Submitted:
13 September 2024
Posted:
17 September 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Post severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2, Covid-19) sequalae’s, immune and RNA based treatments are associated with the development of micro-stones and chronic kidney disease (CKD). In addition, circulating cytokines cocktail and viral envelope spike protein (SP) instigates CKD. During epigenetic regulation, Covid-19 methyltransferase (MT) methylates the DNA/RNA/Proteins/Histones and generates homocysteine (Hcy). Although cysteine promotes the growth and aggregation of calcium oxalate crystals in normal undiluted human urine. However, during elevated levels of Hcy (i.e., hyperhomocysteinemia, HHcy) due in part to the impaired transsulfuration (i.e., mitochondrial sulfur metabolism) pathways, homocysteine is the only sulfur-denoting non-protein amino acid may promote crystals. HHcy is associated with severity of Covid-19 infection and pneumonia post Covid-19. It is unclear however whether the HHcy facilitates homocysteine crystals. Although an E. coli strain degrades tricarboxylic acid (TCA, citric acid), and disrupts mitochondrial TCA cycle, to dicarboxylic acid (DCA, oxalic acid), DCA also promotes calcium oxalate crystals and may contributes to kidney stones, it is unclear whether gut-dysbiosis by Covid-19 infection contributes to kidney stones. There is robust succinylation of enzymes in TCA cycle which causes dysfunctional mitochondrial bioenergetics during Covid-19 infection. In this regards interaction between SIRT5 and mitochondrial transsulfuration during Covid-19 infection attenuates TCA cycle. Interestingly, the mitochondrial bioenergetics though sulfur metabolism participate in TCA cycle and epigenetic acetylation/succinylation, therefore, Covid-19 infection transition acute kidney injury (AKI) to CKD by defective mitochondria sulfur metabolism (transsulfuration) pathways, forming uricemia micro stones.
Keywords:
Kidney
; Covod-19
; AKI
; CKD
- Key Contributions: The acute kidney injury post Covid-19 can lead to CKD by depletion of renal microvascular nitric oxide, leading to kidney stones
- Running Title: Covid-19 AKI to CKD
- *A part of this study was supported by NIH grants AR-71789; HL139047; and DK116591
- DISCLOSURES: No conflicts of interest; financial or otherwise; are declared by the author
Introduction
Although the acute respiratory syndrome coronavirus-2 (SARS-CoV-2, Covid-19, lethal or non-lethal/asymptomatic) causes infection. However, only the immune and RNA based therapies are the current options. Post Covid-19 sequalae’s are associated with chronic kidney disease (CKD) [1,2,3,4,5]. However, the mechanisms are far from understanding. The objective of this review is to postulate a mechanism by which acute kidney injury (AKI) leads to chronic kidney disease CKD. Interestingly, beside the viral envelope proteins, the antibody treatments for Covid-19 infection can also increase the possibility of the development of CKD by increasing micro-stones [6,7,8]. Mechanistically, the entry of the various Covid-19 variants to the host is caused by viral envelope spike protein (Figure 1), therefore, it is important to determine the mechanism-based therapy for Covid-19 infection.
Discussion
The epigenetic contribution to uricemia is regulated primarily by methylation (Figure 2). In this regard the levels of methionine adenosyl transferase (MAT) is important and regulates the functional regenerative capacity [9]. Interestingly, the SARS-Co-V2 methyltransferase (MT) methylates the DNA/RNA/Protein/histone and generates homocysteine (Hcy) [10]. It is known that cysteine promotes the growth and aggregation of calcium oxalate crystals in normal undiluted human urine [11,12]. The mechanism(s) are largely unknow. Interestingly, during elevated levels of Hcy (i.e., hyperhomocysteinemia, HHcy) due in part to the impaired epigenetic rhythmic methylation/de-methylation and transsulfuration (i.e., mitochondrial sulfur metabolism) pathways (Figure 2), homocysteine is the only sulfur-denoting non-protein amino acid. Although HHcy is associated with severity of Covid-19 infection and pneumonia post Covid-19 [13], the mechanisms are unclear. Also, it is unclear whether the HHcy facilitates homocysteine crystals and kidney stones post Covid-19.
SARS-CoV-2 enters the cell by spike protein (SP) and causes renal injury and mask ACE2 and increases availability of Ang II, 1-8 amino acid peptide and decreases Ang-II, 1-7 amino acid peptide, suggesting a role in hypertension [3,14,15,16]. However, there are post Covid-19 sequelae those are associated with acute kidney injury (AKI) [17,18,19,20], leading to chronic kidney disease (CKD) [21,22,23,24,25] and vascular coagulopathy/thromboembolism [26]. Although vascular coagulopathy and venous thromboembolism are the hallmark of Covid-19 associated morbidity and mortality, however, the rate and coagulation time, and the platelet counts are unchanged. This suggests the role of other coagulation pathways, than thrombosis during Covid-19. Interestingly, a disintegrin and metalloproteinase thrombospondin domain 13 (ADAMTS13), a metalloproteinase instigates thrombolysis, is decreased in Covid-19 patients [27,28]. Also, there is endothelial dysfunction [29,30], vascular stiffness, perivascular fibrosis [31], and extracellular matrix (ECM) fragmentation [32,33,34,35,36,37] post Covid-19, the underlying mechanisms are unclear.
Previously, we showed activation of inflammatory M1 macrophages with renal infiltrates in the humanized angiotensin converting enzyme 2 (hACE2) mice transfected with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2 or Covid-19-spike protein (SP)), intranasally [17]. We suggested that occurrence of Covid-19 associated coagulopathy (CAC) is influenced by TMPRSS2, ADAMTS13, NGAL and MMP- 2, and -9 factors, and an intervention with iNOS blocker may mitigate the CAC [26].
