
Submitted:
09 September 2024
Posted:
11 September 2024
You are already at the latest version
Abstract
We report the synthesis and characterization of Mn(III) chloride (MnIIICl3) complexes coordinated with N-oxide ylide ligands, namely trimethyl-N-oxide (Me3NO) and pyridine-N-oxide (PyNO). The compounds are reactive and, while isolable in the solid-state at room temperature, readily decompose into Mn(II). For example, “[MnIIICl3(ONMe3)n]” decomposes into the 2D polymeric network compound complex salt [MnII(µ-Cl)3MnII(µ-ONMe3)]n[MnII(µ-Cl)3]n•(Me3NO•HCl)n (4). The reaction of MnIIICl3 with PyNO forms varied Mn(III) compounds with PyNO coordination and these react with hexamethylbenzene (HMB) to form the chlorinated organic product 1-cloromethyl-2,3,4,5,6-pentamethylbenzene (8). In contrast to N-oxide coordination to Mn(III), the reaction between [MnIIICl3(OPPh3)2] and 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) resulted in electron transfer forming d5 manganate of the [TEMPO] cation instead of TEMPO–Mn(III) adducts. The reactivity affected by N-oxide coordination is discussed through comparisons with other L–MnIIICl3 complexes within the context of reduction potential.
Keywords:
Mn(III)
; N-oxide ligands
; coordination chemistry
; C–H chlorination
1. Introduction
The manganese (III) ion is important in environmental and biological processes[i] and is used for various applications in organic synthesis [ii,iii,iv]. Its importance stems from its potent reactive properties, but as a result, much of the chemistry surrounding Mn(III) halides is limited due to lack of stable molecular precursors. For instance, MnIIICl3 is typically prepared by treating manganese oxides with ethereal hydrochloric acid generating deep purple-brown solutions whose contents have been demonstrated to contain solvated forms of MnIIICl3 [v]. These solutions are unstable above temperatures of -35 °C and are sensitive to reaction conditions. A room temperature (r.t.) meta-stable solution containing MnCl3 was prepared by Christou and coworkers using Mn12 ([Mn12Ol2(OAc)16(H2O)4]•2HOAc•4H2O) as the starting material [vi]. We used “Christou’ solution” of MnCl3 to prepare the complex [MnIIICl3(OPPh3)2] (1), which is bench stable and can be stored indefinitely as a solid open to air [vii]. We hypothesized that the pnictogen-oxide ylide ligand triphenylphosphine oxide (PPh3O) is responsible for the stabilization of solvated MnIIICl3 complexes. As an extension of this hypothesis, we have begun to explore other pnictogen-oxide ylide ligands to determine how differences affect the chemical properties of MnIIICl3.
For this study, we chose to explore the synthesis of Mn(III) chloride complexes with N-oxide ylide ligands pyridine-N-oxide (PyNO), trimethylamine-N-oxide (Me3NO), and 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO). Me3NO has rarely been used as a ligand for metal complexes [viii], with only one example for Mn previously reported by us [ix]. A few Mn(III) chloride complexes with PyNO ligand have been reported [x]. However, their syntheses involve the noted cumbersome low temperature procedures, and they have not been characterized with crystallography, nor have their chemical properties been studied. We recently reported the first example of a structurally characterized PyNO ligation to Mn(III) [xi]. Therefore, in this current study we continue the coordination chemistry of N-oxide ligands to Mn(III) chloride complexes.
Scheme 1.
Synthesis of Mn(III) chloride complexes.
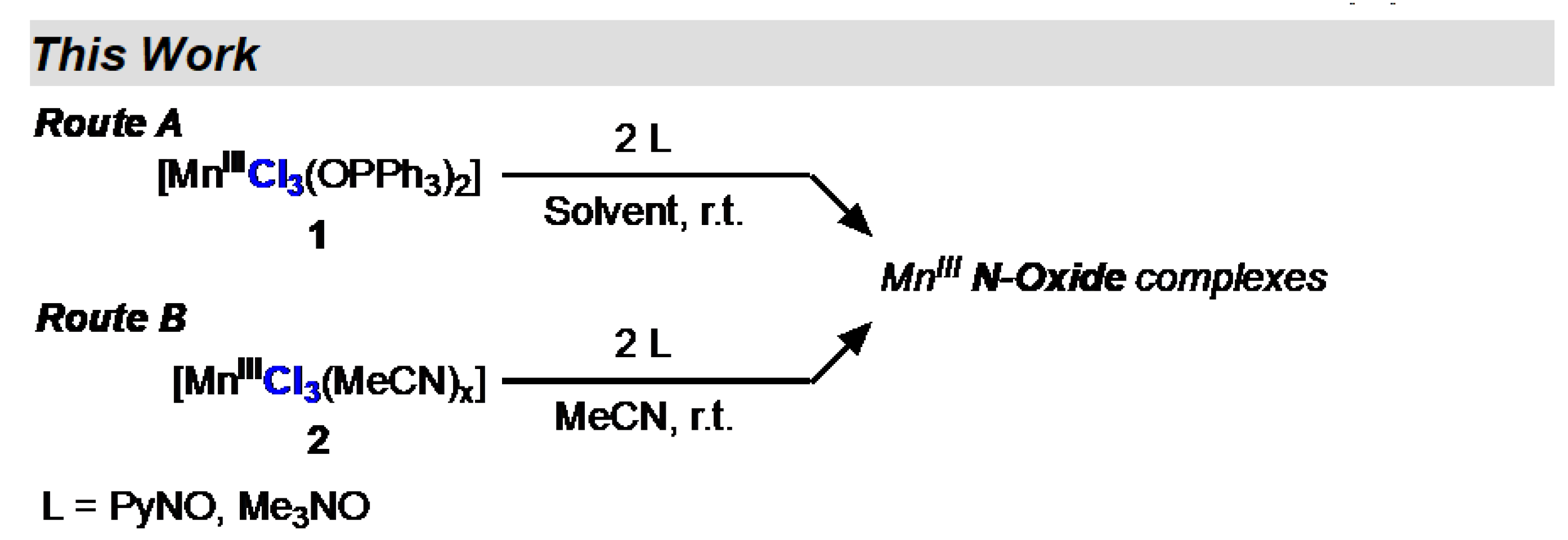
2. Results and Discussion
2.1. Description of starting materials
Our initial success with preparing Mn(III) complexes utilized in-situ generated MnIIICl3(MeCN)x (x = 2, 3) (2) prepared from treating Mn12 with an excess of Me3SiCl in MeCN [7]; we refer to this solution as “Christou’s solution” and contains 2, Me3Si-derived byproducts, water, and acetic acid. While Christou’s solution is convenient, it has non-precise stoichiometry and the noted byproducts may interfere with desired chemistry. Furthermore, 2 is not a thermally stable compound and needs to be used soon after generation (within an hour after generation at r.t. or one day if stored at -35 °C). All our attempts at isolation of 2 resulted in the isolation of the known molecular compound [MnIICl2(MeCN)2] [xii].
We initially used 2 to prepare 1, but we also developed a single-pot synthesis [7]. 1 is a convenient MnIIICl3 source as it is an air-stable solid and can be weighed out accurately and can be used in a variety of solvents such as benzene, toluene, DCM, MeCN, and acetone [7, xiii]. As such, 1 is the ideal starting material in most cases, but 2 can be used when Ph3PO interferes with desired coordination chemistry or purification.
2.2. Synthesis of Mn(III) trimethylamine-N-oxide compounds
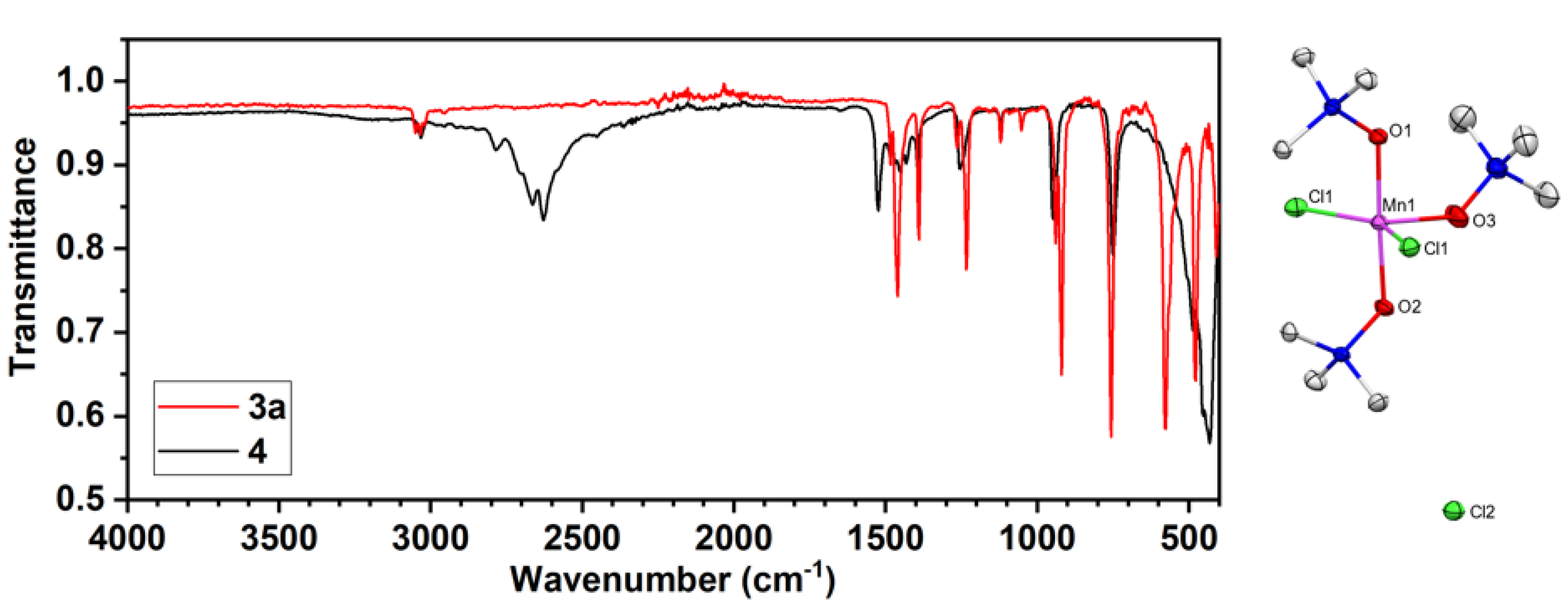
The reaction of Me3NO with 1 or 2 in MeCN produces an insoluble purple solid as the major product which we tentatively assign as [MnIIICl3(Me3NO)2]n (3a) for reasons that are discussed soon (Scheme 2). The FTIR spectrum of the solid shows a shifted N−O stretch of Me3NO at 1234 cm-1 (Figure ); free Me3NO has a stretch at 1254 cm-1. CHN combustion analysis agrees with a composition of Mn3Cl9(Me3NO)5.7.
