
Submitted:
05 September 2024
Posted:
05 September 2024
You are already at the latest version
Abstract
The study of the morphology, thermal, and chemical stability of the fluorinated compounds was performed using Becke’s three-parameter hybrid functional approach with the correlation provided by Lee, Yang, and Parr and the cc-pVTZ basis set aiming to design low sensitivity, toxicity, instability, and proneness to decomposition or degradation over a short time high-energy materials. The most stable conformers of the compounds under study were selected based on their total energy. Their thermal and chemical stability was evaluated based on the binding energy per atom, chemical hardness, and softness. The oxygen-fluorine balance is assessed to evaluate the sensitivity of these new materials. The density, detonation pressure, and velocity of the selected conformers were theoretically obtained to reveal the influence of -CF3, -OCF3, and cyclic -O(CF2)nO- fragments on the energetic properties of nitroaromatics as well as their stability and resistance to shock stimuli. The results allow us to predict new multipurpose energetic materials with a good balance between power and stability. Referring to the results obtained, we recommend CF3N2, OCF3N2, C2F6N2, 1CF2N2/O2CF2N2, and 2CF4N2/O2C2F4N2 for practical usage because these compounds possess greater stability compared to tetryl and better explosive properties than TNT.
Keywords:
nitro compounds
; polynitrobenzenes
; fluorinated high-energy materials (F-HEMs)
; energetic properties
; density
; detonation pressure
; detonation velocity
; oxygen balance
; stability
; sensitivity
1. Introduction
The seeking of new high-energy materials (HEMs) which, from one side, correspond to safety and environment-friendly requirements and, on the opposite side, should be powerful, stable, insensitive to mechanical stimuli, and provide large quantities of energy release during intentional detonation [1,2,3] is a long-lasting process. There are various ways to reach the above goal: incorporating nitro, nitramine, or azido groups into the molecular structure; developing completely new compounds; forming salts, etc. Since the 1960s some fluorinated polymers (Viton-A, Teflon, Kel-F) were widely applied in producing of various polymer-bonded explosives (PBX). These polymers were selected as optimal binders, substantially improving the physicochemical and energetic properties of explosive charges [4].
The first fluorine-containing group, which was introduced in energetic materials (HEMs) during structural design, was -C(NO2)2F [5,6,7,8,9,10,11]. Notably, -C(NO2)3 changing in HEMs with -C(NO2)2F or -C(NO2)F2 groups leads to the development of the energetic materials, possessing markedly higher stability and decreased sensitivity to mechanical impact [12,13].
Currently, known perspective energetic fluorinated groups used in the new energetic structures are -C(NO2)2F and -C(NO2)2F2 [14,15,16,17,18,19,20,21], -NF2 and -C(NO2)2NF2, [22,23,24,25,26,27,28,29,30,31], and -SF5 [32,33,34,35,36]. It was obtained that introducing even the simplest fluorine “C-F” group instead of “C-H” sometimes substantially improves practical HEMs properties, i.e. it lowers sensitivity and increases the density of traditional explosives leading to upgrading energetic properties. For example, the sensitivity to shock stimuli of the 3,5-difluoro-2,4,6-trinitroaniline [37] or 3,5-difluoro-2,4,6-trinitrophenol [38], 3,5-difluoro-2,4,6-trinitrotoluene [39], 3,5-difluoro-2,4,6-trinitroanisole [40,41,42] and 1, 2-difluoro-4, 5-dinitrobenzene [43], are lower than that of trinitroaniline, trinitrophenol, trinitrotoluene, trinitro anisole, and dinitrobenzene compound without F. It is because the fluorine introduction improves the oxygen balance of the corresponding energetic material due to their ability to generate HF (from H) and CF4 (from C) [38,43]. The introduction of fluorine promotes an increase in the density and detonation velocity. Chinese authors, Guo et al. [44], exhibit that the energy of the C-F bond (485 kJ/mol ) is higher than that of the C-H bond (413 kJ/mol ), which means that fluorine-containing energetic materials possess in general higher energy. Moreover, C-F bond can stabilize the corresponding energetic material due to halogen and hydrogen bonds. It implies that fluorine-containing materials could be thermally and chemically stable and possess a high amount of energy to be released during detonation.
The fluorinated high-energy materials also have been reported to have a good potential as perspective melt-castable explosives, and energetic plasticizers since fluorination is beneficial for lowing the melting point and increasing the interval between the melting point and thermal decomposition temperature of the oxidative properties of energetic materials [39,43,45].
It is also reported that typical CHNO-containing explosives deliberate mostly carbon dioxide (CO2), carbon monoxide (CO), nitrogen (N2), and water (H2O) after the explosion, while gaseous products of the explosion of fluorine-containing HEMs (F-HEMs) can include hydrogen fluoride (HF), carbon tetrafluoride (CF4), and minor product, carbon oxodifluoride (COF2) [44,46].
The fluorinated HEMs can be divided into separate classes such as -NF2, -C(NO2)2NF2, SF5, etc. based on the type of fluorine-containing functional groups HEM molecule [44]. Previously widely investigated F-HEMs possessed mostly -NF2 [47,48] or C(NO2)2F [49]. However, other F-groups (such as -CF3, etc.) could also improve the energetic properties and stability of HEMs [50,51,52,53,54,55].
Thus, for this reason, we performed an investigation of polynitrobenzene derivatives containing more rare -CF3, -OCF3, and -O(CF2)nO- functional groups aiming to observe their potential to be used as multifunctional energetic materials possessing better energetic properties than that of currently expended, i.e we proposed and investigated a low sensitivity, toxicity, instability, and proneness to decomposition or degradation over a short time high-energy materials. On the other hand, the results of this study allow us to propose ways to increase the explosion properties of high-energy materials along with high resistance to various environmental impacts.
2. Materials and Methods
Let us remind you that the study was performed aiming to develop new energetic materials and predict which of them could be used practically. We do not change our research methodology provided in [56]:
- Design of the compounds and their conformers for the study. The difference between conformers lies in both: i). the position of NO2 and fluorine containing groups; ii). The cis/trans position of -OCF3 group where it is possible.
- Performance of the Berny optimization without any symmetry constraints (all bond lengths, angles, and dihedral angles are changed) to find an equilibrium point. The vibration frequencies analysis was performed to verify that the energy minima were reached. The total energy of the conformers was compared to selecting the most abundant conformers. The stability and energetic properties of the designated conformers were only investigated.
- Evaluation of the binding energy per atom to compare the thermal stability of the compounds under study. This energy indicates the amount of energy required to separate an atom from a system of particles and its larger value shows higher thermal stability.
- Determination of the stability related to the chemical properties and aging of the compounds. HOMO-LUMO gap, chemical hardness, and softness are calculated for this purpose. It is known that compounds with larger HOMO-LUMO gap and chemical hardness are more resistant to undergoing a chemical reaction or to being transformed by an external perturbation, such as an applied electric field. On the other hand, a low chemical softness value denotes a high tendency of the molecule to degrade [57,58].
- Calculation of hardness index to evaluate the stability of the compounds.
To obtain data necessary to evaluate the above-listened parameters, Becke’s three-parameter hybrid functional approach with non-local correlation provided by Lee, Yang, and Parr (B3LYP), and the cc-pVTZ basis set implemented in a GAUSSIAN package was applied [59,60,61,62]. This approach accurately described the geometric and electronic structure of various molecules and their derivatives [63,64,65,66,67,68,69,70,71,72,73,74,75].
The energetic properties, such as detonation velocity and pressure of the compounds under study, were obtained by approaching a low computational demand-required approach created for the CaHbNcOdFe compound. The reliability of the approach used was checked by comparison of the results of simulations of TNT, APATO (3-amino-5-[(2, 4, 6-trinitrophenyl) amino]-1H-1, 2, 4-triazole), and the known fluorinated compounds with experimental ones. We also checked if the conclusions following the results of the investigations coincided with the common properties of the energetic materials such as the relationship of the number of nitro groups and energetic properties [1].
The oxygen/fluorine balance [57] was calculated to evaluate the resistance to impact. We used oxygen-fluorine balance because of the introduction of another oxidant fluorine element [76].
The main obstacle in this research was the calculation of the density. This is due to several issues. First, the density evaluated as the division of molecular weight from the molar volume calculated by the division of molar from additive increments is, in some cases, significantly larger than that obtained experimentally [77]. Second, the experimental data on the density of the CaHbNcOdFe are scarce, i.e. there is no possibility of evaluating the deviations of the simulations. So, the density of the compounds was calculated by using three approaches: one implemented in ACD/ChemSketch based on the Van der Walls volumes molecular modeling program [78], second one, suggested in [79], i.e., the calculated density is corrected by the obtained dependence; iii) the division of molecular weight from the molar volume without any corrections. In the last cases, the molar volume was calculated by B3LYP/cc-pVTZ approach implemented in the Gaussian program. A comparison of the calculated densities revealed that in many cases, the densities obtained by using ACD/ChemSketch approach are significantly larger than those obtained from the approach including density correction, while in another case, the above density is lower. Nevertheless, the density of CF3N3 following experimental measurement is reported as 1.716 - 1.816 g/cm3, while that calculated by ACD/ChemSketch is equal to 1.77 g/m3 [80]. So, considering the dependence of the energetic parameters on the density of the compound as well as the goal of our investigations, below we presented the energetic parameters obtained with the values of ACD/ChemSketch density of the compounds to avoid unreliable conclusions. However, the possibility to exhibit the dependence of energetic properties on the conformation of compounds is lost. Thus, the density obtained from one of the rest mentioned methods is presented to show that the density of the conformers is different and leads to a variety of energetic properties and difficulties in producing certain materials.
We obtained detonation velocity and pressure to predict the energetic properties of the compound investigated to evaluate the energetic properties of the compounds under study. The detonation velocity was calculated by approaching the equation and pressure presented in [81] and [82].
These approaches are suitable for compounds consisting of F and as exhibited below, give reliable results.
3. Results
The views of the selected compounds are depicted in Appendix 1. The thermal and chemical stability of the compounds under study were evaluated based on the binding energy per atom, HOMO -LUMO gap, chemical hardness, electronegativity, and hardness index calculation. These parameters are presented in Table 1.

Table 1.
The binding energy per atom (BEA), HOMO -LUMO gap (G), chemical hardness (H), chemical softness (S)), and hardness index (Y) evaluated by B3LYP/cc-pVTZ approach.
Table 1.
