
Submitted:
11 September 2024
Posted:
13 September 2024
You are already at the latest version
Abstract
Heusler compounds and alloys represent a rapidly expanding family of materials that exhibit novel properties of significant interest for advanced technological applications. Electronic band structure calculations play a pivotal role in advancing research in this area. In a recent publication [Özdoğan, K.; Galanakis, I. . Crystals 2024, 14, 693], we explored the properties of a new class of Heusler compounds based on Li, referred to as "p0-d Semi-Heusler Compounds." In this study, we take the research a step further by focusing on "d0-d Semi-Heusler Compounds," with the chemical formula KZ(Ga, Ge, As, or Se), where Z represents a transition metal. Our investigation centers on the structural, electronic, and magnetic properties of these compounds, particularly in relation to the three possible C1b structures. Most of these compounds are found to be magnetic and, notably, several among them exhibit half-metallicity making them appealing for applications in spintronics. Our findings provide a foundation for future experimental research on these materials.
Keywords:
Heusler compounds
; Ab-initio calculations
; First-principles
; Electronic structure
; magnetic materials
; Slater-Pauling rule
1. Introduction
In the early 20th century, German metallurgist Heusler made a significant breakthrough while researching ways to improve the electrical conductivity of steel [1,2]. He discovered a novel compound, Cu2MnAl. As scientific instruments advanced over the decades, it was revealed that Cu2MnAl has a face- centered cubic (f.c.c.) lattice structure, similar to well-known semiconductors like Si and GaAs. This same structure was later identified in various intermetallic compounds with unique properties, which became known as "Heusler compounds" or "Heusler alloys" [3,4,5]. Many Heusler compounds are ferromagnetic with high Curie temperatures, and they can be categorized into four main families based on their atomic composition and valence: (a) Semi-Heusler (or half- Heusler) compounds, like NiMnSb, crystallizing in the "" structure, (b) Full-Heusler compounds, such as Co2MnSi, crystallizing in the "" structure, (c) Inverse Heuslers resemble full- Heuslers, like Mn2CoSi with their structure designated as "" or ".", and (d) Ordered equiatomic quaternary Heusler compounds, such as (CoFe)TiSi crystallizing in the "LiMgPdSn" structure [4,6]. Recently there is growing interest in Heusler compounds containing exclusively transition metal atoms, known as all-d-metal Heusler compounds [7,8,9,10].
One of the key properties of Heusler compound is the so called half-metallicity [11]. Half-metallic ferro- or ferri-magnetic materials exhibit metallic behavior for majority spin electrons while acting as semiconductors for minority spin electrons [12]. This results in high spin polarization at the Fermi level, making them highly attractive for spintronics and magnetoelectronics, with the potential to enable novel electronic device functionalities. Heusler compounds have the advantage of combining their half-metallic character with high values of their Curie temperatures and thus have attracted strong interest due to their potential applications [13,14,15,16].
First-principles (or ab initio) calculations have emerged as a powerful tool for understanding material properties and predicting new compounds with tailored characteristics; e.g. spin-gapless semiconducting and spin-filtering behaviors [17].. In recent years, several large databases based on first- principles calculations have been created, covering hundreds of magnetic Heusler compounds [18,19,20,21,22,23,24]. These databases complement more focused studies that investigate the fundamental properties of specific Heusler compounds. In magnetic semi-Heusler compounds having the chemical formula , X is typically a transition metal or rare-earth element. However, in semi-Heusler compounds, X can sometimes be replaced by an alkali or alkaline-earth metal, leading to the formation of "-d" or "-d" Heusler compounds. The term refers to elements like Li, Be, Na, and Mg, while refers to elements such as K, Rb, Cs, Ca, Sr, and Ba, reflecting the nature of the first unoccupied states in the free atom.
Damewood et al. [25] and Dehghan and Davatolhagh [26] conducted ab initio studies on compounds like LiMnPt, SrVSb, and KMnP. In Reference [27], Dehghan and Davatolhagh developed a database of 420 -d Heusler compounds, where X is K, Rb, or Cs, Y is a transition metal, and Z is a group-IV, -V, or -VI element. Of these, 98 were identified as half-metals, following the Slater-Pauling rule: (or when Y is Cu or Zn) [11]. In 2022, the same group expanded their database to include -d semi-Heusler compounds, where X is Li, Be, Na, or Mg, and Z is a group-V or group-VI element [28]. While these databases offer valuable insights into a wide range of compounds, they do not delve into the detailed properties of individual compounds. Concequently, we have presented in Reference [29] a detailed study of the structural, electronic and magnetic properties of the LiYGa and LiYGe compounds, where Y is a 3d transition metal, and their interconnection. Our study revealed that these compounds do not crystallize in the usual variant of the structure and there are a few compounds which are half-metals for all three possible variants of this structure.
In the present study we expand our work on the Li-compounds, to the case of the Heusler compounds. As representative families we have chosen the KZGa, KZGe, KZAs and KZSe compounds where Z varies between Sc and Zn. Such a choice enables us to investigate the effect not only of the Z element but also of the elements on the properties of these compounds. For our first-principles electronic band structure calculations we employed the full-potential nonorthogonal local- orbital minimum-basis band structure approach (FPLO) (version FPLO14.00-47) [30,31] in conjunction with the Perdew-Burke-Ernzerhof (PBE) parameterization to the generalized gradient approximation (GGA) [32]. The computational details of our calculations are identical to the ones in Reference [29].
2. Results and Discussion
2.1. Structural Properties
Semi-Heusler compounds, like those studied here, crystallize in the lattice structure. As shown in Figure 1 of Reference [29], this structure consists of four sites, with one site remaining vacant. Depending on the specific arrangement of the atoms, the structure can exist in three distinct variants, commonly referred to as the , , and phases. The key difference between these phases lies in the chemical nature of the neighboring atoms, which significantly affects the orbital interactions between nearest neighbors. As a result, the physical properties of the same compound can vary greatly across these phases, despite the preservation of the overall tetrahedral symmetry.