From the same reaction mixture, the purple solvated complex salt [MnIIICl2(ONMe3)3]Cl•MeCN (3b) also forms as a minor product. XRD analysis of 3b revealed a Mn(III) center in pseudo trigonal bipyramidal geometry (τ5 = 0.52) with trans-Me3NO ligation. FTIR spectra of (3b) shows an N−O stretch at 1234 cm-1 identical to 3a and a C−N stretch at 2251 cm-1 arising from the free acetonitrile solvate. Suspensions of 3a in MeCN with or without Me3NO did not increase yields of 3b. Considering the low yield and extreme sensitivity to air and moisture and room temperature instability, 3b was not included in the subsequent reactivity studies.
3a is extremely reactive to moisture and immediately changes into an insoluble brown solid, 4; this conversion occurs quickly in air (≤ 1 min) and even occurs in a glovebox with 1 ppm water (≤ 1 week). We were successful at obtaining a molecular structure of 4 using microcrystal electron diffraction (MicroED) [xiv] and this revealed that 4 is an ion pair of two manganese chloride polymers (Figure 1). The cationic polymeric species contains a repeating unit of [MnII(µ-Cl)3MnII(µ-ONMe3)3]+, and the anionic polymeric species has the repeating unit of [MnIICl3]–. The material from which the MicroED structure of 4 was obtained also contained crystals of Me3NO•HCl consistent with the characteristic broad H−Cl stretch at 2650 cm-1 in the FTIR spectrum (Figure 2). These data, in addition to a CHN analysis, enabled us to characterize the brown solid 4 as [MnII(µ-Cl)3MnII(µ-ONMe3)]n[MnII(µ-Cl)3]n•(Me3NO•HCl)n (4). The polymeric [{MnIICl3}–]n entity has been observed before [xv] and has Mn–Cl bond lengths of 2.5 – 2.6 Å, in agreement with the Mn–Cl bond length for the cation (2.6 Å) and anion (2.5 Å) polymer chains in 4, supporting our assignment of the Mn(II) oxidation states in both. Although not pursued in this study due to limited access to the MicroED instrumentation, we assume a similar structure for the insoluble 3a, which has intriguing similarities to certain polymers proposed in the literature to have unique electronic properties [xvi]; the structural elucidation of 3a is being pursued in a separate study.
The instability of 3a and 3b towards reduction to Mn(II) species is characteristic of Mn(III) complexes in general. We have shown that one path for decomposition is reduction of Mn(III)X species by C–H bonds [11,xvii] and test this hypothesis later with hexamethylbenzene (HMB) (vide infra). Two alternative paths are water oxidation or disproportionation. No gas evolution was observed, and we do not see evidence for H2O2 in the FTIR spectrum or in the structure of 4 indicating that water oxidation is unlikely decomposition pathway, albeit rigorous gas evolution studies were not performed. Disproportionation is unlikely too because we do not observe any Mn(IV) species. The structure of 4 rules out pathways involving O-atom transfer from Me3NO. Therefore, in the absence of an external substrate, we presume that the organic solvent or the methyl groups in Me3NO are acting as the reductant for 3a. Its decomposition onset by inclusion of water is not well understood.
2.3. Synthesis of Mn(III) PyNO compounds
Two Mn(III) chloride complexes with PyNO ligands, namely [MnIIICl3(PyNO)2] and [MnIIICl3(PyNO)3], have been reported previously [10]. The synthesis of these complexes uses ethereal solutions of MnIIICl3 that require manipulations below -35 °C and the products of these reactions are only partially characterized. Furthermore, there are some dubious aspects of the assignments that likely arise from the difficulty in assigning the empirical formulas to paramagnetic compounds from CHN analysis alone.
Following a similar procedure from Uson to prepare [MnIIICl3(PyNO)2] [10a], we reacted PyNO with 1 in EtOH. Rather than forming [MnIIICl3(PyNO)2] as noted in the literature [10a], we obtained the complex salt [MnIIICl(H2O)(PyNO)4][MnIICl4] (5) (Scheme 3) and it was characterized by X-ray diffraction (Figure 2); the same product was formed when PyNO was reacted with Christou’s solution, i.e. 2 in MeCN, open to air.
The FTIR spectrum of 5 exhibits a shifted N–O stretch at 1199 cm-1 and O–H stretch at 3396 cm-1; the N–O stretch for free PyNO is 1238 cm-1. The Mn(III) center exhibits an octahedral geometry with trans chlorido and aquo ligands. The complex exhibits Jahn teller distortion with elongation along the Mn–Cl axis (Figure 3a). Bond lengths and angles of the second manganese center is consistent with [MnIICl4]2- [xviii]. The red-brown crystals of 5 dissolve in DCM or MeCN to produce deep green solutions which are stable in air. The UV-vis spectrum of 5 in MeCN exhibits three main bands centered at 332 nm (ε = 7200 M-1cm-1), 375 nm (ε = 6300 M-1cm-1), and 615 nm (ε = 820 M-1cm-1).
When the reaction of 2 with PyNO was performed excluding air and moisture, we instead obtained the composite compound complex salt [MnIII(PyNO)4Cl2]2[MnIICl4]•[MnIII(PyNO)2Cl3] (6•7) (Scheme 3) and was characterized by XRD (Figure 3b). The molecular cation in 6, [MnIII(PyNO)4Cl2]+, exhibits elongated trans-Mn–Cl bonds as a result of Jahn-Teller distortion and is charge balanced with a [MnIICl4]2–. The neutral complex [MnIII(PyNO)2Cl3] (7), has a distorted trigonal bipyramidal geometry (τ5 = 0.55), with the chlorido ligands occupying the three equatorial positions and PyNO ligands occupying the two axial positions (Figure 3c). MeCN solution of 6•7 shows intense green color with similar bands as 5 in the UV-vis spectrum at 340 nm (ε = 17000 M-1cm-1), 375 nm (ε = 15000 M-1cm-1), and 612 nm (1900 M-1cm-1).
The synthesis of 5 and 6•7 show that small changes in the reaction conditions lead to dramatically different results. In our hands, we found that the isolation of [MnIII(PyNO)2Cl3] (7) was only possible by treating 1 with two equivalents of PyNO in THF. Of the compounds described so far, 7 is the only thermally stable Mn(III) complex and thereby enabled us to determine the magnetic properties of the Mn(III) ion in isolation from any other magnetically active contaminant [xix]. Evans method indicates a high spin Mn(III) center (S = 2), consistent with its Mn(III) oxidation state assignment and similar to 1 and other Mn(III) complexes we have prepared [7,11-13, 17]. The FTIR spectrum of 7 exhibits characteristic N–O stretch at 1188 cm-1 enabling a tentative assignment of 1201 cm1 of the N–O stretch in 6 (Figure S14). The complex 7 dissolves in DCM and MeCN to produce intense green solutions with UV-vis bands centered at 340 nm (ε = 5100 M-1cm-1), 375 nm (ε = 4800 M-1cm-1), and 632 nm (ε = 1200 M-1cm-1). The Mn(III) center in 7 has a distorted trigonal bipyramidal geometry (τ5 = 0.70), with chlorido and aquo ligands trans to each other similar to the structure found in 6•7 (Figure c). Under no set of conditions were we able to prepare the complex [MnIIICl3(PyNO)3] [10b].
Although the solid-state structures of 5, 6•7, and 7 are different, the UV-vis features in solution are strikingly similar. Likewise, the reduction potentials obtained from cyclic voltammetry (CV) and differential pulse voltammetry (DPV) are essentially the same (Figure S19-S21), each having a reversible reduction 0.47 ± 0.02 V vs FeCp2 in MeCN (0.5 M [nBu4N][PF6]). This implicates that the differences are mostly due to conditions related to crystallization. We suspect that the solution state behavior is an equilibrated mixture of these species, although we do not know which is the dominant form in solution. This hypothesis is supported by their similar reactivity, which is described next.
2.4. Reactivity of Mn(III) chloride compounds
As noted above, we have shown that C–H bonds can act as reductants toward Mn(III)X3 species [11,17]. Hence, we sought to explore this mode of reactivity of the newly synthesized Mn(III) chloride complexes with hexamethylbenzene (HMB) as the substrate. The mechanism for this reaction is consistent with C–H cleavage by a MnIIICl species to form HCl and a benzylic radical [xx], the latter of which rebounds with a second equivalent of MnIIICl to form the C–Cl bond in the final product [xxi]; this mechanism is being studied in a separate study and not discussed further here. Therefore, two equivalents of the Mn(III) reactant is needed for each C–H bond reacted; yields are reduced by 50% if only one equivalent of Mn(III) reactant is used.
Complexes 3a, 5, and 7 were chosen for exploring C–H chlorination reactivity for their combined ease of synthesis and handling and their well-defined Mn(III) stoichiometry. Each complex reacts with HMB to give the chlorinated product 1-cloromethyl-2,3,4,5,6-pentamethylbenzene (8). Out of all the complexes explored in this study, the pyridine N-oxide complexes produced the highest yield of 8, reaching 88% conversion in 4 h for 7 and 86% in 6 h for 5 (Table 1). The similar reactivity of 5 and 7 supports the noted common solution-state speciation hypothesis. The reaction of HMB with 3a produced 8 in only 40% yield; the Mn product is not 4 but some other Mn(II) byproduct. We suspect that the low yield is due to insolubility of 3a and competing side reactions.
In comparing with the reactivity between 7 and 1, we noted that 1 is far more efficient at the chlorination of HMB. 1 produces near quantitative conversion to 8 at room temperature in 7 h [17]. This is consistent with the higher potential of 1, which is 0.77 V vs FeCp2 compared to 0.47 V for the PyNO complexes. Using the reduction potential of 0.47 V and a pKa of 10.6 for HCl in MeCN [xxii] in the Bordwell equation furnishes an upper limit of C–H cleaving ability (i.e., BDFEMn(II)/X–H [xxiii]) of 78 kcal/mol in MeCN. This is just below the 81 kcal/mol C–H bond strength in HMB [xxiv] and thus consistent with the need for heating the reactions to achieve conversion. By contrast, 1 has a BDFEMn(II)/X–H of 85 kcal/mol and is thus capable of reacting with HMB at r.t. [17].
The reactivity of HMB with 2 was also explored. Although the yields were higher for 2 compared to the other Mn(III) reactants, the product mixture was complicated by several byproducts. Furthermore, the reaction with 2 proceeded quickly at r.t., consistent with the higher degree of reactivity expected of this solvated entity that is not stabilized by a pnictogen-oxide ligand.
2.5. Reaction of 1 with 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO):
The reaction of 1 with TEMPO did not result in coordination of TEMPO to the manganese center but instead caused oxidation of TEMPO to form TEMPO+ ion (Scheme 5). This is not surprising given that the redox potential of 1 (0.77 V vs. FeCp2 in MeCN) is more positive to that of TEMPO (0.22 V vs. FeCp2) [xxv]. The orange product was characterized by XRD to reveal the d8 manganate salt of the TEMPO cation, [TEMPO]2[MnIICl4] (9) (Figure 4). No stretches associated with Ph3PO were observed in the FTIR spectrum of 9. The compound has low solubility, but CHN and pXRD support the assignment of the bulk material as 9. Treatment of this solid with AgBF4 allowed the isolation of known salt [TEMPO]BF4 [xxvi].
