Submitted:
05 September 2024
Posted:
09 September 2024
You are already at the latest version
Abstract
The global prevalence of cancer is on the rise, and while targeted therapies, early diagnosis, im-munotherapies, and conventional personalized chemotherapy have significantly upgraded out-comes for cancer patients, there is still a significant likelihood that cancer cannot be fully eradi-cated. Cancer stem-like cells (CSCs) and Epithelial-mesenchymal transition (EMT) exhibit pivotal roles in cancer cell metastasis, tumor recurrence, chemotherapy resistance, and cancer cell death, among other processes. This review aims to elucidate the molecular mechanisms underlying EMT and CSCs, their interplay, associated signaling pathways, and regulation of the EMT process and the initiation of CSCs. EMT induction confers mesenchymal properties and the potential of cancer cells to adopt the CSC state. These processes are intricately associated with the regulation of Notch and Wnt signaling pathways, transforming growth factor-β (TGF-β), as well as the expres-sion of miRNAs. Further exploring the relationship between EMT and CSCs is essential for the development of new chemotherapeutic strategies and the identification of novel therapeutic tar-gets, and hence, the development of therapeutic strategies targeting EMT, or CSCs holds promise for improving cancer therapy.
Keywords:
Epithelial-mesenchymal transition (EMT)
; Cancer stem cells (CSCs)
; Hypoxia
; Plasticity
; Cancer signaling pathways
; miRNA
; Therapeutic avenues
1. Introduction
Epithelial-to-mesenchymal transition (EMT) is the process by which epithelial cells no longer have their defining characteristics and transform into mesenchymal cells under certain conditions [1]. This transformation is crucial in normal embryonic development, tissue fibrogenesis, and tumor progression [2]. Cancer stem/stem-like cells (CSCs) exhibit the ability to self-renew and differentiate without limitation, contributing to metastasis. These cells have a decisive role in tumorigenesis, metastasis, recurrence, drug-efflux, and resistance to drugs [3]. During EMT, epithelial cells lose their expression markers such as E-cadherin and obtain mesenchymal phenotypes by showing the expressions of N-cadherin, fibronectin, and vimentin, facilitating detachment from the primary tumor and promoting invasiveness [4]. Cells undergoing EMT acquire stemness or stem cell-like features, while cancer cells with stem cell characteristics display EMT biomarkers [5]. EMT, as a prime regulator of stemness phenotype of cancer cells, influences on the biological behavior of CSCs. Additionally, EMT also plays an important role in resistance to apoptosis and immune evasion, making it a key contributor to cancer aggressiveness and poor prognosis [4]. Various cancer, including pancreatic, colorectal, and breast cancers, show EMT-driven progression, revealing its universal role across malignancies.
Conventional cancer therapies, such as chemotherapy, radiotherapy, and surgical resection, etc. primarily target rapidly dividing tumor cells. While these treatments can eliminate some cancer cells, they fail to eradicate CSCs persuaded by EMT, which ultimately leads to persistent tumor recurrence and metastasis [6]. EMT-induced cancer cells are associated with cancer invasion, and EMT-induced CSCs aid to tumor recurrence and resistance to chemoradiotherapy. Therefore, targeting both CSCs and EMT is essential for improving the existing cancer treatments. Additionally, exploring the relationship between EMT and CSC could lead to the development of novel cancer therapeutic strategies.
2. CSCs and Cancer
CSCs were first hinted at in Rudolf Virchow’s cytopathological study [7,8]. Later, Paget proposed that cancer cells have an innate propensity to metastasize to distant organs [9]. The CSC hypothesis suggests the self-renew potentiality of certain cancer cells, which assists the sustenance of tumor growth and differentiation into multiple cell types [10,11,12]. Both genetic and epigenetic alterations endow these cells with immense proliferative ability, self-renewal capability, and the potential for multidirectional differentiation, which make them a determining factor in cancer development, metastasis, and further recurrence. Dick’s team first discovered CSCs [13], identifying that a small subset of leukemia cells expressing CD34+ and CD38+, could influence heterogeneity in acute myeloid leukemia when transplanted into mice [14,15,16]. Ponti et al. further revealed that breast cancer cells with CD44+/CD24- markers were endowed with self-renewal proficiency and could form new tumors in vitro, impersonating the progression of normal breast stem cells [17]. Gene expression profiling of CD44+/CD24- breast cancer cells revealed invasive signatures compared to normal breast epithelial cells [18], which further implies the CSCs-mediated tumor invasiveness. Breast stem cells with this phenotype are indicative for cancer cell spread [19]. In addition, ALDH+ breast CSCs exhibit self-renewal potentiality, with as few as CD44+/CD24- and ALDH+ cells able to form new tumors [20]. Thus, generation of CSC is a prime factor in metastasis.
CSCs are pivotal in cancer recurrence after treatment [21], their self-renewal capacity [22] significantly contributes to tumor relapse [23]. Distant CSCs can elicit the regrowth of metastatic tumors [24,25], stipulating that tumors with higher CSC numbers have poorer prognosis. Traditional chemotherapy and radiotherapy kill apoptosis-sensitive cancer cells, however, often fail to banish the tumor-resistant CSCs, which ultimately leads to the tumor recurrence [26,27]. The resistance of CSCs against existing cancer treatments arises from both intrinsic and extrinsic factors. There is an intrinsic connection of EMT to the stemness of CSCs [28], empowering immune evasion and resistance to chemotherapy and radiotherapy [29]. EMT programs can be prompted in non-CSCs, modifying them into apoptosis-resistant CSCs. Hypoxic conditions within tumors moreover potentiate CSC resistance to drugs [30], and reduce their responsiveness to both radiotherapy and chemotherapy [31]. Hypoxia not only triggers CSC self-renewal [30] but also fosters late-stage migration and invasion. CSCs also display highly efficient DNA repair mechanisms [32], aiding them foil DNA damage from reactive oxygen species accumulation [13]. These robust repair systems enable CSCs to dodge apoptotic signals, contributing to their resistance to radiotherapy [33]. Moreover, CSCs were reported to be augmenting ATP-binding cassette (ABC) transport proteins, which actively pump chemotherapy drugs out of the cell, taking the edge off drug efficacy and concentration [12]. For example, the ABC-G2 transport protein exhibits an essential role in the development of multidrug resistance (MDR) in tumor cells [34]. Furthermore, tumor microenvironment (TME) or tumor immune microenvironment (TIME) is another extrinsic factor governing CSCs-mediated drug resistance. TIME encompasses tumor cells, cytokines, immune cells, and other components, and their interplay shape the antitumor immunity [35]. These various constituents showed an impact on CSC characteristics, with the tumor-stroma interlinkage being important in developing chemoresistance in CSCs. Additionally, conditions such as hypoxia, acid-base imbalances, and nutrient deprivation within the TIME can provoke adaptive responses in CSCs [36].
3. EMT and Cancer
EMT is vitally important for various biological processes. Initially proposed by Hay [37], EMT was first noted during embryogenesis. Epithelial cells, which are usually considered fully differentiated and closely connected, can lose their epithelial characteristics under certain factors, allowing them to migrate and invade, thereby transforming into mesenchymal-like cells [2,38]. This transformation implies a shift from a polygonal to a spindle-shaped morphology, loss of cell polarity and contacts, elevated resistance to apoptosis, and increased migratory and invasive capabilities [39,40,41]. EMT can be triggered by specific extracellular stimuli during cancer progression [42]. Growth factors such as TGF-β, epidermal growth factor, and fibroblast growth factors, released in the TME during hypoxic conditions, can induce the expression of transcription factors like zinc-finger homologs, basic helix-loop-helix transcription factors, and the ZEB family [43]. These transcription factors were reported to suppress E-cadherin expression, hence promoting EMT and facilitating tumor progression [42].
EMT is crucial for the normal embryonic development, such as the formation of the primitive mesoderm and the differentiation of neural structures into somites, muscles, and bones [44,45]. However, abnormal EMT activation can expedite pathological conditions, including organ fibrosis, wound healing, and cancer progression. In cancer, EMT entitles tumor cells to sneak into surrounding tissues and metastasize to distant sites, empowering tumor spread, recurrence, and chemotherapy resistance. Epithelial-origin tumors comprise the majority of cancer types. During transformation, cancer cells express adhesion molecules and create structures that impound them to the primary site [43]. Still, cells that are undergoing EMT near the tumor gain mesenchymal traits, diminishing cell adhesion and promoting motility and extracellular matrix degradation, which eases local and distant invasion (metastasis) [43]. Mesenchymal-epithelial transition (MET) also displays a vital role in tumor metastasis, facilitating cancer cells to go back to an epithelial phenotype at distant sites, enabling metastatic tumor growth [46].
