
Submitted:
17 September 2024
Posted:
18 September 2024
You are already at the latest version
Abstract
Cutaneous Squamous Cell Carcinoma (cSCC) is an increasingly common malignancy of the skin and the leading cause of death from skin cancer in adults over the age of 85. Fibroblast Growth Factor Receptor 2 (FGFR2) has been identified as an important effector of signaling pathways that lead to the growth and development of cSCC. In recent years, there have been numerous studies evaluating the role FGFR2 plays in multiple cancers, its contribution to resistance to anti-cancer therapy, and new drugs that may be used to inhibit FGFR2. This review will provide an overview of our current understanding of FGFR2 and potential mechanisms in which we can target FGFR2 in cSCC. The goals of this review are the following: 1) to highlight our current knowledge on the role of FGFR2 in healthy skin and contrast this with its role in the development of cancer; 2) to further explain the specific molecular mechanisms that FGFR2 uses to promote tumorigenesis; 3) to describe how FGFR2 contributes to more invasive disease, 4) to describe its immunosuppressive effects in skin, and 5) to evaluate its effect on current anticancer therapy and discuss therapies on the horizon to target FGFR2 related malignancy.
Keywords:
Cutaneous Squamous Cell Carcinoma
; Fibroblast Growth Factor Receptor 2
; Treatment Resistance
; Epithelial Mesenchymal Transition
; Metastasis
; Immunosuppression
1. Introduction
Cutaneous squamous cell carcinoma (cSCC) is the second most common skin cancer, representing around 20% of all skin cancer diagnoses [1]. In the United States, it accounts for over 1 million diagnoses and over 9,000 deaths annually and is the leading cause of death from skin cancer in adults over the age of 85 [2,3,4]. The burden of disease of cSCC is growing, with an observed 300% increase in incidence over the last 30 years [4]. There are numerous risk factors for developing cSCC, the most significant being ultraviolet (UV) radiation exposure, increasing age, fair skin, and immunosuppression [2]. Currently, the National Comprehensive Cancer Network (NCCN) recommends peripheral and deep en face margin assessment (PDEMA) as the preferred surgical technique for high-risk local cSCC [2]. PDEMA is attained through either the Mohs or Tubingen technique[5]. Local medical therapy for pre-cancerous lesions include cryotherapy, 5-FU, calcipotriol, topical imiquimod, topical tirbanibulin, and photodynamic therapy[5]. Patients with local disease tend to have more positive outcomes, with the 5-year survival rate exceeding 90% [3,5]
Recent research in cancer biology has highlighted the role that the fibroblast growth factor receptor (FGFR) family plays in tumorigenesis. FGFR are tyrosine kinase receptors with three extracellular immunoglobulin domains, one transmembrane domain, and one intracellular domain that has tyrosine kinase activity [6]. The FGFR family consists of 4 members, FGFR1-4 with unique tissue expression and function[7]. FGFR signaling pathways are responsible for cellular proliferation, survival, angiogenesis, differentiation, and more. FGFRs role in cancer development is notable, with a recent study observing FGFR mutations in 7.1% of tumor cells across all cancer types [6]. The most common aberration of FGFR in cancer is amplification, with FGFR1 as the most altered subtype and FGFR2 as the second most altered subtype [8].
In this review, we will examine the current body of literature regarding FGFR2 and its role in the development and course of cSCC. The topics discussed will focus on the role of FGFR2 in tumorigenesis and immune cell function, and the currently developing therapies to target FGFR2. The goal of this review is to inform clinicians and scientists of an important genetic marker in an increasingly common cutaneous malignancy.
2. FGFR2 Expression in Skin and cSCC
Fibroblast growth factor receptors (FGFRs) are a family of transmembrane receptor tyrosine kinases belonging to the immunoglobulin superfamily [7]. In humans, four genes encode these closely related receptors. Ligand binding to FGFRs induces receptor dimerization and subsequent transphosphorylation of tyrosine residues within the intracellular domain, including the kinase domain and C-terminus. This activation triggers diverse cellular responses such as proliferation, differentiation, and motility. Further, alternative splicing of FGFR transcripts generates IIIb and IIIc isoforms, further diversifying signaling outcomes. Additionally, ligand redundancy adds complexity to the FGFR signaling network [9]. Recently, Thakur et al. demonstrated basal expression of FGFR1, 2, and 3 proteins in the mouse epidermis [10]. However, no appreciable expression of FGFR4 protein was detected in mouse epidermis. FGFR2 is involved in skin development during embryogenesis and postnatal growth by regulating the proliferation and differentiation of various cell types within the skin, ensuring proper formation and maintenance of the skin’s structure. FGFR2, particularly the epithelial isoform FGFR2b, promotes the differentiation of keratinocytes, the predominant cell type in the epidermis essential for forming the skin’s protective barrier [11]. Further, FGFR2 promoted re-epithelialization process during wound healing. Recently, study published by Thakur et al. demonstrated basal expression of FGFR1, 2, and 3 proteins in the mouse epidermis [10]. However, no appreciable expression of FGFR4 protein was detected in mouse epidermis.
Aberrations in FGFR2 have been found to play an important role in the development of cSCC. It has been reported that FGFR2 has both oncogenic and tumor suppressive activity. Tumor suppressive activity was postulated from a study that observed increased inflammation and papilloma formation in mice with FGFR2 knocked out exposed to a two-stage chemical carcinogenesis protocol [12]. However, more recent studies have presented strong evidence to suggest that FGFR2 signaling is pro-tumorigenic, noting that its activation/upregulation leads to tumor cell proliferation and survival through various downstream pathways [13,14]. Using a keratinocyte specific, tamoxifen inducible mouse model of FGFR2 deficiency, FGFR2 was determined to play a causative role in UVB-induced epidermal hyperproliferation, hyperplasia, and inflammation and development of cSCC [15]. In case of human cSCC, increased expression of FGFR2 has been found in both local and metastatic cSCC, without a significant difference in the rate of expression between groups [14]. Strong expression of FGFR2 is observed in cSCC compared to actinic keratosis (AK), suggesting a relationship between FGFR2 presence and disease severity [14]. FGFR2’s responsiveness to UV-B radiation closely links it to a well-known risk factor for developing cSCC. Mouse models have demonstrated that exposure to UV-B radiation leads to the phosphorylation and activation of FGFR2 due to upregulation of specific FGFs, activating multiple downstream signaling pathways such as phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT), mitogen-activated protein kinase (MAPK), and signal transducer and activator of transcription 3 (STAT3) that promote tumorigenesis [14].
3. Genetic Diversity and Genomic Alterations of FGFR2 Connected with cSCC
Various mutations of FGFR2 have been documented across a wide variety of tumor types. In cSCC, the most common aberration in FGFR2 has been observed to be a missense mutation [16]. Cell-based assays demonstrate that this mutation confers oncogenic properties to the Fgfr2 protein through a gain-of-function mechanism. In the presence of ligand, mutant Fgfr2 exhibits increased kinase activity and drives enhanced cell proliferation, ultimately leading to cellular transformation [14,16]. Two common FGFR2 mutations affect the genes S252W and P253R, which regulate the extracellular portion of the transmembrane receptor [17]. They are thought to increase the ligand binding affinity differentially, increasing the signaling activity of FGFR2 [17]. Another common mutation affects the N549 residue, which inhibits kinase activity [17]. Mutations to this residue result in the loss of the inhibitory activity, increasing signaling [17]. Other mutations in FGFR2 that lead to cell transformation include those at residues C382R and K659E [17].