Others have shown elevation in the levels of neopterin (NPT) post Covid-19 infection [38,39]. Because NPT is generated by activated pro-inflammatory M1 macrophages in response to Covid-19 infection via inducible nitric oxide synthase (iNOS) and tetrahydrobiopterin (BH4) via peroxinitrite (ONOO-) [ROS and RNS} consequencely, thisactivates the proteinases. This causes to generate neopterin (NPT) by exhausting the levels of BH4 and reduces availability of BH4 to eNOS . The generating ONOO- (ROS and RNS) activates latent resident metalloproteinase including the adisintegrin and metalloproteinase thrombospondin-13 domain (ADAMTS13, anti-thrombosis/anti-coagulant [40] and urinary neutrophil gelatinase associated lipocalin (NGAL, pro-thrombosis/pro-coagulant), and transmembrane serine proteinase S2 (TMPRSS2, proteolytic factor facilitates Covid-19) entry [26,41,42] and the matrix metalloproteinases (MMP), leading to shedding of the glycocalyx, endothelial dysfunction [43,44], glomerular leakage and fibrosis.
NGAL and FGF23 (hypertrophic factor) are increased during post Covid-19 sequelae [45,46,47,48,49. Interestingly, Covid-19 is associated with coagulopathy in glomerular capillaries-microvascular wall, the interaction between macrophages, neutrophil and immune cells, causes build-up of damage endothelial via NETs, NGAL, iNOS, and ADAMTS, and sets the stage for pro-thrombotic and pro-coagulant [24] states. Single cell analysis also revealed activation of ADAM17 post Covid-19 invasion [50]. This also contributes to focal glomerulosclerosis lesions [51,52].
Because there was greater COVID-19 severity and mortality in males compared with females [53]. The blocking of ADAM17 and TMPRSS2 offered potential disease-modifying approaches to ameliorate enhanced severity in males [53,54]. Therefore, it is novel to measure ADAMTS and TMPRSS2 in both the males versus and females.
In addition to cellular 1-carbon metabolism, the ions and solute cellular co-transporter [55,56] SLC22A17 contributes to osmotic stress adaptation, protection against urinary tract infection, and renal carcinogenesis in part by transporting transition metals (TMs), Fe, Cd, Zn, and others [55,56]. This transporter complexing with metalloproteins cause nephrotoxicity [57,58,59]. In this regard, its interaction with NGAL- a metalloprotein is crucial in the sense that metalloproteinase activity of NGAL may cause tubular leakage, endocytosis, glycocalyx shedding and increase in neutrophil extracellular traps (NETs). Therefore, a treatment with DNaase I may be beneficial [60,61].
The epigenetic DNA methylation, activation of transmembrane serine protease 2 (TMPRSS2) and a disintegrin and metalloproteinase thrombospondin domain 13 (ADAMTS13) have been associated of SARS-CoV-2 entry into the cells [62,63,64,65]. The mechanism(s) are unclear. TMPRSS2 processes the Covid-19 viral protein secretion. The ADAMTS13 facilitates coagulation. Because the levels of transmembrane (TMPRSS2), EMMPRIN (CD147) and ECM proteinases are elevated post Covid-19 [26,41,42]. These proteinases are associated with collagen/elastin breakdown during renal glomerular remodeling [66]. However, because the turnover of collagen is rapid than elastin [67], the degraded elastin is replaced by collagen, causing fibrosis, stiffness, and thickening of the basement membrane in the glomeruli, instigating impaired glomerular filtration rate (GFR).
We conclude that it is important to test the hypothesis if the increase in M1 macrophages iNOS decreases BH4 bioavailability to eNOS, causing glomerular capillary microvascular endothelial dysfunction. The peroxinitrite (ONOO-) activates NGAL and FGF23 (released by the kidney) and ADAMTS. This causes podocyte glycocalyx shedding, leading to glomerular leakage [68,69]. These are novel aspects of Covid-19 induced CKD.
In addition, recently mitochondrial miR-2392 was shown to drive downstream suppression of mitochondrial gene expression, increasing inflammation, glycolysis, and hypoxia, as well as promoting symptoms associated with corona virus disease (COVID-19) infection, such as respiratory congestion [70]. Interestingly, an iNOS inhibitor (aminoguanidine, AG) attenuated inflammatory miRNA-21 levels [71].
Because post Covid-19, circulating cytokine cocktail inflammatory response and viral envelope proteins, such as spike protein (SP) mimics post Covid-19 sequalae [72,73,74,75,76], we injected SP protein in hACE2 mice [26]. Also, co-morbidity associated with Covid-19 infection increases microparticles and further the inflammatory response [80]. The Covid-19 infection causes disruption in blood vascular endothelial layer [77] by activation of MMP8 (a neutrophil-derived MMP) and creates enlarged perivascular space (EPVS) and fibrosis [78]. We have suggested increase in inflammatory infiltrate and neutrophil gelatinase associated lipocalin (NGAL) in renal vascular coagulopathy, leading to CKD, post Covid-19 sequalae [26]. In this regard it is novel to measure NGAL activation by spike protein and its mitigation by iNOS blocker (Figure 3).
Kidney Stones
Although an E. coli strain degrades tricarboxylic acid (TCA, citric acid), and disrupts mitochondrial TCA cycle, to dicarboxylic acid (DCA, oxalic acid) [79,80], DCA also promotes calcium oxalate crystals and may contributes to kidney stones, it is unclear whether gut-dysbiosis contributes to kidney stones post-Covid-19.
Although the severity of Covid-19 infection and pneumonia are associated with the increase levels of homocysteine (Hcy, i.e., hyperhomocysteinemia (HHcy) [81,82], the mechanisms are unclear. The fucus of this article is to review the role of Hcy metabolic transsulfuration (mitochondrial sulfur metabolism and bioenergetics) pathways in the severity of long Covid-19. The 3-mercaptopyruvate sulfur transferase (3MST) converts homocysteine into hydrogen sulfide and improves endothelial function. The pyruvate supplement suppresses viral replication via induction of TCA cycle [83] and this may improve endothelial dysfunction post Covid-19 infection.
