
Submitted:
22 September 2024
Posted:
24 September 2024
You are already at the latest version
Abstract
The rat osteosarcoma cell line UMR-106 is widely used for the study of bone cancer biology. We have characterized UMR-106 with a combination of optical genome mapping (OGM), long-read sequencing (for identification of variants and 5mC methylation), and short-read RNA sequencing for expression. Genome sequence was compared to a Sprague-Dawley control animal, the strain from which UMR-106 was derived, and expression data were compared to a public rat osteoblast dataset. Using the COSMIC database to identify the most affected genes in human osteosarcomas (OS) we found somatic mutations in Tp53 and H3f3a in UMR-106. OGM identified a relatively small number of differences between the cell line and a strain-matched control animal but did detect a ~45 Mb block of amplification that included Myc on chromosome 7 which was confirmed by long-read sequencing. The amplified region showed several blocks of non-contiguous rearranged sequence implying complex rearrangements during their formation and included 14 genes reported as biomarkers in human osteosarcoma, many of which also showed increased transcription. A comparison of 5mC methylation from the nanopore reads of tumor and control identified several genes with distinct differences including the OS marker Cdkn2a. This dataset illustrates the value of long DNA methods for the characterization of cell lines and how inter-species analysis can inform about the genetic nature underlying mutations that underpin specific tumor types. The data should be a valuable resource for investigators studying osteosarcoma, in general, and specifically the UMR-106 model.
Keywords:
optical mapping
; nanopore sequencing
; osteosarcoma
; Myc amplification
1. Introduction
Cancers can result from a variety of genomic errors including chromosomal events (translocations, amplifications, deletions, etc.), somatic mutations and epigenetic changes all of which may be reflected in altered gene expression. Osteosarcoma (OS) is a rare largely pediatric cancer of the bone ([1,2,3]). Cell lines made from OS and other cancers are often created to better understand and model disease. During passage genetic changes can accumulate [4] and it is essential to characterize these to understand how faithful the model is to the disease of interest and to understand the experimental behavior of such cell lines. UMR-106 is widely used in studies of OS ([5,6,7,8,9,10,11,12,13,14,15,16]) and serves as a model of pediatric cancer such as the characterization of the tumor microenvironment following implantation into rat tibias [14]. A thorough characterization of UMR-016 is necessary to determine how similar the line is to human OS and if we are to use this model to explore potential targets for drug therapy.
At present no single technology permits a thorough characterization of cancer at all genomic scales. But newer long DNA techniques ([17,18]) suggest that they may offer improved insights into the tumor genome. Here, we chose to evaluate two long DNA technologies, optical genome mapping (OGM) as developed by Bionano Genomics and nanopore sequencing as implemented by Oxford Nanopore Technologies (ONT), as both assess the genome at larger scales than typical short-read methods. Although OGM has been used to study human sarcomas [19] it has not yet been applied to characterizing osteosarcoma genomes. Likewise, long nanopore reads have been shown to be a valuable tool in cancer research (e.g., [20,21]) but only a few studies have compared OGM to ONT [22]. In this study we evaluated how well both long read technologies compare and what new insights they provide over traditional methods. We also provide the community with the resources produced for subsequent studies.
2. Methods
2.1. UMR-106 Cell Culture
Rat osteosarcoma UMR-106 cells (CRL-1661) were purchased from ATCC and expanded for three passages. Cells were grown in DMEM media supplemented with 10% (v/v) FBS (Fetal Bovine Serum), penicillin (10U/mL streptomycin (10U/ml at 37o C in a humidified 5% CO2 atmosphere. Cells used for characterization were within passages 3-6.
2.2. Bionano OGM Methods
DNA from UMR-106 cells and from WBCs of the control rat were isolated. Optical Genome Mapping (OGM) methods included ultrahigh-molecular-weight (UHMW) genomic DNA (gDNA) isolation from UMR-106 cells and from WBCs of the control animal isolated via the blood and cell culture DNA Isolation Kit according to manufacturers’ instructions (Bionano Genomics, San Diego, CA, USA). Briefly, cells were treated with lysis-and-binding buffer (LBB) to release gDNA, which was bound to a Nanobind disk before it was washed and resuspended in the elution buffer. The Direct Label and Stain (DLS) DNA Labeling Kit (Bionano Genomics, San Diego, CA, USA) was used to label UHMW gDNA molecules. 750 ng of gDNA was labeled with Direct Label Enzyme (DLE-1) and DL-green fluorophores. The G3.3 chips were used and samples were processed on a Bionano Saphyr instrument (San Diego, CA, USA). OGM analysis was performed using the Rare Variant Analysis (RVA) pipelines [23], and the Bionano AccessTM software v1.8.2 [24]. CNVs and SVs were manually determined using 1 Mb as a size cutoff for SVs and 5 Mb as a size cutoff for CNVs. OGM was performed on UMR-106 cells and WBC DNA from a Sprague-Dawley (SD) rat control. In the absence of an SD reference sequence, maps were compared to the rat genome reference rn7.2.
2.3. Nanopore Sequencing (ONT)
HMW DNA was isolated from freshly expanded rat osteosarcoma UMR-106 cells using the NEB Monarch kit (NEB #T3060) and an ultralong ONT library was prepared using the SQK-ULK114 kit. A Qiagen DNAeasy Blood and Tissue kit (#69506) was used to isolate DNA from blood of a female Sprague-Dawley rat and a standard ONT library was made (SQK-LSK114). The ULK library was made on the cell line in anticipation of likely structural variants. Both libraries were run on 10.4.1 PromethION flow cells with triple loading. The UMR-106 produced 45.3 Gb of aligned bases (to the rat rn7.2 reference) with an N50 of 78 kb. Data were aligned to reference using minimap2 [25]. Methylation calls were generated using modbam2bed [26]. Modbamtools was also used to plot methylation data. Summary statistics for the runs were generated with Nanoplot [27].
2.4. RNASeq Methods
Isolated RNA from UMR-106 was poly-A selected using the NEBNExt Poly(A) isolation module (NEB #E7490) and libraries were made using NEBNext Ultra II RNA Library Prep Kit for Illumina (Cat# E7775). Sequencing was performed on a NovaSeq 6000 SP 2x50bp run. FASTQ data were aligned to reference using the STAR aligner [28]. Control osteoblast data were from SRR16368266. 800 million reads were obtained from UMR-106 and compared to 45 million available from the osteoblast control. UMR-106 data were downsampled to match read length and approximate depth using samtools [29]. RNAseq reads were normalized between osteoblast and UMR-106 reads using GAPDH. Replicates were not available.
2.5. Mutation Identification
We focused on genes identified in the COSMIC database ([76]; https://cancer.sanger.ac.uk/cosmic). A detailed analysis was limited to the top twenty genes most commonly associated with human osteosarcoma. We also submitted variant call files for the UMR and control rat genomes to the Variant Effect Predictor ([30]; https://useast.ensembl.org/info/docs/tools/vep/index.html) and searched the output files for variants with “high” predicted designations in the tumor cells but which were absent in the control genome.
3. Results
3.1. Optical Genome Mapping
Optical mapping identified differences in UMR-106 relative to a healthy Sprague-Dawley (SD) strain sex-matched control animal principally in large blocks of amplified DNA including Myc. A tumor/normal variant analysis was performed using the Bionano AccessTM software and the NCBI rn7.2 rat genome reference sequence. Figure 1 shows differences in copy number across chromosome 7.
3.2. Correlation between Optical Mapping and Nanopore Sequence Depth
The region of significantly increased copy number by OGM was compared to ONT read depth. We normalized the nanopore sequence reads for UMR-106 and the SD control and compared read depth for the region showing relative amplification between UMR-106 and the control. Figure 2 shows that both optical mapping and nanopore sequence depth identify significantly similar amplification across the regions identified above.
3.3. Osteosarcoma Mutation Analysis
ONT sequence was analyzed for candidate genes from human studies of osteosarcoma reported in COSMIC, the Catalogue of Somatic Mutations in Cancer database ([31]; cancer.sanger.ac.uk/cosmic). Because a Sprague-Dawley rat genome reference was not available at the time of analysis and because some variants might reflect strain differences, we chose to sequence an SD control for comparison. To increase the likelihood of identifying structural variants (SVs) relevant to tumorgenicity, we used an ultralong read library for the UMR-106 cells (N50 ~78 kb). A standard ONT library (~20 kb N50) was generated for the control animal.
