCl, 4.7 KCl, 2.0 CaCl2, 1.2 MgSO4, 20.0 NaHCO3) kept at 35°C and equilibrated with 95% N2- 5% CO2. The preparation was allowed to stabilize for 45-60 min before starting the experiment. Supplemental doses of dilute anaesthetic in isotonic saline (10 mg/Kg, i.p.) were administrated as appropriate throughout the experiment. At the end of the experiment the animals were euthanized by application of an anaesthetic overdose.
Vessel Diameters
The cremaster muscle was transilluminated, and the microscope image was acquired through a video camera (Dage-MTI Series 65, IN) and displayed on a monitor (Dage-MTI Model HR1000, IN). The inner diameters of the arterioles were continuously measured using Diamtrak software (5,24).
Arterioles were stimulated focally with a pressure-pulse ejection (10-15 psi) of 10 µM ACh, 10 µM S-nitroso-N-acetylpenicillamine (SNAP) or 1 µM calcitonin gene-related peptide (alpha isoform, α-CGRP) using a micropipette (inner diameter 3 to 4 µm). The duration of the pressure-pulse ejection of ACh and α-CGRP was set to induce a local vasodilation of ~50% (ACh: 400 ms, α-CGRP: 500 ms) and also 30% in one group of arterioles stimulated with ACh (200 ms). In the case of SNAP, the stimulation period was adjusted to evoke a response of a similar magnitude to the NO-mediated vasodilator component activated by ACh (pressure-pulse duration 300 ms) and, in one group of experiments, the length of the pulse of SNAP was extended to 700 ms to evaluate the conduction of a larger vasodilation, as that attained in response to ACh.
Experimental Protocols
Changes in diameter were measured first at the stimulation site (local), and then, at locations 500, 1000 and 2000 µm upstream in four separate stimuli. Maximal diameter was estimated during superfusion of 1 mM adenosine after finishing the experimental protocol and variations in diameter were expressed as percentage of the maximal dilation possible (% Maximum), using the following equation: (Dst - Dcont)/(Dmax - Dcont) x 100, where Dst is the diameter after the stimulation, Dcont is the diameter before stimulation (control diameter), and Dmax is the maximal diameter.
Focal Application of Tetraethylammonium (TEA)
The tip (inner diameter ~10 µm) of a micropipette filled with MOPS-buffered saline solution containing 100 mM TEA was positioned above the stimulation site of the arteriole (local) and the blocker was ejected by pressure during 10-15 min previous to the stimulation with ACh.
Blockade of NO Production
Two NOS blockers were used, NG-nitro-L-arginine methyl ester (L-NAME) and NG-nitro-L-arginine (L-NA), which were applied through the superfusion solution. The responses were assessed under control conditions and after topical application for 45 min of 100 µM L-NAME, 100 µM L-NA or the combination of both blockers (100 µM each).
Chemicals
All chemicals of analytical grade were obtained from Merck (Darmstadt, Germany). In addition, adenosine, ACh, L-NAME, L-NA, TEA and MOPS were purchased from Sigma Chemical Co., (St. Louis, MO, USA). SNAP was obtained from Calbiochem (La Jolla, CA, USA) and α-CGRP from Bachem (Torrance, CA, USA). SNAP was dissolved in DMSO, and then, diluted in the buffer solution to the final working concentration. Control experiments confirmed that application of the vehicle of SNAP (DMSO) did not have effect per se (data not shown).
Statistical Analysis
Results are presented as mean ± s.e.m.. Comparisons between groups were made using paired Student’s t-test or one-way ANOVA plus Newman-Keuls post-hoc test, as appropriate. p<0.05 was considered significant.
Results
Cremasteric arterioles of second and third branching order were analyzed. The maximum diameter of these arterioles ranged from 25.2 to 56.6 µm and the mean resting diameter was 20.5 ± 1.2 µm (n = 28), reflecting the prominent degree of vasomotor tone developed by these arterioles (49.1 ± 2.5%), which remains stable along the time in resting conditions. The level of vasomotor tone in vivo is tonically controlled by the endothelium through NO production (25–27), but the magnitude of the NO-dependent vasodilator component in resting conditions is controversial.
Contribution of Tonic NO Production to Vasomotor Tone
To evaluate the importance of the endothelium-mediated NO signaling in the tonic control of vasomotor tone, we measured the changes in basal diameter observed after the treatment for 45 min with the NOS blockers NG-nitro-L-arginine methyl ester (L-NAME), NG-nitro-L-arginine (L-NA) or the combination of both. Application of either 100 µM L-NAME or 100 µM L-NA elicited a small, but consistent, reduction in diameter (Figure 1A). Interestingly, in contrast to the modest effect of L-NAME or L-NA alone, combined application of both blockers resulted in a dramatic decrease in the diameter of the arterioles (Figure 1A) that was almost three times more prominent than that observed with each blocker separately (Figure 1B), indicating the presence of a synergistic effect between these two inhibitors and highlighting the relevance of NO production in the control of vascular function.
Contribution of NO Production to ACh-Activated Conducted Vasodilation
Stimulation of a short arteriolar segment with a pulse of ACh induced a rapid and transient vasodilator response that reached a peak after ~5 s and gradually returned to control diameter within 10 to 20 s (Figure 2). The response to ACh was not restricted to the stimulation site (i.e., local site), but it was propagated along the entire arteriole showing only a slight decay in magnitude (Figure 2), mainly during the first 500 µm and beyond the 1000 µm conducted site (Figure 2 and Figure 3), and the mechanical length constant (8) of the response (11.1 ± 2.5 mm) was much higher than the electrical length constant (0.9 –1.6 mm) determined by current injection in arterioles in vitro or in vivo (28–30). The slow decay of the ACh-activated vasodilator response along the length of the arteriole is not consistent with a simple electrotonic conduction of a hyperpolarizing signal (5) and rather suggests that a regenerative mechanism is involved in the process, as further supported by the blockade of NO production. Interestingly, blockade of NO production with the combined application of L-NAME and L-NA not only reduced the magnitude of the ACh-induced vasodilation at the local site, as expected, but also inhibited the response achieved at the conducted sites (Figure 2). In addition, in contrast to the decay with distance observed in control conditions, the conduction of the vasodilator response initiated by ACh was enhanced in the presence of L-NAME and L-NA (Figure 2 and 3), and consequently, the vasodilation recorded at 2000 µm was higher than that measured at the stimulation site (Figure 3B), suggesting that NO works as a negative feedback signaling of the regenerative vasodilator mechanism activated by ACh. Nevertheless, the effect of the inhibition of NO production on the response may be related to the decrease in the magnitude of the ACh-induced vasodilation. Then, to evaluate this possibility, we reduced the pressure-pulse ejection of ACh to elicit a response of similar magnitude to that observed after blocking NO production. In these conditions, the propagation of the vasodilation decayed along the length of the arterioles showing exactly the same characteristics of that observed with the higher ACh stimulation in control conditions (Figure 3A) and the magnitude of the decay in the vasodilation from the ACh application site to the 2000 µm conducted site was similar (Figure 3B), which confirms the participation of NO in the coordination of the changes in vessel diameter.