Fusion of FGFR2 genes has also been frequently observed. The most common fusion partner is TACC3, followed by NPM1, TACC2, and BICC1[17]. These fusions are frequently caused by chromosomal translocations, which lead to the overexpression of FGFR2 [17]. Overexpression of FGFR2 increases the activity of many downstream signaling pathways, including PI3k/AKT via mTOR, MAPK, STAT3, and phospholipase C (PLC) [13]. These pathways have been shown to contribute to cSCC development through the promotion of inflammation, hyperplasia, and hyperproliferation in the epidermis [13].
4. FGF-FGFR2 Interactions in the Development and Progression of cSCC
4.1. FGF-FGFR2 Interactions in the Development and Progression of cSCC
Figure 1: FGFR2 activation leads to downstream signaling via the MAPK (RAS), PI3K/AKT, JAK/STAT, and PLC pathways to promote cell survival, proliferation and migration. When activated, these pathways can lead to changes in gene expression and the associated cellular changes that promote tumor development and progression.
Fibroblast growth factors (FGF) bind FGFR within a heparin sulfate complex, which protects it from degradation and stabilizes the interaction [18]. Binding of FGF ligands to FGFR2 results in the dimerization of the receptor, leading to transphosphorylation of the intracellular tyrosine kinase domains [7]. This transphosphorylation activates the four downstream signaling pathways, MAPK, PI3K-AKT (mTOR), STAT3, and PLC shown in Figure 1 and their effects on cellular growth [7,19]. Both ligand-independent and ligand-dependent signaling contribute to carcinogenesis, with ligand-dependent pathways utilizing both autocrine and paracrine signaling mechanisms [7]. This increased pattern of endocrine signaling leads to the early development of cancer through STAT3, a keratinocyte survival factor, by promoting cellular proliferation and migration [18,20]. The downstream signaling via PLC activates protein kinase C (PKC), which further promotes cellular proliferation, migration, and dedifferentiation [18]. The PI3K-AKT pathway uses mTOR, which contributes to carcinogenesis by inhibiting apoptosis and promoting proliferation[21]. Increased mTOR activity has been observed in cSCC and AK compared to normal skin, highlighting an important downstream effector of FGFR2[21]. These pathways work in parallel to promote carcinogenesis, hyperproliferation, and tumor cell expansion into the extracellular matrix.
4.2. Role of FGFR2 in Epithelial Mesenchymal Transition (EMT) and Enrichment of Cancer Stem Cells (CSCs)
Epithelial Mesenchymal Transition (EMT) is the process by which epithelial cells acquire the properties of mesenchymal cells. EMT plays a central role in the metastasis of many carcinomas [22]. The process of EMT enables epithelial cells to acquire fibroblast like properties, exhibit reduced cell-cell adhesion, and increase cellular motility [23,24]. This facilitates the escape of tumor cells from the primary tumor site. EMT relies on an intricate signaling network that controls the expression of transcription factors like Snail, Slug, and Twist. These factors suppress epithelial markers such as E-cadherin and activate mesenchymal markers like N-cadherin and vimentin. The FGFR2 gene encodes for the FGFR2b and FGFR2c isoforms through alternative splicing. The alternative splicing process for FGFR2 is regulated by a highly conserved GCAUG element, Fox protein family members, and Epithelial Splicing Regulatory Protein (ESRP) 1 and 2 [25,26,27]. Fox-2 protein enables the switching of FGFR2c to FGFR2b, which helps retain the epithelial phenotype and transforms mesenchymal cells to epithelial cells. Likewise, ESRP 1 and 2 enable splicing of FGFR2 pre-mRNA to the epithelial isoform FGFR2b [25]. FGFR2b, also named keratinocyte growth factor receptor KGFR, plays a crucial role in maintaining epithelial properties and promoting early differentiation of keratinocytes in the skin [28]. The epithelial isoform FGFR2b acts as a tumor suppressor in vitro and in vivo and the mesenchymal isoform FGFR2c exerts oncogenic activity in different types of cancer [29,30]. Aberrant switching of FGFR2 to the FGFR2c isoform triggers EMT, promoting cell migration [31,32]. Expression of FGFR2c impairs the basal layer formation in the skin due to altered actin cytoskeletal organization and down-regulation of cell adhesion molecules (CAMs), such as E-cadherin and the desmosomal component DSG1 [33]. The FGFR2 isoform switch to FGFR2c leads to PKCε activation, which in turn induces a cascade of EMT-related transcription factors, including STAT3, Snail1, and FRA1. These factors likely interact and coordinate with each other to trigger and maintain EMT [34]. PKCε demonstrates the highest oncogenic activity of the PKC family and has been consistently overexpressed in various carcinomas [35,36]. EMT has also been implicated in the development of cancer stem cells (CSCs), which possess the ability to initiate new tumors [37]. Notably, a study on esophageal squamous cell carcinoma has shown that EMT induction, resulting from FGFR2 knockdown, leads to an enrichment of CSCs through the activation of the AKT signaling pathway, suggesting the importance of FGFR2 in cancer plasticity [38].
4.3. FGFR2’s role in Promoting Angiogenesis and Metastatic Spread
Angiogenesis and metastasis are two intricately linked processes that play a pivotal role in cancer progression and dissemination. The angiogenic process forms new blood vessels within the tumor mass, supplying nutrients for the growth and progression of malignancy [39]. Angiogenesis also connects the primary malignancy to the broader blood supply, which can facilitate extravasation of cancer cells into the blood stream and increase the likelihood of distant metastasis. cSCC is typically a localized cancer that metastasizes rarely. Prior studies have demonstrated a metastasis rate of 0.1 to 12.5% in patients with cSCC [23,40,41,42] . The American Joint Committee on Cancer (AJCC) and NCCN have identified risk factors for cSCC metastasis as immunosuppression, depth, diameter, location of tumor, and certain genetic mutations [43]. Dysregulation of FGFR2 has been seen to promote angiogenesis and metastasis. FGFR2 signaling facilitates the progression of cSCC by activating pro-survival pathways, indirectly enabling cancer cells’ ability to become invasive [44]. FGFR2 signaling promotes cancer metastasis mainly by promoting EMT, modifying the extracellular matrix, enhancing cell motility and invasion, and facilitating angiogenesis.
