Submitted:
18 September 2024
Posted:
19 September 2024
Read the latest preprint version here
Abstract
Myalgic Encephalomyelitis (ME), sometimes called Chronic Fatigue Syndrome (CFS), is a complex, debilitating condition characterised by symptoms such as profound fatigue, autonomic dysfunction, gastrointestinal problems, unrefreshing sleep, cognitive problems, exercise intolerance and post-exertional malaise (PEM) (a worsening of symptoms after mild physical or mental effort). The pathophysiology of ME remains poorly understood. This paper proposes a novel hypothesis centred on norepinephrine dysregulation and insulin signaling as the underlying mechanisms contributing to distinct subtypes of ME. I hypothesise that ME can be categorised into three subtypes based on variations in norepinephrine transport and insulin signaling: ME1, ME2, and ME3. In ME1, a higher than normal rate of insulin secretion during the first phase insulin response and/or hypersensitive insulin receptors leads to reduced neuronal norepinephrine reuptake, resulting in elevated extracellular norepinephrine and subsequent down-regulation of beta-2 adrenergic receptors. ME2 is characterised by prolonged insulin release and insulin resistance, which may contribute to the development of postural orthostatic tachycardia syndrome (POTS) and microclot formation. ME3 is marked by reduced norepinephrine synthesis and a resultant deficiency of norepinephrine within neurons. All three subtypes involve a deficiency of norepinephrine inside the noradrenergic neuron which is worsened by physical and mental exertion. There are many similarities between ME and Long COVID. Both have their onset after a virus and have a similar symptom profile, as well as having the same co-occurring conditions such as mast cell activation and POTS. It is quite possible that a subset of Long COVID patients have ME, and so I will be referring to both ME and Long COVID research papers throughout this hypothesis paper. This hypothesis integrates genetic, metabolic, and neuroendocrine data to delineate these subtypes and suggests tailored diagnostic and therapeutic approaches for each. This hypothesis aims to guide future research into the distinct pathophysiological mechanisms of ME and enhance understanding and management of this debilitating condition.
Keywords:
Myalgic Encephalomyelitis
; Chronic Fatigue Syndrome
; ME/CFS
; Norepinephrine
; Insulin
; Neuron
Introduction
Myalgic Encephalomyelitis (ME), also known as Chronic Fatigue Syndrome (CFS), is a multi-system illness characterised by profound and disabling fatigue that is not alleviated by rest and is often exacerbated by physical or mental exertion. The etiology of ME remains elusive, and the condition is associated with a range of symptoms including cognitive dysfunction, orthostatic intolerance, sleep disturbances, gastrointestinal dysfunction, widespread pain and post-exertional malaise (PEM). Despite extensive research, the pathophysiological mechanisms underlying ME are not fully understood, leading to challenges in diagnosis and treatment (1).
Recent advances have highlighted the role of neuroendocrine dysregulation in ME, with evidence suggesting abnormalities in autonomic nervous system function and hypothalamic-pituitary-adrenal (HPA) axis regulation (2,3). One potential area of interest is the interplay between norepinephrine dysregulation and insulin signaling.
The neurotransmitter norepinephrine plays a critical role in modulating autonomic function, mood, and cognitive processes and emerging evidence suggests that altered norepinephrine signaling could be a fundamental feature of ME(4). Metabolic and genetic research has suggested that insulin signaling, which regulates glucose metabolism and has widespread effects on cellular function, may be playing a part in the pathophysiology of the disease(5,6).
This hypothesis proposes that ME may encompass distinct subtypes based on the interplay between norepinephrine dysregulation and insulin signaling abnormalities. Specifically, it suggests that there are three subtypes of ME—ME1, ME2, and ME3—each characterised by different patterns of norepinephrine transport and insulin response. Understanding these subtypes could provide valuable insights into the underlying mechanisms of ME and lead to more targeted and effective treatment strategies.
The Sympathetic Nervous System and Noradrenergic Neurons
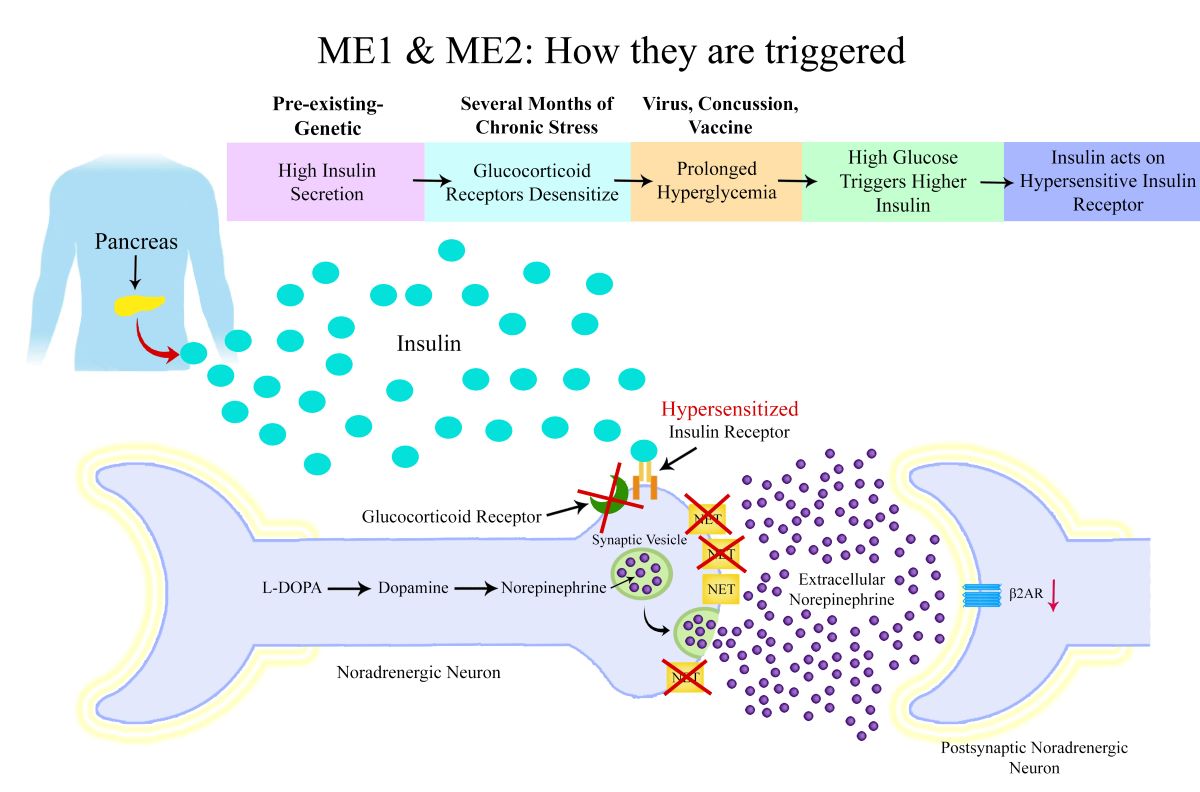
The synthesis of norepinephrine begins with the amino acid phenylalanine, which is first converted into tyrosine by the enzyme phenylalanine hydroxylase. Tyrosine is then processed by tyrosine hydroxylase to form L-DOPA, which subsequently undergoes decarboxylation to become dopamine. Finally, dopamine is converted into norepinephrine by the enzyme dopamine beta-hydroxylase.
Once synthesised, norepinephrine is stored in synaptic vesicles and released into the synaptic cleft upon neuronal stimulation. It then acts on postsynaptic noradrenergic receptors to modulate synaptic activity. The reuptake of norepinephrine into the neuron via the norepinephrine transporter (NET) is a critical process that regulates neurotransmitter levels and neuronal signaling. In the brain, norepinephrine is predominantly released from the locus coeruleus and has widespread effects on various neural circuits. Norepinephrine exerts its effects through noradrenergic receptors, including alpha and beta receptors, which modulate synaptic transmission and neuronal excitability.
The sympathetic nervous system (SNS) is a critical component of the autonomic nervous system, responsible for the ‘fight or flight’ response. It orchestrates physiological changes in response to stress and maintains homeostasis by modulating cardiovascular, respiratory, and metabolic functions. The SNS is made up of noradrenergic neurons which release norepinephrine as its primary neurotransmitter, which is released from sympathetic nerve terminals to exert its effects on target tissues throughout the body.