Interestingly, there is robust succinylation of enzymes in TCA cycle which causes dysfunctional mitochondrial bioenergetics pathways during Covid-19 infection [84]. In this regards interaction between SIRT5 and mitochondrial bioenergetics during Covid-19 infection [84] is suggested with the focus target for the treatment post Covid-19.
What are the underlying mechanisms for COVID-19 infection to kidney stone formation:
Interestingly, the mitochondrial bioenergetics though sulfur metabolism by 3-mercaptopyruvate sulfur transferase (3MST) participate in TCA cycle and epigenetic acetylation/succinylation (Figure 2). The dysfunctional sulfur metabolism enzymes and renal transulfuration may increase homocysteine. This may lead to accumulate homocysteine crystal.. Thus the mitochondrial bioenergetics though sulfur metabolism participate in TCA cycle and epigenetic acetylation/succinylation, therefore, Covid-19 transition acute kidney injury (AKI) to chronic kidney disease (CKD) by defective mitochondria sulfur metabolism (transsulfuration) pathways, forming kidney uricemia micro stones. Defective sulfur transferase activity i.e., mitochondrial transsulfuration pathways, in the kidney causes kidney stones in chronic kidney disease [75,86].
Conclusions and Future Direction:
Although current knowledge in Covid-19 research focus only on the infection, very little is known about the spike-protein induced viral entry and alteration in gut-dysbiosis and renal-axis-remodeling leading to CKD. Therefore, it is important to test the hypothesis that the spike protein induces iNOS, exhausts 169 BH4, increases NPT, NETs, NGAL, oxidative/peroxynitrite, activates ADAMTS, MMPs and transmembrane serine proteinase S2 (TMPRSS2) causing glycocalyx shedding, tubular epithelial leakage, and leading CKD. To address this hypothesis, it is novel to determine whether the blockade of inflammatory M1 macrophage iNOS attenuates SARS CoV-2 spike protein-induced activation NETs, NGAL, TMPRSS2, ADAMTS, glycocalyx shedding, endothelial dysfunction, collagen deposits, glomerulosclerosis, tubular interstitial fibrosis, and leakage. Interestingly, because there was greater COVID-19 severity and mortality in males compared with females. The blocking of ADAM17 and TMPRSS2 offered potential disease-modifying approaches to ameliorate enhanced severity in males.
References
- Martin de Francisco, A. & Fernandez Fresnedo, G. Long COVID-19 renal disease: A present medical need for nephrology. Nefrologia (Engl Ed) 43, 1-5 (2023). [CrossRef]
- Schiffl, H. & Lang, S. M. Long-term interplay between COVID-19 and chronic kidney disease. Int Urol Nephrol 55, 1977-1984 (2023). [CrossRef]
- Teng, L. et al. The pattern of cytokines expression and dynamic changes of renal function at 6 months in patients with Omicron COVID-19. J Med Virol 95, e28477 (2023). [CrossRef]
- Atiquzzaman, M. et al. Long-term effect of COVID-19 infection on kidney function among COVID-19 patients followed in post-COVID recovery clinic in British Columbia, Canada. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association (2023). [CrossRef]
- Chancharoenthana, W. et al. Gastrointestinal manifestations of long-term effects after COVID-19 infection in patients with dialysis or kidney transplantation: An observational cohort study. World J Gastroenterol 29, 3013-3026 (2023). [CrossRef]
- Wang CS, Glenn DA, Helmuth M, Smith AR, Bomback AS, Canetta PA, Coppock GM, Khalid M, Tuttle KR, Bou-Matar R, Greenbaum LA, Robinson BM, Holzman LB, Smoyer WE, Rheault MN, Gipson D, Mariani LH; Cure Glomerulonephropathy (CureGN) Study Consortium. Association of COVID-19 Versus COVID-19 Vaccination With Kidney Function and Disease Activity in Primary Glomerular Disease: A Report of the Cure Glomerulonephropathy Study. Am J Kidney Dis. 2024 Jan;83(1):37-46. Epub 2023 Aug 31. [CrossRef]
- Bai S, Zhan Y, Pan C, Liu G, Li J, Shan L. Prospective comparison of extracorporeal shock wave lithotripsy versus flexible ureterorenoscopy in patients with non-lower pole kidney stones under the COVID-19 pandemic. Urolithiasis. 2023 Feb 16;51(1):38. [CrossRef] [PubMed]
- Spooner J, Masoumi-Ravandi K, MacNevin W, Ilie G, Skinner T, Powers AL. Septic and febrile kidney stone presentations during the COVID-19 pandemic What is the effect of reduced access to care during pandemic restrictions? Can Urol Assoc J. 2024 Jan;18(1):E19-E25. [CrossRef] [PubMed]
- Rajabian N, Ikhapoh I, Shahini S., et al, has shown that the Methionine adenosyltransferase2A (MAT) inhibition restores metabolism to improve regenerative capacity and strength of aged skeletal muscle, Nature Communications, 14, article number: 886; Feb 16, 2023.