The reduction in the magnitude of the ACh-induced vasodilation observed at the conducted sites after the treatment with NOS blockers revealed that NO contributes to the propagation of the response by the spread of the signaling triggered at the local sites or by the activation of eNOS along the vessel length (Figure 2 and Figure 3A). To discern between these two possibilities, we stimulated a short vessel segment through the application via micropipette of a pressure-ejection pulse (300 ms) of SNAP, a NO donor, to induce a vasodilator response comparable to the NO-dependent vasodilator component activated by ACh (i.e., difference between Control and L-NAME+L-NA observed in Figure 2). As anticipated, SNAP elicited a rapid vasodilation at the stimulation site, which showed a time course similar to that achieved in response to ACh (Figure 4). However, the vasodilator response induced by SNAP declined very fast with distance, showing a mechanical length constant of 0.2 ± 0.04 mm (Figure 4A and 4B). As the magnitude of the dilation may have not reached the threshold for triggering a regenerative-like propagation of the response, we extended the pressure-pulse ejection of SNAP from 300 to 700 ms to elicit a larger vasodilation. Although the increase in the intensity of the stimulation resulted in a vasodilation like that induced by ACh at the application site (Figure 4A), the response decayed along the vessel length just as observed with the lower SNAP stimulation (Figure 4B), and the mechanical length constants observed in these arterioles was 0.36 ± 0.06 mm. In contrast, the longitudinal propagation of the NO-dependent vasodilator component of the ACh-induced vasodilation was much stronger than the SNAP-initiated conducted response, specially from the 500 µm to the 1000 µm conducted sites (Figure 4C). Consistent with this, the mechanical length constant (2.4 ± 0.3 mm) was significantly higher than that attained with the SNAP stimulation pulse of 300 ms or 700 ms (P<0.0001 by one-way ANOVA plus Newman-Keuls post hoc test), which suggests that the ACh-elicited NO-dependent conducted vasodilation represents the activation of eNOS along the vessel length.
EDH-Independent Regenerative Conducted Vasodilation
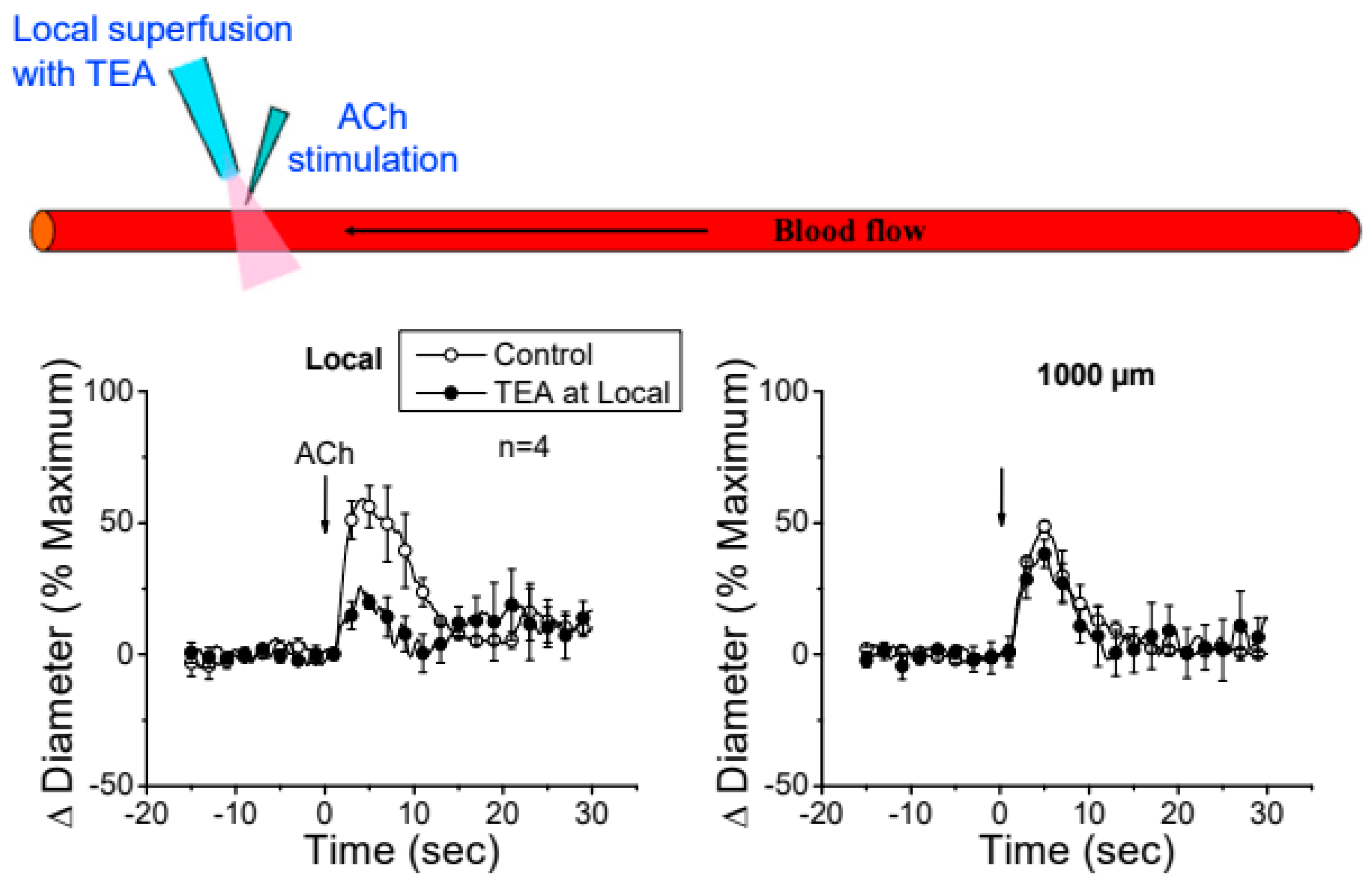
The apparent contribution of a locally activated NO-mediated vasodilator component to the conducted response initiated by ACh suggests that the regenerative-like conduction of the vasodilator response does not depend on the simple spread of the EDH signaling activated at the local site. To test this hypothesis, we evaluated the conduction of CGRP-induced vasodilation. CGRP is the main neurotransmitter of perivascular sensory nerve endings and is a potent vasodilator. Interestingly, although the vasodilation evoked by CGRP is associated with the hyperpolarization of the vessel wall (31,32), the response activated at the stimulation site declined rapidly along the vessel length (Figure 5). In addition, the contribution of EDH signaling to the conducted vasodilation was further tested by application via micropipette of 100 mM TEA to inhibit the activation of KCa channels exclusively at the stimulation site (Figure 6). TEA did not change the resting diameter of the vessel segment treated with this inhibitor (14.3±0.3 vs. 16.6±3.8), suggesting that KCa channels are not relevant for the tonic control of vasomotor tone. However, the ACh-induced vasodilation was strongly reduced in the TEA application site and, despite the clear effect on the direct response to ACh, this treatment did not affect the conducted vasodilation recorded 1000 µm from the local site (Figure 6), which is consistent with the propagation of a vasodilator signal, independent of KCa channels opening, that is coupled to the activation of the EDH signaling and NO production along the vessel length.