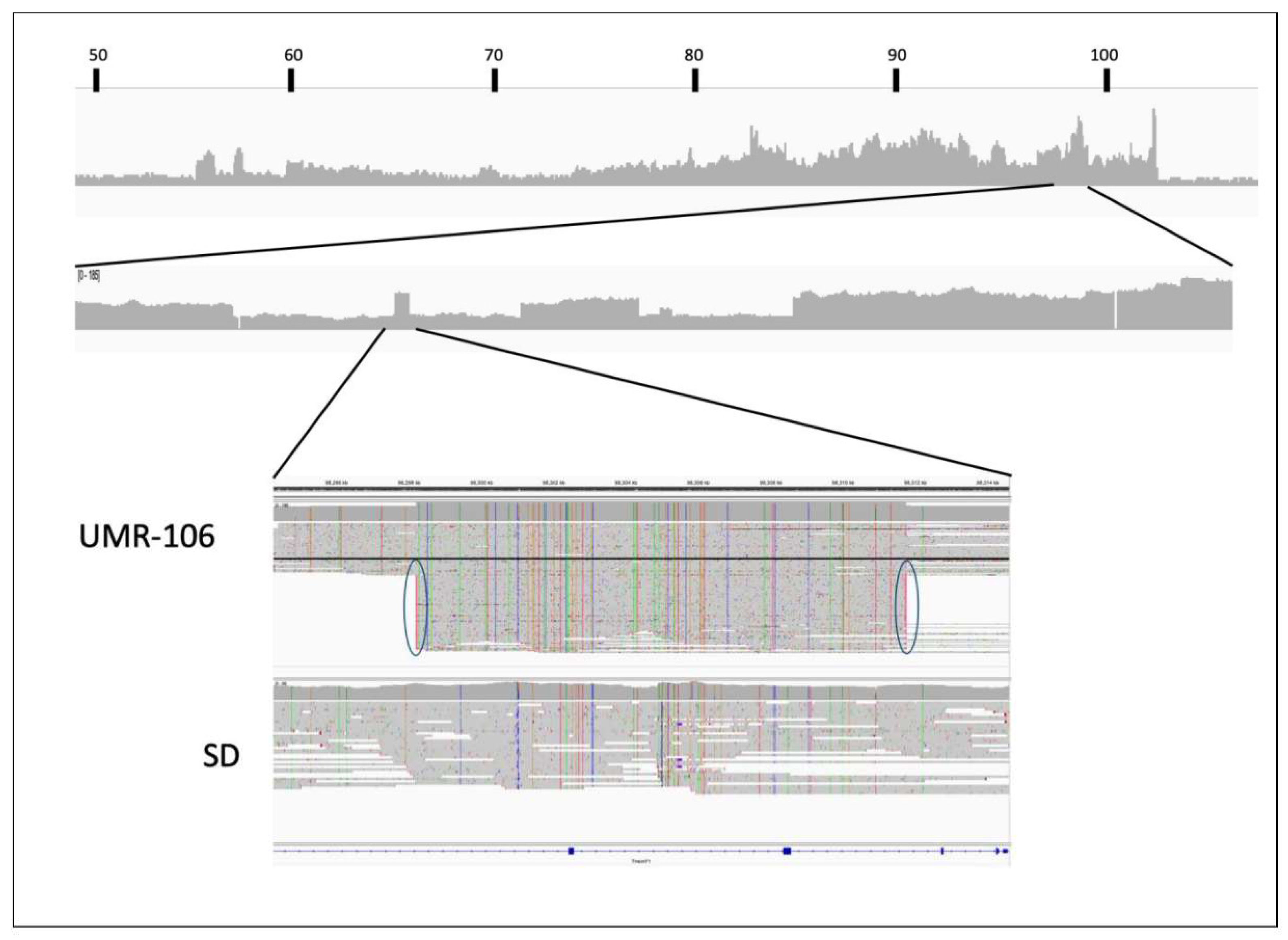
Using the top 20 mutated genes in human osteosarcoma listed in COSMIC, we did a manual review of UMR-106 versus the SD control using the Integrated Genomics Viewer (IGV) [32]. We also reviewed other reported cancer genes for copy number, somatic mutations and changes in 5mC methylation. In the Tp53 gene, UMR-106 DNA had a Leu to Phe change at codon 192 (chr10:54,308,638 C>T; VAF 1.0) which has been reported as likely pathogenic. This mutation has also been seen in a rat endometrial tumor [33]. In human cancers, the orthologous codon 194 position is reported in ClinVar ([34]; (https://www.ncbi.nlm.nih.gov/clinvar/RCV000417813.1/) and in chronic lymphocytic leukemia [35]). Manual inspection of data generated with the Variant Effect Predictor [30] tool comparing UMR-106 to the control genome did not identify any obvious pathogenic single nucleotide or small indel mutations in other cancer-related genes. However, a large deletion was detected in one-fourth of UMR-106 reads for H3f3a, a gene listed among the top twenty COSMIC loci associated with human OS. As shown in Figure 3, we observed a deletion of 10,995 bases in the affected reads. The figure highlights the utility of long reads in identification of structural variants that might otherwise be missed.
3.4. Expression Data
Short read RNA sequencing was done on poly-A RNA from the UMR-106 cell line and compared to a public dataset from SD rat osteoblasts ([36]; run SRR16368266). We downsampled and normalized the UMR reads relative to GAPDH in the osteoblast data. Relative expression was used to compare genes in UMR-106 from the amplified region identified by OGM and ONT. As shown in Table S1 we found that most genes in the region showed significantly increased expression relative to the osteoblast data.
3.5. Methylation
Because 5mC status can be directly measured from ONT sequencing we explored the UMR-106 data for genes that differed in epigenetic patterns from the SD control. The human tumor suppressor CDKN2A has been reported to be more methylated in OS and to be predictive of progression ([37,38]). Analysis of the ultralong UMR-106 Cdkn2a data showed hypermethylation at its 3’ end in exons 2 and 3 (Figure 4a). For Lsamp, there was notably less methylation in UMR-106 than in the SD control sample (Figure 4b). Down regulation of Lsamp has been implicated in lung cancer progression and poor prognosis ([39,40]).
3.6. Chromosome 7 Amplified Gene Region
Based on the OGM data showing amplification of the region around Myc we further explored this part of chromosome 7 with both ONT and RNA sequencing. Genes were examined for normalized read depth between UMR-106 and the SD control and for expression for UMR-106 versus an osteoblast dataset available at NCBI [36]. To reduce skewness [41] log(10) values were calculated for the ratios of tumor to control for OGM molecule depth, ONT sequence depth and TPM relative expression.
For Myc, we observed an approximately 7X-fold increase in sequence copy number and a 3.5X increase in the number of RNA reads (Table S1). For the adjacent Ndrg1 gene, a reported biomarker for human osteosarcoma ([42,43]), we observed about a 7.5-fold increase in copy number and a 3.2 increase in transcription. Other genes reported as markers of human OS are listed in Table 1. Of note, a large number of genes in this block of about 45 Mb have been reported as markers in OS or in other cancers. Myc amplification has been well known in cancer for over 40 years, is often associated with chromothripsis and with generally poorer prognosis [44]. While chromosome breakage and the coamplification of other genes near Myc have been reported, to our knowledge, there have not been comprehensive studies of the sub-chromosomal amplicons due to the absence of methods to characterize long DNA molecules other than cytogenetically. We looked at additional genes associated with OS in humans and dogs not within the amplified block including Pten [45], Magi2 [45], Rb1 [45], Dst [45], Dlg2 [45], Dmd [45] and Wwox [46]. None showed evidence of somatic mutations, copy number, methylation or significant expression differences relative to control data.

Table 1.
Genes in or near the chromosome 7 region identified by OGM (Figure 1). The log(10) ratios of normalized UMR to SD control are calculated from Table S1. RNA expression was normalized for UMR and SSR osteoblast data relative to GAPDH. The log(10) Tumor:Normal (T:N) ratios for OGM and ONT were compared using a Pearson correlation test and gave an r=0.957 and a p value of 1.38E-18 (see Figure 2). The log(10) of the ONT read depth to the expression TPM ratio gave an r=0.411 and a p-value of 0.018 excluding the low expressing Csmd3, Gsdmc, Kcnq3, Col21a1 and Mfng genes. Genes reported as osteosarcoma markers in the literature (OS marker) or otherwise associated with cancer are indicated. TAF2 has been reported to be amplified in breast cancer and CSMD3 as mutated in Esophageal Squamous Cell Carcinoma. N.B., Gene symbols are capitalized when referring to human genes and with a leading capital letter followed by lower case when referring to rat genes ([74,75]). .