- Kottur J, White KM, Rodriguez ML, Rechkoblit O, Quintana-Feliciano R, Nayar A, García-Sastre A, Aggarwal AK. Structures of SARS-CoV-2 N7-methyltransferase with DOT1L and PRMT7 inhibitors provide a platform for new antivirals. PLoS Pathog. 2023 Jul 31;19(7):e1011546. eCollection 2023 Jul. [CrossRef] [PubMed]
- Martins MC, Meyers AA, Whalley NA, Rodgers AL. Cystine: a promoter of the growth and aggregation of calcium oxalate crystals in normal undiluted human urine. J Urol, 2002, Jan;167(1):317-321. [CrossRef]
- Wallace B, Chmiel JA, Al KF, Bjazevic J, Burton JP, Goldberg HA, Razvi H. The Role of Urinary Modulators in the Development of Infectious Kidney Stones. J Endourol. 2023 Mar;37(3):358-366. Epub 2023 Jan 20. [CrossRef] [PubMed]
- Kalan Sarı I, Keskin O, Seremet Keskin A, Elli Dağ HY, Harmandar O. Is Homocysteine Associated with the Prognosis of Covid-19 Pneumonia. Int J Clin Pract. 2023 Mar 2;2023:9697871. eCollection 2023. Free PMC article. [CrossRef] [PubMed]
- Govender, N., Khaliq, O., Moodley, J. & Naicker, T. Unravelling the Mechanistic Role of ACE2 and TMPRSS2 in Hypertension: A Risk Factor for COVID-19. Curr Hypertens Rev 18, 130-137 (2022). [CrossRef]
- South, A. M., Diz, D. I. & Chappell, M. C. COVID-19, ACE2, and the cardiovascular consequences. American journal of physiology. Heart and circulatory physiology 318, H1084-H1090 (2020). [CrossRef]
- South, A. M. et al. Fetal programming and the angiotensin-(1-7) axis: a review of the experimental and clinical data. Clin Sci (Lond) 133, 55-74 (2019). [CrossRef]
- Singh, M. et al. Simulation of COVID-19 symptoms in a genetically engineered mouse model: implications for the long haulers. Mol Cell Biochem 478, 103-119 (2023). [CrossRef]
- Srivastava, S. P., Srivastava, R., Chand, S. & Goodwin, J. E. Coronavirus Disease (COVID)-19 and Diabetic Kidney Disease. Pharmaceuticals (Basel) 14 (2021). [CrossRef]
- Borczuk, A. C. & Yantiss, R. K. The pathogenesis of coronavirus-19 disease. J Biomed Sci 29, 87 (2022). [CrossRef]
- Sachetto, A. T. A. & Mackman, N. Tissue Factor and COVID-19: An Update. Curr Drug Targets 23, 1573-1577 (2022). [CrossRef]
- Altillero, M., Jr., Danguilan, R. & Arakama, M. H. Incidence of, and Risk Factors and Outcomes Associated with, Acute Kidney Injury in COVID-19 at the National Kidney and Transplant Institute, Philippines. Trop Med Infect Dis 8 (2023). [CrossRef]
- Kim, I. S. et al. Role of increased neutrophil extracellular trap formation on acute kidney injury in COVID-19 patients. Front Immunol 14, 1122510 (2023). [CrossRef]
- Zhang, W. et al. Identification of common molecular signatures of SARS-CoV-2 infection and its influence on acute kidney injury and chronic kidney disease. Front Immunol 14, 961642 (2023). [CrossRef]
- Narayanan, A., Cunningham, P., Mehta, M., Lang, T. & Hammes, M. Acute Kidney Injury in Coronavirus Disease and Association with Thrombosis. American journal of nephrology 54, 156-164 (2023). [CrossRef]
- Pickkers, P. et al. Study protocol of a randomised, double-blind, placebo-controlled, two-arm parallel-group, multi-centre phase 3 pivotal trial to investigate the efficacy and safety of recombinant human alkaline phosphatase for treatment of patients with sepsis-associated acute kidney injury. BMJ Open 13, e065613 (2023). [CrossRef]
- Singh, M. et al. Novel mechanism of the COVID-19 associated coagulopathy (CAC) and vascular thromboembolism. npj Viruses 1, 3 (2023). [CrossRef]
- AbdelHamid, S. G. et al. Deciphering epigenetic(s) role in modulating susceptibility to and severity of COVID-19 infection and/or outcome: A systematic rapid review. Environ. Sci. Pollut. Res. Int. 28, 54209–54221 (2021). [CrossRef]
- Chlamydas, S., Papavassiliou, A. G. & Piperi, C. Epigenetic mechanisms regulating COVID-19 infection. Epigenetics 16, 263–270 (2021). [CrossRef]
- Jadali Z. Double-edged sword effect of platelets in COVID-19. J Vasc Bras. 2023 Mar 6;22:e20220101. eCollection 2023. Free PMC article. No abstract available. [CrossRef] [PubMed]
- Vuorio A, Raal F, Kovanen PT. Familial hypercholesterolemia: The nexus of endothelial dysfunction and lipoprotein metabolism in COVID-19. Curr Opin Lipidol. 2023 Mar 10. Online ahead of print. [CrossRef] [PubMed]
- Zhou S, Yu Z, Chen Z, Ning F, Hu X, Wu T, Li M, Xin H, Reilly S, Zhang X. Olmesartan alleviates SARS-CoV-2 envelope protein induced renal fibrosis by regulating HMGB1 release and autophagic degradation of TGF-beta1. Front Pharmacol. 2023 May 15;14:1187818. eCollection 2023. [CrossRef] [PubMed]
- Tudoran, C. et al. Correspondence between Aortic and Arterial Stiffness, and Diastolic Dysfunction in Apparently Healthy Female Patients with Post-Acute COVID-19 Syndrome. Biomedicines 11 (2023). [CrossRef]
- Wierzbicki, T. & Bai, Y. Finite element modeling of alpha-helices and tropocollagen molecules referring to spike of SARS-CoV-2. Biophys J 121, 2353-2370 (2022). [CrossRef]
- Brennan, G. T. COVID-19-Induced Collagenous Colitis. Gastro Hep Adv 1, 976 (2022). [CrossRef]
- Gutman, H. et al. Matrix Metalloproteinases Expression Is Associated with SARS-CoV-2-Induced Lung Pathology and Extracellular-Matrix Remodeling in K18-hACE2 Mice. Viruses 14 (2022). [CrossRef]
- Brusa, S. et al. Circulating tissue inhibitor of metalloproteinases 1 (TIMP-1) at COVID-19 onset predicts severity status. Front Med (Lausanne) 9, 1034288 (2022). [CrossRef]
- Belen Apak, F. B. et al. Coagulopathy is Initiated with Endothelial Dysfunction and Disrupted Fibrinolysis in Patients with COVID-19 Disease. Indian J Clin Biochem, 1-11 (2023). [CrossRef]
- Chauvin, M. et al. Elevated Neopterin Levels Predict Fatal Outcome in SARS-CoV-2-Infected Patients. Front Cell Infect Microbiol 11, 709893 (2021). [CrossRef]
- Al-Kuraishy, H. M., Al-Gareeb, A. I., Alzahrani, K. J., Cruz-Martins, N. & Batiha, G. E. The potential role of neopterin in Covid-19: a new perspective. Mol Cell Biochem 476, 4161-4166 (2021). [CrossRef]
- Zhang, Q. et al. Effects of convalescent plasma infusion on the ADAMTS13-von Willebrand factor axis and endothelial integrity in patients with severe and critical COVID-19. Res Pract Thromb Haemost 7, 100010 (2023). [CrossRef]
- Hoffmann, M. et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 181, 271-280 e278 (2020). [CrossRef]
- Matthew D. Cheung, Elise N. Erman, Shanrun Liu, Nathaniel B. Erdmann, Gelare Ghajar-Rahimi, Kyle H. Moore, Jeffrey C. Edberg, James F. George, and Anupam Agarwal. Single-Cell RNA Sequencing of Urinary Cells Reveals Distinct Cellular Diversity in COVID-19–Associated AKI. American Society of Nephrology www.kidney360.org Vol 3 January, 2022.