3. Conclusions
In this work, we set out to synthesize Mn(III) chloride complexes stabilized by ligands with N-oxide functional groups. The coordination of N-oxide ligands is rare, and we formed the first examples of Mn(III) stabilized by Me3NO ligands through the characterization of 3a and 3b. The species in 3a was not characterized by diffraction techniques due to its instability. However, it decomposed into a unique molecular polymer salt, where one polymer was a cationic species with Me3NO coordination to Mn(II) centers in 4. Hence, it is apparent that Me3NO can as act as a ligand and not just as an O-atom transfer reagent as it is typically employed.
The synthesis and characterization of the PyNO complexes 5, 6•7, and 7 was also performed. Although the solid-state characterization for each revealed different molecular structures, their solution-state properties were essentially identical. As a result, their reactivity in solution with HMB was also nearly identical. They were, however, diminished in reactivity compared to 1 and this is rationalized by the lower reduction potentials for the PyNO complexes. In contrast, the highly reactive solvated entity in Christou’s solution (i.e., 2) reacted with HMB rapidly at r.t., indicating that the coordination of PyNO remains in solution and has a stabilizing affect. Finally, when the potential of the ligand is low enough, electron transfer occurs as opposed to coordination. This was demonstrated with the stable radical TEMPO, which furnished outer sphere [TEMPO]+ ions rather than MnIII–TEMPO adducts.
4. Materials and Methods
General Considerations: All chemicals were purchased from chemical vendors and used as received unless otherwise noted. Anhydrous Me3NO was obtained by refluxing the dihydrate in toluene using a Dean-Stark apparatus on a Schlenk line overnight and subsequently isolating the material by removal of toluene. The solid thus obtained was washed with hexane, filtered and dried under vacuum inside a glovebox. Material obtained this way contained no evidence of water by NMR or FTIR. Dry, oxygen free solvents were obtained from a PPT solvent purification system, were stored over 3 Å molecular sieves prior to use. Unless otherwise stated, synthetic manipulations of air sensitive compounds were performed in a nitrogen filled VAC glovebox or on a Schlenk line. NMR experiments were carried out on Bruker Neo-400 MHz or Bruker Neo-500 MHz spectrometers. ATR-FTIR spectra were collected using a Bruker Alpha IR spectrometer with the “ATR Platinum” insert adapter (diamond crystal) stored inside a nitrogen filled VAC Atmospheres glovebox. UV-vis experiments were performed using an 8154 Agilent Spectrophotometer equipped with an Unisoku cryostat. CHN combustion analyses were performed using a Thermo Scientific FlashEA1112 CHNS analyzer. Electrochemistry was performed on a SP-200 Bio-Logic potentiostat. The following compounds were prepared according to literature:[Mn12Ol2(OAc)16(H2O)4]•2HOAc•4H2O (Mn12) [xxvii], [MnCl3(OPPh3)2] (1) [7].
Crystallographic methods: Low-temperature X-ray diffraction data for [MnIIICl2(Me3NO)3]Cl•MeCN (3b), [MnIIICl(H2O)(PyNO)4][MnIICl4] (5), [MnIIICl2(PyNO)4]2[MnIICl4]•[MnIIICl3(PyNO)2] (6•7), [MnIIICl3(PyNO)2] (7), and (TEMPO)2[MnIICl4] (9) were collected on a Rigaku XtaLAB Synergy diffractometer coupled to a Rigaku Hypix detector with either Cu Kα radiation (λ = 1.54184 Å) or Ag Kα radiation (λ = 0.56087 Å, for 9), from PhotonJet micro-focus X-ray sources at 100 K. The diffraction images were processed and scaled using the CrysAlisPro software [xxviii]. The structures were solved through intrinsic phasing using SHELXT[xxix] and refined against F2 on all data by full-matrix least squares with SHELXL[xxx] following established refinement strategies [xxxi]. All non-hydrogen atoms were refined anisotropically. All hydrogen atoms bound to carbon were included in the model at geometrically calculated positions and refined using a riding model. The isotropic displacement parameters of all hydrogen atoms were fixed to 1.2 times the Ueq value of the atoms they are linked to.
Electron diffraction measurements were performed on 4, using a Rigaku XtalAB Synergy-ED equipped with a Rigaku HyPix-ED detector optimized for operation in the Micro-ED experimental setup1. Datasets were collected at 100 K with a wavelength of 0.0251 Å using the Rigaku program CrysAlisPro-ED for simultaneous sample measurement and data processing [14b]. Samples were prepared by grinding the powder between two glass slides to reduce particle size and then sweeping a 3mm Cu TEM grid with lacey carbon film into the ground powder. The grid was then mounted on an Elsa 698 cryo-sample holder from Gatan and cooled to 100 K. Finally, the sample holder was inserted into the diffractometer for analysis.
The crystallite used for data collection for both samples was a thin particle with the other two dimensions ranging between 0.3 µm and 1 µm. Using Olex2 [xxxii], crystal structures were readily solved with the ShelXT [29] structure solution program, using Intrinsic Phasing and refined kinematically with the ShelXL [30] refinement package, using Least Squares minimization. All atoms were refined in anisotropic approximation. The hydrogen atoms were placed at their idealized position and refined as riding atoms. Hydrogen bond distances from neutron diffraction data were used for final refinements in ShelXL (using the command ‘neutronHdist’), as hydrogen bond distances observed from electron diffraction are closer to those observed from neutron diffraction than from X-ray diffraction.
Electrochemistry experiments: The MnIII/MnII reduction potentials of Mn(III) complexes were determined using cyclic voltammetry and differential pulse voltammetry experiments. The electrochemical cell was equipped with a glassy carbon working electrode, Ag/Ag+ (4 mM AgNO3 in MeCN, 0.5 M [nBu4N][PF6]) reference electrode with a CoralPorTM separator, and a platinum auxiliary electrode. 0.5 M solution of [nBu4N][PF6] in MeCN was used as the supporting electrolyte. Scans were performed with internal resistance compensation (85%). The analyte concentration was 4 mM for 5 and 7 and 1.33 mM for 6. Scan direction was cathodic (scan rate: 200 mV/s). The reference electrode was externally referenced to ferrocene at the beginning and end of the experiment. For differential pulse voltammetry, the step height was set to 10 mV, the pulse height was 100 mV, the pulse width was 25 ms, and the step width was 100 ms. Scan direction was cathodic.
Synthesis:
Synthesis of [MnIIICl3(ONMe3)2]n (3a) and [MnCl2(Me3NO)3]Cl•MeCN (3b). Route A: In 50 mL round bottom flask equipped with a stir bar, 1 (800 mg, 1.14 mmol, 1 eq.) was stirred in 25 mL MeCN to give a navy-blue suspension. Me3NO (209 mg, 2.78, 2.5 eq.) was added as a solid in under vigorous stirring to form a purple precipitate. The reaction mixture was allowed to stir at room temperature for 30 minutes. Upon completion of the reaction, the reaction mixture was filtered through a medium porosity glass fritted funnel (medium frit) to obtain a purple solid (3a) and a deep blue filtrate. The solid 3a washed with 3 x 2 mL MeCN, followed by 2 x 2 mL diethyl ether, and dried under vacuum (178.3 mg, 51%). Storage of the deep blue filtrate at -35 °C overnight produced 48 mg (10%) of deep purple crystals of 3b.
Route B:Mn12 (100.0 mg, 0.048 mmol, 1 eq.) was dissolved in 10 mL MeCN in a 20 mL scintillation vial equipped with a stir bar and stirred to give an intense coffee brown mixture. Me3SiCl (0.220 mL, 1.75 mmol, 36 eq.) was added dropwise via syringe with vigorous stirring to give a deep purple solution containing 2. Me3NO (87.5 mg, 1.16 mmol, 24 eq.) was added to the reaction mixture. The reaction mixture immediately turned deep green and formation of a purple precipitate was observed. After 30 minutes of stirring at room temperature, the reaction mixture was filtered through a medium frit and the solid was washed with MeCN (2 x 1 mL) and 1 mL diethyl ether, then dried under vacuum to yield the product 3a as a purple solid (77.8 mg, 43%). Spectroscopic characterization matched the product obtained from route A.
ATR-FTIR (cm-1) of 3a : 3050, 3033, 3019, 1483, 1460, 1390, 1265, 1234, 1118, 1052, 939, 921, 756, 579, 479.
ATR-FTIR (cm-1) of 3b :3033, 3015, 2961, 2916, 2251, 1493, 1464, 1433, 1382, 1267, 1238, 1124, 968, 939, 931, 760, 581, 533, 484, 472.
CHN-Analysis [Calc. (found)] for 3a, [Mn3Cl9(Me3NO)5.7]: %C22.09 (22.04), %H 5.56 (5.28), %N 8.59 (8.49). The formula is presented as [MnCl3(Me3NO)2]n throughout the manuscript for clarity.
Synthesis of [MnII(µ-Cl)3MnII(µ-ONMe3)]n[MnII(µ-Cl)3]n•(Me3NO•HCl)n (4). 3a was added to a 20 mL scintillation vial and exposed to air overnight. The solid immediately turns brown within a minute of exposure to air to form 4 almost quantitatively. The same process occurs more slowly upon storage in the glovebox and could not be prevented, even at -35 °C.
ATR-FTIR (cm-1): 3033, 2785, 2704,2663, 2626, 1522, 1485, 1468, 1452, 1432, 1394, 1256, 1124, 950, 750, 486, 453, 430.
CHN-Analysis [Calc. (found)] for [MnCl2(ONMe3)]•(Me3NO•HCl): %C 23.06 (23.58), %H 6.13 (6.38), %N 8.96 (8.83).
Synthesis of [MnCl(H2O)(PyNO)4][MnCl4] (5). Route A: Mn12 (100 mg, 0.048 mmol, 1 eq.) was dissolved in 20 mL MeCN in a schlenk flask under Ar atmosphere and stirred to give an intense coffee brown solution. Me3SiCl (0.220 mL, 1.75 mmol, 36 eq.) was added dropwise via syringe with vigorous stirring to give a deep purple mixture containing 2. PyNO (110 mg, 1.16 mmol, 24 eq.) was added in one portion to the reaction mixture. The reaction mixture immediately turns deep emerald green. After 30 minutes of stirring at room temperature, the reaction mixture was reduced to 5 mL in vacuo. The solution was filtered through a medium frit, transferred (open to air) into a 25 mL Erlenmeyer flask, and left undisturbed for slow evaporation at room temperature for 5 days, from which 109 mg (56% yield) of deep red-brown crystals of 5 was isolated.
Route B: This procedure is performed open to air. In a 20 mL scintillation vial, PyNO (212 mg, 2.23 mmol, 2 eq.) was dissolved in 40 mL 200 proof ethanol at room temperature. Under vigorous stirring, 1 (800 mg, 1.11 mmol, 1 eq.) was added as a solid to cause an immediate formation of a green reaction mixture and formation of a brown precipitate. The reaction mixture was allowed to stir for 30 minutes. The mixture was then filtered through a medium frit to isolate a brown-black residue. The residue was extracted with 3 x 10 mL EtOH. The EtOH washings were combined and poured into 70 mL pet. ether to precipitate 5 as a red-brown solid. The solid was washed with 10 mL pet. ether and dried open to air (185.6 mg, 51%). Spectroscopic characterization matches the product obtained via route A.
ATR-FTIR (cm-1): 3112, 3051, 1464,1200, 1171, 1095, 1070, 1023, 830, 770, 667.