Discussion
Control of blood flow distribution depends on the coordination of the changes in vasomotor tone along the length of the arterioles and among the resistance vessels in the microcirculation (1,4,33). NO production plays a critical role in the tonic regulation of vasomotor tone and conduction of vasomotor responses provides the bases for timing coupling of vascular function between different arteriolar segments and among arterioles with feed arteries (1,4,6). It is thought that conducted vasomotor responses rely on electrotonic spread of the changes in membrane potential observed at the stimulation site through gap junctions connecting cells of the vessel wall (4,7,10). However, conduction of endothelium-dependent vasodilation does not seem to be consistent with the electrotonic mechanism, since these responses have been observed to propagate along the entire vessel without apparent decay (5,24) and NO has been reported to inhibit the longitudinal conduction of vasodilator signals (23), suggesting that a NO-sensitive regenerative mechanism is involved in the propagation of the response. Consistent with this hypothesis, our results indicate that the vasodilation activated by ACh is propagated for, at least, 2000 µm showing a much smaller decay in magnitude than that anticipated by an electrotonic conduction. Interestingly, inhibition of NO production with simultaneous application of L-NAME and L-NA converted the slightly decaying conducted response activated by ACh into a vasodilation that progressively increased along the vessel length, unveiling the presence of a NO-sensitive regenerative vasodilator mechanism.
NO production plays a critical role in the response to endothelium-dependent vasodilators, but also in the control of vasomotor tone in resting conditions (13,26,27). In this line, although the treatment with L-NAME or L-NA alone evoked a modest reduction in resting diameter, simultaneous application of both inhibitors resulted in a synergistic increase in vasomotor tone (i.e., vasoconstriction), highlighting the importance of NO in the regulation of microvascular function (Fig. 1). As eNOS function depends on the subcellular location of the enzyme (34–36), the synergistic effect observed with L-NAME and L-NA may be related to the uptake mechanisms and further intracellular distribution of the inhibitors. In endothelial cells, NO production is coordinated by dynamic sub-cellular targeting of eNOS between two functional pools of the enzyme: one associated with the trans-Golgi network and other located at caveolae, which are invaginated plasmalemmal rafts that function as signaling microdomains (35,37). Interestingly, function of the trans-Golgi-associated eNOS pool depends on intracellular L-arginine, whereas substrate supply of the caveolae-located pool of the enzyme is directly provided by L-arginine influx (38–40). In this context, L-NAME can get into the cell through plasma membrane, which may provide a preferential access to the trans-Golgi-associated eNOS pool. In contrast, L-NA uptake relies on the same amino acid transporter systems involved in L-arginine uptake, which in addition to reduce the substrate supply to the caveolae-located eNOS pool, may also favor the direct access of the inhibitor to the environment of the enzyme in this microdomain (41–43). Therefore, we hypothesize that simultaneous inhibition of these two complementary eNOS pools potentiates the inhibition of NO production, but the mechanisms involved in this process required further investigation.
The most relevant endothelium-derived vasodilator signals in resistance arteries are NO and EDH (4,19,44). Although the importance of NO production is widely recognized, the involvement of this signaling molecule in the generation of conducted vasodilation is controversial (21,45–47). Conduction of the changes in diameter initiated by endothelium-dependent vasodilators, such ACh, is thought to rely on the electrotonic spread of the hyperpolarization of the vessel wall observed in response to EDH signaling at the stimulation site (6,7,10). However, the vasodilation evoked by ACh in control conditions was propagated over distances much longer than those predicted by the electrotonic model and, in addition, the magnitude of the response increased along the vessel length after blocking NO production (Fig. 2 and 3). It must be noted that the increase in the conducted vasodilation was not related to the reduction of the local response attained in the presence of L-NAME and L-NA, since the longitudinal decay of an ACh-elicited vasodilation of a similar magnitude showed exactly the same characteristics to those exhibited by the control response observed before the application of the NOS blockers (Fig. 3). Therefore, these results suggest that, in addition to the EDH signaling-initiated conducted vasodilation, ACh also activates a NO-sensitive regenerative vasodilator mechanism.
Although NO has been frequently thought to contribute only to the local response activated by endothelium-dependent vasodilators (21,46,48), Budel et al. (47) found that the generation of a NO wave along the endothelium was unmasked by a focal smooth muscle damage in the middle of the conduction pathway of the vasodilation initiated by ACh. In this line, our results show that blockade of NO production not only inhibited the vasodilation activated by ACh at the stimulation site, as expected, but also reduced the conducted response (Fig. 2 and 3). In this context, it is important to note that the vasodilation induced directly by NO (i.e., SNAP) decayed very rapidly with distance, in contrast to the NO-mediated vasodilator component that was propagated in response to ACh (Fig. 4). Therefore, the reduction in the magnitude of the vasodilation observed at the conducted sites in the presence of L-NAME and L-NA suggests that the regenerative vasodilator signal initiated by ACh is coupled to NO production along the vessel length.
Interestingly, eNOS is a Ca2+ dependent enzyme, and then, the increment in NO production at a remote vessel segment from the stimulation site implies that the propagation of the ACh-induced vasodilator signal is associated with a mechanism that mediates an increase in [Ca2+]i, which, in addition to eNOS activation, may also trigger the myoendothelial signaling through the EDH pathway (4). It should be noted that endothelial cell hyperpolarization does not promote an increase in [Ca2+]i in intact vessels, unlike what has been reported in the case of cell cultures (49–52). Therefore, the increase in [Ca2+]i attained at the conducted sites must be activated by a mechanism different than the simple membrane hyperpolarization. Consistent with this hypothesis, inhibition of EDH signaling activation at the ACh application site by micropipette-mediated focal superfusion of TEA did not affect the conducted vasodilation generated 1000 µm upstream from the stimulation site (Fig.6). In this context, it must be noted that the expression of voltage-dependent Na+ and Ca2+ channels has been detected in resistance arteries and sequential activation of these channels may support the regenerative propagation of a vasodilator signal, as previously demonstrated in the case of the conducted vasodilation activated by focal electrical field stimulation (24,53). However, the activation of this mechanism in response to ACh remains to be determined.
In summary, the results of the present study are consistent with the hypothesis denoting that ACh, in addition to the local response, triggers the regenerative propagation of a vasodilator signal coupled to a mechanism that leads to NO production and activation of the EDH signaling along the length of the arterioles. Interestingly, the ACh-elicited regenerative vasodilator mechanism is sensitive to NO, which appears to function as a negative feedback signaling of the conducted response and, consequently, the magnitude of the vasodilator responses increases after blocking eNOS activity. Coordination of the changes in diameter among different segments of resistance arteries plays a central role in the control of blood flow distribution and arterial blood pressure, and then, these findings may contribute to the understanding of the mechanism involved in the vascular dysfunction typically associated with the progress of cardiovascular-related diseases, such as hypertension and diabetes.
Authors’ Contributions
NPB Contributed to the analysis and interpretation of data and to the manuscript writing and editing. MM and MM-U Contribute to data analysis and the preparation of the manuscript. XFF Designed the experimental protocols, contributed to collect and analyze the data and was the major contributor in writing and editing the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by Grants ANID/ACT210057 and Fondecyt Regular 1,211,060 from Agencia Nacional de Investigación y Desarrollo (ANID).