The precise mechanisms by which EMT promotes chemotherapy resistance are still unclear. Transcriptional regulators of EMT (such as Snail, Twist, and Slug, etc.) uplift tumor invasiveness and survival in unfavorable microenvironments by resisting apoptosis [43]. In addition, EMT is associated with resistance to targeted therapies, as reported in non-small cell lung cancer (NSCC) studies showing reduced sensitivity to erlotinib in EMT-like phenotypes [10,47]. Table 1 summarizes the importance of the EMT process in the onset and progression of cancer.
4. EMT-CSCs Nexus
The EMT-CSCs association is firmly established, with many researchers exploring how EMT contributes to the initiation of CSCs and the progression of cancer metastasis. The seminal works by Hanahan and Weinberg provide a comprehensive framework for understanding cancer mechanisms. Their reports outline key hallmarks and emerging aspects of cancer biology, which are important for placing EMT and CSCs in the broader context of cancer research [135,136]. Becoming resistant to chemotherapy and adjuvant drugs after EMT is quite common, which ultimately leads to tumor relapse. This chemotherapy resistance is a hallmark of CSCs, as documented by CD44+/CD24- breast CSCs undergoing EMT [49]. EMT markers have been identified in stem cells from mice and human mammary glands, and breast epithelial cells underwent EMT expressed elevated efficiency in forming tumors and soft agar colonies [49]. Therefore, EMT and CSCs are closely linked; EMT entitles cancer cells to gain mesenchymal properties and stem-cell-related traits, leading to more aggressive and metastatic tumor cell types [137,138].
For tumor cells to metastasize, they trigger the EMT program, transforming into metastatic CSCs (MCSCs). These MCSCs enter the bloodstream, disseminate, extravasate, and colonize distant sites, accomplishing the metastatic process [139]. The level of EMT transcription factors is hyper activated in cancers such as urinary liver cancer, bladder carcinoma, and lung cancer, stipulating their omnipresent role in cancer cell metastasis and in triggering the differentiation of cancer stem cells into more aggressive and treatment-resistant traits. Major EMT signaling pathways, including TGF-β, Wnt, and Notch, collectively initiate the EMT program in CSCs through a series of cell signaling mechanisms [140]. Nevertheless, EMT can be served both an instigator and a consequence of CSCs induction. EMT can facilitate the acquisition of CSC-like properties in cancer cells, potentiating their migratory and invasive properties through the activation of prime signaling pathways (such as TGF-β and Wnt), which are known to ultimately promote stemness [2,49]. On the other hand, CSCs can also induce EMT as an adaptive response to their environment cues, forming a feedback loop that increases tumor progression and resistance to therapeutics [141,142].
4.1. Mechanisms Governing EMT-CSC Pathways
Both EMT and CSC are modulated at the genetic level through various pathways such as TGFβ-SMAD, MAPK/ERK, JAK/STAT, WNT/β-catenin, and PI3K-AKT-NFκB [143]. Figure 1 provides a pictorial representation of the EMT-CSC linkage. Numerous genes contribute to cancer phenotypes and heterogeneity, elucidating breast cancer subtypes [144]. EMT and CSCs impact immune modulation, which is vital for cancer immunity in response to immunotherapy [145,146,147]. EMT and CSC traits also favor the resistance against cytotoxic T lymphocytes [148]. Tumor-associated macrophages (TAMs) in the TME foster EMT characteristics [149]. EMT and CSCs are also related to cellular senescence, which can be earmarked to address stemness in cancer therapy [150,151]. CSC-allied senescence is a notable factor in anticancer treatments that impede cell division [150,151]. Tumor dormancy, governed by CSCs, contributes to resistance against chemotherapy [152]. Dormant tumor cells with EMT traits may elevate the metastatic proliferation [115]. Cytoskeletal changes programmed by TGFβ signaling are pivotal for cell metastasis [153]. EMT phenotype cells display unclasped tight junctions and decreased cell-to-cell adhesion, which are involved in facilitating migration [153,154].
4.2. Involvement of EMT and CSCs in Hypoxia
Hypoxic condition triggers the formation and aggressiveness of solid tumors, with hypoxia-inducible factor (HIF) modulating hypoxia-responsive genes that boost proliferation, survival, invasion, angiogenesis, metastasis, and therapy resistance. EMT and CSCs are linked to hypoxia, maintaining the CSC phenotype. In vitro studies show that hypoxia induces EMT in human carcinoma cell lines through HIF-1 activation [155,156,157]. HIF-1 promotes EMT in carcinoma cells, suppressing E-cadherin via ZEB1, ZEB2, and E12/E47 induction [158,159]. Recently, the cell-cell adhesion molecule NECTIN-4 has been identified as a marker for both cancer cells and cancer stem cells, playing a role in EMT promotion [70]. Moreover, reports suggest that NECTIN-4 is activated by HIF-1α [71,72], stipulating a strong connection between hypoxia, EMT, and CSCs. Collectively, these findings put a spotlight towards the complexity of factors driving EMT in carcinoma cells, posing challenges for therapeutic approaches.
4.3. Plasticity in EMT and CSCs
CSCs exhibit substantial heterogeneity, both intra- and intertumorally [160,161], complicating targeted therapy development. CSC heterogeneity arises from a combination of genetic and epigenetic alterations, mutations, and changes in microenvironmental, including different factors like cytokines, cell-cell interactions, hypoxia, etc. [160,161]. Data shows CSCs exist in specialized tumor niches supporting their survival [162,163]. CSCs rely on niche signals across different tumors [162,164] and can modulate their niche, using signaling pathways to maintain homeostasis, including inflammation, hypoxia, EMT, and angiogenesis [162,164]. These niches adapt with tumor progression and treatment, potentially drifting back non-tumorigenic cells into CSCs via EMT, elevating tumor invasion and metastasis [11,162,164,165]. This dynamic interchange revealed that therapies targeting CSCs alone may result in tumor relapse if residual cancer cells repopulate CSC niches. Cells with a mesenchymal phenotype get into circulation as CTCs, potentially undergoing MET to express EPCAM, an epithelial cell adhesion molecule in blood. The relationship between high EPCAM level in CTCs and survival remains debated [166], and not to be mentioned, CTCs also have diverse subpopulations [167].
4.4. EMT and CSCs and Their Involvement in Chemoresistance
EMT has been reported to be strongly associated with cancer drug resistance, especially multidrug resistance and radioresistance, likely due to elevated cancer cell survival, cell fate transitions, and hyperactivation of drug resistance-related genes [116,117]. SPARC levels help in identifying aggressive breast cancers with increased EMT, treatment resistance, and poor outcomes. Amino-bisphosphonates may reverse EMT by blocking the suppressive activity of certain stem cells [123]. EMT also bridges the CSC phenotype to resistance to traditional therapeutics across various carcinomas, with EMT programs upregulating genes associated with resistance in breast cancer [124] and shifting cell dependency from EGFR to AXL in NSCLC and ovarian cancer, which ultimately, develops resistance to EGFR-targeted therapies [125,126]. Additionally, EMT and CSCs also regulate ATP-binding cassette (ABC) transporters, leading to the drug-efflux-mediated development of chemoresistance [168,169]. Furthermore, EMT has a contribution in immunosuppressive tumor microenvironment, further influencing resistance to immunotherapies [127,128].
5. Signaling Pathways in CSCs and EMT
The signaling pathways involved in CSCs are thoroughly detailed by Sinha et al. [170], elucidating pathways crucial for cancer stem cell growth, survival, differentiation, and so on. This current work, however, briefly describes the signaling cascades associated with both CSCs and EMT.