- Jadali Z. Double-edged sword effect of platelets in COVID-19. J Vasc Bras. 2023 Mar 6;22:e20220101. eCollection 2023. Free PMC article. No abstract available. [CrossRef] [PubMed]
- Vuorio A, Raal F, Kovanen PT. Familial hypercholesterolemia: The nexus of endothelial dysfunction and lipoprotein metabolism in COVID-19. Curr Opin Lipidol. 2023 Mar 10. Online ahead of print. [CrossRef] [PubMed]
- Zhou, S. et al. ADAMTS13 protects mice against renal ischemia-reperfusion injury by reducing inflammation and improving endothelial function. American journal of physiology. Renal physiology 316, F134-F145 (2019). [CrossRef]
- Henry, B. M. et al. Cell-Free DNA, Neutrophil extracellular traps (NETs), and Endothelial Injury in Coronavirus Disease 2019- (COVID-19-) Associated Acute Kidney Injury. Mediators Inflamm 2022, 9339411 (2022). [CrossRef]
- Li, H., Yu, Z., Gan, L., Peng, L. & Zhou, Q. Serum NGAL and FGF23 may have certain value in early diagnosis of CIN. Ren Fail 40, 547-553 (2018). [CrossRef]
- Faul, C. et al. FGF23 induces left ventricular hypertrophy. The Journal of clinical investigation 121, 4393-4408 (2011). [CrossRef]
- Zheng, X. L. ADAMTS13 and von Willebrand factor in thrombotic thrombocytopenic purpura. Annu Rev Med 66, 211-225 (2015). [CrossRef]
- Chen, C., Wang, J., Liu, Y. M. & Hu, J. Single-cell analysis of adult human heart across healthy and cardiovascular disease patients reveals the cellular landscape underlying SARS-CoV-2 invasion of myocardial tissue through ACE2. J Transl Med 21, 358 (2023). [CrossRef]
- Bolignano, D. et al. Neutrophil gelatinase-associated lipocalin (NGAL) and progression of chronic kidney disease. Clin J Am Soc Nephrol 4, 337-344 (2009). [CrossRef]
- Zhang, Q. et al. Clinical Significance of Urinary Biomarkers in Patients With Primary Focal Segmental Glomerulosclerosis. Am J Med Sci 355, 314-321 (2018). [CrossRef]
- Viveiros, A. et al. Sex differences in COVID-19: candidate pathways, genetics of ACE2, and sex hormones. American journal of physiology. Heart and circulatory physiology 320, H296-H304 (2021). [CrossRef]
- Viveiros A, Gheblawi M, Aujla PK, Sosnowski DK, Seubert JM, Kassiri Z, Oudit GY. Sex- and age-specific regulation of ACE2: Insights into severe COVID-19 susceptibility. J Mol Cell Cardiol. 2022 Mar;164:13-16. Epub 2021 Nov 11. Free PMC article. [CrossRef] [PubMed]
- Schroder, S. K., Gasterich, N., Weiskirchen, S. & Weiskirchen, R. Lipocalin 2 receptors: facts, fictions, and myths. Front Immunol 14, 1229885 (2023). [CrossRef]
- Thevenod, F. et al. Role of the SLC22A17/lipocalin-2 receptor in renal endocytosis of proteins/metalloproteins: a focus on iron- and cadmium-binding proteins. American journal of physiology. Renal physiology 325, F564-F577 (2023). [CrossRef]
- Cabedo Martinez, A. I. et al. Biochemical and Structural Characterization of the Interaction between the Siderocalin NGAL/LCN2 (Neutrophil Gelatinase-associated Lipocalin/Lipocalin 2) and the N-terminal Domain of Its Endocytic Receptor SLC22A17. The Journal of biological chemistry 291, 2917-2930 (2016). [CrossRef]
- Langelueddecke, C., Roussa, E., Fenton, R. A. & Thevenod, F. Expression and function of the lipocalin-2 (24p3/NGAL) receptor in rodent and human intestinal epithelia. PloS one 8, e71586 (2013). [CrossRef]
- Langelueddecke, C. et al. Lipocalin-2 (24p3/neutrophil gelatinase-associated lipocalin (NGAL)) receptor is expressed in distal nephron and mediates protein endocytosis. The Journal of biological chemistry 287, 159-169 (2012). [CrossRef]
- Kim, J. K. et al. Prognostic role of circulating neutrophil extracellular traps levels for long-term mortality in new end-stage renal disease patients. Clin Immunol 210, 108263 (2020). [CrossRef]
- Veras, F. P. et al. Targeting neutrophils extracellular traps (NETs) reduces multiple organ injury in a COVID-19 mouse model. Respiratory research 24, 66 (2023). [CrossRef]
- AbdelHamid, S. G. et al. Deciphering epigenetic(s) role in modulating susceptibility to and severity of COVID-19 infection and/or outcome: A systematic rapid review. Environ. Sci. Pollut. Res. Int. 28, 54209–54221 (2021). [CrossRef]
- Chlamydas, S., Papavassiliou, A. G. & Piperi, C. Epigenetic mechanisms regulating COVID-19 infection. Epigenetics 16, 263–270 (2021). [CrossRef]