ME3 Subtype: Norepinephrine Deficiency
This hypothesis suggests that one subtype of ME involves deficiency of norepinephrine caused by reduced norepinephrine synthesis. This deficiency could arise from several potential disruptions in the noradrenergic system. Possible causes include reduced synthesis of norepinephrine due to impaired tyrosine hydroxylase activity or a deficiency of tetrahydrobiopterin (BH4). This can result in a reduced ability to maintain adequate norepinephrine levels within the neuron, impacting overall neurotransmission and cognitive functions.
A 2024 NIH Intramural ME study found low Dihydroxyphenylglycol (DHPG) levels in ME patients(4). DHPG is a metabolite made from the breakdown of neuronal norepinephrine. This means that it can be a good indicator of the level of norepinephrine inside the neuron, and low levels could indicate a neuronal norepinephrine deficiency. There are two types of dysregulation that could lead to reduced norepinephrine in the synaptic vesicles of noradrenergic neurons- a reduction in synthesis, and a reduction in reuptake of norepinephrine from the synaptic cleft.
ME1 & ME2 Subtype: Reduced Norepinephrine Transporters and Beta-2 Adrenergic Receptor Down-Regulation
The proposed mechanism for ME1 and ME2 subtypes is that reduced NET expression leads to elevated extracellular norepinephrine. Reduced expression or dysfunction of these transporters can lead to increased extracellular norepinephrine levels, while depleting intracellular norepinephrine stores. Research has found high plasma norepinephrine levels in ME patients and one particular study found there were subgroups of high and low norepinephrine which indicates subtypes of different noradrenergic dysfunction (7,8).
As well as causing a deficiency of norepinephrine in the synaptic vesicle of the neuron, prolonged elevation of extracellular norepinephrine can trigger down-regulation of the β2-adrenergic receptor (β2-AR) on target cells, which may diminish noradrenergic signaling. β2-ARs are critical in mediating physiological responses such as vasodilation, bronchodilation, and metabolic regulation, and their down-regulation can contribute to problems such as increased blood pressure, dysregulated glucose metabolism, reduced lipolysis, reduced vasodilation of blood vessels, altered neurotransmission, reduced gastrointestinal motility and a dysregulated immune response. Research has found ME patient immune cell β2-ARs to be less responsive to β2-AR agonists(9).
The reduced expression of NET in ME1 and ME2 subtypes would reduce norepinephrine reuptake into the neuron and could result in a reduction of norepinephrine levels within the neuron. This would explain the low levels of DHPG found in ME patients in the NIH Intramural Study.
Overtraining Syndrome
Overtraining syndrome (OTS) is a condition resulting from excessive exercise without adequate rest, leading to a persistent decline in performance, fatigue, and autonomic dysfunction. One of the key findings in OTS is a reduced density of β2-ARs on cell surfaces, which impairs SNS function, reducing the body’s ability to respond to stress and physical exertion(10). This downregulation of β2-ARs is hypothesised to contribute to the chronic fatigue, impaired exercise tolerance, and autonomic symptoms seen in OTS. Interestingly, there is a striking overlap between the symptoms of OTS and ME, including profound fatigue, post-exertional malaise, and autonomic dysregulation, such as orthostatic intolerance. Furthermore, a significant number of ME patients report being avid exercisers or athletes prior to the onset of illness, suggesting that excessive physical activity or overtraining may be a predisposing factor for the development of ME in some individuals. The similarities in symptoms and underlying mechanisms, such as adrenergic receptor downregulation, highlight potential shared pathways in the pathophysiology of OTS and ME/CFS, although the precise triggers and disease course differ between the two conditions.
Chronotropic incompetence (CI) is the inability to increase heart rate commensurate with metabolic demand and can contribute to exertion intolerance. Research has shown that when undertaking a cardiopulmonary exercise test (CPET) test on two concurrent days, ME patients don’t perform as well on the second day as healthy controls, and appear to have increased CI on day two. Research shows that CI involves cardiac β-adrenergic receptor down-regulation, and so this down-regulation of beta adrenergic receptors on the second day CPET test could be due to excessive and prolonged norepinephrine release during the CPET test on day one causing down-regulation of adrenergic receptors(11). However it should also be considered that the exercise on day one could worsen a norepinephrine deficiency, affecting the performance on day two.
Negative Feedback Mechanism of Norepinephrine Regulation
The alpha 2 adrenergic autoreceptor feedback mechanism plays a critical role in maintaining norepinephrine homeostasis. High extracellular norepinephrine levels activate alpha 2 adrenergic autoreceptors on presynaptic neurons, which inhibits further norepinephrine synthesis and release. In ME1 and ME2 patients, reduced NET expression can lead to sustained high norepinephrine levels, disrupting this feedback mechanism and causing a cyclical pattern of excessive norepinephrine release followed by excessive activation of autoreceptors and inhibition of norepinephrine synthesis, which would worsen the neuronal norepinephrine deficiency. This may explain the boom and bust pattern of symptom severity that many ME patients experience, although it is also possible that the boom and bust pattern of symptoms could actually be due to excessive norepinephrine release down-regulating postsynaptic receptors, or possibly a combination of both.
Insulin’s Role in Regulating Norepinephrine Transporters
Insulin plays a key role in the regulation of NETS. Research has shown that insulin reduces norepinephrine reuptake by reducing NET expression on noradrenergic neurons(12). In ME1 and ME2 patients, insulin dysregulation, specifically in the form of high insulin levels, likely exacerbates NET dysfunction. Insulin reduces the expression of NETS on noradrenergic neurons, and in the setting of prolonged hyperinsulinemia, this would lead to increased extracellular norepinephrine levels.
Recent studies have highlighted insulin dysregulation in ME patients, with genetic research identifying variants associated with insulin signaling and secretion. In particular gene variants for the ATP9A, KCNB1 and CLOCK genes have been found to be risk factors for ME, and these genes are involved in insulin secretion from the pancreas(6). The insulin receptor INSR gene has also been found to be a risk factor for both ME and Long COVID(13). Hypersensitivity of the neuronal insulin receptor could also cause a reduction in NETS.
A large metabolic study of ME patients identified three subtypes of ME, with the ME2 subtype showing considerably increased insulin levels. It is this metabolic study that has inspired the usage of the terms ME1, ME2 and ME3 used in this paper, as the metabolic signatures match with the hypothesis suggested here(5).
It should be considered that the root cause of increased pancreatic insulin secretion may be elsewhere in the body. For example, a genetic dysregulation that leads to increased free fatty acids could result in increased pancreatic insulin secretion(14).
Distinguishing ME1 and ME2 Subtypes Based on Insulin Dynamics
The key difference between ME1 and ME2 subtypes could be the type of dysregulation of insulin release and action. In ME1, individuals could experience rapid and excessive insulin release during the first-phase insulin secretion in response to glucose intake. This subtype is characterised by a fast and high insulin spike within approximately 3-20 minutes of glucose intake, which may result in frequent episodes of reactive hypoglycaemia(15). In these patients, despite the elevated insulin levels, there is no insulin resistance, leading to a rapid glucose uptake by tissues. The sudden drop in glucose levels can have significant cognitive effects, including confusion, brain fog, and fatigue, which are common symptoms in ME patients. Moreover, the rapid rise in insulin could lead to over-activation of insulin receptors on noradrenergic neurons, causing NET downregulation.
It’s also possible that rather than a high and fast pancreatic insulin secretion, the ME1 subtype could be caused by a very high sensitivity of insulin receptors to insulin, as this would have the same effect as a high and fast insulin release. These patients may actually have low insulin levels when tested, as the fast glucose uptake due to high insulin sensitivity would lead to the pancreas not needing to release as much insulin.
Conversely, the ME2 subtype is characterised by prolonged insulin release and insulin resistance. These patients may develop systemic insulin resistance, except in the neurons, where insulin sensitivity remains high. The chronic hyperinsulinemia in ME2 patients could lead to similar effects on neurons as seen in ME1, such as a reduction of NETS. However, the insulin resistance elsewhere in the body introduces additional complications, such as metabolic disturbances, microclot formation, and postural orthostatic tachycardia syndrome (POTS). The combination of insulin resistance and prolonged high insulin levels likely exacerbates both the metabolic and neurological symptoms in the ME2 subtype.
ME1: Does It Have a Prodrome Phase?
The ME1 subtype, characterised by early-phase hyperinsulinemia and/or hypersensitive insulin receptors, may have two stages to the disease. There may be a prodrome phase which precedes the onset of full-blown ME. The evidence suggests that prolonged exposure to high extracellular norepinephrine and SNS hyperactivity could lead to gradual down-regulation of the β2ARs.