Table 1.
Genes in or near the chromosome 7 region identified by OGM (Figure 1). The log(10) ratios of normalized UMR to SD control are calculated from Table S1. RNA expression was normalized for UMR and SSR osteoblast data relative to GAPDH. The log(10) Tumor:Normal (T:N) ratios for OGM and ONT were compared using a Pearson correlation test and gave an r=0.957 and a p value of 1.38E-18 (see Figure 2). The log(10) of the ONT read depth to the expression TPM ratio gave an r=0.411 and a p-value of 0.018 excluding the low expressing Csmd3, Gsdmc, Kcnq3, Col21a1 and Mfng genes. Genes reported as osteosarcoma markers in the literature (OS marker) or otherwise associated with cancer are indicated. TAF2 has been reported to be amplified in breast cancer and CSMD3 as mutated in Esophageal Squamous Cell Carcinoma. N.B., Gene symbols are capitalized when referring to human genes and with a leading capital letter followed by lower case when referring to rat genes ([74,75]). .
| OGM T:N | ONT T:N | TPM T:N | Comments, references | ||||
|---|---|---|---|---|---|---|---|
| Gene | Chr | Start | End | Ratio log(10) | Ratio log(10) | Ratio log(10) | |
| Mdm2 | 7 | 53290660 | 53315205 | 0.167 | 0.037 | -0.209 | OS amp; [47] |
| Mdm1 | 7 | 53729603 | 53766034 | 0.207 | 0.064 | 0.419 | |
| Oxr1 | 7 | 72528750 | 72965666 | 0.167 | 0.127 | -0.109 | |
| Angpt1 | 7 | 73528345 | 73783953 | 0.196 | 0.170 | -0.703 | OS marker; [48] |
| Csmd3 | 7 | 78747322 | 80066466 | 0.500 | 0.473 | mut in ESCC; [49] | |
| Trps1 | 7 | 81916668 | 82142733 | 0.541 | 0.544 | 0.374 | OS marker; [50] |
| Eif3h | 7 | 83091037 | 83174451 | 0.753 | 0.737 | 0.887 | OS marker; [51] |
| Taf2 | 7 | 86422613 | 86479616 | 0.706 | 0.639 | 0.952 | BRC amp; [52] |
| Deptor | 7 | 86514859 | 86668817 | 0.721 | 0.604 | -0.707 | OS marker; [53] |
| Has2 | 7 | 88113326 | 88139337 | 0.649 | 0.515 | -1.152 | OS marker; [54] |
| Zhx2 | 7 | 89226358 | 89374266 | 0.744 | 0.719 | -0.222 | [55] |
| Fam91a1 | 7 | 89969558 | 90007546 | 0.758 | 0.727 | 0.796 | OS marker: [56] |
| Tmem65 | 7 | 90336997 | 90378930 | 0.751 | 0.776 | 0.838 | OS marker; [57] |
| Rnf139 | 7 | 90439726 | 90450911 | 0.793 | 0.767 | 0.928 | OS marker; [58] |
| Myc | 7 | 93593705 | 93598633 | 0.759 | 0.859 | 0.544 | OS marker; [59] |
| Gsdmc | 7 | 95594015 | 95606106 | 0.412 | 0.531 | [60] | |
| Cyrib | 7 | 95633876 | 95760588 | 0.389 | 0.511 | 0.719 | [61] |
| Asap1 | 7 | 95786130 | 96093111 | 0.562 | 0.512 | 0.580 | [62] |
| Adcy8 | 7 | 96417310 | 96665911 | 0.457 | 0.508 | 0.699 | [63] |
| Efr3a | 7 | 97552677 | 97633369 | 0.696 | 0.695 | 0.755 | [64] |
| Kcnq3 | 7 | 97730219 | 98025652 | 0.680 | 0.744 | ||
| Phf20l1 | 7 | 98330580 | 98396526 | 0.552 | 0.496 | 0.089 | [65] |
| Ccn4 | 7 | 98645238 | 98677253 | 0.558 | 0.464 | 0.748 | OS marker; [66] |
| Ndrg1 | 7 | 98684487 | 98725869 | 0.708 | 0.873 | 0.508 | OS marker; [42] |
| St3gal1 | 7 | 98845270 | 98913409 | 0.922 | 0.882 | 0.236 | OS marker; [67] |
| Zfat | 7 | 99886954 | 100054288 | 0.624 | 0.641 | 0.637 | [68] |
| Khdrbs3 | 7 | 100837707 | 100995644 | 0.636 | 0.604 | 1.655 | [69] |
| Col22a1 | 7 | 103730939 | 103968452 | 0.021 | 0.037 | OS marker; [70] | |
| Trappc9 | 7 | 104521593 | 104998352 | 0.068 | -0.092 | -0.073 | [71] |
| Chrac1 | 7 | 105013047 | 105016435 | 0.095 | 0.185 | 0.091 | [72] |
| Mfng | 7 | 110310810 | 110328653 | 0.096 | 0.111 | [73] |
The fine structure of the chromosome 7 amplified region is complex as suggested by both ONT sequence and OGM molecule depth across the region. Figure 5 shows a plot for the ONT ultralong reads along with a detailed view of one region to illustrate local complexity and sequence scrambling. Examination of the sequence along the amplified region found many instances of abrupt transitions in UMR-106 relative to the reference but not in the control animal. Because many of the long UMR-106 ONT reads extended beyond the highlighted block we could confidently map the locations of secondary alignments against the reference. For example, the ~15kb block near 98.3 Mb has reads that both align well to the reference and also near 98.7 and 57.4 Mb (Figure 5). Similar instances can be seen throughout the amplified region implying a history of rearrangements and other events in the formation of UMR-106.
Overall ONT sequence depth from ~50 Mb to ~107 Mb along the rat 7.2 reference genome is shown with increased detail from 98 to 99.1 Mb. The bottom IGV image shows alignments at ~98.3 Mb for UMR-106 vs. the SD control. The black highlighted sequence shows a single read that extends across the block and agrees with the reference while the ovals highlight reads that have supplementary matches to regions near 98.7 and 57.4 Mb.
4. Discussion
As OGM software, by default, ignores molecules smaller than 150 kb, it will identify large scale structural variants orders of magnitude greater than can be seen with short-read sequencing. The resolution of OGM is reported to be from 5 to 10 Mb down to a few kb [76]. Because nanopore sequencing can now routinely produce reads with N50s of 20-100 kb or longer it can effectively bridge the scale afforded by OGM for analysis of smaller SVs and SNVs. Pei et al. [22] recently compared both long DNA methods and concluded that the precision of OGM was very high and that ONT sequencing outperformed short reads for SV detection. In this manuscript we have combined both long DNA approaches to evaluate their utility in characterizing a widely used osteosarcoma cell line. But, because we anticipated many cancer-related structural anomalies in the UMR-106 cells we used ONT transposase-based ultralong reads on freshly grown cells isolated to minimize DNA fragmentation. The sequencing produced reads with an N50 of ~78kb with many exceeding 100 kb and a few outliers over 200 kb.
Both OGM and ONT identified excess molecules and sequence depth, respectively, corresponding to large portions of chromosome 7 that included the Myc gene, a likely driver of oncogenicity for UMR-106. The correspondence between OGM and ONT depth measurements agreed well (Figure 2). When we explored the region in greater depth using the ultralong reads we concluded that the fine structure of the region was more complicated than simple alignments show. In Figure 5 the sequence depth can be seen to vary across a large portion of chromosome 7 and some regions showed what initially appeared to be local amplification. But, because of the ultralong reads we were able to identify that many of these had secondary alignments elsewhere on chromosome 7, often tens of Mb away. The example shown (Figure 5) from a region near 98.3 Mb includes both reads that are in agreement with the reference and others that were non-contiguous with long secondary alignments mapping near 98.7 and 57.4 Mb. This is best explained as sequence scrambling followed by amplification. We conclude that the amplification of genes along chromosome 7 is not the result of a simple duplication or multiplication event but more likely from a series of events involving complex rearrangements and, possibly, the formation of abnormal chromosomes such as rings, double-minutes or others. Other than chromosome 7, both OGM and nanopore sequencing identified a relatively small number of genomic changes suggesting that the UMR-106 cell line has been relatively stable despite its many years in culture although this requires additional analysis.