5.1. Wnt Signaling
Wnt/β-catenin signaling, a major signaling for stem cell proliferation, activates EMT in tumor cells [171]. Upregulation of Six1 induces EMT in mouse breast tumor cells, initiates Wnt signaling, and promotes stem-like traits in vivo [172]. Lipoprotein receptor-related protein 6 downregulates Wnt/β-catenin signaling in breast carcinoma, inhibiting Slug and Twist expression and prohibiting cancer cell self-renewal [173]. In breast carcinoma, upregulated HER2 leads to Wnt-3 hyperactivation, triggering Wnt/β-catenin signaling and EMT, with elevated β-catenin nuclear expression and N-cadherin, Twist, and Slug levels [174]. Consequently, in breast cancer cells, expressing higher level of HER2, exhibit resistance to trastuzumab [174]. miR-1 reduces proliferation, stemness, and migration of breast CSCs by aiming at Frizzled-7 and tankyrase-2, downregulating the Wnt/β-catenin pathway [175]. In colorectal cancer (CRC) patients, miR-146a reduces Numb expression, which leads to inhibition of β-catenin, and moreover, repressing Wnt signaling, and cancer stemness [176,177].
5.2. Notch Signaling
Increasing evidence have established an important association between Notch-regulated transcription factors and the pathways that regulate stem cells, pinpointing that Notch serves as a common signaling pathway linking tumor EMT to CSCs [72]. In breast cancer cells, Notch signaling potentiates EMT, invasion, and migration by activating Slug expression [56]. In addition, Notch1 stimulates the phosphorylation of transcription activator 3, facilitating the acquisition of EMT and CSC traits [73]. In the same study, quercetin-3-methyl ether was shown to prohibit sphere formation (three-dimensional clusters of cancer cells that are enriched for CSCs) and the expression of stemness-related genes such as SRY-box2 and Nanog, while also downregulating Notch1 expression in breast cancer patients [74]. Moreover, doxorubicin stimulates the Notch pathway, with a marked expression of target genes observed at cytostatic doses [75]. Thus, doxorubicin may induce stemness in osteosarcoma cells through Notch pathway activation. Taken together, this evidence underscores the relationship between EMT and CSCs, indicating that Notch signaling potentiates CSC stemness, thereby contributing to cancer metastasis and treatment resistance.
5.3. Hedgehog Signaling
Hedgehog signaling is pivotal in CSCs formation and EMT process [171]. In pancreatic cancer, silencing the smoothened gene impedes Hedgehog signaling, which in turn hinders EMT and the invasion of pancreatic CSCs. As a result, Hedgehog signaling modulates the stemness and chemoresistance of pancreatic CSCs, stimulating carcinogenesis and metastasis [178]. Similarly, Hedgehog signaling inhibition through cyclopamine treatment diminishes Snail expression while elevating E-cadherin expression, thereby repressing stemness and EMT induction, eventually bringing down the pancreatic cancer metastasis [179]. Moreover, Hedgehog signaling is associated with the self-renewal of breast stem cells [180,181]. Stimulation of the RAS-mitogen-activated protein kinase pathway in a breast tumor progression model leads to the differentiation of human mammary epithelial cells into CSCs and the induction of EMT, which eases the acquisition of stem cell properties [55]. In PTEN-deficient cells, RAS signaling activation elicits EMT, endowing these cells with CSC characteristics [182]. These signaling pathways illuminate the correlation between EMT and CSCs, specifying that cells undergoing EMT are more susceptible to invasion and the acquisition of stem cell-like traits.
5.4. TGF-β Signaling
TGF-β, a multifunctional cytokine, primarily contributes to tumor metastasis [183,184]. Supporting this, van der Horst et al. reported that the tumor-initiating stem cell (TISC) properties in mesenchymal liver cancers were associated with EMT [185]. TGF-β aids EMT and TISC traits by uplifting Snail and Nanog expressions [186]. In hepatoma cells, miR-148A weakens cancer stemness traits by suppressing TGF-β/Smad2 signaling [187,188]. Obstructing EMT in breast cancer averts the progression of breast CSC characteristics [189,190]. Report also suggests that TGF-β type I receptor kinase inhibitor, EW-719, downregulates Snail expression, hence, reducing EMT and breast CSC characteristics. Similarly, miR-190 hinders breast cancer metastasis by targeting the TGF-β pathway and Smad2 expression [187]. Inhibition of miR-138-5p triggers TGF-β1, potentiating EMT and CSC traits, and declining lung cancer cell resistance to chemoradiotherapy [191]. Moreover, miR-495 suppresses TGF-β signaling; its hyperactivation backpedals CSC formation and EMT in oral squamous cell carcinoma, restricting the tumor growth in vivo [192]. Thus, TGF-β signaling plays a key role in cancer initiation, migration, and the EMT and CSC transformation processes.
5.5. MicroRNAs
MicroRNAs (miRNAs) also play a central role in modulating EMT-induced CSCs. These small, non-coding RNA molecules foil translation or lessen the firmness of their target mRNAs by binding to their 3′-untranslated regions [193]. Findings indicate that certain miRNAs directly modulate EMT transcription factors, influencing the EMT process and enduring CSC functionality, thereby assisting tumorigenesis. The miR-200 family, highly conserved and widely studied, is known for its role in modulating EMT and stemness properties by tempering the expression of Bmi1 and Notch1 [194]. Members of the miR-200 family and their targets form a regulatory loop that directs EMT [140]. This loop involves in negative regulation of ZEB1 and ZEB2, which leads to downregulation of Notch signaling components such as Jag1, Maml2, and Maml3. By targeting ZEB1 and ZEB2, miR-200 family members bring down their expression, thereby maintaining stemness by forbidding the expression of the miR-200 family [183]. In addition, miR-200 stamps out BMI1 expression, which in turn suppresses the proliferation and oncogenicity of breast cancer cells [183].
6. EMT as Therapeutic Target against CSCs
The obligatory role of EMT in acquiring CSC characteristics indicates that targeting EMT could be a potent strategy to hinder CSC growth. For instance, targeting EMT-associated pathways to combat CSCs represents an encouraging therapeutic strategy. Wang et al. developed nanoparticle drug delivery systems containing salinomycin, which targeted ZEB1 and ZEB2, significantly reducing their expression and repressing the EMT process, thereby inhibiting CSC formation [195,196]. Chatterjee and Kundu also utilized nano-formulated quinacrine in highly metastatic cervical cancer stem cells to battle against NECTIN-4, which directly associated with EMT and CSCs [197]. Hyperactivation of miR-495 hinders homeobox C6-mediated TGF-β signaling, therefore holding up the EMT progression and repressing the proliferation, metastasis, and invasion of oral squamous CSCs [192]. Metformin interrupts the mixed E/M stem-like state by enhancing Notch-Jagged signaling, which negatively regulates EMT and CSC invasiveness [187,198]. MiR-612 modulates EMT-linked stem cell-like traits via the Wnt pathway, with its elevation resulting in declined tumor sphere size [199]. Curcumin impedes epithelial transformation and CSC property progression in bladder cancer by inactivating Wnt/β-catenin signaling [200].
In addition, targeting the lncRNA HOX transcript antisense intergenic RNA can hinder CSC formation in CRC by regulating the EMT process, marked by enhanced E-cadherin and lessen vimentin expression [201]. Again, knocking down TROY can resist gefitinib resistance and moderately reverse EMT and CSC formation in NSCC [202]. Further, downregulating miR-21 expression reverses EMT and CSC phenotypes by disabling the Akt and extracellular-regulated kinase 1/2 pathways and targeting PTEN [203]. Furthermore, the miRNA-200 family was found to be involved in inhibiting EMT and CSC formation. Liu et al. reported that nanoparticles co-delivering miR-200c and docetaxel promote docetaxel's cytotoxicity and repress CSC expression [204]. Nevertheless, hindering miR-429 modulates its downstream target ZEB1, therefore reducing EMT as well as the tumorigenicity of osteosarcoma stem cells [205,206,207].
7. Conclusions
The surge in cancer incidence poses a significant threat to humanity, with tumors striking various organs and tissues. While advancements in therapies have extended the lives of cancer patients, complete eradication of tumors remains grueling. Conventional treatments often fail to eliminate cancer cells in their activated state, particularly those that have undergone EMT and acquired cancer stemness traits. Thus, understanding the contributions of EMT and CSCs in tumor biology has shed light on the mechanisms underlying treatment resistance. The gene expression profiles in EMT and CSCs are interrelated, stipulating a major connection. This study highlighted the interaction between EMT and CSCs through various signaling pathways, confirming their complex association. EMT can promote CSC formation, endowing them with elevated metastatic potential due to their mesenchymal phenotype. The exact mechanism by which CSCs arise from the primary tumor and expedite metastasis remains elusive. A deeper understanding of this mechanism could lead to develop therapeutics not only to prevent tumor growth and recurrence but also to inhibit metastasis. Targeting the signaling pathways associated with EMT and CSCs holds promise for upgrading cancer therapy, with miRNAs appearing as potential therapeutic targets due to their regulatory effects on EMT and CSC phenotype. Future research endeavors should delve into the reciprocation between EMT and CSCs to illustrate the mechanisms driving CSC stemness and strengthen treatment strategies. Therefore, advancing tailored therapies that target EMT or CSCs could revolutionize cancer treatment and pave the way for novel therapeutic interventions.