During the prodrome phase, ME1 individuals may experience a prolonged period of sympathetic dominance. This state of over-activation has been documented in several chronic conditions, including Long COVID, where patients show increased SNS tone, as evidenced by elevated heart rate variability and other autonomic markers(16). In such a state, symptoms such as tachycardia, anxiety, sleep disturbances, and heightened stress responses might predominate, signaling an overactive SNS. This hyperactive phase could last for months, with the body trying to maintain homeostasis in the face of continuous high extracellular norepinephrine.
Prolonged norepinephrine stimulation leads to β2-AR down-regulation, a process that takes time and repeated exposure to high levels of norepinephrine. It is plausible that this process of receptor down-regulation is gradual and may take many months to happen. Patients in the prodrome phase may not yet exhibit full ME symptoms including PEM but may experience signs of SNS over-activation such as palpitations, gastrointestinal dysregulation and cognitive dysfunction.
Some patients with Long COVID manage to recover within 6-12 months, potentially because their autonomic dysregulation resolves before β2-ARs undergo significant down-regulation. Early treatment interventions aimed at rebalancing autonomic function or reducing norepinephrine levels may help prevent progression to receptor down-regulation and, consequently, ME. However, for individuals who suffer a second immune challenge—such as a viral infection, vaccination, or physical injury—this may hasten the β2-AR down-regulation process. The introduction of another stressor could amplify norepinephrine release, leading to an accelerated decline in receptor sensitivity and the rapid onset of ME.
The Effect of Metformin on Long COVID Risk: Implications for the ME2 Subtype
A recent randomised clinical trial published in The Lancet Infectious Diseases demonstrated that metformin, an anti-diabetic medication, reduced the incidence of Long COVID by 41% when administered early in the course of SARS-CoV-2 infection(17). Metformin’s primary mechanism of action is the reduction of hepatic glucose production and improvement in insulin sensitivity, making it an essential treatment for insulin resistance. This finding is of particular interest in the context of the ME2 subtype, which is hypothesised to involve insulin resistance and prolonged hyperinsulinemia.
Research has shown that SARS-CoV-2 infection can induce hyperglycaemia and insulin resistance for at least two months post-infection(18). It is not completely clear if this is due to the immune response or whether it could be a consequence of viral persistence, as research has shown that SARS-CoV-2 is capable of infecting hepatocytes and stimulating these cells to produce glucose through gluconeogenesis(19).
Metformin’s ability to lower both blood glucose levels and insulin resistance could be the key to preventing the metabolic dysregulation that predisposes some individuals to the ME2 subtype of Long COVID. Reduced blood glucose levels would prevent an increase in insulin levels and the overstimulation of insulin receptors, which would help reduce the excessive activation of the sympathetic nervous system. As a result, patients at risk of developing the ME2 subtype of Long COVID may avoid developing the disease if they take metformin during SARS-CoV-2 infection.
Postural Orthostatic Tachycardia Syndrome (POTS)
Postural Orthostatic Tachycardia Syndrome (POTS) is a condition characterised by an excessive increase in heart rate upon standing, often accompanied by a range of debilitating symptoms including dizziness, lightheadedness, palpitations and fatigue. It is classified as a form of orthostatic intolerance, wherein the autonomic nervous system fails to properly regulate blood pressure and heart rate when transitioning from a supine to upright posture. This results in a range of cardiovascular, neurological, and gastrointestinal symptoms that can severely impact daily functioning and quality of life.
The underlying pathophysiology of POTS is multifactorial, and various subtypes of the condition have been proposed, including hyperadrenergic POTS, neuropathic POTS, and hypovolemic POTS, each associated with distinct autonomic and vascular dysfunctions. A common feature across many POTS patients is peripheral vasodilation and blood pooling, which contributes to the inadequate return of blood to the heart when standing, thereby triggering a compensatory increase in heart rate to maintain cardiac output and cerebral perfusion.
Vascular endothelial dysfunction may play a role in POTS, particularly through dysregulation of nitric oxide production. Nitric oxide is synthesised by endothelial nitric oxide synthase (eNOS) in response to various stimuli, including insulin, and serves as a potent vasodilator that helps regulate vascular tone by relaxing the smooth muscle cells in blood vessels. Under normal conditions, the release of nitric oxide ensures appropriate vasodilation, maintaining blood flow and pressure throughout the circulatory system.
However, in ME2 POTS patients, overproduction of nitric oxide in response to high insulin levels may contribute to excessive vasodilation, particularly in the peripheral vasculature. In individuals with ME2 POTS, this insulin-mediated vasodilation could exacerbate peripheral pooling of blood, especially in the lower extremities, when transitioning to an upright posture.
As blood pools in the legs and splanchnic region due to excessive vasodilation, venous return to the heart is compromised, leading to a reduction in stroke volume and cardiac output. To compensate for the decreased venous return, the heart increases its rate of contraction, thereby manifesting as tachycardia upon standing. This increased heart rate represents the body’s attempt to maintain adequate cerebral and systemic perfusion in the face of poor vascular tone and excessive peripheral vasodilation.
When POTS occurs as a result of another disease, it is called secondary POTS, and diabetes mellitus, particularly in its chronic forms, is a leading cause of secondary POTS. This could be due to the increased insulin levels in Type 2 diabetes patients.
Microclots in ME/CFS and Long COVID
In ME/CFS and Long COVID patients, microclots have been detected in both the plasma and endothelium, where they appear to interfere with normal blood flow and oxygen exchange(20,21). These microclots are often resistant to breakdown by the body’s natural fibrinolytic processes, contributing to microvascular obstruction and chronic tissue hypoxia. As a result, many patients experience profound fatigue, cognitive impairment, and a range of other symptoms consistent with poor blood flow and cellular energy deficits.
Interestingly, microclots have also been found in patients with Type 2 diabetes, where chronic hyperglycaemia and insulin resistance may play a role in their formation(22). Prolonged high levels of insulin can lead to insulin resistance on platelets, causing them to become hyper-activated. Hyper-activated platelets are more prone to aggregating and forming clots, which can obstruct blood flow in the microvasculature, particularly in the capillaries and small arterioles.
The same platelet hyper-activation and clotting dysfunction found in diabetes patients may also play a role in the formation of microclots in ME/CFS and Long COVID patients, particularly in individuals who experience prolonged episodes of high insulin levels.
Glucocorticoid Receptor Resistance
Glucocorticoid receptor (GR) resistance refers to a condition in which tissues become less responsive to the effects of glucocorticoids, despite normal or elevated levels of circulating hormones such as cortisol. The human GR is encoded by the NR3C1 gene, which produces two main isoforms, GR-α and GR-β, via alternative splicing. GR-α is the classic isoform responsible for mediating the anti-inflammatory and metabolic effects of glucocorticoids, while GR-β acts as a dominant-negative inhibitor of GR-α, reducing its transcriptional activity. Dysregulation in the balance between these two isoforms can lead to GR resistance.
Evidence suggests that ME patients may exhibit GR resistance. A study by Kavelaars et al. (2000) demonstrated the maximal effect of dexamethasone on T-cell proliferation is significantly reduced in ME patients as compared with controls, which may indicate a reduced responsiveness to cortisol’s regulatory effects on inflammation and metabolic homeostasis(9). It could be possible that some ME patients have genetic variants that make their glucocorticoid receptors more vulnerable to developing resistance.
GR resistance also has significant implications for insulin signaling. Cortisol normally promotes insulin resistance by antagonising insulin receptor signaling, reducing glucose uptake in peripheral tissues. However, in the context of GR resistance, tissues may exhibit heightened sensitivity to insulin due to diminished cortisol-mediated insulin antagonism. This effect could be particularly pronounced in noradrenergic neurons. Reduced glucocorticoid signaling may lead to increased insulin receptor sensitivity on these neurons, amplifying the effects of insulin and leading to disruptions in norepinephrine homeostasis. Research shows that corticosterone up-regulates the expression and function of the NET(23).
It’s also possible that some ME patients may have low cortisol levels instead of glucocorticoid resistance, as this would have the same effect on insulin signaling and norepinephrine transport.
The Sequence of Events Leading to ME
The onset of ME1 and ME2 subtypes is hypothesised to follow a multistep process, starting with a genetic predisposition to insulin dysregulation. In these individuals, genetic variants affecting insulin secretion and insulin receptor sensitivity predispose them to abnormal insulin dynamics. Under normal conditions, this genetic predisposition may remain latent. However, the triggering of ME is initiated by stress, inflammation, or conditions that reduce GR responsiveness.