The FGF/FGFR system is identified as one of the putative regulators of angiogenesis, although angiogenesis is primarily mediated by the vascular endothelial growth factor (VEGF)/VEGF receptor (VEGFR) system [45]. FGF/FGFR signaling stimulates the proliferation and migration of endothelial cells via chemotaxis, playing a very important role in angiogenic process [46,47]. FGFR2 signaling triggers the angiogenic process by multiple mechanisms. Activation of FGFR2 leads to phosphorylation/activation of Akt, STAT3, c-Jun and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [48]. FGFR2-AKT-STAT3 signaling influences the expression of matrix metalloproteinases (MMPs), leading to the remodeling of the extracellular matrix, migration of endothelial cells, and formation of new capillaries within the tumor vicinity [49]. Activation of FGFR2b by FGF7 also initiates the extracellular signal-regulated kinases, ERK1/2 signaling pathway, contributing to the proliferation and migration of epithelial cells [50]. FGF7 binding triggers FGFR2 dimerization and activates several signal transduction pathways which play fundamental roles in tumor progression [7]. FGF7/FGFR2 binding has been seen to inhibit the ubiquitination and degradation of hypoxia inducible factor (HIF-1α), posing a significant risk of angiogenesis and the spread of tumor cells [51]. Similarly, FGF7/FGFR2 interaction has been seen to promote the invasion and migration of gastric cancer by upregulating the protein thrombospondin-1 (TSP-1) [52]. Thrombospondins are a family of homologous proteins that regulate cellular phenotypes and extracellular structure during tissue genesis and remodeling. TSP-1 has been observed to increase the migration of invasive breast cancer cells and stimulate invasion and chemotaxis in squamous cell carcinoma cells [53,54]. Through a wide variety of effector pathways, FGFR2 has a significant influence on the ability of cSCC to utilize the blood supply to metastasize.
4.4. FGFR2 in the Development of Therapeutic Resistance Against Current Treatments
The emergence of resistance to currently available therapeutics is one of the significant challenges in cancer treatment. Despite the remarkable progress in medical therapies and increasing availability of anti-cancer drugs, the incidence of cancer recurrence and relapse are increasing. Relapsed tumors are often more aggressive and tend to respond poorly to treatment, increasing the morbidity and mortality they pose for patients [55]. The FGF/FGFR signaling complex drives the growth and progression of many cancers and contributes to the development of chemoresistance, limiting the efficacy of current treatment strategies. The role of FGFs and FGFRs in the development of resistance to various anti-cancer drugs has been extensively reviewed, highlighting a new understanding of their role in treatment failure [56].
FGFR2 signaling has been identified as a key contributor to resistance against many widely used anticancer drugs like cetuximab, tamoxifen and cisplatin, suggesting a crucial role in cancer treatment failure [57,58,59]. The two major causes recognized for the development of FGFR2 mediated drug resistance are the overexpression of FGFR2 and gene mutations. FGFR2 gene amplification and overexpression have been seen to transform cetuximab-sensitive esophageal squamous cell carcinoma into a resistant phenotype. However, inhibition of FGFR2 with pan-FGFR inhibitor NVP-BGJ398 inhibits the growth of tumor xenografts in mice treated with cetuximab. This stalling of tumor growth indicates the restoration of cetuximab sensitivity in these cancer cells when FGFR2 is no longer active. In breast cancer, the FGF2, FGF7/FGFR2 interaction activates the PI3K/AKT pathway, inducing the phosphorylation and proteasomal degradation of the estrogen receptor (ER) [58]. This degradation reverses tamoxifen-driven ER stabilization, hindering its efficacy. FGFR2 has also been identified as a key regulator of drug-resistant CSCs [58]. A recent study evaluated cisplatin-resistant head and neck cancer stem cells (HNCSCs) with substantially higher FGFR2 and ALDHhighCD44high expression [59]. Treatment with a pan-FGFR small-molecule inhibitor led to a decrease in ALDHhighCD44high cells, indicating a crucial role for FGFR2 in maintenance of drug-resistant CSCs [59]. Mutations in the FGFR2 kinase domain have been identified to confer resistance to FGFR inhibitor drugs. The N550K/H mutation in FGFR2 resists the pan-FGFR inhibitor PD173074, dovitinib, and ponatinib, suggesting a potential limitation of FGFR-targeted therapy [60]. Similarly, the V564M mutation of FGFR2 confers the ability to resist dovitinib and the pan FGFR-inhibitor BGJ398 [61]. To understand how the FGF/FGFR2 interaction contributes to chemoresistance, we focused on FGFR2-dependent signaling pathways and their downstream targets. FGFR2 transduces FGF signals to Ras-extracellular signal-regulated kinase (ERK), PI3K-AKT, Ca2+, and diacylglycerol (DAG) signaling cascades [62]. Malfunctions in the FGFR2 guidance of downstream signaling cascades have the potential to disrupt several beneficial homeostatic processes, including cytoprotective mechanisms against environmental insults such as UV irradiation. The existing information on FGFR2-mediated chemoresistance in cSCC is insufficient to provide a comprehensive understanding. As a result, future preclinical studies should employ bioinformatics and proteomics tools to investigate the complex interactions between FGF and FGFR2, shedding light on their contributions to cSCC development and progression.