Availability of Data and Materials
This work was supported by Grants ANID/ACT210057 and Fondecyt Regular 1211060 from Agencia Nacional de Investigación y Desarrollo (ANID).
Competing Interests
The authors declare that they have no competing interests.
Ethics Approval
All studies and experimental procedures were approved by the Institutional Bioethics Committee (protocol ID 210422002).
References
- Segal SS: Regulation of blood flow in the microcirculation. Microcirculation 12: 33–45, 2005.
- Segal SS, John T and Haven N: Integration of blood flow control to skeletal muscle: key role of feed arteries. Acta Physiol Scand 168: 511–518, 2000.
- Mulvany MJ and Aalkjaer C: Structure and function of small arteries. Physiol Rev 70: 921–961, 1990.
- Figueroa XF and Duling BR: Gap Junctions in the Control of Vascular Function. Antioxid Redox Signal 11: 251–266, 2009.
- Figueroa XF and Duling BR: Dissection of two Cx37-independent conducted vasodilator mechanisms by deletion of Cx40: electrotonic versus regenerative conduction. Am J Physiol Heart Circ Physiol 295: H2001–H2007, 2008.
- Gustafsson F and Holstein-Rathlou NH: Conducted vasomotor responses in arterioles: Characteristics, mechanisms and physiological significance. Acta Physiol Scand 167: 11–21, 1999.
- Emerson GG and Segal SS: Electrical Coupling Between Endothelial Cells and Smooth Muscle Cells in Hamster Feed Arteries: Role in Vasomotor Control. Circ Res 87: 474–479, 2000.
- Xia J and Duling BR: Electromechanical coupling and the conducted vasomotor response. Am J Physiol 269: H2022-30, 1995.
- Figueroa XF, Isakson BE and Duling BR: Connexins: Gaps in Our Knowledge of Vascular Function. Physiology 19: 277–284, 2004.
- Welsh DG and Segal SS: Endothelial and smooth muscle cell conduction in arterioles controlling blood flow. Am J Physiol Heart Circ Physiol 274: H178–H186, 1998.
- Lillo MA, Gaete PS, Puebla M, et al.: Critical contribution of Na+-Ca2+ exchanger to the Ca2+-mediated vasodilation activated in endothelial cells of resistance arteries. The FASEB Journal 32: 2137–2147, 2018.
- Dora KA, Doyle MP, Duling BR and Berne RM: Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles. Proc Natl Acad Sci USA 94: 6529–6534, 1997.
- Moncada S and Higgs EA: Molecular mechanisms and therapeutic strategies related to nitric oxide. The FASEB Journal 9: 1319–1330, 2018.
- Félétou M and Vanhoutte PM: EDHF: an update. Clin Sci 117: 139–155, 2009.
- Félétou M and Vanhoutte PM: Endothelium-Dependent Hyperpolarization: No Longer an F-Word! J Cardiovasc Pharmacol 61: 91–92, 2013.
- Crane GJ, Gallagher N, Dora KA and Garland CJ: Small- and intermediate-conductance calcium-activated K + channels provide different facets of endothelium-dependent hyperpolarization in rat mesenteric artery. 183–189, 2003.
- Doughty JM, Plane F and Langton PD: Charybdotoxin and apamin block EDHF in rat mesenteric artery if selectively applied to the endothelium. Am J Physiol Heart Circ PhysiolH 276: H1107–H1112, 1999.
- Grgic I, Eichler I, Heinau P, Si H, Brakemeier S, Hoyer J and Köhler R: Selective blockade of the intermediate-conductance Ca2+ - activated K+ channel suppresses proliferation of microvascular and macrovascular endothelial cells and angiogenesis in vivo. Arterioscler Thromb Vasc Biol 25: 704–709, 2005.
- Bolz SS, De Wit C and Pohl U: Endothelium-derived hyperpolarizing factor but not NO reduces smooth muscle Ca2+ during acetylcholine-induced dilation of microvessels. Br J Pharmacol 128: 124–134, 1999.
- Bolz S, Vogel L, Sollinger D, Derwand R, Wit C De, Loirand G and Pohl U: Nitric Oxide – Induced Decrease in Calcium Sensitivity of Resistance Arteries Is Attributable to Activation of the Myosin Light Chain Phosphatase and Antagonized by the RhoA/Rho Kinase Pathway. Circulation 107: 3081–3087, 2003.
- Welsh DG and Segal SS: Role of EDHF in conduction of vasodilation along hamster cheek pouch arterioles in vivo. Am J Physiol Heart Circ Physiol 278: H1832–H1839, 2017.
- Hoepfl B, Rodenwaldt B, Pohl U and de Wit C: EDHF, but not NO or prostaglandins, is critical to evoke a conducted dilation upon ACh in hamster arterioles. Am J Physiol Heart Circ Physiol 283: H996–H1004, 2002.
- Payne GW, Madri JA, Sessa WC and Segal SS: Histamine inhibits conducted vasodilation through endothelium-derived NO production in arterioles of mouse skeletal muscle.
- Figueroa XF, Chen C-C, Campbell KP, Damon DN, Day KH, Ramos S and Duling BR: Are voltage-dependent ion channels involved in the endothelial cell control of vasomotor tone? Am J Physiol Heart Circ Physiol 293: H1371–H1383, 2007.
- De Wit C, Jahrbeck B, Schafer C, Bolz S-S and Pohl U: Nitric oxide opposes myogenic pressure responses predominantly in large arterioles in vivo. Hypertension 31: 787–794, 1998.
- de Wit C, Schäfer C, von Bismarck P, Bolz S-S and Pohl U: Elevation of plasma viscosity induces sustained NO-mediated dilation in the hamster cremaster microcirculation in vivo. Pflügers Arch – Eur J Physiol 434: 354–361, 1997.
- Figueroa XF, Martínez AD, González DR, et al.: In vivo assessment of microvascular nitric oxide production and its relation with blood flow. Am J Physiol Heart Circ Physiol 280: H1222-31, 2001. [CrossRef]
- Emerson GG, Neild TO and Segal SS: Conduction of hyperpolarization along hamster feed arteries: augmentation by acetylcholine. Am J Physiol Heart Circ Physiol 283: H102–H109, 2002.
- Hirst GD, Edwards FR, Gould DJ, Sandow SL and Hill CE: Electrical properties of iridial arterioles of the rat. Am J Physiol 273: H2465--H2472, 1997.
- Hirst GD and Neild TO: An analysis of excitatory junctional potentials recorded from arterioles. J Physiol 280: 87–104, 1978.
- Brain SD and Grant AD: Vascular Actions of Calcitonin Gene-Related Peptide and Adrenomedullin. Physiol Rev 84: 903–934, 2004.
- Nelson MT, Huang Y, Brayden JE, Hescheler J and Standen NB: Arterial dilations in response to calcitonin gene-related peptide involve activation of K+ channels. Nature 344: 770–773, 1990.
- Segal SS: Integration and Modulation of Intercellular Signaling Underlying Blood Flow Control. J Vasc Res 52: 136–157, 2015.
- Church JE and Fulton D: Differences in eNOS Activity Because of Subcellular Localization Are Dictated by Phosphorylation State Rather than the Local Calcium Environment *. Journal of Biological Chemistry 281: 1477–1488, 2006.