References
- Acloque, H., et al., Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest, 2009. 119(6): p. 1438-49. [CrossRef]
- Thiery, J.P., et al., Epithelial-mesenchymal transitions in development and disease. Cell, 2009. 139(5): p. 871-90. [CrossRef]
- Huang, T., et al., Stem cell programs in cancer initiation, progression, and therapy resistance. Theranostics, 2020. 10(19): p. 8721-8743. [CrossRef]
- Ribatti, D., R. Tamma, and T. Annese, Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl Oncol, 2020. 13(6): p. 100773. [CrossRef]
- Saitoh, M., Involvement of partial EMT in cancer progression. J Biochem, 2018. 164(4): p. 257-264. [CrossRef]
- Clevers, H., The cancer stem cell: premises, promises and challenges. Nat Med, 2011. 17(3): p. 313-9. [CrossRef]
- Greaves, M. and C.C. Maley, Clonal evolution in cancer. Nature, 2012. 481(7381): p. 306-13. [CrossRef]
- .
- Paget, S., The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev, 1989. 8(2): p. 98-101.
- Yauch, R.L., et al., Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res, 2005. 11(24 Pt 1): p. 8686-98. [CrossRef]
- Visvader, J.E. and G.J. Lindeman, Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer, 2008. 8(10): p. 755-68. [CrossRef]
- Alison, M.R., S.M. Lim, and L.J. Nicholson, Cancer stem cells: problems for therapy? J Pathol, 2011. 223(2): p. 147-61.
- Chatterjee, S., C.R. Patil, and C.N. Kundu, An Overview of Antioxidative Anticancer Therapies with Reference to the Cancer Stem Cells, in Handbook of Oxidative Stress in Cancer: Therapeutic Aspects, S. Chakraborti, Editor. 2021, Springer Singapore: Singapore. p. 1-23.
- Bonnet, D. and J.E. Dick, Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med, 1997. 3(7): p. 730-7. [CrossRef]
- Lapidot, T., et al., A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature, 1994. 367(6464): p. 645-8. [CrossRef]
- Huang, F., et al., Oncolytic viruses against cancer stem cells: A promising approach for gastrointestinal cancer. World J Gastroenterol, 2016. 22(35): p. 7999-8009. [CrossRef]
- Dontu, G., et al., In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev, 2003. 17(10): p. 1253-70. [CrossRef]
- Liu, R., et al., The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med, 2007. 356(3): p. 217-26.
- Zheng, X., et al., Communication Between Epithelial-Mesenchymal Plasticity and Cancer Stem Cells: New Insights Into Cancer Progression. Front Oncol, 2021. 11: p. 617597. [CrossRef]
- Ginestier, C., et al., ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell, 2007. 1(5): p. 555-67.
- Chou, M.Y., et al., Sox2 expression involvement in the oncogenicity and radiochemoresistance of oral cancer stem cells. Oral Oncol, 2015. 51(1): p. 31-9. [CrossRef]
- Yeh, D.W., et al., Interplay between Inflammation and Stemness in Cancer Cells: The Role of Toll-Like Receptor Signaling. J Immunol Res, 2016. 2016: p. 4368101.
- Baumann, M., M. Krause, and R. Hill, Exploring the role of cancer stem cells in radioresistance. Nat Rev Cancer, 2008. 8(7): p. 545-54. [CrossRef]
- Dhawan, A., et al., Mathematical modelling of phenotypic plasticity and conversion to a stem-cell state under hypoxia. Sci Rep, 2016. 6: p. 18074. [CrossRef]
- Chen, X., et al., Epithelial mesenchymal transition and hedgehog signaling activation are associated with chemoresistance and invasion of hepatoma subpopulations. J Hepatol, 2011. 55(4): p. 838-45.
- Najafi, M., K. Mortezaee, and J. Majidpoor, Cancer stem cell (CSC) resistance drivers. Life Sci, 2019. 234: p. 116781.
- Wang, L., et al., Enrichment of prostate cancer stem-like cells from human prostate cancer cell lines by culture in serum-free medium and chemoradiotherapy. Int J Biol Sci, 2013. 9(5): p. 472-9.
- Wu, M.J., et al., Epithelial-Mesenchymal Transition Directs Stem Cell Polarity via Regulation of Mitofusin. Cell Metab, 2019. 29(4): p. 993-1002 e6.
- Caramel, J., M. Ligier, and A. Puisieux, Pleiotropic Roles for ZEB1 in Cancer. Cancer Res, 2018. 78(1): p. 30-35. [CrossRef]
- Lee, I.C., Y.C. Wu, and W.S. Hung, Hyaluronic Acid-Based Multilayer Films Regulate Hypoxic Multicellular Aggregation of Pancreatic Cancer Cells with Distinct Cancer Stem-Cell-like Properties. ACS Appl Mater Interfaces, 2018. 10(45): p. 38769-38779.
- Zhang, Z., et al., Hypoxia potentiates gemcitabine-induced stemness in pancreatic cancer cells through AKT/Notch1 signaling. J Exp Clin Cancer Res, 2018. 37(1): p. 291. [CrossRef]
- Todaro, M., et al., Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell, 2007. 1(4): p. 389-402.
- D'Andrea, F.P., et al., Cancer stem cell overexpression of nicotinamide N-methyltransferase enhances cellular radiation resistance. Radiother Oncol, 2011. 99(3): p. 373-8. [CrossRef]
- Prochazkova, J., M. Lanova, and J. Pachernik, Multidrug resistance-associated ABC transporters - too much of one thing, good for nothing. Biomol Concepts, 2012. 3(4): p. 319-31.
- Balkwill, F.R., M. Capasso, and T. Hagemann, The tumor microenvironment at a glance. J Cell Sci, 2012. 125(Pt 23): p. 5591-6.
- Jaggupilli, A. and E. Elkord, Significance of CD44 and CD24 as cancer stem cell markers: an enduring ambiguity. Clin Dev Immunol, 2012. 2012: p. 708036.
- Greenburg, G. and E.D. Hay, Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J Cell Biol, 1982. 95(1): p. 333-9.
- Kalluri, R. and R.A. Weinberg, The basics of epithelial-mesenchymal transition. J Clin Invest, 2009. 119(6): p. 1420-8. [CrossRef]
- Abdullah, A., et al., Epigenetic targeting of neuropilin-1 prevents bypass signaling in drug-resistant breast cancer. Oncogene, 2021. 40(2): p. 322-333.
- Babaei, G., S.G. Aziz, and N.Z.Z. Jaghi, EMT, cancer stem cells and autophagy; The three main axes of metastasis. Biomed Pharmacother, 2021. 133: p. 110909. [CrossRef]
- Sinha, D., et al., Emerging Concepts of Hybrid Epithelial-to-Mesenchymal Transition in Cancer Progression. Biomolecules, 2020. 10(11). [CrossRef]
- Moustakas, A. and C.H. Heldin, Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci, 2007. 98(10): p. 1512-20.
- Hollier, B.G., K. Evans, and S.A. Mani, The epithelial-to-mesenchymal transition and cancer stem cells: a coalition against cancer therapies. J Mammary Gland Biol Neoplasia, 2009. 14(1): p. 29-43.
- Turley, E.A., et al., Mechanisms of disease: epithelial-mesenchymal transition--does cellular plasticity fuel neoplastic progression? Nat Clin Pract Oncol, 2008. 5(5): p. 280-90.
- Baum, B., J. Settleman, and M.P. Quinlan, Transitions between epithelial and mesenchymal states in development and disease. Semin Cell Dev Biol, 2008. 19(3): p. 294-308.
- Garg, M., Epithelial Plasticity, Autophagy and Metastasis: Potential Modifiers of the Crosstalk to Overcome Therapeutic Resistance. Stem Cell Rev Rep, 2020. 16(3): p. 503-510.
- Thomson, S., et al., Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res, 2005. 65(20): p. 9455-62. [CrossRef]
- Ye, X. and R.A. Weinberg, Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol, 2015. 25(11): p. 675-686.