In Table 1, we present the structural properties for all the compounds analyzed in this study. To determine the equilibrium lattice parameter for each compound and for all three phases, we conducted total energy calculations across a wide range of lattice parameters, with the equilibrium value being the one that minimizes the total energy. The calculated equilibrium lattice constants range from approximately 6.5 Å to 7.5 Å, which is consistent with other Heusler compounds and semiconductors, an important factor for applications involving multilayer heterostructures. In most cases, the lattice constants follow the general trend of the atomic radii. Specifically, the atomic radii slightly decrease as we move from Ga→Ge→As→Se in the periodic table. Similarly, among the 3d transition metals, the atomic radii decrease progressively from Sc to V. This pattern also appeared in the case of the Li-based -d compounds discussed in Reference [29]. However, a notable difference arises when comparing the Li-based and K-based compounds. For the Li compounds in Reference [29], the phase generally had the smallest equilibrium lattice constants, while no consistent trend emerged between the and phases—though the phase typically had the larger constants. In contrast, for the K-based compounds, the phase consistently exhibits the largest lattice constant, except in the cases of KScGa and KScGe. The origin of this differing behavior is difficult to pinpoint, as both the chemical composition and the magnetic properties of the compounds significantly affect the lattice constants (magnetic and non-magnetic phases of the same compound often exhibit different equilibrium lattice constants)
In Table 1, we also present the total energy differences between the three phases, calculated at their respective equilibrium lattice constants. These energy differences are reported per formula unit in units of eV. A negative value indicates that the phase corresponding to the first total energy is more stable than the other phase in the comparison. In the Li-based compounds studied in Reference [29], the phase was generally the most stable for most compounds, while the phase became the ground state for those with heavier transition metals. The energy differences between the three phases were relatively small, with absolute values always below 1 eV. However, in the K-based compounds examined here, the energy differences between the phase and the / phases are significantly larger, exceeding 2 eV in many cases, as shown in Table 1. Consequently, the phase—where each K atom is at the center of a cube surrounded by four transition metal atoms and four atoms as nearest neighbors—emerges as the least stable phase across all forty compounds. In contrast, the energy difference between the and phases remains small, consistently less than 1 eV. Generally, the phase tends to be the ground state for lighter transition metals, while the phase becomes the ground state for heavier transition metals, with both phases being nearly degenerate for intermediate cases
2.2. Electronic Properties
For all forty compounds in our study and across all three phases, we conducted electronic band structure calculations at the equilibrium lattice constants and subsequently extracted the total density of states (DOS) per formula unit (f.u.). The results for the KZGa and KZGe compounds are shown in Figure 1, while those for the KZAs and KZSe compounds are presented in Figure 2. To ensure clarity, we have chosen to display the DOS in the energy range of -5 eV to 2 eV for all compounds, with the vertical DOS axis ranging from -4.5 to 4.5 states per eV per spin per f.u. This standardization facilitates easier comparison between different compounds. Additionally, there is a small DOS contribution in both spin directions below the energy range shown in Figure 1 and Figure 2, originating from the s states of the K atoms.