5. Deregulated FGFR2 Activity in Immune Cell Function and Adverse Immune Outcomes in cSCC
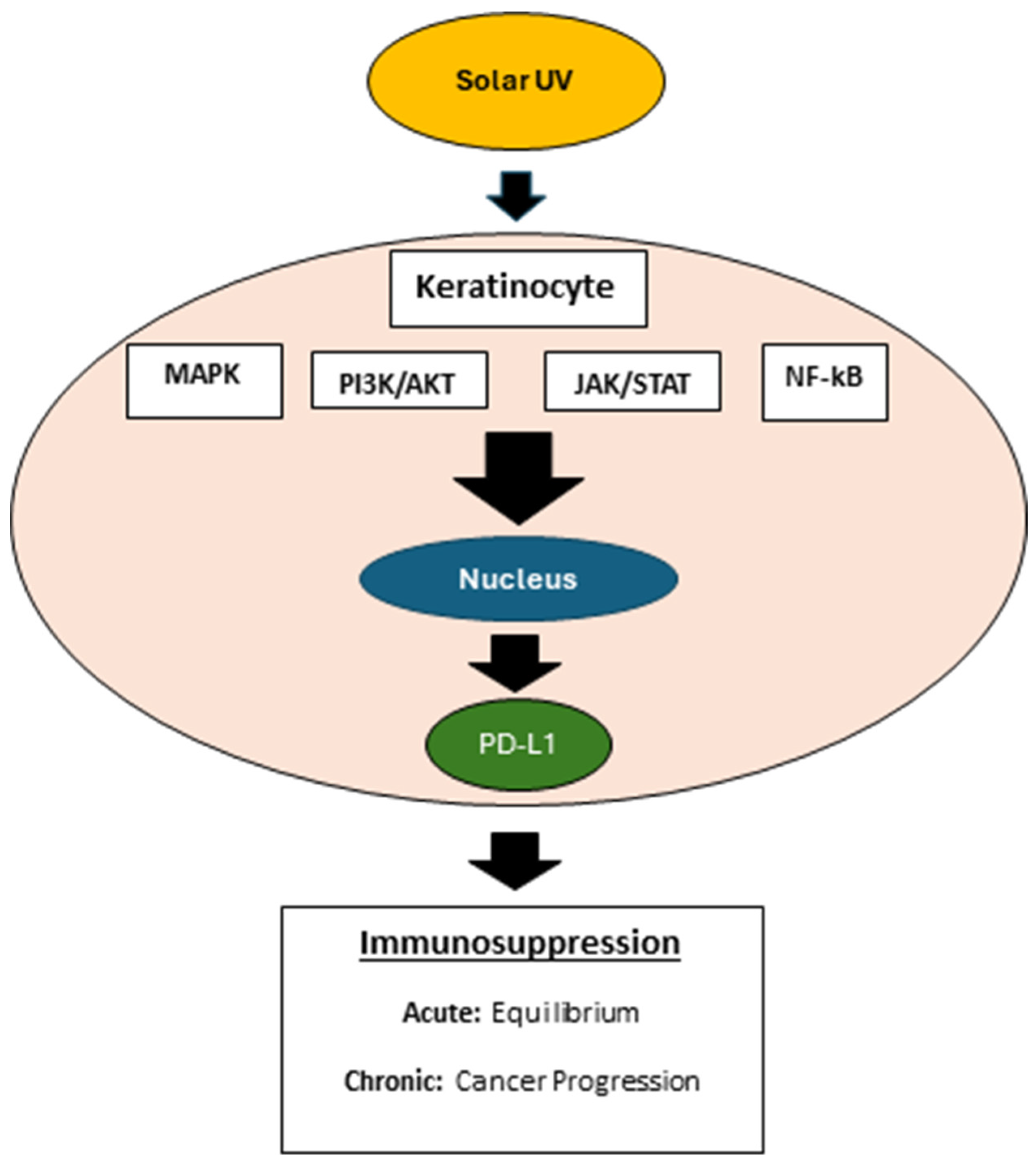
Recent studies have highlighted the use of solar UV (SUV) radiation to mimic the pathogenies of human cSCC in murine models [63,64]. UV radiation has been linked to increased transcription of PD-L1, leading to immunosuppression (Figure 2) [65]. A seminal study analyzed the expression of immune cells at all stages and locations of cSCC, including in situ and invasive stages of tumor draining lymph node (TDLN) and non-TDLN [66]. Compared to in situ cSCC, invasive cSCC expressed a greater number of myeloid-derived suppressor cells (MDSC). However, invasive cSCC expressed fewer MDSCs than many subpopulations of T cells, tumor-associated macrophages (TAM’s) and tumor infiltrating dendritic cells (TIDC). Furthermore, invasive tumors expressed lower levels of tumor infiltrating dendritic cells (TIDC), CD4+IFN-γ+ T cells, CD8+IFN-γ+ T cells, CD8+granzyme B (GzmB)+ T cells, and a reduced ratio of CD8+ T cells to regulatory T cells compared to in situ tumors. IFN-γ-producing T cells correlate with more potent antitumor immune responses and GzmB+ T cells are capable of lysing tumor cells. Consequently, in situ tumors maintain an immune-activating environment, whereas invasive tumors present an immunosuppressive environment. The immunosuppressive environment comprises low levels of CD4+IFN-γ+, CD8+IFN-γ+, and CD8+GzmB+ T cells [67]. Moreover, the invasive cSCC tumors express higher levels of PDL-1, which binds to the inhibitory immune checkpoint PD-1 on infiltrating lymphocytes and on tumor cells [68]. Low recognition of neoantigens by T cells is observed in invasive cSCC compared to AK [69]. The importance of these constrained T cells in cSCC is underscored by the successful treatment of advanced cases with cemiplimab, a PD-1 blocker.
Figure 2: The binding of PD-1 and PD-L1 in healthy cells serves to downregulate the immune system. Cancer cells upregulate PD-L1 to evade immune system surveillance, preventing cytotoxic T cells from killing tumor cells.
FGFR2 signaling potentially impacts various types of immune cells, each playing a crucial role in maintaining immune homeostasis and responding to tumorigenesis. Erdafitinib, a fibroblast growth factor receptor (FGFR) inhibitor, combined with a PD-1 blockade in an autochthonous FGFR2K660N/p53mut lung cancer mouse model increased T- cell infiltration activity and reduced Treg cell population [70]. In addition, Erdafinitib combined with anti-PD-1 decreased infiltration of immunosuppressive TAM’s, increased NK and B cell abundance, and a higher proliferative, activated state of T and NK cells relative to erdafitinib alone. Macrophages can exhibit pro-inflammatory (M1) or anti-inflammatory (M2) phenotypes. The macrophages present in the cSCC microenvironment predominantly exhibit the M2 phenotype and promote tumor invasion and metastasis through producing MMPs and lymphangiogenic mediators [71]. FGFR2 signaling influences macrophage polarization, and deregulated FGFR2 activity in cSCC can skew macrophages towards the M2 phenotype, supporting tumor growth, angiogenesis, and metastasis [72]. Dendritic cells are essential for antigen presentation and the initiation of adaptive immune responses. To date, no studies exist evaluating the effect of FGFR2 in dendritic cell function in the context of cSCC. Nevertheless, FGFR2 positive cancer associated fibroblasts secrete WNT proteins that play a critical role in dendritic cell (DC) differentiation and maturation [73]. This phenomenon suggests a paracrine role of FGFR2 in modulating dendritic cell function [73]. Deregulated FGFR2 activity in cSCC can impair DC maturation and antigen-presenting capability, leading to suboptimal T cell activation and immune surveillance. In recently published studies, keratinocyte specific deletion of FGFR2 was associated with a significant reduction in UVB-induced PD-L1 expression, mast cell and macrophages infiltration in mouse epidermis [20].
6. Current Progress in the Development of FGFR2-Oriented Therapies
The development of FGFR inhibitors has significantly advanced, targeting various cancers where FGFR2 overexpression or mutations are implicated. Key drugs in this category include ponatinib, dovitinib, and BGJ398 (infigratinib), which are designed to specifically inhibit the growth of tumors with FGFR2 gene mutations, amplifications or rearrangement [74,75,76,77]. The design of these agents is to inhibit tumor growth and proliferation. These drugs work by blocking the phosphorylation and activation of receptors, which is crucial for the FGFR-driven signaling pathways that promote cellular division and survival. Combining FGFR inhibitors with chemotherapy or radiotherapy has been explored as a strategy to enhance the efficacy of cancer treatment [78]. The rationale is that while FGFR inhibitors can suppress the proliferation of cancer cells by targeting specific signaling pathways, chemotherapy and radiotherapy can broadly kill both dividing and non-dividing cells. This combination aims to reduce the development of resistance and to achieve a more comprehensive tumor response. Studies have shown that the synergistic effects of combining FGFR inhibitors with other forms of therapy can lead to improved outcomes in various cancers. For instance, the use of FGFR inhibitors alongside DNA-damaging agents, like cisplatin or radiation therapy, has been shown to enhance the cytotoxic effects on cancer cells [79]. This synergy may be due to the inhibition of DNA repair pathways by FGFR inhibitors, making cancer cells more susceptible to damage induced by chemotherapy or radiotherapy. Integrating FGFR inhibitors with other targeted therapies such as EGFR inhibitors, VEGF blockers, or PD-1/PD-L1 checkpoint inhibitors is a promising area of research [70,80,81]. The goal is to block multiple pathways that the cancer cell uses for growth and survival, potentially leading to more effective treatment strategies. For example, the combined inhibition of FGFR2 and VEGF has shown potential in reducing angiogenesis and tumor growth more effectively than targeting either pathway alone [82]. This multi-pronged approach reflects the complexity and adaptability of cancer, requiring a comprehensive strategy to combat the disease effectively. The integration of FGFR2-oriented therapies into broader cancer treatment regimens continues to be a focus of clinical research, aiming to improve patient outcomes through targeted and synergistic treatment strategies.