- Mani, S.A., et al., The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell, 2008. 133(4): p. 704-15. [CrossRef]
- May, C.D., et al., Epithelial-mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res, 2011. 13(1): p. 202.
- Guo, W., et al., Slug and Sox9 cooperatively determine the mammary stem cell state. Cell, 2012. 148(5): p. 1015-28.
- Scheel, C. and R.A. Weinberg, Phenotypic plasticity and epithelial-mesenchymal transitions in cancer and normal stem cells? Int J Cancer, 2011. 129(10): p. 2310-4.
- Korpal, M., et al., Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med, 2011. 17(9): p. 1101-8. [CrossRef]
- Wang, Z., et al., Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res, 2009. 69(6): p. 2400-7.
- Morel, A.P., et al., Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One, 2008. 3(8): p. e2888. [CrossRef]
- Fan, F., et al., Overexpression of snail induces epithelial-mesenchymal transition and a cancer stem cell-like phenotype in human colorectal cancer cells. Cancer Med, 2012. 1(1): p. 5-16.
- Kong, D., et al., Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS One, 2010. 5(8): p. e12445. [CrossRef]
- Long, H., et al., CD133+ ovarian cancer stem-like cells promote non-stem cancer cell metastasis via CCL5 induced epithelial-mesenchymal transition. Oncotarget, 2015. 6(8): p. 5846-59. [CrossRef]
- Rasheed, Z.A., et al., Prognostic significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. J Natl Cancer Inst, 2010. 102(5): p. 340-51. [CrossRef]
- Chen, Y., et al., CCL21/CCR7 interaction promotes EMT and enhances the stemness of OSCC via a JAK2/STAT3 signaling pathway. J Cell Physiol, 2020. 235(9): p. 5995-6009.
- Akbar, M.W., et al., A Stemness and EMT Based Gene Expression Signature Identifies Phenotypic Plasticity and is A Predictive but Not Prognostic Biomarker for Breast Cancer. J Cancer, 2020. 11(4): p. 949-961.
- Rhim, A.D., et al., EMT and dissemination precede pancreatic tumor formation. Cell, 2012. 148(1-2): p. 349-61.
- Ding, Q., et al., CD133 facilitates epithelial-mesenchymal transition through interaction with the ERK pathway in pancreatic cancer metastasis. Mol Cancer, 2014. 13: p. 15.
- Oskarsson, T., E. Batlle, and J. Massague, Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell, 2014. 14(3): p. 306-21. [CrossRef]
- Sanchez-Garcia, I., The crossroads of oncogenesis and metastasis. N Engl J Med, 2009. 360(3): p. 297-9. [CrossRef]
- Rojas-Puentes, L., et al., Epithelial-mesenchymal transition, proliferation, and angiogenesis in locally advanced cervical cancer treated with chemoradiotherapy. Cancer Med, 2016. 5(8): p. 1989-99. [CrossRef]
- Holderfield, M.T. and C.C. Hughes, Crosstalk between vascular endothelial growth factor, notch, and transforming growth factor-beta in vascular morphogenesis. Circ Res, 2008. 102(6): p. 637-52.
- Desai, S., S. Laskar, and B.N. Pandey, Autocrine IL-8 and VEGF mediate epithelial-mesenchymal transition and invasiveness via p38/JNK-ATF-2 signalling in A549 lung cancer cells. Cell Signal, 2013. 25(9): p. 1780-91.
- Gonzalez-Moreno, O., et al., VEGF elicits epithelial-mesenchymal transition (EMT) in prostate intraepithelial neoplasia (PIN)-like cells via an autocrine loop. Exp Cell Res, 2010. 316(4): p. 554-67. [CrossRef]
- Yang, A.D., et al., Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res, 2006. 66(1): p. 46-51.
- Chatterjee, S., S. Sinha, and C.N. Kundu, Nectin cell adhesion molecule-4 (NECTIN-4): A potential target for cancer therapy. Eur J Pharmacol, 2021. 911: p. 174516. [CrossRef]
- Kang, Y. and J. Massague, Epithelial-mesenchymal transitions: twist in development and metastasis. Cell, 2004. 118(3): p. 277-9.
- Wick, W., M. Platten, and M. Weller, Glioma cell invasion: regulation of metalloproteinase activity by TGF-beta. J Neurooncol, 2001. 53(2): p. 177-85.
- Ocana, O.H., et al., Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell, 2012. 22(6): p. 709-24.
- Tsai, J.H., et al., Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell, 2012. 22(6): p. 725-36. [CrossRef]
- Shibue, T., M.W. Brooks, and R.A. Weinberg, An integrin-linked machinery of cytoskeletal regulation that enables experimental tumor initiation and metastatic colonization. Cancer Cell, 2013. 24(4): p. 481-98.
- Adams, D.L., et al., Mitosis in circulating tumor cells stratifies highly aggressive breast carcinomas. Breast Cancer Res, 2016. 18(1): p. 44.
- Yu, M., et al., Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science, 2013. 339(6119): p. 580-4. [CrossRef]
- Grunert, S., M. Jechlinger, and H. Beug, Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol, 2003. 4(8): p. 657-65.
- Hay, E.D., An overview of epithelio-mesenchymal transformation. Acta Anat (Basel), 1995. 154(1): p. 8-20.
- Huber, M.A., N. Kraut, and H. Beug, Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol, 2005. 17(5): p. 548-58. [CrossRef]
- Yaguchi, T., et al., The mechanisms of cancer immunoescape and development of overcoming strategies. Int J Hematol, 2011. 93(3): p. 294-300.
- Fernando, R.I., et al., IL-8 signaling plays a critical role in the epithelial-mesenchymal transition of human carcinoma cells. Cancer Res, 2011. 71(15): p. 5296-306. [CrossRef]
- Sullivan, N.J., et al., Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene, 2009. 28(33): p. 2940-7.
- Wu, Y., et al., Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell, 2009. 15(5): p. 416-28.
- Kachroo, P., et al., IL-27 inhibits epithelial-mesenchymal transition and angiogenic factor production in a STAT1-dominant pathway in human non-small cell lung cancer. J Exp Clin Cancer Res, 2013. 32(1): p. 97. [CrossRef]
- Tlsty, T.D. and L.M. Coussens, Tumor stroma and regulation of cancer development. Annu Rev Pathol, 2006. 1: p. 119-50.
- Kong, D., et al., Platelet-derived growth factor-D overexpression contributes to epithelial-mesenchymal transition of PC3 prostate cancer cells. Stem Cells, 2008. 26(6): p. 1425-35. [CrossRef]
- Thompson, E.W., D.F. Newgreen, and D. Tarin, Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition? Cancer Res, 2005. 65(14): p. 5991-5; discussion 5995.
- Thiery, J.P., Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer, 2002. 2(6): p. 442-54.
- Hass, R., J. von der Ohe, and H. Ungefroren, Potential Role of MSC/Cancer Cell Fusion and EMT for Breast Cancer Stem Cell Formation. Cancers (Basel), 2019. 11(10).
- Giannoni, E., et al., Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res, 2010. 70(17): p. 6945-56.
- Yu, Y., et al., Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-beta signalling. Br J Cancer, 2014. 110(3): p. 724-32.
- Bonde, A.K., et al., Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer, 2012. 12: p. 35.
- Akalay, I., et al., Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res, 2013. 73(8): p. 2418-27.
- Hamilton, D.H., et al., Immunological targeting of tumor cells undergoing an epithelial-mesenchymal transition via a recombinant brachyury-yeast vaccine. Oncotarget, 2013. 4(10): p. 1777-90. [CrossRef]
- Ricciardi, M., et al., Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br J Cancer, 2015. 112(6): p. 1067-75. [CrossRef]
- Mak, M.P., et al., A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin Cancer Res, 2016. 22(3): p. 609-20.
- Lu, Y., et al., CXCL1-LCN2 paracrine axis promotes progression of prostate cancer via the Src activation and epithelial-mesenchymal transition. Cell Commun Signal, 2019. 17(1): p. 118.
- Pelizzo, G., et al., Microenvironment in neuroblastoma: isolation and characterization of tumor-derived mesenchymal stromal cells. BMC Cancer, 2018. 18(1): p. 1176.
- Gao, D., et al., Microenvironmental regulation of epithelial-mesenchymal transitions in cancer. Cancer Res, 2012. 72(19): p. 4883-9.