1H-NMR (CD2Cl2): 1.7, 10.4, 14.7, 20.0.
CHN-Analysis [Calc. (found)] for [MnCl(H2O)(PyNO)4][MnCl4]•H2O: %C 34.14 (33.82), %H 3.44 (3.38), %N 7.96 (7.81).
UV-vis λmax [MeCN, nm (ε, M-1cm -1)]: 332 (7200), 375 (6300), 615 (820).
Synthesis of [MnCl2(PyNO)4]2[MnCl4]•[Mn(PyNO)2Cl3] (6•7). Mn12 (100 mg, 0.048 mmol, 1 eq.) was dissolved in 10 mL MeCN in a 20 mL scintillation vial equipped with a stir bar and stirred to give an intense coffee brown mixture. Me3SiCl (0.220 mL, 1.75 mmol, 36 eq.) was added dropwise via syringe with vigorous stirring to give a deep purple solution containing 2. PyNO (110 mg, 1.16 mmol, 24 equiv.) was added in one portion to the reaction mixture. The reaction mixture immediately turned deep emerald green. After 30 minutes of stirring at room temperature, the reaction mixture reduced to 5 mL under vacuum. The solution was filtered through a medium frit and stored at -35 °C for slow evaporation, from which 84.0 mg (36% yield) of deep green crystals of 6•7 was isolated.
ATR-FTIR (cm-1): 3112, 3051, 1469,1198, 1174, 1095, 1071, 1022, 834, 824, 774, 667.
1H-NMR (CD2Cl2):102, 13.2, 18.0.
CHN-Analysis [Calc. (found)] for [MnCl2(PyNO)4]2[MnCl4]•[Mn(PyNO)2Cl3]: %C 38.48 (38.18), %H 3.23 (3.31), %N 8.97 (8.98).
UV-vis λmax [MeCN, nm (ε, M-1cm -1)]: 340 (17000), 375 (15000), 612 (1900).
Synthesis of [MnCl3(PyNO)2] (7). In 20 mL scintillation vial equipped with a stir bar, 1 (200 mg, 0.279 mmol, 1 eq.) was stirred in 12 mL THF to give a purple suspension. PyNO (53 mg, 0.56, 2 eq.) was added as a solid under vigorous stirring to cause a color change to green. The reaction mixture was allowed to stir at room temperature overnight (~16 h). Upon completion of the reaction, the reaction mixture was filtered through a medium frit and the product was obtained as green solid that was washed with 2 x 1 mL THF, followed by 2 x 1 mL pentane, and dried under vacuum (94 mg, 95 %). Crystals of 7 were obtained from slow diffusion of a saturated DCM solution of 7 with pet. ether at -35 °C.
ATR-FTIR (cm-1): 3116, 3077, 3050, .1606, 1464, 1244, 1188, 1166, 1096, 1073, 1042, 1026, 933.
1H-NMR (CD2Cl2):11.8, 17.9, 25.4.
Evans method (CD3CN, 500 MHz, 298 K) μeff = 4.96 μB.
CHN [Calc. (found)] for [MnCl3(PyNO)2]•CH2Cl2 : %C 30.27 (30.70), %H 2.77 (2.62), %N 6.42 (6.82).
UV-vis λmax [DCM, nm (ε, M-1cm -1)]: 340 (5100), 375 (4800), 632 (1200).
General procedure for chlorination of hexamethylbenzene using Mn(III)−Cl complexes: This procedure is modified after a literature report [17]. Mn(III) complex (0.0813 mmol, 2.2 eq of Mn(III)) was weighed out into a 25 mL bomb flask with a stir bar and 4 mL MeCN was added. A stock solution of HMB was prepared in DCM (74 mM). The HMB solution (0.5 mL or 6 mg, 0.0369 mmol, 1.0 eq.) was added to the reaction mixture and sealed and left to stir at 60 °C until the deep coloration of the solution disappeared. Upon completion of the reaction, the reaction mixture was reduced to ≈0.2 mL under vacuum and loaded onto a plug of silica (pipet, 1.5 inches) to remove metal containing byproducts. The plug was eluted with 15 mL DCM. To the combined DCM washings, an internal standard (10 mg of 2-nitro benzaldehyde) was added and then the solvent was removed under vacuum. The solid residue obtained was dissolved in CDCl3 to prepare an NMR sample. Average yields were calculated from duplicated trials.
Reaction of 2 with hexamethylbenzene: In 20 mL scintillation vial equipped with a stir bar, Mn12 (31.7 mg, 0.0154 mmol, 1 eq.) was stirred in 5 mL MeCN to give coffee brown mixture. Me3SiCl (0.070 mL, 0.554 mmol, 36 eq.) was added dropwise via syringe with vigorous stirring to give a deep purple solution containing 2 and left to stir for 5 mins at room temperature. HMB (15.0 mg, 0.0924 mmol, 6.0 eq.) was added as a solid to the reaction mixture. The reaction vessel was sealed and left to stir for one hour until the deep coloration of the solution disappeared. Upon completion of the reaction, the reaction mixture was reduced to ≈0.2 mL under vacuum and loaded onto a plug of silica (pipet, 1.5 inches) to remove metal containing byproducts. The plug was eluted with 20 mL DCM. To the combined DCM washings, internal standard (10 mg, 2-nitro benzaldehyde) was added and then the solvent was removed under vacuum. The solid residue obtained was dissolved in CDCl3 to prepare NMR sample.
Synthesis of (TEMPO)2[MnCl4] (9): In a 20 mL scintillation vial, 1 (300 mg, 0.418 mmol, 1eq.) was stirred in 2 mL DCM to give deep blue solution. A solution of TEMPO (65.3 mg, 0.418 mmol, 1 eq.) in DCM was added to the reaction mixture under vigorous stirring. The reaction mixture immediately turned red and gradually formed an orange precipitate. The reaction mixture was left to stir for 30 minutes at room temperature, then filtered through a medium frit to isolate 9 as an orange precipitate (56.6 mg, 53%). The precipitate was washed with 1mL DCM, 1 mL pentane and dried under vacuum. The deep orange filtrate was reduced to ≈2 mL under vacuum and stored for slow diffusion with pentane at -35 °C to obtain a few orange crystals of 9. 9 was recrystallized from a solution of 9 in MeCN by slow diffusion of diethyl ether at -35 °C to obtain crystals suitable for XRD.
ATR-FTIR (cm-1) of 9: 2991, 2945, 2877, 1608, 1460, 1394, 1380, 1332, 1293, 1239, 1215, 1211, 1147, 1116, 1098, 1067, 980, 941, 881, 863, 857, 764, 725, 702.
ATR-FTIR (cm-1) of [TEMPO]BF4: 3002, 2965, 2939, 1627, 1473, 1460, 1398, 1382, 1289, 1240, 1213, 1098, 1044, 974, 945, 900, 877, 856, 762,704.
CHN [Calc. (found)] for {(TEMPO)2[MnCl4]}n•0.1CH2Cl2 : %C 41.99 (41.74), %H 7.05 (6.86), %N 5.41 (5.83).
NMR (CD3CN) of [TEMPO]BF4: 1H (400 MHz) 2.41, 2.14, 1.65 ppm; 19F-{1H} (376 MHz) -151.85 ppm.
Reaction of 9 with AgBF4. A stirring suspension of 9 in DCM was treated with AgBF4 (80 mg, 0.411 mmol, 1 eq.), which caused immediate formation of a white solid (AgCl and MnCl2) and a yellow solution. The reaction mixture was filtered and the yellow filtrate was collected. Yellow crystals of [TEMPO]BF4 were obtained after removing volatiles from the filtrate under vacuum (30.0 mg, 30%). The spectroscopic data agree with the literature [26].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: ATR-FTIR spectra of 3a and 3b. Figure S2: ATR-FTIR spectrum of 4. Figure S3: PXRD spectra of 4 and authentic Me3NO•HCl. Figure S4: 1H-NMR spectrum of 5 in CD2Cl2. Figure S5: ATR-FTIR spectrum of 5. Figure S6: UV-vis spectrum of 5 in MeCN at various concentration with corresponding Beer’s law plot for extinction coefficient. Figure S7: Molecular structure of 5 determined with XRD. Figure S8: 1H-NMR spectrum of 6•7 in CD2Cl2. Figure S9: ATR-FTIR spectrum of 6•7. Figure S10: UV-vis spectrum of 6•7 in MeCN at various concentration with corresponding Beer’s law plot for extinction coefficient. Figure S11: Molecular structure of 6•7 determined with XRD. Figure S12: 1H-NMR spectrum of 7 in CD2Cl2. Figure S13: ATR-FTIR spectrum of 7. Figure S14: UV-vis spectrum of 7 in DCM at various concentration with corresponding Beer’s law plot for extinction coefficient. Figure S15: ATR-FTIR spectra of 9, TEMPO, and (TEMPO)BF4. Figure S16: ATR-FTIR spectra of 6•7 and 7. Figure S17: PXRD spectra of 9. Figure S18: 1H-NMR spectrum of [TEMPO]BF4 in CD3CN. Figure S19: 19F-NMR spectrum of [TEMPO]BF4 in CD3CN. Figure S20: Overlaid differential pulse voltammograms of 5, 6•7, and 7. Figure S21: The cyclic voltammogram of 5 and 7 overlaid with the MnIII/MnII reversible event. Figure S22: The cyclic voltammogram of 6•7 overlaid with the MnIII/MnII reversible event. Figure S23: Cyclic voltammograms of 5 at varying scan rates and peak current vs. (scan rate)1∕2 with linear fit. Figure S24: Cyclic voltammograms of 6•7 at varying scan rates and peak current vs. (scan rate)1∕2 with linear fit. Figure S25: Cyclic voltammograms of 7 at varying scan rates and peak current vs. (scan rate)1∕2 with linear fit.
Author Contributions
Conceptualization, DCL and AS; methodology, all authors; investigation, AS, SNM, MRC; writing—original draft preparation, AS; writing—review and editing, DCL and AS; visualization, DCL and AS; supervision, DCL; project administration, DCL; funding acquisition, DCL. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NSF CHE-1847933 (DCL) and NIH-R21-GM141685-01 (DCL).
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data supporting the conclusions of this article are available by the authors on request.
Acknowledgments
University at Buffalo (UB) provided support. X-ray diffraction (XRD) System Rigaku XtaLAB Synergy-S was purchased with NSF award CHE-2216151. The Bruker Ascend-500 NMR spectrometer in the UB Magnetic Resonance Center was purchased with NSF CHE-2018160.
