
Submitted:
08 September 2024
Posted:
09 September 2024
You are already at the latest version
Abstract
Infection-related cardiovascular diseases (CVDs) represent a significant health challenge, necessitating novel therapeutic strategies targeting key receptors involved in inflammation and infection. Antimicrobial peptides (AMPs) offer a promising approach, potentially disrupting pathogenic processes. This study aimed to investigate the efficacy of AMPs as therapeutic agents by examining their interactions with critical CVD-related receptors. A comprehensive computational approach was utilized to assess the interactions between AMPs and receptors associated with CVDs. Molecular docking studies were conducted to evaluate AMP binding to target receptors: ACE2, CRP, MMP9, NLRP3, and TLR4. The top-performing AMPs were further analyzed through 100 ns molecular dynamics (MD) simulations, and their binding affinities were quantified using MM/PBSA calculations. The analysis revealed that Tachystatin, Pleurocidin, and Subtilisin A exhibit strong binding affinities to key CVD-related receptors, including ACE2, CRP, and MMP9. These AMPs demonstrated the potential for disrupting receptor-peptide interactions critical to infection and inflammation. MD simulations confirmed the stability of AMP-receptor complexes, with MM/PBSA calculations showing significant binding energies. Future directions include conducting in vitro and in vivo studies to validate the therapeutic efficacy and safety of these AMPs in clinical settings.
Keywords:
antimicrobial peptides
; cardiovascular disease
; molecular docking
; molecular dynamics
1. Introduction
Infections and cardiovascular diseases (CVDs) share a complex, bidirectional relationship. Pathogenic microorganisms can initiate or exacerbate CVDs through direct infection of cardiovascular tissues, immune system dysregulation, or the induction of chronic inflammatory states [1,2]. For instance, respiratory pathogens such as influenza virus and bacterial pathogens like Chlamydia pneumoniae and Helicobacter pylori have been associated with an increased risk of myocardial infarction and atherosclerosis [3,4]. The chronic inflammation induced by these pathogens accelerates the formation of atherosclerotic plaques, a major cause of coronary artery disease. Among the most prominent mechanisms is the activation of the innate immune system via receptors such as Toll-Like Receptors (TLRs), which recognize pathogen-associated molecular patterns (PAMPs) [5,6,7]. TLRs initiate signaling cascades upon activation that produce pro-inflammatory cytokines, resulting in endothelial dysfunction and plaque instability. Another key player in this inflammatory process is the NLRP3 inflammasome, which responds to infectious and non-infectious stimuli, including viral RNA, bacterial toxins, and cholesterol crystals [8,9]. Once activated, the NLRP3 inflammasome promotes the secretion of IL-1β, a potent pro-inflammatory cytokine implicated in the progression of atherosclerosis [10,11]. Furthermore, angiotensin-converting enzyme 2 (ACE2), a receptor known for regulating blood pressure, has gained considerable attention due to its interaction with the SARS-CoV-2 virus [12,13]. The binding of the virus to ACE2 receptors impairs its physiological functions, leading to cardiovascular complications, including myocardial injury and arrhythmias [14]. Matrix metalloproteinases (MMPs), a family of proteolytic enzymes involved in extracellular matrix remodeling, also play a critical role in CVD progression, particularly in the degradation of the fibrous cap of atherosclerotic plaques, leading to plaque rupture and subsequent cardiovascular events [15,16].
Treating infections related to cardiovascular diseases typically involves using antimicrobial agents such as antibiotics, antivirals, and antifungals, depending on the pathogen involved. For bacterial infections, antibiotics such as macrolides and β-lactams are commonly prescribed. For example, azithromycin is often used to treat Chlamydia pneumoniae infections associated with atherosclerosis [17,18]. However, the use of antibiotics poses several challenges, including the emergence of antibiotic resistance, which has become a significant global health threat. The overuse and misuse of antibiotics have led to the development of multidrug-resistant (MDR) strains, which are not only harder to treat but also contribute to higher mortality rates in patients with CVDs [19,20,21]. Moreover, the long-term use of antibiotics has been associated with adverse cardiovascular outcomes, including arrhythmias and QT interval prolongation [22]. As a result, there is growing interest in exploring alternative therapeutic strategies, such as the use of antimicrobial peptides (AMPs), which have the potential to overcome the limitations of current therapies and provide more targeted interventions. One of the significant advantages of AMPs is their ability to selectively target microbial membranes, which reduces the likelihood of developing resistance compared to traditional antibiotics [23,24]. Unlike conventional antimicrobial agents that often target specific bacterial proteins or enzymes, AMPs disrupt microbial membranes by interacting with their lipid bilayers, leading to cell lysis and death [25]. This mode of action is less prone to resistance, as microbes would need to undergo significant changes in membrane composition to evade AMP activity [26,27].
This study aimed to explore the therapeutic potential of AMPs in targeting key receptors involved in infection-related CVDs. Using a comprehensive approach that combined molecular docking and molecular dynamics simulations, we assessed the binding affinity, stability, and interaction dynamics of various AMPs with critical receptors. This study offered a novel, in-depth understanding of how AMPs can modulate critical receptors in infection-driven CVDs by employing these cutting-edge techniques. The findings from this study contributed to the growing field of peptide-based therapies, offering new perspectives on how AMPs could be utilized to combat cardiovascular diseases linked to infections, especially in cases where conventional treatments have limitations. This research underscored the therapeutic potential of AMPs and laid the groundwork for future investigations into peptide-based drug design for complex cardiovascular and inflammatory conditions.
2. Materials and Methods
2.1. Selection and Preparation of the Receptors Associated with Infection-Related CVDs
In this study, five key receptors associated with infection-related CVDs were selected for analysis: Angiotensin-Converting Enzyme 2 (ACE2), C-reactive protein (CRP), Matrix Metalloproteinase-9 (MMP9), NLRP3 Inflammasome, and Toll-Like Receptor-4 (TLR4) (Table 1). To ensure accurate molecular docking and dynamics simulations, these proteins were chosen based on their well-characterized functions and the availability of high-resolution 3D structures in the Protein Data Bank (PDB). The preparation process involved retrieving the structural data, optimizing the protein structures, and refining them using Swiss-PdbViewer [28] to correct potential issues and enhance model accuracy. This refinement process included adjusting side-chain conformations, adding missing residues, and validating structural quality, ensuring the proteins were suitable for precise interaction modeling. Additionally, their active sites were defined using CASTp 3.0 [29], facilitating detailed docking studies with the selected antimicrobial peptides.
2.2. Selection and Preparation of Antimicrobial Peptides (AMPs)
Specific criteria were employed to guide the selection of AMPs for this study, ensuring the relevance and reliability of the molecular docking and dynamics simulations. Only peptides with available 3D structures in the PDB were considered, guaranteeing the availability of accurate structural data necessary for effective modeling and simulation. The peptides chosen were classified under the SCOP (Structural Classification of Proteins) as “peptides.” The resolution of the structural data was restricted to 0.5 - 2.5 Å, as high-resolution structures are preferred for their detailed and accurate depiction of peptide conformation, which is crucial for precise docking simulations. The active sites of the AMPs were analyzed using CASTp 3.0 [29]. The complete dataset, including type, PDB ID, sequence, and active residues of the selected AMPs, is provided in Supplementary Data S1.

Table 2.
Selected antimicrobial peptides (AMPs) based on the employed criterion.
| Antimicrobial Peptide | PDB ID | Active Site (Number of Residues) | Ref |
|---|---|---|---|
| Aurein | 1VM5 | 1, 2, 5 | [35] |
| Beta-defensin 2 | 1FD4 | 6, 7, 9, 10, 11, 12 | [36] |
| Bombinin | 2AP7 | 1, 3, 4, 6, 7 | [37] |
| Cathelicidin | 2K6O | 1, 4, 5, 8 | [38] |
| Cecropin | 1D9J | 7, 9, 10, 11, 12 | [39] |
| Chim2 | 8EB1 | 10, 11, 14, 15 | [40] |
| Dermcidin | 2NDK | 18, 19, 22, 25, 26, 29 | [41] |
| Esculentin | 5XDJ | 2, 3, 6 | [42] |
| Exendin-4 | 3C59 | 26, 27, 28, 29, 32, 33 | [43] |
| Hepcidin | 3H0T | 13, 14, 16, 18, 19, 20, 21, 22 | [44] |
| Hs05 | 6VLA | 5, 8, 9 | [45] |
| Indolicidin | 1HR1 | 9, 10, 11, 12, 13 | [46] |
| Lactoferrin | 1LFC | 1, 23, 25 | [47] |
| Lavracin | 2N8D | 1, 3, 4, 6 | [48] |
| Magainin | 2MAG | 1, 2, 6, 9, 17, 21 | [49] |
| Melittin | 2MLT | 13, 16, 17, 20 | [50] |
| Microcin J25 | 4CU4 | 9, 10, 19, 20, 21 | [51] |
| Nisin | 1WCO | 9, 12, 17, 19, 20, 21 | [52] |
| Pardaxin | 2KNS | 2, 3, 5, 6, 9, 15, 22, 23, 26, 27, 29, 30, 33 | [53] |
| Piscidin | 6PEZ | 3, 4, 7 | [54] |
| Pleurocidin | 2LS9 | 10, 13, 14, 17, 20, 23, 24 | [55] |
| Polyphemusin I | 1RKK | 7, 9, 12, 14 | [56] |
| Protegrin-1 | 1PG1 | 5, 6, 7, 14, 15, 16 | [57] |
| PvHCt | 2N1C | 14, 15, 16, 17, 18, 19, 20, 22, 23 | [58] |
| Subtilisin A | 1PXQ | 1, 4, 5, 7, 9, 10, 24, 25, 29, 30, 33 | [59] |
| Tachyplesin-1 | 2RTV | 1, 2, 3, 16, 17 | [60] |
| Tachystatin | 1CIX | 5, 11, 12, 15, 18, 22, 23, 29 | [61] |
| Temporin-L | 6GS5 | 4, 7 | [62] |
| Thanatin | 8TFV | 11, 13, 16, 17, 18 | [63] |
| Thermolysin | 6FHP | 258, 263, 267, 305, 306, 309, 310 | [64] |
Based on these criteria, a set of 30 AMPs was chosen (Table 2), including Exendin-4, a notable AMP with similarities to GLP-1, and has been investigated for its potential to influence ACE2 activity [65]. CRP is a marker of inflammation often elevated in cardiovascular diseases [66]. Lactoferrin, an iron-binding glycoprotein known for its antimicrobial properties, has been shown to affect CRP levels [67], thus, Lactoferrin potentially influences cardiovascular outcomes and highlights its role in controlling inflammation in cardiovascular diseases. Cathelicidin, another well-studied AMP, has been shown to modulate the NLRP3 inflammasome, a critical component of the inflammatory response linked to cardiovascular diseases [68]. These examples illustrate how AMPs interact with key receptors involved in cardiovascular diseases, offering valuable insights into their potential therapeutic applications for modulating receptor activity and managing disease progression.
2.3. Molecular Docking Simulations
In this phase, molecular docking simulations were meticulously performed to investigate the interactions between a selected set of AMPs and specific receptors implicated in cardiovascular diseases. These receptors are critical targets in cardiovascular diseases and infections, influencing disease progression and inflammatory responses. The simulations were executed using the stand-alone version of HADDOCK (High Ambiguity Driven protein-protein DOCKing) [69], a robust and versatile docking software that enables detailed exploration of binding modes and energetic interactions between complex biomolecules. HADDOCK is well-regarded for incorporating experimental data into the docking process, allowing for a more accurate prediction of the possible binding conformations between AMPs and target receptors. The DX600 peptide (sequence: GDYSHCSPLRYYPWWKCTYPDPEGGG) was used as a reference standard due to its known interaction with ACE2 (IC50: 10.1 μM) and therapeutic implications in cardiovascular conditions [70]. The 3D structure of peptide 35409 was generated using AlphaFold [71], providing a precise model for further analysis. DX600 peptide was utilized as a benchmark to evaluate the efficacy of other AMPs in binding to the target receptors. Using this standard peptide allowed for a comparative analysis of the binding efficiency and inhibitory potential of other AMPs. The insights gained from these simulations are intended to guide the selection of AMPs with the highest potential for therapeutic applications. To further refine and validate the docking results, PRODIGY (PROtein binDIng enerGY prediction) [72] was employed to predict the binding affinity of the AMP-adhesion protein complexes. PRODIGY is an advanced computational tool that leverages state-of-the-art algorithms to estimate the binding affinity between interacting proteins and ligands based on structural data [73]. This prediction of binding free energy is critical for ranking the AMP candidates, as it provides a quantitative measure of how strongly an AMP binds to a target receptor. By identifying the AMPs with the most favorable binding affinities, PRODIGY helps narrow down the candidates most likely to impact cardiovascular disease receptors effectively. All molecular docking simulations were conducted on a high-performance computing workstation with an Intel® Core™ i7-12650H processor, an NVIDIA™ RTX 4060 graphics card with 8 GB VRAM, and 16 GB of DDR5 RAM.