- Glucocorticoid Receptor Resistance or Low Cortisol: Glucocorticoids, such as cortisol, regulate insulin sensitivity, particularly in neurons. Due to GR resistance or low cortisol levels, there is a reduction in GR activity. This leads to increased insulin receptor sensitivity in neurons, as cortisol normally acts to promote insulin resistance in the body. The reduced GR function or low cortisol levels amplify insulin’s effect on neurons, making them more susceptible to hyperinsulinemia.
- Hyperglycaemia Trigger: The second major step in the sequence is the occurrence of a triggering event—such as a viral infection, vaccination, concussion, or physical injury—that induces a period of hyperglycemia. This period of high blood sugar levels, combined with the genetically predisposed insulin dysregulation, leads to elevated insulin secretion from the pancreas.
- Sympathetic Nervous System Over-Activation: In individuals with genetic dysregulation of insulin secretion and increased insulin receptor sensitivity on noradrenergic neurons, the hyperinsulinemia results in over-activation of the SNS. The insulin receptors on noradrenergic neurons, now hypersensitive due to the reduced GR function, become over-stimulated by the excess insulin. This triggers down-regulation of NETS and reduced reuptake of norepinephrine, contributing to a state of sympathetic overdrive.
- Downstream Effects of Norepinephrine Dysregulation: Over time, the NET down-regulation and chronic SNS over-activation leads to impaired norepinephrine reuptake, norepinephrine depletion in neurons and eventual down-regulation of adrenergic receptors.
Autonomic Dysfunction Across ME Subtypes
Each ME subtype demonstrates distinct autonomic profiles, reflecting differences in norepinephrine dysregulation and adrenergic receptor responses(24).
ME2: Hypovolemic Postural Orthostatic Tachycardia Syndrome (POTS)
Patients in the ME2 subtype, characterised by insulin dysregulation and hyperinsulinemia, are likely to experience hypovolemic POTS. In this form of POTS, excessive peripheral vasodilation occurs due to the over-activation of eNOS, leading to increased nitric oxide production in the vascular endothelium. Nitric oxide induces vasodilation, which, in ME2 patients, contributes to blood pooling in the lower extremities when standing. This results in reduced venous return to the heart, causing compensatory tachycardia to maintain blood pressure.
ME3: Orthostatic Intolerance (OI) from Norepinephrine Deficiency
In contrast, ME3 patients, who are hypothesised to suffer from norepinephrine deficiency, are more likely to experience orthostatic intolerance (OI) without significant tachycardia. In these individuals, the insufficient norepinephrine release upon standing impairs the body’s ability to maintain adequate blood pressure and vascular tone, leading to symptoms such as dizziness, lightheadedness, and fatigue. OI in ME3 is distinct from POTS in that the heart rate increase upon standing is not as pronounced, and the underlying issue is primarily related to the inability to sustain vascular resistance due to the norepinephrine deficit.
ME1: Initial Orthostatic Intolerance (IOI)
ME1 patients generally exhibit less obvious autonomic dysfunction. However, a subset may report episodes of dizziness or lightheadedness when standing after being supine for an extended period. This phenomenon, termed Initial Orthostatic Intolerance (IOI) involves a temporary drop in blood pressure upon standing, which may self-correct too quickly to be detected by standard blood pressure monitors. One hypothesis is that in ME1, down-regulation of adrenergic receptors delays the vasoconstrictive response normally mediated by norepinephrine. As more extracellular norepinephrine is required to achieve the necessary vascular response, a temporary delay in peripheral vasoconstriction may lead to an initial drop in blood pressure.
Muscle contraction before standing, particularly in the lower body, can alleviate IOI symptoms, and this could be by inducing norepinephrine release before standing, as contracting muscles releases norepinephrine(25). This could raise the norepinephrine levels higher to act upon adrenergic receptors and initiate vasoconstriction before standing.
In summary, the varying autonomic dysfunction across ME subtypes—ranging from POTS in ME2, OI in ME3, to IOI in ME1—reflects the complex interactions between insulin signaling, norepinephrine regulation, and adrenergic receptor responsiveness.
The Effect of Testosterone on Symptom Profile and Illness Risk
Testosterone’s role in increasing the expression of the β2AR may provide protective effects in males, particularly in subtypes ME1 and ME2, which are characterised by higher extracellular NE and receptor down-regulation(26). This may explain the lower prevalence of ME in males as compared to females.
Moreover, testosterone enhances the activity of catechol-O-methyltransferase (COMT), an enzyme that breaks down extracellular norepinephrine into normetanephrine, thereby reducing norepinephrine levels in synaptic clefts(27). Enhanced COMT activity, by reducing norepinephrine accumulation, may prevent over-activation of adrenergic receptors, particularly in the sympathetic nervous system, further decreasing the risk of receptor down-regulation. These factors may contribute to a lower risk of illness onset in males and milder symptom severity in male ME1 and ME2 patients. Research supporting this role of testosterone includes studies showing that female Long COVID patients with lower testosterone levels tend to exhibit more severe symptoms, indicating that testosterone may provide some protection against autonomic dysregulation(28).
However, this protective effect may not extend to ME3, where norepinephrine deficiency is thought to play a central role. In ME3, the up-regulation of COMT by testosterone may exacerbate norepinephrine deficiency by accelerating extracellular norepinephrine breakdown, leading to worsened symptoms in male patients. This subtype may thus present with more severe autonomic dysfunction, cognitive impairments, and fatigue in males due to testosterone’s impact on norepinephrine metabolism. Furthermore, ME3 may be more likely to manifest or worsen during adolescence, a period characterised by increased testosterone levels. The accelerated breakdown of norepinephrine during this developmental stage could predispose ME3 patients to worsening of symptoms such as orthostatic intolerance, cognitive deficits, and fatigue.
Is There a Cholinergic ME Subtype of Noradrenergic Dysfunction?
There may be a possibility of a fourth ME subtype, driven by pathways not previously discussed. While the focus of this hypothesis paper has been on norepinephrine deficiency and NET downregulation caused by insulin dysregulation, there is another potential mechanism of noradrenergic neuron dysfunction: an increase in tyrosine hydroxylase (TH) activity, the rate-limiting enzyme for norepinephrine synthesis. Over-activation of TH could lead to excessive norepinephrine release and disrupt the normal autoreceptor feedback inhibition that regulates its activity. Elevated extracellular norepinephrine could contribute to chronic sympathetic overdrive and postsynaptic adrenergic down-regulation.
One factor that could up-regulate TH activity is increased acetylcholine levels. Acetylcholine can stimulate nicotinic receptors on noradrenergic neurons, leading to up-regulation of TH and heightened norepinephrine production. This mechanism is similar to the effects of nicotine patches, which enhance NE release by acting on nicotinic receptors and increasing TH activity. If this represents a distinct ME subtype, these patients might be distinguished by having high normetanephrine levels without insulin dysregulation or carbohydrate sensitivity. Additionally, if patients have adverse reactions to cholinergic medications like pyridostigmine or nicotine patches, but benefit from anticholinergic medications such as phenergan or cyproheptadine, this may point to high acetylcholine. This subtype could also present with more frequent migraines, potentially managed with anticholinergic treatment strategies.
Potential Links to Endometriosis and Polycystic Ovary Syndrome
Endometriosis
Endometriosis is a chronic gynaecological condition characterised by the presence of endometrial-like tissue outside the uterus, primarily on the pelvic organs. This condition affects approximately 10% of women of reproductive age and is associated with debilitating symptoms such as chronic pelvic pain, heavy menstrual bleeding, and infertility. Emerging evidence suggests a potential overlap between endometriosis and ME, with one research paper indicating that approximately one-third of ME patients are also diagnosed with endometriosis(29). This co-occurrence suggests a possible shared pathophysiology between the two conditions.
One notable feature of endometriosis is the presence of reactive hypoglycaemia in affected individuals. Reactive hypoglycaemia refers to episodes of low blood sugar following a meal, typically a result of excessive insulin release. In a study published by Miles et al. (2001), it was demonstrated that patients with endometriosis exhibit abnormal insulin responses, characterised by heightened insulin sensitivity in peripheral tissues, including nerves(30). The authors found that the cellular membranes of endometriosis patients’ peripheral nerves were hyper-responsive to insulin. This finding is particularly relevant to the hypothesis presented in this paper, which posits that the ME1 and ME2 subtypes may involve increased sensitivity of noradrenergic neuron insulin receptors.