Figure 1.
Total density of states (DOS) for the KZGa (left pannel) and KZGe (right panel) compounds for all three phases. The zero energy has been assigned to the Fermi level. Positive(Negative) DOS values correspond to the spin-up(spin-down) electrons.
Figure 1.
Total density of states (DOS) for the KZGa (left pannel) and KZGe (right panel) compounds for all three phases. The zero energy has been assigned to the Fermi level. Positive(Negative) DOS values correspond to the spin-up(spin-down) electrons.
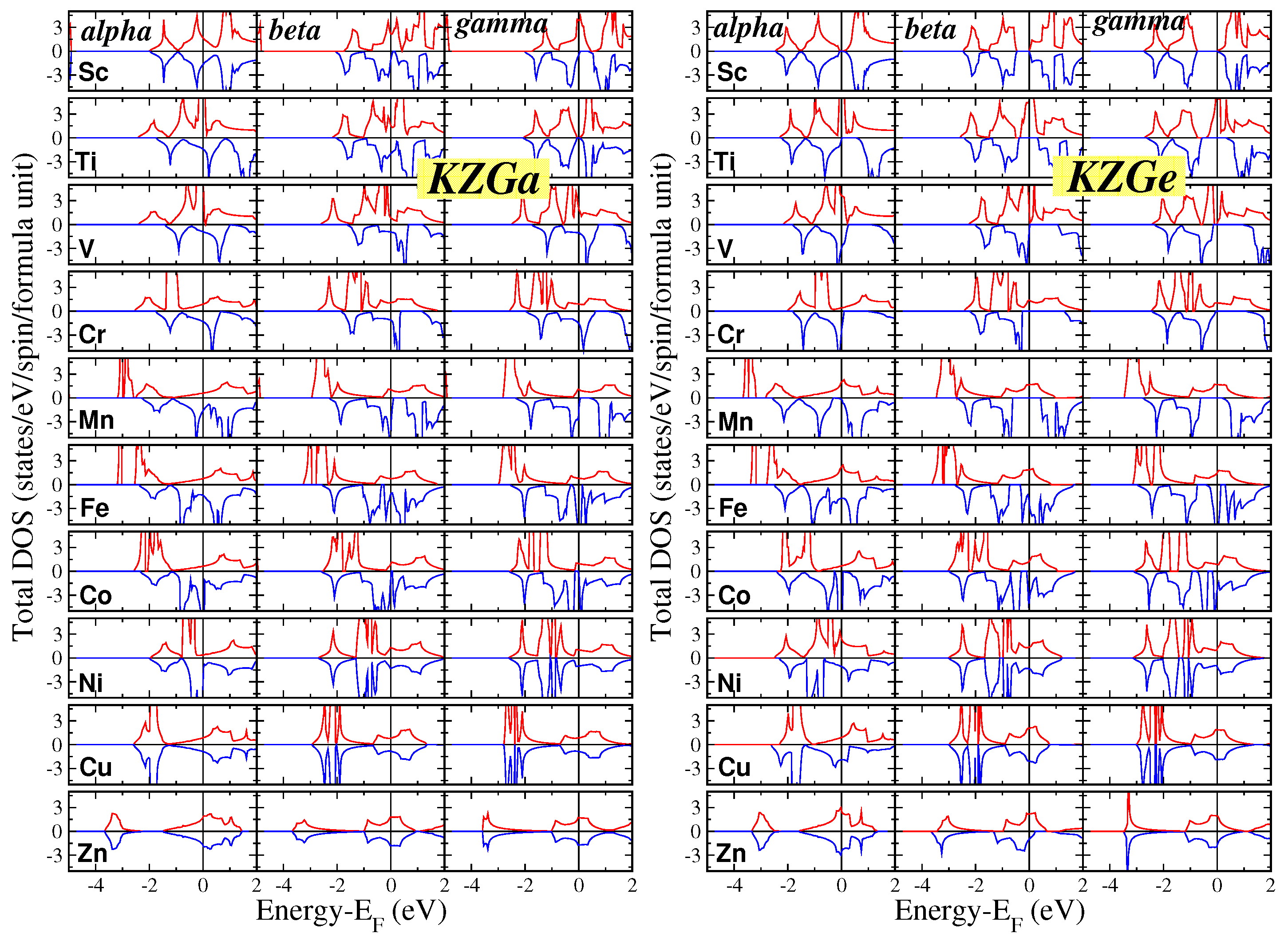
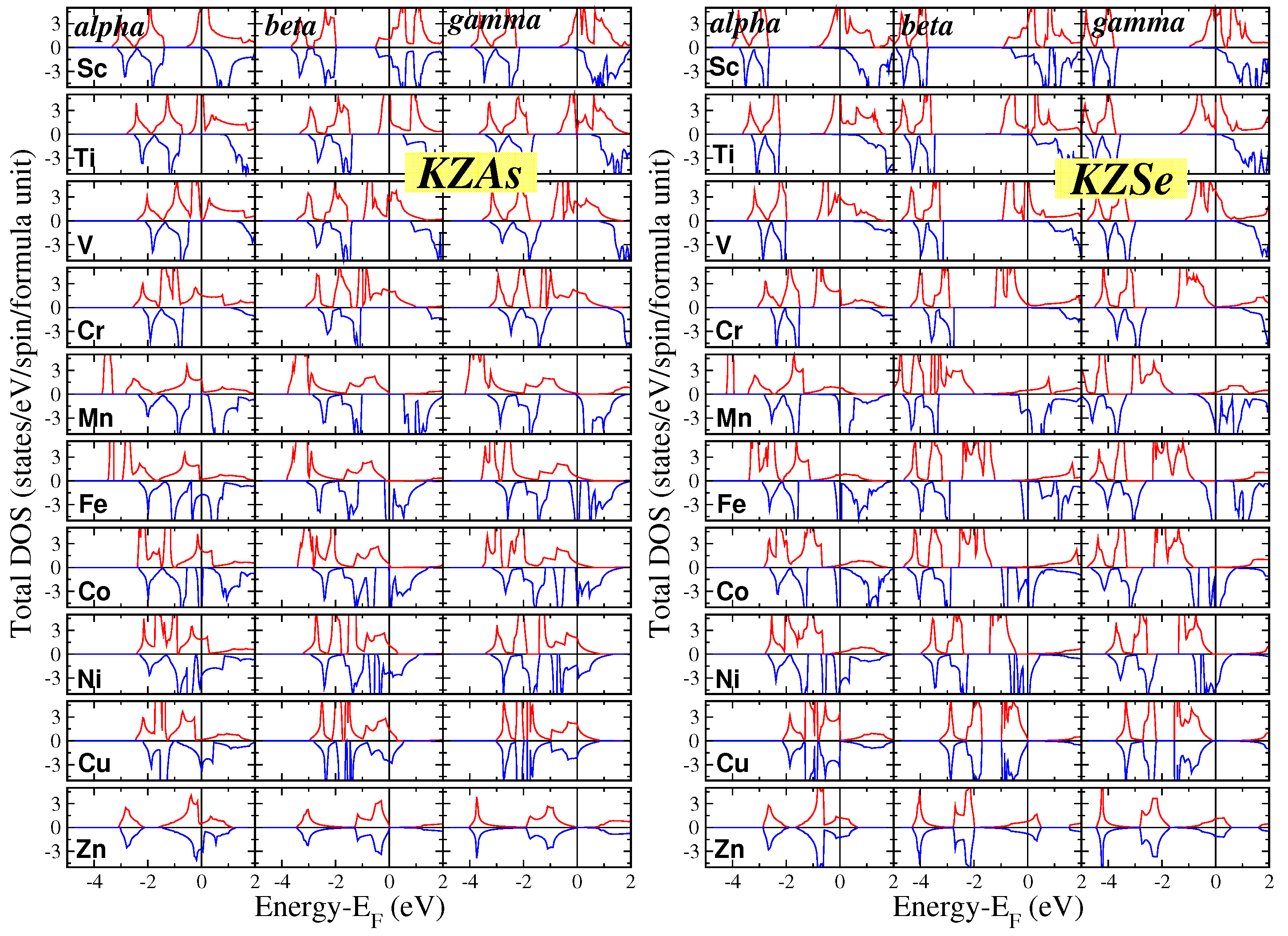
The general characteristics of the DOS are comparable to those observed in the Li compounds discussed in Reference [29]. The DOS presented in the figures is primarily due to the p-d hybridization between the p-states of the Ga/Ge/As/Se atoms and the d-states of the transition metal atoms. In the context of tetrahedral symmetry, the d-states split into two subgroups: (a) the triple-degenerate states, which are symmetrically allowed to hybridize with the p-states of the Ga/Ge/As/Se atoms, forming hybrids that extend across both the transition metal and the Ga/Ge/As/Se atoms, and (b) the double-degenerate d-states, which, due to symmetry restrictions, do not hybridize and remain localized at the transition metal atoms. When Z is one of the heavier elements like Cu or Zn, all valence 3d states are fully occupied. For these cases, the 3d-DOS for both spin directions is concentrated in a narrow, high-intensity peak located deep in energy (for Zn compounds, this peak is outside the energy range displayed in the figures).
In contrast to the Li compounds from Reference [29], the K-based semi-Heusler compounds exhibit predominantly magnetic behavior, as shown in Table 2 and Table 3 and discussed further in the next subsection. Only a few of these compounds are metals or semiconductors, with the transition metal atoms being either Sc (the lightest transition metal considered) or Ni/Cu/Zn (the heaviest transition metals considered), where the 3d states are nearly or completely occupied. Many of the magnetic compounds studied are either perfect or almost half-metals, displaying metallic behavior for spin-up electrons and semiconducting behavior for spin-down electrons across all three phases: KTiGe, KMnGe, KTiAs, KVAs, KCrAs, KMnAs, KTiSe, KVSe, and KCrSe. Some compounds exhibit (almost) half-metallic properties in only one or two of the three phases: KScGa, KCoGa, KVGe, KCrGe, KScAs, and KScSe. Among these, the compounds that are nearly half-metallic have total spin magnetic moments slightly below the ideal integer value, as will be discussed in the next subsection, and the Fermi level marginally crosses the band just below the spin-down energy gap
Examining the key characteristics of the total DOS for all forty compounds in this study, it is evident that the total DOS for the and phases generally exhibits a similar shape, while the DOS for the phase displays a distinct behavior. This difference can be attributed to symmetry considerations. In the and phases, the Z and Ga/Ge/As/Se atoms are nearest neighbors, leading to similar bonding interactions and, consequently, similar DOS profiles for these phases. In contrast, in the phase, these atoms are next- nearest neighbors, resulting in significantly weaker orbital interactions. This weaker interaction helps explain why the phase is the least stable, as indicated in Table 1.