2.4. Molecular Dynamics (MD) Simulations
Molecular dynamics (MD) simulations were utilized to explore the dynamics and stability of AMP-receptor complexes, specifically focusing on interactions between AMPs and receptors implicated in infection-related CVDs. The simulations were conducted using GROMACS 2022.5 [74], a highly regarded tool known for its accuracy and efficiency in modeling biomolecular systems. The Optimized Potentials for Liquid Simulations (OPLS-AA/L) force field was employed to accurately represent molecular interactions within these complexes [75]. Simulation boxes were configured using default cubic box parameters to accommodate the AMP-receptor complexes effectively. Standard procedures were followed, including adding water molecules via the Single Point Charge Extended (SPCE) model and incorporating counterions to ensure system neutrality [76]. Energy minimization was conducted using the steepest-descent method to eliminate steric clashes and stabilize the system. Equilibration was performed in two stages: first, in the number of particles, volume, and temperature (NVT) ensemble to stabilize temperature and system conditions, and second, in the number of particles, pressure, and temperature (NPT) ensemble to maintain constant pressure and temperature [77,78]. Following equilibration, production MD simulations were conducted for 100 nanoseconds to observe the long-term dynamics of the AMP-receptor complexes. During the simulations, parameters such as Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), Radius of Gyration (RoG), potential energies, and intermolecular hydrogen bonding interactions were monitored and analyzed to evaluate the stability and conformational dynamics of the complexes. Molecular visualization software, including PyMOL [79] and UCSF Chimera [80], was used to visualize critical residues and intermolecular interactions within the simulated complexes. This analysis provided valuable insights into the binding mechanisms and stability of the AMPs with the cardiovascular disease receptors.
2.5. Molecular Mechanics/Poisson–Boltzmann Surface Area (MM/PBSA) Calculations
The Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) method evaluated the peptide-receptor interactions involving AMPs and receptors implicated in infection-related CVDs. This method utilizes MD simulation data to calculate the binding free energy of the AMP-receptor complexes, providing insights into the strength and stability of these interactions. The MD simulations generated a variety of receptor conformations, and representative snapshots from these simulations were selected for detailed analysis [81]. Comprehensive energy computations were performed for each snapshot, including gas-phase energy calculations, solvation energy estimation using a continuum solvent model, and entropy calculations. These components were integrated to determine the overall binding free energy of the AMP-receptor complex [82,83]. The calculations used the gmx_MMPBSA module available within the GROMACS simulation package [84,85]. It is well-regarded for its precision and efficiency in calculating binding free energies for biomolecular complexes. The MM/PBSA method is precious for predicting binding affinities, as it accounts for the energetic and solvation contributions to the binding process [86]. The binding free energy (ΔG_binding) was computed using the following equation:
ΔG_binding = ΔG_complex - ΔG_peptide - ΔG_protein
where:
ΔG_binding: the binding free energy associated with forming the peptide-protein complex.
ΔG_complex: the free energy of the fully solvated peptide-protein complex.
ΔG_peptide: the free energy of peptide in its solvated state when unbound.
ΔG_protein: the free energy of protein in its solvated state when unbound.
By calculating the difference between the free energy of the complex and the combined free energies of the unbound AMP and receptor, this method provided insights into the energetic changes that occur upon complex formation, elucidating the interaction’s strength and stability.
3. Results
3.1. Molecular Docking Simulations of AMPs and Receptors Implicated in Infection-Related CVDs
The molecular docking simulations aimed to investigate the interactions between the selected AMPs and CVD-related receptors associated with infection and inflammation pathways. Key interaction parameters were analyzed to understand better how AMPs bind to these target receptors. The docking results included an evaluation of the HADDOCK score, free binding energy (kcal/mol), van der Waals energy, electrostatic energy, and desolvation energy for various AMP-receptor combinations. The best binding poses of AMPs and ACE2 (as one of the target receptors) are presented in Figure 1. The HADDOCK score provided an overall measure of the docking quality by integrating both the spatial and energetic fit of the AMP to the receptor. Free binding energy, measured in kilocalories per mole, offered a quantitative assessment of the binding strength between the AMPs and the receptors [77]. The van der Waals and electrostatic energies were also analyzed to gain insight into the non-covalent forces driving the interactions, which are crucial for understanding how these peptides interact at the molecular level. Additionally, desolvation energy, which reflects the energy of displacing water molecules from the receptor surface upon peptide binding, was calculated to understand the binding thermodynamics [87]. These results laid the groundwork for further molecular dynamics simulations to explore the stability and dynamics of the AMP-receptor complexes.
Figure 2 presents the free binding energy (kcal/mol) scores for the top-performing AMPs interacting with different target proteins, focusing on Tachystatin, Thermolysin, Pleurocidin, and Subtilisin A as consistent performers. Tachystatin emerged as a potent peptide across multiple targets. When docked with ACE2, Tachystatin achieved a HADDOCK score of -102.0 ± 3.7 a.u. and a binding energy of -10.7 kcal/mol, outperforming the standard inhibitor DX600 (-8.6 kcal/mol). The van der Waals energy was -48.4 ± 4.4 kcal/mol, and the electrostatic energy was -220.9 ± 32.4 kcal/mol, reflecting strong interaction forces, while the RMSD value of 1.5 ± 0.0 Å indicates a relatively stable conformation. Tachystatin similarly showed strong binding with MMP9, achieving a HADDOCK score of -136.4 ± 3.4 a.u. and a binding energy of -12.2 kcal/mol. These results highlight Tachystatin’s consistent performance across different proteins.
Thermolysin was also identified as a highly effective AMP, particularly with ACE2 and NLRP3. In the ACE2 complex, Thermolysin achieved a HADDOCK score of -91.4 ± 2.4 a.u. and a binding energy of -10.7 kcal/mol, comparable to Tachystatin. Its van der Waals energy of -49.2 ± 2.5 kcal/mol and electrostatic energy of -131.8 ± 20.5 kcal/mol indicate a well-balanced interaction. Thermolysin’s performance with MMP9 also stood out, yielding a binding energy of -10.6 kcal/mol with a HADDOCK score of -114.4 ± 3.8 a.u., and it demonstrated a stable RMSD of 1.2 ± 0.2 Å. Pleurocidin, another top-performing peptide, demonstrated robust binding across multiple proteins. In complex with ACE2, Pleurocidin achieved a HADDOCK score of -104.8 ± 1.9 a.u. and a binding energy of -11.2 kcal/mol, with a van der Waals energy of -46.6 ± 6.0 kcal/mol and an electrostatic energy of -220.6 ± 23.5 kcal/mol, indicating strong non-covalent interactions. Similarly, Pleurocidin exhibited a strong interaction with MMP9, with a HADDOCK score of -143.7 ± 4.0 a.u. and a binding energy of -9.7 kcal/mol. The stability of these interactions was underscored by an RMSD of 1.2 ± 0.1 Å. Subtilisin A demonstrated strong binding interactions, particularly with CRP and NLRP3. In the CRP complex, Subtilisin A achieved a HADDOCK score of -128.1 ± 8.3 a.u. and a binding energy of -12.0 kcal/mol, significantly surpassing the binding energy of DX600 (-9.7 kcal/mol). The electrostatic energy was -306.9 ± 57.8 kcal/mol, indicating powerful electrostatic interactions, while the RMSD value of 0.6 ± 0.5 Å reflects a stable interaction. In the NLRP3 complex, Subtilisin A showed similar effectiveness, with a HADDOCK score of -138.7 ± 2.8 a.u. and a binding energy of -12.1 kcal/mol, demonstrating its strong and consistent performance. The results, as summarized in Table 3, demonstrate the strong binding affinity and stability of the top-performing AMPs—particularly Tachystatin, Thermolysin, Pleurocidin, and Subtilisin A—toward key protein targets implicated in infection pathways related to CVDs. These AMPs exhibit the potential to inhibit receptor-mediated adhesion and signaling processes, which play a critical role in infection onset and progression. Their stable interactions with these receptors suggest promising therapeutic applications in preventing CVD-associated infections. Detailed molecular docking results are available in Supplementary Data S2.
The correlation matrix depicted in Figure 3 provides a detailed analysis of the interplay between different energy components—van der Waals energy, electrostatic energy, and desolvation energy—and their contributions to the binding energy of AMP-receptor complexes, specifically in receptors associated with infection-related CVDs. This matrix is essential for understanding the nuances of molecular interactions that govern the stability and affinity of AMP binding to these receptors. The correlation coefficients, ranging from -1 to 1, indicate the strength and direction of the relationships between binding energy and individual energy components. Positive values suggest a direct relationship, while negative values indicate an inverse relationship, offering valuable insights into the binding mechanisms of these AMP-receptor complexes. In the ACE2 complexes, a moderate positive correlation (r = 0.68) between binding energy and van der Waals energy suggests that van der Waals interactions significantly stabilize these complexes. This implies that the physical interactions between the AMP and the ACE2 receptor are primarily driven by non-covalent van der Waals forces, contributing to a strong binding affinity. In contrast, electrostatic energy (r = 0.12) and desolvation energy (r = 0.31) show much weaker correlations, indicating that these energy components have a minimal impact on the overall binding energy in ACE2 complexes. The dominance of van der Waals interactions in these complexes suggests that designing AMP-based therapies targeting ACE2 receptors for preventing infection-related CVDs should prioritize optimizing hydrophobic and steric interactions to enhance binding stability.
For the C-reactive protein (CRP) complexes, van der Waals energy (r = 0.61) also shows a significant positive correlation, reinforcing the importance of these interactions in maintaining strong AMP-receptor binding. However, electrostatic energy exhibits a negative correlation (r = -0.27), suggesting that unfavorable electrostatic interactions may slightly weaken the binding affinity. The relatively modest positive correlation for desolvation energy (r = 0.29) indicates that solvation effects do not play a significant role in these complexes. The results imply that, while van der Waals forces are crucial, electrostatic repulsion may limit the binding efficiency of AMPs to CRP receptors. In the case of MMP9 receptor complexes, van der Waals energy (r = 0.39) shows a weaker correlation with binding energy than the other receptors, suggesting a reduced contribution of hydrophobic interactions to the binding affinity. Both electrostatic energy (r = -0.13) and desolvation energy (r = 0.019) display near-zero correlations, indicating that these forces have a negligible impact on binding stability. This suggests that, for MMP9 complexes, neither van der Waals nor electrostatic interactions are particularly dominant. This may point to other factors, such as peptide conformation or flexibility, playing a more prominent role in binding affinity.
NLRP3 complexes present a more balanced interaction profile, with van der Waals energy (r = 0.44) and electrostatic energy (r = 0.32) showing moderate positive correlations. In contrast, desolvation energy (r = -0.39) exhibits a strong negative correlation. This indicates that while van der Waals and electrostatic interactions contribute to binding stability, desolvation effects may destabilize these complexes. The negative impact of desolvation energy could arise from the displacement of water molecules around the receptor site, destabilizing the AMP-receptor complex. Optimizing AMPs for NLRP3 could involve minimizing the unfavorable desolvation contributions while enhancing van der Waals and electrostatic interactions. Lastly, the TLR4 complexes reveal a strong positive correlation between binding energy and van der Waals energy (r = 0.61), similar to the ACE2 and CRP complexes. This suggests that van der Waals forces are again crucial in stabilizing the AMP-TLR4 complexes. However, electrostatic energy (r = -0.032) shows a near-zero correlation, indicating minimal electrostatic contributions to the overall binding energy. The desolvation energy (r = 0.15) exhibits a weak positive correlation, suggesting that solvation effects play a relatively minor role in these complexes.
Note:
- ICs: Number of intermolecular contacts
- NIS: Non-interacting surface
The intermolecular contact (IC) and non-interacting surface (NIS) data in Table 4 provides a detailed assessment of the molecular interactions between receptors associated with infection-related CVDs and various AMPs, compared to the standard inhibitor DX600. This analysis highlights the unique interaction profiles of several top-performing AMPs, including Tachystatin, Thermolysin, Pleurocidin, and Subtilisin A, across different receptors such as ACE2, CRP, MMP9, NLRP3, and TLR4. These AMPs exhibit consistent binding performance, demonstrated through favorable intermolecular contacts across various energy categories and receptor sites, positioning them as potent inhibitors for preventing infection-related CVDs. For the ACE2 receptor, Pleurocidin, Tachystatin, and Thermolysin demonstrate higher charged-apolar and polar-apolar interactions than standard inhibitor DX600. Pleurocidin, with 23 charged-apolar and 11 polar-apolar contacts, indicates strong hydrophobic and polar interactions, suggesting a robust binding stability with ACE2. Tachystatin also presents significant charged-polar (9) and polar-apolar (12) interactions, enhancing its ability to form stable complexes. Thermolysin, with the highest charged-apolar interactions (29), further highlights its potential to form energetically favorable contacts with the receptor. These enhanced interactions, alongside relatively stable NIS values for all three AMPs, suggest that Pleurocidin, Tachystatin, and Thermolysin are strong candidates for inhibiting ACE2-mediated infection pathways in CVDs. Subtilisin A and Pleurocidin stand out in CRP receptor complexes with substantial intermolecular contacts. Subtilisin A exhibits 29 charged-apolar, 23 polar-apolar, and 14 apolar-apolar interactions, outperforming the standard DX600 in every category. This strong interaction profile and a high NIS apolar value (43.98) suggest that Subtilisin A can efficiently block CRP’s role in infection processes linked to CVDs. Pleurocidin also shows considerable interaction strengths, with 24 charged-apolar and 18 polar-apolar contacts, making it a competitive AMP in the CRP complex. Both peptides demonstrate high binding affinity, which could disrupt CRP’s function in inflammatory responses associated with CVDs.