As noted above, there was excellent correspondence between OGM and ultralong ONT sequence and each has advantages and disadvantages depending on the goals of a particular study. Ultralong ONT reads that approach OGM in length provide base level sequence, 5mC status and can detect CNVs and indels below the resolution of OGM. But, OGM can provide a higher-level view of the genome which is more difficult to characterize with sequence data alone. When used in combination they provide complementary data sets. However, ONT sequencing has a clear advantage over optical mapping when exploring genomes in detail since it allows single base resolution. This is illustrated by the detection of the 11 kb deletion in H3f3a (Figure 3) where we found reads that spanned the deletion and were able to precisely map its boundaries. Sequencing also allowed mutation detection and identified the Tp53 variant. But, the relative stability of UMR-106 was confirmed by manual inspection of dozens of known oncogenes by comparing variant call files between the cell line and the control animal. Alignment of both the SD control and UMR-106 also identified shared variants not seen in the reference and which are likely strain-specific (data not shown). The ability to call methylated bases without additional sample manipulation was an ancillary benefit of ONT sequencing. The identification of marked differences in 5mC methylation for Cdkn2a and Lsamp in the tumor were consistent with reports of their role in cancer. Going forward, it will be interesting to reanalyze UMR-106, especially with methylation analysis, as characteristics of the cell line change with passage.
An unexpected observation of this study was how many of the genes in the amplified region are associated with literature implicating their role in this cancer type (Table 1 and Table S1). Oncogene amplification is a well-known phenomenon in cancers especially for Myc [44] and genes flanking Myc can be coamplified. Parris et al. [52] noted that TAF2, NDRG1 and TRPS1 were among genes coamplified in breast cancer along with MYC on human chromosome 8q. The fact that 14 of the genes we identified in the amplified region are already described as OS markers suggests that many of these genes may cooperate in influencing OS progression and could be targets for intervention. Further studies comparing osteosarcomas with Myc amplification might be useful in defining which adjacent genes are most important in progression. Although the RNAseq data from UMR-106 is limited to a single experiment and the publicly available osteoblast data set is fairly small, we did detect a trend for genes in the amplified region as having increased expression (Table S1). But, this observation will require confirmation with further studies especially with additional osteoblast sequence.
This report is a non-exhaustive overview of a single rat osteosarcoma cell line that has been used for over 50 years. The study's purpose was to explore the advantages of long DNA methods to more rapidly, thoroughly and rigorously characterize this cell line. Because we only studied a single osteosarcoma line from one species, we cannot unequivocally conclude which changes are most significant in determining the tumor phenotype. However, the commonality of shared genes from human, rat and canine [45] osteosarcomas and other tumors with Myc amplification suggests shared pathways occur across species. The fact that so many coamplified genes we observed surrounding Myc are reported as OS markers suggests that they are either commonly amplified passengers along with Myc or have functions that augment Myc. The methods we highlight here are rapid and comprehensive but do require high molecular weight DNA so will be limited to fresh samples or established cell lines. We expect that the use of optical mapping and long read sequencing will simplify the study of amplification and chromothripsis and will provide a deeper understanding of genome architecture in cancer, perhaps providing new strategies for treatment.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1 showing the genes from the chromosome 7 amplified block with ratios of OGM and ONT copy numbers and UMR-106 vs. osteoblast TPMs along with 2-sample T-test p values indicating direction of expression.
Data Availability
The resources described here are available for further exploration by the community. Data are available at NCBI under project number PRJNA1148449 and includes fastq files for the UMR-106 and SD control ONT reads, UMR-106 RNAseq fastq reads, and the OGM optical map files.
Institutional Review Board Statement
The animal study protocol (RA23M98) was approved by the Johns Hopkins University Animal Care and Use Committee (ACUC) on 30 March 2023.
Acknowledgments
funding from NIH/NCI R21 CA228582 to KG. Sequencing was performed at the JHU Genetic Resources Core Facility, RRID:SCR_018669. We thank Dr. Ingo Ruczinski (JHU Bloomberg School of Public Health) for statistical advice.
Conflicts of Interest
None.
Author Contributions
Conceptualization: K.G., Y.S.Z., A.F.S.; Optical genome mapping: V.S, Y.S.Z.; ONT library prep: M.K.; ONT sequencing analysis: D.W.M.; NCBI data submission: D.W.M.; RNA isolation and library prep: R.B.; RNAseq analysis: D.W.M.; Sequence data manual review: J.A., J.M., W.A.L., A.F.S.; Writing original draft and statistical analysis: A.F.S.
References
- Eaton, B.R.; Schwarz, R.; Vatner, R.; Yeh, B.; Claude, L.; Indelicato, D.J.; Laack, N. Osteosarcoma. Pediatr Blood Cancer 2021, 68 Suppl 2, e28352.
- Meltzer, P.S.; Helman, L.J. New Horizons in the Treatment of Osteosarcoma. New Engl. J. Med. 2021, 385, 2066–2076. [CrossRef]
- Belayneh, R.; Fourman, M.S.; Bhogal, S.; Weiss, K.R. Update on Osteosarcoma. Curr. Oncol. Rep. 2021, 23, 1–8. [CrossRef]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [CrossRef]
- Martin, T.J.; Ingleton, P.M.; Underwood, J.C.E.; Michelangeli, V.P.; Hunt, N.H.; Melick, R.A. Parathyroid hormone-responsive adenylate cyclase in induced transplantable osteogenic rat sarcoma. Nature 1976, 260, 436–438. [CrossRef]
- Qin, G.; Chen, Y.; Li, H.; Xu, S.; Li, Y.; Sun, J.; Rao, W.; Chen, C.; DU, M.; He, K.; et al. Melittin inhibits tumor angiogenesis modulated by endothelial progenitor cells associated with the SDF-1α/CXCR4 signaling pathway in a UMR-106 osteosarcoma xenograft mouse model. Mol. Med. Rep. 2016, 14, 57–68. [CrossRef]
- Zhang, P.; Dong, L.; Long, H.; Yang, T.-T.; Zhou, Y.; Fan, Q.-Y.; Ma, B.-A. Homologous mesenchymal stem cells promote the emergence and growth of pulmonary metastases of the rat osteosarcoma cell line UMR-106. Oncol. Lett. 2014, 8, 127–132. [CrossRef]
- Yu, Z.; Ma, B.; Zhou, Y.; Zhang, M.; Long, H.; Wang, Y.; Fan, Q. Allogeneic Tumor Vaccine Produced by Electrofusion between Osteosarcoma Cell Line and Dendritic Cells in the Induction of Antitumor Immunity. Cancer Investig. 2007, 25, 535–541. [CrossRef]
- Ma; Yu, Z.; Sun, H.; Fan, Q.; Long, H.; Yang, T.; Ma, B. Establishment of reproducible osteosarcoma rat model using orthotopic implantation technique.. Oncol. Rep. 2009, 21, 1175–1180. [CrossRef]
- Zhu, X.-Z.; Yin, H.-M.; Mei, J. Inhibition of tumors cell growth in osteosarcoma-bearing SD rats through a combination of conventional and metronomic scheduling of neoadjuvant chemotherapy. Acta Pharmacol. Sin. 2010, 31, 970–976. [CrossRef]
- Zhang, L.; Ye, Y.; Yang, D.; Lin, J. Survivin and vascular endothelial growth factor are associated with spontaneous pulmonary metastasis of osteosarcoma: Development of an orthotopic mouse model. Oncol. Lett. 2014, 8, 2577–2580. [CrossRef]
- Wu, D.; Wan, M. Methylene diphosphonate-conjugated adriamycin liposomes: preparation, characteristics, and targeted therapy for osteosarcomas in vitro and in vivo. Biomed. Microdevices 2012, 14, 497–510. [CrossRef]
- Fukumoto, S.; Allan, E.H.; Martin, T. Regulation of plasminogen activator inhibitor-1 (PAI-1) expression by 1,25-dihydroxyvitamin D-3 in normal and malignant rat osteoblasts. Biochim. et Biophys. Acta (BBA) - Gen. Subj. 1994, 1201, 223–228. [CrossRef]
- Ishiyama, S.; Kissel, C.; Guo, X.; Howard, A.; Saeki, H.; Ito, T.; Sysa-Shah, P.; Orita, H.; Sakamoto, K.; Gabrielson, K. A Syngeneic Orthotopic Osteosarcoma Sprague Dawley Rat Model with Amputation to Control Metastasis Rate. J Vis Exp 2021,.
