
Submitted:
17 September 2024
Posted:
17 September 2024
You are already at the latest version
Abstract
The hetero Diels-Alder (HDA) reaction between the ambident heterodiene 3-methylene-2,4-chromandione (MCDO) and the non-symmetric methyl vinyl ether (MVE) is investigated within the Molecular Electron Density Theory (MEDT) at the B3LYP/6-311G(d,p) computational level. The aim of this study is to gain insight into the molecular mechanism and to elucidate the factors that control the selectivity found experimentally. DFT-based reactivity indices reveal that MCDO exhibits strong electrophilic character, while MVE displays a strong nucleophilic character. Meanwhile, Parr function explains the ortho regioselectivity of this HDA reaction. The highly polar nature of this HDA reaction, supported by the high global electron density transfer (GEDT) taking place at the transition state structures (TSs), accounts for the very low activation energy associated with the most favorable TS-4on. The ambident nature of MCDO allows the formation of two constitutional isomeric cycloadduct. In the case of MVE, the pseudocyclic selectivity is attained by a thermodynamic control. This polar HDA reaction displays an endo stereoselectivity and a complete ortho regioselectivity. A comparative Relative Interacting Atomic Energy (RIAE) analysis of the two diastereomeric TS-4on and TS-6on indicates a highly likeness between them, which explains the low pseudocyclic selectivity under kinetic control.
Keywords:
Molecular Electron Density Theory
; Hetero Diels-Alder
; ambident heterodiene
; 3-methylene-2
; 4-chromandione
; methyl vinyl ether
; pseudocyclic selectivity
; Relative Interacting Atomic Energy
1. Introduction
Diels-Alder (DA) reactions between a diene and an ethylene derivative are one of the most studied organic reactions, enabling the construction of any six-membered carbocyclic compound [1-3]. Often, when the diene or the ethylene incorporates a heteroatom, the resulting reaction is called a hetero Diels-Alder (HDA) reaction [4]. This type of reaction is a powerful tool in synthetic organic chemistry as it can be used to construct chemo- regio- and stereoselective heterocycles [5-9]. Consequently, HDA reaction provides an effective method for the synthesis of a diverse array of hetero and polycyclic compounds [10]. The formed heterocycles are of significant biological importance and utility in a multitude of fields. This is evidenced by the fact that they constitute over half of the catalogue of known organic compounds [11].
The majority of naturally compounds, including those belonging to the classes of alkaloids, vitamins, hormones and antibiotics, contain heterocyclic moieties [12]. Coumarin and its derivatives are one of the most important classes of natural compounds widely distributed in the natural kingdom that exhibited various biological activities [13]. A number of processes for the synthesis of these heterocyclic molecules have also been developed [14], and used as precursors in many reactions such as cycloaddition [15].
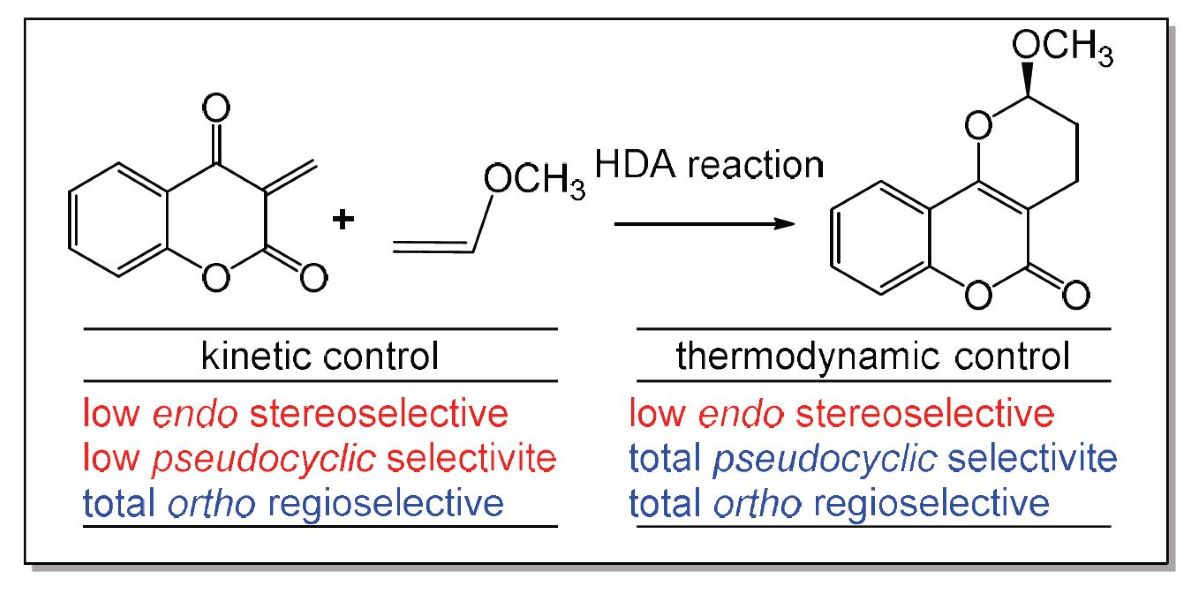
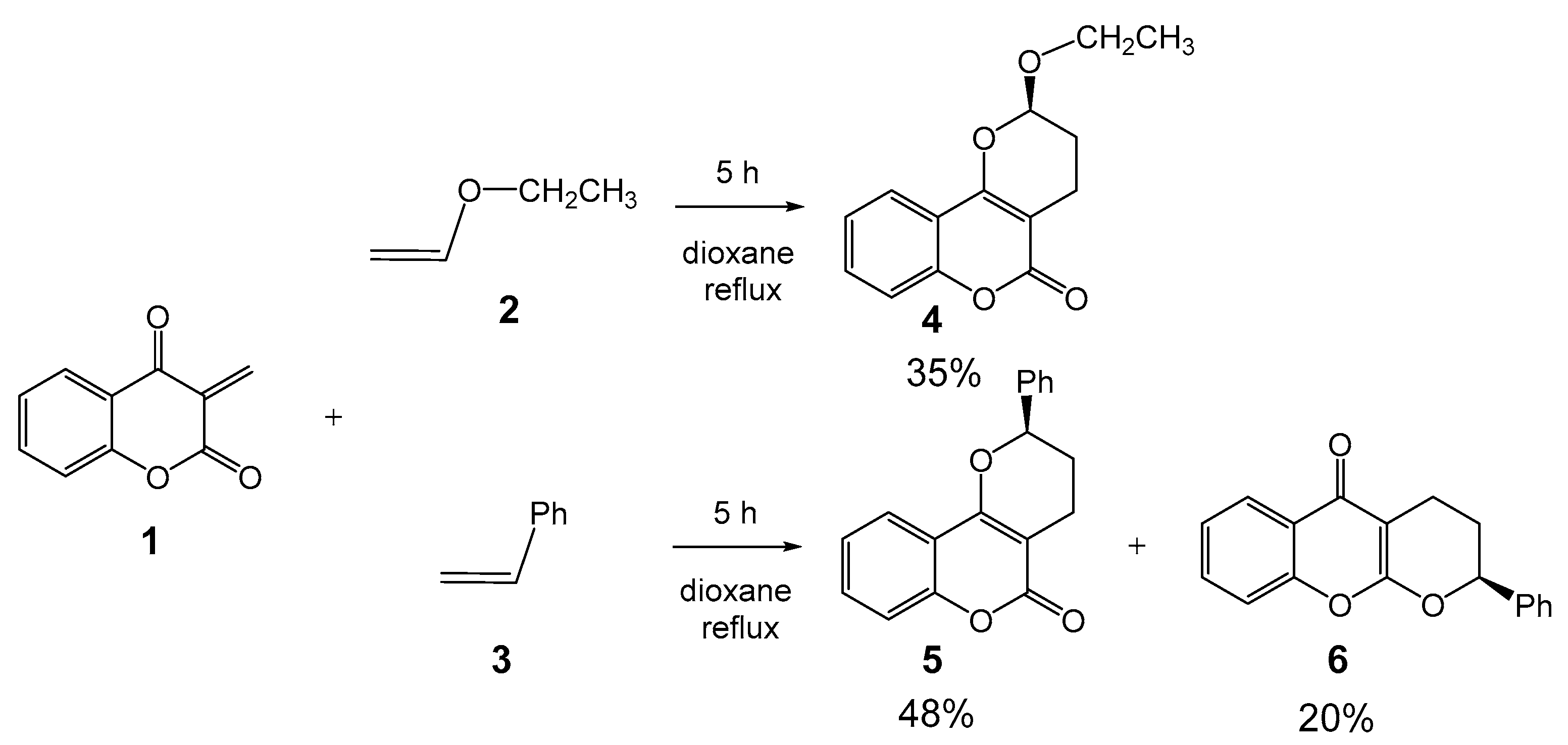
In 1994, Appendino et al. reported the DA trapping of 3-methylene-2,4-chromandione (MCDO) 1 with a series of ethylene derivatives (see Scheme 1) [16]. When a non-symmetric ethylene such as ethyl vinyl ether 2 was used, the corresponding HDA reaction took place yielding only the cycloadduct (CA) 4 in a yield of 35%; the use of styrene 3 yielded a mixture of two constitutional isomeric CAs 5 and 6 in a yield of 48% and 20%, respectively. In both cases, the HDA reactions took place with a total ortho regioselectivity [16].
A theoretical study of the HDA reaction of the model ambident compound 7 with ethylene 8 using AM1 and RHF calculations was performed (see Scheme 2) [16]. The activation enthalpy for the formation of the constitutional isomeric CA 10 was 4.10 kcal·mol-1 higher in energy than that associated to the formation of CA 9, 23.27 kcal·mol-1, in agreement with the selectivity experimentally observed in the HDA reaction with isoprene 11. In order to explain the suggested chemo- and regioselectivity of these HDA reactions, an AM1 analysis of the LUMO coefficients of MCDO 1 and the LUMO coefficients of the non-symmetric ethylenes as ethyl vinyl ether 2 was carried out.
After many theoretical studies of experimental DA reactions, a very good correlation between the activation energies of the DA reactions and the global electron density transfer [17] GEDT taking place at the transition state structures (TSs) was recognized in 2009, thus establishing the general mechanism of polar Diels-Alder (P-DA) reactions [18], which are controlled by the nucleophilic and electrophilic interactions taking place at the TSs. In this context, the analysis of the DFT-based reactivity indices [19,20] has become a powerful tool for studying polar cycloaddition reactions [21].
In 2016, Domingo proposed the Molecular Electron Density Theory [22] (MEDT) to study chemical organic reactivity. This theory states that the energy cost associated with the reorganization of the molecular electron density along a reaction path is what determines the chemical reactivity. In the MEDT studies a wide range of quantum chemical tools that allow the analysis of density functions, such as the DFT-based reactivity indices [19,20], the electron localization function [23] (ELF), the bonding evolution theory [24] (BET), the quantum theory of atoms in molecules [25,26] (QTAIM) and the non-covalent interaction [27] (NCI), are used. Very recently, an energy decomposition analysis based on the Interacting Quantum Atoms [28] (IQA), namely Relative Interacting Atomic Energy [29] (RIAE), has been proposed to analyze the factors controlling the activation energies; i.e., the relative energies of the TSs to respect the reference species, i.e. reagents or molecular complexes. Applying the RIAE analysis in three studies of polar DA reactions [29-31], it has been proved that the GEDT taking place from the nucleophile to the electrophile is the main electronic factor responsible for the reduction of the activation energies in polar reactions [30].