Conflicts of Interest
The authors declare no conflicts of interest.
| 1 | (a) Lingappa, U.F.; Monteverde, D.R.; Magyar, J.S.; Valentine, J.S.; Fischer, W.W. How manganese empowered life with dioxygen (and vice versa). Free Radic. Biol. Med. 2019, 140, 113–125. (b) Zhu, W.; Richards, N.G.J. Biological functions controlled by manganese redox changes in mononuclear Mn-dependent enzymes. Essays Biochem. 2017, 61, 259-270. (c) Li, H.; Santos, F.; Butler, K.; Herndon, E.; A critical review on the multiple roles of manganese in stabilizing and destabilizing soil organic matter. Environ. Sci. Technol. 2021, 55, 12136–12152. |
| 2 | Alkene difunctionalization: (a) Fu, N.; Sauer, G. S.; Saha, A.; Loo, A.; Lin, S. Metal-catalyzed electrochemical diazidation of alkenes. Science 2017, 357, 575–579. (b) Sauer, G. S.; Lin, S. An Electrocatalytic approach to the radical difunctionalization of alkenes. ACS Catal. 2018, 8, 5175–5187. (c) Dong, X.; Roeckl, J. L.; Waldvogel, S. R.; Morandi, B. Merging shuttle reactions and paired electrolysis for reversible vicinal dihalogenations. Science 2021, 371, 507–514. |
| 3 | Alkene hydroxylation and epoxidation: (a) Philip, R. M.; Radhika, S.; Abdulla, C. M. A.; Anilkumar, G. Recent trends and prospects in homogeneous manganese-catalyzed epoxidation. Adv. Synth. Catal. 2021, 363, 1272–1289. (b) Eisink, N. N. H. M.; Browne, W. R. Chapter 10. Manganese-catalyzed dihydroxylation and epoxidation of olefins. In Manganese Catalysis in Organic Synthesis. Wiley-VCH, 2022, 323-343. |
| 4 | Radical functionalizations with Mn(OAc)3. (a) Demir, A.S.; Emrullahoglu, M. Manganese(III) acetate: a versatile reagent in organic chemistry. Curr. Org. Synth. 2007, 4, 321–350. (b) Mondal, M.; Bora, U. Recent advances in manganese(III) acetate mediated organic synthesis. RSC Advances. 2013, 3, 18716-18754. (c) Carney, J. R.; Dillon, B. R.; Thomas, S. P. Recent advances of manganese catalysis for organic synthesis. Eur. J. Org. Chem. 2016, 3912-3929. (d) Snider, B. B. Chapter 9. Manganese(III) acetate-mediated cyclizations. In Manganese Catalysis in Organic Synthesis. Wiley-VCH, 2022, 293-322. |
| 5 | (a) Goodwin, H. A.; Sylva, R. N. The magnetic properties of some complexes of higher-valent manganese. Aust. J. Chem. 1967, 20, 629–637. (b) Funk, H.; Kreis, H. Zur kenntnis des dreiwertigen mangans: verbindungen des mangan(III)-chlorids mit aminen und einigen Äthern. Z. Anorg. Allg. Chem. 1967, 349, 45–49. (c) Davis, T. S.; Fackler, J. P.; Weeks, M. J. spectra of manganese(III) complexes. the origin of the low-energy band. Inorg. Chem. 1968, 7, 1994–2002. (d) Nachtigall, O.; Pataki, A.; Molski, M.; Lentz, D.; Spandl, J. solvates of manganese trichloride revisited - synthesis, isolation, and crystal structure of MnCl3(THF)3. Z. Anorg. Allg. Chem. 2015, 641, 1164–1168. |
| 6 | Perlepes, S. P.; Blackman, A. G.; Huffman, J. C.; Christou, G. Complete carboxylate removal from [Mn12O12(OAc)16(H2O)4]•2HOAc•4H2O with Me3SiCl: synthesis and characterization of polymeric [MnCl3(bipy)]n and an improved Synthesis of (NEt4)2MnCl5. Inorg. Chem. 1991, 30, 1665–1668. |
| 7 | Saju, A.; Griffiths, J. R.; MacMillan, S. N.; Lacy, D. C. Synthesis of a bench-stable manganese(III) chloride compound: coordination chemistry and alkene dichlorination. J. Am. Chem. Soc. 2022, 144, 16761. |
| 8 | ConQuest (CSD version 5.45) search for transition metal complexes with Me3NO ligands produces 18 examples of the following metal ions –Re+1, Mn1+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+, and Y3+. |
| 9 | Kadassery, K.J.; Dey, S.K.; Friedman, A.E.; Lacy, D.C. Exploring the role of carbonate in the formation of an organomanganese tetramer. Inorg. Chem. 2017, 56, 8748–8751. |
| 10 | (a) Uson, R.; Riera, V.; Ciriano, M.A.; Valderrama, M. Pentacoordinate neutral manganese (III) complexes. Transit. Met. Chem. 1976, 1, 122–126. (b) Contreras, E.; Riera, V.; Usón, R. Stable complexes of manganese (III) with oxides of pyridine, phosphine and arsine. Inorg. Nucl. Chem. Lett. 1972, 8, 287–291. |
| 11 | Saju, A.; Crawley, M.R.; MacMillan, S.N.; Lacy, D.C. Manganese(III) nitrate complexes as bench-Stable powerful oxidants. J. Am. Chem. Soc. 2024, 146, 11616–11621. |
| 12 | Pokhodnya, K. I.; Bonner, M.; DiPasquale, A. G.; Rheingold, A. L.; Her, J. H.; Stephens, P. W.; Park, J. W.; Kennon, B. S.; Arif, A. M.; Miller, J. S. Structural and magnetic properties of MCl2 (M = Fe, Mn, Co): acetonitrile solvates. Inorg. Chem. 2007, 46, 2471–2477. |
| 13 | Paul, S.; Saju, A.; Cohen, C.; Crawley, M. R.; MacMillan, S. N.; Lacy, D. C. Synthesis of Mn(III)X3 (X = Cl, Br, I) Compounds with Phosphine (R3P) Ligands. Inorg. Chem. 2024, 34, 15791-15803. |
| 14 | (a) Nannenga, B. L.; Gonen, T. The cryo-EM method microcrystal electron diffraction (MicroED) Nature Methods, 2019, 16, 369-379. (b) Ito, S.; White, F. J.; Okunishi, E.; Aoyama, Y.; Yamano, A.; Hiroyasu, S.; Ferrara, J. D.; Jansnowski, M.; Meyer, M. Structure determination of small molecule compounds by an electron diffractometer for 3D ED/MicroED. CrystEngComm. 2021, 23, 8622-8630. |
| 15 | (a) Caputo, R. E.; Roberts, S.; Willett, R. D.; Gerstein, B. C. Crystal structure and magnetic susceptibility of [(CH3)3NH]3Mn2Cl7. Inorg. Chem. 1976, 15, 820-823. (b) Ravindran, M.; Willey, G. R.; Drew, M. G. B. Reactions of trimethylamine with Mn(II) and Cd(II) chlorides: crystal and molecular structure of [Me3NH][MnCl3]. Inorg. Chimica Acta 1990, 175, 99-103. (c) Naito, T.; Inabe, T. Molecular hexagonal perovskite: a new type of organic-inorganic hybrid conductor. Journal of Solid State Chemistry. 2003, 176, 243-249. (d) Sun, X.-F.; Li, P.-F.; Liao, W.-Q.; Wang, Z.; Gao, J.; Ye, H.-Y.; Zhang, Y. Notable broad dielectric relaxation and highly efficient red photoluminescence in perovskite-type compound: (N-methylpyrrolidinium)MnCl3. Inorg. Chem. 2017, 56, 12193-12198. |
| 16 | (a) Sun, Q.; Kioussis, N.; Prediction of manganese trihalides as two-dimensional Dirac half-metals. Phys. Rev. B, 2018, 97, 094408. (b) Zhou, B.; Li, Z.; Theoretical investigation of nonvolatile electrical control behavior by ferroelectric polarization switching in two-dimensional MnCl3/CuInF2S6 van der Waals heterostructures. J. Mater. Chem. C. 2020, 8, 4534. (c) Guo, T.; Liu, Y.; Sun, Y.; Zhang, S.; Xu, X.; Wang, L.; Zhou, W.; Liu, Y.; Yao, X.; Zhang, X. Insight into tunable electronic and magnetic properties in 2D ferromagnetic/antiferromagnetic van der Waals heterostructure. Appl. Phys. Lett. 2023, 122, 192403. |
| 17 | Saju, A.; Gunasekera, P. S.; Morgante, P.; MacMillan, S. N.; Autschbach, J.; Lacy, D. C. Experimental and computational determination of a M−Cl homolytic bond dissociation free energy: Mn(III)Cl-mediated C−H cleavage and chlorination. J. Am. Chem. Soc. 2023, 145, 13384-13391. |
| 18 | Caputo, R.E.; Roberts, S.; Willett, R.D.; Gerstein, B.C. Crystal structure and magnetic susceptibility of heptachlorotris(trimethylammonium)dimanganese. Inorg. Chem. 1976, 15, 820–823. |
| 19 | The instability of 3, the inclusion of MnII counterions in 5, and the composite nature of 6•7 precluded similar characterization. Therefore, the magnetic properties of the other compounds were not pursued in this study. Generally, mononuclear Mn(III) centers studied by us have had S = 2 ground states. Some exceptions are strong-field six-coordinate cationic Mn(III) complexes that are S = 1 (see [7,11,13,17]). |
| 20 | (a) Mondal, P.; Pirovano, P.; Das, A.; Farquhar, E. R.; McDonald, A. R. Hydrogen atom transfer by a high-valent nickel-chloride complex. J. Am. Chem. Soc. 2018, 140, 1834-1841. (b) Mondal, P.; Lovisari, M.; Twamley, B; McDonald, A. R. Fast hydrocarbon oxidation by a high-valent nickel-fluoride complex. Angew. Chem. Int. Ed. 2020, 59, 13044-13050. (c) Kwon, Y. M.; Lee, Y.; Schmautz, A. K.; Jackson, T. A.; Wang, D. C–H bond activation by a mononuclear nickel(IV)-nitrate complex. J. Am. Chem. Soc. 2022, 144, 12072-12080. (d) Kwon, Y. M.; Lee, Y.; Evenson, G. E.; Jackson, T. A.; Wang, D. Crystal structure and C–H bond cleaving reactivity of a mononuclear CoIV-dinitrate complex. J. Am. Chem. Soc. 2020, 142, 13435-13441. (e) Bower, J. K.; Reese, M. S.; Mazin, I. M.; Zarnitsa, L. M.; Cypcar, A. D.; Moore, C. E.; Sokolov, A. Y.; Zhang, S. C(Sp3)-H cyanation by a formal copper(III) cyanide complex. Chem. Sci. 2023, 14, 1301–1307. |
| 21 | (a) Liu, W.; Huang, X.; Cheng, M.; Nielsen, R. J.; Goddard, W. A.; Groves, J. T. Oxidative Aliphatic C-H Fluorination with Fluoride Ion Catalyzed by a Manganese Porphyrin. Science 2012, 337, 1322–1325. (b) Yadav, V.; Wen, L.; Yadav, S.; Siegler, M. A.; Goldberg, D. P. Selective radical transfer in a series of nonheme iron(III) complexes. Inorg. Chem. 2023, 62, 17830–17842. |
| 22 | Kütt, A.; Rodima, T.; Saame, J.; Raamat, E.; Mäemets, V.; Kaljurand, I.; Koppel, I. A.; Garlyauskayte, R. Y.; Yagupolskii, Y. L.; Yagupolskii, L. M.; Bernhardt, E.; Willner, H.; Leito, I. Equilibrium acidities of superacids. J. Org. Chem. 2011, 76, 391. |
| 23 | Explanation for BDFEMn(II)/XH. (a) In a previous report [11], we used the reduction potential of [MnIII(NO3)3(OPPh3)2] and the pKa of the conjugate acid of dissociated [NO3]– in the Bordwell equation to arrive at a thermodynamic value and referred to it as an effective bond dissociation free energy (BDFEeff). The BDFEeff, described by Mayer [24], uses the reduction potential and pKa of oxidant/base pairs that can combine in a single entity (e.g., through H-bonded adduct) to react in bimolecular C–H bond cleavage. The BDFEeff can be used as an estimate for the upper-limit of C–H bond strength the oxidant/base pair can cleave. However, since the base (X) is coordinated to the Mn(III) center, it is more appropriate to use a {MnIIX–H} BDFE (BDFEMn(II)/XH) like the {MnIIIO–H} BDFE (BDFEO–H) reported in metal-oxo/metal-hydroxo conversions as described by Borovik and others [24]. Therefore, we use the same approach as Borovik except that the pKa of the conjugate acid of the free base is used instead of the pKa of [MnIIX2(HX)]/[MnIIX3]– and refer to it as the BDFEMn(II)/XH. Hence, the BDFEMn(II)/XH is an estimate of the upper limit of C–H bond strength that can be cleaved by a {MnIIIX} reactant. We have performed a systemic analysis of this square scheme approach to estimate C–H cleavage capability in a previous report [17]. (b) Barman, S. K.; Yang, M.-Y.; Parsell, T. H.; Green, M. T.; Borovik, A. S. Semiemperical method for examining asynchronicity in metal-oxo-mediated C–H bond activation. Proc. Natl. Acad. Sci., USA. 2021, 118, e2108648118. |
| 24 | Agarwal, R. G.; Coste, S. C.; Groff, B. D.; Heuer, A. M.; Noh, H.; Parada, G. A.; Wise, C. F.; Nichols, E. M.; Warren, J. J.; Mayer, J. M. Free energies of proton-coupled electron transfer reagents and their applications. Chem. Rev. 2022, 122, 1– 4. |
| 25 | Kadassery, K. J.; Sethi, K.; Fanara, P. M.; Lacy, D. C. CO-Photolysis-induced H-atom transfer from MnIO−H Bonds. Inorg. Chem. 2019, 58, 4679–4685. |
| 26 | Hostmann, T.; Molloy, J. J.; Bussmann, K.; Gilmour, R. Light-enabled enantiodivergence: stereospecific reduction of activated alkenes using a single organocatalyst enantiomer. Org. Lett. 2019, 21, 10164–10168. |
| 27 | Eppley, H. J.; Christou, G., Synthesis of dodecaoxohexadecacarboxylatotetraaquo-dodecamanganese [Mn12O12(O2CR)16(H2O)4] (R = Me, Et, Ph, Cr) complexes, Inorg. Syn. 2002, 33, 61. |
| 28 | CrysAlisPro; Rigaku OD, The Woodlands, TX, 2015. |
| 29 | Sheldrick, G. M., SHELXT – Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. 2015, A71, 3. |
| 30 | Sheldrick, G.M. A Short History of SHELX. Acta Cryst. 2008, A64, 112. |
| 31 | Müller, P. Practical Suggestions for Better Crystal Structures. Crystallogr. Rev. 2009, 15, 57. |
| 32 | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339-341. |
Scheme 2.