- Partridge, N.C.; Frampton, R.J.; Eisman, J.A.; Michelangeli, V.P.; Elms, E.; Bradley, T.R.; Martin, T.J. Receptors for 1,25(OH)2-vitamin D3 enriched in cloned osteoblast-like rat osteogenic sarcoma cells. FEBS Lett. 1980, 115, 139-142.
- Ek, E.T.; Dass, C.R.; Choong, P.F. Commonly used mouse models of osteosarcoma. Crit. Rev. Oncol. Hematol. 2006, 60, 1–8. [CrossRef]
- Kolmogorov, M.; Billingsley, K.J.; Mastoras, M.; Meredith, M.; Monlong, J.; Lorig-Roach, R.; Asri, M.; Alvarez Jerez, P.; Malik, L.; Dewan, R.; Reed, X.; Genner, R.M.; Daida, K.; Behera, S.; Shafin, K.; Pesout, T.; Prabakaran, J.; Carnevali, P.; Yang, J.; Rhie, A.; Scholz, S.W.; Traynor, B.J.; Miga, K.H.; Jain, M.; Timp, W.; Phillippy, A.M.; Chaisson, M.; Sedlazeck, F.J.; Blauwendraat, C.; Paten, B. Scalable Nanopore sequencing of human genomes provides a comprehensive view of haplotype-resolved variation and methylation. Nat Methods 2023, 20, 1483-1492.
- Barford, R.G.; Whittle, E.; Weir, L.; Fong, F.C.; Goodman, A.; Hartley, H.E.; Allinson, L.M.; Tweddle, D.A. Use of Optical Genome Mapping to Detect Structural Variants in Neuroblastoma. Cancers 2023, 15, 5233. [CrossRef]
- Baelen, J.; Dewaele, B.; Debiec-Rychter, M.; Sciot, R.; Schöffski, P.; Hompes, D.; Sinnaeve, F.; Wafa, H.; Bempt, I.V. Optical Genome Mapping for Comprehensive Cytogenetic Analysis of Soft-Tissue and Bone Tumors for Diagnostic Purposes. J. Mol. Diagn. 2024, 26, 374–386. [CrossRef]
- Thibodeau, M.L.; O’neill, K.; Dixon, K.; Reisle, C.; Mungall, K.L.; Krzywinski, M.; Shen, Y.; Lim, H.J.; Cheng, D.; Tse, K.; et al. Improved structural variant interpretation for hereditary cancer susceptibility using long-read sequencing. Anesthesia Analg. 2020, 22, 1892–1897. [CrossRef]
- O’Neill, K.; Pleasance, E.; Fan, J.; Akbari, V.; Chang, G.; Dixon, K.; Csizmok, V.; MacLennan, S.; Porter, V.; Galbraith, A.; Grisdale, C.J.; Culibrk, L.; Dupuis, J.H.; Corbett, R.; Hopkins, J.; Bowlby, R.; Pandoh, P.; Smailus, D.E.; Cheng, D.; Wong, T.; Frey, C.; Shen, Y.; Paulin, L.F.; Sedlazeck, F.J.; Nelson, J.M.T.; Chuah, E.; Mungall, K.L.; Moore, R.A.; Coope, R.; Mungall, A.J.; McConechy, M.K.; Williamson, L.M.; Schrader, K.A.; Yip, S.; Marra, M.A.; Laskin, J.; Jones, S.J.M. Long-read sequencing of an advanced cancer cohort resolves rearrangements, unravels haplotypes, and reveals methylation landscapes medRixiv 2024,.
- Pei, Y.; Tanguy, M.; Giess, A.; Dixit, A.; Wilson, L.C.; Gibbons, R.J.; Twigg, S.R.F.; Elgar, G.; Wilkie, A.O.M. A Comparison of Structural Variant Calling from Short-Read and Nanopore-Based Whole-Genome Sequencing Using Optical Genome Mapping as a Benchmark. Genes 2024, 15, 925. [CrossRef]
- Nilius-Eliliwi, V.; Gerding, W.M.; Schroers, R.; Nguyen, H.P.; Vangala, D.B. Optical Genome Mapping for Cytogenetic Diagnostics in AML Cancers 2023, 15, 1684.
- Genomics, B. Bionano Solve Theory of Operation: Variant Annotation Pipeline 2024,.
- Li, H. Minimap2: pairwise alignment for nucleotide sequences Bioinformatics 2018, 34, 3094-3100.
- Razaghi, R.; Hook, P.W.; Ou, S.; Schatz, M.C.; Hansen, K.D.; Jain, M.; Timp, W. Modbamtools: Analysis of single-molecule epigenetic data for long-range profiling, heterogeneity, and clustering bioRxiv 2022, 2022.07.07.499188.
- De Coster, W.; Rademakers, R. NanoPack2: population-scale evaluation of long-read sequencing data. Bioinformatics 2023, 39, btad311.
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 1–14. Available online: https://grch37.ensembl.org/info/docs/tools/vep/index.html. (Accessed on ).
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [CrossRef]
- Wilkinson, M.; Sinclair, P.; Dellatorre-Teixeira, L.; Swan, P.; Brennan, E.; Moran, B.; Wedekind, D.; Downey, P.; Sheahan, K.; Conroy, E.; et al. The Molecular Effects of a High Fat Diet on Endometrial Tumour Biology. Life 2020, 10, 188. [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [CrossRef]
- De Luca, G.; Cerruti, G.; Lastraioli, S.; Conte, R.; Ibatici, A.; Di Felice, N.; Morabito, F.; Monti, P.; Fronza, G.; Matis, S.; et al. The spectrum of subclonal TP53 mutations in chronic lymphocytic leukemia: A next generation sequencing retrospective study. Hematol. Oncol. 2022, 40, 962–975. [CrossRef]
- Liu, X.; Zhan, Y.; Xu, W.; Liu, L.; Liu, X.; Da, J.; Zhang, K.; Zhang, X.; Wang, J.; Liu, Z.; et al. Characterization of transcriptional landscape in bone marrow-derived mesenchymal stromal cells treated with aspirin by RNA-seq. PeerJ 2022, 10, e12819. [CrossRef]
- Lietz, C.E.; Newman, E.T.; Kelly, A.D.; Xiang, D.H.; Zhang, Z.; Luscko, C.A.; Lozano-Calderon, S.A.; Ebb, D.H.; Raskin, K.A.; Cote, G.M.; et al. Genome-wide DNA methylation patterns reveal clinically relevant predictive and prognostic subtypes in human osteosarcoma. Commun. Biol. 2022, 5, 1–20. [CrossRef]
- Righi, A.; Gambarotti, M.; Sbaraglia, M.; Sisto, A.; Ferrari, S.; Tos, A.P.D.; Picci, P. p16 expression as a prognostic and predictive marker in high-grade localized osteosarcoma of the extremities: an analysis of 357 cases. Hum. Pathol. 2016, 58, 15–23. [CrossRef]
- Chang, C.-Y.; Wu, K.-L.; Chang, Y.-Y.; Liu, Y.-W.; Huang, Y.-C.; Jian, S.-F.; Lin, Y.-S.; Tsai, P.-H.; Hung, J.-Y.; Tsai, Y.-M.; et al. The Downregulation of LSAMP Expression Promotes Lung Cancer Progression and Is Associated with Poor Survival Prognosis. J. Pers. Med. 2021, 11, 578. [CrossRef]
- Fletcher; Yen, C.-C.; Chen, W.-M.; Chen, T.-H.; Chen, W.Y.-K.; Chen, P.C.-H.; Chiou, H.-J.; Hung, G.-Y.; Wu, H.-T.H.; Wei, C.-J.; et al. Identification of chromosomal aberrations associated with disease progression and a novel 3q13.31 deletion involving LSAMP gene in osteosarcoma. Int. J. Oncol. 2009, 35, 775–788. [CrossRef]
- West, R.M. Best practice in statistics: The use of log transformation. Ann. Clin. Biochem. Int. J. Biochem. Lab. Med. 2021, 59, 162–165. [CrossRef]
- Hua, Y.; Jia, X.; Sun, M.; Zheng, L.; Yin, L.; Zhang, L.; Cai, Z. Plasma membrane proteomic analysis of human osteosarcoma and osteoblastic cells: revealing NDRG1 as a marker for osteosarcoma. Tumor Biol. 2011, 32, 1013–1021. [CrossRef]
- Joshi, V.; Lakhani, S.R.; Reed, A.E.M. NDRG1 in Cancer: A Suppressor, Promoter, or Both?. Cancers 2022, 14, 5739. [CrossRef]
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC Deregulation in Primary Human Cancers. Genes 2017, 8, 151. [CrossRef]
- Megquier, K.; Turner-Maier, J.; Morrill, K.; Li, X.; Johnson, J.; Karlsson, E.K.; London, C.A.; Gardner, H.L. The genomic landscape of canine osteosarcoma cell lines reveals conserved structural complexity and pathway alterations. PLOS ONE 2022, 17, e0274383. [CrossRef]
- Yang, J.; Cogdell, D.; Yang, D.; Hu, L.; Li, H.; Zheng, H.; Du, X.; Pang, Y.; Trent, J.; Chen, K.; et al. Deletion of the WWOX gene and frequent loss of its protein expression in human osteosarcoma. Cancer Lett. 2010, 291, 31–38. [CrossRef]
- Sciot, R. MDM2 Amplified Sarcomas: A Literature Review. Diagnostics 2021, 11, 496. [CrossRef]
- Sorenson, L.; Fu, Y.; Hood, T.; Warren, S.; McEachron, T.A. Targeted transcriptional profiling of the tumor microenvironment reveals lymphocyte exclusion and vascular dysfunction in metastatic osteosarcoma. OncoImmunology 2019, 8, e1629779. [CrossRef]
- Mangalaparthi, K.K.; Patel, K.; Khan, A.A.; Manoharan, M.; Karunakaran, C.; Murugan, S.; Gupta, R.; Gupta, R.; Khanna-Gupta, A.; Chaudhuri, A.; et al. Mutational Landscape of Esophageal Squamous Cell Carcinoma in an Indian Cohort. Front. Oncol. 2020, 10. [CrossRef]
- Li, Z.; Jia, M.; Wu, X.; Cui, J.; Pan, A.; Li, L. Overexpression of Trps1 contributes to tumor angiogenesis and poor prognosis of human osteosarcoma. Diagn. Pathol. 2015, 10, 1–7. [CrossRef]
- Hong, S.; Liu, Y.; Xiong, H.; Cai, D.; Fan, Q. Eukaryotic translation initiation factor 3H suppression inhibits osteocarcinoma cell growth and tumorigenesis. Exp Ther Med 2018, 15, 4925-4931.