The binding energy per atom (BEA), HOMO -LUMO gap (G), chemical hardness (H), chemical softness (S)), and hardness index (Y) evaluated by B3LYP/cc-pVTZ approach.
| Compound | BDE, eV | G, eV | H,eV | S, eV | Y |
|---|---|---|---|---|---|
| CF3N2 | 6.00 | 4.94 | 2.47 | 0.20 | 0.92 |
| CF3N3 | 5.43 | 4.73 | 2.36 | 0.21 | 0.91 |
| C2F6N2 | 5.42 | 5.24 | 2.62 | 0.19 | 0.93 |
| C2F6N3 | 5.38 | 5.02 | 2.51 | 0.20 | 0.92 |
| C3F9N2a | 5.39 | 5.16 | -2.58 | 0.19 | 0.92 |
| C3F9N2b | 5.39 | 5.46 | 2.73 | 0.18 | 0.93 |
| C3F9N3 | 5.35 | 5.01 | 2.50 | 0.20 | 0.92 |
| OCF3N2/CF3ON2 | 5.41 | 5.10 | 2.55 | 0.20 | 0.92 |
| OCF3N3/CF3ON3 | 5.37 | 4.94 | 2.47 | 0.20 | 0.92 |
| 1O2C2F6N2a | 5.33 | 4.65 | 2.33 | 0.21 | 0.91 |
| 1O2C2F6N2b | 5.33 | 4.95 | 2.47 | 0.20 | 0.92 |
| 1O3C3F9N2b | 5.27 | 5.14 | 2.57 | 0.19 | 0.92 |
| 1O2C2F6N3a | 5.30 | 4.96 | 2.48 | 0.20 | 0.92 |
| 1O2C2F6N3b | 5.30 | 4.95 | 2.47 | 0.20 | 0.92 |
| 1O2C2F6N4a | 5.27 | 4.85 | 2.43 | 0.21 | 0.92 |
| 1O2C2F6N4b | 5.27 | 4.85 | 2.43 | 0.21 | 0.92 |
| 1O3C3F9N3a | 5.24 | 5.22 | 2.61 | 0.19 | 0.93 |
| 1O3C3F9N3b | 5.24 | 5.23 | 2.62 | 0.19 | 0.93 |
| 1CF2N2/1O2CF2N2 | 5.61 | 4.65 | 2.33 | 0.22 | 0.91 |
| 1CF2N3/1O2CF2N3 | 5.68 | 4.00 | 2.00 | 0.25 | 0.87 |
| 1CF2N4/1O2CF2N4 | 5.48 | 4.48 | 2.24 | 0.22 | 0.90 |
| 2CF4N2/1O2C2F4N2 | 5.66 | 4.73 | 2.37 | 0.21 | 0.91 |
| 12CF4N3 | 5.47 | 4.77 | 2.39 | 0.21 | 0.91 |
| 2CF4N4//O2C2F4N4 | 5.42 | 4.89 | 2.44 | 0.20 | 0.92 |
| 3CF6N2/O2C3F6N2 | 5.43 | 4.93 | 2.46 | 0.20 | 0.92 |
| 3CF6N3 | 5.53 | 4.46 | 2.23 | 0.22 | 0.90 |
| 3CF6N4/1O2C3F6N4 | 5.36 | 4.99 | 2.50 | 0.20 | 0.92 |
| TNT | 5.52 | 4.15 | 2.07 | 0.24 | 0.88 |
| APATO* | 5.70 | 3.21 | 1.60 | 0.31 | 0.81 |
1 There and below a and b letters mark the conformers of the compounds under study. The difference between conformers relies on cis- and trans–position F containing groups or their different positions.
We would like to pay attention to the fact that APATO is not used as a standard to evaluate the energetic properties of new materials. It is included to show the validity of the approach for the simulations, i.e. the parameters of this compound obtained by well-known and widely used equations and that presented here coincide well.
The calculated densities are presented in Table 2. As mentioned above, the density was calculated by approaching the ChemSkech program and as the division of molecular weight from the molar volume obtained by B3LYP/cc-pVTZ approach implemented in the Gaussian09 program package.
Table 2.
The densities obtained by the approach implemented in the ChemSkech program (ACD, ρACD) and as the division of molecular weight from the molar volume obtained (Gaussian ρ).
Table 2.
The densities obtained by the approach implemented in the ChemSkech program (ACD, ρACD) and as the division of molecular weight from the molar volume obtained (Gaussian ρ).
| Compounds | ACD ρ ACD , g/cm3 |
Gaussian ρ , g/cm3 |
|---|---|---|
| CF3N3 | 1.77 | 2.07 |
| C2F6N2 | 1.69 | 2.07 |
| C2F6N3 | 1.82 | 2.34 |
| C3F9N2a | 1.74 | 2.09 |
| C3F9N2b | 1.74 | 1.96 |
| C3F9N3 | 1.85 | 2.01 |
| OCF3N2/CF3ON2 | 1.64 | 1.93 |
| OCF3N3/CF3ON3 | 1.80 | 2.11 |
| O2C2F6N2a | 1.74 | 2.42 |
| O2C2F6N2b | 1.73 | 1.99 |
| O3C3F9N2b | 1.79 | 2.11 |
| O2C2F6N3a | 1.85 | 1.98 |
| O2C2F6N3b | 1.85 | 2.01 |
| O2C2F6N4a | 1.96 | 2.14 |
| O2C2F6N4b | 1.96 | 2.14 |
| O3C3F9N3b | 1.89 | 2.13 |
| O3C3F9N3a | 1.89 | 2.05 |
| 1CF2N2/O2CF2N2 | 1.84 | 1.83 |
| 1CF2N3/O2CF2N3 | 1.95 | 1.80 |
| 1CF2N4/O2CF2N4 | 2.05 | 2.23 |
| 2CF4N2/O2C2F4N2 | 1.85 | 2.09 |
| 2CF4N3/O2C2F4N3 | 1.98 | 1.81 |
| 2CF4N4//O2C2F4N4 | 2.01 | 1.89 |
| 3CF6N2/O2C3F6N2 | 1.85 | 2.19 |
| 3CF6N3/O2C3F6N3 | 1.97 | 2.42 |
| 3*0CF6N4/O2C3F6N4 | 1.98 | 2.34 |
Indeed, in many cases, the densities of the compounds under study obtained by the approaches applied are higher than 1.8 g/cm3, coinciding with the results obtained by L. Wen et al [80]. However, in many cases, the density ρ is significantly higher than that ρACD, although ρ exhibits the influence of compound morphology on the density. Considering good agreement between the density of CF3N3 obtained theoretically ( 1.77 g/cm3) and experimentally(1.716 - 1.816 g/cm3), the energetic parameters are calculated with smaller values of ρACD aiming not to overestimate the energetic properties of the compounds under study. On the other hand, it is very well known that the density could vary depending on its morphology (variety of conformers) and physical forms. Thus, the densities presented in Table 2 could represent the highest and lowest densities of the compounds. We would like to point out, that we do not evaluate precise values of detonation pressure and velocity but exhibit the case of usage of the same approach, one can ensure that the statistical errors in the densities, detonation velocities, and pressure for each molecule will be similar. This allows us to compare the detonation velocities of the compounds studied here, subsequently rank those molecules, and make qualitative conclusions concerning the influence of substitutions on the energetic properties of the compounds under study. Going ahead, we would like to mention that the values of detonation pressure and velocity of the TNT and APATO obtained from the methodology described in this paper coincide well with experimental measures which indicate its reliability.
The oxygen/fluorine balance is calculated to evaluate the resistance to shock stimuli. The results of this simulation along with the dependence of the sensitivity on the number of nitro groups are presented in Fig 1. It is seen that increasing the nitro group in the compounds with F leads to lower resistance to shock stimuli. These results coincide with the previously obtained tendencies for the development of safe energetic materials [1,12,14,19,44] and indicate that some proposed materials could be addressed to high-safety HEMs.
Figure 2.
The oxygen balance and its dependence on the number of nitro groups in the compounds.
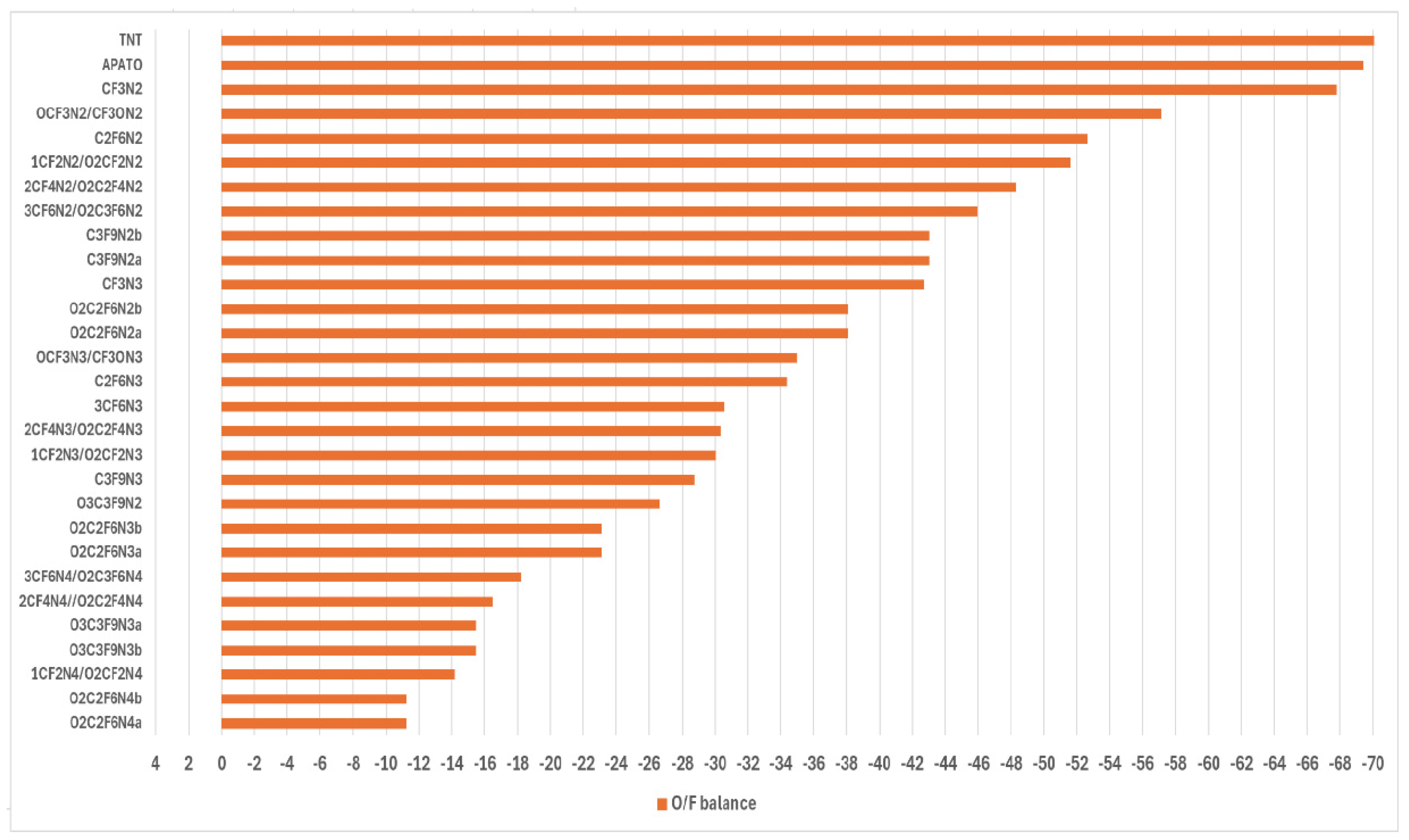
Table 2.