Synthesis of 3a, 3b, and 4.
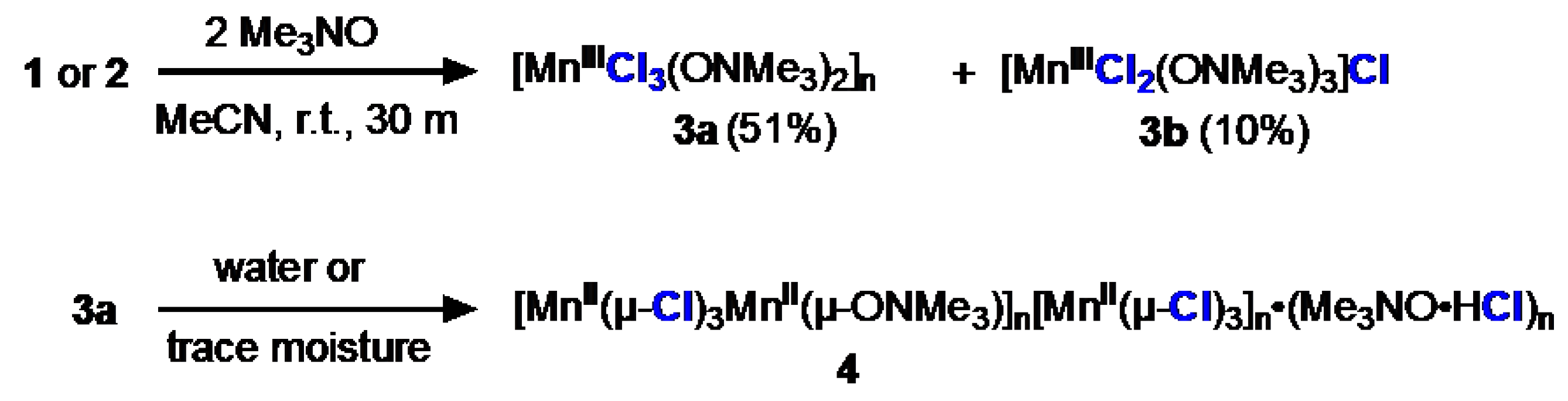
Figure 1.
MicroED structure of [MnII(µ-Cl)3MnII(µ-ONMe3)]n[MnII(µ-Cl)3]n•(Me3NO•HCl)n (4). The grey box contains the unit cell and is viewed down the b-axis (left) and c-axis (right). Selected bond lengths (Å) for the cationic chain: Mn1–Cl1 = 2.56(3); Mn1–O1 = 2.19(3); N1–O1 = 1.397(19); Mn1(µ-Cl)···Mn1(µ-O)···Mn1 = 3.27, 3.11. Selected bond lengths (Å) for the anionic chain: Mn2–Cl2 = 2.54(3); Mn2···Mn2 =3.19. Color scheme: cyan polyhedra = [MnIICl3]–; green polyhedra = [MnII2Cl3(ONMe3)]+; green sphere = Cl; magenta spheres = Mn; blue spheres = N; red spheres = O; grey spheres = C.
Figure 1.
MicroED structure of [MnII(µ-Cl)3MnII(µ-ONMe3)]n[MnII(µ-Cl)3]n•(Me3NO•HCl)n (4). The grey box contains the unit cell and is viewed down the b-axis (left) and c-axis (right). Selected bond lengths (Å) for the cationic chain: Mn1–Cl1 = 2.56(3); Mn1–O1 = 2.19(3); N1–O1 = 1.397(19); Mn1(µ-Cl)···Mn1(µ-O)···Mn1 = 3.27, 3.11. Selected bond lengths (Å) for the anionic chain: Mn2–Cl2 = 2.54(3); Mn2···Mn2 =3.19. Color scheme: cyan polyhedra = [MnIICl3]–; green polyhedra = [MnII2Cl3(ONMe3)]+; green sphere = Cl; magenta spheres = Mn; blue spheres = N; red spheres = O; grey spheres = C.
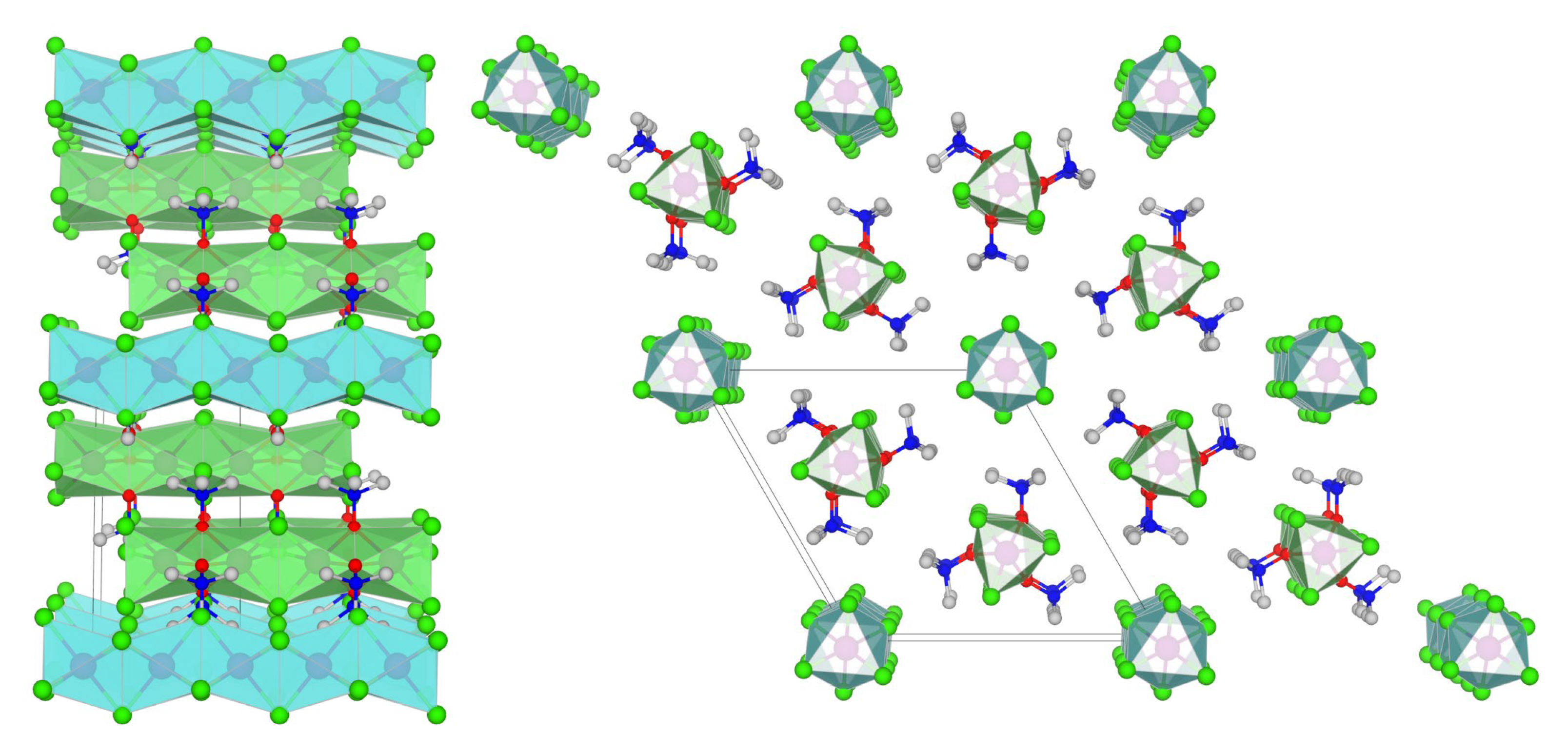
Figure 2.
(Left) FTIR spectra of 3a (red) and 4 (black). (Right) Molecular structure of 3b with the outer sphere Cl counter anion shown (hydrogen atoms and MeCN are omitted for clarity). Selected bond lengths (Å) and angles (deg.) for 3a : Mn1−Cl1 = 2.3225(6); Mn1−O1 = 1.901(2); Mn1−O2 = 1.896(2); Mn1−O3 = 1.991(3); Cl1−Mn1−Cl1 = 145.87(4); O1−Mn1−O2 = 177.33(11).