Topological analyses of the electron density of the TSs involved in polar DA reactions have shown that the formation of the new single bonds has not yet started at the TSs [33]. In 2016, the concept of pseudocyclic TSs was proposed to characterize those involved in DA reactions in which the six associated atomic centers are assembled in a more or less distorted six-membered cyclic rearrangement in which they are not necessarily bonded [34].
In 2018, Domingo et al. studied within MEDT the Lewis acid catalyzed HDA reaction between isoprene 11 and formaldehyde 12 [35]. Together with the expected CA 13, the reaction path yielding the constitutional isomeric Alder-ene adduct 14 was characterized (see Scheme 3). This MEDT study allowed establishing the similarity of the two diastereomeric TSs associated with the formation of the first C–C single bond along the two competitive reaction paths. In this work, the term pseudocyclic selectivity was suggested to connote the selective formation of a constitutional isomer through diastereomeric pseudocyclic TSs [35].
In the present manuscript, a MEDT study of the HDA reaction of MCDO 1 with methyl vinyl ether 15 (MVE), as a model of the experimental ethyl vinyl ether used by Appendino et al. [16] (see Scheme 1), is carried out in order to establish the molecular mechanism of these HDA reactions as well as the origin of the selectivity experimentally observed.
2. Results and Discussion
This MEDT study is divided into five parts: i) first, an ELF topological analysis of MCDO 1 with MVE 15 in their ground state was carried out; ii) next, an analysis of the DFT-based reactivity indices was performed to characterize the reactivity of these species participating on polar HDA reactions, as well as to characterize the specific active sites of each reagent; iii) then, the competitive reaction paths associated with the HDA reaction between MCDO 1 and MVE 15 were studied; iv) the electronic structure of the more favorable TSs associated with for the formation of the CA as 4 and its possible constitutional isomeric CA was characterized by topological ELF and QTAIM analysis in the forty part; and finally, v) the fifth part presents a RIAE analysis of the HDA reactions of MCDO 1 with MVE 15 and with ethylene 8 was performed in order to characterize the electronic factor controlling the activation energies of the HDA reaction.
2.1. ELF Topological Analysis of the ELF of MCDO 1 and MVE 15
Topological analysis of the ELF [23] is a valuable method to study the distribution of electron density in a molecule [36], thereby facilitating the prediction of its chemical reactivity. Thus, an ELF topological analysis of the electronic structures of MCDO 1 and MVE 15 was performed firstly. ELF basin attractor positions, together with the most relevant valence basin populations, are shown in Figure 1.
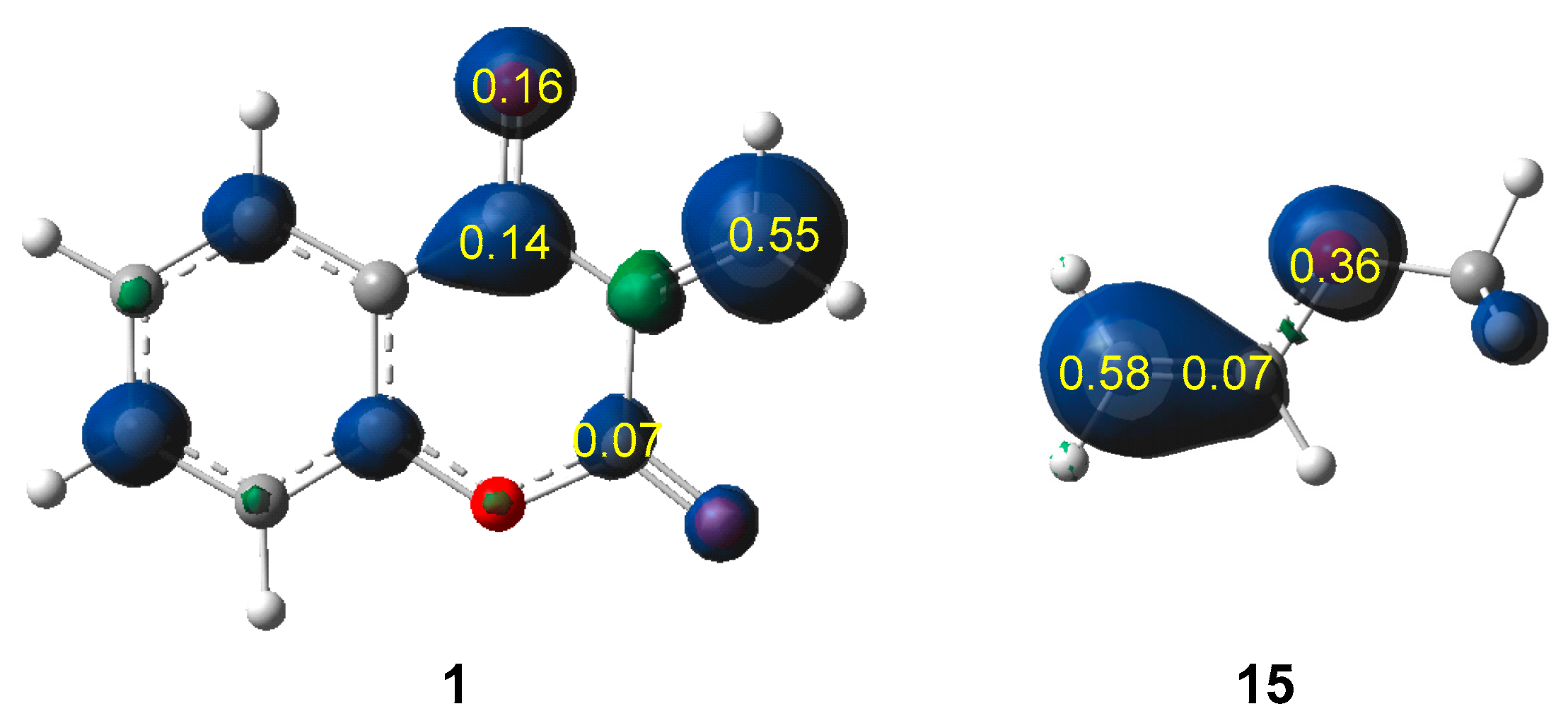
ELF of MCDO 1 shows the presence of two disynaptic basin, V(C1,C2) and V’(C1,C2), integrating a total of 3.28 e, associated with a depopulated C1−C2 double bond, one V(C3,O4) disynaptic basin, integrating 2.21 e, characterizing the C3−O4 bonding region as a single bond, two monosynaptic basins, V(O4) and V’(O4), integrating a total population of 5.92 e, associated with the non-bonding electron density at the O4 oxygen atom, one V(C5,O6) disynaptic basin, integrating 2.49 e, characterizing the C5−O6 bonding region as a single bond, and two monosynaptic basins, V(O6) and V’(O6), integrating a total of 5.24 e, associated with the non-bonding electron density at the O6 oxygen atom. On the other hand, the ELF of MVE 15 reveals the presence of two disynaptic basins, V(C8,C9) and V’(C8,C9), integrating a total of 3.60 e, associated with a depopulated C8−C9 double bond, and one V(C9,O10) disynaptic basin integrating 1.50 e associated with a depopulated C9−O10 single bond.
The Lewis-like structures of MCDO 1 and MVE 15, together with their natural charges [37,38], are shown in Figure 2. The natural charges of MCDO 1 indicate that the olefinic C1 and C2 carbons are charged negatively by −0.16 and −0.27 e, respectively, while carbonyl C3 and C5 carbons are strongly positively charged by 0.53 and 0.81 e due to strong polarization of the carbonyl C−O bonding regions under the effect of the electronegative O4 and O6 oxygen atoms, which are negatively charged by −0.55 e. For the MVE 15, the C8 carbon is negative charged by −0.48 e while the C9 carbon is positive charge by 0.15 e, indicating the strong charge polarization of the C8−C9 single bond.
2.2. Analysis of the DFT-Based Reactivity Indices in the Ground State of the Reagents
The analysis of the DFT-based reactivity indices [19,20] are powerful tools to understand the reactivity in polar cycloaddition reactions [21]. The reactivity indices were calculated at the B3LYP/6-31G(d) computational level since it was used to establish the electrophilicity and nucleophilicity scales [20]. The global reactivity indices, namely, the electronic chemical potential μ, chemical hardness η, global electrophilicity ω, and global nucleophilicity N of MCDO 1 and MVE 15 are given in Table 1.
The electronic chemical potential [39] μ of MCDO 1, μ = −4.88 eV is positioned below to that of MVE 15, μ = −2. 34 eV. The high energy difference between these values indicate that this HDA reaction will have a high polar character, the GEDT [17] taking place from MVE 15 towards MCDO 1.
On one hand, MCDO 1 has an electrophilicity ω index [41] of 2.87 eV, being classified as a strong electrophile within the electrophilicity scale [20], and a nucleophilicity N index[42] of 2.16 eV, being classified as a poor nucleophile within the nucleophilicity scale [20]. On the other hand, MVE 15 as an electrophilicity ω index of 0.42 eV, being classified as a marginal electrophile, and a nucleophilicity N index of 3.20 eV, being classified as a strong nucleophile.
The strong electrophilic character of MCDO 1 combined with the strong nucleophilic character of MVE 15 means that the corresponding HDA reaction will be remarkably polar character, being characterized as forward electron density flux (FEDF) [40].
Ethylene 8 characterized with a ω = 0.73 eV and a N = 1.86 eV is classified as a marginal electrophile and a marginal nucleophile. Consequently, it is expected that ethylene 8 will have a poor tendency to participate in polar cycloaddition reactions [17].
In a polar cycloaddition reaction involving non-symmetric species, the most favorable reaction path takes place at the two-center interaction between the more electrophilic center and the more nucleophilic center of the two interacting reagents [43]. Numerous studies have indicated that the analysis of the Parr functions [44], derived from the excess spin electron density collected by the GEDT, is one of the most effective means of analyzing local reactivity in polar and ionic processes. Consequently, the electrophilic Parr functions of MCDO 1 and the nucleophilic Parr functions of MVE 15 were studied (see Figure 2).
Figure 3.
3D Representations of the Mulliken atomic spin densities of the radical anion of MCDO 1 and of the radical cation of MVE 15, together with the electrophilic Parr functions of MCDO 1 and the nucleophilic functions of MVE 15.
Figure 3.
3D Representations of the Mulliken atomic spin densities of the radical anion of MCDO 1 and of the radical cation of MVE 15, together with the electrophilic Parr functions of MCDO 1 and the nucleophilic functions of MVE 15.
The analysis of the electrophilic Parr functions of the electrophilic MCDO 1 shows that the C1 carbon, = 0.55, is the more electrophilic of this molecule. It is noticed that this carbon has concentrated the 55% of the electrophilicity of this molecule. Similarly, the analysis of the nucleophilic Parr functions of MVE 15 indicates that the C8 carbon, = 0.58, is the more nucleophilic center of this molecule
Therefore, the analysis of the Parr functions predicts that the most likely electrophilic / nucleophilic interaction along the nucleophilic attack of MVE 15 to MCDO 1 will be between the C8 of the former and the C1 carbon of the latter, providing the explanation of the ortho regioselectivity observed in DA reactions [45].