The detonation velocity (v) and detonation pressure (p) calculated by using equations presented by M. H. Keshavarz [82,83].
| Compounds | v,km/s | p,kba |
|---|---|---|
| CF3N2 | 7.01 | 189.13 |
| CF3N3 | 7.46 | 308.45 |
| C2F6N2 | 7.31 | 348.83 |
| C2F6N3 | 7.96 | 385.51 |
| C3F9N2a | 7.83 | 432.39 |
| C3F9N2b | 7.84 | 432.63 |
| C3F9N3 | 8.40 | 459.75 |
| OCF3N2/CF3ON2 | 6.91 | 276.32 |
| OCF3N3/CF3ON3 | 7.63 | 322.50 |
| O2C2F6N2a | 7.66 | 383.27 |
| O2C2F6N2b | 7.66 | 371.38 |
| O3C3F9N2b | 8.27 | 458.16 |
| O2C2F6N3a | 8.26 | 452.77 |
| O2C2F6N3b | 8.26 | 408.54 |
| O2C2F6N4a | 8.82 | 483.42 |
| O2C2F6N4b | 8.82 | 447.03 |
| O3C3F9N3b | 8.78 | 448.03 |
| O3C3F9N3a | 8.78 | 449.03 |
| 1CF2N2/O2CF2N2 | 7.34 | 329.08 |
| 1CF2N3/O2CF2N3 | 8.16 | 363.76 |
| 1CF2N4/O2CF2N4 | 8.55 | 400.03 |
| 2CF4N2/O2C2F4N2 | 7.69 | 386.62 |
| 2CF4N3/O2C2F4N3 | 8.36 | 430.63 |
| 2CF4N4//O2C2F4N4 | 8.70 | 436.06 |
| 3CF6N2/O2C3F6N2 | 7.97 | 437.87 |
| 3CF6N3 | 8.60 | 478.82 |
| 3CF6N4/O2C3F6N4 | 8.84 | 471.75 |
| TNT | 6.74 | 172.02 |
| APATO | 7.67 | 253.06 |
We pointed out the coincidence between the experimentally obtained detonation velocity of 6.9 km/s TNT and the 6.8 km/s obtained by us [83]. The calculated value of detonation pressure of 217.15 kbar of this compound fits the reported 210 kbar at 1.63 g/cm3 density in a solid which is used as a standard [85]. Moreover, the detonation velocity of CF3N3 obtained experimentally varies from 6.919 km/s to 8.029 km/s, while that of 7.46 km/s obtained by us coincides with these results. This coincidence proves that the approaches and methodology used give trustworthy results.
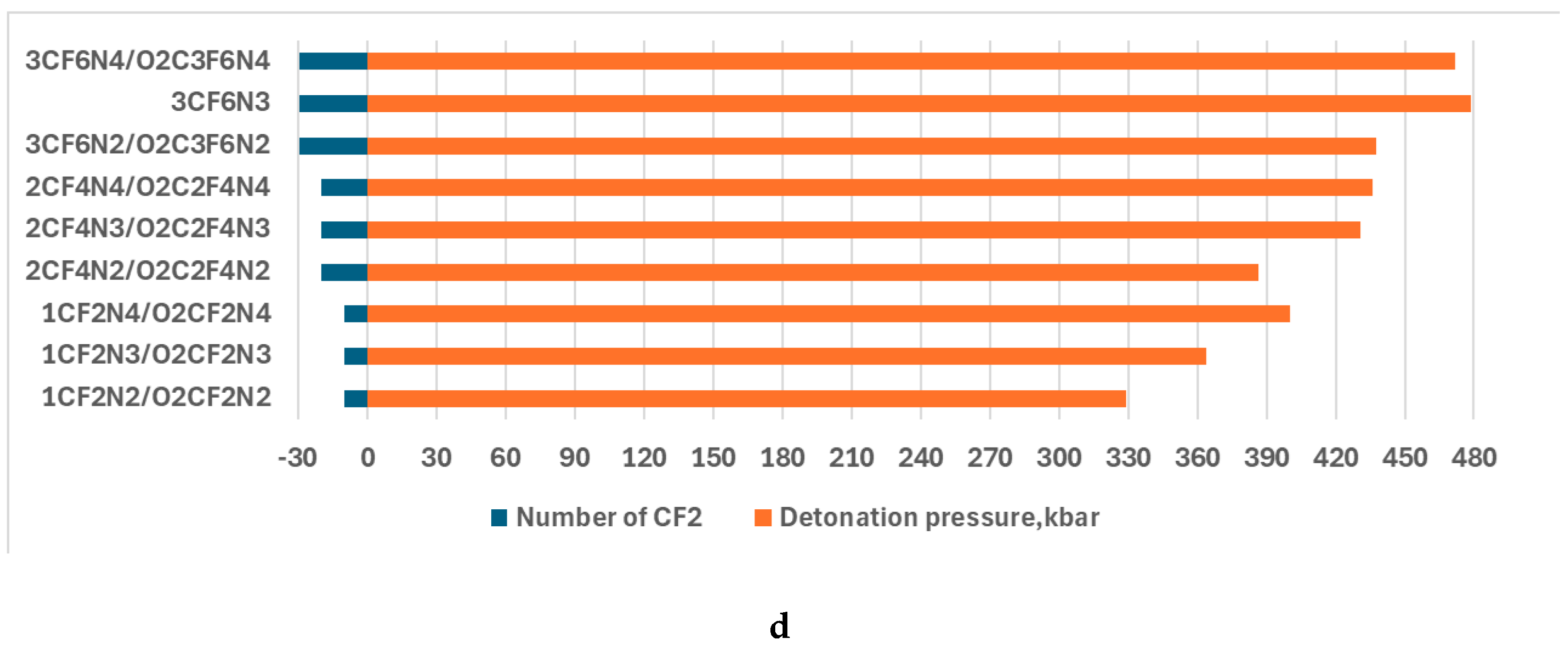
The detonation velocity and detonation process are depicted in Figs. 3 and 4 to exhibit the parameter dependence on the number of nitro, -CF3, and -OCF3 groups as well as cycling O2(CF2)n.
Figure 3.
The detonation velocity dependence on the number of -NO2 group (a), -CF3 (b), -OCF3 (c), and that of CF2 in the -O-(CF2)n-O- (d).
Figure 3.
The detonation velocity dependence on the number of -NO2 group (a), -CF3 (b), -OCF3 (c), and that of CF2 in the -O-(CF2)n-O- (d).
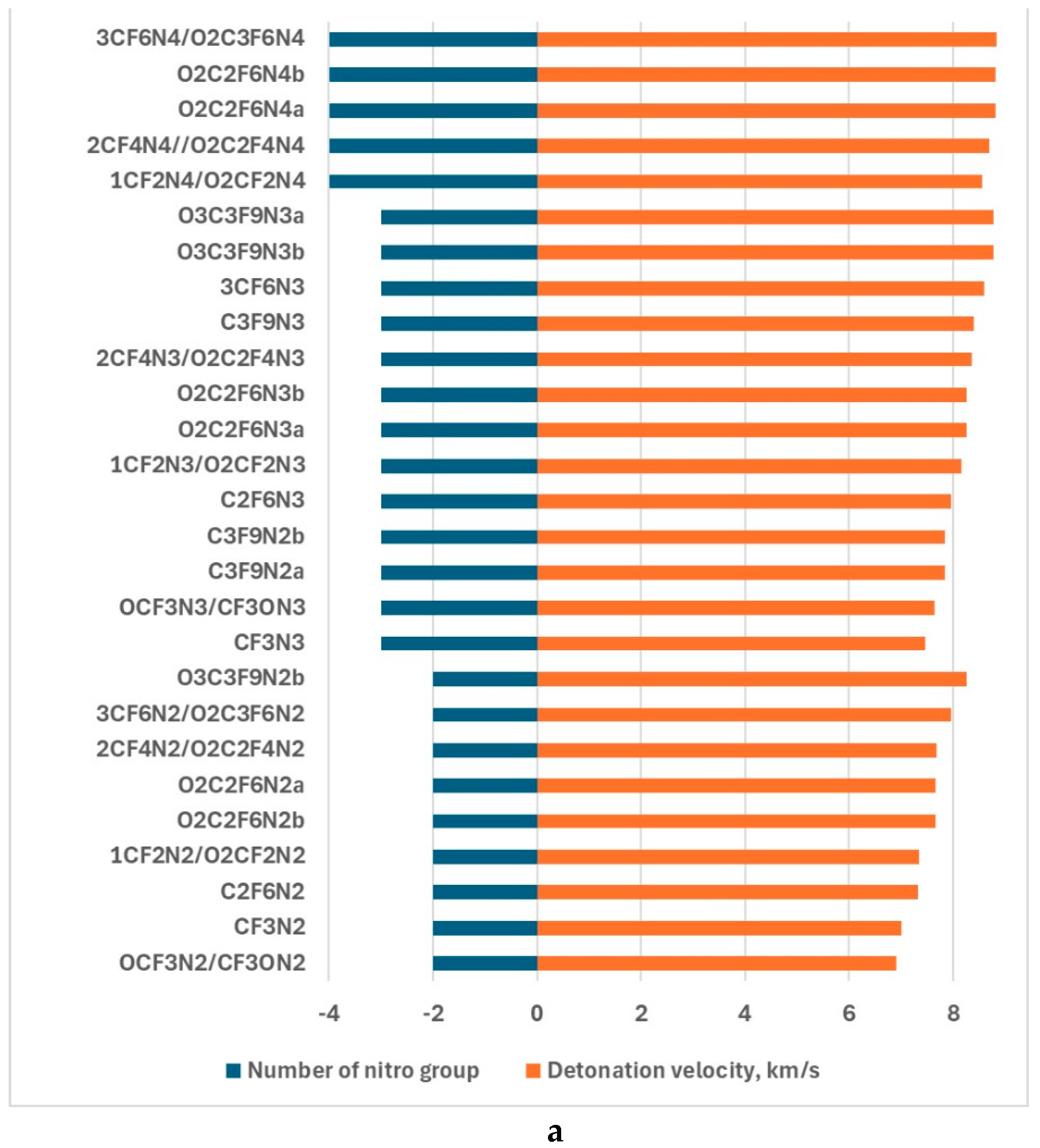
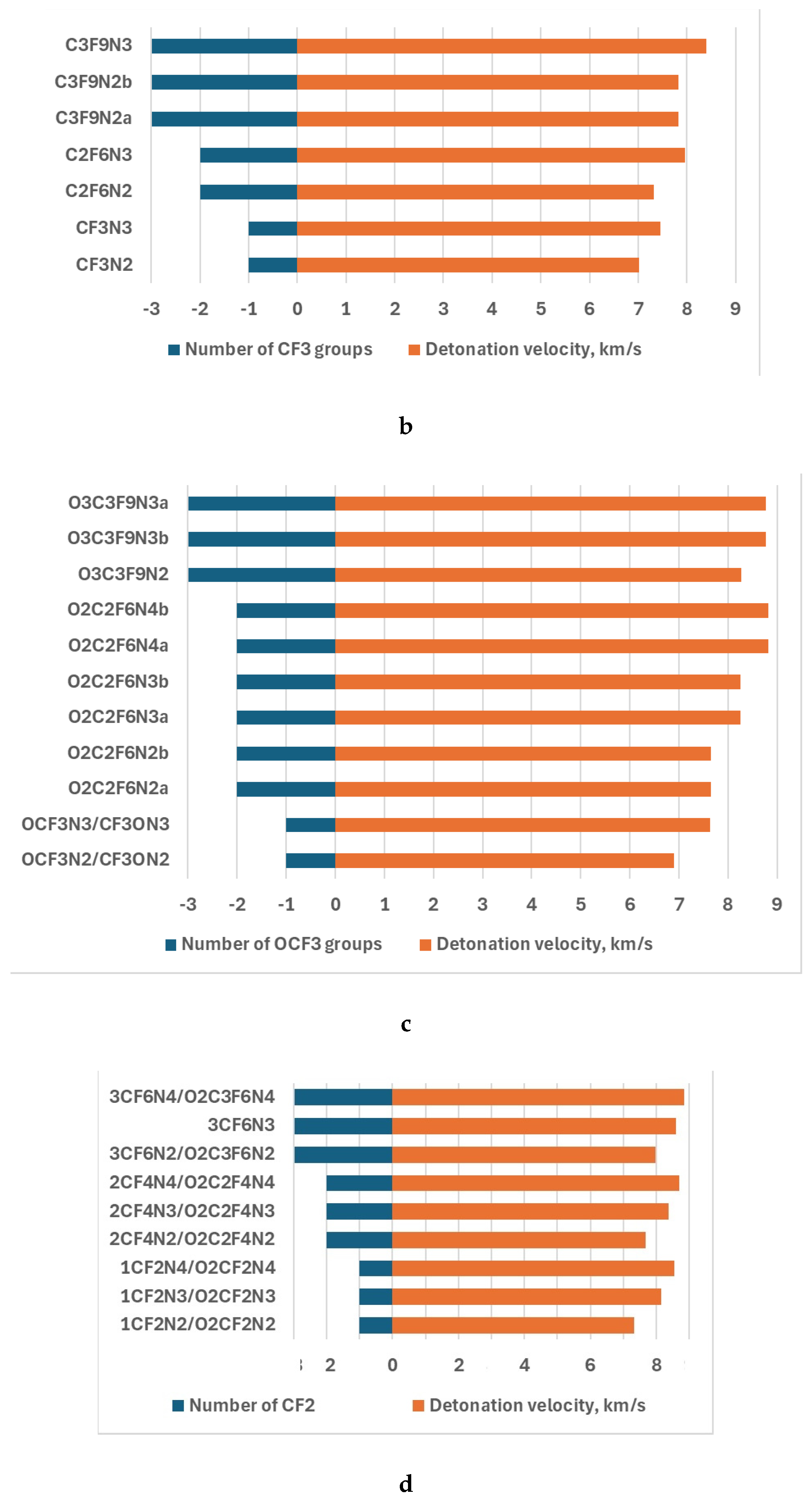
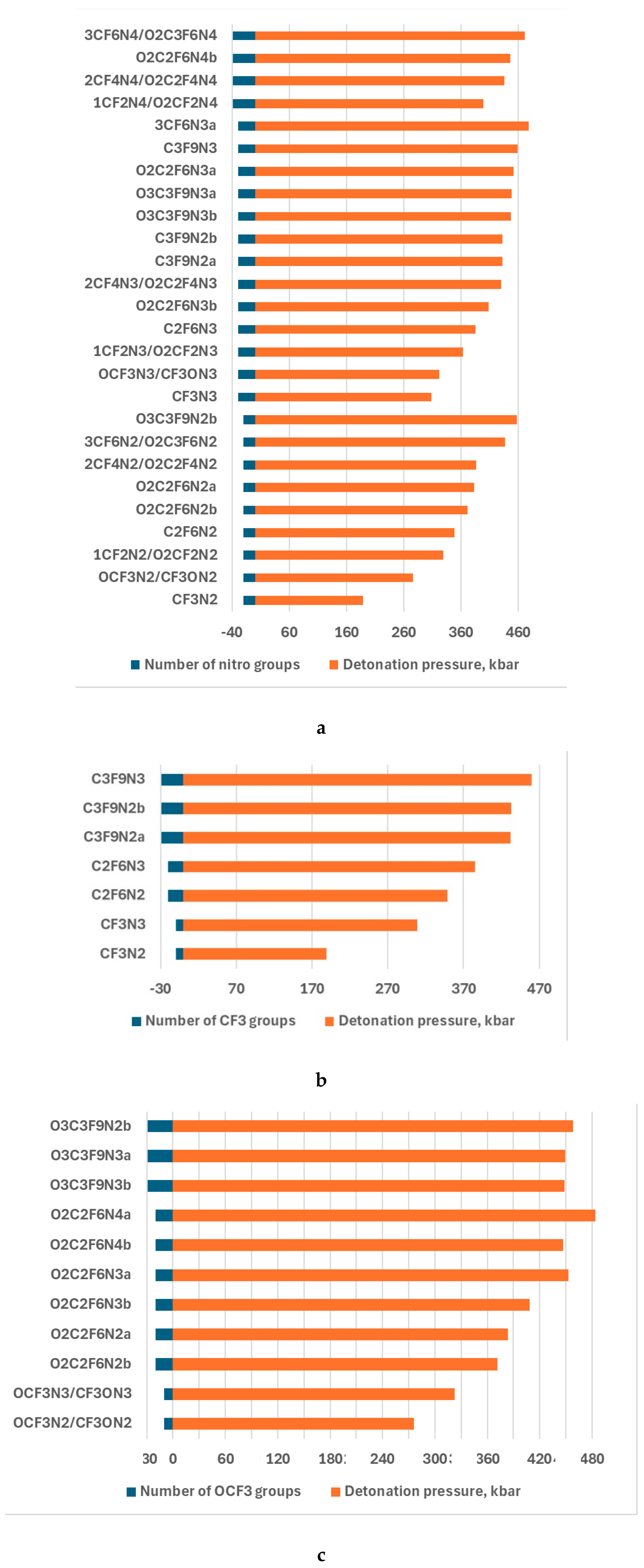
Figure 4.
The detonation pressure dependence on -NO2 group (a), -CF3 (b), -OCF3 (c), and that of CF2 in the -O-(CF2)n-O- ( (d). The numbers of the groups and cycle size are enlarged by 10 for better viewing.
Figure 4.
The detonation pressure dependence on -NO2 group (a), -CF3 (b), -OCF3 (c), and that of CF2 in the -O-(CF2)n-O- ( (d). The numbers of the groups and cycle size are enlarged by 10 for better viewing.
4. Discussion
The evaluation of the stability of the energetic materials is performed by using different methodologies. The investigation of thermal stability could be based on the comparison of the total energy of the conformers or binding energy per atom (BDE) when the stability of different chemical composition compounds is investigated. The chemical stability could be predicted based on the electronic structure, i.e. the HOMO-LUMO gap, and the parameters, such as chemical softness and hardness, etc. The study of the compound behavior under different conditions also gives insight into thermal stability, while calculations of Gibbs Free energy, enthalpy, and entropy are essential to predict different decomposition reactions. We emphasize that at this stage of the research, there is no