2.3. Magnetic Properties
Most of the compounds in this study are magnetic, and Table 2 and Table 3 present the atomic and total spin magnetic moments calculated at their equilibrium lattice constants. For non-magnetic compounds, we provide their electronic classification as either metal, semiconductor, or gapless semiconductor (where the energy gap is negligible). The K atoms generally contribute negligible spin magnetic moments, with a few exceptions, notably when the metalloid is Se, the heaviest atom. As anticipated, the spin magnetic moments are primarily located at the transition metal atoms. However, due to the strong Ga/Ge/As/Se-p – Z- hybridization, significant induced spin magnetic moments are also present on the Ga/Ge/As/Se atoms. For most compounds, these induced moments are antiparallel to the spin magnetic moments of the transition metal atoms. For KScGa, KCuGe, KZnGe, and KCuAs, we have adjusted the signs of the atomic spin magnetic moments to ensure that the total spin magnetic moment is negative. This adjustment is made to maintain consistency with the Slater-Pauling rule, as discussed below.
Next, we focus on the total spin magnetic moment per formula unit. Table 2 and Table 3 display also the total number of valence electrons per unit cell (equivalent to the per formula unit value), , and the total spin magnetic moment predicted by the Slater-Pauling rule, which will be discussed in the following paragraph. All total spin magnetic moments are reported in units. Figure 3 illustrates the total spin magnetic moment as a function of the transition metal atom Z for all four families of compounds under study and for all three phases. The red dashed line represents the Slater-Pauling rule. Magnetic compounds that align with this rule and thus exhibit integer values of their total spin magnetic moments are identified as half-metallic ferromagnets, consistent with their total DOS discussed earlier. In contrast to the Li-based compounds studied in Reference [29], the K-based compounds examined here display half-metallic behavior in several cases, particularly when the metalloid atom is Ge, As, or Se, and the transition metal atom is Ti, V, Cr, or Mn. When the number of valence electrons, as noted in Table 2 and Table 3, equals or exceeds 14, achieving half- metallicity would necessitate a total spin magnetic moment of 6 or more. Such values are energetically unfavorable, leading to the loss of half- metallicity, and the compounds remain merely magnetic. Compounds that exhibit half- metallic behavior across all three phases are especially promising for spintronic and magnetoelectronic applications.
The explanation for the origin of half-metallicity follows the same reasoning as that for the Li-based semi-Heuslers discussed in Reference [29]. Here, we will briefly summarize it for completeness. When Z ranges from Sc to Ni and in the case of half-metallicity, the spin-down electronic band structure reveals exactly four fully occupied states. A low-energy s valence state from the K atom lies low in energy. The gap arises due to the hybridization of the p- states between the Ga/Ge/As/Se and the Z atoms. This leads to three bonding hybrids below the gap which are fully occupied, and three above the gap which are empty. The total spin magnetic moment corresponds to the number of uncompensated spins. For half-metallicity to be achieved, the total spin magnetic moment in the unit cell should adhere to the Slater-Pauling rule: . When Z is Cu or Zn, all d valence states of the transition metal atoms are filled. In this case, the rule adjusts to , provided half-metallicity is present.
3. Summary and conclusions
Half-metallic semi-Heusler compounds are a prominent area of research due to their potential applications in spintronic devices. Unlike other semi-Heuslers, -d and -d compounds can crystallize in three distinct variations of the lattice structure—, , and —each with a different atomic arrangement within the unit cell. Using first-principles electronic band structure calculations, we investigate the KZ(Ga,Ge,As, or Se) -d Heusler compounds, with Z ranging from Sc to Zn. Our study examines the structural, electronic, and magnetic properties of these compounds across the three phases. We find that the and phases are favored, while the phase is consistently the least stable. This behavior is attributed to the variations in local environment and symmetry in each phase. Most of the compounds under study are magnetic and exhibit half-metallicity, in line with the Slater-Pauling rule regarding their total spin magnetic moment. Notably, compounds such as KTiGe, KMnGe, KTiAs, KVAs, KCrAs, KMnAs, KTiSe, KVSe, and KCrSe demonstrate (almost) half-metallic ferromagnetism across all three phases, highlighting their potential for practical applications.
We anticipate that our results will stimulate further experimental and theoretical research on these compounds, which hold significant potential for advancements in spintronics and magnetoelectronics.
Funding
This research received no external funding
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The author declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| DOS | Density of States |
| f.u. | formula unit |
| FPLO | Full-potential nonorthogonal local-orbital minimum- basis band structure approach |
| GGA | Generalized gradient approximation |
| PBE | Perdew Burke Ernzerhof |
References
- Heusler, F. Über magnetische manganlegierungen. Verh. Dtsch. Phys. Ges. 1903, 12, 219. [Google Scholar]
- Heusler, F.; Take, E. The nature of the Heusler alloys. Phys. Z. 1912, 13, 897. [Google Scholar] [CrossRef]
- Webster, P.J. ; Ziebeck., K.R.A. Alloys and Compounds of d-Elements with Main Group Elements. Part 2. In Landolt-Börnstein, New Series, Group III, vol 19c; Editor Wijn, H.R.J.; Springer: Berlin. 1988; pp. 75–184.
- Ziebeck, K.R.A.; Neumann, K.-U. Magnetic Properties of Metals. In Landolt-Börnstein, New Series, Group III, vol 32/c. Editor Wijn, H.R.J.; Springer: Berlin. 2001; pp. 64–414.