For the MMP9 receptor, Thermolysin and Pleurocidin again show superior performance. Thermolysin, with 29 charged-apolar and 11 polar-apolar interactions, demonstrates a clear advantage in forming hydrophobic interactions crucial for MMP9 inhibition. Pleurocidin, with 27 charged-apolar interactions, also shows strong binding potential, supported by this complex’s highest apolar-apolar contact count (34). These AMPs outperform the standard inhibitor DX600, which has only 26 charged-apolar contacts, suggesting that Thermolysin and Pleurocidin can better interfere with MMP9’s role in infection-related tissue damage in CVDs. In the NLRP3 receptor, Tachystatin, Thermolysin, and Pleurocidin again exhibit consistently strong binding characteristics. Tachystatin displays many polar-polar (5) and apolar-apolar (11) contacts, reinforcing its stability within the NLRP3 complex. Thermolysin, with 31 charged-apolar and 8 polar-apolar interactions, showcases its potent hydrophobic interaction profile, while Pleurocidin leads with the highest charged-apolar contact count (31), emphasizing its binding efficiency. These interaction profiles indicate that these AMPs can effectively inhibit NLRP3, potentially reducing its involvement in inflammatory responses during infection-related CVDs. Finally, in TLR4 receptor complexes, Tachystatin, Subtilisin A, and Pleurocidin demonstrate strong intermolecular contacts. Tachystatin, with 24 charged-apolar and 26 polar-apolar contacts, highlights its capacity to engage with both charged and polar regions of the receptor. With 28 apolar-apolar interactions, Subtilisin A presents a solid hydrophobic binding potential. In contrast, Pleurocidin’s interaction profile, including 20 charged-apolar and 25 polar-apolar contacts, shows its versatility in forming intermolecular bonds. These interactions and comparable NIS values suggest that these AMPs can effectively inhibit TLR4-mediated infection pathways, often linked to inflammation and cardiovascular complications.
Table 5 presents a detailed examination of hydrogen bond interactions between infection-related CVD receptor complexes and AMPs compared with the standard inhibitor. For the ACE2 complex, strong interactions were observed, with the shortest hydrogen bond distance being 2.58 Å between Glu23 of ACE2 and Lys24 of Hepcidin. In the CRP complex, notable interactions include the bond between Glu147 of CRP and Lys22 of Nisin at a distance of 2.66 Å. MMP9 shows multiple hydrogen bonds, with a prominent bond between Glu111 of MMP9 and Leu6 of Tachystatin at 2.66 Å, indicating stable interaction. Similarly, the NLRP3 A complex reveals strong binding with the shortest bond between Arg452 of NLRP3 and Glu23 of Subtilisin A, both at 2.61 Å. Finally, TLR4 interactions show consistent hydrogen bonding, particularly between His458 of TLR4 and Val12 of Tachystatin at 2.69 Å, contributing to the peptide’s binding efficacy. These hydrogen bonds indicate that the antimicrobial peptides, particularly Tachystatin and Subtilisin A, form strong and stable interactions with their respective receptors, comparable to or exceeding the standard inhibitor.
3.2. Molecular Dynamics (MD) Simulations
Table 6 overviews the time-averaged structural properties obtained from molecular dynamics (MD) simulations of target receptor-AMP complexes. The data reveal that Tachystatin, Thermolysin, Pleurocidin, and Subtilisin A exhibit notable structural stability and binding characteristics compared to the standard inhibitor, DX600 peptide, across various receptors associated with infection-related CVDs. For ACE2 complexes, Tachystatin shows a higher average RMSD (3.327 Å) and average RMSF (0.747 Å) compared to the standard inhibitor DX600 peptide (RMSD: 3.559 Å, RMSF: 0.792 Å), indicating a slight increase in conformational fluctuations and structural deviation. However, Tachystatin has the highest average number of hydrogen bonds (53) and the most favorable potential energy (-660,317.634 kcal/mol), suggesting that it forms more stable and energetically favorable interactions with ACE2 than DX600 peptide.
In CRP complexes, Subtilisin A and Pleurocidin display better structural stability with average RMSD values of 1.977 Å and 2.313 Å, respectively, compared to the standard inhibitor DX600 peptide (2.402 Å). Subtilisin A also has a comparable number of hydrogen bonds (16) and lower potential energy (-166,979.914 kcal/mol) than DX600 peptide (-184,719.286 kcal/mol), indicating efficient binding and favorable energetics. Pleurocidin exhibits a similar number of hydrogen bonds (16) and lower potential energy (-143,351.144 kcal/mol) than the DX600 peptide. For MMP9 complexes, Tachystatin and Thermolysin demonstrate lower average RMSD values (2.209 Å and 1.992 Å, respectively) compared to the standard inhibitor DX600 peptide (2.555 Å), indicating better structural stability. Tachystatin has a higher average number of hydrogen bonds (13) and more favorable potential energy (-195,363.451 kcal/mol) compared to DX600 peptide (-164,237.839 kcal/mol), suggesting that Tachystatin provides more stable and energetically favorable interactions with MMP9. In NLRP3 complexes, Tachystatin and Subtilisin A exhibit better structural stability with average RMSD values of 3.459 Å and 3.621 Å, respectively, compared to the standard inhibitor DX600 peptide (3.636 Å). Tachystatin also shows a higher number of hydrogen bonds (49) and the most favorable potential energy (-1,467,888.897 kcal/mol) among the peptides tested, indicating that it forms highly stable and energetically favorable interactions with NLRP3 compared to DX600 peptide. Finally, in TLR4 complexes, Tachystatin and Subtilisin A have lower average RMSD values (2.914 Å and 3.106 Å) compared to the standard inhibitor DX600 peptide (2.968 Å). Tachystatin also exhibits a higher number of hydrogen bonds (46) and more favorable potential energy (-780,309.925 kcal/mol) than DX600 peptide (-755,432.195 kcal/mol), suggesting that Tachystatin provides a more stable and energetically favorable binding interaction with TLR4.
RMSF values offer a detailed view of residue flexibility within receptors associated with infection-related CVD-AMP complexes, as depicted in Figure 4. The results indicate that the AMPs exhibit a notable correspondence with the standard inhibitor, DX600 peptide, regarding residue flexibility, suggesting that these AMPs can disrupt receptor stability similarly to the standard inhibitor. The ACE2 complexes’ RMSF patterns of Pleurocidin, Tachystatin, and Thermolysin closely resemble those of the standard inhibitor, particularly within residues Asn330 to Asp355. In CRP complexes, the RMSF values of these AMPs show a strong correlation with the standard inhibitor around residues Ala55 to Ile65 and Glu130 to Asp155. For MMP9 complexes, the RMSF profiles of Pleurocidin, Tachystatin, and Thermolysin match those of the standard inhibitor in residues Ile125 to Asp138. In the NLRP3 and TLR4 complexes, the RMSF values of the AMPs align closely with those of the standard inhibitor in crucial binding regions, including residues Ser161 to His175 and Lys375 to Asn400 for NLRP3, and Ser360 to Leu380 and Gln510 to Leu535 for TLR4. Overall, the RMSF data highlight that the AMPs can disrupt receptor stability in a manner similar to the standard inhibitor. The ability of these AMPs to induce comparable flexibility in critical binding regions underscores their potential as effective disruptors of receptor stability, akin to the DX600 peptide.
3.3. Molecular Mechanics/Poisson–Boltzmann Surface Area (MM/PBSA) Calculations
The binding affinities of selected AMPs for target receptors were assessed using MM/PBSA calculations (based on the MD simulation), with results in Table 7. Among the peptides evaluated, Tachystatin, Pleurocidin, and Subtilisin A emerged as the most consistent in exhibiting favorable binding energies. For ACE2 complexes, Tachystatin stands out with an average binding energy of -61.58 kcal/mol, significantly more favorable than the standard inhibitor DX600 peptide, which has an average binding energy of -22.28 kcal/mol. Pleurocidin also shows strong binding with an average energy of -46.58 kcal/mol, while Subtilisin A’s binding affinity is slightly less favorable at -44.82 kcal/mol. These results suggest that Tachystatin and Pleurocidin exhibit superior binding capabilities compared to the standard inhibitor, with Tachystatin showing the most significant potential. In CRP complexes, Subtilisin A exhibits the most favorable binding energy with an average of -70.71 kcal/mol, followed by Protegrin-1 at -67.56 kcal/mol and Nisin at -38.73 kcal/mol. This contrasts with the standard inhibitor DX600 peptide, which has an average binding energy of -27.99 kcal/mol. The superior binding energy of Subtilisin A and Protegrin-1 in CRP complexes underscores their effectiveness compared to the standard inhibitor.
For MMP9 complexes, Tachystatin again demonstrates the highest binding affinity with an average energy of -96.61 kcal/mol, followed closely by Pleurocidin at -94.24 kcal/mol. These values are significantly lower (more favorable) than the standard inhibitor DX600 peptide, which has an average energy of -52.61 kcal/mol. The binding energies of Tachystatin and Pleurocidin indicate their strong interaction with MMP9, surpassing that of the standard inhibitor. In NLRP3 complexes, Subtilisin A and Tachystatin exhibit comparable binding affinities with averages of -71.12 kcal/mol and -69.20 kcal/mol, respectively, outperforming the standard inhibitor DX600 peptide, which has an average of -44.35 kcal/mol. This highlights the superior binding potential of Subtilisin A and Tachystatin for NLRP3. Finally, in TLR4 complexes, Tachystatin displays a favorable binding energy of -59.69 kcal/mol, more favorable than the standard inhibitor DX600 peptide, with an average energy of -32.82 kcal/mol. Subtilisin A’s average binding energy is -43.24 kcal/mol, indicating that it also binds effectively, though less so than Tachystatin.
4. Discussion
4.1. Key Findings from Molecular Simulations and Their Correlation with Other Research
The molecular docking and dynamics simulations conducted in this study provided significant insights into the interactions between AMPs and receptors implicated in infection-related CVDs. These findings highlight the potential of AMPs as therapeutic agents and contribute to our understanding of their binding mechanisms and stability compared to conventional inhibitors. Our simulations highlight that several AMPs—Tachystatin, Thermolysin, Pleurocidin, and Subtilisin A—demonstrate significant binding affinity towards key receptors such as ACE2, MMP9, CRP, NLRP3, and TLR4. These peptides exhibited strong binding interactions, as indicated by favorable HADDOCK scores and binding energies. Notably, Tachystatin and Thermolysin showed particularly strong binding with ACE2, and Pleurocidin performed well with multiple receptors, including ACE2 and MMP9. These findings are consistent with previous studies that have demonstrated the potential of AMPs to modulate receptor activity in various disease contexts [88,89]. In relation to prior research, Tachystatin’s superior binding affinity for ACE2 aligns with a finding that peptides targeting ACE2 could modulate its activity and influence cardiovascular disease outcomes [90]. Similarly, the strong binding of Thermolysin and Pleurocidin to MMP9 and CRP reflects their potential to target inflammation-related pathways. These corroborating studies emphasize the role of peptides in controlling matrix metalloproteinase activity and inflammatory responses [16,91,92]. MD simulations provided insights into the stability and conformational dynamics of AMP-receptor complexes. Tachystatin and Pleurocidin, in particular, displayed favorable structural stability with lower RMSD and RMSF values compared to the standard inhibitor DX600. These observations are supported by previous studies showing the importance of peptide stability in maintaining therapeutic efficacy [93,94]. The consistent hydrogen bonding patterns and favorable potential energies observed in our simulations underscore the potential of these AMPs to form stable and energetically favorable interactions with their targets. Furthermore, our results suggest that van der Waals interactions are crucial for binding stability, as corroborated by studies that emphasize the role of non-covalent interactions in peptide-receptor binding [95,96]. Our molecular simulations provide valuable insights into the binding mechanisms and stability of AMPs interacting with infection-related CVD receptors. The results validate previous research on peptide-receptor interactions and highlight the potential of these AMPs as therapeutic candidates for modulating infection-related pathways in CVDs.
4.2. Clinical Implications and Limitations
Exploring peptide-receptor interactions has significant clinical implications, particularly in developing targeted therapies and precision medicine. Understanding the fundamental mechanisms by which peptides bind to their receptors provides crucial insights for designing novel therapeutics that target specific biological pathways more effectively. For instance, inhibiting MMPs by peptides has been shown to play a critical role in managing cardiovascular diseases by preventing the degradation of extracellular matrix components, thereby improving vascular stability and function [92,97]. Similarly, peptides that target ACE2 could potentially modulate cardiovascular disease outcomes, offering new avenues for treatment [90]. These findings underscore the importance of peptide-receptor interactions in crafting targeted therapeutic interventions. However, several limitations associated with peptide-based therapies must be addressed. One primary challenge is the stability of peptides in physiological conditions, which often impacts their efficacy. For example, peptides are susceptible to rapid degradation by proteolytic enzymes, reducing their therapeutic potential and necessitating the development of strategies to enhance their stability [98,99]. Additionally, the specificity of peptide-receptor interactions is crucial for minimizing off-target effects and maximizing therapeutic outcomes [100]. While advances in computational modeling and simulations have improved our understanding of these interactions, translating these findings into clinical applications remains challenging [101]. Another limitation is the potential for adverse immune responses to peptide-based therapies. Peptides can sometimes trigger immune reactions, leading to reduced patient efficacy or adverse effects [102,103]. Furthermore, the cost and complexity of developing and producing peptide-based drugs can be significant, potentially limiting their accessibility and widespread use [104,105]. Therefore, while peptide-based therapeutics offer promising prospects, ongoing research and development are needed to address these limitations and optimize their clinical application.