- Parris, T.Z.; Kovács, A.; Hajizadeh, S.; Nemes, S.; Semaan, M.; Levin, M.; Karlsson, P.; Helou, K. Frequent MYC coamplification and DNA hypomethylation of multiple genes on 8q in 8p11-p12-amplified breast carcinomas. Oncogenesis 2014, 3, e95–e95. [CrossRef]
- Hu, B.; Lv, X.; Gao, F.; Chen, S.; Wang, S.; Qing, X.; Liu, J.; Wang, B.; Shao, Z. Downregulation of DEPTOR inhibits the proliferation, migration, and survival of osteosarcoma through PI3K/Akt/mTOR pathway. OncoTargets Ther. 2017, ume 10, 4379–4391. [CrossRef]
- Zhao, Z.; Liang, T.; Feng, S. Silencing of HAS2-AS1 mediates PI3K/AKT signaling pathway to inhibit cell proliferation, migration, and invasion in glioma. J. Cell. Biochem. 2019, 120, 11510-11516.
- Ding, R.; Shi, H.; Guo, Y.; Zeng, W.; Fan, J. Zinc fingers and homeoboxes 2 inhibition could suppress the proliferation of ovarian cancer cells by apoptosis pathway. Clin. Transl. Oncol. 2023, 25, 2116–2126. [CrossRef]
- Lin, C.; Miao, J.; He, J.; Feng, W.; Chen, X.; Jiang, X.; Liu, J.; Li, B.; Huang, Q.; Liao, S.; Liu, Y. The regulatory mechanism of LncRNA-mediated ceRNA network in osteosarcoma. Sci Rep 2022, 12, 8756.
- Jia, Y.; Liu, Y.; Han, Z.; Tian, R. Identification of potential gene signatures associated with osteosarcoma by integrated bioinformatics analysis. PeerJ 2021, 9, e11496. [CrossRef]
- Pandya, P.H.; Cheng, L.; Saadatzadeh, M.R.; Bijangi-Vishehsaraei, K.; Tang, S.; Sinn, A.L.; Trowbridge, M.A.; Coy, K.L.; Bailey, B.J.; Young, C.N.; et al. Systems Biology Approach Identifies Prognostic Signatures of Poor Overall Survival and Guides the Prioritization of Novel BET-CHK1 Combination Therapy for Osteosarcoma. Cancers 2020, 12, 2426. [CrossRef]
- Pandya, P.H.; Cheng, L.; Saadatzadeh, M.R.; Bijangi-Vishehsaraei, K.; Tang, S.; Sinn, A.L.; Trowbridge, M.A.; Coy, K.L.; Bailey, B.J.; Young, C.N.; et al. Systems Biology Approach Identifies Prognostic Signatures of Poor Overall Survival and Guides the Prioritization of Novel BET-CHK1 Combination Therapy for Osteosarcoma. Cancers 2020, 12, 2426. [CrossRef]
- Li, M.; Jiang, Q.; Liu, X.; Han, L.; Chen, S.; Xue, R. The Pyroptosis-Related Signature Composed of GSDMC Predicts Prognosis and Contributes to Growth and Metastasis of Hepatocellular Carcinoma. Front. Biosci. 2023, 28, 235. [CrossRef]
- Pereira, B.S.; Wisnieski, F.; Calcagno, D.Q.; Santos, L.C.; Gigek, C.O.; Chen, E.S.; Rasmussen, L.T.; Payão, S.L.M.; Almeida, R.S.; Pinto, C.A.; et al. Genetic and Transcriptional Analysis of 8q24.21 Cluster in Gastric Cancer. Anticancer. Res. 2022, 42, 4381–4394. [CrossRef]
- Müller, T.; Stein, U.; Poletti, A.; Garzia, L.; Rothley, M.; Plaumann, D.; Thiele, W.; Bauer, M.; Galasso, A.; Schlag, P.; et al. ASAP1 promotes tumor cell motility and invasiveness, stimulates metastasis formation in vivo, and correlates with poor survival in colorectal cancer patients. Oncogene 2010, 29, 2393–2403. [CrossRef]
- El-Kafrawy, S.A.; El-Daly, M.M.; Bajrai, L.H.; Alandijany, T.A.; Faizo, A.A.; Mobashir, M.; Ahmed, S.S.; Ahmed, S.; Alam, S.; Jeet, R.; et al. Genomic profiling and network-level understanding uncover the potential genes and the pathways in hepatocellular carcinoma. Front. Genet. 2022, 13, 880440. [CrossRef]
- Zhou, D.; Yang, L.; Zheng, L.; Ge, W.; Li, D.; Zhang, Y.; Hu, X.; Gao, Z.; Xu, J.; Huang, Y.; et al. Exome Capture Sequencing of Adenoma Reveals Genetic Alterations in Multiple Cellular Pathways at the Early Stage of Colorectal Tumorigenesis. PLOS ONE 2013, 8, e53310. [CrossRef]
- Hou, Y.; Liu, W.; Yi, X.; Yang, Y.; Su, D.; Huang, W.; Yu, H.; Teng, X.; Yang, Y.; Feng, W.; et al. PHF20L1 as a H3K27me2 reader coordinates with transcriptional repressors to promote breast tumorigenesis. Sci. Adv. 2020, 6, eaaz0356. [CrossRef]
- Tsai, H.-C.; Tzeng, H.-E.; Huang, C.-Y.; Huang, Y.-L.; Tsai, C.-H.; Wang, S.-W.; Wang, P.-C.; Chang, A.-C.; Fong, Y.-C.; Tang, C.-H. WISP-1 positively regulates angiogenesis by controlling VEGF-A expression in human osteosarcoma. Cell Death Dis. 2017, 8, e2750–e2750. [CrossRef]
- Zou, Y.; Guo, S.; Liao, Y.; Chen, W.; Chen, Z.; Chen, J.; Wen, L.; Xie, X. Ceramide metabolism-related prognostic signature and immunosuppressive function of ST3GAL1 in osteosarcoma. Transl. Oncol. 2024, 40, 101840. [CrossRef]
- Tsunoda, T.; Shirasawa, S. Roles of ZFAT in haematopoiesis, angiogenesis and cancer development.. 2013, 33, 2833–7.