- Kawasaki, J.K.; Chatterjee, S.; Canfield, P.C. Full and half-Heusler compounds. MRS Bulletin 2022, 47, 555. [Google Scholar] [CrossRef]
- Graf, T.; Felser, C.; Parkin, S.S.P. Simple rules for the understanding of Heusler compounds. Progress in Solid State Chemistry 2011, 39, 1. [Google Scholar] [CrossRef]
- Bachagha, T; Sunol, J. -J. All-d-Metal Heusler Alloys: A Review. Metals 2023, 13, 111. [Google Scholar] [CrossRef]
- de Paula, V. G.; Reis, M. S. All-d-Metal Full Heusler Alloys: A Novel Class of Functional Materials. Chem. Mater. 2021, 33, 5483. [Google Scholar] [CrossRef]
- Tas, M; Özdoğan, K. ; Şaşıoğlu, E.; Galanakis,I. High Spin Magnetic Moments in All-3d-Metallic Co-Based Full Heusler Compounds. Materials 2023, 16, 7543. [Google Scholar] [CrossRef]
- Fortunato, N. M.; Li, X.; Schönecker, S.; Xie, R.; aubel, A.; Scheibel, F.; Opahle, I.; Gutfleisch, O.; Zhang, H. High-Throughput Screening of All-d-Metal Heusler Alloys for Magnetocaloric Applications. Chem. Mater. 2024, 36, 6765. [Google Scholar] [CrossRef]
- Galanakis, I. Slater–Pauling Behavior in Half-Metallic Heusler Compounds. Nanomaterials 2023, 13, 2010. [Google Scholar] [CrossRef]
- Katsnelson, M.I.; Irkhin, V. Yu; Chioncel, L.; Lichtenstein, A.I.; de Groot, R.A. Half-metallic ferromagnets: From band structure to many-body effects. Rev. Mod. Phys. 2008, 80, 315. [Google Scholar] [CrossRef]
- Hirohata, A.; Takanashi, K. Perspectives of Heusler compounds. J. Phys. D: Appl. Phys. 2014, 47, 193001. [Google Scholar] [CrossRef]
- Spintronics. From Materials to Devices. Editors Felser, C.; Fecher, G.H. Spintronics. From Materials to Devices. Editors Felser, C.; Fecher, G.H., Springer. 2013.
- Half-metallic Materials and Their Properties. In Series on Materials for Engineering, vol. 2. Editors Fong, C.Y.; Pask, J.E.; Yang, L.H. Imperial College Press. 2013.
- Heusler Alloys. Properties, Growth, Applications. In Springer Series in Materials Science, vol. 222. Editors Felser, C.; Hirohata, A. Springer International Publishing. 2018.
- Galanakis,I. ; Özdoğan, K.; Şaşıoğlu, E. Spin-filter and spin-gapless semiconductors: The case of Heusler compounds. AIP Adv. 2016, 6, 055606. [Google Scholar] [CrossRef]
- Gillessen, M.; Dronskowski, R. A combinatorial study of full Heusler alloys by first-principles computational methods. J. Comput. Chem. 2009, 30, 1290. [Google Scholar] [CrossRef] [PubMed]
- Gillessen, M.; Dronskowski, R. A combinatorial study of inverse Heusler alloys by first-principles computational methods. J. Comput. Chem. 2010, 31, 612. [Google Scholar] [CrossRef]
- Ma, J.; Hegde, V.I.; Munira, K.; Xie, Y.; Keshavarz, S.; Mildebrath, D.T.; Wolverton, C.; Ghosh, A.W.; Butler, W.H. Computational investigation of half-Heusler compounds for spintronics applications. Phys. Rev. B 2017, 95, 024411. [Google Scholar] [CrossRef]
- Sanvito, S.; Oses, C.; Xue, J.; Tiwari, A.; Zic, M.; Archer, T.; Tozman, P.; Venkatesan, M.; Coey, M.; Curtarolo, S. Accelerated discovery of new magnets in the Heusler alloy family. Sci. Adv. 2017, 3, e1602241. [Google Scholar] [CrossRef]
- Faleev, S.V.; Ferrante, Y.; Jeong, J.; Samant, M.G.; Jones, B.; Parkin, S.S.P. Unified explanation of chemical ordering, the Slater-Pauling rule, and half-metallicity in full Heusler compounds. Phys. Rev. B 2017, 95, 045140. [Google Scholar] [CrossRef]
- Faleev, S.V.; Ferrante, Y.; Jeong, J.; Samant, M.G.; Jones, B.; Parkin, S.S.P. Heusler compounds with perpendicular magnetic anisotropy and large tunneling magnetoresistance. Phys. Rev. Mater. 2017, 1, 024402. [Google Scholar] [CrossRef]
- He, J.; Rabe, K. M; Wolverton, C. Computationally accelerated discovery of functional and structural Heusler materials. MRS Bulletin 2022, 47, 559. [Google Scholar] [CrossRef]
- Damewood, L.; Busemeyer, B.; Shaughnessy, M.; Fong, C. Y, ; Yang, L. H.; Felser, C. Stabilizing and increasing the magnetic moment of half-metals: The role of Li in half-Heusler LiMnZ (Z=N,P,Si). Phys. Rev. B 2015, 91, 064409. [Google Scholar] [CrossRef]
- Dehghan, A.; Davatolhagh, S. d0-d half-Heusler alloys: A potential class of advanced spintronic materials. J. All. Comp. 2019, 773, 132. [Google Scholar] [CrossRef]
- Dehghan, A.; Davatolhagh, S. First principles study of d0-d half-Heusler alloys containing group-IV, -V, and -VI sp atoms as prospective half-metals for real spintronic applications. Mat. Chem. Phys. 2021, 273, 125064. [Google Scholar] [CrossRef]
- Javari, A. R.; Davatolhagh, S; Dehghan, A. Half-metallic p0-d half-Heusler alloys with stable structure in ferromagnetic state. J. Phys. Chem. Solids 2022, 166, 110702. [Google Scholar] [CrossRef]
- Özdoğan, K.; Galanakis, I. Interplay between Structural, Electronic, and Magnetic Properties in the p0-d Semi-Heusler Compounds: The Case of Li-Based Compounds. Crystals 2024, 14, 693. [Google Scholar] [CrossRef]
- Koepernik, K.; Eschrig, H. Full-potential nonorthogonal local-orbital minimum-basis band-structure scheme. Phys. Rev. B 1999, 59, 1743. [Google Scholar] [CrossRef]
- Kopernik, K. Full Potential Local Orbital Minimum Basis Bandstructure Scheme User’s Manual (http: //www.fplo.de/download/doc.pdf).
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
Figure 3.
Total spin magnetic per formula unit in units as a function of the Z chemical element ( Sc → Ni) for all four studied families of K-based compounds. The red lines represent the ideal Slater Pauling rules for half-metallicity: =-8.
Figure 3.
Total spin magnetic per formula unit in units as a function of the Z chemical element ( Sc → Ni) for all four studied families of K-based compounds. The red lines represent the ideal Slater Pauling rules for half-metallicity: =-8.