7. The Scope of Immune Targeting of cSCC Together with FGFR2 Inhibition Therapy
Combining immunotherapy with FGFR2 inhibitors (e.g., RLY-4008) presents a potentially promising strategy for enhancing treatment efficacy in cSCC. Immunotherapies, such as immune checkpoint inhibitors targeting PD-1/PD-L1, can unleash the immune system’s ability to attack cancer cells. When used with FGFR2 inhibitors, this combined approach could overcome the immunosuppressive tumor microenvironment, improving anti-tumor immune responses. In our recent studies, mice exposed to UVB demonstrated a significant induction of epidermal PD-L1 mRNA and protein [13,20,63]. Furthermore, PD-L1 expression is increased in chronically sun-damaged, noncancerous human skin, underscoring its potential as a target for skin cancer prevention [83]. PD-L1 expression is known to be regulated by IFNγ signaling, which is pivotal in limiting the action of cytotoxic T-cells [84,85]. The binding of PD-L1 to its receptor, PD-1, on T-cells culminates in T-cell exhaustion and diminished function. We have previously demonstrated an increase in PD-L1 mRNA expression following exposure to solar UV, was contingent on the active IFNγ/pSTAT1/IRF-1 signaling pathway in keratinocytes [63] as well as FGFR2 signaling (11,13). Animal studies have shown that UV-induced systemic immunosuppression results in the emergence of specific regulatory T-cells (CD4+CD25+FOXP3+cells), transferable to unaffected animals [86]. Additional research is required, however, the increased levels of PD-L1 observed post-UV exposure in epidermis might significantly influence local immune suppression during skin carcinogenesis [63]. AZD4547, a pan FGFR inhibitor, and keratinocyte FGFR2 deficiency counteracted UVB induced PD-L1 expression in epidermal keratinocytes in vivo [13,20]. These data suggest that PD-L1 expression is downstream of several pathways, including FGFR2 signaling. The FGFR pathway inhibitors have been in clinical trials for many cancers including lung and GI cancers but have not yet been explored for UVB-induced cSCC in humans [87,88,89]. The FGFR2 signaling pathway has emerged as a significant player in skin biology, impacting various cellular processes including differentiation, proliferation, and wound repair as noted above. FGFR signaling, particularly that of FGFR2, may have a broader implication in the skin’s immune milieu, potentially influencing T-cell dynamics. Given the critical role of PD-L1 in cSCC progression and immune evasion, targeting PD-L1 in combination with FGFR2 inhibitors may provide a synergistic approach for the prevention and treatment of cSCC, addressing both the inflammatory and proliferative pathways involved in skin carcinogenesis. Although clinically relevant monoclonal antibodies targeting PD-L1 are effective for systemic tumor treatment, they are less suitable for preventive strategies or topical application [64]. Recently, a topical small-molecule inhibitor for PD-L1 was reported (BMS-202) to protect against UV-induced inflammatory stress in skin and associated with downregulation of keratinocytic PD-L1 expression [64]. The availability of such selective small molecular weight compounds will allow for the evaluation of a combinatorial approach targeting both PD-L1 and FGFR2 for prevention and treatment of cSCC.
8. Conclusions and Future Directions
FGFR2 plays an important role in the maintenance of healthy skin and dysregulation of FGFR2 is closely linked to the pathogenesis and progression of cSSC. Some of the emerging trends in FGFR2 research include the development of more selective and potent inhibitors against FGFR2, exploration of biomarkers that may better stratify patients for these treatments, and mechanisms of resistance to FGFR2-targeted therapies. Next-generation inhibitors and combination therapies are being designed with the discovery of novel FGFR2 mutations and alterations by new genomic and proteomic technologies. Potential advances in cSCC treatment with FGFR2 targeting are of major consequence. A better understanding of the role of FGFR2 in cSCC tumor biology is most likely to be linked to the development of new therapeutic strategies. These would comprise combination therapies with other targeted agents, immunotherapies, and personalized treatment approaches based on a patient’s tumor genetic makeup. The combination of FGFR2 inhibitors with local application for either prevention or therapy of cSCC, associated use of more sophisticated drug delivery systems and nanotechnology, might be developed for better accuracy and effect of treatment.
Author Contributions
Conceptualization; C.N. Writing – original draft preparation; E.K., A.K., P.N, J.D. Writing – reviewing and editing; C.N., S.A.N., J.D. Supervision; C.N., S.A.N.
Funding
The authors have received no funding for this study and have no funding to disclose.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Waldman, A.S., C, Cutaneous Squamous Cell Carcinoma. Hematology/Oncology Clinics of North America, 2019. 33(1): p. 1-12.
- Schmults, C.D., et al., NCCN Guidelines® Insights: Squamous Cell Skin Cancer, Version 1.2022. Journal of the National Comprehensive Cancer Network, 2021. 19(12): p. 1382-1394.
- Schmults, A.W.a.C., Cutaneous Squamous Cell Carcinoma. Hematology/Oncology Clinics of North America, 2019. 33(1): p. 1-12.
- Corchado-Cobos, R., et al., Cutaneous Squamous Cell Carcinoma: From Biology to Therapy. International Journal of Molecular Sciences, 2020. 21(8): p. 2956. [CrossRef]
- Schmults, C.D., et al., NCCN Guidelines® Insights: Squamous Cell Skin Cancer, Version 1.2022. Journal of the National Comprehensive Cancer Network, 2021. 19(12): p. 1382-1394.
- Porta, R., et al., FGFR a promising druggable target in cancer: Molecular biology and new drugs. Critical Reviews in Oncology/Hematology, 2017. 113: p. 256-267. [CrossRef]
- Grose, R. and C. Dickson, Fibroblast growth factor signaling in tumorigenesis. Cytokine Growth Factor Rev, 2005. 16(2): p. 179-86. [CrossRef]
- De Luca, A., et al., FGFR Fusions in Cancer: From Diagnostic Approaches to Therapeutic Intervention. International Journal of Molecular Sciences, 2020. 21(18): p. 6856.