It is worth emphasizing that the most electrophilic center of MCDO 1 is the C1 carbon, which is negatively charged, −0.16 e, while the two carbonyl C3 and C4 carbons are positively charged (see Figure 1). This finding supports the Domingo’s proposal made in 2014 where the local electrophilic/nucleophilic behaviors of organic molecules are not simply caused by charges [46]. The most electrophilic center of a molecule is the one that accepts the highest amount of electron density resulting from the nucleophilic/electrophilic interactions taking place along the approach of the two reagents [46].
2.3. Analysis of Competitive Reaction Paths Associated with the HDA Reaction of MCDO 1 with MVE 15
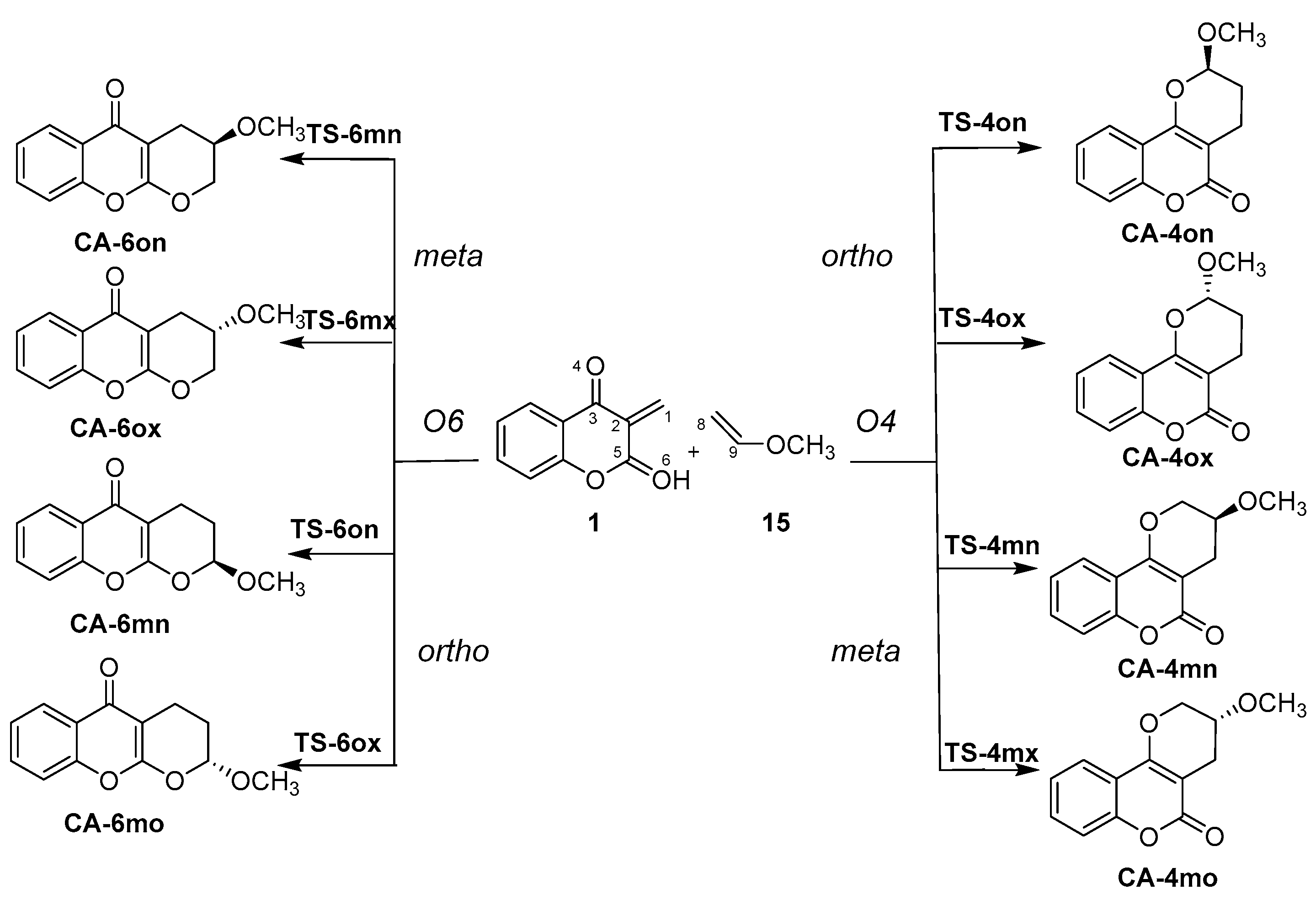
Due to the non-symmetric character of the two reagents and the presence of two heterodiene frameworks in MCDO 1, eight competitive reaction paths are feasible along the nucleophilic attack of MVE 15 on the C1 carbon of MCDO 1 (see Scheme 3). They are related to the pseudocyclic selectivity [35] associated with the participation of the carbonyl O4 or O6 oxygen atom of the heterodiene, named as O4 and O6, respectively, the regioisomeric approach mode of the C8−C9 double bond of MVE 15 to the C1−C2 double bond of MCDO 1, named as ortho and meta, and the stereoisomeric approach mode of the ether O19 oxygen atom of MVE 15 with respect to the heterodiene system, named as endo and exo. The eight competitive reaction paths were analyzed (see Scheme 3). The analysis of the stationary points located along the eight competitive reaction paths indicates that this HDA reaction proceeds according to a non-concerted one-step mechanism. Table 2 shows the B3LYP/6-3111G(d,p) relative electronic energies in the gas phase and in dioxane. Table S1 in the Supplementary Materials presents the corresponding total electronic energies.
Scheme 2.
The eight competitive reaction paths associated with the HDA reaction of MCDO 1 with MVE 15.
Scheme 2.
The eight competitive reaction paths associated with the HDA reaction of MCDO 1 with MVE 15.
Some appealing conclusion can be obtained from the gas phase relative energies given in Table 2: i) the activation energies associated to the eight competitive reaction paths vary from 4.21 kcal·mol-1 (TS-4on) to 25.69 kcal·mol-1 (TS-6mx), while the exothermic character of these reaction paths vary from −18.46 kcal·mol-1 (CA-6mx) to −35.39 kcal·mol-1 (CA-4ox); ii) the activation energy associated to this HDA reaction is 13.05 kcal·mol-1 which is lower in energy than that of the HDA reaction of MCDO 1 with ethylene 8 as a consequence of the strong nucleophilic character od MVE 15 (see Scheme S1 in the Supplementary Material); iii) the most favorable TS-4on is associated to the orto/endo approach mode of MVE 15 to the C1−C2−C3−O4 heterodiene system MCDO 1; iv) this HDA reaction is endo stereoselective as TS-4ox is located at 0.67 kcal·mol-1 above TS-4on; v) this HDA reaction is low pseudocyclic selective as TS-6on is located only at 0.05 kcal·mol-1 above TS-4on; v) this HDA reaction is completely ortho regioselective as TS-4mn is located at 15.55 kcal·mol-1 above TS-4on [45]; vi) formation of CA-4on along the most favorable reaction path is strongly exothermic by −34.12 kcal·mol-1. Consequently, formation of CA-4on can be considered irreversible (see later). Inclusion of solvent effect of dioxane do not produces remarkable changes on the gas phase energies (see Table 3).
The relative Gibbs free energies of the stationary points associated the more favorable regioisomeric ortho reaction paths associated with HDA reaction of MCDO 1 with MVE 15 were computed at 101.1 ºC in 1,4-dioxane. The high activation energy of the meta TSs, more than 15.55 (TS-4mn) kcal·mol-1, discards the thermodynamic study of these non-competitive reaction paths. The thermodynamic data are given in Table Sx in the Supplementary Material, while the corresponding Gibbs free energy profiles are given in Figure 4.
The inclusion of the thermal correction, the entropies and the temperature rise the activation Gibbs free energies of the most favorable reactions path via TS-4on to 20.93 kcal·mol-1. The reaction is endo stereoselective as TS-4ox is 0.85 kcal·mol-1 higher in Gibbs free energy than TS-4on, and very low O6 pseudocyclic selective as TS-6ox is only 0.01 kcal·mol-1 higher in Gibbs free energy than TS-4on. Interestingly, at this computational level, while O4 reaction paths are exergonic by more than 11.4 kcal·mol-1, while the O6 reaction paths are exergonic by only 3.7 kcal·mol-1. Consequently, in dioxane under reflux condition, the O6/ortho/endo reaction path can be reversible, yielding the thermodynamically more stable O4/ortho/endo CA-4on (see 8 in Scheme 1).
The geometries of the most favourable regioisomeric ortho TSs associated with HDA reaction of MCDO 1 with MVE 15 are given in Figure 5, while the geometries of the regioisomeric meta TSs are given in Figure S3 in the Supplementary Material. Regarding the four TSs, the distance between the two interacting carbons involving the most electrophilic C1 carbon of MCDO 1 and the most nucleophilic C8 carbon of MVE 15 are found in the narrow range of 1.97 – 2.05 Å, while the distances between the C9 carbon of MVE 15 and the O4 and O6 oxygen atoms of MCDO 1 are found between 2.8 - 3.2 Å. These geometrical parameters indicate that this HDA reaction takes place through very high asynchronous TSs. The IRC analysis of the most favourable TS-4on indicates that this HDA reaction takes place trough non-concerted one-step two-stage mechanism [47] in which the formation of the second O4−C9 single bond begin when the formation of the C1−C8 is practically completed. The addition of dioxane as solvent does not produce remarkable changes on the geometries (see Figure 5). In fact, in the presence of dioxane, the TSs are slightly more delayed.
Figure 6 displays a representation of the stereoisomeric TS-4on and TS-6ox along the axis formed by the C1 and C8 interacting carbons. Similar to the endo and exo TSs, these TSs have a diastereomeric relationship. After passing these stereoisometic TSs, and once the formation of the first new C1−C8 single bond is fully completed, the formation of the second O4−C9 or C6−C19 single bond occurs at the end of the corresponding reaction path, permitting the formation of the constitutional isomers CA-4on and CA-6ox.
The GEDT analysis [17] at the TSs involved in this HDA reaction permits quantifying the polar character of this reaction [18]. In polar reactions, the GEDT happening at the TSs, which changes from the neutrophilic towards the electrophilic species, is the main factor responsible for the stabilization of the TSs [30]. GEDT values lower than 0.05 e are in correspondence with non-polar processes, while values higher than 0.20 e correspond to polar processes. The GEDT values at the two more favourable ortho/endo TSs are 0.38 e at TS-4on and 0.39 e at TS-6on. These uppermost values illustrate the highly polar character of these HDA reactions, and consequently of the lowest activation energies associated with this TSs.
The flux of the electron density at these polar TSs, which goes form strong nucleophilic MVE 15 towards the strong electrophilic MCDO 1, classifies this HDA reaction as FEDF [40], which is in complete agreement with the previous analysis of the DFT-based reactivity indices.