Figure 2.
(Left) FTIR spectra of 3a (red) and 4 (black). (Right) Molecular structure of 3b with the outer sphere Cl counter anion shown (hydrogen atoms and MeCN are omitted for clarity). Selected bond lengths (Å) and angles (deg.) for 3a : Mn1−Cl1 = 2.3225(6); Mn1−O1 = 1.901(2); Mn1−O2 = 1.896(2); Mn1−O3 = 1.991(3); Cl1−Mn1−Cl1 = 145.87(4); O1−Mn1−O2 = 177.33(11).
Scheme 3.
Synthesis of 5, 6, and 7. Small changes in reaction conditions give different products.
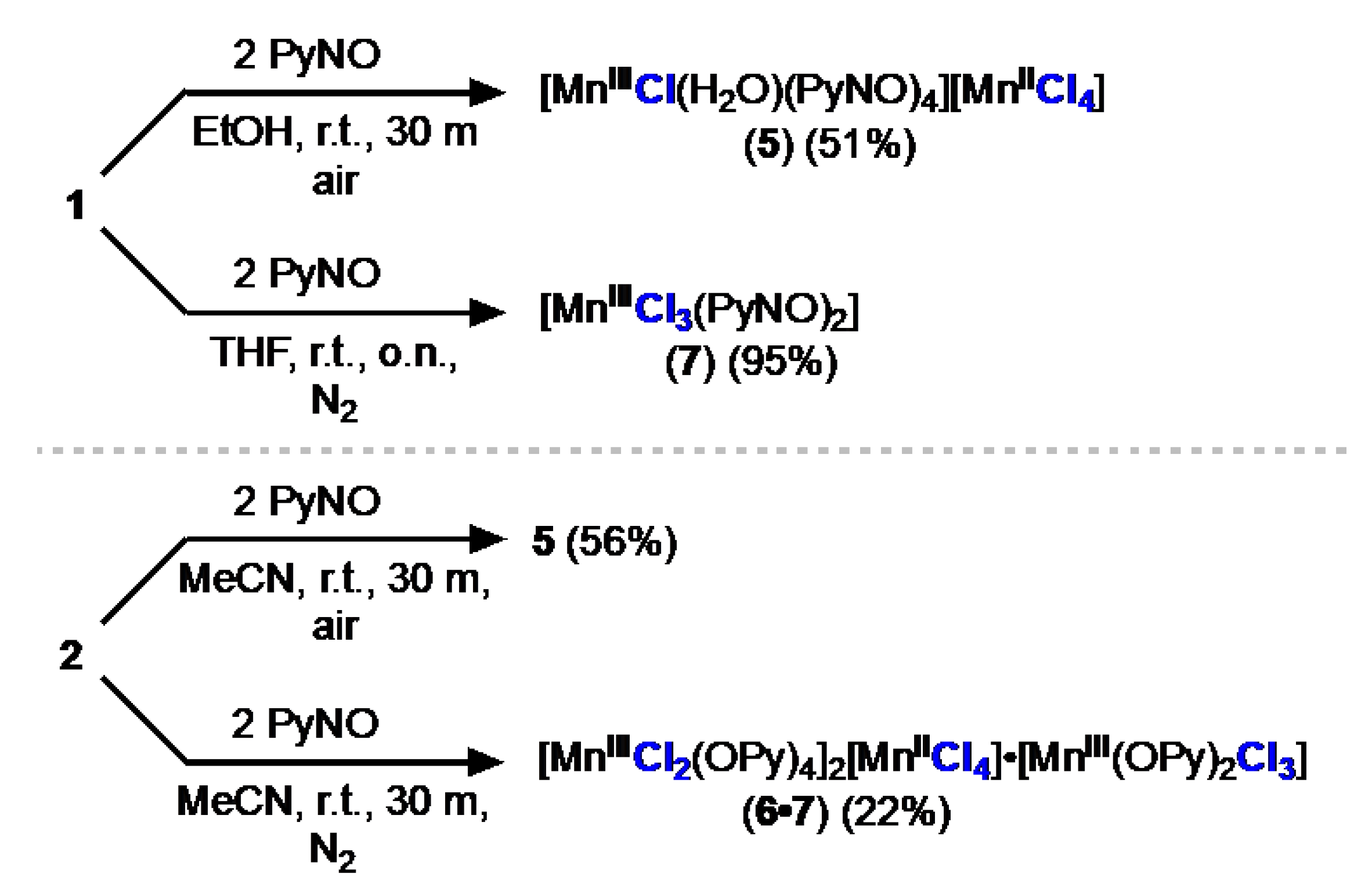
Figure 3.
Molecular structures of Mn(III) centers in (a) 5, (b) 6•7, and (c) 7 (Full crystal structures are presented in the SI). Selected bond lengths (Å) and angles (deg.) for 5: Mn1−Cl1 = 2.5535(4); Mn1−O1 = 1.9389(12); Mn1−O2 = 1.9293(11); Mn1−O3 = 1.9301(12); Mn1−O4 = 1.9255(11); Mn1−O5 = 2.2340(13); O1−Mn1−Cl1 = 93.54(4); O1−Mn1−O4 = 92.03(5); O4−Mn1−Cl1 = 89.60(4). Selected bond lengths (Å) and angles (deg.) for 6•7: Mn1−Cl1 = 2.2804(6); Mn1−Cl2 = 2.3687(6); Mn1−Cl3 = 2.2790(6); Mn1−O1 = 1.9218(15); Mn1−O2 = 1.9194(16); Mn2−Cl4 = 2.5172(6); Mn2−Cl5 = 2.5390(6); Mn2−O3 = 1.9397(15); Mn2−O4 = 1.9309(15); Mn2−O5 = 1.9493(15); Mn2−O6 = 1.9465(15); Cl1−Mn1−Cl2 = 116.23(2); Cl2−Mn1−Cl3 = 105.08(2); Cl1−Mn1−Cl3 = 138.69(3); O1−Mn1−Cl1 = 90.58(5); O2−Mn1−Cl1 = 84.49(5); O3−Mn2−Cl4 = 91.04(5); O3−Mn2−O4 = 89.02(6); O4−Mn2−Cl4 = 91.28(5). Selected bond lengths (Å) and angles (deg.) for 7: Mn1−Cl1 = 2.3172(8); Mn1−Cl2 = 2.3091(8); Mn1−Cl3 = 2.2961(8), Mn1−O1 = 1.912(2); Mn1−O2 = 1.916(2); Cl1−Mn1−Cl2 = 109.02(3); O1−Mn1−O2 = 168.97(9).
Figure 3.
Molecular structures of Mn(III) centers in (a) 5, (b) 6•7, and (c) 7 (Full crystal structures are presented in the SI). Selected bond lengths (Å) and angles (deg.) for 5: Mn1−Cl1 = 2.5535(4); Mn1−O1 = 1.9389(12); Mn1−O2 = 1.9293(11); Mn1−O3 = 1.9301(12); Mn1−O4 = 1.9255(11); Mn1−O5 = 2.2340(13); O1−Mn1−Cl1 = 93.54(4); O1−Mn1−O4 = 92.03(5); O4−Mn1−Cl1 = 89.60(4). Selected bond lengths (Å) and angles (deg.) for 6•7: Mn1−Cl1 = 2.2804(6); Mn1−Cl2 = 2.3687(6); Mn1−Cl3 = 2.2790(6); Mn1−O1 = 1.9218(15); Mn1−O2 = 1.9194(16); Mn2−Cl4 = 2.5172(6); Mn2−Cl5 = 2.5390(6); Mn2−O3 = 1.9397(15); Mn2−O4 = 1.9309(15); Mn2−O5 = 1.9493(15); Mn2−O6 = 1.9465(15); Cl1−Mn1−Cl2 = 116.23(2); Cl2−Mn1−Cl3 = 105.08(2); Cl1−Mn1−Cl3 = 138.69(3); O1−Mn1−Cl1 = 90.58(5); O2−Mn1−Cl1 = 84.49(5); O3−Mn2−Cl4 = 91.04(5); O3−Mn2−O4 = 89.02(6); O4−Mn2−Cl4 = 91.28(5). Selected bond lengths (Å) and angles (deg.) for 7: Mn1−Cl1 = 2.3172(8); Mn1−Cl2 = 2.3091(8); Mn1−Cl3 = 2.2961(8), Mn1−O1 = 1.912(2); Mn1−O2 = 1.916(2); Cl1−Mn1−Cl2 = 109.02(3); O1−Mn1−O2 = 168.97(9).
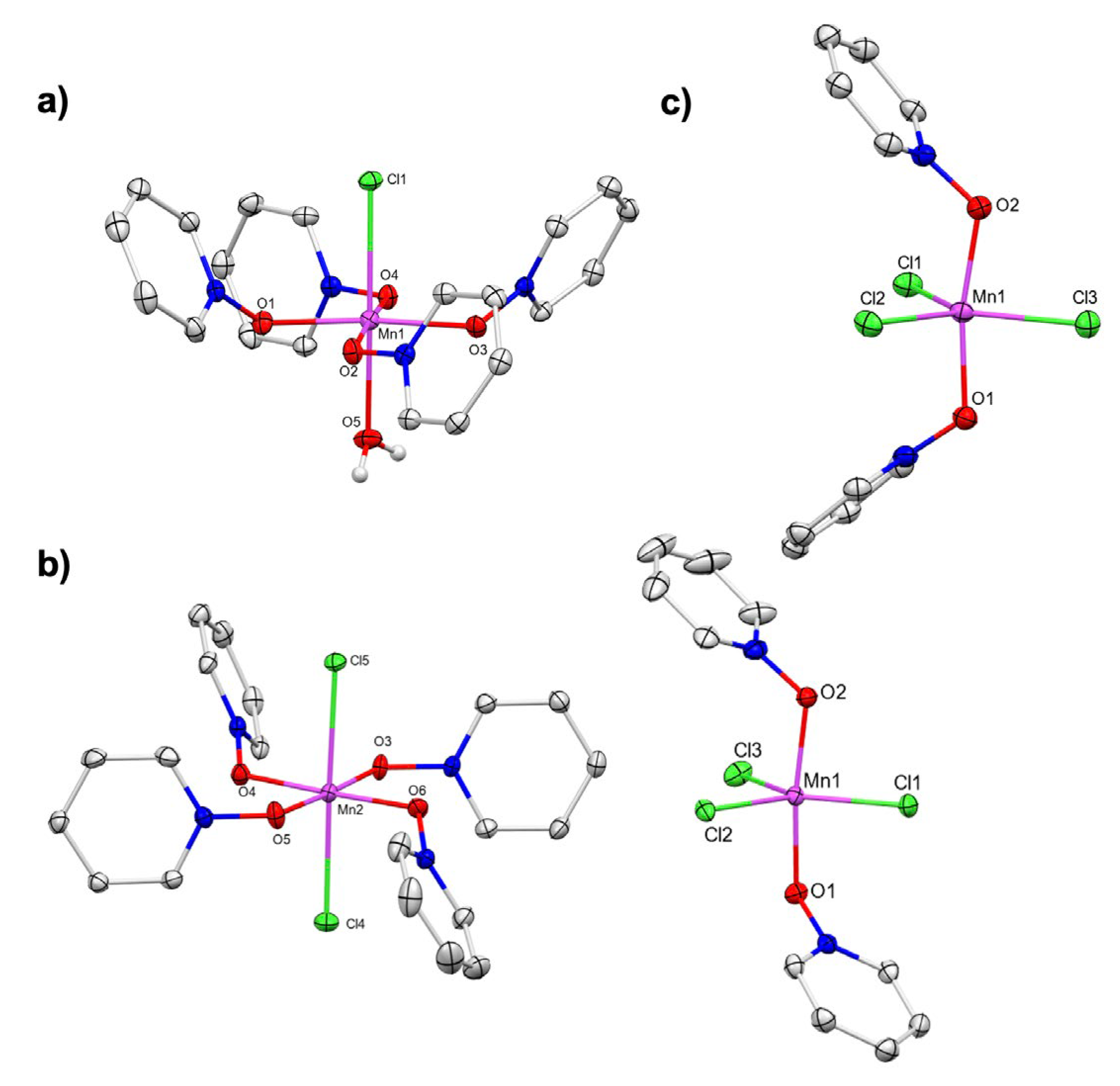
Scheme 4.