- Zhao, M.; Zhang, Y.; Li, L.; Liu, X.; Zhou, W.; Wang, C.; Tang, Y. KHDRBS3 accelerates glycolysis and promotes malignancy of hepatocellular carcinoma via upregulating 14-3-3ζ. Cancer Cell Int. 2023, 23, 1–13. [CrossRef]
- Pan, R.; Pan, F.; Zeng, Z.; Lei, S.; Yang, Y.; Yang, Y.; Hu, C.; Chen, H.; Tian, X. A novel immune cell signature for predicting osteosarcoma prognosis and guiding therapy. Front. Immunol. 2022, 13, 1017120. [CrossRef]
- Zhang, Y.; Liu, S.; Wang, H.; Yang, W.; Li, F.; Yang, F.; Yu, D.; Ramsey, F.V.; Tuszyski, G.P.; Hu, W. Elevated NIBP/TRAPPC9 mediates tumorigenesis of cancer cells through NFκB signaling. Oncotarget 2015, 6, 6160–6178. [CrossRef]
- Wang, M.; Li, S.; Guo, W.; Wang, L.; Huang, J.; Zhuo, J.; Lai, B.; Liao, C.; Ge, T.; Nie, Y.; et al. CHRAC1 promotes human lung cancer growth through regulating YAP transcriptional activity. Carcinog. 2021, 43, 264–276. [CrossRef]
- Gao, Y.; Luo, L.; Qu, Y.; Zhou, Q. MFNG is an independent prognostic marker for osteosarcoma. Eur. J. Med Res. 2023, 28, 1–8. [CrossRef]
- Sundberg, J.P.; Schofield, P.N. Commentary: Mouse Genetic Nomenclature. Veter- Pathol. 2010, 47, 1100–1104. [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [CrossRef]
- Vieler, L.-M.; Nilius-Eliliwi, V.; Schroers, R.; Ben Vangala, D.; Nguyen, H.P.; Gerding, W.M. Optical Genome Mapping Reveals and Characterizes Recurrent Aberrations and New Fusion Genes in Adult ALL. Genes 2023, 14, 686. [CrossRef]
Figure 1.
OGM copy number along chromosome 7 with the position of Myc indicated.
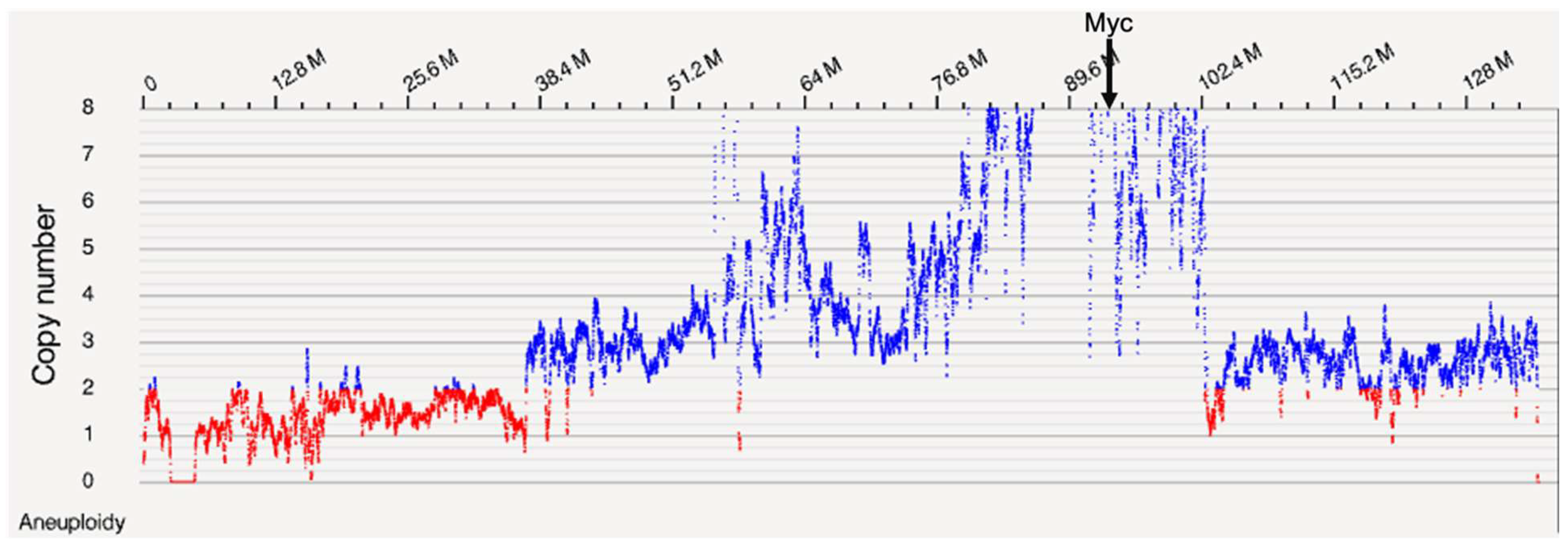
Figure 2.
Plot of log10 values for the ratios of OGM and ONT depth (molecules or sequence reads) of UMR to the control SD animal across genes in the amplified region shown in Figure 1. Data is from Table 1 below. .
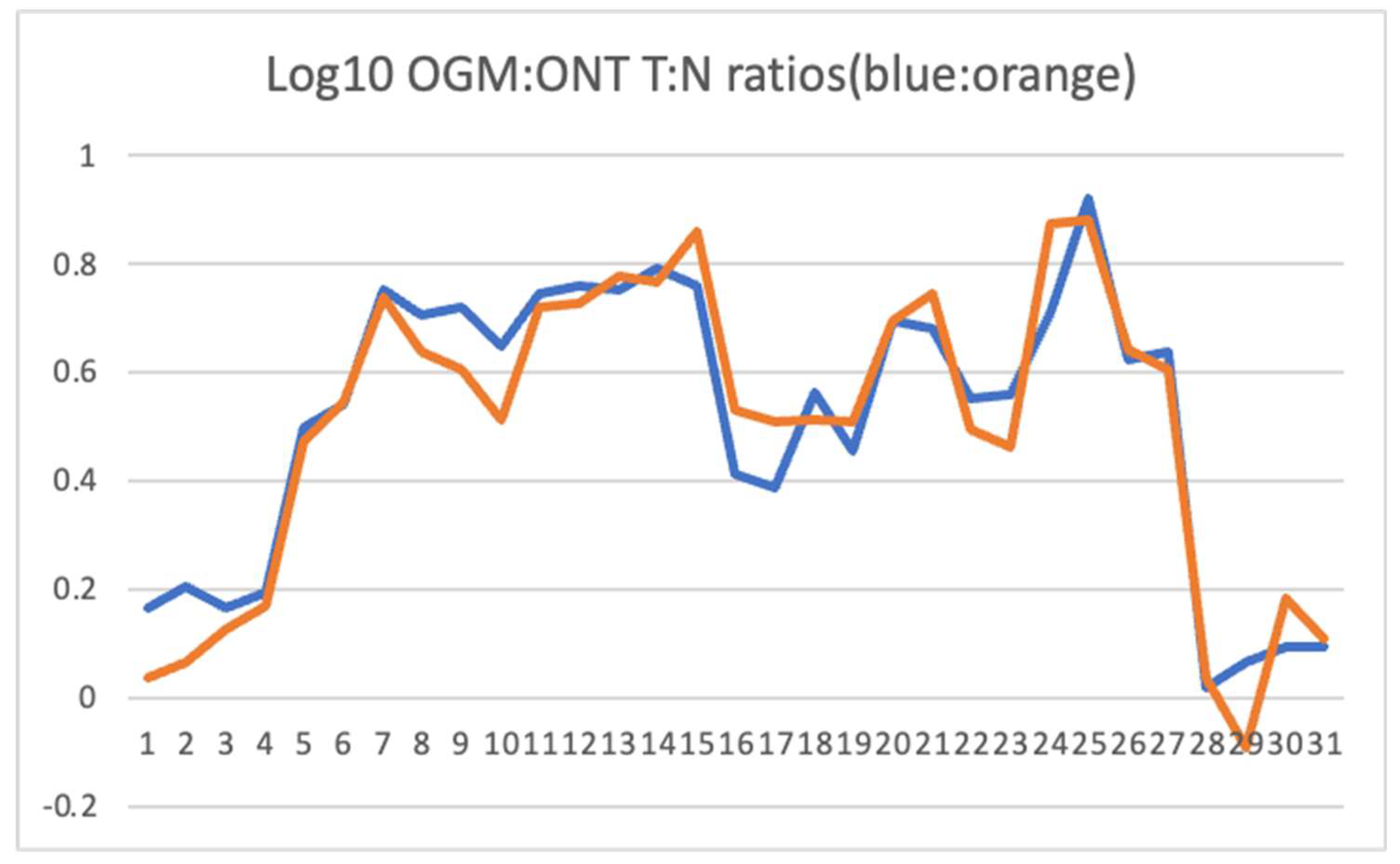
Figure 3.