Polycystic Ovary Syndrome (PCOS)
Polycystic Ovary Syndrome (PCOS) is a common endocrine disorder that affects approximately 6-12% of women of reproductive age and is characterised by a combination of hyperandrogenism, ovulatory dysfunction, and polycystic ovaries. One of the key features of PCOS is insulin resistance, where the body’s cells become less responsive to insulin, requiring the pancreas to produce more insulin to maintain normal blood glucose levels. This condition is frequently associated with metabolic disturbances, including obesity, type 2 diabetes, and cardiovascular disease.
Insulin resistance in PCOS has been well-documented in both lean and obese women, with many studies showing that PCOS patients have higher levels of circulating insulin compared to healthy controls. Given the hypothesis of insulin dysregulation in ME, particularly in the ME2 subtype where insulin resistance is a prominent feature, PCOS could be more prevalent among ME2 patients. The insulin resistance in PCOS fits the profile of prolonged and elevated insulin secretion, a characteristic proposed to be part of the ME2 subtype.
PCOS serves as a clear example of how insulin secretion from the pancreas can be influenced by other disorders. In PCOS, the insulin resistance triggers compensatory hyperinsulinemia, where the pancreas releases excessive amounts of insulin in response to the body’s impaired ability to use glucose. This further insulin release not only worsens the metabolic profile in PCOS patients but could also contribute to the noradrenergic neuronal dysregulation hypothesised in ME2.
Dopamine Deficiency in ME
Dopamine is a critical neurotransmitter in the brain and body, playing an essential role in motor control, motivation, reward processing, cognition, and the regulation of the sleep-wake cycle. The dopaminergic system is responsible for transmitting signals between different parts of the brain, particularly the prefrontal cortex, striatum, and substantia nigra. In the brain, dopamine is synthesised from the amino acid tyrosine, which is converted into L-DOPA and subsequently into dopamine. Any disruption in dopamine synthesis or regulation can have widespread physiological and neurocognitive effects, many of which overlap with the symptoms observed in ME.
In the prefrontal cortex, dopamine plays a critical role in higher-order cognitive functions, such as working memory, attention, and executive function. A deficiency in dopamine in this region can lead to cognitive impairments, including difficulties with concentration, mental fatigue, and memory problems—symptoms that are frequently reported by ME patients. Dopamine also regulates the sleep-wake cycle, particularly through its modulation of arousal and alertness. The prefrontal cortex interacts with brain regions like the hypothalamus to maintain circadian rhythms, and a deficiency in dopamine in this region could contribute to the disordered sleep patterns commonly seen in ME, such as unrefreshing sleep and insomnia.
Beyond the brain, dopamine also exerts effects on the peripheral nervous system. It is involved in regulating cardiovascular function, renal activity, and the gastrointestinal system. Dopamine acts as a vasodilator, helping to regulate blood pressure by widening blood vessels, and it promotes sodium excretion in the kidneys, helping to control fluid balance.
Evidence of Dopamine Deficiency in ME
Several lines of evidence suggest that dopamine deficiency may play a role in the pathophysiology of ME. A recent intramural study by the National Institutes of Health (NIH), found reduced levels of L-DOPA in the cerebrospinal fluid of ME patients(4). As L-DOPA is the immediate precursor to dopamine, this finding suggests that ME patients may have impaired dopamine synthesis.
Additionally, clinical observations indicate that many ME patients respond favourably to medications that modulate dopamine activity. A small trial found that 74% of ME patients experienced improvement when treated with low-dose aripiprazole, a partial agonist at dopamine 2 receptors(31). This suggests that targeting the dopaminergic system may alleviate some symptoms of ME, particularly in patients with dopamine dysregulation.
Another key piece of evidence supporting dopamine deficiency in ME is the abnormal prolactin response seen in patients. Prolactin is a hormone whose release is inhibited by dopamine, and elevated prolactin levels could be an indirect marker of dopamine deficiency. During a buspirone challenge test, which assesses the body’s response to serotonin agonism, ME patients released significantly more prolactin than healthy controls(32). This exaggerated prolactin response could suggest impaired dopaminergic inhibition of prolactin secretion, reinforcing the hypothesis that dopamine levels are deficient in ME patients. Elevated prolactin levels may also serve as a potential biomarker for identifying ME patients who are likely to respond to dopaminergic treatments, such as low-dose aripiprazole.
Mechanisms of Dopamine Deficiency in ME
The underlying mechanisms leading to dopamine deficiency in ME are likely multifactorial, involving several interacting processes related to the dysregulation of the noradrenergic and metabolic systems.
Firstly, the dysfunction of noradrenergic neurons, as discussed in previous sections, could directly impair dopamine synthesis. Tyrosine, the precursor for both norepinephrine and dopamine, may be depleted due to noradrenergic dysfunction, reducing the availability of tyrosine for dopamine synthesis. As a result, ME patients, particularly those with the ME3 subtype characterised by norepinephrine deficiency, may also experience reduced dopamine production.
Secondly, insulin dysregulation, which is more pronounced in ME1 and ME2 subtypes, could further exacerbate dopamine deficiency. High insulin levels have been shown to increase the expression of dopamine transporters in dopamine neurons of the striatum(33). This leads to increased reuptake of dopamine from the synaptic cleft, resulting in reduced extracellular dopamine levels. In ME1 and ME2 patients, who may experience hyperinsulinemia, this mechanism could contribute to the depletion of extracellular dopamine, particularly in brain regions involved in reward and motivation, such as the striatum.
Lastly, glucocorticoid receptor resistance could impair dopamine regulation in the prefrontal cortex. Glucocorticoids, such as cortisol, play a key role in stimulating dopamine release in this region, and adequate glucocorticoid signaling is crucial for maintaining cognitive function. In the context of glucocorticoid receptor resistance, the ability of cortisol to modulate dopamine release may be diminished, leading to reduced dopamine levels in the prefrontal cortex. This would further exacerbate cognitive deficits and disturbances in the sleep-wake cycle, which are prominent features of ME.
In summary, dopamine deficiency in ME likely results from a combination of noradrenergic dysfunction, insulin dysregulation, and glucocorticoid receptor resistance. These interacting factors may impair dopamine synthesis, increase dopamine reuptake, and reduce dopamine release. This growing body of evidence underscores the importance of targeting dopaminergic pathways in potential therapeutic strategies for ME.
Mast Cell Activation
Mast cells are immune cells that are predominantly located in tissues near blood vessels and nerves, particularly in the skin, lungs, gastrointestinal tract, and brain. These cells play a crucial role in the body’s immune response, especially in allergic reactions and inflammation. Mast cells contain granules that are rich in histamine, heparin, proteases, and various cytokines. Upon activation, mast cells undergo a process called degranulation, where they release these stored mediators into the surrounding tissue, initiating a cascade of immune responses.
Mast cell activation can occur through various stimuli, including allergens, stress, infection, and neuropeptides. Degranulation is a key event in this process, leading to the release of pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interleukin-1 beta (IL-1β). These cytokines contribute to inflammation and tissue damage, which, when dysregulated, can exacerbate chronic conditions like ME. Excessive mast cell activation and cytokine release are known to cause widespread immune activation, leading to symptoms such as fatigue, pain, cognitive dysfunction, and autonomic disturbances, which are characteristic of ME.
Glucocorticoids, produced by the adrenal glands, are potent anti-inflammatory agents that regulate immune responses, including mast cell activity. They exert their effects by binding to the glucocorticoid receptors (GR) on immune cells, inhibiting cytokine production, and reducing mast cell degranulation. However, in ME patients, GR resistance has been proposed as a contributing factor to immune dysregulation. Resistance to glucocorticoids may reduce their inhibitory effects on mast cells, leading to increased degranulation and chronic inflammation.
Norepinephrine, a key neurotransmitter of the sympathetic nervous system, is also involved in modulating mast cell activity. β2ARs on mast cells typically mediate the anti-inflammatory effects of norepinephrine. However, in ME patients, chronic high levels of norepinephrine, combined with the down-regulation of β2ARs, could impair this protective mechanism, resulting in increased mast cell activation. This dysregulation may contribute to the chronic inflammatory state observed in ME.
The HPA axis normally maintains homeostasis through a negative feedback mechanism. Corticotropin-releasing hormone (CRH), secreted by the hypothalamus, stimulates the release of adrenocorticotropic hormone (ACTH) from the pituitary, which in turn triggers cortisol release from the adrenal glands. Cortisol acts to inhibit further CRH release, completing the feedback loop. However, in ME, glucocorticoid receptor resistance could impair this feedback mechanism, leading to elevated CRH levels. CRH is known to activate mast cells directly, exacerbating mast cell degranulation and perpetuating a cycle of inflammation(34). This interplay between HPA axis dysregulation and mast cell activation may explain the heightened immune activation and systemic inflammation observed in ME patients.