2.4. ELF and QTAIM Analyses of the of the Structure Electronic of the Stereoisomeric TS-4on and TS-6on
With the aim of understanding the similitude of the diastereomeric ortho/endo TS-4on and TS-6on, an ELF [23] and QTAIM [25,26] topological analysis of the two TSs were performed. The ELF basin attractor positions, together with the most relevant valence basin populations at the two TSs are shown in Figure 7.
The analysis of the ELF in both TSs displays a great similarity between them. In fact, they show the presence of two monosynaptic basins, V(C2) and V(C8), integrating ca. 0.29 and 0.65 e, respectively; one V(C1,C2) disynaptic basin, integrating ca. 2.53 e, and one V(C8,C9) disynaptic basin, integrating ca. 2.83 e. The two V(C2) and V(C8) monosynaptic basins are associated with the two C2 and C8 pseudoradical centers [48] created along the reaction paths. The C8 pseudoradical center is required for the subsequent creation of the first new C1−C2 single bond in a further step of the reaction path [17]. The electron density of these C2 and C8 pseudoradical centers comes from the depopulation of the C1−C2 and C8−C9 bonding regions at MCDO 1 and MVE 15, respectively.
Next, a QTAIM analysis of the electronic structure of TS-4on and TS-6on was performed. The positions of the (3, +1) and (3, −1) critical points (CPs) in both TSs are represented in Figure 8, while the calculated QTAIM parameters of the (3, −1) CPs characterizing the C1−C8 and C9−O regions are given in Table 4.
A comparative QTAIM analysis of the C1−C8 and C9−O interacting regions in TS-4on and TS-6on shows a high degree of electronic similarity between the two TSs (see Figure 8). The three selected CP, CP1-CP3, show a positive Laplacian ∇2ρ(r) value indicating the absence of any covalent interaction (see Table 4). TS-4on does not have any CP in the C9−O4 interacting region due to the large distance between these centers, while TS-6on shows the presence of a CP3 but with a very low electron density of 0.0078 e, not having any chemical meaning.
Consequently, both ELF and QTAIM topological analysis of the electron density of the diastereomeric TS-4on and TS-6on shows a high degree of electronic similarity between them. This behaviour is a consequence of the fact that the two TS are only associated with the formation of the C1−C8 single bond. These finding accounts for the closer total electronic energy calculated between the two TSs, explaining the low pseudocyclic selectivity [35] of this HDA reaction under kinetic control.
2.5. RIAE Analysis of the HDA Reactions of MCDO 1 with MVE 15 and with Ethylene 8.
Finally, to define the electronic effects responsible for the activation energies of the polar HDA reaction of MCDO 1 with MVE 15, as well as the low pseudocyclic selectivity under kinetic control of this HDA reaction, a RIAE analysis [29] on the most favourable gas phase TS-4on and TS-6on was performed. The HDA reaction of MCDO 1 with ethylene 8 as the reaction reference is also studied (see Section 1 in the Material Supplementary). The B3LYP/6-311G(d,p) ξ total, ξ intra-atomic and ξ interatomic energies of the MCDO and the MVE and ethylene (Et) frameworks of the TSs with respect to the separated reagents are given in Table 5.
RIAE analysis of TS-4 and TS-6 associated with the HDA reaction of MCDO 1 with ethylene 8 shows a close similarity between the two diastereomeric TSs. The ξ energies analysis of the two interacting frameworks at the most favourable TS-4 shows that while the Et framework is destabilized, ξ = 41.06 kcal·mol-1, the electrophilic MCDO one is stabilized, ξ = −23.87 kcal·mol-1. The ξ intra-atomic and ξ interatomic energies analysis at the two frameworks indicates that the intramolecular interactions at the electrophilic MCDO framework is the most stabilizing factor, ξ= −83.78 kcal·mol-1, while those at the Et framework are destabilizing, ξ= 44.92 kcal·mol-1. The ξ intramolecular energies analysis of the ethylene framework at TS-6, which are 2.33 kcal·mol-1 more energetic than those at TS-4, shows that these intramolecular energies are the main factor responsible for the pseudocyclic selectivity of this HDA reaction. Note that the difference between the two RIAE activation energies is 1.83 kcal·mol-1.
RIAE analysis of TS-4on and TS-6on associated with the HDA reaction of MCDO 1 with MVE 15 newly shows a great similarity between the two diastereomeric TSs (see Table 5), in complete agreement with the previous topological analysis of their electronic structure.
The ξ energies analysis of the two frameworks at the most favorable TS-4on shows that while the nucleophilic MVE framework is destabilized, ξ = 41.56 kcal·mol-1, the electrophilic MCDO framework is stabilized, ξ = −37.60 kcal·mol-1. Interestingly, while the ξ and ξ total energies in the HDA reactions with ethylene 8 and MVE 15 are very similar, the ξ total energy in the reaction involving MVE 15 is ca. 14 kcal·mol-1 more negative than that in the reaction involving ethylene 8. This behaviour is a consequence of the higher GEDT happening at TS-4on, 0.38 e, that at the TS-4, 0.23 e (see Section 1 in the Supplementary Material).
The ξ intra-atomic and ξ interatomic energies analysis at the two frameworks indicates that the intramolecular interactions at the electrophilic MCDO framework is the more stabilizing factor, ξ= −66.20 kcal·mol-1, while the intramolecular interactions at the MVE framework are destabilizing, ξ= 71.48 kcal·mol-1. Although the ξ intramolecular energies in the reaction involving MVE 15 are lesser stabilizing than those in the reaction involving ethylene 8, the unfavorable ξ inter-molecular energies in the reaction with MVE 15 are half that those in the reaction with ethylene 8. These behaviours are responsible of the strong reduction of the activation energies of the polar HDA reaction involving the nucleophilic MVE 15 with respect to that of ethylene 8.
A similar result is obtained in the RIAE analysis of the stereoisomeric TS-6on. The very low difference of the RIAE activation energies associated with TS-4on and TS-6on, ΔΔEact = 0.06 kcal·mol-1, accounts for the very low pseudocyclic selectivity of this HDA reaction under kinetic control.
Finally, Figure 9 shows a graphical representation of the ξ energies of the MCDO framework and the ethylene and MVE frameworks at the two pairs of diasteromeric TSs, in blue and red, respectively. The ξ energies, shown in black, correspond to the RIAE activation energies of these DA reactions. As can be see, the stabilization of the MCDO framework with the increase of GEDT is the main factor responsible for the reduction of the RIAE activation energies; note that at the four TSs the ξ is ca. 40 kcal·mol-1.
The ξ total energies associated to the MCDO framework of TS-6 and TS-6on are more stabilized than those at TS-4 and TS-4on as a consequence of the slightly more polar character of the former, but the ξ total energies associated to the Et and MVE frameworks of the former TSs are more destabilized. As a consequence, these HDA reactions as some O4 pseudocyclic are selective under kinetic control (see Scheme 1).
3. Conclusion
The HDA reaction between MCDO 1 and the non-symmetric MVE 15, experimentally studied by Appendino et al. [16], has been investigated within MEDT at the B3LYP/6-311(d,p) computational level. DFT-based reactivity indices analysis of the two reagents indicates that HDA reaction, classified as REDF, will have a high polar character as a consequence of the strong electrophilic character MCDO 1 and the strong nucleophilic character of MVE 15. The electrophilic Parr function analysis indicates that the methylene C1 carbon of MCDO 1 concentrates the 55% of the electrophilic character of this molecule, justifying the total ortho regioselectivity experimentally observed.
The eight competitive reaction path analysis indicates that this HDA reaction is totally ortho regioselective, endo selective, and very low pseudocyclic selective. Due to the high polar character of this HDA reaction, it presents a very low activation energy of only 4.21 kcal·mol-1. The relative Gibb free energies analysis involved in the more favourable ortho reaction paths indicate that the complete pseudocyclic selectivity experimentally observed can be only explained by a thermodynamic control yielding the thermodynamically most stable CA 4 in Scheme 1.
The geometrical parameter analysis of the four ortho TSs shows the similitude between them, having a diastereomeric relationship. These TSs are characterized by a highly asynchronous single bond formation process associated with a two-center interaction between the most nucleophilic C8 carbon of MVE 15 and the most electrophilic C1 carbon of MCDO 1.
ELF and QTAIM topological analyses of the electronic structure of the diastereomeric TS-4on and TS-6on show the high electron density similarity between them. Finally, a RIAE analysis of these TSs indicates that the stabilization of the electrophilic MCDO at these polar TSs, resulting of the high GEDT, is the main factor responsible for the acceleration found in this HDA reaction. A comparative RIAE analysis of the two diastereomeric TS-4on and TS-6on shows the great similarity between them, accounting for a low pseudocyclic selectivity under kinetic control.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Study of the HDA reaction between of MCDO 1 and ethylene 8. Computational Details. References. Figure with the geometries of the regioisomeric meta TSs associated with HDA reaction of MCDO 1 with MVE 15. Table with total electronic energies of the stationary points associated with HDA reaction between MCDO 1 and ethylene 8. Table with the total electronic energies, in gas phase and in dioxane, of the stationary points associated with HDA reaction between MCDO 1 and MVK 15. Table with the thermodynamic data of the stationary points associated with the ortho reaction paths of the HDA reaction between MCDO 1 and MVK 15.
Author Contributions
A.B. and A.M. co-wrote the main manuscript and carried out the calculations; L.B. revised and edited the manuscript; L.B. and L.R.D. co-wrote the main manuscript and analyzed the results and supervised the final version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors are thankful to the Sultan Moulay Slimane University, Morocco.
References
- Diels, O.; Alder, K. Synthesen in der hydroaromatischen Reihe. Justus Liebigs Ann. Chem. 1928, 460, 98–122. [Google Scholar] [CrossRef]
- Carruthers, W. Some Modern Methods of Organic Synthesis, 2nd ed.; Cambridge University Press: Cambridge, UK, 1978. [Google Scholar]
- Carruthers, W. Cycloaddition Reactions in Organic Synthesis; Pergamon: Oxford, UK, 1990. [Google Scholar]
- Tietze, L.F.; Kettschau, G. In Stereoselective Heterocyclic Synthesis I. (Eds.: Metz, P.), Springer-Verlag Berlin Heidelberg, 1997, 2, 1–120.