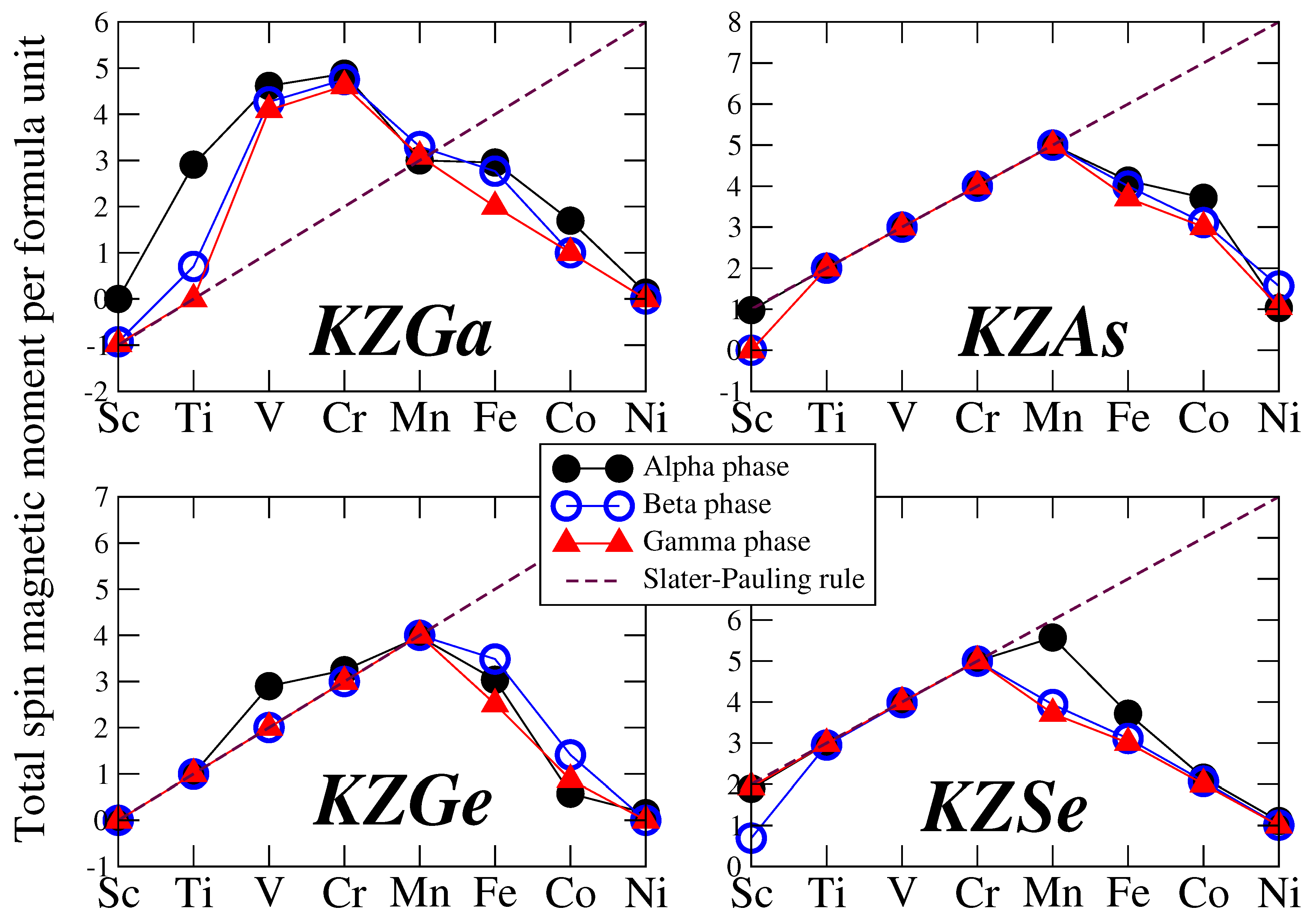

Table 1.
We provide the equilibrium lattice constants for all compounds under study in all three , and phases. The next three columns present the total energy difference between the two phases using for each one the equilibrium lattice constant. In the last two columns the most and least stable phases are depicted.
Table 1.
We provide the equilibrium lattice constants for all compounds under study in all three , and phases. The next three columns present the total energy difference between the two phases using for each one the equilibrium lattice constant. In the last two columns the most and least stable phases are depicted.
| Compound | Lattice constant a in Å | Energy difference in eV | Most stable | Least stable | ||||
| -phase | -phase | -phase | phase | phase | ||||
| KScGa | 7.21 | 7.31 | 7.17 | -1.54 | -0.51 | 1.03 | ||
| KTiGa | 7.44 | 7.04 | 6.85 | -1.73 | -0.17 | 1.55 | ||
| KVGa | 7.47 | 7.14 | 7.07 | -1.47 | -0.20 | 1.27 | ||
| KCrGa | 7.58 | 7.24 | 7.17 | -1.07 | -0.07 | 1.00 | ||
| KMnGa | 7.44 | 7.12 | 6.96 | -1.23 | 0.003 | 1.23 | ||
| KFeGa | 7.39 | 6.95 | 6.80 | -1.23 | 0.01 | 1.25 | ||
| KCoGa | 7.29 | 6.85 | 6.60 | -1.42 | 0.34 | 1.77 | ||
| KNiGa | 7.12 | 6.75 | 6.63 | -1.34 | 0.44 | 1.78 | ||
| KCuGa | 7.40 | 7.02 | 6.95 | -0.86 | 0.29 | 1.15 | ||
| KZnGa | 7.64 | 7.26 | 7.23 | -0.82 | 0.11 | 0.93 | ||
| KScGe | 7.01 | 7.14 | 6.93 | -2.07 | -0.37 | 1.70 | ||
| KTiGe | 7.00 | 6.94 | 6.74 | -2.30 | -0.18 | 2.11 | ||
| KVGe | 7.12 | 6.92 | 6.66 | -2.08 | 0.0001 | 2.08 | ||
| KCrGe | 7.31 | 6.99 | 6.78 | -1.62 | 0.03 | 1.65 | ||
| KMnGe | 7.20 | 7.00 | 6.84 | -1.80 | -0.01 | 1.79 | ||
| KFeGe | 7.17 | 6.87 | 6.65 | -1.76 | 0.15 | 1.91 | ||
| KCoGe | 7.17 | 6.75 | 6.46 | -1.87 | 0.39 | 2.25 | ||
| KNiGe | 6.96 | 6.69 | 6.52 | -1.72 | 0.55 | 2.27 | ||
| KCuGe | 7.24 | 6.91 | 6.77 | -1.33 | 0.38 | 1.70 | ||
| KZnGe | 7.41 | 7.06 | 6.95 | -1.38 | 0.16 | 1.53 | ||
| KScAs | 7.03 | 6.98 | 6.88 | -2.39 | -0.71 | 1.68 | ||
| KTiAs | 6.98 | 6.92 | 6.71 | -2.48 | -0.30 | 2.19 | ||
| KVAs | 6.99 | 6.85 | 6.66 | -2.24 | -0.14 | 2.10 | ||
| KCrAs | 7.10 | 6.93 | 6.74 | -1.92 | -0.07 | 1.85 | ||
| KMnAs | 7.08 | 6.92 | 6.75 | -2.26 | -0.12 | 2.15 | ||
| KFeAs | 7.09 | 6.80 | 6.60 | -2.15 | 0.15 | 2.30 | ||
| KCoAs | 7.03 | 6.76 | 6.51 | -2.35 | 0.20 | 2.55 | ||
| KNiAs | 6.94 | 6.70 | 6.47 | -2.01 | 0.43 | 2.44 | ||
| KCuAs | 7.27 | 6.82 | 6.63 | -1.72 | 0.33 | 2.05 | ||
| KZnAs | 7.29 | 6.88 | 6.74 | -1.97 | 0.01 | 1.98 | ||
| KScSe | 7.29 | 6.99 | 7.01 | -2.00 | -0.90 | 1.10 | ||
| KTiSe | 7.21 | 6.97 | 6.84 | -2.01 | -0.62 | 1.39 | ||
| KVSe | 7.14 | 6.97 | 6.80 | -1.69 | -0.26 | 1.43 | ||
| KCrSe | 7.19 | 7.03 | 6.86 | -1.65 | -0.20 | 1.45 | ||
| KMnSe | 7.32 | 7.03 | 6.76 | -1.41 | -0.22 | 1.19 | ||
| KFeSe | 7.11 | 6.87 | 6.57 | -2.07 | 0.07 | 2.14 | ||
| KCoSe | 7.07 | 6.76 | 6.50 | -1.72 | 0.10 | 1.82 | ||
| KNiSe | 7.02 | 6.75 | 6.51 | -1.77 | 0.25 | 2.03 | ||
| KCuSe | 7.12 | 6.84 | 6.61 | -1.77 | 0.16 | 1.93 | ||
| KZnSe | 7.36 | 7.25 | 7.09 | -1.05 | -0.19 | 0.85 | ||
Table 2.