- Labelle, M., S. Begum, and R.O. Hynes, Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell, 2011. 20(5): p. 576-90.
- Techasen, A., et al., Cytokines released from activated human macrophages induce epithelial mesenchymal transition markers of cholangiocarcinoma cells. Asian Pac J Cancer Prev, 2012. 13 Suppl: p. 115-8.
- Lopez-Novoa, J.M. and M.A. Nieto, Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med, 2009. 1(6-7): p. 303-14. [CrossRef]
- Yan, Y., et al., High tumor-associated macrophages infiltration is associated with poor prognosis and may contribute to the phenomenon of epithelial-mesenchymal transition in gastric cancer. Onco Targets Ther, 2016. 9: p. 3975-83.
- Hu, Y., et al., Tumor-associated macrophages correlate with the clinicopathological features and poor outcomes via inducing epithelial to mesenchymal transition in oral squamous cell carcinoma. J Exp Clin Cancer Res, 2016. 35: p. 12.
- Fu, X.T., et al., Macrophage-secreted IL-8 induces epithelial-mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int J Oncol, 2015. 46(2): p. 587-96.
- Deng, Y.R., et al., Sorafenib inhibits macrophage-mediated epithelial-mesenchymal transition in hepatocellular carcinoma. Oncotarget, 2016. 7(25): p. 38292-38305. [CrossRef]
- Fan, Q.M., et al., Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett, 2014. 352(2): p. 160-8. [CrossRef]
- Long, X., et al., IL-8, a novel messenger to cross-link inflammation and tumor EMT via autocrine and paracrine pathways (Review). Int J Oncol, 2016. 48(1): p. 5-12.
- Cohen, E.N., et al., Inflammation Mediated Metastasis: Immune Induced Epithelial-To-Mesenchymal Transition in Inflammatory Breast Cancer Cells. PLoS One, 2015. 10(7): p. e0132710. [CrossRef]
- Vega, S., et al., Snail blocks the cell cycle and confers resistance to cell death. Genes Dev, 2004. 18(10): p. 1131-43.
- Emanuele, M.J., et al., Proliferating cell nuclear antigen (PCNA)-associated KIAA0101/PAF15 protein is a cell cycle-regulated anaphase-promoting complex/cyclosome substrate. Proc Natl Acad Sci U S A, 2011. 108(24): p. 9845-50. [CrossRef]
- Blick, T., et al., Epithelial mesenchymal transition traits in human breast cancer cell lines. Clin Exp Metastasis, 2008. 25(6): p. 629-42.
- Weidenfeld, K., et al., Dormant tumor cells expressing LOXL2 acquire a stem-like phenotype mediating their transition to proliferative growth. Oncotarget, 2016. 7(44): p. 71362-71377.
- Fischer, K.R., et al., Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature, 2015. 527(7579): p. 472-6.
- Zheng, X., et al., Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature, 2015. 527(7579): p. 525-530.
- Bao, Y., et al., Eukaryotic translation initiation factor 5A2 (eIF5A2) regulates chemoresistance in colorectal cancer through epithelial mesenchymal transition. Cancer Cell Int, 2015. 15: p. 109.
- Hu, T., et al., Mechanisms of drug resistance in colon cancer and its therapeutic strategies. World J Gastroenterol, 2016. 22(30): p. 6876-89.
- Tato-Costa, J., et al., Therapy-Induced Cellular Senescence Induces Epithelial-to-Mesenchymal Transition and Increases Invasiveness in Rectal Cancer. Clin Colorectal Cancer, 2016. 15(2): p. 170-178 e3.
- Tsoumas, D., et al., ILK Expression in Colorectal Cancer Is Associated with EMT, Cancer Stem Cell Markers and Chemoresistance. Cancer Genomics Proteomics, 2018. 15(2): p. 127-141.
- Yang, Y., et al., Epithelial-mesenchymal transition and cancer stem cell-like phenotype induced by Twist1 contribute to acquired resistance to irinotecan in colon cancer. Int J Oncol, 2017. 51(2): p. 515-524.
- Sangaletti, S., et al., Mesenchymal Transition of High-Grade Breast Carcinomas Depends on Extracellular Matrix Control of Myeloid Suppressor Cell Activity. Cell Rep, 2016. 17(1): p. 233-248. [CrossRef]
- Farmer, P., et al., A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med, 2009. 15(1): p. 68-74.
- Byers, L.A., et al., An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res, 2013. 19(1): p. 279-90.
- Gjerdrum, C., et al., Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc Natl Acad Sci U S A, 2010. 107(3): p. 1124-9. [CrossRef]
- Terry, S., et al., New insights into the role of EMT in tumor immune escape. Mol Oncol, 2017. 11(7): p. 824-846.
- Dongre, A., et al., Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res, 2017. 77(15): p. 3982-3989.
- Ansieau, S., et al., Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell, 2008. 14(1): p. 79-89.
- Liu, Y., et al., Zeb1 links epithelial-mesenchymal transition and cellular senescence. Development, 2008. 135(3): p. 579-88.
- Ohashi, S., et al., Epidermal growth factor receptor and mutant p53 expand an esophageal cellular subpopulation capable of epithelial-to-mesenchymal transition through ZEB transcription factors. Cancer Res, 2010. 70(10): p. 4174-84.
- Coppe, J.P., et al., Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol, 2008. 6(12): p. 2853-68.
- Yuan, A., et al., The role of interleukin-8 in cancer cells and microenvironment interaction. Front Biosci, 2005. 10: p. 853-65.
- Laberge, R.M., et al., Epithelial-mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron, 2012. 5(1): p. 39-44.
- Hanahan, D. and R.A. Weinberg, Hallmarks of cancer: the next generation. Cell, 2011. 144(5): p. 646-74. [CrossRef]
- Hanahan, D., Hallmarks of Cancer: New Dimensions. Cancer Discov, 2022. 12(1): p. 31-46.
- Lu, H., et al., A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol, 2014. 16(11): p. 1105-17.
- Brown, R.L., et al., CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J Clin Invest, 2011. 121(3): p. 1064-74.
- Brabletz, T., et al., Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer, 2005. 5(9): p. 744-9.
- Roy, S., et al., EMT imparts cancer stemness and plasticity: new perspectives and therapeutic potential. Front Biosci (Landmark Ed), 2021. 26(2): p. 238-265.
- Scheel, C. and R.A. Weinberg, Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol, 2012. 22(5-6): p. 396-403. [CrossRef]
- Brabletz, T., et al., EMT in cancer. Nat Rev Cancer, 2018. 18(2): p. 128-134.
- Loret, N., et al., The Role of Epithelial-to-Mesenchymal Plasticity in Ovarian Cancer Progression and Therapy Resistance. Cancers (Basel), 2019. 11(6).
- Sorlie, T., et al., Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A, 2001. 98(19): p. 10869-74. [CrossRef]
- Deng, Z., et al., Adoptive T-cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC Immunol, 2015. 16(1): p. 1.
- da Silveira, W.A., et al., Transcription Factor Networks derived from Breast Cancer Stem Cells control the immune response in the Basal subtype. Sci Rep, 2017. 7(1): p. 2851.
- Saygin, C., et al., Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem Cell, 2019. 24(1): p. 25-40. [CrossRef]
- Terry, S. and S. Chouaib, EMT in immuno-resistance. Oncoscience, 2015. 2(10): p. 841-2.
- Li, S., et al., Tumor-associated macrophages remodeling EMT and predicting survival in colorectal carcinoma. Oncoimmunology, 2018. 7(2): p. e1380765. [CrossRef]
- Del Barco, S., et al., Metformin: multi-faceted protection against cancer. Oncotarget, 2011. 2(12): p. 896-917.
- Olivos, D.J. and L.D. Mayo, Emerging Non-Canonical Functions and Regulation by p53: p53 and Stemness. Int J Mol Sci, 2016. 17(12).
- Steinbichler, T.B., et al., Therapy resistance mediated by cancer stem cells. Semin Cancer Biol, 2018. 53: p. 156-167. [CrossRef]
- Tsubakihara, Y. and A. Moustakas, Epithelial-Mesenchymal Transition and Metastasis under the Control of Transforming Growth Factor beta. Int J Mol Sci, 2018. 19(11).