5. Conclusions
In conclusion, the molecular docking and dynamics simulations provide compelling evidence of the efficacy of AMPs in targeting receptors implicated in infection-related CVDs. Our simulations highlight that Tachystatin, Thermolysin, Pleurocidin, and Subtilisin A exhibit superior binding affinities and stability with key receptors such as ACE2, CRP, MMP9, NLRP3, and TLR4 compared to the standard inhibitor DX600. Tachystatin consistently strongly binds with multiple receptors, including ACE2 and MMP9, showing favorable HADDOCK scores and binding energies. Thermolysin and Pleurocidin also emerged as potent candidates, with notable interaction profiles and stability across different receptor complexes. While showing strong performance with CRP and NLRP3, Subtilisin A also demonstrated impressive binding characteristics with TLR4. The detailed analysis of energy components, intermolecular contacts, and hydrogen bonds further underscores the potential of these AMPs to disrupt receptor-mediated infection pathways effectively. The MD simulations corroborate these findings, showing that AMPs like Tachystatin and Subtilisin A exhibit better structural stability and favorable energetics compared to DX600. These results pave the way for future therapeutic applications of AMPs in preventing and treating CVDs associated with infection and inflammation, highlighting their promise as potent candidates for further development and clinical evaluation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: Antimicrobial peptides and receptors dataset; Table S2: Complete molecular docking results; Table S3: Molecular interaction results.
Author Contributions
Conceptualization, Doni Dermawan and Nasser Alotaiq; Data curation, Nasser Alotaiq; Formal analysis, Doni Dermawan; Funding acquisition, Nasser Alotaiq; Investigation, Doni Dermawan; Methodology, Doni Dermawan and Nasser Alotaiq; Project administration, Nasser Alotaiq; Resources, Nasser Alotaiq; Software, Doni Dermawan; Supervision, Nasser Alotaiq; Validation, Doni Dermawan and Nasser Alotaiq; Visualization, Doni Dermawan; Writing – original draft, Doni Dermawan and Nasser Alotaiq; Writing – review & editing, Doni Dermawan and Nasser Alotaiq.
Funding
This work was supported and funded by the Deanship of Scientific Research at Imam Mohammad Ibn Saud Islamic University (IMSIU) (grant number IMSIU-RG23154).
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 2018;9(6):7204-18. Epub 20171214. PubMed PMID: 29467962; PubMed Central PMCID: PMC5805548. [CrossRef]
- Yang T-H, Gao W-C, Ma X, Liu Q, Pang P-P, Zheng Y-T, et al. A Review on The Pathogenesis of Cardiovascular Disease of Flaviviridea Viruses Infection. Viruses. 2024;16(3):365. PubMed PMID:. [CrossRef]
- Liu C, Waters DD. Chlamydia pneumoniae and atherosclerosis: from Koch postulates to clinical trials. Prog Cardiovasc Dis. 2005;47(4):230-9. PubMed PMID: 15991152; PubMed Central PMCID: PMC7118749. [CrossRef]
- Jung SH, Lee KT. Atherosclerosis by Virus Infection-A Short Review. Biomedicines. 2022;10(10). Epub 20221019. PubMed PMID: 36289895; PubMed Central PMCID: PMC9599298. [CrossRef]
- Wolf D, Ley K. Immunity and Inflammation in Atherosclerosis. Circ Res. 2019;124(2):315-27. PubMed PMID: 30653442; PubMed Central PMCID: PMC6342482. [CrossRef]
- Laera N, Malerba P, Vacanti G, Nardin S, Pagnesi M, Nardin M. Impact of Immunity on Coronary Artery Disease: An Updated Pathogenic Interplay and Potential Therapeutic Strategies. Life (Basel). 2023;13(11). Epub 20231027. PubMed PMID: 38004268; PubMed Central PMCID: PMC10672143. [CrossRef]
- Jaén RI, Val-Blasco A, Prieto P, Gil-Fernández M, Smani T, López-Sendón JL, et al. Innate Immune Receptors, Key Actors in Cardiovascular Diseases. JACC: Basic to Translational Science. 2020;5(7):735-49. [CrossRef]
- Kircheis R, Planz O. The Role of Toll-like Receptors (TLRs) and Their Related Signaling Pathways in Viral Infection and Inflammation. Int J Mol Sci. 2023;24(7). Epub 20230404. PubMed PMID: 37047674; PubMed Central PMCID: PMC10095430. [CrossRef]
- Goulopoulou S, McCarthy CG, Webb RC. Toll-like Receptors in the Vascular System: Sensing the Dangers Within. Pharmacol Rev. 2016;68(1):142-67. PubMed PMID: 26721702; PubMed Central PMCID: PMC4709508. [CrossRef]
- Tanase DM, Valasciuc E, Gosav EM, Ouatu A, Buliga-Finis ON, Floria M, et al. Portrayal of NLRP3 Inflammasome in Atherosclerosis: Current Knowledge and Therapeutic Targets. Int J Mol Sci. 2023;24(9). Epub 20230503. PubMed PMID: 37175869; PubMed Central PMCID: PMC10179095. [CrossRef]
- Karasawa T, Takahashi M. Role of NLRP3 Inflammasomes in Atherosclerosis. J Atheroscler Thromb. 2017;24(5):443-51. Epub 20170304. PubMed PMID: 28260724; PubMed Central PMCID: PMC5429158. [CrossRef]
- Bourgonje AR, Abdulle AE, Timens W, Hillebrands JL, Navis GJ, Gordijn SJ, et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J Pathol. 2020;251(3):228-48. Epub 20200610. PubMed PMID: 32418199; PubMed Central PMCID: PMC7276767. [CrossRef]
- Liu LP, Zhang XL, Li J. New perspectives on angiotensin-converting enzyme 2 and its related diseases. World J Diabetes. 2021;12(6):839-54. PubMed PMID: 34168732; PubMed Central PMCID: PMC8192247. [CrossRef]
- Cao Q, Lei H, Yang M, Wei L, Dong Y, Xu J, et al. Impact of Cardiovascular Diseases on COVID-19: A Systematic Review. Med Sci Monit. 2021;27:e930032. Epub 20210406. PubMed PMID: 33820904; PubMed Central PMCID: PMC8035813. [CrossRef]
- Olejarz W, Łacheta D, Kubiak-Tomaszewska G. Matrix Metalloproteinases as Biomarkers of Atherosclerotic Plaque Instability. Int J Mol Sci. 2020;21(11). Epub 20200531. PubMed PMID: 32486345; PubMed Central PMCID: PMC7313469. [CrossRef]
- Liu P, Sun M, Sader S. Matrix metalloproteinases in cardiovascular disease. Can J Cardiol. 2006;22 Suppl B(Suppl B):25b-30b. PubMed PMID: 16498509; PubMed Central PMCID: PMC2780831. [CrossRef]
- Porritt RA, Crother TR. Chlamydia pneumoniae Infection and Inflammatory Diseases. For Immunopathol Dis Therap. 2016;7(3-4):237-54. PubMed PMID: 30687565; PubMed Central PMCID: PMC6345537. [CrossRef]
- Muhlestein JB, Anderson JL, Hammond EH, Zhao L, Trehan S, Schwobe EP, Carlquist JF. Infection With Chlamydia pneumoniae Accelerates the Development of Atherosclerosis and Treatment With Azithromycin Prevents It in a Rabbit Model. Circulation. 1998;97(7):633-6. [CrossRef]
- Llor C, Bjerrum L. Antimicrobial resistance: risk associated with antibiotic overuse and initiatives to reduce the problem. Ther Adv Drug Saf. 2014;5(6):229-41. PubMed PMID: 25436105; PubMed Central PMCID: PMC4232501. [CrossRef]
- Muteeb G, Rehman MT, Shahwan M, Aatif M. Origin of Antibiotics and Antibiotic Resistance, and Their Impacts on Drug Development: A Narrative Review. Pharmaceuticals (Basel). 2023;16(11). Epub 20231115. PubMed PMID: 38004480; PubMed Central PMCID: PMC10675245. [CrossRef]
- Ahmed SK, Hussein S, Qurbani K, Ibrahim RH, Fareeq A, Mahmood KA, Mohamed MG. Antimicrobial resistance: Impacts, challenges, and future prospects. Journal of Medicine, Surgery, and Public Health. 2024;2:100081. [CrossRef]
- Heianza Y, Zheng Y, Ma W, Rimm EB, Albert CM, Hu FB, et al. Duration and life-stage of antibiotic use and risk of cardiovascular events in women. Eur Heart J. 2019;40(47):3838-45. PubMed PMID: 31216010; PubMed Central PMCID: PMC6911167. [CrossRef]
- Luong HX, Thanh TT, Tran TH. Antimicrobial peptides - Advances in development of therapeutic applications. Life Sci. 2020;260:118407. Epub 20200912. PubMed PMID: 32931796; PubMed Central PMCID: PMC7486823. [CrossRef]
- Xuan J, Feng W, Wang J, Wang R, Zhang B, Bo L, et al. Antimicrobial peptides for combating drug-resistant bacterial infections. Drug Resistance Updates. 2023;68:100954. [CrossRef]
- Benfield AH, Henriques ST. Mode-of-Action of Antimicrobial Peptides: Membrane Disruption vs. Intracellular Mechanisms. Front Med Technol. 2020;2:610997. Epub 20201211. PubMed PMID: 35047892; PubMed Central PMCID: PMC8757789. [CrossRef]
- Gong H, Hu X, Zhang L, Fa K, Liao M, Liu H, et al. How do antimicrobial peptides disrupt the lipopolysaccharide membrane leaflet of Gram-negative bacteria? Journal of Colloid and Interface Science. 2023;637:182-92. [CrossRef]
- Zhang Q-Y, Yan Z-B, Meng Y-M, Hong X-Y, Shao G, Ma J-J, et al. Antimicrobial peptides: mechanism of action, activity and clinical potential. Military Medical Research. 2021;8(1):48. [CrossRef]
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18(15):2714-23. PubMed PMID: 9504803. [CrossRef]
- Tian W, Chen C, Lei X, Zhao J, Liang J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 2018;46(1):363-7. [CrossRef]
- Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215-20. [CrossRef]
- Thompson D, Pepys MB, Wood SP. The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure. 1999;7(2):169-77. [CrossRef]
- Rowsell S, Hawtin P, Minshull CA, Jepson H, Brockbank SMV, Barratt DG, et al. Crystal Structure of Human MMP9 in Complex with a Reverse Hydroxamate Inhibitor. Journal of Molecular Biology. 2002;319(1):173-81. [CrossRef]
- Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature. 2019;570(7761):338-43. [CrossRef]
- Park BS, Song DH, Kim HM, Choi B-S, Lee H, Lee J-O. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature. 2009;458(7242):1191-5. [CrossRef]
- Wang G, Li Y, Li X. Correlation of Three-dimensional Structures with the Antibacterial Activity of a Group of Peptides Designed Based on a Nontoxic Bacterial Membrane Anchor *. Journal of Biological Chemistry. 2005;280(7):5803-11. [CrossRef]
- Hoover DM, Rajashankar KR, Blumenthal R, Puri A, Oppenheim JJ, Chertov O, Lubkowski J. The Structure of Human β-Defensin-2 Shows Evidence of Higher Order Oligomerization *. Journal of Biological Chemistry. 2000;275(42):32911-8. [CrossRef]
- Zangger K, Gößler R, Khatai L, Lohner K, Jilek A. Structures of the glycine-rich diastereomeric peptides bombinin H2 and H4. Toxicon. 2008;52(2):246-54. [CrossRef]
- Wang G. Structures of Human Host Defense Cathelicidin LL-37 and Its Smallest Antimicrobial Peptide KR-12 in Lipid Micelles *<sup></sup>. Journal of Biological Chemistry. 2008;283(47):32637-43. [CrossRef]