- Fulton D, Babbitt R, Zoellner S, et al.: Targeting of endothelial nitric-oxide synthase to the cytoplasmic face of the Golgi complex or plasma membrane regulates Akt- versus calcium-dependent mechanisms for nitric oxide release. Journal of Biological Chemistry 279: 30349–30357, 2004. [CrossRef]
- Figueroa XF, González DR, Martínez AD, Durán WN and Boric MP: ACh-induced endothelial NO synthase translocation, NO release and vasodilation in the hamster microcirculation in vivo. Journal of Physiology 544: 883–896, 2002.
- Jagnandan D, Sessa WC and Fulton D: Intracellular location regulates calcium-calmodulin-dependent activation of organelle-restricted eNOS. American Journal of Physiology-Cell Physiology 289: C1024–C1033, 2005.
- Zani BG and Bohlen HG: Transport of extracellular L -arginine via cationic amino acid transporter is required during in vivo endothelial nitric oxide production Transport of extracellular L -arginine via cationic amino acid transporter is required during in vivo endothelial nit. Am J Physiol Heart Circ Physiol 289: H1381–H1390, 2005.
- Hardy TA and May JM: Coordinate regulation of L-arginine uptake and nitric oxide synthase activity in cultured endothelial cells. Free Radic Biol Med 32: 122–131, 2002.
- Mcdonald KK, Zharikov S, Block ER and Kilberg MS: A Caveolar Complex between the Cationic Amino Acid Transporter 1 and and Endothelial Nitric-oxide Synthase May Explain the “Arginine Paradox.” J Biol Chem 272: 31213–31216, 1997.
- Boer R, Udiger W, Klein T, Mirau B, Haas S and Baur I: The Inhibitory Potency and Selectivity of Arginine Substrate Site Nitric-Oxide Synthase Inhibitors Is Solely Determined by Their Affinity toward the Different Isoenzymes. Mol Pharmacol 58: 1026–1034, 2000.
- Edwards RM, Stack EJ and Trizna W: Interaction of L -Arginine Analogs with L -Arginine Uptake in Rat Renal Brush Border Membrane Vesicles. J Pharmacol Exp Ther 285: 1019–1022, 1998.
- Schmidt K, Klatt P and Mayer B: Characterization of endothelial cell amino acid transport systems involved in the actions of nitric oxide synthase inhibitors. Mol Pharmacol 44: 615–621, 1993.
- Bakker ENTP and Sipkema P: Components of acetylcholine-induced dilation in isolated rat arterioles. American Journal of Physiology-Heart and Circulatory Physiology 273: H1848–H1853, 1997.
- Segal SS, Welsh DG and Kurjiaka DT: Spread of vasodilatation and vasoconstriction along feed arteries and arterioles of hamster skeletal muscle. J Physiol 516: 283–291, 1999.
- Hungerford JE, Sessa WC and Segal SS: Vasomotor control in arterioles of the mouse cremaster muscle. 197–207.
- Budel S, Bartlett IS and Segal SS: Homocellular conduction along endothelium and smooth muscle of arterioles in hamster cheek pouch: Unmasking an NO wave. Circ Res 93: 61–68, 2003.
- de Wit C, Griffith TM and Park H: Connexins and gap junctions in the EDHF phenomenon and conducted vasomotor responses. Pflugers Arch 459: 897–914, 2010.
- Ghisdal P and Morel N: Cellular target of voltage and calcium-dependent K+channel blockers involved in EDHF-mediated responses in rat superior mesenteric artery. Br J Pharmacol 134: 1021–1028, 2001.
- Stankevičius E, Lopez-Valverde V, Rivera L, Hughes AD, Mulvany MJ and Simonsen U: Combination of Ca2+-activated K+ channel blockers inhibits acetylcholine-evoked nitric oxide release in rat superior mesenteric artery. Br J Pharmacol 149: 560–572, 2006.
- Cohen KD and Jackson WF: Membrane hyperpolarization is not required for sustained muscarinic agonist-induced increases in intracellular Ca2+ in arteriolar endothelial cells. Microcirculation 12: 169–182, 2005.
- McSherry IN, Spitaler MM, Takano H and Dora KA: Endothelial cell Ca2+increases are independent of membrane potential in pressurized rat mesenteric arteries. Cell Calcium 38: 23–33, 2005.
- Lillo MA, Gaete PS, Puebla M, Burboa PC, Poblete I and Figueroa XF: Novel Pannexin-1-Coupled Signaling Cascade Involved in the Control of Endothelial Cell Function and NO-Dependent Relaxation. Oxid Med Cell Longev 2021: Article 2678134, 2021.
Figure 1.
Reduction in diameter of cremasteric arterioles observed in response to the blockade of NO production. Cremaster muscle microcirculation was treated for 45 min with 100 µM L-NAME, 100 µM L-NA or the combination of both blockers (100 µM each) of the enzyme NOS and the changes in diameter were evaluated 45 min thereafter (A). In addition, the percentage of reduction in vessel diameter attained in the presence of the blockers is also shown (B). Note that the combined application of L-NAME and L-NA evoked an effect almost three-fold larger than that observed with each blocker alone. *, P<0.05 and ***, P<0.001 vs Control by paired Student’s t test. †, P<0.05 vs L-NAME or L-NA by one-way ANOVA plus Newman-Keuls post hoc test.
Figure 1.
Reduction in diameter of cremasteric arterioles observed in response to the blockade of NO production. Cremaster muscle microcirculation was treated for 45 min with 100 µM L-NAME, 100 µM L-NA or the combination of both blockers (100 µM each) of the enzyme NOS and the changes in diameter were evaluated 45 min thereafter (A). In addition, the percentage of reduction in vessel diameter attained in the presence of the blockers is also shown (B). Note that the combined application of L-NAME and L-NA evoked an effect almost three-fold larger than that observed with each blocker alone. *, P<0.05 and ***, P<0.001 vs Control by paired Student’s t test. †, P<0.05 vs L-NAME or L-NA by one-way ANOVA plus Newman-Keuls post hoc test.