- Iqbal, W., et al., Targeting signal transduction pathways of cancer stem cells for therapeutic opportunities of metastasis. Oncotarget, 2016. 7(46): p. 76337-76353. [CrossRef]
- Imai, T., et al., Hypoxia attenuates the expression of E-cadherin via up-regulation of SNAIL in ovarian carcinoma cells. Am J Pathol, 2003. 163(4): p. 1437-47. [CrossRef]
- Lester, R.D., et al., uPAR induces epithelial-mesenchymal transition in hypoxic breast cancer cells. J Cell Biol, 2007. 178(3): p. 425-36. [CrossRef]
- Yang, M.H., et al., Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol, 2008. 10(3): p. 295-305.
- Esteban, M.A., et al., Regulation of E-cadherin expression by VHL and hypoxia-inducible factor. Cancer Res, 2006. 66(7): p. 3567-75. [CrossRef]
- Krishnamachary, B., et al., Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res, 2006. 66(5): p. 2725-31.
- Magee, J.A., E. Piskounova, and S.J. Morrison, Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell, 2012. 21(3): p. 283-96.
- Meacham, C.E. and S.J. Morrison, Tumour heterogeneity and cancer cell plasticity. Nature, 2013. 501(7467): p. 328-37.
- Plaks, V., N. Kong, and Z. Werb, The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell, 2015. 16(3): p. 225-38. [CrossRef]
- Brozovic, A., The relationship between platinum drug resistance and epithelial-mesenchymal transition. Arch Toxicol, 2017. 91(2): p. 605-619.
- Borovski, T., et al., Cancer stem cell niche: the place to be. Cancer Res, 2011. 71(3): p. 634-9.
- Iliopoulos, D., et al., Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A, 2011. 108(4): p. 1397-402. [CrossRef]
- de Wit, S., et al., EpCAM(high) and EpCAM(low) circulating tumor cells in metastatic prostate and breast cancer patients. Oncotarget, 2018. 9(86): p. 35705-35716.
- Onidani, K., et al., Monitoring of cancer patients via next-generation sequencing of patient-derived circulating tumor cells and tumor DNA. Cancer Sci, 2019. 110(8): p. 2590-2599.
- Begicevic, R.R. and M. Falasca, ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int J Mol Sci, 2017. 18(11).
- Jiang, Z.S., et al., Epithelial-mesenchymal transition: potential regulator of ABC transporters in tumor progression. J Cancer, 2017. 8(12): p. 2319-2327. [CrossRef]
- Sinha, S., K.C. Hembram, and S. Chatterjee, Targeting signaling pathways in cancer stem cells: A potential approach for developing novel anti-cancer therapeutics. Int Rev Cell Mol Biol, 2024. 385: p. 157-209.
- Wang, S.S., et al., Links between cancer stem cells and epithelial-mesenchymal transition. Onco Targets Ther, 2015. 8: p. 2973-80.
- McCoy, E.L., et al., Six1 expands the mouse mammary epithelial stem/progenitor cell pool and induces mammary tumors that undergo epithelial-mesenchymal transition. J Clin Invest, 2009. 119(9): p. 2663-77.
- DiMeo, T.A., et al., A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res, 2009. 69(13): p. 5364-73.
- Wu, Y., et al., Expression of Wnt3 activates Wnt/beta-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol Cancer Res, 2012. 10(12): p. 1597-606.
- Liu, T., et al., MicroRNA-1 down-regulates proliferation and migration of breast cancer stem cells by inhibiting the Wnt/beta-catenin pathway. Oncotarget, 2015. 6(39): p. 41638-49.
- Hwang, W.L., et al., MicroRNA-146a directs the symmetric division of Snail-dominant colorectal cancer stem cells. Nat Cell Biol, 2014. 16(3): p. 268-80.
- Wei, X., et al., Activation of the JAK-STAT3 pathway is associated with the growth of colorectal carcinoma cells. Oncol Rep, 2014. 31(1): p. 335-41. [CrossRef]
- Wang, F., et al., Hedgehog Signaling Regulates Epithelial-Mesenchymal Transition in Pancreatic Cancer Stem-Like Cells. J Cancer, 2016. 7(4): p. 408-17. [CrossRef]
- Feldmann, G., et al., Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res, 2007. 67(5): p. 2187-96.
- Liu, S., et al., Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res, 2006. 66(12): p. 6063-71.
- Rangwala, F., A. Omenetti, and A.M. Diehl, Cancer stem cells: repair gone awry? J Oncol, 2011. 2011: p. 465343.
- Mulholland, D.J., et al., Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res, 2012. 72(7): p. 1878-89.
- Shao, S., et al., Notch1 signaling regulates the epithelial-mesenchymal transition and invasion of breast cancer in a Slug-dependent manner. Mol Cancer, 2015. 14(1): p. 28.
- Dian, L., et al., Berberine alkaloids inhibit the proliferation and metastasis of breast carcinoma cells involving Wnt/beta-catenin signaling and EMT. Phytochemistry, 2022. 200: p. 113217.
- van der Horst, G., et al., Targeting of alpha(v)-integrins in stem/progenitor cells and supportive microenvironment impairs bone metastasis in human prostate cancer. Neoplasia, 2011. 13(6): p. 516-25.
- Dang, H., et al., Snail1 induces epithelial-to-mesenchymal transition and tumor initiating stem cell characteristics. BMC Cancer, 2011. 11: p. 396. [CrossRef]
- Bocci, F., et al., A mechanism-based computational model to capture the interconnections among epithelial-mesenchymal transition, cancer stem cells and Notch-Jagged signaling. Oncotarget, 2018. 9(52): p. 29906-29920. [CrossRef]
- Cao, W., et al., Dynamics of Proliferative and Quiescent Stem Cells in Liver Homeostasis and Injury. Gastroenterology, 2017. 153(4): p. 1133-1147. [CrossRef]
- Park, S.Y., et al., Combinatorial TGF-beta attenuation with paclitaxel inhibits the epithelial-to-mesenchymal transition and breast cancer stem-like cells. Oncotarget, 2015. 6(35): p. 37526-43.
- Sun, Y., et al., Jatrorrhizine inhibits mammary carcinoma cells by targeting TNIK mediated Wnt/beta-catenin signalling and epithelial-mesenchymal transition (EMT). Phytomedicine, 2019. 63: p. 153015.
- Gupta, P.B., et al., Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell, 2011. 146(4): p. 633-44.
- You, X., et al., MicroRNA-495 confers inhibitory effects on cancer stem cells in oral squamous cell carcinoma through the HOXC6-mediated TGF-beta signaling pathway. Stem Cell Res Ther, 2020. 11(1): p. 117.
- Hao, J., et al., MicroRNA control of epithelial-mesenchymal transition in cancer stem cells. Int J Cancer, 2014. 135(5): p. 1019-27.
- Shimono, Y., et al., Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell, 2009. 138(3): p. 592-603.
- Wang, Q., et al., Enhanced and Prolonged Antitumor Effect of Salinomycin-Loaded Gelatinase-Responsive Nanoparticles via Targeted Drug Delivery and Inhibition of Cervical Cancer Stem Cells. Int J Nanomedicine, 2020. 15: p. 1283-1295.
- Wang, Q., et al., Combined delivery of salinomycin and docetaxel by dual-targeting gelatinase nanoparticles effectively inhibits cervical cancer cells and cancer stem cells. Drug Deliv, 2021. 28(1): p. 510-519.
- Chatterjee, S. and C.N. Kundu, Nanoformulated quinacrine regulates NECTIN-4 domain specific functions in cervical cancer stem cells. Eur J Pharmacol, 2020. 883: p. 173308.
- Cheng, K. and M. Hao, Metformin Inhibits TGF-beta1-Induced Epithelial-to-Mesenchymal Transition via PKM2 Relative-mTOR/p70s6k Signaling Pathway in Cervical Carcinoma Cells. Int J Mol Sci, 2016. 17(12).
- Tang, J., et al., MiR-612 suppresses the stemness of liver cancer via Wnt/beta-catenin signaling. Biochem Biophys Res Commun, 2014. 447(1): p. 210-5.
- Liang, Z., et al., Curcumin reversed chronic tobacco smoke exposure induced urocystic EMT and acquisition of cancer stem cells properties via Wnt/beta-catenin. Cell Death Dis, 2017. 8(10): p. e3066.
- Dou, J., et al., Decreasing lncRNA HOTAIR expression inhibits human colorectal cancer stem cells. Am J Transl Res, 2016. 8(1): p. 98-108.