- Oh D, Shin SY, Kang JH, Hahm KS, Kim KL, Kim Y. NMR structural characterization of cecropin A(1-8) - magainin 2(1-12) and cecropin A (1-8) - melittin (1-12) hybrid peptides. J Pept Res. 1999;53(5):578-89. PubMed PMID: 10424354. [CrossRef]
- Viana de Freitas T, Karmakar U, Vasconcelos AG, Santos MA, Oliveira do Vale Lira B, Costa SR, et al. Release of immunomodulatory peptides at bacterial membrane interfaces as a novel strategy to fight microorganisms. Journal of Biological Chemistry. 2023;299(4). [CrossRef]
- Nguyen VS, Tan KW, Ramesh K, Chew FT, Mok YK. Structural basis for the bacterial membrane insertion of dermcidin peptide, DCD-1L. Scientific Reports. 2017;7(1):13923. [CrossRef]
- Loffredo MR, Ghosh A, Harmouche N, Casciaro B, Luca V, Bortolotti A, et al. Membrane perturbing activities and structural properties of the frog-skin derived peptide Esculentin-1a(1-21)NH2 and its Diastereomer Esc(1-21)-1c: Correlation with their antipseudomonal and cytotoxic activity. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2017;1859(12):2327-39. [CrossRef]
- Runge S, Thøgersen H, Madsen K, Lau J, Rudolph R. Crystal Structure of the Ligand-bound Glucagon-like Peptide-1 Receptor Extracellular Domain * . Journal of Biological Chemistry. 2008;283(17):11340-7. [CrossRef]
- Jordan JB, Poppe L, Haniu M, Arvedson T, Syed R, Li V, et al. Hepcidin Revisited, Disulfide Connectivity, Dynamics, and Structure. Journal of Biological Chemistry. 2009;284(36):24155-67. [CrossRef]
- Mariano GH, Gomes de Sá LG, Carmo da Silva EM, Santos MA, Cardozo Fh JL, Lira BOV, et al. Characterization of novel human intragenic antimicrobial peptides, incorporation and release studies from ureasil-polyether hybrid matrix. Materials Science and Engineering: C. 2021;119:111581. [CrossRef]
- Friedrich CL, Rozek A, Patrzykat A, Hancock REW. Structure and Mechanism of Action of an Indolicidin Peptide Derivative with Improved Activity against Gram-positive Bacteria *. Journal of Biological Chemistry. 2001;276(26):24015-22. [CrossRef]
- Hwang PM, Zhou N, Shan X, Arrowsmith CH, Vogel HJ. Three-Dimensional Solution Structure of Lactoferricin B, an Antimicrobial Peptide Derived from Bovine Lactoferrin. Biochemistry. 1998;37(12):4288-98. [CrossRef]
- Pillong M, Hiss JA, Schneider P, Lin Y-C, Posselt G, Pfeiffer B, et al. Rational Design of Membrane-Pore-Forming Peptides. Small. 2017;13(40):1701316. [CrossRef]
- Gesell J, Zasloff M, Opella SJ. Two-dimensional 1H NMR experiments show that the 23-residue magainin antibiotic peptide is an α-helix in dodecylphosphocholine micelles, sodium dodecylsulfate micelles, and trifluoroethanol/water solution. Journal of Biomolecular NMR. 1997;9(2):127-35. [CrossRef]
- Terwilliger TC, Weissman L, Eisenberg D. The structure of melittin in the form I crystals and its implication for melittin’s lytic and surface activities. Biophys J. 1982;37(1):353-61. PubMed PMID: 7055627; PubMed Central PMCID: PMC1329151. [CrossRef]
- Mathavan I, Zirah S, Mehmood S, Choudhury HG, Goulard C, Li Y, et al. Structural basis for hijacking siderophore receptors by antimicrobial lasso peptides. Nature Chemical Biology. 2014;10(5):340-2. [CrossRef]
- Hsu S-TD, Breukink E, Tischenko E, Lutters MAG, de Kruijff B, Kaptein R, et al. The nisin–lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nature Structural & Molecular Biology. 2004;11(10):963-7. [CrossRef]
- Bhunia A, Domadia PN, Torres J, Hallock KJ, Ramamoorthy A, Bhattacharjya S. NMR Structure of Pardaxin, a Pore-forming Antimicrobial Peptide, in Lipopolysaccharide Micelles: MECHANISM OF OUTER MEMBRANE PERMEABILIZATION 2. Journal of Biological Chemistry. 2010;285(6):3883-95. [CrossRef]
- Comert F, Greenwood A, Maramba J, Acevedo R, Lucas L, Kulasinghe T, et al. The host-defense peptide piscidin P1 reorganizes lipid domains in membranes and decreases activation energies in mechanosensitive ion channels. Journal of Biological Chemistry. 2019;294(49):18557-70. [CrossRef]
- Amos ST, Vermeer LS, Ferguson PM, Kozlowska J, Davy M, Bui TT, et al. Antimicrobial Peptide Potency is Facilitated by Greater Conformational Flexibility when Binding to Gram-negative Bacterial Inner Membranes. Sci Rep. 2016;6:37639. Epub 20161122. PubMed PMID: 27874065; PubMed Central PMCID: PMC5118786. [CrossRef]
- Powers J-PS, Rozek A, Hancock REW. Structure–activity relationships for the β-hairpin cationic antimicrobial peptide polyphemusin I. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 2004;1698(2):239-50. [CrossRef]
- 57. Fahrner RL, Dieckmann T, Harwig SS, Lehrer RI, Eisenberg D, Feigon J. Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes. Chem Biol. 1996;3(7):543-50. PubMed PMID: 8807886. [CrossRef]
- Petit VW, Rolland J-L, Blond A, Cazevieille C, Djediat C, Peduzzi J, et al. A hemocyanin-derived antimicrobial peptide from the penaeid shrimp adopts an alpha-helical structure that specifically permeabilizes fungal membranes. Biochimica et Biophysica Acta (BBA) - General Subjects. 2016;1860(3):557-68. [CrossRef]
- Kawulka KE, Sprules T, Diaper CM, Whittal RM, McKay RT, Mercier P, et al. Structure of Subtilosin A, a Cyclic Antimicrobial Peptide from Bacillus subtilis with Unusual Sulfur to α-Carbon Cross-Links: Formation and Reduction of α-Thio-α-Amino Acid Derivatives. Biochemistry. 2004;43(12):3385-95. [CrossRef]
- Kushibiki T, Kamiya M, Aizawa T, Kumaki Y, Kikukawa T, Mizuguchi M, et al. Interaction between tachyplesin I, an antimicrobial peptide derived from horseshoe crab, and lipopolysaccharide. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 2014;1844(3):527-34. [CrossRef]
- Fujitani N, Kawabata S-i, Osaki T, Kumaki Y, Demura M, Nitta K, Kawano K. Structure of the Antimicrobial Peptide Tachystatin A *. Journal of Biological Chemistry. 2002;277(26):23651-7. [CrossRef]
- Manzo G, Ferguson PM, Hind CK, Clifford M, Gustilo VB, Ali H, et al. Temporin L and aurein 2.5 have identical conformations but subtly distinct membrane and antibacterial activities. Scientific Reports. 2019;9(1):10934. [CrossRef]
- Mandard N, Sodano P, Labbe H, Bonmatin J-M, Bulet P, Hetru C, et al. Solution structure of thanatin, a potent bactericidal and fungicidal insect peptide, determined from proton two-dimensional nuclear magnetic resonance data. European Journal of Biochemistry. 1998;256(2):404-10. [CrossRef]
- Fiebig D, Storka J, Roeder M, Meyners C, Schmelz S, Blankenfeldt W, et al. Destructive twisting of neutral metalloproteases: the catalysis mechanism of the Dispase autolysis-inducing protein from Streptomyces mobaraensis DSM 40487. The FEBS Journal. 2018;285(22):4246-64. [CrossRef]
- Mehdi SF, Pusapati S, Anwar MS, Lohana D, Kumar P, Nandula SA, et al. Glucagon-like peptide-1: a multi-faceted anti-inflammatory agent. Front Immunol. 2023;14:1148209. Epub 20230517. PubMed PMID: 37266425; PubMed Central PMCID: PMC10230051. [CrossRef]
- Amezcua-Castillo E, González-Pacheco H, Sáenz-San Martín A, Méndez-Ocampo P, Gutierrez-Moctezuma I, Massó F, et al. C-Reactive Protein: The Quintessential Marker of Systemic Inflammation in Coronary Artery Disease-Advancing toward Precision Medicine. Biomedicines. 2023;11(9). Epub 20230902. PubMed PMID: 37760885; PubMed Central PMCID: PMC10525787. [CrossRef]
- Sortino O, Hullsiek KH, Richards E, Rupert A, Schminke A, Tetekpor N, et al. The Effects of Recombinant Human Lactoferrin on Immune Activation and the Intestinal Microbiome Among Persons Living with Human Immunodeficiency Virus and Receiving Antiretroviral Therapy. J Infect Dis. 2019;219(12):1963-8. PubMed PMID: 30721997; PubMed Central PMCID: PMC6784498. [CrossRef]
- Agier J, Efenberger M, Brzezińska-Błaszczyk E. Cathelicidin impact on inflammatory cells. Cent Eur J Immunol. 2015;40(2):225-35. Epub 20150803. PubMed PMID: 26557038; PubMed Central PMCID: PMC4637384. [CrossRef]
- Dominguez C, Boelens R, Bonvin AMJJ. HADDOCK: A Protein−Protein Docking Approach Based on Biochemical or Biophysical Information. Journal of the American Chemical Society. 2003;125(7):1731-7. [CrossRef]
- Huang L, Sexton DJ, Skogerson K, Devlin M, Smith R, Sanyal I, et al. Novel peptide inhibitors of angiotensin-converting enzyme 2. J Biol Chem. 2003;278(18):15532-40. Epub 20030226. PubMed PMID: 12606557. [CrossRef]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583-9. [CrossRef]
- Vangone A, Bonvin A. PRODIGY: A Contact-based Predictor of Binding Affinity in Protein-protein Complexes. BIO-PROTOCOL. 2017;7. [CrossRef]
- Grassmann G, Miotto M, Desantis F, Di Rienzo L, Tartaglia GG, Pastore A, et al. Computational Approaches to Predict Protein–Protein Interactions in Crowded Cellular Environments. Chemical Reviews. 2024;124(7):3932-77. [CrossRef]
- Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845-54. [CrossRef]
- Robertson MJ, Tirado-Rives J, Jorgensen WL. Improved Peptide and Protein Torsional Energetics with the OPLSAA Force Field. J Chem Theory Comput. 2015;11(7):3499-509. Epub 2015/07/21. PubMed PMID: 26190950; PubMed Central PMCID: PMC4504185. [CrossRef]
- Yuet P, Blankschtein D. Molecular Dynamics Simulation Study of Water Surfaces: Comparison of Flexible Water Models. The journal of physical chemistry B. 2010;114:13786-95. [CrossRef]
- Dermawan D, Sumirtanurdin R, Dewantisari D. Simulasi dinamika molekular reseptor estrogen alfa dengan andrografolid sebagai anti kanker payudara. Indones J Pharm Sci Technol. 2019;6(2):65-76.
- Saini RS, Binduhayyim RIH, Gurumurthy V, Alshadidi AAF, Aldosari LIN, Okshah A, et al. Dental biomaterials redefined: molecular docking and dynamics-driven dental resin composite optimization. BMC Oral Health. 2024;24(1):557. [CrossRef]
- Schrödinger. The PyMOL Molecular Graphics System. 2.4 ed2020.
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605-12. PubMed PMID: 15264254. [CrossRef]
- Tian S, Sun H, Pan P, Li D, Zhen X, Li Y, Hou T. Assessing an ensemble docking-based virtual screening strategy for kinase targets by considering protein flexibility. J Chem Inf Model. 2014;54(10):2664-79. Epub 20140929. PubMed PMID: 25233367. [CrossRef]
- Yuan Z, Chen X, Fan S, Chang L, Chu L, Zhang Y, et al. Binding Free Energy Calculation Based on the Fragment Molecular Orbital Method and Its Application in Designing Novel SHP-2 Allosteric Inhibitors. Int J Mol Sci [Internet]. 2024; 25(1):[1-24 pp.].