Norepinephrine Dysregulation in the Brain and Its Impact on Astrocyte Function
Norepinephrine is a key neurotransmitter involved in a variety of brain functions, including arousal, attention, cognition, and the regulation of the sleep-wake cycle. However, dysregulation of norepinephrine signaling, particularly chronic elevations in extracellular norepinephrine, can have detrimental effects on brain function and may contribute to the pathophysiology of ME. One critical aspect of norepinephrine dysregulation is it’s impact on astrocytes, the glial cells that play a crucial role in regulating brain energy metabolism, neurotransmitter balance, cerebral blood flow, and waste clearance through the glymphatic system. In ME, norepinephrine dysregulation could lead to astrocytic dysfunction, resulting in a cascade of metabolic and neuronal disturbances.
Norepinephrine Dysregulation of Astrocytes and Glucose Uptake
Astrocytes are vital for maintaining brain energy homeostasis, in part by regulating glucose uptake from the bloodstream. Norepinephrine acts on astrocytic β2ARs, which are known to stimulate glucose uptake via increased expression of glucose transporters, such as GLUT1. While this process is essential for meeting the energy demands of the brain during periods of heightened activity, chronic norepinephrine stimulation, as may occur in ME, could lead to excessive glucose uptake by astrocytes, particularly in the hypothalamus and cortex. This overactivity of astrocytes, driven by continuous norepinephrine signaling, has been proposed to lead to an overproduction of lactate(35).
Excessive astrocytic lactate production may contribute to the elevated brain lactate levels observed in ME patients, as demonstrated by magnetic resonance spectroscopy studies(36). Elevated lactate is a marker of metabolic dysfunction and is associated with increased fatigue, cognitive impairment, and altered sleep patterns, all of which are characteristic symptoms of ME.
Impact of High Lactate Levels on Sleep
The accumulation of lactate in the brain has significant implications for sleep regulation. Lactate is thought to act as a signaling molecule that modulates sleep-wake cycles, particularly slow-wave sleep (SWS), which is the deepest and most restorative phase of sleep. Under normal conditions, the brain’s lactate levels decrease during sleep, particularly during SWS, as the brain clears out metabolic byproducts. However, elevated lactate levels, driven by chronic norepinephrine activation of astrocytes, may interfere with this process, leading to disrupted sleep architecture and unrefreshing sleep, which are hallmark features of ME.
Lactate as a Preferential Fuel
Continuous norepinephrine stimulation of astrocytes may also shift the brain’s neuronal fuel preference from glucose to lactate. This metabolic adaptation is similar to what has been observed in patients with diabetes, who experience repetitive hypoglycaemic episodes. In these patients, the brain adapts to rely more heavily on lactate as an energy source, rather than glucose, during periods of hypoglycaemia(37). In ME, chronic norepinephrine activation may cause a similar shift in metabolic substrate utilisation, particularly in astrocytes of the hypothalamus, which plays a crucial role in energy balance and homeostasis. Moreover, prolonged reliance on lactate as a fuel may impair neuronal function, as neurons require a delicate balance between glucose and lactate metabolism to maintain optimal function.
Sex Differences in Astrocytic Response to Norepinephrine Dysregulation
Emerging evidence suggests that astrocytes may respond differently to chronic norepinephrine dysregulation in males and females, with distinct implications for metabolic and neuronal function. A study by Rahman et al. (2020) highlighted these sex-specific differences in astrocytic metabolism and signaling in response to elevated norepinephrine levels(38). In male rats, chronic norepinephrine stimulation led to reduced astrocyte β2-AR expression, which could result in decreased glucose uptake and lactate production in astrocytes. This suggests that males may have a protective down-regulation of the β2-AR pathway in response to high norepinephrine, which helps to moderate the metabolic impact.
In contrast, female rats exhibited increased β2-AR expression in response to high norepinephrine, which could amplify glucose uptake and lactate production by astrocytes. The up-regulation of β2-ARs in females means that they may be more vulnerable to the effects of high extracellular norepinephrine in the brain, as the overstimulation of astrocytes can lead to excessive lactate accumulation and more pronounced metabolic disturbances.
Norepinephrine, Astrocytes and the Glymphatic System
Astrocytes play a central role in regulating the glymphatic system, a waste clearance pathway in the brain that is responsible for removing metabolic byproducts and toxins, particularly during sleep. Norepinephrine is a key regulator of the glymphatic system, as it modulates astrocytic aquaporin-4 (AQP4) channels, which facilitate the movement of cerebrospinal fluid through the brain parenchyma. Under normal conditions, norepinephrine levels decrease during sleep, allowing the glymphatic system to function optimally and clear out waste products such as amyloid-beta and tau proteins. However, in ME, chronic high extracellular norepinephrine may impair glymphatic function by interfering with AQP4 channel regulation, leading to the accumulation of toxic metabolic byproducts in the brain. This could further exacerbate cognitive dysfunction and contribute to the development of neuroinflammatory processes in ME.
Norepinephrine, Astrocytes and Neurotransmitter Regulation
Astrocytes play a key role in regulating neurotransmitter levels, particularly glutamate and gamma-aminobutyric acid (GABA), both of which are crucial for maintaining proper excitatory and inhibitory balance in the brain. The neurotransmitter norepinephrine has a significant impact on astrocyte-mediated glutamate clearance, and its dysregulation could contribute to neurotransmitter imbalances in ME patients. In healthy states, norepinephrine enhances astrocytic glutamate uptake by increasing the expression of glutamate transporters, thereby reducing extracellular glutamate and preventing excitotoxicity.
In ME subtypes characterised by high norepinephrine levels (ME1 and ME2), this increased glutamate uptake by astrocytes could lead to lower extracellular glutamate. Although this might protect against excitotoxicity, a persistent reduction in glutamate levels may disrupt normal synaptic transmission, contributing to cognitive dysfunction and fatigue in these patients. Glutamate is critical for excitatory signaling and cognitive processes, so chronically reduced levels could impair memory formation, learning, and overall brain function. This mechanism could help explain the cognitive impairments often reported in ME patients.
Conversely, in the ME3 subtype, where norepinephrine levels are deficient, astrocytes may take up less glutamate, leading to an accumulation of extracellular glutamate. Elevated glutamate can result in excitotoxicity, which is harmful to neurons and may lead to neuroinflammation, further exacerbating symptoms of cognitive dysfunction, pain sensitivity, and fatigue.
This dysregulation of glutamate and GABA signaling is likely to have widespread effects on brain function, affecting not only cognition but also the sleep-wake cycle, which is commonly disrupted in ME patients.
Astrocytic Regulation of Cerebral Blood Flow
Astrocytes are key mediators of cerebral blood flow, as they respond to neuronal activity by releasing vasoactive substances that dilate or constrict blood vessels. Norepinephrine plays a role in modulating this astrocytic response through its actions on adrenergic receptors. In ME, chronic norepinephrine dysregulation may impair astrocytic regulation of blood flow, leading to reduced oxygen and nutrient delivery to neurons. This could contribute to the cognitive deficits and fatigue commonly experienced by ME patients, as impaired cerebral blood flow can compromise brain function and exacerbate metabolic dysfunction.
Ehlers-Danlos Syndrome and Compensatory POTS: Potential Overlap with Myalgic Encephalomyelitis
Ehlers-Danlos Syndrome (EDS), particularly its hypermobility subtype (hEDS), is characterised by defects in connective tissue, including abnormalities in collagen synthesis. These defects compromise the structural integrity of blood vessels, leading to increased vascular laxity. Vascular laxity results in an inability of blood vessels to constrict effectively, which is necessary to maintain blood pressure when a person stands. The failure of blood vessels to constrict leads to blood pooling in the lower extremities, reducing venous return to the heart and, consequently, reducing stroke volume. As a compensatory mechanism, the heart increases its rate (tachycardia) in an attempt to maintain adequate blood pressure and perfusion. This physiological response underpins POTS that occurs in EDS patients.
The excessive vasodilation seen in EDS, and the subsequent compensatory POTS, leads to symptoms such as dizziness, fatigue, lightheadedness, and palpitations—symptoms that can overlap with those observed in ME. Moreover, EDS patients may experience cognitive dysfunction (“brain fog”) and exercise intolerance, which are also prominent features in ME. Both conditions share autonomic dysfunction as a key element, complicating differential diagnosis between POTS secondary to EDS and autonomic disturbances in ME.