aim to predict the products of the decomposition of the energetic materials, thus we limit ourselves to the studies of total energies and electronic structure. So, we calculated the Binding energy per atom (BDE), HOMO–LUMO gap, chemical hardness, and softness. It is known that compounds with a larger HOMO–LUMO gap and chemical hardness are more resistant to undergoing chemical reactions or to being transformed by an external perturbation, such as an applied electric field. On the other hand, a low chemical softness value denotes a high tendency of the molecule to degrade [76,77]. The values BDA shed some light on the thermal stability of the compounds.
We would like to pay attention to the values of the hardness indexes that are larger than 0.9 indicating high thermal and chemical stability. These indexes of TNT and APATO calculated by the same approach are equal to 0.88 and 0.81 exhibiting their slightly lower stability than that of the compounds under study (Table 1). The higher chemical stability of the compounds under study than that of TNT and APATO is also confirmed by other parameters presented in Table 1, although the thermal stability and resistance to degradation of APATO could be higher and comparable with that of TNT. Overall, we may predict that the new high-energy materials could be more stable thermally or/and chemically than the above-mentioned known ones.
Referring to BDE, we may state that compounds consisting of 1CF2N3/O2CF2N3 and 1CF2N2/O2CF2N2 are slightly more thermally stable than TNT, but less stable than APATO. The thermal resistance of CF3N2 is higher than that of APATO.
The high chemical stability of the compounds under study represents values of HOMO-LUMO gaps as well as chemical hardness (Table 1). The chemical stability of 1CF2N4/O2CF2N4 is higher than that of APATO and lower than TNT, while the rest compounds considering our results are more chemically stable than TNT. The analysis of chemical hardness and softness confirms these findings. Additionally, our study reveals, that increasing nitro groups in the compounds does not significantly influence the thermal stability, or the resistance to degradation, and that undergoing chemical reactions to being transformed by an external perturbation of the compound consisting of fluorine.
The increase of the -CF3 or -OCF3 groups as well as the inclusion of -CF2 in the -O-(CF2)n- cycle does not significantly influence the chemical and thermal stability of the compound of the study – the parameters presented in Table 1 are comparable to the results presented [84,85,86,87,88,89].
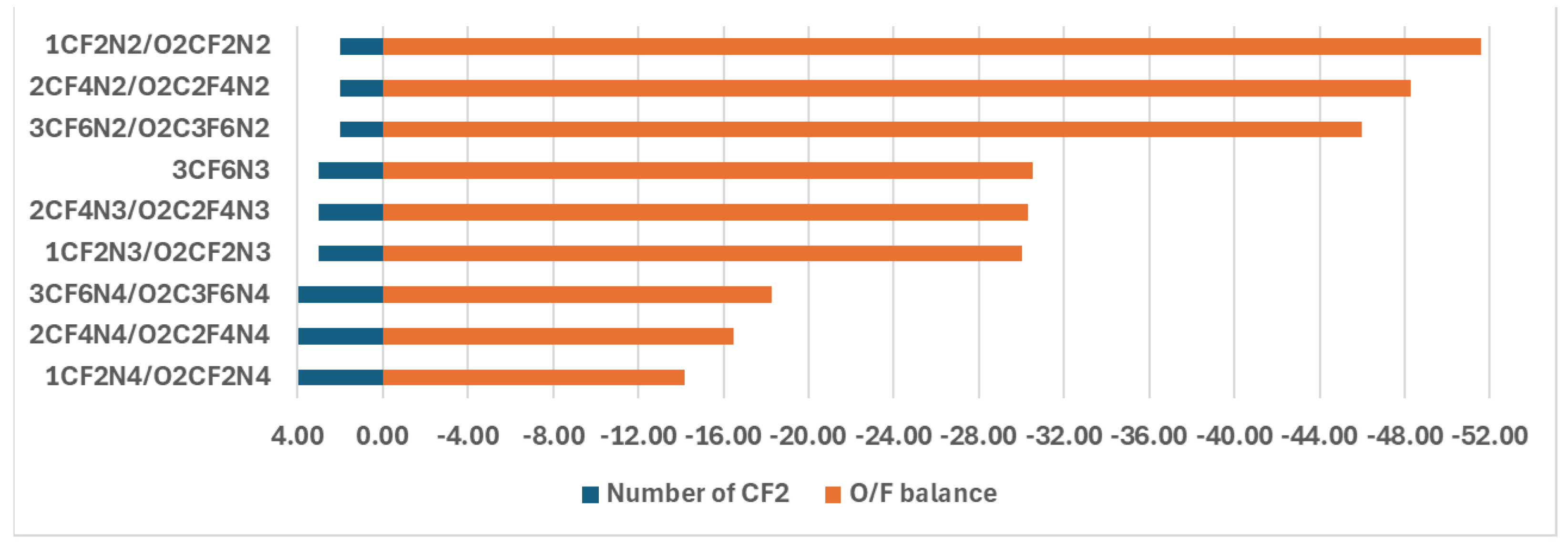
The investigation of the energetic properties exhibited significant effects of the nitro groups on the energetic properties of the compounds under study. The -CF3, or -OCF3 groups as well as -O-(CF2)n-O cycle also change the energetic properties of the compounds under study, but their influence is nitro group number dependent. For example, the enlargement of the above cycle due to the inclusion of CF2 leads to an increase in the sensitivity to the shock stimuli of this group materials under study when the nitro group in the compound is two (Fig. 5). The opposite effect is obtained for the compounds with four nitro groups. In this case, the inclusion of -CF2 leads to a decrease in this sensitivity. When this type of compound consists of three nitro groups, the above enlargement does not change the sensitivity to shock stimuli (Fig. 5).