- Ornitz, D.M. and N. Itoh, The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol, 2015. 4(3): p. 215-66. [CrossRef]
- Thakur, M.A., et al., Inhibition of Fibroblast Growth Factor Receptor Attenuates UVB-Induced Skin Carcinogenesis. J Invest Dermatol, 2022. 142(11): p. 2873-2884 e7. [CrossRef]
- Yang, J., et al., Fibroblast growth factor receptors 1 and 2 in keratinocytes control the epidermal barrier and cutaneous homeostasis. J Cell Biol, 2010. 188(6): p. 935-52.
- Grose, R., et al., The role of fibroblast growth factor receptor 2b in skin homeostasis and cancer development. The EMBO Journal, 2007. 26(5): p. 1268-1278. [CrossRef]
- Thakur, M., et al., Inhibition of Fibroblast Growth Factor Receptor Attenuates UVB-Induced Skin Carcinogenesis. Journal of Investigative Dermatology, 2022. 142: p. 2873-2884. [CrossRef]
- Khandelwal, A.R., et al., Fibroblast growth factor receptor promotes progression of cutaneous squamous cell carcinoma. Molecular Carcinogenesis, 2019. 58(10): p. 1715-1725. [CrossRef]
- Thakur, M., et al., Inducible Keratinocyte Specific FGFR2 Deficiency Inhibits UVB-Induced Signaling, Proliferation, Inflammation, and Skin Carcinogenesis. J Invest Dermatol, 2024. 144(2): p. 341-350 e7. [CrossRef]
- Zilberg, C., et al., Analysis of clinically relevant somatic mutations in high-risk head and neck cutaneous squamous cell carcinoma. Modern Pathology, 2018. 31(2): p. 275-287. [CrossRef]
- Helsten, T., et al., The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clinical Cancer Research, 2016. 22(1): p. 259-267. [CrossRef]
- Babina, I.S. and N.C. Turner, Advances and challenges in targeting FGFR signalling in cancer. Nature Reviews Cancer, 2017. 17(5): p. 318-332. [CrossRef]
- Czyz, M., Fibroblast Growth Factor Receptor Signaling in Skin Cancers. Cells, 2019. 8(6): p. 540. [CrossRef]
- Thakur, M., et al., Inducible Keratinocyte Specific FGFR2 Deficiency Inhibits UVB-Signaling, Proliferation, Inflammation, and Skin Carcinogensis. Journal of Investigative Dermatology, 2024. 144: p. 341-350.
- Carr, T.D., et al., Inhibition of mTOR Suppresses UVB-Induced Keratinocyte Proliferation and Survival. Cancer Prevention Research, 2012. 5(12): p. 1394-1404. [CrossRef]
- Thiery, J.P., Epithelial–mesenchymal transitions in tumour progression. Nature Reviews Cancer, 2002. 2(6): p. 442-454. [CrossRef]
- Kalluri, R. and R.A. Weinberg, The basics of epithelial-mesenchymal transition. Journal of Clinical Investigation, 2009. 119(6): p. 1420-1428. [CrossRef]
- Wells, A., Epithelial and mesenchymal phenotypic switchings modulate cell motility in metastasis. Frontiers in Bioscience, 2011. 16(1): p. 815. [CrossRef]
- Warzecha, C.C., et al., ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell, 2009. 33(5): p. 591-601. [CrossRef]
- Baraniak, A.P., J.R. Chen, and M.A. Garcia-Blanco, Fox-2 mediates epithelial cell-specific fibroblast growth factor receptor 2 exon choice. Mol Cell Biol, 2006. 26(4): p. 1209-22. [CrossRef]
- Baraniak, A.P., et al., A stem structure in fibroblast growth factor receptor 2 transcripts mediates cell-type-specific splicing by approximating intronic control elements. Mol Cell Biol, 2003. 23(24): p. 9327-37. [CrossRef]
- Belleudi, F., V. Purpura, and M.R. Torrisi, The Receptor Tyrosine Kinase FGFR2b/KGFR Controls Early Differentiation of Human Keratinocytes. PLoS ONE, 2011. 6(9): p. e24194. [CrossRef]
- Zhang, Y., et al., Growth inhibition by keratinocyte growth factor receptor of human salivary adenocarcinoma cells through induction of differentiation and apoptosis. Proceedings of the National Academy of Sciences, 2001. 98(20): p. 11336-11340. [CrossRef]
- Haugsten, E.M., et al., Roles of Fibroblast Growth Factor Receptors in Carcinogenesis. Molecular Cancer Research, 2010. 8(11): p. 1439-1452. [CrossRef]
- Ranieri, D., et al., Expression of the FGFR2 mesenchymal splicing variant in epithelial cells drives epithelial-mesenchymal transition. Oncotarget, 2016. 7(5): p. 5440-5460. [CrossRef]
- Shih, W. and S. Yamada, N-cadherin-mediated cell–cell adhesion promotes cell migration in a three-dimensional matrix. Journal of Cell Science, 2012. 125(15): p. 3661-3670. [CrossRef]
- Ranieri, D., et al., Expression of the FGFR2c mesenchymal splicing variant in human keratinocytes inhibits differentiation and promotes invasion. Molecular Carcinogenesis, 2018. 57(2): p. 272-283. [CrossRef]
- Ranieri, D., et al., Role of PKCε in the epithelial-mesenchymal transition induced by FGFR2 isoform switch. Cell Communication and Signaling, 2020. 18(1). [CrossRef]
- Isakov, N., Protein kinase C (PKC) isoforms in cancer, tumor promotion and tumor suppression. Semin Cancer Biol, 2018. 48: p. 36-52. [CrossRef]
- Gorin, M.A. and Q. Pan, Protein kinase C epsilon: an oncogene and emerging tumor biomarker. Mol Cancer, 2009. 8: p. 9.
- Gupta, P.B., C.L. Chaffer, and R.A. Weinberg, Cancer stem cells: mirage or reality? Nature Medicine, 2009. 15(9): p. 1010-1012.
- Maehara, O., et al., FGFR2 maintains cancer cell differentiation via AKT signaling in esophageal squamous cell carcinoma. Cancer Biology & Therapy, 2021. 22(5-6): p. 372-380. [CrossRef]
- Majidpoor, J. and K. Mortezaee, Angiogenesis as a hallmark of solid tumors - clinical perspectives. Cell Oncol (Dordr), 2021. 44(4): p. 715-737. [CrossRef]
- Brantsch, K.D., et al., Analysis of risk factors determining prognosis of cutaneous squamous-cell carcinoma: a prospective study. Lancet Oncol, 2008. 9(8): p. 713-20. [CrossRef]
- Brougham, N.D., et al., The incidence of metastasis from cutaneous squamous cell carcinoma and the impact of its risk factors. J Surg Oncol, 2012. 106(7): p. 811-5. [CrossRef]
- Nelson, T.G. and R.E. Ashton, Low incidence of metastasis and recurrence from cutaneous squamous cell carcinoma found in a UK population: Do we need to adjust our thinking on this rare but potentially fatal event? J Surg Oncol, 2017. 116(6): p. 783-788.