Conditions for C–H chlorination reactivity of HMB with Mn(III) chloride compounds.
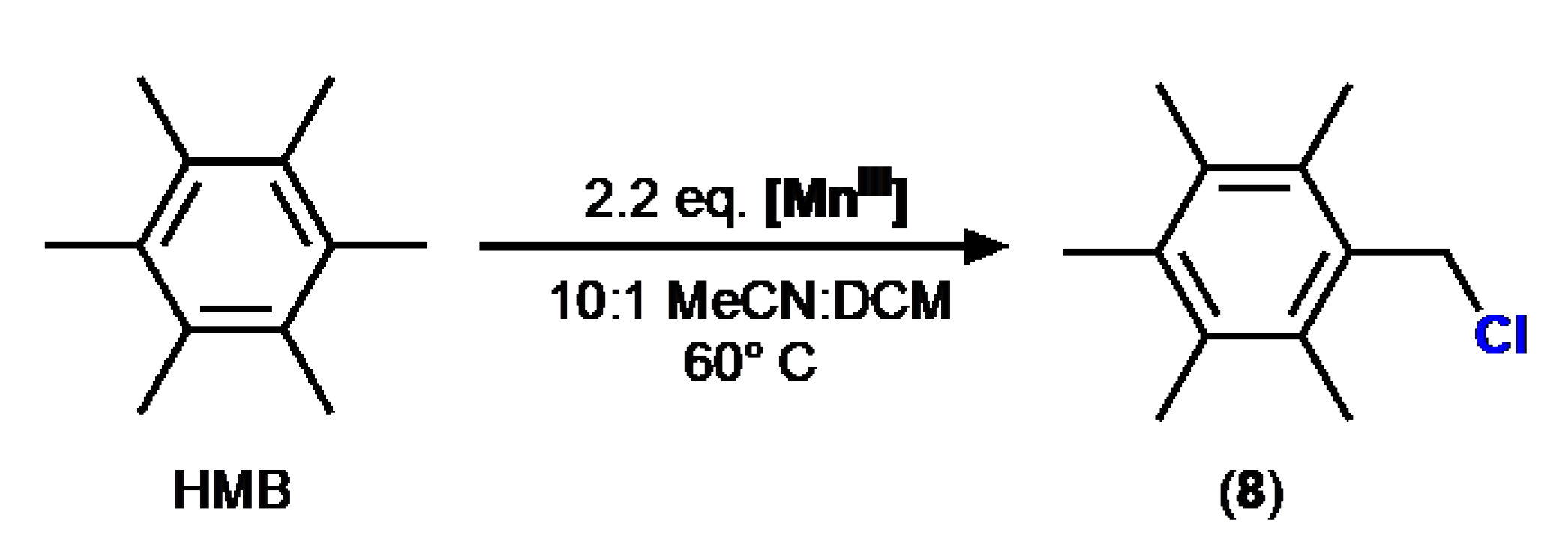
Scheme 5.
Reaction of 1 with TEMPO.
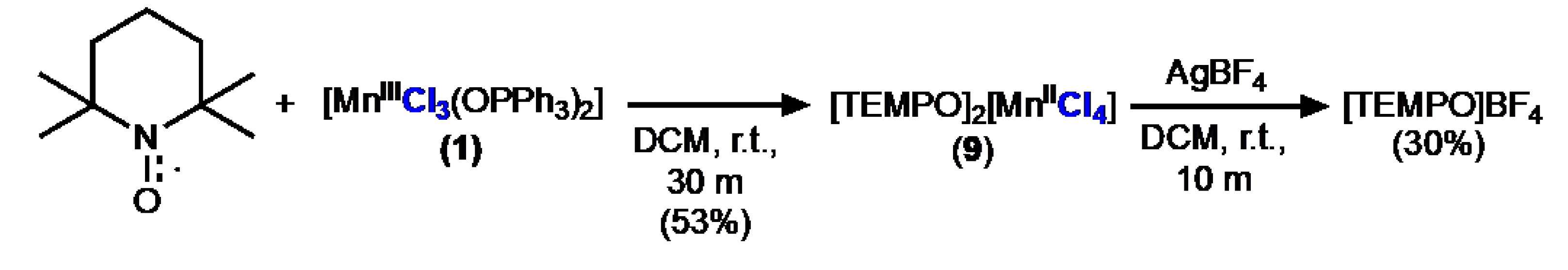
Figure 4.
Molecular structure (ellipsoids 50%) of 9 determined with XRD (H atoms and one part of disorder omitted, only one of the three identical subunits in the unit cell shown for clarity) Selected bond lengths (Å) and angles (deg.) for 9: Mn1−Cl1 = 2.3737(5); Mn1−Cl2A = 2.3549(18); Mn1−Cl3A = 2.3728(10); Mn1−Cl4A = 2.4025(15); N1−O1 = 1.1922(17); N6−O6 = 1.191(2); Cl1−Mn1−Cl2A = 108.62(6); Cl3A−Mn1−Cl4A = 103.78(6).
Figure 4.
Molecular structure (ellipsoids 50%) of 9 determined with XRD (H atoms and one part of disorder omitted, only one of the three identical subunits in the unit cell shown for clarity) Selected bond lengths (Å) and angles (deg.) for 9: Mn1−Cl1 = 2.3737(5); Mn1−Cl2A = 2.3549(18); Mn1−Cl3A = 2.3728(10); Mn1−Cl4A = 2.4025(15); N1−O1 = 1.1922(17); N6−O6 = 1.191(2); Cl1−Mn1−Cl2A = 108.62(6); Cl3A−Mn1−Cl4A = 103.78(6).
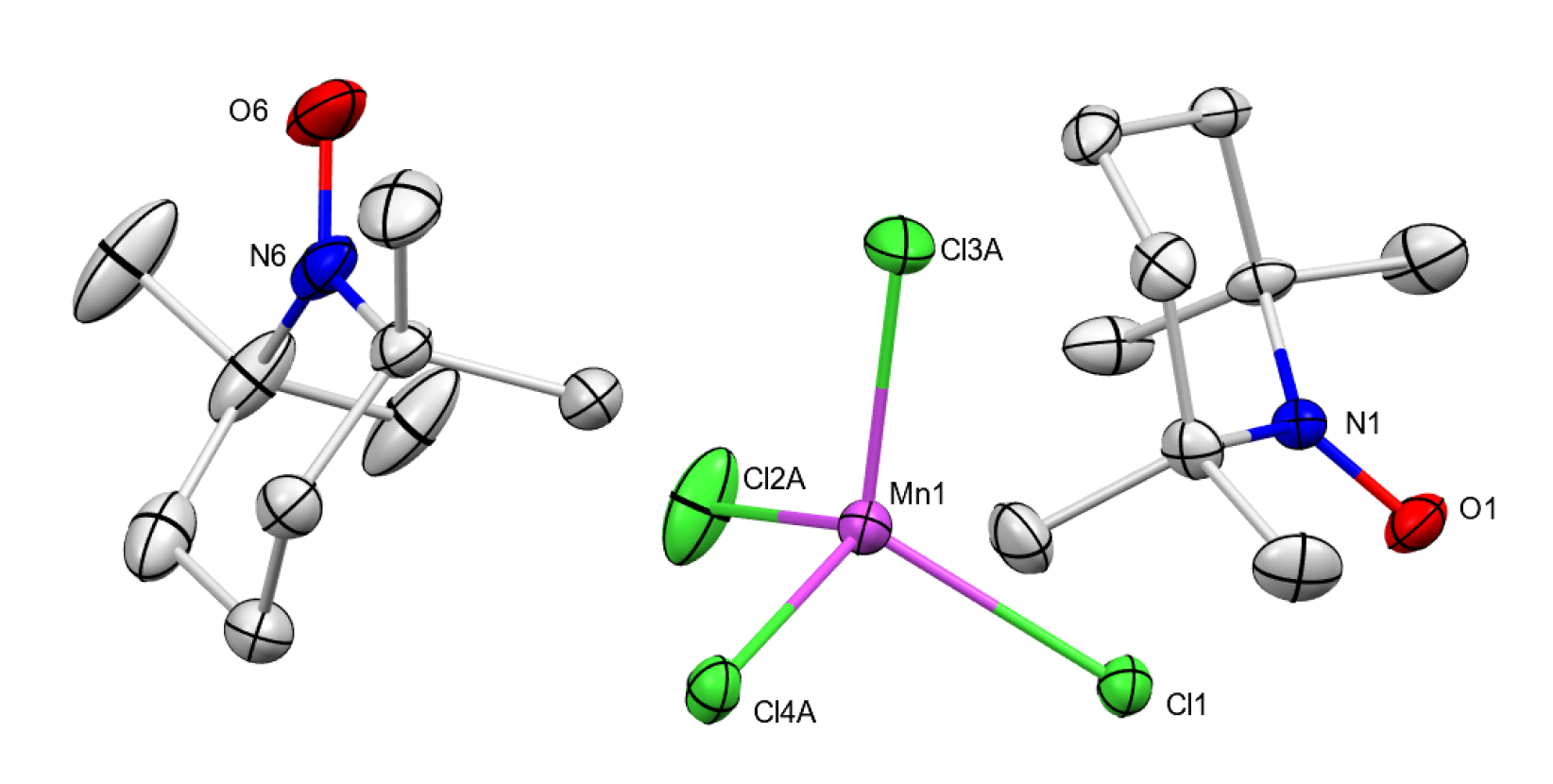

Table 1.
C–H chlorination reactivity of HMB with Mn(III) chloride compounds.
| Complex | Time | % Yielda |
|---|---|---|
| [MnIIICl3(Me3NO)2]n (3a) | 5.0 h | 45 |
| [MnIIICl(H2O)(PyNO)4][MnIICl4] (5) | 6.5 h | 86 |
| [MnIIICl3(PyNO)2] (7) | 4.0 h | 88 |
| [MnIIICl3(MeCN)x] (2) | 1.0 h at r.t.(b) | 78 |
See Scheme 4 for conditions. (a) Yields determined by 1H-NMR, average yields from duplicate runs reported; (b) reaction performed in MeCN without DCM added.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