- Waldmann, H. Asymmetric Hetero Diels-Alder Reactions. Synthesis 1994, 535–551. [Google Scholar] [CrossRef]
- Schaus, S.E.; Brånalt, J.; Jacobsen, E.N. Asymmetric hetero-Diels-Alder reactions catalyzed by chiral (salen)chromium(III) complexes. J. Org. Chem. 1998, 63, 403–405. [Google Scholar] [CrossRef]
- Maruoka, K.; Itoh, T.; Shirasaka, T.; Yamamoto, H. Asymmetric Hetero-Diels-Alder Reaction Catalyzed by Organo-Aluminum Reagents. J. Am. Chem. Soc. 1988, 110, 310–312. [Google Scholar] [CrossRef]
- Juhl, K.; Jorgensen, K.A. The first organocatalytic enantioselective inverse-electron-demand hetero-Diels-Alder reaction. Angew. Chem., Int. Ed. Engl. 2003, 42, 1498–1501. [Google Scholar] [CrossRef]
- Oehlenschlaeger, K.K.; Mueller, J.O.; Brandt, J.; Hilf, S.; Lederer, A.; Wilhelm, M.; Graf, R.; Coote, M.L.; Schmidt, F.G.; Barner-Kowollik, C. Adaptable Hetero Diels-Alder Networks for Fast Self-Healing under Mild Conditions. Adv. Mater. 2014, 26, 3561–3566. [Google Scholar] [CrossRef]
- Lozynskyi, A.; Zimenkovsky, B.; Karkhut, A.; Polovkovych, S.; Gzella, A.K.; Lesyk, R. Application of the 2(5H)furanone Motif in the Synthesis of New Thiopyrano[2,3-d]thiazoles via the Hetero-Diels-Alder reaction and Related Tandem Processes. Tetrahedron Lett. 2016, 57, 3318–3321. [Google Scholar] [CrossRef]
- Hossain, M. A Review on Heterocyclic: Synthesis and Their Application in Medicinal Chemistry of Imidazole Moiety. Sci. J. Chem. 2018, 6, 83. [Google Scholar] [CrossRef]
- Shukla, P.K.; Verma, A.; Mishra, P. Significance of Nitrogen Heterocyclic Nuclei in the Search of Pharmacologically Active Compounds. New Perspective. Agric. Hum. Med. 2017, 100–126. [Google Scholar]
- Annunziata, F.; Pinna, C.; Dallavalle, S.; Tamborini, L.; Pinto, A. An Overview of Coumarin as a Versatile and Readily Accessible Scaffold with Broad-Ranging Biological Activities. Int. J. Mol. Sci. 2020, 21, 1–83. [Google Scholar] [CrossRef]
- Bouhaoui, A.; Eddahmi, M.; Dib, M.; Khouili, M.; Aires, A.; Catto, M.; Bouissane, L. Synthesis and Biological Properties of Coumarin Derivatives. A Review. ChemistrySelect 2021, 6, 5848–5870. [Google Scholar] [CrossRef]
- Soliman, A.Y.; Mohamed, F.K.; Abdel-Motaleb, R.M.; Abdel-Rahman, R.M.; Abdel-Mohsen, A.M.; Fouda, M.M.G.; Mohamed, A.S. Synthesis of New Coumarin Derivatives Using Diels-Alder Reaction. Life Sci. J. 2013, 10, 846–850. [Google Scholar]
- Appendino, G.; Cravotto, G.; Toma, L.; Annunziata, R.; Palmisano, G. The Chemistry of Coumarin Derivatives. Part VI. Diels-Alder Trapping of 3-Methylene-2,4-chromandione. A New Entry to Substituted Pyrano[3,2-c]coumarins. J. Org. Chem. 1994, 59, 5556–5564. [Google Scholar] [CrossRef]
- Domingo, L.R. A new C-C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A. Understanding the mechanism of polar Diels-Alder reactions. Org. Biomol. Chem. 2009, 7, 3576–3583. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Yang, W. Density functional theory of atoms and molecules. Oxford University Press, New York, 1989.
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the conceptual density functional indices to organic chemistry reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M. In Application of Reactivity Indices in the Study of Polar Diels–Alder Reactions. Conceptual Density Functional Theory: Towards a New Chemical Reactivity Theory, Ed. Shubin Liu. WILEY-VCH GmbH. 2022, Vol. 2, pp. 481–502.
- Domingo, L.R. Molecular Electron Density Theory: A Modern View of Reactivity in Organic Chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization n atomic and molecular-systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Krokidis, X.; Noury, S.; Silvi, B. Characterization of elementary chemical processes by catastrophe theory J. Phys. Chem. A 1997, 101, 7277–7282. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Tang, Y.H.; Tal, Y.; Biegler-König, F.W. Properties of atoms and bonds in hydrocarbon molecules. J. Am. Chem. Soc. 1982, 104, 946–952. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory. Oxford University Press, Oxford, New York, 1994.
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.A.; Martín Pendás, A.; Francisco, E. Interacting Quantum Atoms: A Correlated Energy Decomposition Scheme Based on the Quantum Theory of Atoms in Molecules. J. Chem. Theory Comput. 2005, 1, 1096–1109. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. An Interacting Quantum Atoms Analysis of the Electronic Effects of Lewis Acid Catalysts in Accelerating Polar Diels-Alder Reactions. J. Org. Chem. 2024, 89, 12349–12359. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Pérez, P.; Ríos-Gutiérrez, M.; Aurell, M.J. A Molecular Electron Density Theory Study of Hydrogen Bond Catalysed Polar Diels–Alder Reactions of α,β-unsaturated Carbonyl Compounds. Tetrahedron Chem. 2024, 10, 100064. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M. Revealing the Decisive Role of Global Electron Density Transfer in the Reaction Rate of Polar Organic Reactions within Molecular Electron Density Theory. Molecules 2024, 29, 1870. [Google Scholar] [CrossRef]
- Domingo, L.R. 1999 – 2024, a Quarter Century of the Parr’s Electrophilicity ω Index. Sci. Rad. 2024, 3, 157–186. [Google Scholar] [CrossRef]
- Berski, S.; Andrés, J.; Silvi, B.; Domingo, L.R. New Finding on the Diels-Alder Reactions. An Analysis Based on the Bonding Evolution Theory. J. Phys. Chem. A 2006, 110, 13939–13947. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. A Molecular Electron Density Theory Study of the Competitiveness of Polar Diels-Alder and Polar Alder Ene Reactions. Molecules 2018, 23, 1913. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Chamorro, E.; Pérez, P. Aromaticity in Pericyclic Transition State Structures? A Critical Rationalisation Based on the Topological Analysis of Electron Density. ChemistrySelect 2016, 1, 6026–6039. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions Nature 1994, 371, 683–686.
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M. A Useful Classification of Organic Reactions Bases on the Flux of the Electron Density. Sci. Rad. 2023, 2, 1. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpaly, L.v.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef]
- Aurell, M.J.; Domingo, L.R.; Pérez, P.; Contreras, R. A theoretical study on the regioselectivity of 1,3-dipolar cycloadditions using DFT-based reactivity indexes. Tetrahedron 2004, 60, 11503–11509. [Google Scholar] [CrossRef]
- Domingo, L. R.; Pérez, P.; Sáez, J. A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P.; Sáez, J.A. Understanding the regioselectivity in hetero Diels-Alder reactions. An ELF analysis of the reaction between nitrosoethylene and 1-vinylpyrrolidine. Tetrahedron 2013, 69, 107–114. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Pérez, P.; Sáez, J.A. Understanding the Origin of the Asynchronicity in Bond-Formation in Polar Cycloaddition Reactions. A DFT Study of the 1,3-Dipolar Cycloaddition Reaction of Carbonyl Ylides with 1,2-Benzoquinones. RSC Adv. 2012, 2, 1334–1342. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A. , Zaragozà, J.R.; Arnó, M. Understanding the Participation of Quadricyclane as Nucleophile in Polar [2σ+2σ+2π] Cycloadditions toward Electrophilic π Molecules. J. Org. Chem. 2008, 73, 8791–8799. [Google Scholar] [CrossRef]
Scheme 1.
HDA Reactions of MCDO 1 with olefins 2-3.
Scheme 2.
HDA Reactions of the ambident compound 7 with ethylene 8.
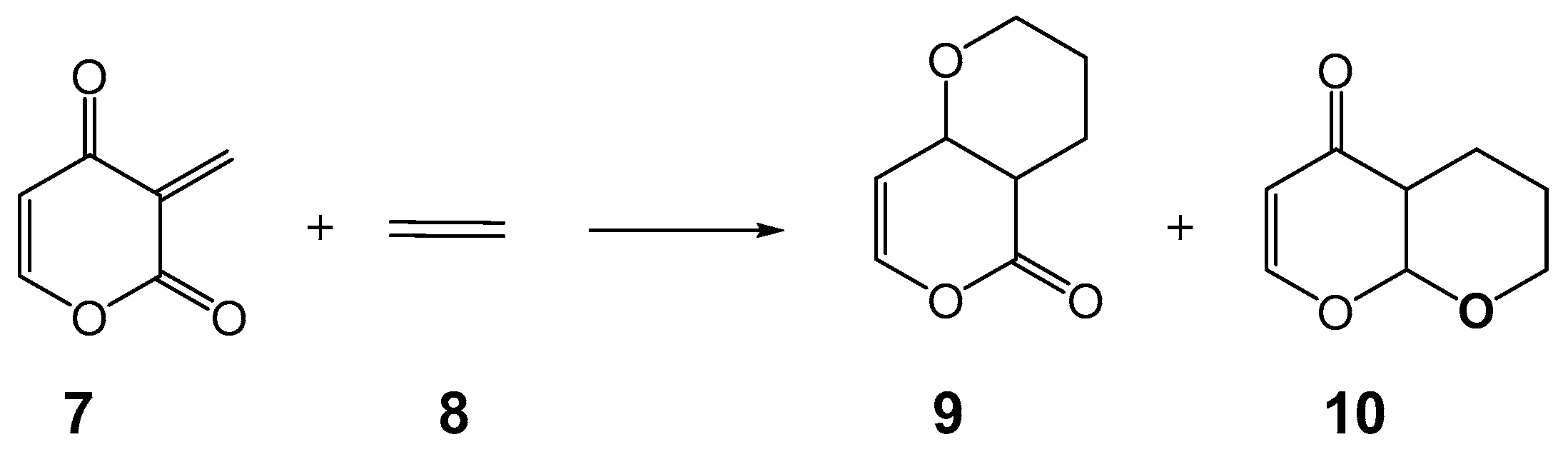
Scheme 3.
Competitive HDA and Alder-ene reactions in the Lewis acid catalyzed reaction between isoprene 11 and formaldehyde 12.
Scheme 3.
Competitive HDA and Alder-ene reactions in the Lewis acid catalyzed reaction between isoprene 11 and formaldehyde 12.