Categorising and Naming Subtypes
Fundamentally, if we consider ME to be a deficiency of norepinephrine in the noradrenergic neuron due to reduced NET reuptake or reduced synthesis, if neither of these processes are happening in EDS patients, would we consider EDS patients to simply have secondary POTS rather than ME? Or would the development of postsynaptic adrenergic receptor down-regulation (which may result from POTS) qualify these EDS patients for an ME diagnosis? Once a clear pathophysiology for these disease subtypes is ascertained, it will be necessary to clarify how they are named and categorised.
One way to categorise these subtypes would be to name the ME3 subtype as Norepinephrine Deficiency Disorder (NDD), with subtypes based on what the cause of the deficiency is. The ME1 and ME2 subtypes could be called Hyperinsulinemic Noradrenergic Disorder (HND), with ME1 being HND Type 1 and ME2 being HND Type 2. If postsynaptic receptor down-regulation has occurred, this could be considered as having moved to Stage 2 HND. All of these diseases could be categorised as a type of Systemic Exertional Intolerance Disease (SEID), as they all have common symptoms of physical and mental exertion exacerbating symptoms.
Potential Treatments
Pharmacological Interventions
Statins, typically used for their cholesterol-lowering properties, can increase insulin receptor resistance. By increasing the insulin receptor resistance of ME1 patients, statins may prevent postprandial hypoglycaemia and increase the expression of the NET on neurons. This should reduce extracellular norepinephrine and, over the course of a few months, up-regulate adrenergic receptors. Additionally, nimodipine, a calcium channel blocker, may be effective in reducing insulin secretion of ME1 patients.
Metformin, a first-line treatment for insulin resistance, could be a cornerstone of therapy in ME2. By improving insulin sensitivity and reducing hyperinsulinemia, metformin could help to prevent down-regulation of NETS and reduce microclots and POTS severity. Nimodipine may be co-prescribed to further reduce insulin secretion, while beta-blockers are often used to manage symptoms of POTS by reducing heart rate and alleviating excessive sympathetic activity.
In ME3, the primary treatment goal is to increase norepinephrine availability. Levodopa and droxidopa, synthetic precursors to norepinephrine, can help raise norepinephrine levels, thus alleviating symptoms of orthostatic intolerance and cognitive dysfunction. Additionally, MAO inhibitors (Monoamine oxidase inhibitors) may prevent the breakdown of norepinephrine from inside the synaptic vesicles, ensuring higher levels of this neurotransmitter in the synaptic vesicles of the neurons.
Non-Pharmacological Interventions
A low-glycemic index diet helps stabilise blood glucose levels, thereby preventing spikes in insulin that could otherwise exacerbate the sympathetic over-activation. Supplementation with norepinephrine precursors, such as L-Tyrosine and L-Phenylalanine, alongside essential cofactors like folate, may support norepinephrine synthesis. Additionally, anti-histamines may be useful in addressing mast cell activation, which is often a comorbid condition in ME, providing relief from inflammatory and allergic symptoms. ME2 patients may benefit from taking the supplement Tauroursodeoxycholic Acid (TUDCA) as it can increase insulin sensitivity and increase insulin clearance. The supplement nattokinase may help with reducing microclots in ME2 patients. ME3 patients may benefit from using the nicotine patch, as it can up-regulate tyrosine hydroxylase and increase norepinephrine synthesis and release, although whether a patient benefits may depend on what is causing their reduced norepinephrine synthesis.
Testing for Subtypes
To differentiate ME subtypes, several tests can be employed to assess norepinephrine and insulin dynamics. First, measuring levels of norepinephrine metabolites, such as DHPG and normetanephrine, could help distinguish ME3 (NE deficiency subtype) from ME1 and ME2. In ME3, both of these metabolites would likely be reduced, reflecting a deficiency in norepinephrine metabolism. However ME1 and ME2 subtypes are more likely to involve low DHPG and high normetanephrine levels.
For ME1 and ME2, an Oral Glucose Tolerance Test (OGTT) with insulin measurements at multiple time points (15 minutes, 1 hour, 2 hours, and 3 hours post-ingestion) could differentiate the two subtypes. ME1 is marked by either an early spike in insulin due to hypersecretion or low levels of insulin secretion due to hyper-sensitised insulin receptors. ME2 is indicated by prolonged insulin elevation, indicating insulin resistance. If low insulin secretion is found, but the patient does notice a strong sensitivity to carbohydrates, genetic testing for insulin receptor variants may indicate that the patient has a hypersensitive insulin receptor response to insulin.
Conclusion
In conclusion, this paper proposes that distinct subtypes of ME are driven by different underlying mechanisms, primarily involving norepinephrine dysregulation and insulin signaling abnormalities. ME1 and ME2 appear to be associated with hyperinsulinemia-induced noradrenergic dysfunction, while ME3 is characterised by norepinephrine deficiency. The unique metabolic and autonomic features of each subtype suggest that targeted treatment approaches, such as insulin modulation, norepinephrine precursors, and autonomic interventions, could be more effective than generalised therapies. By refining diagnostic criteria based on insulin response and norepinephrine levels, these subtypes can be better identified, allowing for more personalised and effective management strategies.
Author Contributions
TC confirmed sole responsibility for the following: theory/hypothesis conception, research, analysis and interpretation of research, and manuscript preparation.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Conflicts of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Arron HE, Marsh BD, Kell DB, Khan MA, Jaeger BR, Pretorius E. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: The Biology of a Neglected Disease. Front Immunol. 2024;15:1386607. DOI: 10.3389/fimmu.2024.1386607.
- Słomko J, Estévez-López F, Kujawski S, Zawadka-Kunikowska M, Tafil-Klawe M, Klawe JJ, et al. Autonomic Phenotypes in Chronic Fatigue Syndrome (CFS) Are Associated with Illness Severity: A Cluster Analysis. J. Clin. Med. 2020, 9(8), 2531. DOI: 10.3390/jcm9082531.
- Morris G, Anderson G, Maes M. Hypothalamic-Pituitary-Adrenal Hypofunction in Myalgic Encephalomyelitis (ME)/Chronic Fatigue Syndrome (CFS) as a Consequence of Activated Immune-Inflammatory and Oxidative and Nitrosative Pathways. Mol Neurobiol. 2016; 2017 Nov;54(9):6806-6819. DOI: 10.1007/s12035-016-0170-2.
- Walitt B, Singh K, LaMunion SR, Hallett M, Jacobson S, Chen K, et al. Deep Phenotyping of Post-Infectious Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Nat Commun. 2023; 15(1):907. DOI: 10.1038/s41467-024-45107-3.
- Hoel F, Hoel A, Pettersen IK, Rekeland IG, Risa K, Alme K, et al. A Map of Metabolic Phenotypes in Patients with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. JCI Insight. 2021;6(16):e149217. DOI: 10.1172/jci.insight.149217.
- Das S, Taylor K, Kozubek J, Sardell J, Gardner S. Genetic Risk Factors for ME/CFS Identified Using Combinatorial Analysis. J Transl Med. 2022;20(1):598. DOI: 10.1186/s12967-022-03815-8.
- Wyller VB, Vitelli V, Sulheim D, Fagermoen E, Winger A, Godang K, et al. Altered Neuroendocrine Control and Association to Clinical Symptoms in Adolescent Chronic Fatigue Syndrome: A Cross-Sectional Study. J Transl Med. 2016;14(1):121. DOI: 10.1186/s12967-016-0873-1.
- Nguyen CC, Kumar S, Zucknick M, Kristensen VN, Gjerstad J, Nilsen H, Wyller VB. Associations Between Clinical Symptoms, Plasma Norepinephrine, and Deregulated Immune Gene Networks in Subgroups of Adolescents with Chronic Fatigue Syndrome. Brain Behav Immun. 2019;76:82-96. DOI: 10.1016/j.bbi.2018.11.008.
- Kavelaars A, Kuis W, Knook LME, Sinnema G, Heijnen CJ. Disturbed Neuroendocrine-Immune Interactions in Chronic Fatigue Syndrome. J Clin Endocrinol Metab. 2000;85(2):692-696. DOI: 10.1210/jcem.85.2.6379.
- Fry AC, Schilling BK, Weiss LW, Chiu LZF. β2-Adrenergic Receptor Downregulation and Performance Decrements During High-Intensity Resistance Exercise Overtraining. J Appl Physiol. 2006;101(6):1664-72. DOI: 10.1152/japplphysiol.01599.2005.