- Cherpelis, B.S., C. Marcusen, and P.G. Lang, Prognostic factors for metastasis in squamous cell carcinoma of the skin. Dermatol Surg, 2002. 28(3): p. 268-73.
- Ramsey, M.R., et al., FGFR2 signaling underlies p63 oncogenic function in squamous cell carcinoma. J Clin Invest, 2013. 123(8): p. 3525-38. [CrossRef]
- Takano, S., Glioblastoma angiogenesis: VEGF resistance solutions and new strategies based on molecular mechanisms of tumor vessel formation. Brain Tumor Pathology, 2012. 29(2): p. 73-86. [CrossRef]
- Cross, M.J. and L. Claesson-Welsh, FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol Sci, 2001. 22(4): p. 201-7. [CrossRef]
- Gillis, P., et al., Keratinocyte growth factor induces angiogenesis and protects endothelial barrier function. Journal of Cell Science, 1999. 112(12): p. 2049-2057. [CrossRef]
- Dudka, A.A., S.M. Sweet, and J.K. Heath, Signal transducers and activators of transcription-3 binding to the fibroblast growth factor receptor is activated by receptor amplification. Cancer Res, 2010. 70(8): p. 3391-401.
- Quintero-Fabián, S., et al., Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Frontiers in Oncology, 2019. 9.
- Finch, P.W. and J.S. Rubin, Keratinocyte Growth Factor⧸Fibroblast Growth Factor 7, a Homeostatic Factor with Therapeutic Potential for Epithelial Protection and Repair. 2004, Elsevier. p. 69-136.
- Feng, S., et al., Cancer-associated fibroblast-secreted FGF7 as an ovarian cancer progression promoter. Journal of Translational Medicine, 2024. 22(1). [CrossRef]
- Huang, T., et al., FGF7/FGFR2 signal promotes invasion and migration in human gastric cancer through upregulation of thrombospondin-1. International Journal of Oncology, 2017. 50(5): p. 1501-1512.
- Wang, T.N., et al., Thrombospondin-1 (TSP-1) promotes the invasive properties of human breast cancer. J Surg Res, 1996. 63(1): p. 39-43. [CrossRef]
- Wang, T.N., et al., The effect of thrombospondin on oral squamous carcinoma cell invasion of collagen. Am J Surg, 1995. 170(5): p. 502-5. [CrossRef]
- Housman, G., et al., Drug resistance in cancer: an overview. Cancers (Basel), 2014. 6(3): p. 1769-92. [CrossRef]
- Szymczyk, J., et al., FGF/FGFR-Dependent Molecular Mechanisms Underlying Anti-Cancer Drug Resistance. Cancers (Basel), 2021. 13(22). [CrossRef]
- Zhang, Y., et al., Resistance to Cetuximab in EGFR-Overexpressing Esophageal Squamous Cell Carcinoma Xenografts Due to FGFR2 Amplification and Overexpression. Journal of Pharmacological Sciences, 2014. 126(1): p. 77-83. [CrossRef]
- Turczyk, L., et al., FGFR2-Driven Signaling Counteracts Tamoxifen Effect on ERα-Positive Breast Cancer Cells. Neoplasia, 2017. 19(10): p. 791-804. [CrossRef]
- McDermott, S.C., et al., FGFR signaling regulates resistance of head and neck cancer stem cells to cisplatin. Oncotarget, 2018. 9(38): p. 25148-25165.
- Byron, S.A., et al., The N550K/H mutations in FGFR2 confer differential resistance to PD173074, dovitinib, and ponatinib ATP-competitive inhibitors. Neoplasia, 2013. 15(8): p. 975-88. [CrossRef]
- Goyal, L., et al., Polyclonal Secondary <i>FGFR2</i> Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion–Positive Cholangiocarcinoma. Cancer Discovery, 2017. 7(3): p. 252-263.
- Katoh, M., FGFR2 Abnormalities Underlie a Spectrum of Bone, Skin, and Cancer Pathologies. Journal of Investigative Dermatology, 2009. 129(8): p. 1861-1867. [CrossRef]
- Blazanin, N., et al., Activation of a protumorigenic IFNγ/STAT1/IRF-1 signaling pathway in keratinocytes following exposure to solar ultraviolet light. Mol Carcinog, 2019. 58(9): p. 1656-1669.
- Dickinson, S.E., et al., Inhibition of UV-Induced Stress Signaling and Inflammatory Responses in SKH-1 Mouse Skin by Topical Small-Molecule PD-L1 Blockade. JID Innov, 2024. 4(2): p. 100255. [CrossRef]
- Vaishampayan, P., C. Curiel-Lewandrowski, and S.E. Dickinson, Review: PD-L1 as an emerging target in the treatment and prevention of keratinocytic skin cancer. Mol Carcinog, 2023. 62(1): p. 52-61. [CrossRef]
- Adams, A.C., et al., Solar Simulated Light Induces Cutaneous Squamous Cell Carcinoma in Inbred Mice: A Clinically Relevant Model to Investigate T-Cell Responses. J Invest Dermatol, 2021. 141(12): p. 2990-2993.e6.
- Huang, S.J., et al., Imiquimod enhances IFN-gamma production and effector function of T cells infiltrating human squamous cell carcinomas of the skin. J Invest Dermatol, 2009. 129(11): p. 2676-85.
- Gambichler, T., et al., Expression of PD-L1 in keratoacanthoma and different stages of progression in cutaneous squamous cell carcinoma. Cancer Immunol Immunother, 2017. 66(9): p. 1199-1204. [CrossRef]
- Borden, E.S., et al., Neoantigen Fitness Model Predicts Lower Immune Recognition of Cutaneous Squamous Cell Carcinomas Than Actinic Keratoses. Front Immunol, 2019. 10: p. 2799. [CrossRef]
- Palakurthi, S., et al., The Combined Effect of FGFR Inhibition and PD-1 Blockade Promotes Tumor-Intrinsic Induction of Antitumor Immunity. Cancer Immunol Res, 2019. 7(9): p. 1457-1471. [CrossRef]
- Pettersen, J.S., et al., Tumor-associated macrophages in the cutaneous SCC microenvironment are heterogeneously activated. J Invest Dermatol, 2011. 131(6): p. 1322-30. [CrossRef]
- Li, Y., et al., FGFR2 upregulates PAI-1 via JAK2/STAT3 signaling to induce M2 polarization of macrophages in colorectal cancer. Biochim Biophys Acta Mol Basis Dis, 2023. 1869(4): p. 166665.
- Huang, T.X., et al., Targeting cancer-associated fibroblast-secreted WNT2 restores dendritic cell-mediated antitumour immunity. Gut, 2022. 71(2): p. 333-344. [CrossRef]
- Javle, M., et al., Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma. J Clin Oncol, 2018. 36(3): p. 276-282.
- Gozgit, J.M., et al., Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol Cancer Ther, 2012. 11(3): p. 690-9.