For the KZGa and KZGe compounds we provide the atom-resolved spin magnetic moments in units as well as the total spin magnetic moment per formula unit which coincides with the per unit cell value. The last two columns are the total number of valence electrons in the unit cell, , as well as the ideal total spin magnetic moment if the Slater-Pauling rules were valid, . For their non magnetic compounds we just present their electronic character.
Table 2.
For the KZGa and KZGe compounds we provide the atom-resolved spin magnetic moments in units as well as the total spin magnetic moment per formula unit which coincides with the per unit cell value. The last two columns are the total number of valence electrons in the unit cell, , as well as the ideal total spin magnetic moment if the Slater-Pauling rules were valid, . For their non magnetic compounds we just present their electronic character.
| Comp. | phase | Comp. | phase | ||||||||||||
| KScGa | Metal | 7 | -1 | KScGe | Gapless semiconductor | 8 | 0 | ||||||||
| 0.031 | -0.436 | -0.523 | -0.928 | 7 | -1 | Semiconductor | 8 | 0 | |||||||
| 0.013 | -0.537 | -0.448 | -0.972 | 7 | -1 | Semiconductor | 8 | 0 | |||||||
| KTiGa | 0.136 | 2.667 | 0.106 | 2.909 | 8 | 0 | KTiGe | 0.022 | 1.805 | -0.827 | 1.000 | 9 | 1 | ||
| 0.010 | 0.789 | -0.096 | 0.703 | 8 | 0 | 0.108 | 1.374 | -0.482 | 1.000 | 9 | 1 | ||||
| Gapless semiconductor | 8 | 0 | 0.032 | 1.339 | -0.371 | 1.000 | 9 | 1 | |||||||
| KVGa | 0.168 | 4.146 | 0.301 | 4.615 | 9 | 1 | KVGe | 0.062 | 3.635 | -0.797 | 2.900 | 10 | 2 | ||
| 0.058 | 3.764 | -0.448 | 4.270 | 9 | 1 | 0.091 | 2.937 | -1.025 | 2.003 | 10 | 2 | ||||
| 0.041 | 3.855 | 0.198 | 4.094 | 9 | 1 | 0.023 | 2.828 | -0.851 | 2.000 | 10 | 2 | ||||
| KCrGa | 0.052 | 5.089 | -0.261 | 4.880 | 10 | 2 | KCrGe | 0.007 | 4.991 | -1.754 | 3.244 | 11 | 3 | ||
| -0.011 | 4.814 | -0.052 | 4.751 | 10 | 2 | 0.110 | 4.438 | -1.548 | 3.000 | 11 | 3 | ||||
| -0.011 | 4.851 | -0.225 | 4.615 | 10 | 2 | 0.034 | 4.280 | -1.314 | 3.000 | 11 | 3 | ||||
| KMnGa | -0.086 | 4.624 | -0.875 | 3.663 | 11 | 3 | KMnGe | 0.010 | 4.870 | -0.912 | 3.968 | 12 | 4 | ||
| -0.078 | 4.492 | -1.113 | 3.301 | 11 | 3 | 0.120 | 4.756 | -0.876 | 4.000 | 12 | 4 | ||||
| -0.119 | 4.301 | -1.092 | 3.090 | 11 | 3 | 0.080 | 4.574 | -0.654 | 4.000 | 12 | 4 | ||||
| KFeGa | -0.029 | 3.383 | -0.399 | 2.955 | 12 | 4 | KFeGe | -0.005 | 3.526 | -0.481 | 3.040 | 13 | 5 | ||
| -0.023 | 3.311 | -0.519 | 2.769 | 12 | 4 | 0.059 | 3.573 | -0.145 | 3.487 | 13 | 5 | ||||
| -0.037 | 3.115 | -0.566 | 2.512 | 12 | 4 | 0.034 | 3.072 | -0.593 | 2.513 | 13 | 5 | ||||
| KCoGa | -0.044 | 2.022 | -0.281 | 1.697 | 13 | 5 | KCoGe | -0.051 | 2.112 | -1.484 | 0.577 | 14 | 6 | ||
| -0.060 | 1.765 | -0.705 | 1.000 | 13 | 5 | 0.043 | 2.012 | -0.643 | 1.412 | 14 | 6 | ||||
| -0.001 | 1.388 | -0.387 | 1.000 | 13 | 5 | 0.023 | 1.189 | -0.337 | 0.875 | 14 | 6 | ||||
| KNiGa | -0.020 | 0.315 | -0.159 | 0.136 | 14 | 6 | KNiGe | 0.024 | -0.460 | 0.593 | 0.157 | 15 | 7 | ||
| Metal | 14 | 6 | Metal | 15 | 7 | ||||||||||
| Metal | 14 | 6 | Metal | 15 | 7 | ||||||||||
| KCuGa | Metal | 15 | -3 | KCuGe | -0.029 | 0.036 | -1.112 | -1.105 | 16 | -2 | |||||
| Metal | 15 | -3 | Metal | 16 | -2 | ||||||||||
| Metal | 15 | -3 | Metal | 16 | -2 | ||||||||||
| KZnGa | Metal | 16 | -2 | KZnGe | Metal | 17 | -1 | ||||||||
| Metal | 16 | -2 | 0.016 | -0.062 | -0.695 | -0.741 | 17 | -1 | |||||||
| Metal | 16 | -2 | -0.002 | -0.015 | -0.096 | -0.103 | 17 | -1 | |||||||
Table 3.