- Rifai EA, Ferrario V, Pleiss J, Geerke DP. Combined Linear Interaction Energy and Alchemical Solvation Free-Energy Approach for Protein-Binding Affinity Computation. J Chem Theory Comput. 2020;16(2):1300-10. [CrossRef]
- Valdés-Tresanco MS, Valdés-Tresanco ME, Valiente PA, Moreno E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. Journal of Chemical Theory and Computation. 2021;17(10):6281-91. [CrossRef]
- Miller BR, 3rd, McGee TD, Jr., Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. Journal of chemical theory and computation. 2012;8(9):3314-21. [CrossRef]
- Panday SK, Alexov E. Protein-Protein Binding Free Energy Predictions with the MM/PBSA Approach Complemented with the Gaussian-Based Method for Entropy Estimation. ACS Omega. 2022;7(13):11057-67. Epub 2022/04/14. PubMed PMID: 35415339; PubMed Central PMCID: PMC8991903. [CrossRef]
- Nnyigide OS, Lee SG, Hyun K. In Silico Characterization of the Binding Modes of Surfactants with Bovine Serum Albumin. Sci Rep. 2019;9(1):10643. Epub 20190723. PubMed PMID: 31337814; PubMed Central PMCID: PMC6650617. [CrossRef]
- Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415(6870):389-95. [CrossRef]
- Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol. 2003;3(9):710-20. PubMed PMID: 12949495. [CrossRef]
- Festa M, Sansone C, Brunet C, Crocetta F, Di Paola L, Lombardo M, et al. Cardiovascular Active Peptides of Marine Origin with ACE Inhibitory Activities: Potential Role as Anti-Hypertensive Drugs and in Prevention of SARS-CoV-2 Infection. Int J Mol Sci. 2020;21(21). Epub 20201107. PubMed PMID: 33171852; PubMed Central PMCID: PMC7664667. [CrossRef]
- Lee RT. Matrix metalloproteinase inhibition and the prevention of heart failure. Trends Cardiovasc Med. 2001;11(5):202-5. PubMed PMID: 11597832. [CrossRef]
- Ndinguri MW, Bhowmick M, Tokmina-Roszyk D, Robichaud TK, Fields GB. Peptide-based selective inhibitors of matrix metalloproteinase-mediated activities. Molecules. 2012;17(12):14230-48. Epub 20121130. PubMed PMID: 23201642; PubMed Central PMCID: PMC3678729. [CrossRef]
- Al Musaimi O, Lombardi L, Williams DR, Albericio F. Strategies for Improving Peptide Stability and Delivery. Pharmaceuticals (Basel). 2022;15(10). Epub 20221019. PubMed PMID: 36297395; PubMed Central PMCID: PMC9610364. [CrossRef]
- Hossain MS, Shovon MTI, Hasan MR, Hakim FT, Hasan MM, Esha SA, et al. Therapeutic Potential of Antiviral Peptides against the NS2B/NS3 Protease of Zika Virus. ACS Omega. 2023;8(38):35207-18. Epub 20230913. PubMed PMID: 37779969; PubMed Central PMCID: PMC10536883. [CrossRef]
- Kastritis PL, Bonvin AM. On the binding affinity of macromolecular interactions: daring to ask why proteins interact. J R Soc Interface. 2013;10(79):20120835. Epub 20121212. PubMed PMID: 23235262; PubMed Central PMCID: PMC3565702. [CrossRef]
- Lin D, Sun L-C, Huo W-S, Zhang L-J, Chen Y-L, Miao S, Cao M-J. Improved functionality and safety of peptides by the formation of peptide-polyphenol complexes. Trends in Food Science & Technology. 2023;141:104193. [CrossRef]
- Fan Z, Fu M, Xu Z, Zhang B, Li Z, Li H, et al. Sustained Release of a Peptide-Based Matrix Metalloproteinase-2 Inhibitor to Attenuate Adverse Cardiac Remodeling and Improve Cardiac Function Following Myocardial Infarction. Biomacromolecules. 2017;18(9):2820-9. [CrossRef]
- Evans BJ, King AT, Katsifis A, Matesic L, Jamie JF. Methods to Enhance the Metabolic Stability of Peptide-Based PET Radiopharmaceuticals. Molecules. 2020;25(10). Epub 20200514. PubMed PMID: 32423178; PubMed Central PMCID: PMC7287708. [CrossRef]
- Böttger R, Hoffmann R, Knappe D. Differential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serum. PLoS One. 2017;12(6):e0178943. Epub 20170602. PubMed PMID: 28575099; PubMed Central PMCID: PMC5456363. [CrossRef]
- Cavallaro PA, De Santo M, Belsito EL, Longobucco C, Curcio M, Morelli C, et al. Peptides Targeting HER2-Positive Breast Cancer Cells and Applications in Tumor Imaging and Delivery of Chemotherapeutics. Nanomaterials (Basel). 2023;13(17). Epub 20230901. PubMed PMID: 37686984; PubMed Central PMCID: PMC10490457. [CrossRef]
- Ahn WY, Busemeyer JR. Challenges and promises for translating computational tools into clinical practice. Curr Opin Behav Sci. 2016;11:1-7. PubMed PMID: 27104211; PubMed Central PMCID: PMC4834893. [CrossRef]
- Mahadik R, Kiptoo P, Tolbert T, Siahaan TJ. Immune Modulation by Antigenic Peptides and Antigenic Peptide Conjugates for Treatment of Multiple Sclerosis. Med Res Arch. 2022;10(5). Epub 20220601. PubMed PMID: 36381196; PubMed Central PMCID: PMC9648198. [CrossRef]
- Jawa V, Terry F, Gokemeijer J, Mitra-Kaushik S, Roberts BJ, Tourdot S, De Groot AS. T-Cell Dependent Immunogenicity of Protein Therapeutics Pre-clinical Assessment and Mitigation-Updated Consensus and Review 2020. Front Immunol. 2020;11:1301. Epub 20200630. PubMed PMID: 32695107; PubMed Central PMCID: PMC7338774. [CrossRef]
- Rossino G, Marchese E, Galli G, Verde F, Finizio M, Serra M, et al. Peptides as Therapeutic Agents: Challenges and Opportunities in the Green Transition Era. Molecules. 2023;28(20). Epub 20231019. PubMed PMID: 37894644; PubMed Central PMCID: PMC10609221. [CrossRef]
- Wang L, Wang N, Zhang W, Cheng X, Yan Z, Shao G, et al. Therapeutic peptides: current applications and future directions. Signal Transduction and Targeted Therapy. 2022;7(1):48. [CrossRef]
Figure 1.
Molecular docking simulations illustrate the optimal binding orientations for interactions between AMPs and ACE2. (a) ACE2:DX600 peptide (standard antagonist) complex. (b) ACE2:Hepcidin complex. (c) ACE2:Pleurocidin complex. (d) ACE2:Tachystatin complex. (e) ACE2:Thermolysin complex. (f) ACE2:PvHCt complex.
Figure 1.
Molecular docking simulations illustrate the optimal binding orientations for interactions between AMPs and ACE2. (a) ACE2:DX600 peptide (standard antagonist) complex. (b) ACE2:Hepcidin complex. (c) ACE2:Pleurocidin complex. (d) ACE2:Tachystatin complex. (e) ACE2:Thermolysin complex. (f) ACE2:PvHCt complex.
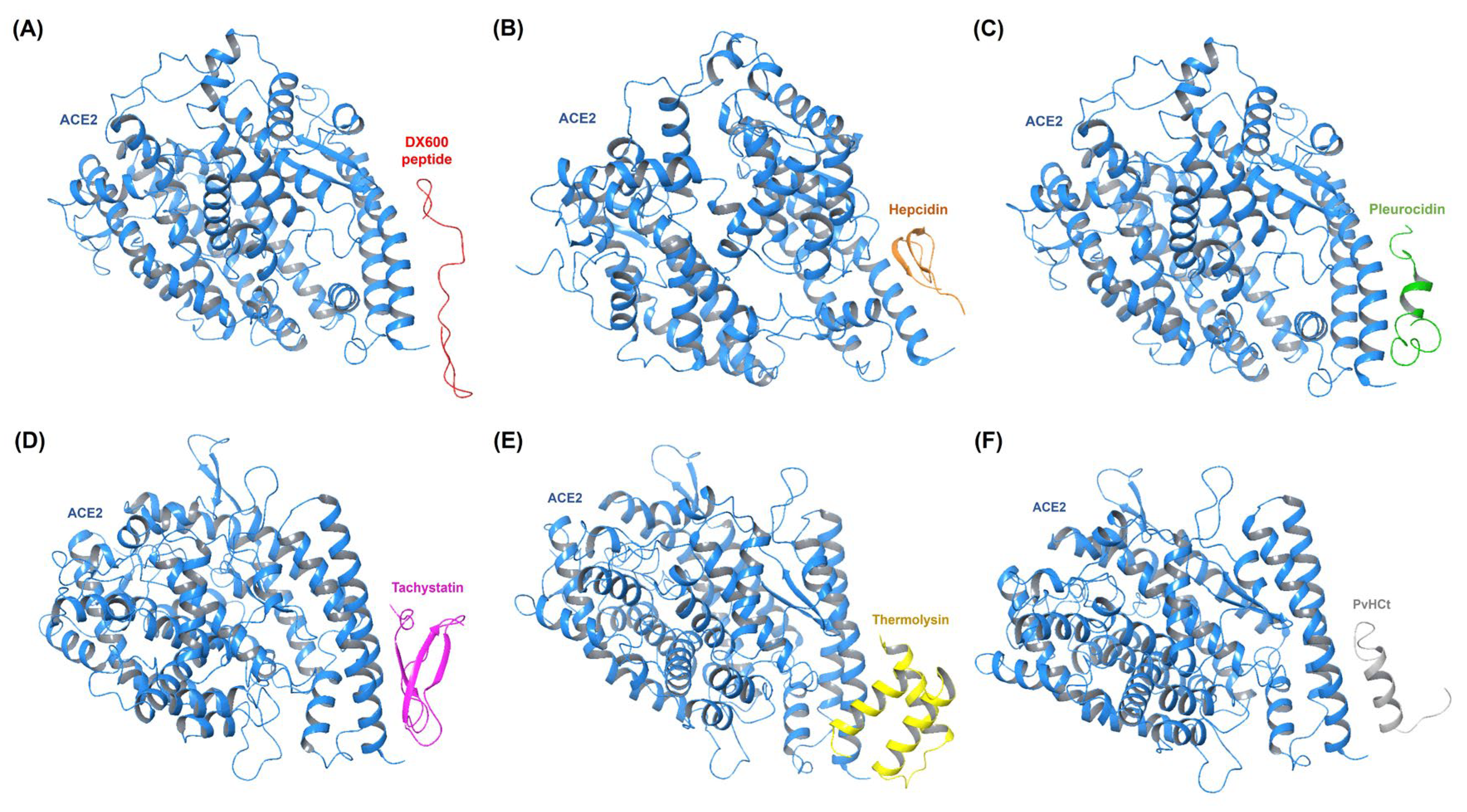
Figure 2.
Molecular docking results of optimal target receptor-AMP complexes, highlighting the lowest binding energy values indicative of a superior affinity. (A) ACE2 complexes. (B) CRP complexes. (C) MMP9 complexes. (D) NLRP3 complexes. (E) TLR4 complexes.
Figure 2.
Molecular docking results of optimal target receptor-AMP complexes, highlighting the lowest binding energy values indicative of a superior affinity. (A) ACE2 complexes. (B) CRP complexes. (C) MMP9 complexes. (D) NLRP3 complexes. (E) TLR4 complexes.
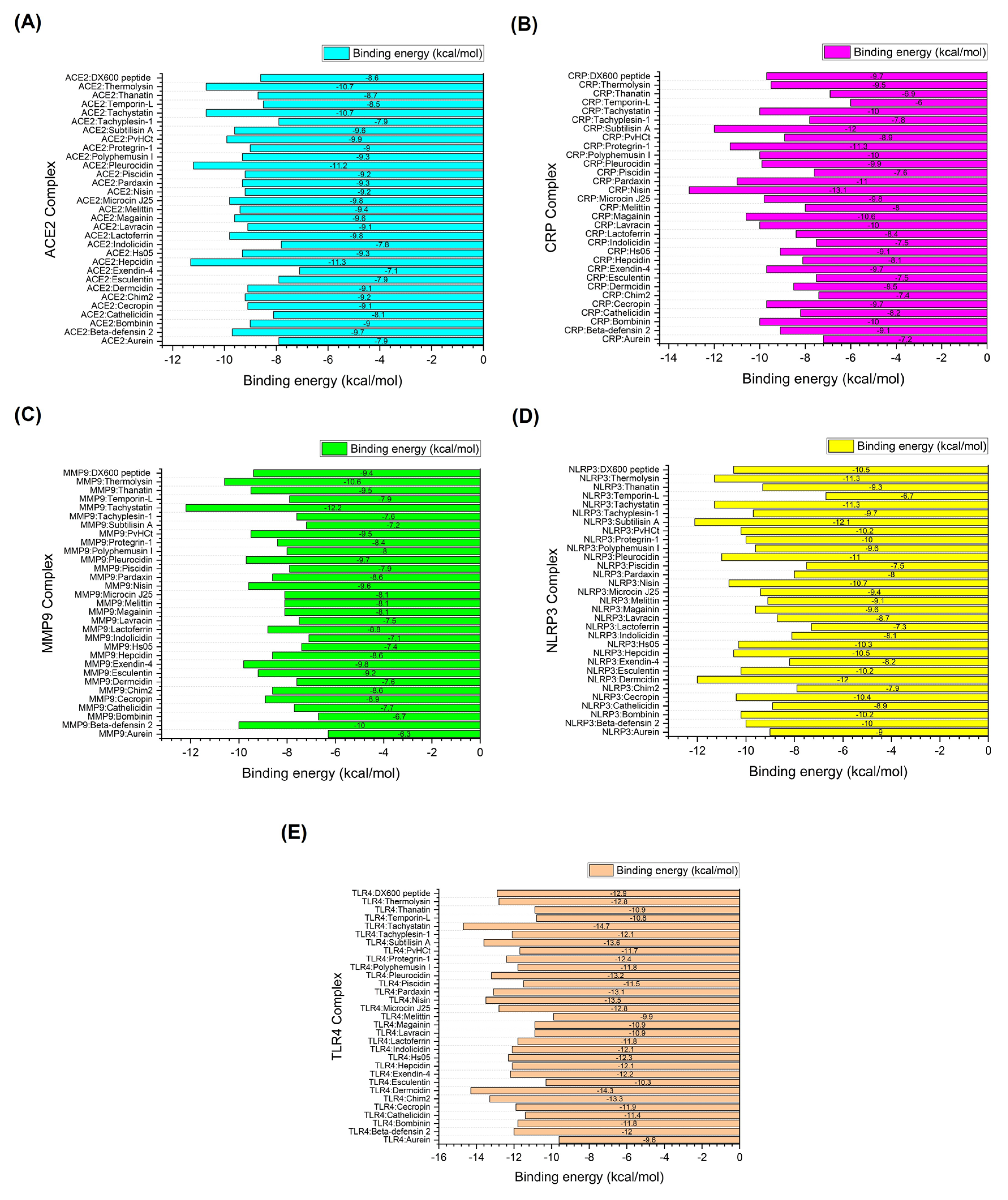
Figure 3.