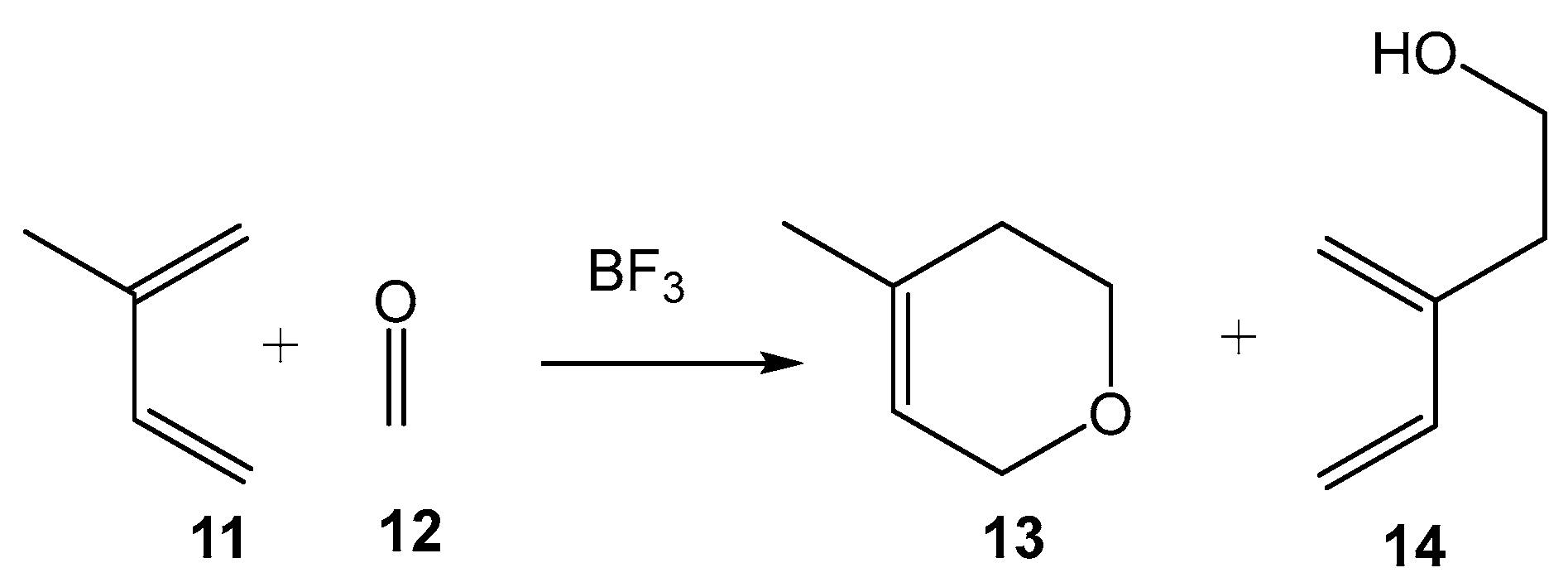
Figure 2.
B3LYP/6-311G(d,p) ELF Basin attractor positions, populations of the most relevant valence basins, and natural atomic charges of MCDO 1 and MVE 15. Valence basin populations and natural atomic charges are given in average number of electrons, e. Negative charges are colored in red, and positive charges in blue.
Figure 2.
B3LYP/6-311G(d,p) ELF Basin attractor positions, populations of the most relevant valence basins, and natural atomic charges of MCDO 1 and MVE 15. Valence basin populations and natural atomic charges are given in average number of electrons, e. Negative charges are colored in red, and positive charges in blue.
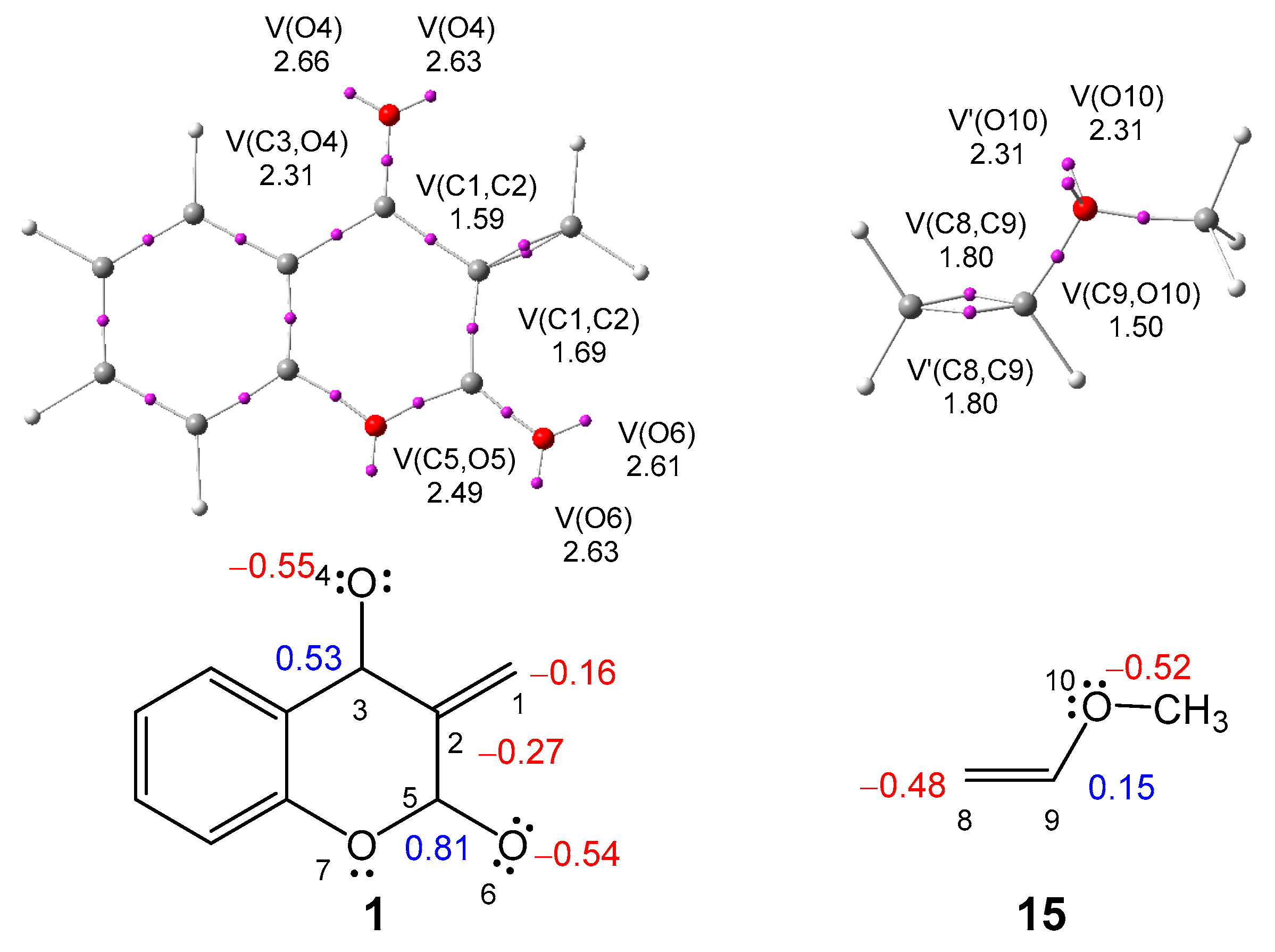
Figure 4.
B3LYP/6-311G(d,p) Gibbs free energy profiles, ΔG in kcal·mol-1, computed at 101.1 °C in dioxane, for the more favorable regioisomeric ortho reaction paths associated with HDA reaction of MCDO 1 with MVE 15.
Figure 4.
B3LYP/6-311G(d,p) Gibbs free energy profiles, ΔG in kcal·mol-1, computed at 101.1 °C in dioxane, for the more favorable regioisomeric ortho reaction paths associated with HDA reaction of MCDO 1 with MVE 15.
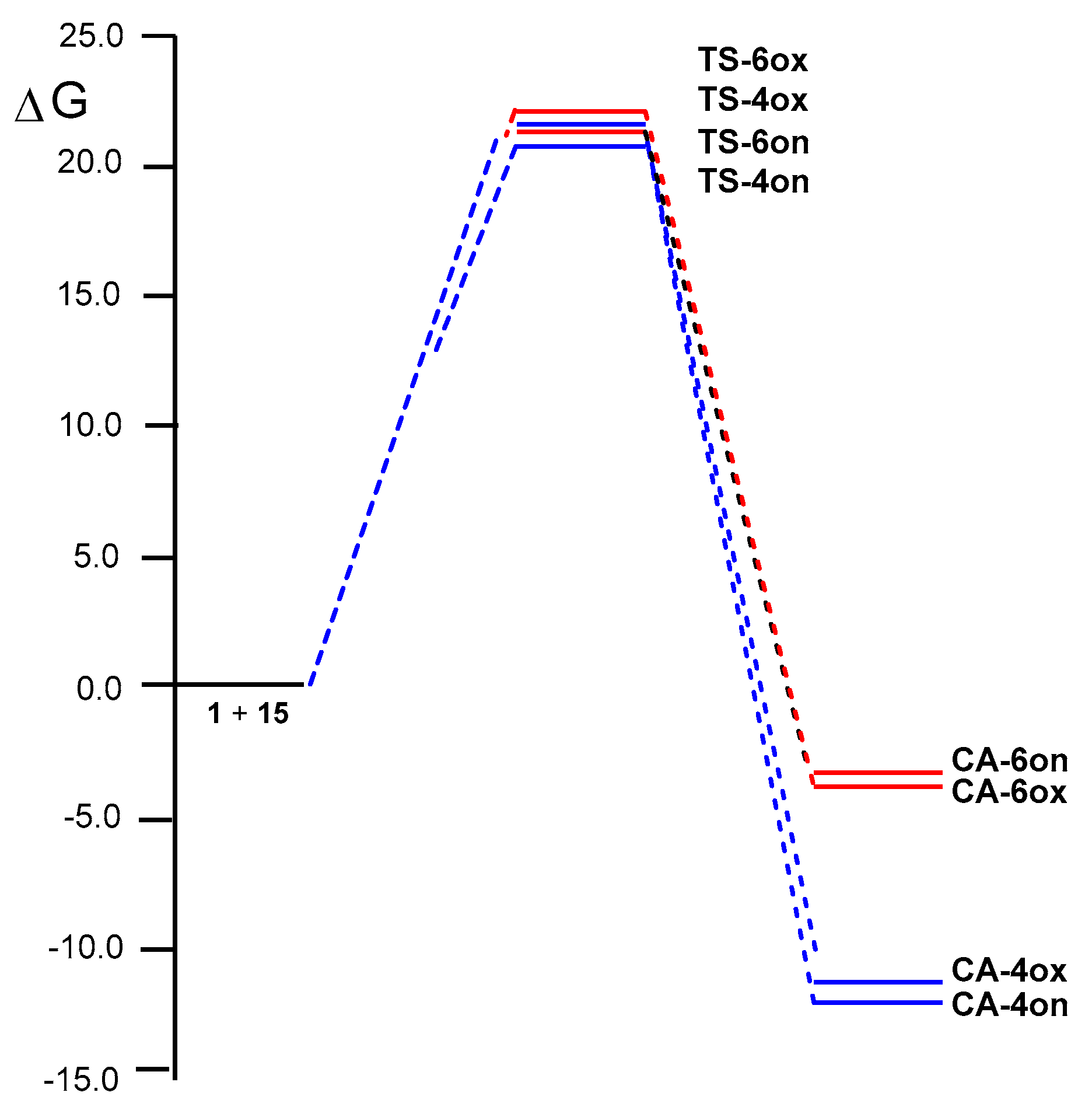
Figure 5.
B3LYP/6-311G(d,p) Geometries of the most favourable regioisomeric ortho TSs associated with HDA reaction of MCDO 1 with MVE 15. Values in dioxane are given in parenthesis. Distances are given in Angstroms Å.
Figure 5.
B3LYP/6-311G(d,p) Geometries of the most favourable regioisomeric ortho TSs associated with HDA reaction of MCDO 1 with MVE 15. Values in dioxane are given in parenthesis. Distances are given in Angstroms Å.