- Michael S Xydakis, Mark W Albers, Eric H Holbrook, Dina M Lyon, Robert Y Shih, Johannes A Frasnelli, Axel Pagenstecher, Alexandra Kupke, Lynn W Enquist, Stanley Perlman. Post-viral effects of COVID-19 in the olfactory system and their implications. Lancet Neurol. 2021 Sep;20(9):753-761. Epub 2021 Jul 30. [CrossRef] [PubMed] [PubMed Central]
- Eileen P Scully, Jenna Haverfield , Rebecca L Ursin, Cara Tannenbaum, Sabra L Klein. Considering how biological sex impacts immune responses and COVID-19 outcomes. Nat Rev Immunol. 2020 Jul;20(7):442-447. Epub 2020 Jun 11. [CrossRef] [PubMed] [PubMed Central]
- Camp, T. M., Smiley, L. M., Hayden, M. R. & Tyagi, S. C. Mechanism of matrix accumulation and glomerulosclerosis in spontaneously hypertensive rats. Journal of hypertension 21, 1719-1727 (2003). [CrossRef]
- Rucklidge, G. J., Milne, G., McGaw, B. A., Milne, E. & Robins, S. P. Turnover rates of different collagen types measured by isotope ratio mass spectrometry. Biochimica et biophysica acta 1156, 57-61 (1992). [CrossRef]
- Ramnath RD, Butler MJ, Newman G, Desideri S, Russell A, Lay AC, Neal CR, Qiu Y, Fawaz S, Onions KL, Gamez M, Crompton M, Michie C, Finch N, Coward RJ, Welsh GI, Foster RR, Satchell SC. Blocking matrix metalloproteinase-mediated syndecan-4 shedding restores the endothelial glycocalyx and glomerular filtration barrier function in early diabetic kidney disease. Kidney Int. 2020 May;97(5):951-965. Epub 2019 Nov 2. Free PMC article. [CrossRef] [PubMed]
- Sarah Fawaz, Aldara Martin Alonso, Yan Qiu, Raina Ramnath, Holly Stowell-Connolly, Monica Gamez, Carl May, Colin Down, Richard J Coward, Matthew J Butler, Gavin I Welsh, Simon C Satchell, Rebecca R Foster. Adiponectin reduces glomerular endothelial glycocalyx disruption and restores glomerular barrier function in a mouse model of type 2 diabetes, Diabetes, 2024 Mar 26:db230455. [CrossRef]
- McDonald, J. T. et al. Role of miR-2392 in driving SARS-CoV-2 infection. Cell Rep 37, 109839 (2021). [CrossRef]
- Jiang, Q. et al. Ursolic acid induced anti-proliferation effects in rat primary vascular smooth muscle cells is associated with inhibition of microRNA-21 and subsequent PTEN/PI3K. Eur J Pharmacol 781, 69-75 (2016). [CrossRef]
- Del Nogal Avila M, Das R, Kharlyngdoh J, Molina-Jijon E, Donoro Blazquez H, Gambut S, Crowley M, Crossman DK, Gbadegesin RA, Chugh SS, Chugh SS, Avila-Casado C, Macé C, Clement LC, Chugh SS. Cytokine storm-based mechanisms for extrapulmonary manifestations of SARS-CoV-2 infection. JCI Insight. 2023 May 22;8(10):e166012. [CrossRef] [PubMed]
- Carreño JM, Alshammary H, Singh G, Raskin A, Amanat F, Amoako A, Gonzalez-Reiche AS, van de Guchte A, Study Group P, Srivastava K, Sordillo EM, Sather DN, van Bakel H, Krammer F, Simon V. Evidence for retained spike-binding and neutralizing activity against emerging SARS-CoV-2 variants in serum of COVID-19 mRNA vaccine recipients. EBioMedicine. 2021 Nov;73:103626. Epub 2021 Oct 20. [CrossRef] [PubMed]
- Dan JM, Mateus J, Kato Y, Hastie KM, Yu ED, Faliti CE, Grifoni A, Ramirez SI, Haupt S, Frazier A, Nakao C, Rayaprolu V, Rawlings SA, Peters B, Krammer F, Simon V, Saphire EO, Smith DM, Weiskopf D, Sette A, Crotty S. Immunological memory to SARS-CoV-2 assessed for up to eight months after infection. bioRxiv [Preprint]. 2020 Dec 18:2020.11.15.383323. [CrossRef] [PubMed]
- Thomas S Metkus, Lori J Sokoll, Andreas S Barth, Matthew J Czarny, Allison G Hays, Charles 419 J Lowenstein, Erin D Michos, Eric P Nolley, Wendy S Post, Jon R Resar, David R 420 BioTech 2024, 5, x FOR PEER REVIEW 12 of 13; Thiemann, Jeffrey C Trost, Rani K Hasan. Myocardial Injury in Severe COVID-19 421 Compared with Non-COVID-19 Acute Respiratory Distress Syndrome, Circulation. 2021 422 Feb 9;143(6):553-565. Epub 2020 Nov 13. 423. [CrossRef] [PubMed] [PubMed Central]
- Zhou S, Yu Z, Chen Z, Ning F, Hu X, Wu T, Li M, Xin H, Reilly S, Zhang X. 425 Olmesartan alleviates SARS-CoV-2 envelope protein induced renal fibrosis by regulating 426 HMGB1 release and autophagic degradation of TGF-beta1. Front Pharmacol. 2023 May 427 15;14:1187818. eCollection 2023. [CrossRef] [PubMed]
- Greene C, Connolly R, Brennan D, Laffan A, O'Keeffe E, Zaporojan L, O'Callaghan J, 429 Thomson B, Connolly E, Argue R, Martin-Loeches I, Long A, Cheallaigh CN, Conlon N, 430 Doherty CP, Campbell M. Blood-brain barrier disruption and sustained systemic 431 inflammation in individuals with long COVID-associated cognitive impairment. Nat 432 Neurosci. 2024 Feb 22. Online ahead of 433 print. [CrossRef] [PubMed]