Correlation matrices illustrate the relationships between binding energy (kcal/mol) and individual energy components for each target receptor-AMP complex. These include (A) ACE2 complexes, (B) CRP complexes, (C) MMP9 complexes, (D) NLRP3 complexes, and (E) TLR4 complexes. The matrices measure the degree of association between binding energy, van der Waals energy, electrostatic energy, and desolvation energy. The correlation values range from -1 to 1, where 1 denotes a perfect positive correlation, -1 indicates a perfect negative correlation, and 0 signifies no correlation.
Figure 3.
Correlation matrices illustrate the relationships between binding energy (kcal/mol) and individual energy components for each target receptor-AMP complex. These include (A) ACE2 complexes, (B) CRP complexes, (C) MMP9 complexes, (D) NLRP3 complexes, and (E) TLR4 complexes. The matrices measure the degree of association between binding energy, van der Waals energy, electrostatic energy, and desolvation energy. The correlation values range from -1 to 1, where 1 denotes a perfect positive correlation, -1 indicates a perfect negative correlation, and 0 signifies no correlation.
Figure 4.
Molecular dynamics (MD) simulation results illustrating the Root Mean Square Fluctuation (RMSF) profiles, highlighting the flexibility of residues within different complexes: (A) ACE2 complexes, (B) CRP complexes, (C) MMP9 complexes, (D) NLRP3 complexes, and (E) TLR4 complexes.
Figure 4.
Molecular dynamics (MD) simulation results illustrating the Root Mean Square Fluctuation (RMSF) profiles, highlighting the flexibility of residues within different complexes: (A) ACE2 complexes, (B) CRP complexes, (C) MMP9 complexes, (D) NLRP3 complexes, and (E) TLR4 complexes.

Table 1.
Overview of target receptors, their PDB IDs, and active sites.
| Receptors Associated with Infection-related CVDs | PDB ID | Active Site (Number of Residues) |
Ref |
|---|---|---|---|
| Angiotensin-Converting Enzyme 2 (ACE2) | 6M0J | 24, 30, 35, 38, 41, 42, 83, 353 | [30] |
| C-reactive protein (CRP) | 1B09 | 58, 60, 61, 66, 74, 81, 138, 139, 140, 147, 150 | [31] |
| Matrix Metalloproteinase-9 (MMP9) | 1GKC | 131, 149, 165, 175, 177, 182, 183, 185, 186, 187, 188, 189, 190, 197, 199, 201, 203, 205, 206, 208, 211, 212, 213, 214, 215, 393, 401, 402, 405, 421, 422, 423 | [32] |
| NLRP3 Inflammasome | 6NPY | 165, 166, 167, 229, 230, 231, 232, 379, 411, 414, 520 | [33] |
| Toll-Like Receptor-4 (TLR4) | 3FXI | 363, 364, 365, 386, 409, 410, 411, 433, 458, 507, 533 | [34] |
Table 3.
Molecular docking results of top 5 performing target protein-AMP complexes compared to the standard inhibitor (DX600 peptide).
Table 3.
Molecular docking results of top 5 performing target protein-AMP complexes compared to the standard inhibitor (DX600 peptide).
| Complex | HADDOCK score (a.u.) | Binding energy (kcal/mol) | Van der Waals energy | Electrostatic energy | Desolvation energy | RMSD |
|---|---|---|---|---|---|---|
| ACE2 Complexes | ||||||
| ACE2:DX600 peptide (standard inhibitor) | -76.8 +/- 0.9 | -8.6 | -38.7 +/- 2.9 | -79.0 +/- 28.3 | -26.1 +/- 4.2 | 1.4 +/- 0.2 |
| ACE2:Hepcidin | -96.3 +/- 7.2 | -11.3 | -44.7 +/- 1.2 | -250.1 +/- 24.0 | -3.1 +/- 4.7 | 0.5 +/- 0.3 |
| ACE2:Pleurocidin | -104.8 +/- 1.9 | -11.2 | -46.6 +/- 6.0 | -220.6 +/- 23.5 | -18.0 +/- 2.2 | 1.5 +/- 0.1 |
| ACE2:Tachystatin | -102.0 +/- 3.7 | -10.7 | -48.4 +/- 4.4 | -220.9 +/- 32.4 | -20.0 +/- 4.3 | 1.5 +/- 0.0 |
| ACE2:Thermolysin | -91.4 +/- 2.4 | -10.7 | -49.2 +/- 2.5 | -131.8 +/- 20.5 | -19.0 +/- 1.1 | 0.7 +/- 0.5 |
| ACE2:PvHCt | -94.0 +/- 2.8 | -9.9 | -44.8 +/- 2.2 | -117.5 +/- 15.6 | -28.6 +/- 1.0 | 0.6 +/- 0.4 |
| CRP Complexes | ||||||
| CRP:DX600 peptide (standard inhibitor) | -90.7 +/- 8.8 | -9.7 | -40.8 +/- 4.6 | -171.9 +/- 13.0 | -15.7 +/- 2.1 | 0.8 +/- 0.5 |
| CRP:Nisin | -79.3 +/- 4.1 | -13.1 | -43.0 +/- 4.4 | -198.5 +/- 14.5 | 3.1 +/- 1.3 | 2.6 +/- 0.6 |
| CRP:Subtilisin A | -128.1 +/- 8.3 | -12.0 | -46.5 +/- 6.1 | -306.9 +/- 57.8 | -22.6 +/- 7.6 | 0.6 +/- 0.5 |
| CRP:Protegrin-1 | -92.7 +/- 4.0 | -11.3 | -37.5 +/- 3.9 | -330.3 +/- 21.8 | 9.7 +/- 2.4 | 1.3 +/- 0.1 |
| CRP:Pardaxin | -102.4 +/- 2.9 | -11.0 | -50.1 +/- 0.3 | -202.1 +/- 37.0 | -16.5 +/- 5.7 | 1.7 +/- 0.2 |
| CRP:Magainin | -108.2 +/- 5.6 | -10.6 | -25.2 +/- 4.2 | -352.4 +/- 45.0 | -14.6 +/- 1.7 | 1.0 +/- 0.2 |
| MMP9 Complexes | ||||||
| MMP9:DX600 peptide (standard inhibitor) | -98.3 +/- 2.5 | -9.4 | -55.5 +/- 3.8 | -205.3 +/- 34.7 | -22.0 +/- 2.3 | 1.4 +/- 0.3 |
| MMP9:Tachystatin | -136.4 +/- 3.4 | -12.2 | -85.2 +/- 7.8 | -235.8 +/- 14.4 | -28.6 +/- 3.1 | 1.6 +/- 0.0 |
| MMP9:Thermolysin | -114.4 +/- 3.8 | -10.6 | -70.3 +/- 6.6 | -173.2 +/- 46.2 | -27.6 +/- 3.6 | 1.2 +/- 0.2 |
| MMP9:Beta-defensin 2 | -93.6 +/- 4.7 | -10.0 | -55.2 +/- 3.8 | -285.5 +/- 37.5 | -3.9 +/- 1.5 | 0.5 +/- 0.3 |
| MMP9:Exendin-4 | -48.2 +/- 3.2 | -9.8 | -47.3 +/- 5.7 | -55.4 +/- 20.3 | -14.5 +/- 1.5 | 0.9 +/- 0.5 |
| MMP9:Pleurocidin | -143.7 +/- 4.0 | -9.7 | -62.7 +/- 6.7 | -406.5 +/- 40.9 | -27.8 +/- 2.8 | 1.2 +/- 0.1 |
| NLRP3 Complexes | ||||||
| NLRP3:DX600 peptide (standard inhibitor) | -87.9 +/- 2.7 | -10.5 | -50.1 +/- 6.8 | -237.3 +/- 44.7 | -5.9 +/- 4.6 | 0.9 +/- 0.5 |
| NLRP3:Subtilisin A | -138.7 +/- 2.8 | -12.1 | -75.8 +/- 4.3 | -271.9 +/- 59.6 | -31.2 +/- 6.0 | 0.7 +/- 0.4 |
| NLRP3:Dermcidin | -98.6 +/- 14.7 | -12.0 | -45.9 +/- 4.1 | -374.3 +/- 87.9 | 11.9 +/- 4.2 | 1.0 +/- 0.6 |
| NLRP3:Tachystatin | -100.1 +/- 8.7 | -11.3 | -50.4 +/- 9.3 | -281.3 +/- 36.1 | -0.2 +/- 5.0 | 1.4 +/- 0.2 |
| NLRP3:Thermolysin | -82.3 +/- 4.9 | -11.3 | -51.1 +/- 4.7 | -193.6 +/- 19.4 | 0.3 +/- 2.8 | 1.2 +/- 0.9 |
| NLRP3:Pleurocidin | -110.5 +/- 9.2 | -11.0 | -63.0 +/- 6.9 | -253.8 +/- 21.7 | -15.0 +/- 6.3 | 1.1 +/- 0.7 |
| TLR4 Complexes | ||||||
| TLR4:DX600 peptide (standard inhibitor) | -78.4 +/- 4.4 | -12.9 | -41.0 +/- 7.0 | -111.4 +/- 34.1 | -22.9 +/- 5.1 | 1.3 +/- 0.2 |
| TLR4:Tachystatin | -125.7 +/- 2.9 | -14.7 | -71.8 +/- 3.2 | -172.7 +/- 15.0 | -23.1 +/- 2.0 | 0.4 +/- 0.2 |
| TLR4:Dermcidin | -91.1 +/- 8.6 | -14.3 | -57.5 +/- 3.3 | -137.5 +/- 37.3 | -10.2 +/- 1.9 | 1.0 +/- 0.6 |
| TLR4:Subtilisin A | -108.7 +/- 3.4 | -13.6 | -59.9 +/- 2.4 | -70.5 +/- 21.6 | -40.4 +/- 3.5 | 1.3 +/- 0.1 |
| TLR4:Nisin | -94.6 +/- 6.3 | -13.5 | -55.8 +/- 5.0 | -153.0 +/- 28.9 | -13.6 +/- 3.1 | 1.3 +/- 0.1 |
| TLR4:Chim2 | -93.3 +/- 4.4 | -13.3 | -47.7 +/- 5.0 | -119.0 +/- 23.8 | -27.3 +/- 5.7 | 0.4 +/- 0.3 |
Table 4.
Intermolecular contacts and non-interacting surface areas for receptors associated with infection-related CVD complexes with standard inhibitor and antimicrobial peptides.
Table 4.
Intermolecular contacts and non-interacting surface areas for receptors associated with infection-related CVD complexes with standard inhibitor and antimicrobial peptides.
| Complex | ICs charged-charged | ICs charged-polar | ICs charged-apolar | ICs polar-polar | ICs polar-apolar | ICs apolar-apolar | NIS charged | NIS apolar |
|---|---|---|---|---|---|---|---|---|
| ACE2 Complexes | ||||||||
| ACE2:DX600 peptide (standard inhibitor) | 3 | 3 | 14 | 0 | 5 | 6 | 27.88 | 33.63 |
| ACE2:Hepcidin | 12 | 3 | 28 | 0 | 7 | 7 | 27.54 | 33.86 |
| ACE2:Pleurocidin | 8 | 5 | 23 | 0 | 11 | 4 | 27.87 | 34.61 |
| ACE2:Tachystatin | 5 | 9 | 20 | 2 | 12 | 8 | 26.88 | 33.76 |
| ACE2:Thermolysin | 4 | 4 | 29 | 2 | 9 | 4 | 27.33 | 34.11 |
| ACE2:PvHCt | 14 | 3 | 16 | 2 | 7 | 2 | 27.35 | 34.30 |
| CRP Complexes | ||||||||
| CRP:DX600 peptide (standard inhibitor) | 4 | 7 | 16 | 2 | 15 | 7 | 26.83 | 40.24 |
| CRP:Nisin | 1 | 3 | 27 | 1 | 26 | 19 | 24.71 | 42.35 |
| CRP:Subtilisin A | 5 | 4 | 29 | 4 | 23 | 14 | 25.9 | 43.98 |
| CRP:Protegrin-1 | 9 | 11 | 19 | 0 | 18 | 11 | 29.03 | 40.00 |
| CRP:Pardaxin | 2 | 10 | 24 | 3 | 21 | 18 | 25.29 | 44.12 |
| CRP:Magainin | 8 | 5 | 20 | 2 | 18 | 10 | 28.12 | 42.50 |
| MMP9 Complexes | ||||||||
| MMP9:DX600 peptide (standard inhibitor) | 6 | 2 | 26 | 0 | 7 | 30 | 23.08 | 43.36 |
| MMP9:Tachystatin | 4 | 10 | 22 | 0 | 20 | 28 | 22.29 | 41.40 |
| MMP9:Thermolysin | 6 | 3 | 29 | 0 | 11 | 33 | 24.36 | 42.31 |
| MMP9:Beta-defensin 2 | 11 | 3 | 28 | 0 | 10 | 24 | 23.33 | 47.33 |
| MMP9:Exendin-4 | 1 | 4 | 19 | 0 | 16 | 22 | 26.06 | 43.66 |
| MMP9:Pleurocidin | 12 | 0 | 27 | 1 | 8 | 34 | 22.3 | 46.04 |
| NLRP3 Complexes | ||||||||
| NLRP3:DX600 peptide (standard inhibitor) | 5 | 7 | 23 | 2 | 15 | 8 | 23.84 | 42.36 |
| NLRP3:Subtilisin A | 1 | 10 | 37 | 2 | 19 | 23 | 23.74 | 44.13 |
| NLRP3:Dermcidin | 14 | 8 | 28 | 0 | 15 | 14 | 25.24 | 42.86 |
| NLRP3:Tachystatin | 8 | 11 | 22 | 5 | 15 | 11 | 23.99 | 41.14 |
| NLRP3:Thermolysin | 6 | 11 | 31 | 0 | 8 | 4 | 24 | 41.93 |
| NLRP3:Pleurocidin | 13 | 6 | 31 | 1 | 10 | 20 | 23.97 | 42.84 |
| TLR4 Complexes | ||||||||
| TLR4:DX600 peptide (standard inhibitor) | 4 | 6 | 15 | 0 | 23 | 8 | 24.43 | 30.77 |
| TLR4:Tachystatin | 5 | 10 | 24 | 4 | 26 | 10 | 23.90 | 31.58 |
| TLR4:Dermcidin | 10 | 2 | 26 | 1 | 21 | 16 | 25.49 | 33.26 |
| TLR4:Subtilisin A | 4 | 5 | 16 | 2 | 24 | 28 | 23.57 | 32.82 |
| TLR4:Nisin | 6 | 9 | 20 | 7 | 25 | 14 | 23.87 | 31.98 |
| TLR4:Chim2 | 6 | 8 | 16 | 1 | 21 | 14 | 25.34 | 31.51 |
Table 5.