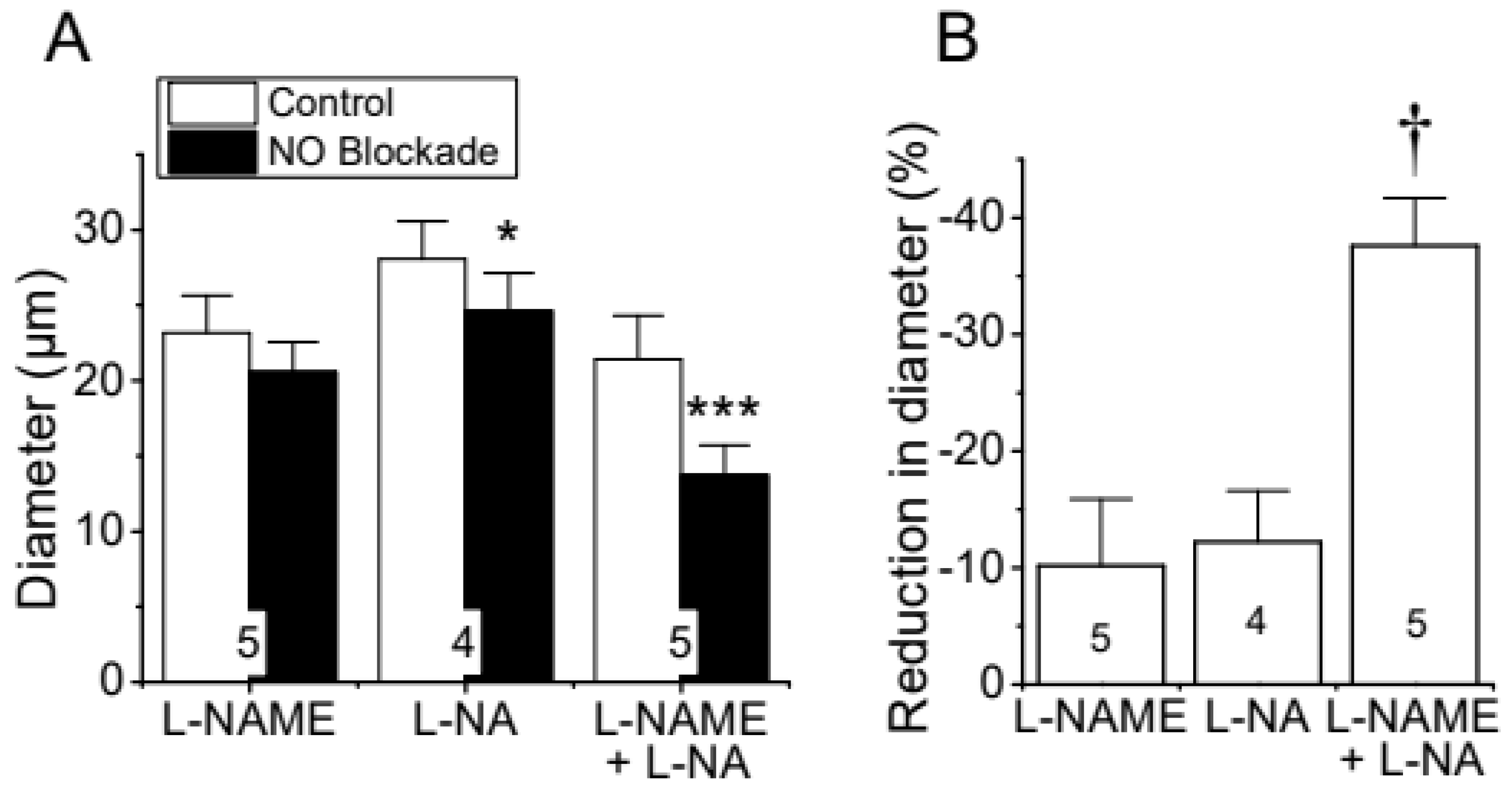
Figure 2.
Time course of the local and conducted vasodilation induced by ACh in control conditions and after blocking NO production. ACh was ejected by a pressure pulse via a micropipette to stimulate a short segment of the cremasteric arterioles and the vasodilator response was analyzed at the stimulation site (local) and at locations at 500, 1000, 2000 µm upstream. The vasodilator responses initiated by ACh was evaluated before and after blocking NO production through the application of the combination of L-NAME and L-NA. Note that the changes in diameter do not decay along the vessel length in the presence of L-NAME plus L-NA. Arrows indicate the time at which the stimulus was applied. *, P<0.05 vs Control by one-way ANOVA plus Newman-Keuls post hoc test.
Figure 2.
Time course of the local and conducted vasodilation induced by ACh in control conditions and after blocking NO production. ACh was ejected by a pressure pulse via a micropipette to stimulate a short segment of the cremasteric arterioles and the vasodilator response was analyzed at the stimulation site (local) and at locations at 500, 1000, 2000 µm upstream. The vasodilator responses initiated by ACh was evaluated before and after blocking NO production through the application of the combination of L-NAME and L-NA. Note that the changes in diameter do not decay along the vessel length in the presence of L-NAME plus L-NA. Arrows indicate the time at which the stimulus was applied. *, P<0.05 vs Control by one-way ANOVA plus Newman-Keuls post hoc test.
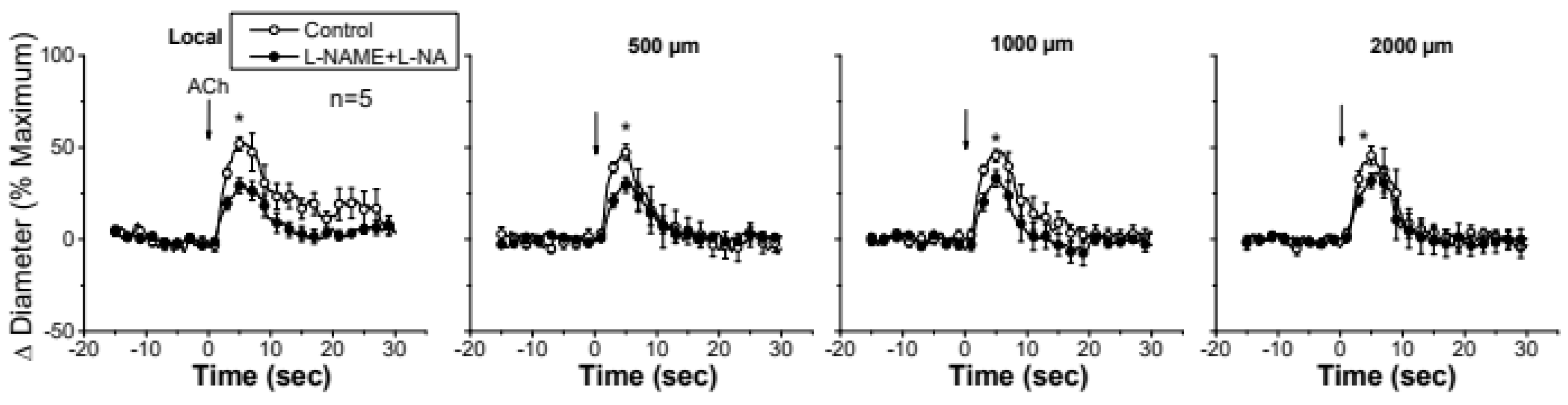
Figure 3.
Analysis of the effect of NO production blockade on the conducted vasodilation activated by ACh. A short segment of the arteriole was stimulated with a pulse (400 ms) of 10 µM ACh ejected by pressure from a micropipette. A, The maximal vasodilator response induced by ACh was evaluated at the stimulation pipette site (local) and at locations 500, 1,000, and 2,000 µm upstream before (Control) and after the blockade of NO production with the combination of 100 µM L-NAME and 100 µM L-NA. Analysis of the conduction of the response induced by a shorter pressure pulse of ACh (ACh-200 ms) is also shown. B, Change in the magnitude of the vasodilation evoked by ACh in the stimulation site (Local) as compared with that observed 2000 µm upstream. Note that in the presence of L-NAME and L-NA the vasodilation increased, instead of decay, along the vessel length. *, P<0.05 vs L-NAME+L-NA by one-way ANOVA plus Newman-Keuls post hoc test. †, P<0.05 vs Local by paired Student’s t test.
Figure 3.