Identification of an H3f3a 11 kb deletion in marked reads from ultralong ONT sequencing. Direction of transcription is from right to left. In the affected reads exons 1-3 are deleted. .
Figure 3.
Identification of an H3f3a 11 kb deletion in marked reads from ultralong ONT sequencing. Direction of transcription is from right to left. In the affected reads exons 1-3 are deleted. .
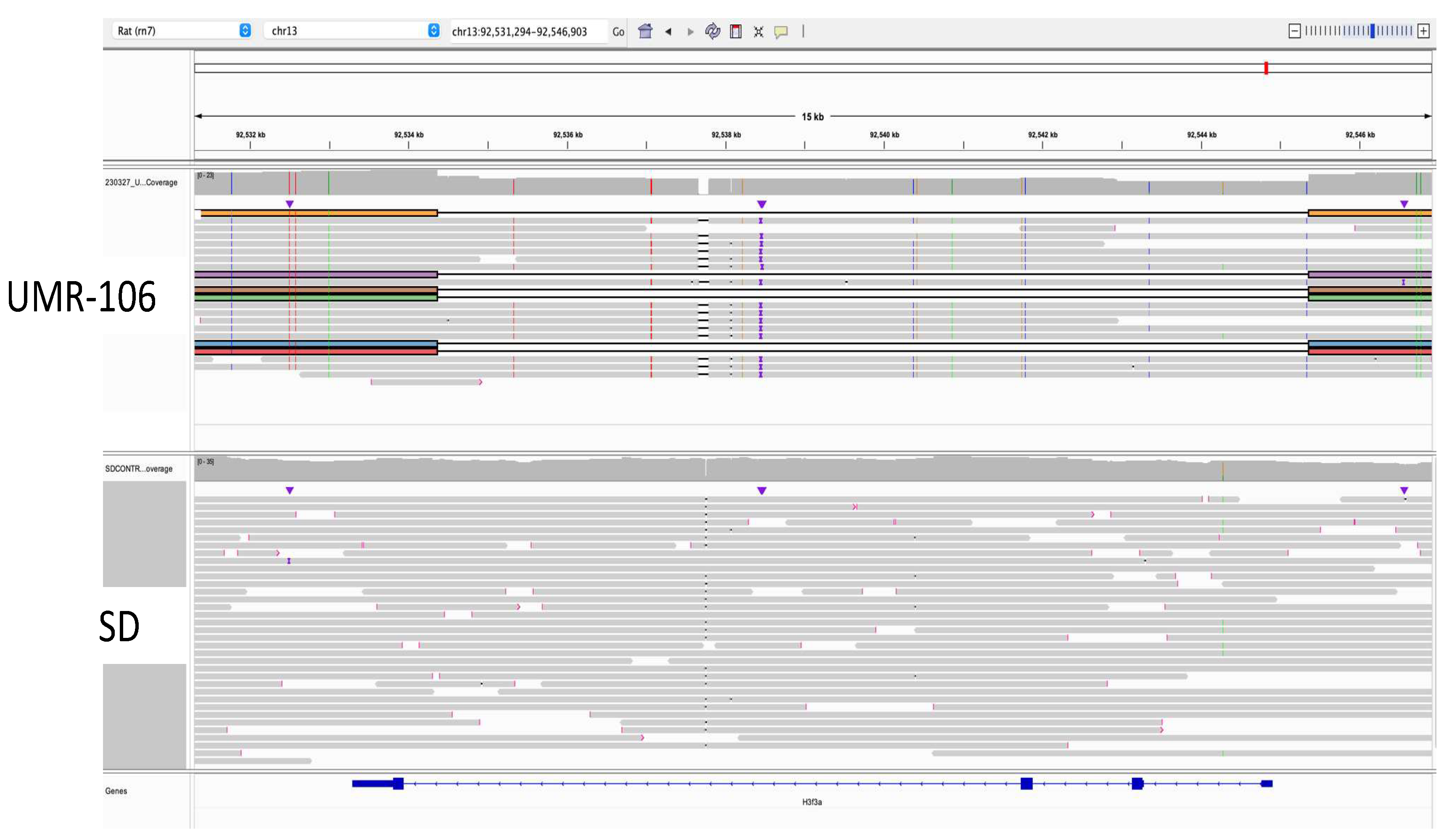
Figure 4.
a. IGV display showing hypermethylation (red) of the 3’ end of Cdkn2A including exons 2 and 3. Figure 4b. Lsamp methylation plots comparing UMR-106 to SD. Direction of transcription for both genes is right to left. .
Figure 4.
a. IGV display showing hypermethylation (red) of the 3’ end of Cdkn2A including exons 2 and 3. Figure 4b. Lsamp methylation plots comparing UMR-106 to SD. Direction of transcription for both genes is right to left. .
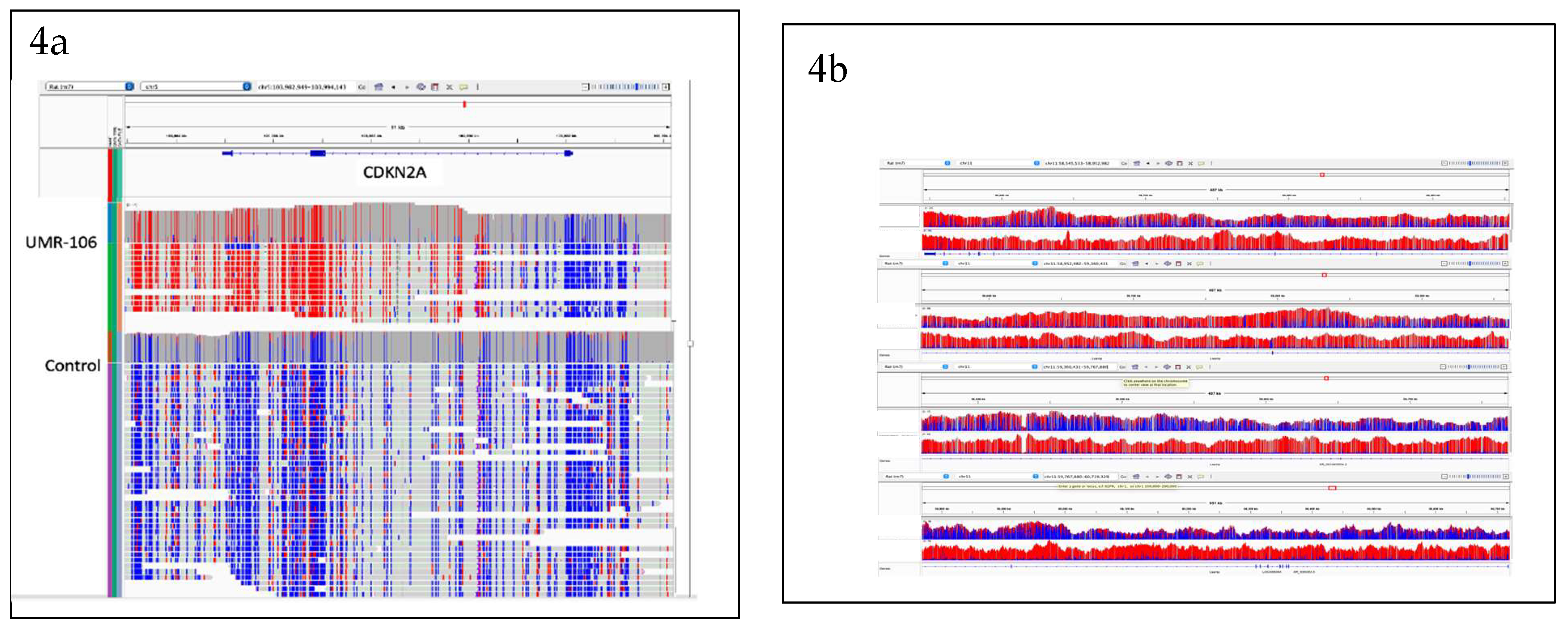
Figure 5.
Complexity of the chromosome 7 amplified region as demonstrated by ONT sequence. .
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