- Cathomas F, Lin HY, Chan KL, Li L, Parise LF, Alvarez J, Durand-de Cuttoli R, Aubry AV, 435 Muhareb S, Desland F, Shimo Y, Ramakrishnan A, Estill M, Ferrer-Pérez C, Parise EM, 436 Wilk CM, Kaster MP, Wang J, Sowa A, Janssen WG, Costi S, Rahman A, Fernandez 437 N, Campbell M, Swirski FK, Nestler EJ, Shen L, Merad M, Murrough JW, Russo SJ. 438 Circulating myeloid-derived MMP8 in stress susceptibility and depression. Nature. 2024 439 Feb 7. Online ahead of print. [CrossRef] [PubMed]
- Valle A, Soto Z, Muhamadali H, Hollywood KA, Xu Y, Lloyd JR, Goodacre R, Cantero D, 441 Cabrera G, Bolivar J. Metabolomics for the design of new metabolic engineering strategies 442 for improving aerobic succinic acid production in Escherichia coli. Metabolomics. 2022 Jul 443 20;18(8):56. Free PMC article. 444 RESULTS: Most of the 65 identified metabolites showed lower relative levels in the M4-445 deltaiclR and M4-deltagnd mutants than those of the M4. However, fructose 1,6-446 biphosphate, trehalose, isovaleric acid and mannitol relative concentrations were increased 447 in M4-deltaiclR a … 448. [CrossRef] [PubMed]
- Tong W, Hannou SA, Wang Y, Astapova I, Sargsyan A, Monn R, Thiriveedi V, Li D, McCann 449 JR, Rawls JF, Roper J, Zhang GF, Herman MA. The intestine is a major contributor to 450 circulating succinate in mice. FASEB J. 2022 Oct;36(10):e22546. The tricarboxylic acid (TCA) cycle is the 452 epicenter of cellular aerobic metabolism. ...Despite the importance of circulating TCA cycle 453 metabolites as signaling molecules, the source of circulating TCA cycle intermediates 454 remains uncertain. ... 455. [CrossRef] [PubMed]
- Kalan Sarı I, Keskin O, Seremet Keskin A, Elli Dağ HY, Harmandar O. Is Homocysteine 456 Associated with the Prognosis of Covid-19 Pneumonia. Int J Clin Pract. 2023 Mar 457 2;2023:9697871. eCollection 2023. Free 458 PMC article. 459. [CrossRef] [PubMed]
- Giovanni Carpenè, Davide Negrini, Brandon M Henry, Martina Montagnana, Giuseppe Lippi 460 Homocysteine in coronavirus disease (COVID-19): a systematic literature review, 461 BioTech 2024, 5, x FOR PEER REVIEW 13 of 13.
- Diagnosis (Berl) 2022 Jun 16;9(3):306-310. doi: 10.1515/dx-2022-0042. eCollection 2022 462 Aug 1. [CrossRef] [PubMed]
- Sang R. Lee, Jeong Yeon Roh, Jihoon Ryu, Hyun-Jin Shin & Eui-Ju Hong, Activation of TCA 464 cycle restrains virus-metabolic hijacking and viral replication in mouse hepatitis 465 virus-infected cells, Cell & Bioscience volume 12, Jan 18, 2022; Article number: 7 (2022). 466.
- Liu Q, Wang H, Zhang H, Sui L, Li L, Xu W, Du S, Hao P, Jiang Y, Chen J, Qu X, Tian M, 467 Zhao Y, Guo X, Wang X, Song W, Song G, Wei Z, Hou Z, Wang G, Sun M, Li X, Lu H, 468 Zhuang X, Jin N, Zhao Y, Li C, Liao M. The global succinylation of SARS-CoV-2-infected 469 host cells reveals drug targets. Proc Natl Acad Sci U S A. 2022 Jul 470 26;119(30):e2123065119. Epub 2022 Jul 471 12. Free PMC article. SARS-CoV-2 infection promotes succinylation of 472 several key enzymes in the TCA, leading to inhibition of cellular metabolic pathways. We 473 demonstrated that host protein succinylation is regulated by viral nonstructural protein 474 (NSP14) through interaction with sirtuin 5 (SIRT … 475. [CrossRef] [PubMed]
- Ruyu Tan, Santao Ou, Ting Kang, Weihua Wu, Lin Xiong, Tingting Zhu and Liling Zhang. 476 Altered serum metabolome associated with vascular calcification developed from CKD and 477 the critical pathways. Frontiers in Cardiovascular Medicine, TYPE Original Research 478 PUBLISHED 11 April 2023 | DOI 10.3389/fcvm.2023.1114528. 479. [CrossRef]
- Cui J, Hong P, Li Z, Lin J, Wu X, Nie K, Zhang X, Wan J. Chloroquine inhibits NLRP3 481 inflammasomes activation and alleviates renal fibrosis in mouse model of 482 hyperuricemic nephropathy with aggravation by a high-fat-diet. Int Immunopharmacol. 483 2023 Jun 3;120:110353. Online ahead of 484 print. Numerous epidemiological studies have demonstrated that 485 hyperuricemia (HUA) is a risk factor for renal diseases and renal fibrosis. Dietary patterns 486 can influence serum urate levels and hyperuricemic nephropathy (HN). ...Additionally, the 487 HN + HFD group displ …. [CrossRef] [PubMed]
Figure 1.
The primary entry of the Covid-19 and its variants, into the cell, though viral envelope spike protein (SP). Therefore, it is important to understand the mechanism by which virus enters the cell and transforms the cell metabolism causing infection.