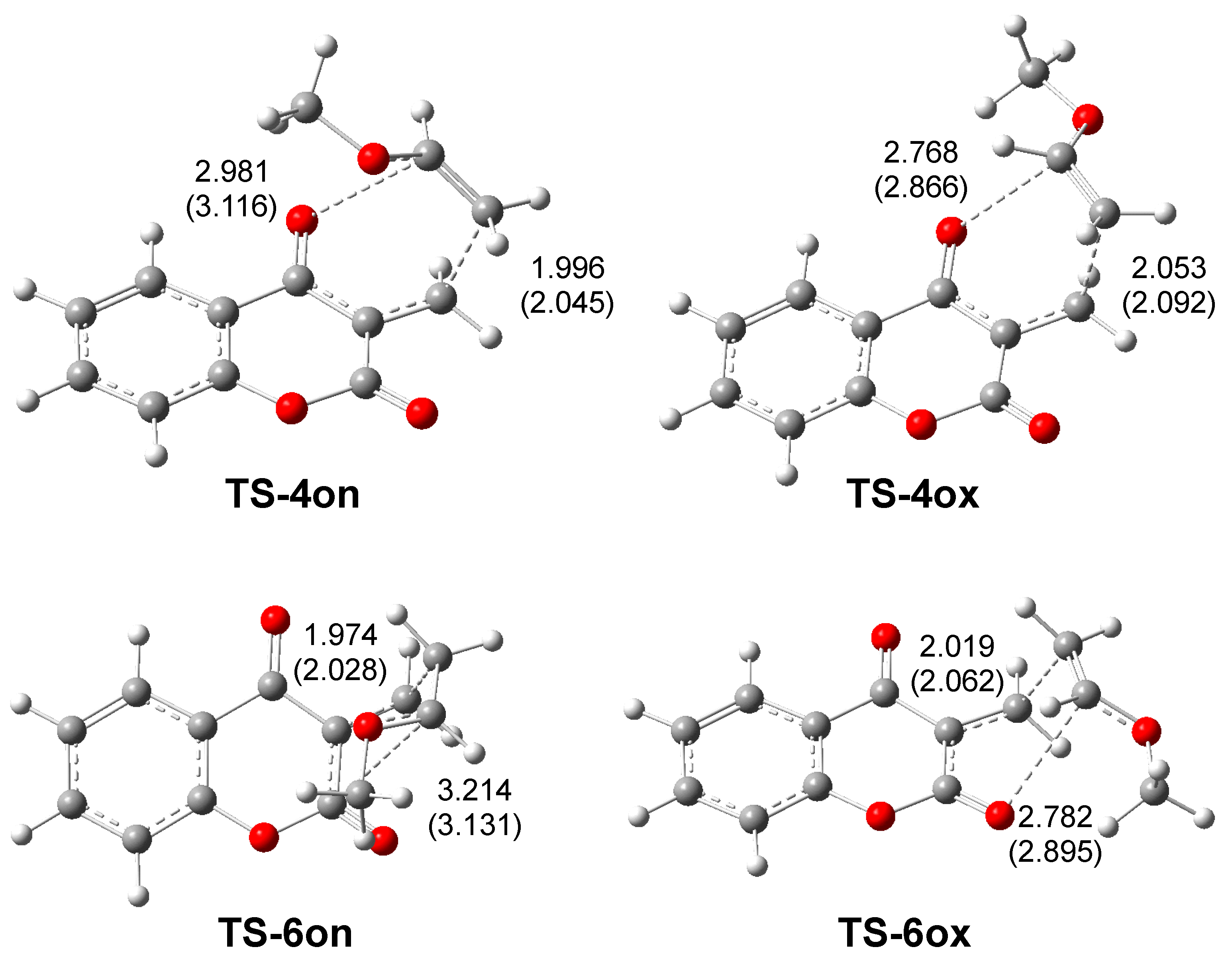
Figure 6.
View of the diastereomeric TS-4on and TS-6ox along the axis formed by the interacting C1 and C8 carbons. The C2−C1−C8−C9 dihedral angles are given in degree.
Figure 6.
View of the diastereomeric TS-4on and TS-6ox along the axis formed by the interacting C1 and C8 carbons. The C2−C1−C8−C9 dihedral angles are given in degree.
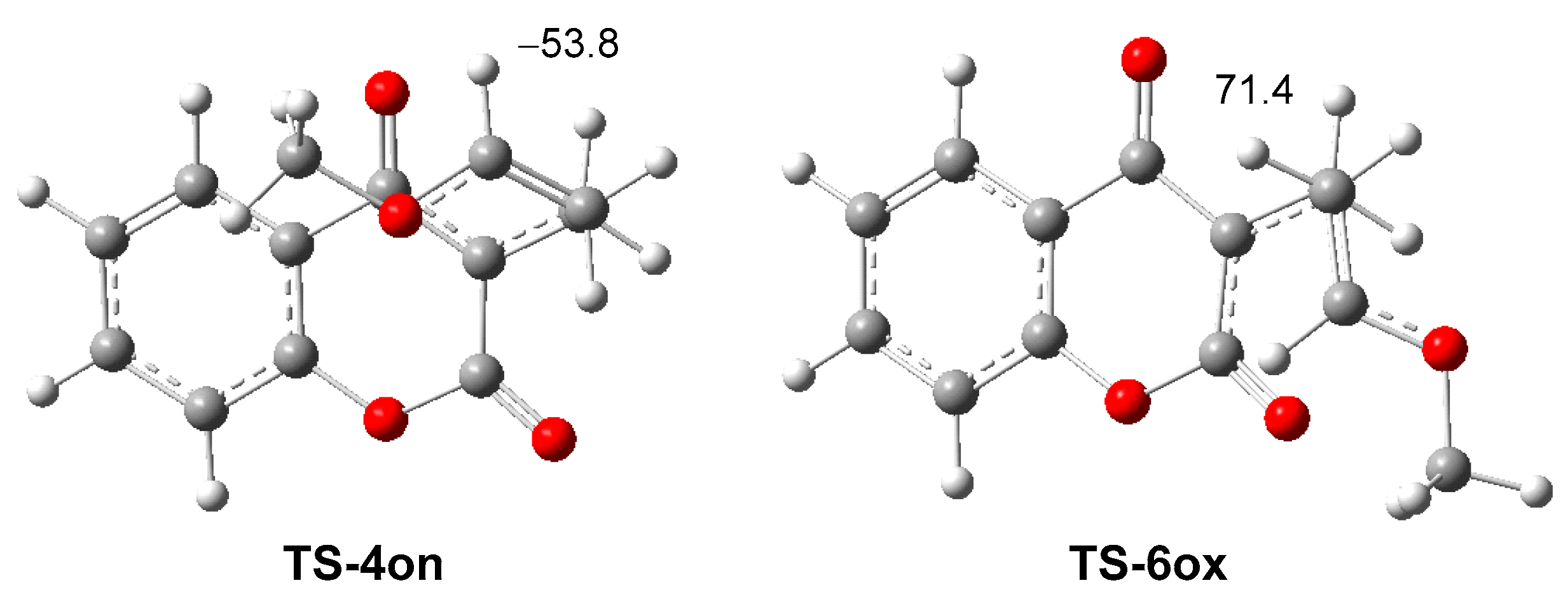
Figure 7.
ELF Basin attractor positions and populations of the most relevant valence basins of TS-4on and TS-6on. Valence basin populations are given in average number of electrons, e.
Figure 7.
ELF Basin attractor positions and populations of the most relevant valence basins of TS-4on and TS-6on. Valence basin populations are given in average number of electrons, e.
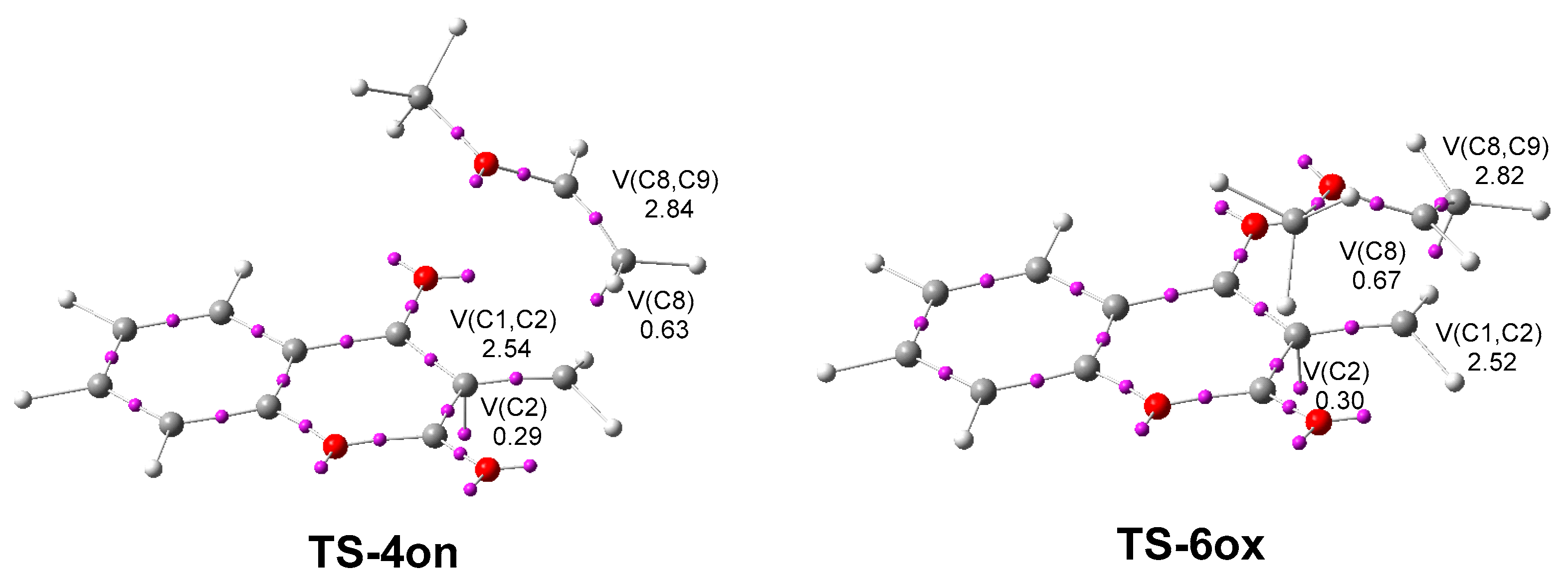
Figure 8.
Positions of the (3,+1), in gray, and (3, −1), in pink, CPs in TS-4on and TS-6on.