- Goto T, Kikuchi S, Mori K, Nakayama T, Fukuta H, Seo Y, et al. Cardiac β-Adrenergic Receptor Downregulation, Evaluated by Cardiac PET, in Chronotropic Incompetence. J Nucl Med. 2021;62(7): 996–998. DOI: 10.2967/jnumed.120.253419.
- Robertson SD, Matthies HJG, Owens WA, Sathananthan V, Christianson NSB, Kennedy JP, et al. Insulin Reveals Akt Signaling as a Novel Regulator of Norepinephrine Transporter Trafficking and Norepinephrine Homeostasis. J Neurosci. 2010;30(34):11305-16. DOI: 10.1523/JNEUROSCI.0126-10.2010.
- Taylor K, Pearson M, Das S, Sardell J, Chocian K, Gardner S. Genetic risk factors for severe and fatigue dominant long COVID and commonalities with ME/CFS identified by combinatorial analysis. J Transl Med. 2023;21(1):775. 2023. DOI: 10.1186/s12967-023-04588-4.
- Cen J, Sargsyan E, Bergsten P. Fatty acids stimulate insulin secretion from human pancreatic islets at fasting glucose concentrations via mitochondria-dependent and -independent mechanisms. Nutrition & Metabolism. 2016;13(59) DOI: 10.1186/s12986-016-0119-5.
- Yuan T, Song S, Zhao T, Duo Y, Wang S, Gao J, Liu S, Dong Y, Li R, Fu Y, Zhao W. Patterns of Insulin Secretion During First-Phase Insulin Secretion in Normal Chinese Adults. Front Endocrinol. 2021;12:738427. DOI: 10.3389/fendo.2021.738427.
- Giunta S, Giordani C, De Luca M, Olivier F. Long-COVID-19 autonomic dysfunction: An integrated view in the framework of inflammaging. Mech Ageing Dev. 2024;218:111915. DOI: 10.1016/j.mad.2024.111915.
- Bramante CT, Buse JB, Liebovitz DM, Nicklas JM, Puskarich MA, Cohen K, et al. Outpatient treatment of COVID-19 and incidence of post-COVID-19 condition over 10 months (COVID-OUT): a multicentre, randomised, quadruple-blind, parallel-group, phase 3 trial. Lancet Infect Dis. 2023;23(10):1119-1129. DOI: 10.1016/S1473-3099(23)00299-2.
- Montefusco L, Ben Nasr M, D’Addio F, Loretelli C, Rossi A, Pastore I, et al. Acute and long-term disruption of glycometabolic control after SARS-CoV-2 infection. Nat Metab. 2021;3(6):774-785. DOI: 10.1038/s42255-021-00407-6.
- Barreto EA, Cruz AS, Veras FP, Leiria LO, et al. COVID-19-related hyperglycemia is associated with infection of hepatocytes and stimulation of gluconeogenesis. Proc Natl Acad Sci U S A. 2023;120(21): e2217119120. DOI: https://doi.org/10.1073/pnas.2217119120.
- Nunes JM, Kruger A, Proal A, Kell DB, Pretorius E. The Occurrence of Hyperactivated Platelets and Fibrinaloid Microclots in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Pharmaceuticals. 2022;15(8):931. DOI: 10.3390/ph15080931.
- Pretorius E, Vlok M, Venter C, Bezuidenhout JA, Laubscher GJ, Steenkamp J, Kell DB. Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc Diabetol. 2021;20(1):172. DOI: 10.1186/s12933-021-01359-7.
- Pretorius E, Venter C, Laubscher GJ, Lourens PJ, Steenkamp J, Kell DB. Prevalence of readily detected amyloid blood clots in ‘unclotted’ Type 2 Diabetes Mellitus and COVID-19 plasma: a preliminary report. Cardiovasc Diabetol. 2020;19(1):193. DOI: 10.1186/s12933-020-01165-7.
- Sun Z, Fan Y, Zha Q, Zhu MY. Corticosterone up-regulates expression and function of norepinephrine transporter in SK-N-BE(2)C cells. J Neurochem. 2010;113(1): 105–116. DOI: 10.1111/j.1471-4159.2010.06587.x.
- Fedorowski A, Fanciulli A, Raj SR, Sheldon R, Shibao CA, Sutton R. Cardiovascular autonomic dysfunction in post-COVID-19 syndrome: a major health-care burden. Nat Rev Cardiol. 2024;21(6):379-395. DOI: 10.1038/s41569-023-00962-3.
- Sheikh NA, Phillips AA, Ranada S, Lloyd M, Kogut K, Bourne KM, Jorge JG, Lei LY, Sheldon RS, Exner DV, Runte M, Raj SR. Mitigating Initial Orthostatic Hypotension: Mechanistic Roles of Muscle Contraction Versus Sympathetic Activation. Hypertension. 2022;79(3):638-647. DOI: 10.1161/HYPERTENSIONAHA.121.18580.
- Carbajal-García A, Reyes-García J, Casas-Hernández MF, Flores-Soto E, Díaz-Hernández V, Solís-Chagoyán H, Sommer B, Montaño LM. Testosterone augments β2 adrenergic receptor genomic transcription increasing salbutamol relaxation in airway smooth muscle. Mol. Cell. Endocrinol. 2020;510:110801 DOI: 10.1016/j.mce.2020.110801.
- Scardapane L, Cardinali DP. Effect of estradiol and testosterone on catechol-O-methyl transferase activity of rat superior cervical ganglion, pineal gland, anterior hypophysis and hypothalamus. J Neural Transm. 1977;40(1):81-6. DOI: 10.1007/BF01250282.
- Boneva RS, Lin JM, Wieser F, Nater UM, Ditzen B, Taylor RN, Unger ER. Endometriosis as a comorbid condition in chronic fatigue syndrome (CFS): secondary analysis of data from a CFS case-control study. Front. Pediatr. 2019; 7: 195. DOI: 10.3389/fped.2019.00195.
- Mathias JR, Franklin RR. Neural dysfunction of the gastrointestinal tract associated with endometriosis: a disease of insulin sensitivity. Fertil Steril. 2002;77(25) DOI: 10.1016/S0015-0282(01)03088-6.
- Crosby LD, Kalanidhi S, Bonilla A, Subramanian A, Ballon JS, Bonilla H. Off-label use of Aripiprazole shows promise as a treatment for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): a retrospective study of 101 patients treated with a low dose of Aripiprazole. J Transl Med. 2021;19(1):50. DOI: 10.1186/s12967-021-02721-9.
- Sharpe M, Clements A, Hawton K, Young AH, Sargent P, Cowen PJ. Increased prolactin response to buspirone in chronic fatigue syndrome. J. Affect. Disord. 1996;41(1):71-76. DOI: https://doi.org/10.1016/0165-0327(96)00075-4.
- Figlewicz DP, Szot P, Chavez M, Woods SC, Veith RC. Intraventricular insulin increases dopamine transporter mRNA in rat VTA/substantia nigra. Brain Res. 1994;644(2):331-4. DOI: 10.1016/0006-8993(94)91698-5.
- Theoharides TC, Singh LK, Boucher W, Pang X, Letourneau R, Webster E, Chrousos G. Corticotropin-releasing hormone induces skin mast cell degranulation and increased vascular permeability: A possible explanation for its proinflammatory effects. Endocrinology. 1998;139(1):403-13. DOI: 10.1210/endo.139.1.5660.
- Fink, K., Velebit, J., Vardjan, N., Zorec, R., & Kreft, M. (2021). Noradrenaline-induced l-lactate production requires d -glucose entry and transit through the glycogen shunt in single-cultured rat astrocytes. Journal of Neuroscience Research, 99(4), 1084–1098. https://doi.org/10.1002/jnr.24783.
- Lee JS, Sato W, Son CG. Brain-regional characteristics and neuroinflammation in ME/CFS patients from neuroimaging: A systematic review and meta-analysis. Autoimmun. Rev. 2024;23:2. DOI: https://doi.org/10.1016/j.autrev.2023.103484.
- Litvin M, Clark AL, Fisher SJ. Recurrent hypoglycemia: boosting the brain’s metabolic flexibility. J Clin Invest. 2013;123(5): 1922–1924. DOI: 10.1172/JCI69796.
- Ibrahim MMH, Bheemanapally K, Briski KP. Norepinephrine regulation of adrenergic receptor expression, 5′ AMP-activated protein kinase activity, and glycogen metabolism and mass in male versus female hypothalamic primary astrocyte cultures. ASN Neuro. 2020;12:17. DOI: 10.1177/1759091420974134.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