- Konecny, G.E., et al., Second-line dovitinib (TKI258) in patients with FGFR2-mutated or FGFR2-non-mutated advanced or metastatic endometrial cancer: a non-randomised, open-label, two-group, two-stage, phase 2 study. Lancet Oncol, 2015. 16(6): p. 686-94. [CrossRef]
- Du, S., Y. Zhang, and J. Xu, Current progress in cancer treatment by targeting FGFR signaling. Cancer Biol Med, 2023. 20(7): p. 490-9. [CrossRef]
- SenthilKumar, G., et al., FGFR Inhibition Enhances Sensitivity to Radiation in Non-Small Cell Lung Cancer. Mol Cancer Ther, 2020. 19(6): p. 1255-1265. [CrossRef]
- Weeden, C.E., et al., Cisplatin Increases Sensitivity to FGFR Inhibition in Patient-Derived Xenograft Models of Lung Squamous Cell Carcinoma. Mol Cancer Ther, 2017. 16(8): p. 1610-1622. [CrossRef]
- Wu, Q., et al., EGFR Inhibition Potentiates FGFR Inhibitor Therapy and Overcomes Resistance in FGFR2 Fusion-Positive Cholangiocarcinoma. Cancer Discov, 2022. 12(5): p. 1378-1395. [CrossRef]
- Peng, M., et al., Dual FGFR and VEGFR inhibition synergistically restrain hexokinase 2-dependent lymphangiogenesis and immune escape in intrahepatic cholangiocarcinoma. J Gastroenterol, 2023. 58(9): p. 908-924. [CrossRef]
- Shen, G., et al., Anlotinib: a novel multi-targeting tyrosine kinase inhibitor in clinical development. J Hematol Oncol, 2018. 11(1): p. 120. [CrossRef]
- Dickinson, S.E., et al., Increased PD-L1 Expression in Human Skin Acutely and Chronically Exposed to UV Irradiation. Photochem Photobiol, 2021. 97(4): p. 778-784. [CrossRef]
- Garcia-Diaz, A., et al., Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep, 2017. 19(6): p. 1189-1201. [CrossRef]
- Mimura, K., et al., PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci, 2018. 109(1): p. 43-53. [CrossRef]
- Rana, S., et al., Ultraviolet B suppresses immunity by inhibiting effector and memory T cells. Am J Pathol, 2008. 172(4): p. 993-1004. [CrossRef]
- Schuler, M., et al., Rogaratinib in patients with advanced cancers selected by FGFR mRNA expression: a phase 1 dose-escalation and dose-expansion study. Lancet Oncol, 2019. 20(10): p. 1454-1466. [CrossRef]
- Subbiah, V., et al., FIGHT-101, a first-in-human study of potent and selective FGFR 1-3 inhibitor pemigatinib in pan-cancer patients with FGF/FGFR alterations and advanced malignancies. Ann Oncol, 2022. 33(5): p. 522-533. [CrossRef]
- Van Cutsem, E., et al., A randomized, open-label study of the efficacy and safety of AZD4547 monotherapy versus paclitaxel for the treatment of advanced gastric adenocarcinoma with FGFR2 polysomy or gene amplification. Ann Oncol, 2017. 28(6): p. 1316-1324. [CrossRef]
Figure 1.
Intracellular signaling pathways of FGFR2 and their contribution to tumorigenesis.
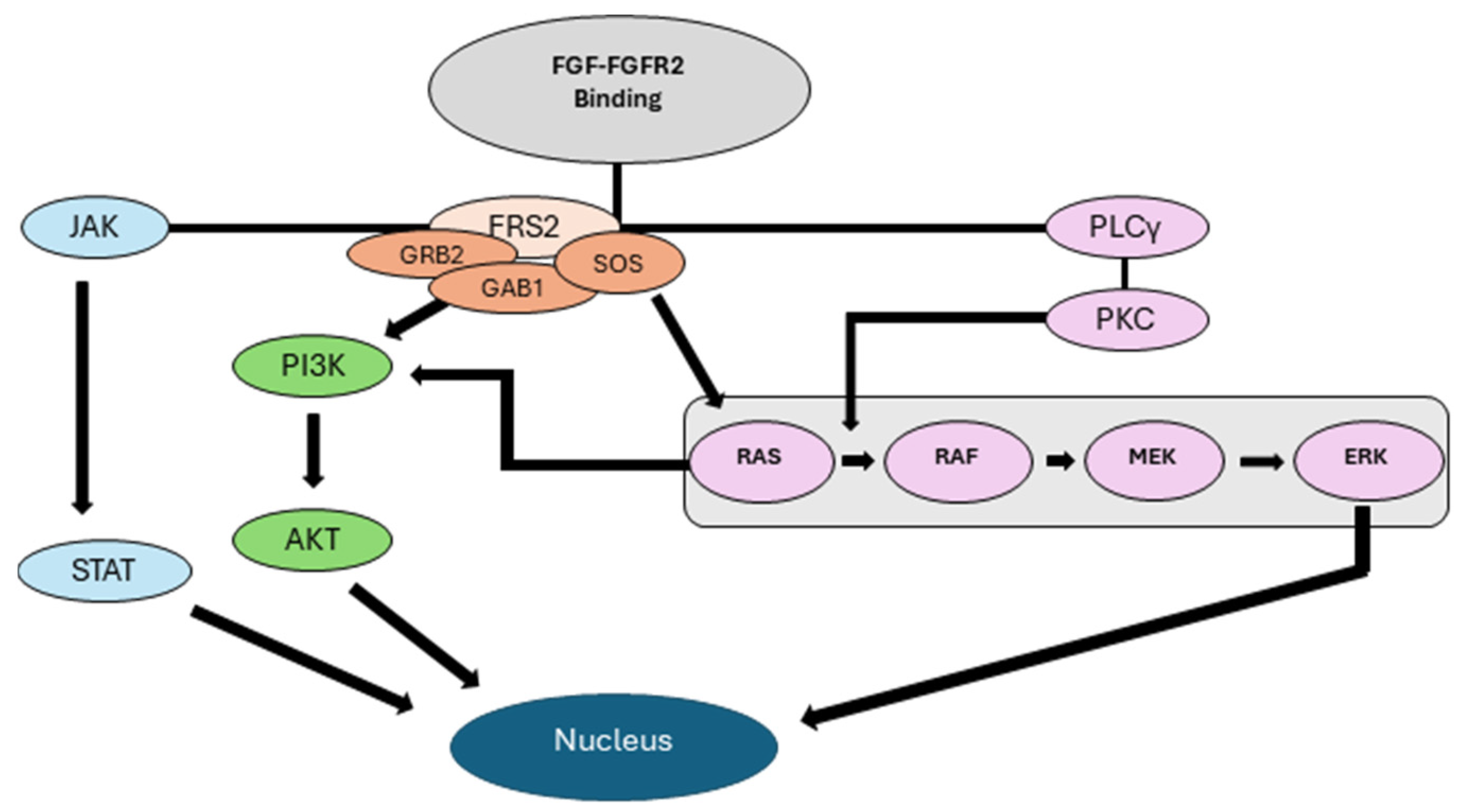
Figure 2.
Interactions between CD8+ T cells and Tumor Cells.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