Figure 1.
The primary entry of the Covid-19 and its variants, into the cell, though viral envelope spike protein (SP). Therefore, it is important to understand the mechanism by which virus enters the cell and transforms the cell metabolism causing infection.
Figure 2.
The viral causes gut dysbiosis (i.e., loss of taste) and therefore alters the epigenetic regulation of metabolic genes. The schematics of epigenetic 1-carbon metabolism regulation by gene writer, eraser and the RNA editor in renal remodeling and the development of chronic kidney disease and uricemia micro stones formation, during Covid-19 infection induced gut-dysbiosis.
Figure 2.
The viral causes gut dysbiosis (i.e., loss of taste) and therefore alters the epigenetic regulation of metabolic genes. The schematics of epigenetic 1-carbon metabolism regulation by gene writer, eraser and the RNA editor in renal remodeling and the development of chronic kidney disease and uricemia micro stones formation, during Covid-19 infection induced gut-dysbiosis.

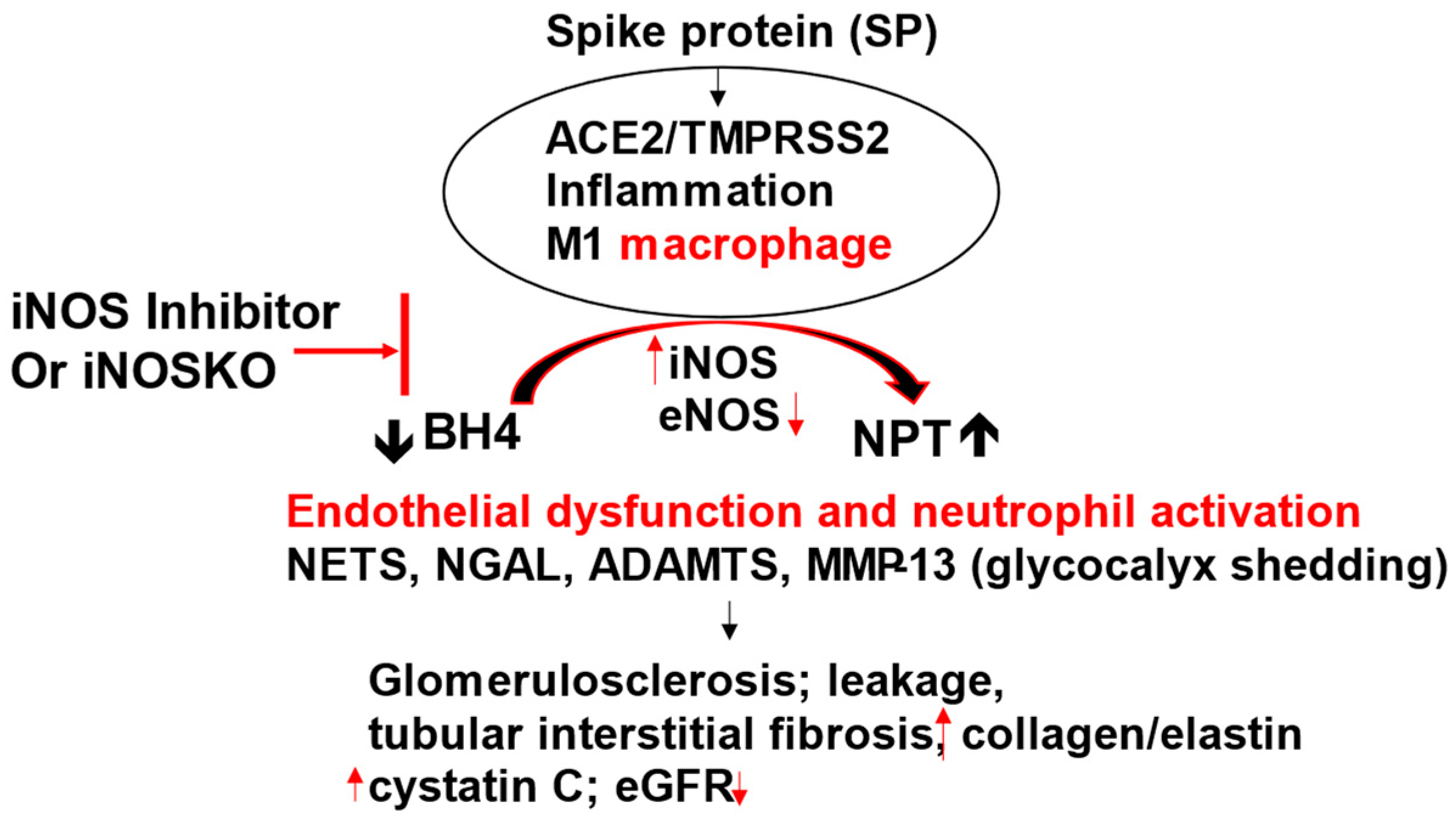
Figure 3.
SARS-CoV-2 spike protein primes ACE2/TMPRSS2, causes M1 macrophage activation, increases iNOS and generates neopterin (NPT) by depleting tetrahydrobiopterin (BH4), leading to endothelial dysfunction. The activation of NETS, NGAL, ADAMTS and MMPs/TIMPs cause proximal tubular epithelial cell and vascular injury, leakage, and CKD. The iNOS blockade or iNOSKO attenuates SARS-CoV-2 induced CKD. “Neopterin: marker to mechanism to medicine”.
Figure 3.
SARS-CoV-2 spike protein primes ACE2/TMPRSS2, causes M1 macrophage activation, increases iNOS and generates neopterin (NPT) by depleting tetrahydrobiopterin (BH4), leading to endothelial dysfunction. The activation of NETS, NGAL, ADAMTS and MMPs/TIMPs cause proximal tubular epithelial cell and vascular injury, leakage, and CKD. The iNOS blockade or iNOSKO attenuates SARS-CoV-2 induced CKD. “Neopterin: marker to mechanism to medicine”.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