Figure 5.
The O/F balance dependence on the CF2 in the -O-(CF2)n-O- cycle.
The increase in -CF3 or -OCF3 number in the compounds with the same nitro group number also leads to a decrease in the resistance to shock stimuli (Fig. 2). Moreover, the compounds with -OCF3 are more sensitive to shock than those with CF3 when the nitro group number in the compound is the same. To exhibit this conclusion evidently, we present O/F balance separately in Table 3.
Table 3.
The O/F balance dependence on OCF3 and CF3 and nitro group number.
| N | m | OnCnF3nNm | CnF3nNm |
|---|---|---|---|
| 1 | -57.12 | -67.77 | |
| 2 | 2 | -38.08 | -52.61 |
| 3 | -26.66 | -43.00 | |
| 1 | -35.00 | -42.69 | |
| 2 | 3 | -23.09 | -34.37 |
| 3 | -15.48 | -28.77 |
Hence, we obtain that increasing the nitro group leads to lower resistance to shock stimuli which indicates the results of the analysis of the O/F balance (OB) ( Fg.1). The increase of CF3 or -OCF3 number also leads to an increase of the sensitivity to stimuli. However, there is no clear influence of the -O-(CF2)n-O- cycle on the sensitivity of shock stimuli.
The oxygen balance of TNT, a standard reference for many purposes, is equal to −74%, and that of tetryl is −47.36%. Referring to our results of simulations, the O balance of TNT is -72.05% and that of APATO is -68.30%. The O/F balance of many of the compounds is higher than -47%. It means that their resistance to stimuli is lower than that of tetryl. Nevertheless, the above parameters of 1CF2N2/O2CF2N2, 2CF4N2/O2C2F4N2, C2F6N2, OCF3N2, and CF3N2 are lower than tetryl, but higher APATO. They can be ranged considered increased resistance to stimuli followingly
TNT<APATO<CF3N2<OCF3N2<C2F6N2<1CF2N2<2CF4N2<Tetryl
The detonation velocity of the compounds under study varies from 6.67 to 8.84 km/s thus they could be addressed to the high explosives whose detonation velocity lies in the range 1,01 km/s to 9.89 km/s. The detonation velocity of OCF3N2/CF3ON2 is lower than the experimentally obtained detonation velocity of 6.9 km/s of TNT [83]. So, only this compound's energetic properties are worse than TNT. On the other hand detonation velocity of the C3F9N2, C2F6N3, 1CF2N3mp/O2CF2N3, O2C2F6N3, 2CF4N3/O2C2F4N3, C3F9N3, 3CF6N, O3C3F9N3, 1CF2N4/O2CF2N4, 2CF4N4/O2C2F4N4, O2C2F6N4, and 3CF6N4/O2C3F6N4 are larger than that of APATO indicating better energetic properties of them.
The investigation of the dependence of the detonation velocity on the number of the -NO2, -CF3,-OCF3, and -O-(CF2)n-O-cycle revealed some interesting features (Fig. 3). The enlargement of the above cycle leads to increasing the detonation velocity of this type compounds under study with the same number of nitro groups. Similar results are obtained in the case of the increase of -OCF3 and -CF3 numbers. Comparison of the detonation velocity of the same type of compounds with different nitro and fluorine-containing groups allows us to predict that the influence of the nitro group competes with that of the F number, i.e. only a certain proportion of various components leads to significantly improving energetic properties. For example, the detonation velocity of CF3N2 and CF3N3 is equal to 7.01 km/s and 7.46 km/s respectively. These compounds consist of different numbers of nitro groups, while the -CF3 number is the same. So, the improvement in the energetic properties is related to the nitro group inclusion. In the case when the nitro group number is the same, the detonation velocity of 7.96 km/s of C2F6N3 is larger than that of CF3N3, which exhibited improving energetic properties due to the bonding of -CF3. The detonation velocity of C3F9N3 formed due to C2F6N3 joining of -CF3, also increased to 8.40 km/s. This value of the detonation velocity is the largest for this type of compound and allows us to speculate that the nitro group/-CF3 rate could be properly chosen to improve energetic properties.
A similar tendency takes place in the case of -OCF3 and -O-(CF2)n-O- cycle. The largest values of detonation velocity are 8.82 and 8.84 of O2C2F6N4 and 3CF6N4/O2C3F6N4 respectively when the nitro/fluorine group rate is the largest. The results of the study of the detonation pressure exhibited the worse energetic properties of CF3N2 than APATO, and better than TNT. That of the rest compounds is better even than the properties of APATO (Fig.4). The results followed from the analysis of the detonation pressure confirm that.
The importance of the rate of nitro/fluorine groups for the energetic properties of the fluorine-containing compounds was followed by the results of the investigation of detonation velocity. The study of the detonation presser supports this finding. For example, the largest detonation pressure of 483,42 is O2C2F6N4a, consisting of four nitro and two -OCF3 groups. Adding -OCF3 and -CF2 in the -O-(CF2)n-O- ring is more effective than -CF3 for energetic properties improvement. For example, the detonation pressure of 348.83 km/s, 383.27 km/s or 371.38 km/s, and 386.62 km/s possesses C2F6N2, O2C2F6N2a/b, and 2CF4N2/O2C2F4N2, respectively.
We also considered that the steric hindrance leads to density increase and improvement of energetic performances ( Fig. 6).
Figure 6.
The detonation pressure of the selected conformers to exhibit the importance of the steric hindrance.
Figure 6.
The detonation pressure of the selected conformers to exhibit the importance of the steric hindrance.
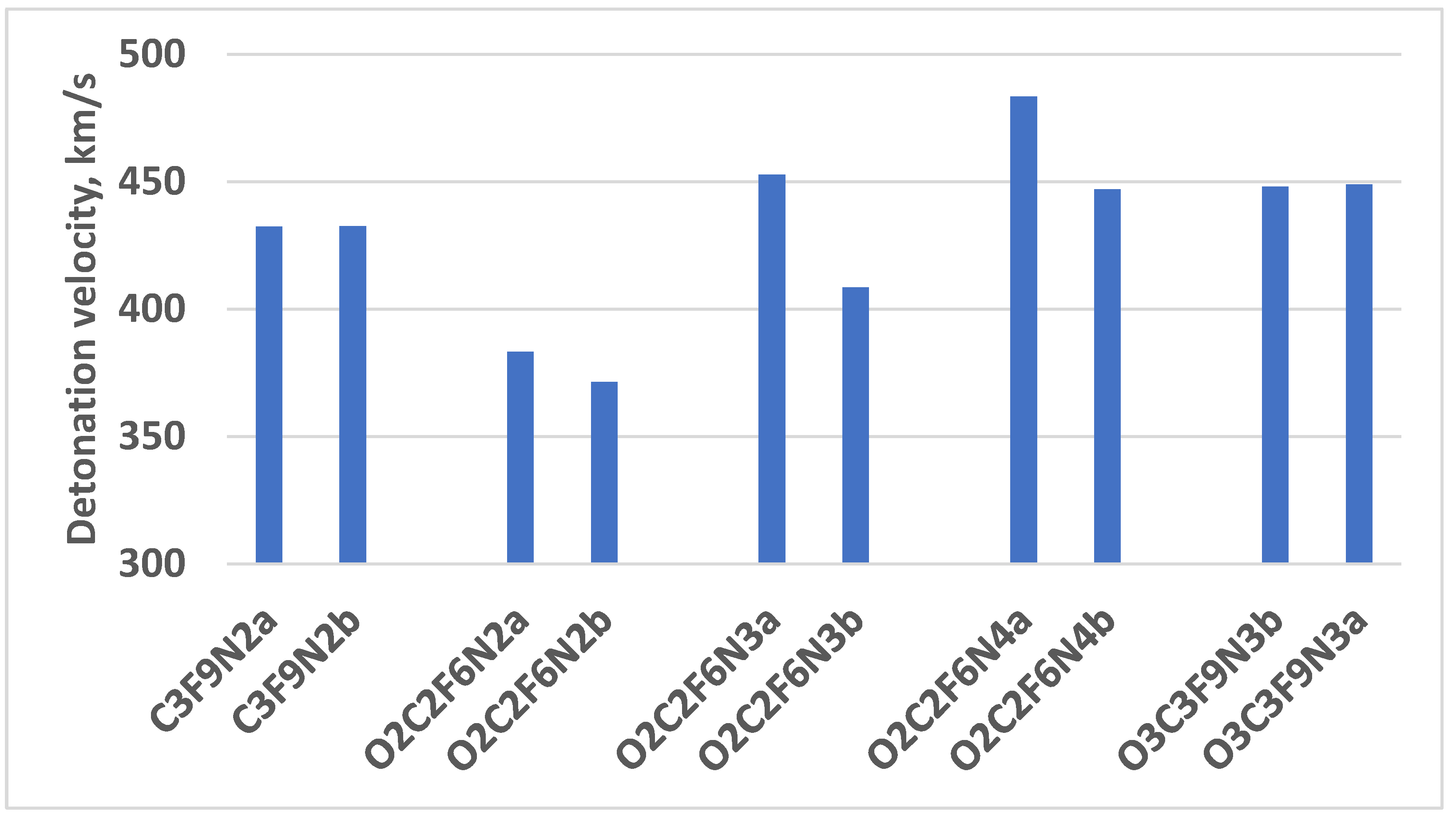
The last statement is based on the comparison of the detonation pressure of compounds which is conformer morphology dependent. The detonation pressure of the conformers, whose morphology is different only due to the position of the -CF3 group in the core of the molecule, is unlike by 0.1 % (Appendix B). The parameters of the cis and trans conformers (O2C2F6N2, O2C2F6N3, and O2C2F6N4) are distinct 3.2 %-10.8 %. The cis and trans conformers of O3C3F9N3 are also mentioned as the most stable compounds, but their detonation pressure is dissimilar only by 0.2%. It is because this compound consists of three -OCF3 that are out of the plane of the core molecule, thus the trans or cis position of one of them does not significantly influence the volume of this molecule, i.e. the density of the compound as well as the detonation pressure. To prove the above statements, we present the molar volume of the above-mentioned conformers (Table 4). It is also reflected by the different densities of the conformers presented in Table 2.