Detailed examination of hydrogen bond interactions between receptors associated with infection-related CVD complexes with standard inhibitor and antimicrobial peptides.
Table 5.
Detailed examination of hydrogen bond interactions between receptors associated with infection-related CVD complexes with standard inhibitor and antimicrobial peptides.
| Complex | Residue (Receptor) | Protein Atom (Receptor) |
Residue (Interacting Peptide) |
Protein Atom (Interacting Peptide) |
Interaction Distance (Å) |
|---|---|---|---|---|---|
| ACE2:Hepcidin | Ser19 | OG | Lys24 | NZ | 2.81 |
| Glu23 | OE2 | Lys24 | NZ | 2.58 | |
| Asp30 | OD1 | Arg16 | NH1 | 2.71 | |
| Asp30 | OD2 | Arg16 | NH2 | 2.59 | |
| Asp38 | OD2 | Lys18 | NZ | 2.61 | |
| CRP:Nisin | Asn61 | OD1 | Cys19 | SG | 2.94 |
| Glu147 | OE1 | Lys22 | NZ | 2.66 | |
| Glu147 | OE2 | Asn20 | ND2 | 2.93 | |
| Gln150 | NE2 | Gly18 | O | 2.97 | |
| Gln150 | NE2 | Cys19 | O | 2.86 | |
| MMP9:Tachystatin | Glu111 | OE2 | Leu6 | N | 2.66 |
| Tyr179 | OH | Arg3 | NE | 2.90 | |
| Pro180 | O | Thr20 | OG1 | 2.64 | |
| Asp182 | O | Arg14 | NH2 | 2.79 | |
| Gly183 | O | Arg14 | NH1 | 2.88 | |
| Asp185 | O | Arg14 | NH1 | 2.81 | |
| Leu188 | N | Tyr38 | OH | 2.85 | |
| Gln199 | OE1 | Arg3 | NH1 | 3.11 | |
| Gln199 | OE1 | Arg3 | NH2 | 2.93 | |
| Tyr393 | OH | Thr37 | OG1 | 2.89 | |
| His411 | O | Arg40 | NH2 | 2.86 | |
| His411 | ND1 | Asn10 | N | 3.27 | |
| Ser412 | O | Arg40 | NH1 | 3.28 | |
| Ser412 | O | Arg40 | NH2 | 2.75 | |
| NLRP3:Subtilisin A | Gln147 | OE1 | Lys2 | NZ | 2.70 |
| Glu150 | OE2 | Ala5 | N | 2.95 | |
| Glu150 | OE2 | Thr6 | N | 3.17 | |
| Glu150 | OE2 | Cys7 | SG | 2.95 | |
| Lys164 | O | Trp34 | NE1 | 2.77 | |
| Glu425 | OE2 | Cys13 | N | 2.68 | |
| Arg452 | NE | Glu23 | OE2 | 2.61 | |
| Arg452 | NH2 | Glu23 | OE1 | 2.61 | |
| Arg502 | NH1 | Thr6 | O | 2.78 | |
| TLR4:Tachystatin | Asn383 | O | Arg30 | NH1 | 3.27 |
| Asn383 | O | Arg30 | NH2 | 2.94 | |
| Ser386 | O | Tyr44 | OH | 2.78 | |
| Lys435 | NZ | Cys23 | O | 2.74 | |
| Lys435 | NZ | Cys24 | O | 2.94 | |
| Lys435 | NZ | Leu27 | O | 2.71 | |
| His458 | NE2 | Val12 | O | 2.69 | |
| Arg460 | NH2 | Gly17 | O | 2.73 |
Table 6.
Time-averaged structural properties obtained from the MD simulations of target receptor-AMP complexes.
Table 6.
Time-averaged structural properties obtained from the MD simulations of target receptor-AMP complexes.
| Complex | Average RMSD (Å) | Average RMSF (Å) | Average RoG (Å) | Number of Hydrogen Bonds Between the Two Proteins | Potential Energy (kcal/mol) |
|---|---|---|---|---|---|
| ACE2 Complexes | |||||
| ACE2 (apo-protein) | 2.870 | 0.772 | 2.501 | N/A | -440,543.758 |
| ACE2:DX600 peptide (standard inhibitor) | 3.559 | 0.792 | 2.597 | 50 | -583,916.418 |
| ACE2:Hepcidin | 3.245 | 0.822 | 2.548 | 49 | -477,259.112 |
| ACE2:Pleurocidin | 3.218 | 0.791 | 2.572 | 50 | -466,473.033 |
| ACE2:Tachystatin | 3.327 | 0.747 | 2.658 | 53 | -660,317.634 |
| ACE2:Thermolysin | 3.340 | 0.789 | 2.703 | 54 | -572,443.081 |
| ACE2:PvHCt | 3.493 | 0.795 | 2.614 | 51 | -603,748.294 |
| CRP Complexes | |||||
| CRP (apo-protein) | 2.010 | 0.726 | 1.608 | N/A | -136,101.530 |
| CRP:DX600 peptide (standard inhibitor) | 2.402 | 0.958 | 1.735 | 14 | -184,719.286 |
| CRP:Nisin | 2.313 | 0.787 | 1.737 | 16 | -288,567.674 |
| CRP:Subtilisin A | 1.977 | 0.757 | 1.704 | 16 | -166,979.914 |
| CRP:Protegrin-1 | 2.416 | 0.951 | 1.653 | 17 | -179,352.210 |
| CRP:Pardaxin | 2.355 | 0.864 | 1.687 | 16 | -143,351.144 |
| CRP:Magainin | 2.192 | 0.814 | 1.688 | 17 | -145,486.514 |
| MMP9 Complexes | |||||
| MMP9 (apo-protein) | 2.078 | 1.094 | 1.495 | N/A | -111,366.323 |
| MMP9:DX600 peptide (standard inhibitor) | 2.555 | 1.267 | 1.610 | 11 | -164,237.839 |
| MMP9:Tachystatin | 2.209 | 1.359 | 1.631 | 13 | -195,363.451 |
| MMP9:Thermolysin | 1.992 | 1.295 | 1.666 | 15 | -131,816.946 |
| MMP9:Beta-defensin 2 | 2.498 | 1.330 | 1.641 | 13 | -163,272.695 |
| MMP9:Exendin-4 | 2.820 | 1.461 | 1.661 | 12 | -191,827.586 |
| MMP9:Pleurocidin | 2.274 | 1.095 | 1.573 | 11 | -119,726.773 |
| NLRP3 Complexes | |||||
| NLRP3 (apo-protein) | 3.176 | 0.924 | 3.614 | N/A | -900,750.476 |
| NLRP3:DX600 peptide (standard inhibitor) | 3.636 | 1.056 | 3.652 | 45 | -1,127,743.577 |
| NLRP3:Subtilisin A | 3.621 | 1.044 | 3.667 | 48 | -1,070,921.883 |
| NLRP3:Dermcidin | 3.501 | 1.048 | 3.728 | 50 | -1,370,475.374 |
| NLRP3:Tachystatin | 3.459 | 0.972 | 3.678 | 49 | -1,467,888.897 |
| NLRP3:Thermolysin | 3.362 | 0.941 | 3.638 | 53 | -1,239,438.186 |
| NLRP3:Pleurocidin | 3.207 | 1.051 | 3.587 | 49 | -977,382.530 |
| TLR4 Complexes | |||||
| TLR4 (apo-protein) | 2.642 | 0.720 | 3.201 | N/A | -718,765.881 |
| TLR4:DX600 peptide (standard inhibitor) | 2.968 | 0.775 | 3.268 | 38 | -755,432.195 |
| TLR4:Tachystatin | 2.914 | 0.878 | 3.221 | 46 | -780,309.925 |
| TLR4:Dermcidin | 2.938 | 0.900 | 3.234 | 43 | -708,531.477 |
| TLR4:Subtilisin A | 3.106 | 0.831 | 3.194 | 45 | -711,005.102 |
| TLR4:Nisin | 2.962 | 0.815 | 3.193 | 44 | -712,561.934 |
| TLR4:Chim2 | 3.143 | 0.918 | 3.220 | 45 | -743,878.601 |
Table 7.
Time-averaged structural properties obtained from the MD simulations of target receptor-AMP complexes.
Table 7.
Time-averaged structural properties obtained from the MD simulations of target receptor-AMP complexes.
| Complex | MM/PBSA Calculation Results ΔGbinding (kcal/mol) | Average (kcal/mol) | ||
|---|---|---|---|---|
| I | II | III | ||
| ACE2 Complexes | ||||
| ACE2:DX600 peptide (standard inhibitor) | -22.07 | -22.51 | -22.27 | -22.28 |
| ACE2:Hepcidin | -53.81 | -53.35 | -52.56 | -53.24 |
| ACE2:Pleurocidin | -46.94 | -46.17 | -46.64 | -46.58 |
| ACE2:Tachystatin | -62.34 | -60.47 | -61.93 | -61.58 |
| ACE2:Thermolysin | -44.14 | -45.63 | -44.70 | -44.82 |
| ACE2:PvHCt | -31.42 | -31.08 | -31.18 | -31.22 |
| CRP Complexes | ||||
| CRP:DX600 peptide (standard inhibitor) | -28.23 | -27.82 | -27.93 | -27.99 |
| CRP:Nisin | -38.81 | -38.65 | -38.75 | -38.73 |
| CRP:Subtilisin A | -70.24 | -70.99 | -70.92 | -70.71 |
| CRP:Protegrin-1 | -67.52 | -67.86 | -67.31 | -67.56 |
| CRP:Pardaxin | -60.72 | -56.91 | -60.72 | -59.45 |
| CRP:Magainin | -53.41 | -53.47 | -53.35 | -53.41 |
| MMP9 Complexes | ||||
| MMP9:DX600 peptide (standard inhibitor) | -53.14 | -53.07 | -51.63 | -52.61 |
| MMP9:Tachystatin | -96.59 | -96.7 | -96.55 | -96.61 |
| MMP9:Thermolysin | -66.02 | -68.04 | -65.97 | -66.67 |
| MMP9:Beta-defensin 2 | -78.69 | -78.72 | -77.75 | -78.38 |
| MMP9:Exendin-4 | -23.95 | -23.51 | -23.60 | -23.68 |
| MMP9:Pleurocidin | -94.82 | -94.15 | -93.77 | -94.24 |
| NLRP3 Complexes | ||||
| NLRP3:DX600 peptide (standard inhibitor) | -43.87 | -45.63 | -43.57 | -44.35 |
| NLRP3:Subtilisin A | -69.45 | -71.10 | -72.81 | -71.12 |
| NLRP3:Dermcidin | -61.90 | -62.54 | -62.43 | -62.29 |
| NLRP3:Tachystatin | -69.04 | -69.56 | -69.02 | -69.20 |
| NLRP3:Thermolysin | -28.14 | -28.87 | -28.77 | -28.59 |
| NLRP3:Pleurocidin | -60.13 | -55.24 | -60.83 | -58.73 |
| TLR4 Complexes | ||||
| TLR4:DX600 peptide (standard inhibitor) | -33.05 | -32.48 | -32.93 | -32.82 |
| TLR4:Tachystatin | -59.73 | -59.72 | -59.62 | -59.69 |
| TLR4:Dermcidin | -45.92 | -46.23 | -44.51 | -45.55 |
| TLR4:Subtilisin A | -43.62 | -42.51 | -43.61 | -43.24 |
| TLR4:Nisin | -56.90 | -57.13 | -56.90 | -56.97 |
| TLR4:Chim2 | -57.95 | -58.19 | -58.45 | -58.19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