Similar to Table 2 for the compounds where Z is As or Se.
Table 3.
Similar to Table 2 for the compounds where Z is As or Se.
| Comp. | phase | Comp. | phase | ||||||||||||
| KScAs | 0.050 | 1.146 | -0.199 | 0.997 | 9 | 1 | KScSe | 0.090 | 1.920 | -0.118 | 1.892 | 10 | 2 | ||
| Metal | 9 | 1 | 0.072 | 0.674 | -0.054 | 0.692 | 10 | 2 | |||||||
| 0.072 | 1.030 | -0.133 | 0.969 | 9 | 1 | 0.259 | 1.793 | -0.118 | 1.934 | 10 | 2 | ||||
| KTiAs | 0.044 | 2.371 | -0.415 | 2.000 | 10 | 2 | KTiSe | 0.096 | 3.062 | -0.174 | 2.984 | 11 | 3 | ||
| 0.140 | 2.193 | -0.333 | 2.000 | 10 | 2 | 0.294 | 2.889 | -0.230 | 2.953 | 11 | 3 | ||||
| 0.042 | 2.248 | -0.290 | 2.000 | 10 | 2 | 0.263 | 2.915 | -0.193 | 2.985 | 11 | 3 | ||||
| KVAs | 0.039 | 3.659 | -0.698 | 3.000 | 11 | 3 | KVSe | 0.090 | 4.167 | -0.698 | 4.000 | 12 | 4 | ||
| 0.150 | 3.431 | -0.581 | 3.000 | 11 | 3 | 0.252 | 4.014 | -0.581 | 3.986 | 12 | 4 | ||||
| 0.092 | 3.461 | -0.553 | 3.000 | 11 | 3 | 0.236 | 4.018 | -0.553 | 4.000 | 12 | 4 | ||||
| KCrAs | 0.028 | 4.988 | -1.016 | 4.000 | 12 | 4 | KCrse | 0.042 | 5.200 | -1.016 | 5.000 | 13 | 5 | ||
| 0.104 | 4.652 | -0.756 | 4.000 | 12 | 4 | 0.132 | 5.097 | -0.756 | 4.997 | 13 | 5 | ||||
| 0.071 | 4.582 | -0.653 | 4.000 | 12 | 4 | 0.137 | 5.060 | -0.653 | 5.000 | 13 | 5 | ||||
| KMnAs | 0.057 | 4.963 | -0.020 | 5.000 | 13 | 5 | KMnSe | 0.039 | 5.306 | -0.020 | 5.572 | 14 | 6 | ||
| 0.094 | 4.911 | -0.005 | 5.000 | 13 | 5 | -0.034 | 4.290 | -0.005 | 3.940 | 14 | 6 | ||||
| 0.123 | 4.801 | 0.064 | 4.988 | 13 | 5 | 0.089 | 4.054 | 0.035 | 3.717 | 14 | 6 | ||||
| KFeAs | 0.018 | 3.641 | 0.477 | 4.136 | 14 | 6 | KFeSe | 0.004 | 3.678 | -0.048 | 3.717 | 15 | 7 | ||
| 0.032 | 3.699 | 0.269 | 4.000 | 14 | 6 | 0.006 | 3.154 | -0.011 | 3.112 | 15 | 7 | ||||
| 0.070 | 3.486 | 0.151 | 3.707 | 14 | 6 | 0.033 | 2.985 | 0.151 | 3.007 | 15 | 7 | ||||
| KCoAs | -0.007 | 2.189 | -0.712 | 3.717 | 15 | 7 | KCoSe | -0.004 | 2.167 | -0.004 | 2.158 | 16 | 8 | ||
| 0.055 | 2.519 | 0.412 | 3.112 | 15 | 7 | -0.049 | 2.046 | 0.075 | 2.072 | 16 | 8 | ||||
| 0.078 | 2.252 | 0.371 | 3.007 | 15 | 7 | 0.005 | 1.893 | 0.106 | 2.004 | 16 | 8 | ||||
| KNiAs | 0.001 | 0.840 | 0.188 | 1.029 | 16 | 8 | KNiSe | -0.010 | 0.947 | 0.170 | 1.107 | 17 | 9 | ||
| 0.022 | 1.017 | 0.528 | 1.567 | 16 | 8 | -0.014 | 0.900 | 0.122 | 1.008 | 17 | 9 | ||||
| 0.029 | 0.645 | 0.382 | 1.056 | 16 | 8 | 0.006 | 0.827 | 0.167 | 1.000 | 17 | 9 | ||||
| KCuAs | -0.004 | 0.060 | -1.132 | -1.076 | 17 | -1 | KCuSe | Gapless semiconductor | 18 | 0 | |||||
| -0.001 | -0.081 | -0.423 | 0.505 | 17 | -1 | Semiconductor | 18 | 0 | |||||||
| Metal | 17 | -1 | Semiconductor | 18 | 0 | ||||||||||
| KZnAs | 0.001 | -0.020 | 0.324 | 0.305 | 18 | 0 | KZnSe | Metal | 19 | 1 | |||||
| Semiconductor | 18 | 0 | Metal | 19 | 1 | ||||||||||
| Semiconductor | 18 | 0 | Metal | 19 | 1 | ||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