Table 4.
The molar volume of the conformers to point out the reason for discrimination of their energetic properties.
Table 4.
The molar volume of the conformers to point out the reason for discrimination of their energetic properties.
| Compounds | Conformer a | Conformer, b | |
| cm3/mol | cm3/mol | ||
| C3F9N2 | 177.79 | 190.26 | |
| O2C2F6N2 | 167.02 | 174.62 | |
| O2C2F6N3 | 203.04 | 189.67 | |
| O2C2F6N4 | 166.04 | 198.60 | |
| O3C3F9N3 | 217.75 | 226.03 | |
As mentioned above the detonation pressure of the compounds under study is better than that of TNT and, in many cases, APATO. Considering resistance to shock stimuli and the energetic properties of the compound under study, we concluded that CF3N2, OCF3N2, C2F6N2, 1CF2N2m/O2CF2N2, and 2CF4N2m/O2C2F4N2 could be practically used. These findings indicate the indirect advantage of -OCF3 and adding of -CF2 in the -O-(CF2)n-O- cycle* for –CF3.
5. Conclusions
Our research provides a theoretical foundation for developing advanced, highly energetic, and stable fluorine-containing materials. We focused on the -O(CF2)nO- cycle,-OCF3, and -CF3 as scarce groups investigated whose presence in the compounds indicates a way to improve the energetic properties of the known materials.
Our study has demonstrated that the incorporation of cyclic -O(CF2)nO-, -OCF3, and -CF3 groups in fluorine-containing compounds significantly enhances their energetic properties. The investigation revealed that the positioning and morphology of these groups play a crucial role in determining the compounds' thermal and chemical stability as well as energetic properties.
Our findings indicate that compounds under study are overall stabler than TNT, although only 1CF2N3/O2CF2N3 and 1CF2N2/O2CF2N2 exhibit slightly higher thermal stability compared to TNT. However, only 1CF2N4/O2CF2N4 compounds demonstrate lower chemical stability than TNT, while other compounds surpass TNT in that. Interestingly, increasing the number of nitro groups along with the enlargement of -CF3 or -OCF3 groups, and with the inclusion of -CF2 in the -O(CF2)nO- cycle in the compounds does not markedly affect their thermal stability or resistance to degradation. Nonetheless, the nitro group number substantially impacts the energetic properties, with -CF3, -OCF3, and the -O(CF2)nO- cycle contributing variably depending on their quantity.
Referring to the results of the O/F balance investigation, we found compounds with -OCF3 groups more shock-sensitive than those with CF3 when the nitro group count is identical. An increase in CF3 or -OCF3 groups also heightens sensitivity to stimuli, although the -O(CF2)nO- cycle's influence on shock sensitivity remains inconclusive. However, 1CF2N2/O2CF2N2, 2CF4N2/O2C2F4N2, C2F6N2, OCF3N2, and CF3N2 exhibit resistance to stimuli lower than TNT, but higher than that tetryl,
Furthermore, comparing the detonation velocity of compounds with varying nitro and fluorine-containing groups revealed that the energetic properties improve significantly with the optimal proportion of these components. Notably, the addition of -OCF3 and -CF2 in the -O(CF2)nO- ring proved more effective in enhancing energetic performance than -CF3. This improvement is attributed to steric hindrance, which increases density and, consequently, energetic efficiency.
In summary, the compounds CF3N2, OCF3N2, C2F6N2, 1CF2N2/O2CF2N2, and 2CF4N2/O2C2F4N2 demonstrate potential for practical application, highlighting the advantages of -OCF3 and the incorporation of -CF2 in the -O(CF2)nO- cycle over -CF3.
Author Contributions
Conceptualization, J.T. and J.S.; methodology, J.T., and J.S; formal analysis, J.T, and J.S.; investigation, J.T. and J.S.; writing—original draft preparation, J.T. and J.S.; writing—review and editing, J.T. and J.S All authors have read and agreed to the published version of the manuscript
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available on request from the corresponding author due to access restriction.
Acknowledgments
The numerical calculations with the GAUSSIAN09 package were performed using the resources of the Information Technology Research Center of Vilnius University and the supercomputer "VU HPC" of Vilnius University in the Faculty of Physics location.
Conflicts of Interest
The funders had no role in the design of the study: in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Appendix A
Table A1.
The views of the compounds are defined as the most stable between developed and investigated.
Table A1.
The views of the compounds are defined as the most stable between developed and investigated.
| No. | Structural formula | Compound Abbreviation |
|---|---|---|
| 1. | 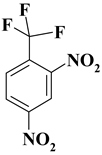 |
CF3N2 |
| 2. | 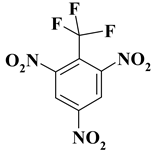 |
CF3N3 |
| 3. | 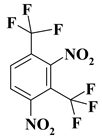 |
C2F6N2 |
| 4. | 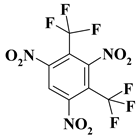 |
C2F6N3 |
| 5. | 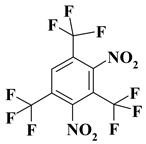 |
C3F9N2 |
| 6. | 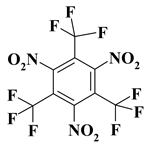 |
C3F9N3 |
| 7. | 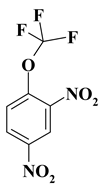 |
CF3ON2/OCF3N2 |
| 8. | 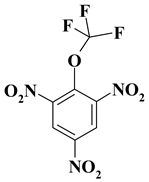 |
CF3ON3/OCF3N3 |
| 9. | 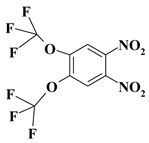 |
O2C2F6N2 |
| 10. | 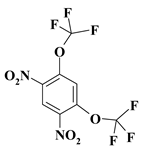 |
O2C2F6N2b/m |
| 11. | 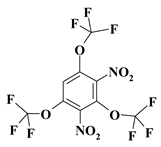 |
O3C3F9N2a |
| 12. | 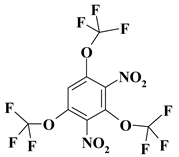 |
O3C3F9N2b |
| 13. | 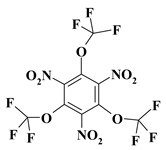 |
O3C3F9N3a |
| 14. |  |
O3C3F9N3b |
| 15. |  |
O2C2F6N2a |
| 16. |  |
O2C2F6N2b |
| 17. |  |
O2C2F6N3 |
| 18. | 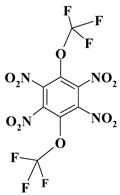 |
O2C2F6N4 |
| 19. | 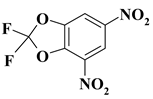 |
1CF2N2/O2CF2N2 |
| 20. | 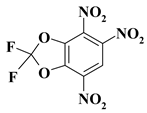 |
1CF2N3/O2CF2N3 |
| 21. | 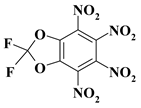 |
1CF2N4/O2CF2N4 |
| 22. | 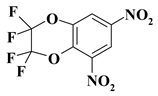 |
2CF4N2/O2C2F4N2 |
| 23. | 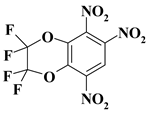 |
2CF4N3/O2C2F4N3 |
| 24. | 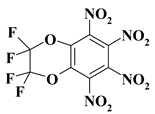 |
2CF4N4/O2C2F4N4 |
Appendix B
The views of the conformers. Borrow color denotes carbon, yellow – fluorine, red – oxygen, blue – nitrogen, and grey – hydrogen.
| Compound | Conformer | ||
| a | B | ||
| C3F6N2 | 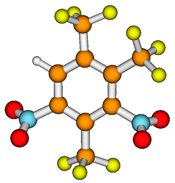 |
 |
|
| O2C2F6N2 |  |
 |
|
| O2C2F6N3 |  |
 |
|
| O2C2F6N4 |  |
 |
|
| O3C3F9N3 |  |
 |
|
References
- Koch, E.C. High Explosives, Propellants, Pyrotechnics; Walter de Gruyter GmbH: Berlin, Germany; Boston, MA, USA, 2021; pp. 1-759. ISBN: 978-3-11-066052-4.
- Agrawal, J.P. High Energy Materials: Propellants, Explosives and Pyrotechnics. Wiley-VCH: Weinheim, Germany, 2010; pp.1-498. ISBN: 978-3-527-32610-5.
- Olah, G.A.; Squire, D.R. (Eds.). Chemistry of Energetic Materials; Academic Press: Cambridge, MA, US, 2012; pp. 1-212. ISBN: 978-0-1239-5897-6.
- Kominia, A.; Smith, J.L.; Sheehan, P.; Oxley, J.C. Thermal decomposition of fluorinated polymers used in plasticized explosives and munitions. Propellants, Explos. Pyrotech. 2024, 49, e202300207. [Google Scholar] [CrossRef]
- Grakauskas, V.; Baum, K. Aqueous fluorination of nitronate salts. J. Org. Chem. 1968, 33, 3080–3082. [Google Scholar] [CrossRef]
- Grakauskas, V. Direct liquid-phase fluorination of methyl trichloroacetate and acetic anhydride. J. Org. Chem. 1969, 34, 963–965. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Adolph, H.G. Fluoronitro aliphatics. II. Fluorodinitromethyl compounds. Synthetic approaches and general properties. J. Org. Chem. 1968, 33, 3073–3080. [Google Scholar] [CrossRef]
- Adolph, H.G.; Kamlet, M.J. Fluoronitroaliphatics. IV. Reactions of 2-fluoro-2, 2-dinitroethanol. J. Org. Chem. 1969, 34, 45–50. [Google Scholar] [CrossRef]
- Grakauskas, V.; Albert, A.H. Polynitroalkyltetrazoles. J. Het. Chem. 1981, 18, 1477–1479. [Google Scholar] [CrossRef]
- Pepekin, V.I.; Matyushin, Y.N.; Rozantsev, G.G.; Shevelev, S.A.; Apin, A.Y. Enthalpies of formation of dinitrophenylmethane, trinitrophenylethane, and fluorodinitrophenylmethane. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1972, 21, 2634–2636. [Google Scholar]
- Pepekin, V.I., Natsibullin, F.Y., Eremenko, L.T., Lebedev, Y.A. Enthalpy of formation of fluoronitromethyl radical and dissociation energy of CH and CF bonds in fluorodinitromethane. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1974, 23, 892-893.
- Hurwitz, H. Computation of detonation properties of fluoroexplosives. Technical Report DTIC NOLTR 65-217, Naval Ordnance Laboratory, White Oak, ML, USA. 1966, pp. 1-54. URL: https://apps.dtic.mil/sti/citations/AD0374171 , accessed at 30-05-2024.
- Bogdanova,Yu.A.; Gubin, S.A.; Korsunskii, B.L.; Pepekin, V.I. Detonation Characteristics of Powerful Insensitive Explosives. Combust. Explos. Shock Waves. 2009, 45, 738 –743.
- Politzer, P.; Lane, P. Structures and Properties of Energetic Difluoramines. In: Hargittai, M.;Hargittai, I. Advances in Molecular Structure Research, Vol.3, 1997, pp.269-285. ISBN: 978-0-7623-0208-6.