- Wu, L., et al., TROY Modulates Cancer Stem-Like Cell Properties and Gefitinib Resistance Through EMT Signaling in Non-Small Cell Lung Cancer. Front Genet, 2022. 13: p. 881875.
- Han, M., et al., Antagonism of miR-21 reverses epithelial-mesenchymal transition and cancer stem cell phenotype through AKT/ERK1/2 inactivation by targeting PTEN. PLoS One, 2012. 7(6): p. e39520.
- Liu, Q., et al., Targeted delivery of miR-200c/DOC to inhibit cancer stem cells and cancer cells by the gelatinases-stimuli nanoparticles. Biomaterials, 2013. 34(29): p. 7191-203.
- Deng, Y., et al., MiR-429 suppresses the progression and metastasis of osteosarcoma by targeting ZEB1. EXCLI J, 2017. 16: p. 618-627.
- Liu, X., et al., Tumor-suppressing effects of miR-429 on human osteosarcoma. Cell Biochem Biophys, 2014. 70(1): p. 215-24.
- Tanabe, S., et al., Interplay of EMT and CSC in Cancer and the Potential Therapeutic Strategies. Front Pharmacol, 2020. 11: p. 904. [CrossRef]
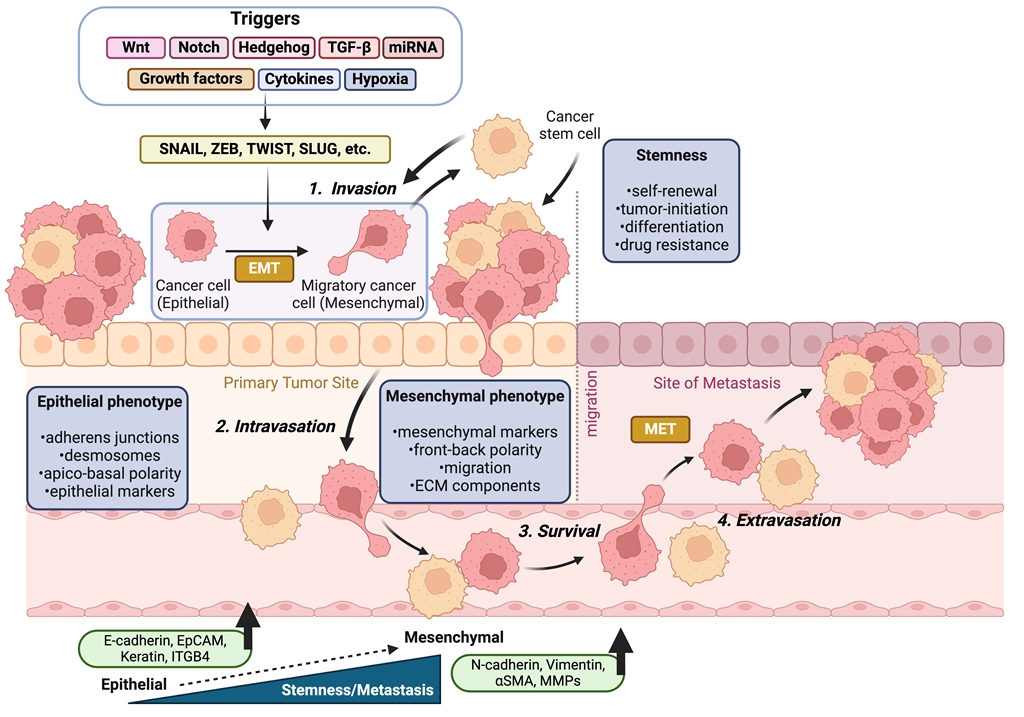
Figure 1.
This schematic model illustrates the linkage between EMT and the CSCs concept in the context of tumor progression. EMT induction in carcinoma cells leads to the acquisition of stem-like properties, characterized by the expression of mesenchymal markers and the loss of epithelial markers. Additionally, CSC subpopulations exhibit EMT phenotypes, with EMT and CSC pathways being regulated at the gene level through various signaling pathways. Cancer cells detach from the basement membrane, intravasate into nearby blood vessels through EMT. Once the cells exit the bloodstream and/or lymphatic system (extravasation), they enter a new microenvironment (secondary site) where they undergo a MET. This transition supports them to colonize the surrounding tissue and initiate the formation of secondary tumors.
Figure 1.
This schematic model illustrates the linkage between EMT and the CSCs concept in the context of tumor progression. EMT induction in carcinoma cells leads to the acquisition of stem-like properties, characterized by the expression of mesenchymal markers and the loss of epithelial markers. Additionally, CSC subpopulations exhibit EMT phenotypes, with EMT and CSC pathways being regulated at the gene level through various signaling pathways. Cancer cells detach from the basement membrane, intravasate into nearby blood vessels through EMT. Once the cells exit the bloodstream and/or lymphatic system (extravasation), they enter a new microenvironment (secondary site) where they undergo a MET. This transition supports them to colonize the surrounding tissue and initiate the formation of secondary tumors.


Table 1.
An overview of the association between EMT and different tumorigenic processes. The table briefly outlines the interconnection of EMT with key aspects of cancer progression, including cancer stemness, angiogenesis, metastasis, CTCs, cytokine involvement, stromal tumor cell interactions, immune evasion, inflammation, tumor dormancy, chemoresistance, and senescence, emphasizing the role of EMT in uplifting tumor cell motility, invasion, survival, immune evasion, and even resistance to therapeutic strategies.
Table 1.
An overview of the association between EMT and different tumorigenic processes. The table briefly outlines the interconnection of EMT with key aspects of cancer progression, including cancer stemness, angiogenesis, metastasis, CTCs, cytokine involvement, stromal tumor cell interactions, immune evasion, inflammation, tumor dormancy, chemoresistance, and senescence, emphasizing the role of EMT in uplifting tumor cell motility, invasion, survival, immune evasion, and even resistance to therapeutic strategies.
| EMT association | Findings | References |
|---|---|---|
| Stemness | EMT activation is closely linked to the generation of CSCs, contributing to tumorigenesis, metastasis, drug resistance, and relapse. EMT transcription factors, such as Zeb1, suppress epithelial differentiation and facilitate stemness, while signaling pathways like TGF-β, Snail1/Twist1, and Notch promote the acquisition of stem-like traits. | [48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64] |
| Tumor Angiogenesis | Angiogenesis and EMT are unified; VEGF, EGF, NECTIN-4 pathways promote EMT and are associated with increased tumor cell motility and invasion. | [65,66,67,68,69,70,71] |
| Metastasis | EMT is also linked with early metastatic processes, which includes cell invasion, cytoskeletal reorganization, and MMPs-mediated basement membrane degradation. | [38,72,73,74,75,76] |
| Circulating Tumor Cells (CTCs) | CTCs show incomplete EMT, express both epithelial and mesenchymal markers, and are involved in metastasis and poor patient prognosis. | [77,78] |
| Cytokine involvement | Cytokines like HGF, FGF, EGF, IL-6, IL-8, TGF-β, TNF-α, and IL-27 play important roles in stimulating or regulating EMT in different cancer types. | [79,80,81,82,83,84,85,86] |
| Stromal Tumor Cells | Cytokines and growth factors from tumor stroma (EGF, HGF, TGF-β, PDGF etc.) activate transcription factors (Snail, Slug, ZEB1, Twist) that induce EMT. | [87,88,89,90,91,92,93,94] |
| Immune interactions | EMT also contributes to immune evasion; a strong association exists between high EMT activity in tumors and the presence of inflammatory cytokines and immune checkpoints (e.g., PD-1, PD-L1). | [95,96,97,98,99,100,101,102,103] |
| Inflammation | Inflammatory mediators (e.g., TNF-α and IL-8) promote EMT in cancer cells, upregulating tumor progression and metastasis, particularly in inflammatory breast cancer. | [104,105,106,107,108,109,110,111] |
| Tumor dormancy | EMT aids in tumor dormancy, with Snail and LOXL2 involved in persevering the mesenchymal phenotype and CSC-like traits. | [112,113,114,115] |
| Chemoresistance | EMT contributes to cancer drug resistance by influencing cell survival, cell fate transition, elevating the drug-resistance-involved genes, promoting stemness, dysregulating transcription factors, and immune suppression. | [116,117,118,119,120,121,122,123,124,125,126,127,128] |
| Senescence | Senescence and EMT are interconnected; EMT can prohibit senescence, promoting tumor progression and invasion. | [129,130,131,132,133,134] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