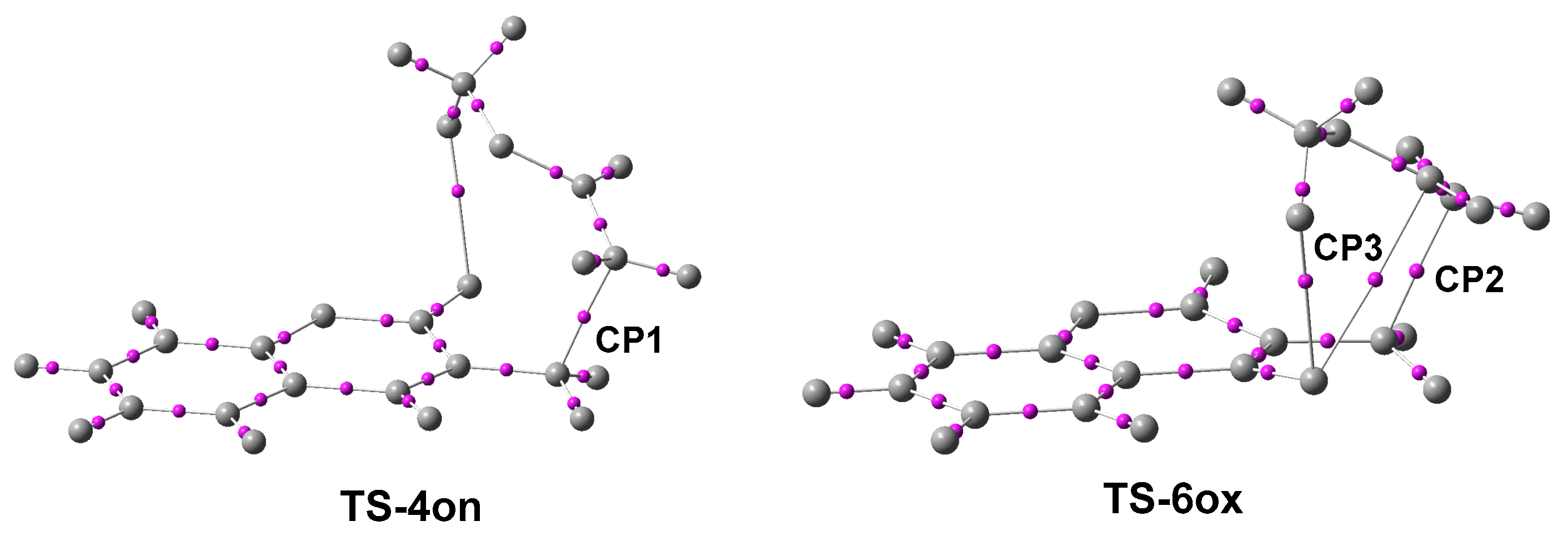
Figure 9.
Graphical representation of the sets of the ξ, ξ, and ξ energies in that order for the TSs associated with the HDA reactions of MCDO 1 with ethylene 8 and MVE 15. The ξ energies correspond to the RIAE activation energies of these HDA reactions. ξ Energies of the Et and MCDO frameworks at the four TSs, in blue and red, respectively. The black bar represents the ξ energies. The GEDT values at the TSs are given in pink. ξ Energies are given in kcal mol-1, and the GEDT in average number of electrons e.
Figure 9.
Graphical representation of the sets of the ξ, ξ, and ξ energies in that order for the TSs associated with the HDA reactions of MCDO 1 with ethylene 8 and MVE 15. The ξ energies correspond to the RIAE activation energies of these HDA reactions. ξ Energies of the Et and MCDO frameworks at the four TSs, in blue and red, respectively. The black bar represents the ξ energies. The GEDT values at the TSs are given in pink. ξ Energies are given in kcal mol-1, and the GEDT in average number of electrons e.
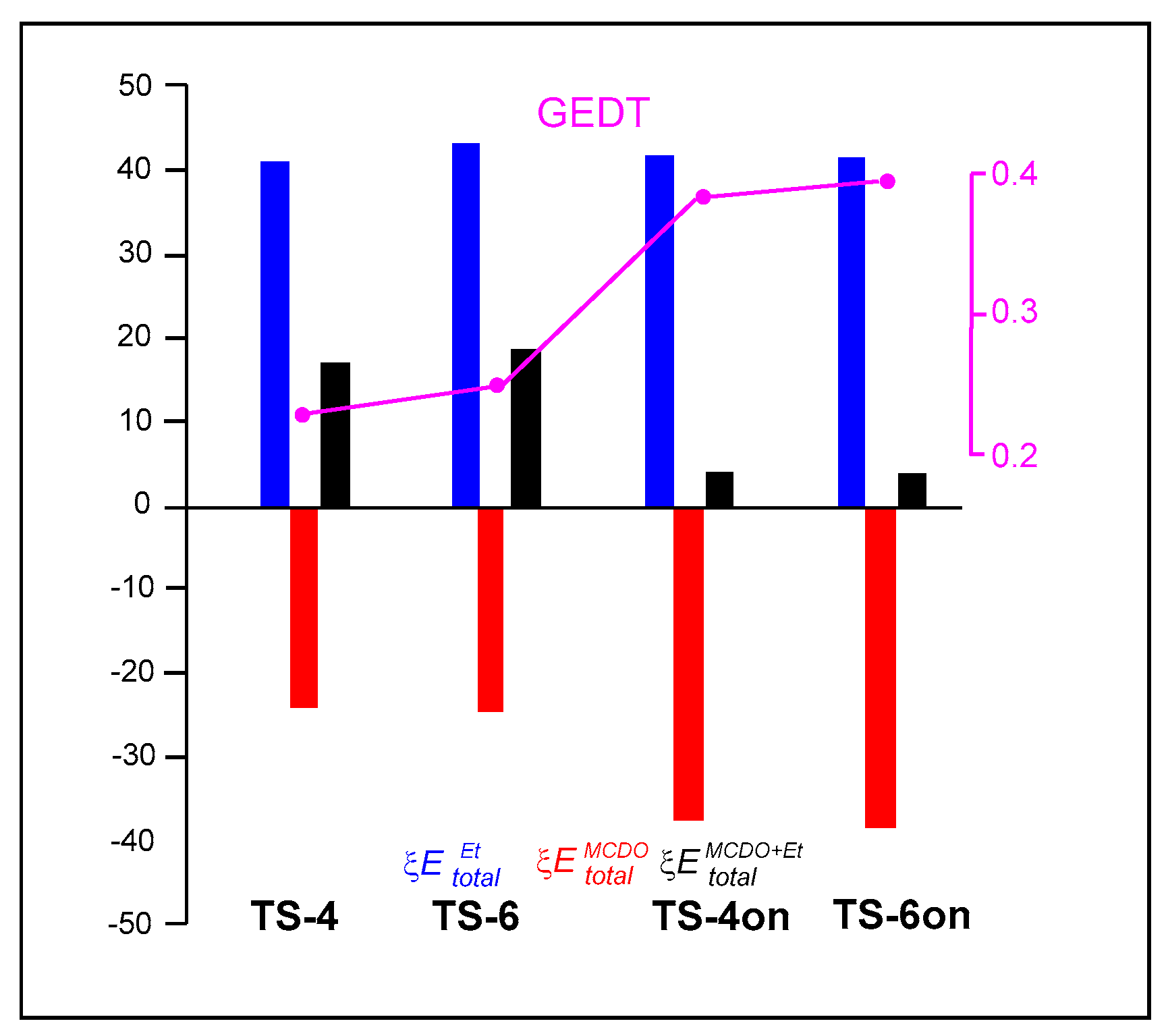

Table 1.
B3LYP/6-31G* Reactivity indices electronic chemical potential μ, chemical hardness η, electrophilicity ω, and nucleophilicity N, in eV, for MCDO 1, MVE 15 and ethylene 8.
Table 1.
B3LYP/6-31G* Reactivity indices electronic chemical potential μ, chemical hardness η, electrophilicity ω, and nucleophilicity N, in eV, for MCDO 1, MVE 15 and ethylene 8.
| Reactants | μ | η | ω | N |
| MCDO 1 | −4.88 | 4.16 | 2.87 | 2.16 |
| ethylene 8 | −3.37 | 7.77 | 0.73 | 1.86 |
| MVE 15 | −2.43 | 6.98 | 0.42 | 3.20 |
Table 3.
B3LYP/6-311G(d,p) Relative energies, ΔE in kcal·mol-1, in gas phase and in dioxane, of the stationary points involved in the HDA reaction of MCDO 1 with MVE 15.
Table 3.
B3LYP/6-311G(d,p) Relative energies, ΔE in kcal·mol-1, in gas phase and in dioxane, of the stationary points involved in the HDA reaction of MCDO 1 with MVE 15.
| Gas phase | 1,4-Dioxane | |
| TS-4on | 4.21 | 3.48 |
| TS-4ox | 4.88 | 4.65 |
| TS-4mn | 19.76 | 20.05 |
| TS-4mx | 23.09 | 23.03 |
| TS-6on | 4.26 | 3.55 |
| TS-6ox | 5.44 | 5.15 |
| TS-6mn | 22.19 | 22.49 |
| TS-6mx | 25.69 | 25.60 |
| CA-4on | -34.12 | -33.62 |
| CA-4ox | -35.39 | -34.79 |
| CA-4mn | -27.26 | -26.61 |
| CA-4mx | -26.64 | -26.35 |
| CA-6on | -19.02 | -18.26 |
| CA-6ox | -18.46 | -18.07 |
| CA-6mn | -26.31 | -25.64 |
| CA-6mx | -27.28 | -26.62 |
Table 4.
The QTAIM parameters of the CPs (3,-1) associated with the C1−C8 and C9−O regions of TS-4on and TS-6on. CPs are given in a.u.
Table 4.
The QTAIM parameters of the CPs (3,-1) associated with the C1−C8 and C9−O regions of TS-4on and TS-6on. CPs are given in a.u.
| TS-4on | TS-6on | ||
| CP1 | CP2 | CP3 | |
| Density ρ(r) | 0.0850 | 0.0810 | 0.0078 |
| Laplacian ∇2ρ(r) | 0.0128 | 0.0194 | 0.0343 |
| G(r) | 0.0309 | 0.0300 | 0.0074 |
| K(r) | 0.0277 | 0.0251 | 0.0012 |
| V(r) | -0.0587 | -0.0552 | -0.0062 |
| E(r) | -0.0277 | -0.0251 | 0.0012 |
Table 5.
B3LYP/6-311G(d,p) Gas-phase ξ total, ξintra-atomic and ξ interatomic energies, in kcal·mol-1, of the MCDO and the ethylene frameworks at the TSs with respect to those at the separated reagents. The sum of the of the ξ energies of the two frameworks, i.e., ξ, yields the RIAE activation energies.
Table 5.
B3LYP/6-311G(d,p) Gas-phase ξ total, ξintra-atomic and ξ interatomic energies, in kcal·mol-1, of the MCDO and the ethylene frameworks at the TSs with respect to those at the separated reagents. The sum of the of the ξ energies of the two frameworks, i.e., ξ, yields the RIAE activation energies.
| ƒ(X) | |||||
|---|---|---|---|---|---|
| TS-4 | MCDO | −83.78 | 59.90 | −23.87 | 17.19 |
| ethylene | 44.92 | −3.87 | 41.06 | ||
| TS-6 | MCDO | −84.44 | 59.89 | −24.55 | 19.02 |
| ethylene | 47.25 | −3.68 | 43.57 | ||
| TS-4on | MCDO | −66.20 | 28.60 | −37.60 | 3.96 |
| MVE | 71.48 | −29.92 | 41.56 | ||
| TS-6on | MCDO | −67.42 | 29.43 | −37.99 | 4.02 |
| MVE | 71.61 | &#;29.59 | 42.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