- Dalinger, I.L.; Kormanov, A. V.; Suponitsky, K.Y.; Muravyev, N.V.; Sheremetev, A.B. Pyrazole–tetrazole hybrid with trinitromethyl, fluorodinitromethyl, or (difluoroamino) dinitromethyl groups: High-performance energetic materials. Chem. Asian J. 2018, 13, 1165–1172. [Google Scholar] [CrossRef]
- Palysaeva, N.V.; Gladyshkin, A.G.; Vatsadze, I.A.; Suponitsky, K.Y.; Dmitriev, D.E.; Sheremetev, A.B. N-(2-Fluoro-2, 2-dinitroethyl) azoles: a novel assembly of diverse explosophoric building blocks for energetic compound design. Org. Chem. Front. 2019, 6, 249–255. [Google Scholar] [CrossRef]
- Kettner, M.A.; Klapötke, T.M. New energetic polynitrotetrazoles. Chem. Eur. J. 2015, 21, 3755–3765. [Google Scholar] [CrossRef]
- Klapötke, T.M.; Krumm, B.; Moll, R.; Rest, S.F.; Schnick, W.; Seibald, M. Asymmetric fluorodinitromethyl derivatives of 2, 2, 2-trinitroethyl N-(2, 2, 2-trinitroethyl) carbamate. J. Fluor. Chem. 2013, 156, 253–261. [Google Scholar] [CrossRef]
- Zhai, L.; Zhang, J.; Wu, M.; Huo, H.; Bi, F.; Wang, B. Balancing good oxygen balance and high heat of formation by incorporating of-C (NO2) 2F Moiety and Tetrazole into Furoxan block. J. Mol. Struct. 2020, 1222, 128934. [Google Scholar] [CrossRef]
- Zhai, L.; Wang, B.; Xu, K.; Huo, H.; Liu, N.; Li, Y.; Li, H.; Lian, P.; Fan, X. A new synthetic route for 3, 3′-bis (fluorodinitromethyl) difurazanyl ether (FOF-13) and its energetic properties. J. Energ. Mater. 2016, 34, 92–102. [Google Scholar] [CrossRef]
- Sheremetev, A.B. 3, 3-Bis(1-fluoro-1, 1-dinitromethyl)difurazanyl ether.In Proc. 29th Int. Ann. Conf. ICT, Karlsruhe, Germany. June 30-July 3. 1998, 58, 1-6.
- Dalinger, I.L.; Vinogradov, V.M.; Shevelev, S.Y.A.; Kuz’min, V.S.; Arnautova, E.A.; Pivina, T.Y.S. Synthesis and Calculation of Properties of N-Difluoroaminoazoles, the Novel Type of Energetic Materials. Propellants Explos. Pyrotech. 1998, 23, 212–217. [Google Scholar] [CrossRef]
- Chapman, R.D. Organic difluoramine derivatives. In: Klapötke, T. M. (Ed.). High energy density materials. Springer Verlag, Berlin Heidelberg. Struct. Bond., Vol. 125. 2007, pp.123–151.
- Reddy, V.P. Synthetic methods for high-energy Organofluorine Compounds. In: Energetic Materials. Advanced Processing Technologies for Next-Generation Materials. Eds. Mezger, M.J.; Tindle, K.J.; Pantoya, M.; Groven, L.J.; Kalyon, D.M. Boca Raton, FL., Taylor&Francis, CRC Press, 2017, pp. 1-19. ISBN 978-1-3516-8125-4.
- Wu, Q.; Li, Q.; Yan, G.; Zhang, Z.; Zhu, W. Molecular design of novel super high energy compounds by incorporating the difluoramino group, N-oxide and different bridge groups into the 1H-tetrazole. J. Fluor. Chem. 2019, 218, 21–26. [Google Scholar] [CrossRef]
- Oxley, J.C.; Smith, J.L.; Zhang, J.; Bedford, C. A comparison of the thermal decomposition of nitramines and difluoramines. J. Phys. Chem. A. 2001, 105, 579–590. [Google Scholar] [CrossRef]
- Ye, C.; Gao, H.; Shreeve, J.M. Synthesis and thermochemical properties of NF2-containing energetic salts. J. Fluor. Chem. 2007, 128, 1410–1415. [Google Scholar] [CrossRef]
- Dalinger, I.L.; Shkineva, T.K.; Sheremetev, A.B. Ethyl butyrates bearing nitro and difluoroamino groups. Mendeleev Commun. 2023, 33, 841–843. [Google Scholar] [CrossRef]
- Ugrak, B.I., Shkineva, T.K., Sheremetev, A.B., Dalinger, I.L. (Difluoroamino) furazans. Russ. Chem. Bull. 2023, 72, 2706-2716.
- Muravyev, N.V., Fershtat, L., Zhang, Q. Synthesis, design and development of energetic materials: Quo Vadis?. J. Chem. Eng. 2024, 150410. [CrossRef]
- Muravyev, N.V.; Meerov, D.B.; Monogarov, K.A.; Melnikov, I.N.; Kosareva, E.K.; Fershtat, L.L.; Sheremetev, A.B.; Dalinger, I.L.; Fomenkov, I.V.; Pivkina, A.N. Sensitivity of energetic materials: Evidence of thermodynamic factor on a large array of CHNOFCl compounds. J. Chem. Eng. 2021, 421, 129804. [Google Scholar] [CrossRef]
- Sitzmann, M.E.; Gilligan, W.H.; Ornellas, D.L.; Thrasher, J.S. Polynitroaliphatic explosives containing the pentafluorosulfanyl (SF5) group: The selection and study of a model compound. J. Energ. Mater. 1990, 8, 352–374. [Google Scholar] [CrossRef]
- Martinez, H.; Zheng, Z.; Dolbier Jr, W.R. Energetic materials containing fluorine. Design, synthesis and testing of furazan-containing energetic materials bearing a pentafluorosulfanyl group. J. Fluor. Chem. 2012, 143, 112–122. [Google Scholar] [CrossRef]
- Gao, H.; Ye, C.; Winter, R.W.; Gard, G.L.; Sitzmann, M.E.; Shreeve, J.N.M. Pentafluorosulfanyl (SF5) containing energetic salts. Eur. J. Inorg. Chem. 2006, 16, 3221–3226. [Google Scholar] [CrossRef]
- Xiao-Hong, L.; Hong-Ling, C.; Wei-Wei, J.; Tong-Wei, L.; Rui-Zhou, Z.; Yong-Liang, Y. Theoretical studies on energetic materials bearing pentaflurosulphyl (SF 5) groups. J. Chem. Sci. 2014, 126, 1163–1172. [Google Scholar]
- Sitzmann, M.E.; Gilardi, R.D. Polynitroalkyl derivatives of SF5N CCl2: nitrations of SF5 imines. J. Fluor. Chem. 1993, 63, 203–215. [Google Scholar] [CrossRef]
- Li, C.; Liu, M.; Li, T.; Wang, L.; Zhang, R.; Jing, S. Fluorine Added to Lead the Way to Future Energetic Materials: 3, 5-difluoro-2, 4, 6-trinitroaniline. J. Energ. Mater. 2022, 1–10. [Google Scholar] [CrossRef]
- Zhu, J.; Li, C.; Jing, S.; Yang, L.; Liu, Y.; Zhang, W.; Zhang, J. 3,5-difluoro-2,4,6-trinitrophenol: A high-energy compound born under the “NO2FNO2” construction strategy. Propellants Explos. Pyrotech. 2024, 49(2), e202300184. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.; Zhou, J.; Bi, F.; Wang, B. Effect of fluoro substituents on polynitroarylenes: design, synthesis and theoretical studies of fluorinated nitrotoluenes. ChemPlusChem 2019, 84, 92–97. [Google Scholar] [CrossRef]
- Hu, F.; Wang, L.J.; Zhao, W.; Liu, Y.C.; Jing, S.M.; Liu, P.; He, J.X. Thermal Decomposition Kinetics and Compatibility of 3, 5-difluoro-2, 4, 6-trinitroanisole (DFTNAN). Materials 2021, 14, 4186. [Google Scholar] [CrossRef]
- Jing, S.; Jiang, Z.; Jiao, Q.; Li, Z.; Liu, Y.; Yang, L. 3, 5-difluoro-2, 4, 6-trinitroanisole: Promising melt-cast insensitive explosives instead of TNT. J. Energ. Mater. 2022, 40, 206–217. [Google Scholar] [CrossRef]
- Jiao, Q.; Li, T.; Ou, Y.; Jing, S.; Wang, F. Probing the Reaction Mechanisms of 3,5-Difluoro-2,4,6-Trinitroanisole (DFTNAN) through a Comparative Study with Trinitroanisole (TNAN). Materials, 2022, 15(7), 2568. [CrossRef]
- Li, Y.; Xue, M.; Sun, B.; Tu, Z.; Wang, X. Regulating the melting point by non-covalent interaction toward a promising insensitive melt-castable energetic material: 1, 2-Difluoro-4, 5-dinitrobenzene. Chinese J. Struc. Chem. 2023, 42(4), 100002. [Google Scholar] [CrossRef]
- Guo, Z.; Yu, Q.; Chen, Y.; Liu, J.; Li, T.; Peng, Y.; Yi, W. Fluorine-Containing Functional Group-Based Energetic Materials. Chem. Rec., 2023, 23(9), e202300108. [CrossRef]
- Fokin, A.V.; Studnev, Y.N.; Rapkin, A.I.; Komarov, V.A.; Verenikin, O.V.; Potarina, T.M. Synthesis and some properties of 5-fluorodinitromethyl- and 5-difluoronitromethyltetrazoles. Izv. Akad. Nauk SSSR Ser. Khim. 1981, 7:1592–1595.
- Chapman, R. D. Halogenated explosives to defeat biological agents. Defense Threat Reduction Agency. Tech. Report DTRA-IR-14-81. 2015 Sept., pp.1-37.
- Muravyev, N.V.; Suponitsky, K.Y.; Fedyanin, I.V.; Fomenkov, I.V.; Pivkina, A.N.; Dalinger, I.L. Bis-(2-difluoroamino-2, 2-dinitroethyl) nitramine–Energetic oxidizer and high explosive. Chem. Eng. 2022, 449, 137816. [Google Scholar] [CrossRef]
- Zhai, L.; Zhang, J.; Zhang, J.; Wu, M.; Bi, F.; Wang, B. Progress in Synthesis and Properties of High Energy Density Compounds Regulated by N—F Bond. Chin. J. Org. Chem. 2020, 40, 1484. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Adolph, H.G. Some comments regarding the sensitivities, thermal stabilities, and explosive performance characteristics of fluorodinitromethyl compounds. In Proc. Seventh Internat. Symp. Detonation, Annapolis, Maryland, USA, Jun. 16-19, 1981, pp.84-92.
- Warner D.A. Bulk synthesis of fluoroexplosives. Tech. Report AFATL-TR-67-154/ DTIC AD0501781. Defense Technical Information Center, Eglin AFB, FL, USA, Oct. 1967, pp.1-35. URL: https://archive.org/details/DTIC_AD0501781, accessed at 30-05-2024.