Analysis of the effect of NO production blockade on the conducted vasodilation activated by ACh. A short segment of the arteriole was stimulated with a pulse (400 ms) of 10 µM ACh ejected by pressure from a micropipette. A, The maximal vasodilator response induced by ACh was evaluated at the stimulation pipette site (local) and at locations 500, 1,000, and 2,000 µm upstream before (Control) and after the blockade of NO production with the combination of 100 µM L-NAME and 100 µM L-NA. Analysis of the conduction of the response induced by a shorter pressure pulse of ACh (ACh-200 ms) is also shown. B, Change in the magnitude of the vasodilation evoked by ACh in the stimulation site (Local) as compared with that observed 2000 µm upstream. Note that in the presence of L-NAME and L-NA the vasodilation increased, instead of decay, along the vessel length. *, P<0.05 vs L-NAME+L-NA by one-way ANOVA plus Newman-Keuls post hoc test. †, P<0.05 vs Local by paired Student’s t test.
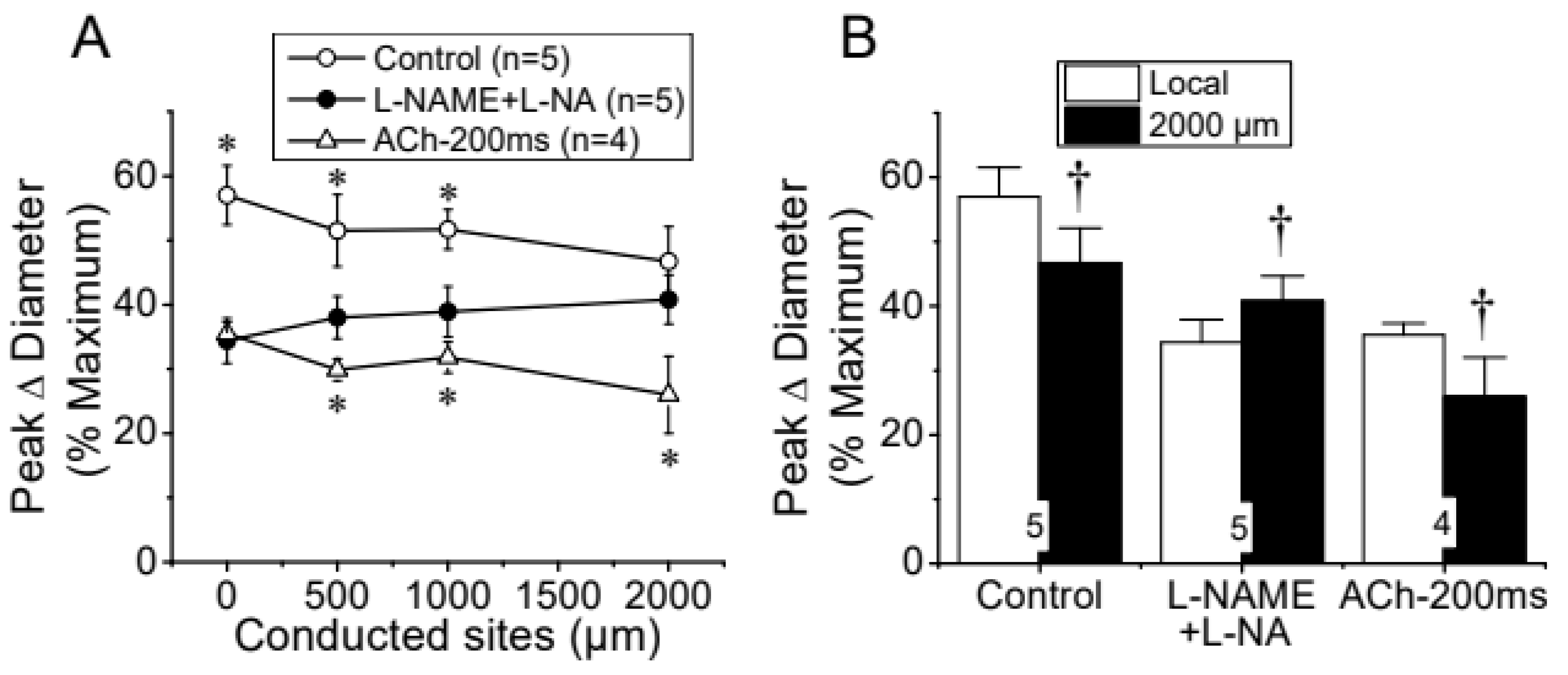
Figure 4.
Analysis of the conducted vasodilation activated by direct stimulation with a pulse of NO. A, Time course of the local and conducted vasodilation induced by 700 ms or 300 ms pressure pulse of 10 µM SNAP, a NO donor. B, Maximal vasodilator response induced by SNAP observed at the stimulation site (Local in panel A) and at locations 500, 1,000, and 2,000 µm upstream. C, Analysis of the conduction of the NO-dependent vasodilator component of the response activated by ACh in control conditions shown in Figure 2. Note that, in contrast to the stimulation with SNAP, the ACh-activated NO-dependent vasodilator component exhibits only a moderate decay along the length of the arterioles and the response can be observed up to the conducted site located at 2000 µm from the stimulation site. *, P<0.05 vs SNAP-700ms by one-way ANOVA plus Newman-Keuls post hoc test.
Figure 4.
Analysis of the conducted vasodilation activated by direct stimulation with a pulse of NO. A, Time course of the local and conducted vasodilation induced by 700 ms or 300 ms pressure pulse of 10 µM SNAP, a NO donor. B, Maximal vasodilator response induced by SNAP observed at the stimulation site (Local in panel A) and at locations 500, 1,000, and 2,000 µm upstream. C, Analysis of the conduction of the NO-dependent vasodilator component of the response activated by ACh in control conditions shown in Figure 2. Note that, in contrast to the stimulation with SNAP, the ACh-activated NO-dependent vasodilator component exhibits only a moderate decay along the length of the arterioles and the response can be observed up to the conducted site located at 2000 µm from the stimulation site. *, P<0.05 vs SNAP-700ms by one-way ANOVA plus Newman-Keuls post hoc test.