- Kumar, A.S.; Kommu, N.; Ghule, V.D.; Sahoo, A.K. Synthesis of trifluoromethyl-substituted N-aryl-poly-1, 2, 3-triazole derivatives. J. Mater. Chem. A. 2014, 2, 7917–7926. [Google Scholar] [CrossRef]
- Yan, Z.; Lu, T.; Liu, Y.; Liu, W.; Zhao, B.; Wang, Y.; Ge, Z. High thermal stability and insensitive fused triazole-triazine trifluoromethyl-containing explosives (TFX). ACS Omega 2021, 6, 18591–18597. [Google Scholar] [CrossRef]
- Chinnam, A.K.; Staples, R.J.; Shreeve, J.N.M. Selective Synthesis of Bis (3-(3-(trifluoromethyl)-1 H-1, 2, 4-triazol-5-yl)-4, 4′-azo-and-azoxyfurazan Derivatives. J. Org. Chem. 2021, 86, 7781–7786. [Google Scholar] [CrossRef]
- Yang, T.; Xu, Z.; Meng, Z.; Zhai, L. A Novel Synthesis, Characterization and Performances of 1, 3, 5-Trinitro-2, 2-bis (trifluoromethyl)-1, 3, 5-triazinane. ChemistrySelect 2019, 4, 6338–6341. [Google Scholar] [CrossRef]
- Liu, Q.; Yuan, M.; He, J.; Yu, P.; Guo, X.; Liu, Y.; Gao, H.; Yin, P. Exchanging of NH2/NHNH2/NHOH groups: An effective strategy for balancing the energy and safety of fused-ring energetic materials. J. Chem. Eng. 2023, 466, 143333. [Google Scholar] [CrossRef]
- Tamuliene, J.; Sarlauskas, J. Impact of Incremental Methylene Groups on the Energetic Properties of Aromatic Nitramines. Energies, 2023,. 16, 3117. [CrossRef]
- Parthasarathi, R.; Padmanabhan, J.; Subramanian, V.; Maiti, B.; Chattaraj, P.K. Toxicity analysis of 3,3’,4,4’,5-pentachloro biphenyl through chemical reactivity and selectivity profiles. Curr. Sci. 2004, 86, 535–542. Available online: https://www.jstor.org/stable/24107906 (accessed on 10 June 2024).
- Kaya, S.; Kaya, C. New equation based on ionization energies and electron affinities of atoms for calculating of group electronegativity. Comput. Theor. Chem. 2015, 1052, 42–46. [Google Scholar] [CrossRef]
- Becke, A.D. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Dunning Jr., T. H, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A.; Bloino, J.; Janesko, B.G.; Gomperts, R.; Mennucci, B.; Hratchian, H.P.; Ortiz, J.V.; Izmaylov, A.F.; Sonnenberg, J.L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V.G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehar, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J.A.Jr., Peralta, J.E.; Ogliaro, F.; Bearpark, M.; Heyd, J.J.; Brothers, E.; Kudin, K.N.; Staroverov, V.N.; Keith, T.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; Millam, J.M.;, Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Farkas, O.; Foresman, J.B., Fox, D.J. Gaussian, Inc., Wallingford CT. 2016, pp. 1-139. URL: https://www.cwu.edu/chemistry/sites/cts.cwu.edu.chemistry/files/documents/Gaussian_09_ReferenceManual.pdf (accessed: 26.05.2024).
- Cardia, R.; Malloci, G.; Mattoni, A.; Cappellini, G. Effects of TIPS-Functionalization and Perhalogenation on the Electronic, Optical, and Transport Properties of Angular and Compact Dibenzochrysene. J. Phys. Chem. A. 2014, 118, 5170–5177. [Google Scholar] [CrossRef]
- Cardia, R.; Malloci, G.; Rignanese, G.M.; Blasé, X.; Molteni, E.; Cappellini, G. Electronic and optical properties of hexathiapentacene in the gas and crystal phases. Phys. Rev. B. 2016, 93, 235132. [Google Scholar] [CrossRef]
- Dardenne, N.; Cardia, R.; Li, J.; Malloci, G.; Cappellini, G.; Blasé, X.; Charlier, J.C.; Rignanese, G. Tuning Optical Properties of Dibenzochrysenes by Functionalization: A Many-Body Perturbation Theory Study. Phys. Chem. C. 2017, 121, 24480–24488. [Google Scholar] [CrossRef]
- Antidormi, A.; Aprile, G.; Cappellini, G.; Cara, E.; Cardia, R.; Colombo, L.; Farris, R.; d’Ischia, M.; Mehrabanian, M.; Melis, C.; Mula, G.; Pezzella, A.; Pinna, E.; Riva, E.R. Physical and Chemical Control of In-terface Stability in Porous Si–Eumelanin Hybrids. J. Phys. Chem. C. 2018, 122, 28405–28415. [Google Scholar] [CrossRef]
- Mocci, P.; Cardia, R.; Cappellini, G. Inclusions of Si-atoms in Graphene nanostructures: A computational study on the ground-state electronic properties of Coronene and Ovalene. J. Phys. Conf. Ser. 2018, 956, 012020. [Google Scholar] [CrossRef]
- Mocci, P.; Cardia, R.; Cappellini, G. Si-atoms substitutions effects on the electronic and optical properties of coronene and ovalene. New J. Phys. 2018, 20, 113008. [Google Scholar] [CrossRef]
- Kumar, A.; Cardia, R.; Cappellini, G. Electronic and optical properties of chromophores from bacterial cellulose. Cellulose. 2018, 25, 2191–2203. [Google Scholar] [CrossRef]
- Szafran, M.; Koput, J. Ab initio and DFT calculations of structure and vibrational spectra of pyridine and its isotopomers. J. Mol. Struct. 2001, 565, 439–448. [Google Scholar] [CrossRef]
- Begue, D.; Carbonniere, P.; Pouchan, C. Calculations of Vibrational Energy Levels by Using a Hybrid ab Initio and DFT Quartic Force Field: Application to Acetonitrile. J. Phys. Chem. A. 2005, 109, 4611–4616. [Google Scholar] [CrossRef]
- Cooper, P.W. Explosives Engineering; Wiley-VCH: New York, USA. 1996; pp.1-480. ISBN: 0-471-18636-8.
- Shevchenko, A.A.; Dolgoborodov, A.Yu.; Brazhnikov, M.A.; Kirilenko, V.G. Pseudoideal detonation of mechanoactivated mixtures of ammonium perchlorate with nanoaluminum. J. Phys. Conf. Ser. (IOP Publ.) 2018, 946, 012055. [Google Scholar] [CrossRef]
- Kozak, G.D. Measurement and calculation of the ideal detonation velocity for liquid nitrocompounds. Combust. Explos. Shock Waves. 1998, 34, 581–586. [Google Scholar] [CrossRef]
- Bolton, O.; Simke, L.R.; Pagoria, P.F.; Matzger, A.J. High Power Explosive with Good Sensitivity: A 2:1 Cocrystal of CL-20:HMX. Cryst. Growth Des. 2012, 12, 4311–4314. [Google Scholar] [CrossRef]
- Viswanath, D.S.; Ghosh, T.K.; Boddu, V.M. 5-Nitro-2,4-dihydro-3H-1,2,4-Triazole-3-one (NTO). In Emerging Energetic Materials: Synthesis, Physicochemical, and Detonation Properties. Springer: Dordrecht, The Netherlands. 2018; pp. 163–211.
- Rajakumar, B.; Arathala, P.; Muthiah, B. Thermal Decomposition of 2-Methyltetrahydrofuran behind Reflected Shock Waves over the Temperature Range of 1179–1361 K. J. Phys. Chem. A. 2021, 125, 5406–54223. [Google Scholar]
- Šarlauskas, J.;Tamulienė, J. Preparation and Characterization of Cationic Energetic Salts of 5-Amino-3-[(2,4,6-trinitrophenyl)amino]-1H-1,2,4-triazole (APATO). Central European Journal of Energetic Materials. 2022, 19(3), 311–325. [CrossRef]
- Free Chemical Drawing Software. ChemSketch. Version 10.0. ACD/Labs. Available online: https://www.acdlabs.com/resources/free-chemistry-software-apps/chemsketch-freeware/ (accessed on 30 January 2023).
- Kaushik, M. A review of innovative chemical drawing and spectra prediction computer software. Mediterr. J. Chem. 2014, 3, 759–766. [Google Scholar] [CrossRef]
- Wen, L.; Wang, B.; Yu, T.; Lai, W.; Shi, J.; Liu, M.; Liu, Y. Accelerating the search of CHONF-containing highly energetic materials by combinatorial library design and high-throughput screening. Fuel 2022, 310, 122241. [Google Scholar] [CrossRef]
- Keshavarz, M.H.; Zamani, A. A simple and reliable method for predicting the detonation velocity of CHNOFCl and aluminized explosives. Cent. Eur. Energ. Mater. 2015, 12, 13–33. [Google Scholar]
- Keshavarz, M.H.; Pouretedal, H.R. An empirical method for predicting detonation pressure of CHNOFCl explosives. Thermochim. Acta 2004, 414, 203–208. [Google Scholar]
- Urizar, M.J.; James, E., Jr.; Smith, L.C. Detonation velocity of pressed TNT. Phys. Fluids. 1961, 4, 262–274. [Google Scholar] [CrossRef]
- Chandler, J.; Ferguson, R.E.; Forbes, J.; Kuhl, A.L.; Oppenheim, A.K.; Spektor, R. Confined combustion of TNT explosion products in air. In Proc. Conf. 8th international Colloquium on Dust Explosions, Schaumburg, IL, US. Tech. Rep. UCRL-JC-131748; ON: DE00003648; OSTI ID; 3648, Sept. 21-25, 1998. Accessed at URL: https://www.osti.gov/biblio/3648 (on 03-07-2024).
- Yang, J.; Bai, T.; Guan, J.; Li, M.; Zhen, Z.; Dong, X.; Wang, Y.; Wang, Y. Novel fluorine-containing energetic materials: how potential are they? A computational study of detonation performance. J. Mol. Model. 2023, 29(8), p.228. [CrossRef]
- Kang, Y.; Dong, Y.; Liu, Y.; Gao, H.; Wang, Y.; Shreeve, J.M. Halogen bonding (CF··· X) and its effect on creating ideal insensitive energetic materials. J. Chem. Eng. 2022, 440, 135969. [Google Scholar] [CrossRef]
- Wu, W.; You, Y.; Weng, Z. Recent advances in the synthesis of fluoroalkylated compounds using fluoroalkyl anhydrides. Chin. Chem. Lett. 2022, 33, 4517–4530. [Google Scholar] [CrossRef]
- Balachandar, K.G.; Thangamani, A. Novel high performance energetic materials of fluorine-containing 2, 6-dinitro-4-(trifluoromethyl) phenol derivatives with substituted azoles. J. Fluor. Chem. 2021, 247, 109801. [Google Scholar] [CrossRef]
- Koch, E.C.; Contini, A. Metal Fluorocarbon Pyrolants, XVI: Theoretical and Experimental Investigation of Poly [bis (2, 2, 2-trifluoroethoxy) phosphazene] (PTFEP) as Oxidizer in Magnesium Based Pyrolants. Propellants Explos. Pyrotech. 2014, 39, 761–767. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