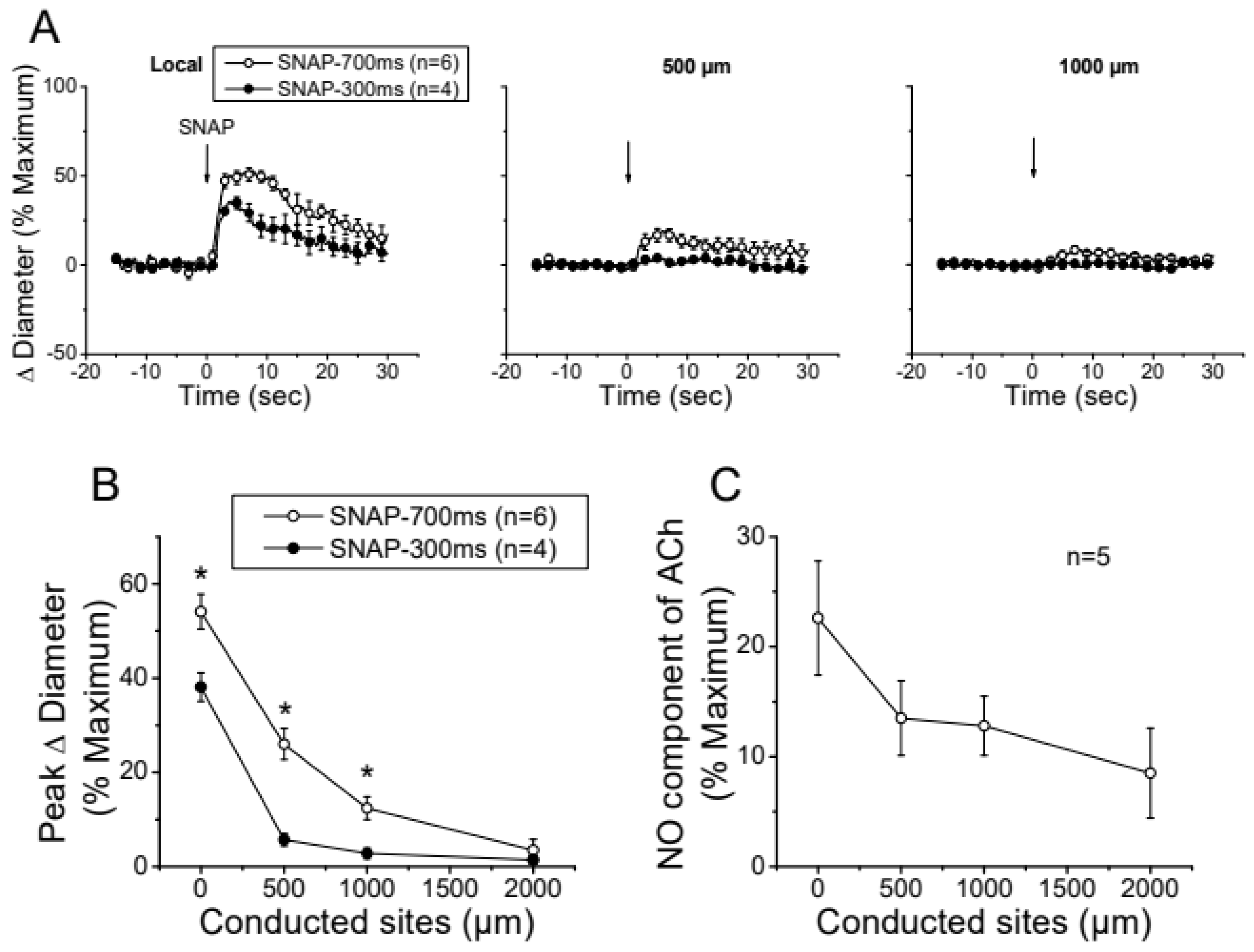
Figure 5.
Time course of local and conducted vasodilation induced by the alpha isoform of calcitonin gene-related peptide (α-CGRP). A short segment of the cremasteric arterioles was stimulated with a pressure pulse-ejection via micropipette of 1 µM α-CGRP and the resultant vasodilator responses were observed at the stimulation pipette site (local) and at locations 500, 1000 and 2000 µm upstream. Arrows indicate the time at which the stimulus was applied.
Figure 5.
Time course of local and conducted vasodilation induced by the alpha isoform of calcitonin gene-related peptide (α-CGRP). A short segment of the cremasteric arterioles was stimulated with a pressure pulse-ejection via micropipette of 1 µM α-CGRP and the resultant vasodilator responses were observed at the stimulation pipette site (local) and at locations 500, 1000 and 2000 µm upstream. Arrows indicate the time at which the stimulus was applied.
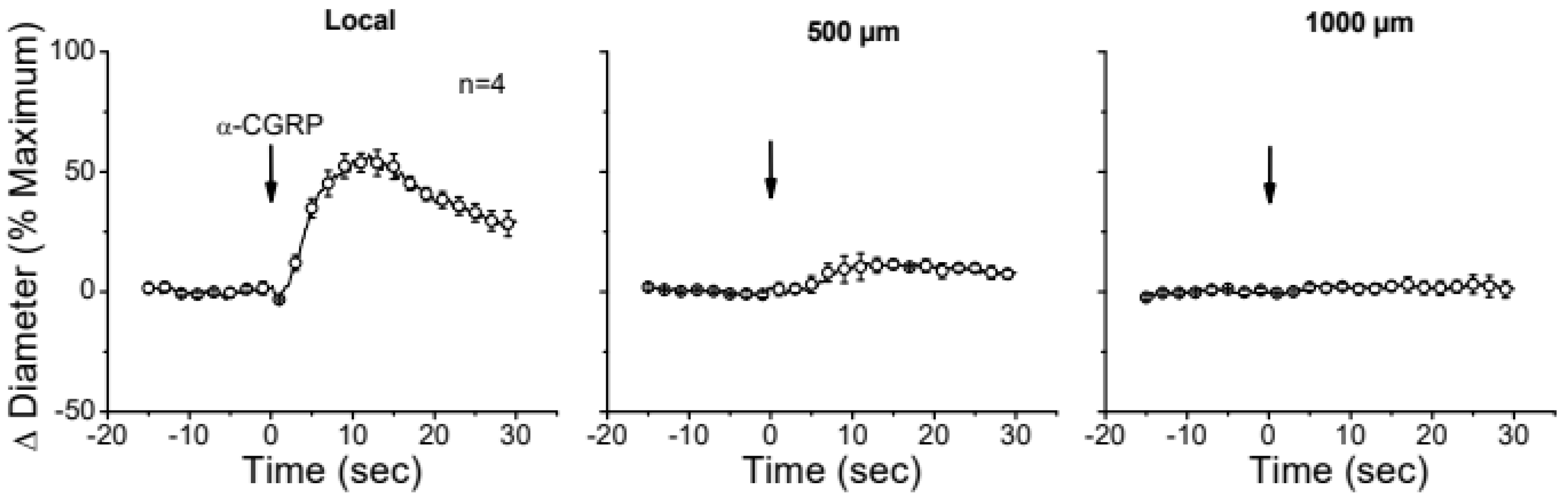
Figure 6.
The conducted vasodilator response activated by ACh does not depend on the direct membrane hyperpolarization of endothelial cells generated at the stimulation site. The time course of the ACh-evoked vasodilation attained at the stimulation site (Local) and 1000 µm up stream were recorded in control conditions and during local application of 100 mM tetraethylammonium (TEA) via micropipette to prevent the Ca2+-activated K+ channel-mediated endothelial cell hyperpolarization. Note that the blockade of the EDH signaling-dependent vasodilation at the local site did not affect the conducted response observed 1000 µm up stream. *, P<0.05 vs Control by one-way ANOVA plus Newman-Keuls post hoc test.
Figure 6.
The conducted vasodilator response activated by ACh does not depend on the direct membrane hyperpolarization of endothelial cells generated at the stimulation site. The time course of the ACh-evoked vasodilation attained at the stimulation site (Local) and 1000 µm up stream were recorded in control conditions and during local application of 100 mM tetraethylammonium (TEA) via micropipette to prevent the Ca2+-activated K+ channel-mediated endothelial cell hyperpolarization. Note that the blockade of the EDH signaling-dependent vasodilation at the local site did not affect the conducted response observed 1000 µm up stream. *, P<0.05 vs Control by one-way ANOVA plus Newman-Keuls post hoc test.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



