
Submitted:
19 September 2024
Posted:
23 September 2024
You are already at the latest version
Abstract
Peptidergic GPCR systems are broadly distributed in the human body and regulate numerous physiological processes by activating complex networks of intracellular biochemical events responsible for cell regulation and survival. Their failure due to overaction, ill-function, or blockade gives rise to cell disturbances, eventually causing disease if compensatory mechanisms do not suffice. Revision of updated experimental research provided an evident relationship associating peptidergic GPCR malfunction with tumor formation and maintenance resulting from uncontrolled cell proliferation and migration, colonization, inhibition of apoptosis or altered metabolism, and increased angiogenesis in tumoral tissues. Determination of the implication of GPCR peptide signaling in specific neoplasia is crucial to designing tailored pharmacological treatments to counteract or dismantle the origin of the signaling circuitry causing cellular disruption. In some cases, particular ligands for these receptors may serve as concomitant treatments to aid other pharmacological or physical approaches to eradicate neoplasias.
Keywords:
G-protein-coupled receptors (GPCR)
; Peptide
; Cell growth
; Apoptosis
; Peptide antagonists
; Peptide signaling pathways
; Cancer
; Cancer therapy
; Drug design
1. Introduction
The word "cancer" refers to several complex diseases with various etiological factors, acting alone or combined, including genetic background/predisposition, environmental influences, aging, hormonal factors, and lifestyle choices. These factors can trigger abnormal biochemical processes and disrupt the cell's available compensatory mechanisms against damage. If the cells cannot recover from these disruptions, tumors can be generated and progress. Each neoplasia has distinct characteristics based on the type of cell and tissue involved and the specific molecular events and transcriptional programs affected. By studying the biochemical processes for every kind of tumor, we may uncover new biochemical incidences and new opportunities for developing targeted therapies to combat these diseases [1,2,3,4,5,6].
G-protein-coupled receptors (GPCRs) form an extensive family of proteins (more than 800) that convey extracellular chemical or sensory information to trigger intracellular changes affecting short-term intermediary metabolic events and long-term gene expression programs through signaling cascades. GPCRs participate in numerous biochemical events associated with physiopathological processes, including cell transformation, apoptosis, growth, and migration [7].
Different classification criteria categorize GPCRs into several families or groups. Approximately 350 GPCRs are non-sensory receptors (processing amines, peptides, proteins, lipids, ions, nucleotides, or hydroxycarboxylic acids), whereas the rest (approximately 450) respond to sensory (odors, tastes, photons, or pheromones) stimuli. Based on sequence homology and functional resemblance, GPCRs are included in eight classes, six of which are present in humans. These include class A (rhodopsin), class B (secretin and adhesion), class C (glutamate), class T (taste), class O (orphans), class E (frizzled/smoothened), and classes D (fungal mating pheromone receptors), and E (cAMP receptors) absent in vertebrates [8,9].
Several GPCRs are presently druggable targets for numerous pathological conditions, including cardiovascular, chronic inflammation, pain, or cancer pathologies [10,11]. The weaponry of therapeutic compounds includes synthetic ligands acting on the receptor as molecules displaying full agonism (triggering maximal signal), partial agonism (eliciting activity below the maximum response), inverse antagonism (inhibition of constitutive receptor activity), and neutral antagonism (the constitutive receptor activity is not changed). Other drugs not binding at the orthosteric but at an allosteric site (outside or within the transmembrane helices) are PAMS (positive allosteric modulators) and NAMS (negative allosteric modulators), which activate or inhibit, respectively, the responses raised by orthosteric ligands. Furthermore, the list grows with the so-called bitopic ligands (binding both the orthosteric and the allosteric sites within the GPCR) [12,13].
Approximately 90 class A and 20 class B GPCRs bind and process around 180 peptide signals. The majority of GPCRs binding peptides belong to class A (for example, receptors for opioids, tachykinins, galanin, neuropeptide Y, orexin, cholecystokinin, bradykinin, somatostatin, endothelin, neurotensin, bombesin, vasopressin, kisspeptin, thyrotropin-releasing hormone, melanocortin, or apelin). A few GPCR class B (secretin) include receptors for glucagon, secretin, corticotropin-releasing factor, parathyroid, vasoactive intestinal peptide, calcitonin or pituitary adenylate cyclase-activating peptide [9].
The interaction between peptides and GPCRs is intricate. It encompasses various possibilities, such as one peptide binding to one GPCR, one peptide binding to multiple GPCRs, or multiple peptide ligands binding to multiple GPCRs [14,15]. The complexity is compounded by many GPCRs being labeled orphans, and their peptide activation awaits identification and corroboration [11,13].
This report reviews recent evidence and findings concerning the fine structure, dynamic conformations, and plausible mechanistic involvement of eight peptidergic GPCRs in tumorigenesis. It also discusses the examination of these systems as pharmacological objects.
2. GPCR Peptides and Their Receptors
Several natural peptides bind with different affinities and activate specific GPCRs with varied localization, triggering a complex network of intracellular signals that control essential cell activity in short, mid, and long-term periods. Peptidergic GPCR systems share common properties concerning their broad distribution and shared signaling pathways regulating an array of pleiotropic cell responses affecting vital physiology [14,16,17]. We focus on advances concerning eight GPCR peptidergic systems and their relationship with tumorigenesis and expansion. Additionally, we describe some synthetic and specific activating or silencing compounds modulating peptidergic GPCR signaling that may be valuable tools for treating some types of cancer alone or as concomitant measures [18]. The systems comprising the main endogenous ligands and their respective GPCRs are schematically shown in Figure 1.
Several opioid peptides (for instance, enkephalins, endorphins, or dynorphins) and three main types of opioid receptors, mu (MOPR), delta (DOPR), and kappa (KOPR) [19,20] form the endogenous opioid system, a primary regulator of intrinsic analgesia, cardiovascular responses, gastrointestinal and hepatic function, respiration, thermoregulation, or immunological reactions (see the recent review series by [21]).
The tachykinin system comprises 3 receptors, namely neurokinin 1, 2, and 3 (NK-1R, NK-2R, and NK-3R), and three main natural ligands, substance P (SP), neurokinin A and neurokinin B, showing different affinities for the receptors. Also, hemokinin-1 is a natural ligand for NK-1R in peripheral tissues [22]. The tachykinin system is widely distributed in the nervous system and other non-neural cells and tissues and influences numerous physiological functions, such as smooth muscle contraction, cell proliferation, pain, inflammation, or tissue regeneration [23,24].
Four neuropeptide Y (NPY) receptors, NPYR 1, 2, 4, and 5, and a handful of peptide ligands with different affinities for the receptors coordinate multiple activities (analgesia, angiogenesis, allergy responses, inflammation mechanisms, or energy homeostasis) from their ubiquitous presence within the human body [25,26,27].
The galanin and GPR151 systems include two ligands, galanin and galanin-like peptide activating three galanin receptor types: GALR1, GALR2, and GALR3 [28]. The galanin system is distributed widely in neural and non-neural tissues and impacts many physiological processes, from pain to metabolism, innate immunity, and inflammation coordination, involved in brain and gastrointestinal function [29,30]. A galanin-binding receptor, GPR151 [14,31], previously considered an orphan GPCR, exhibits high structural similarity with galanin receptors and influences metabolic routes controlling glucose homeostasis [32] and trigeminal neuropathic pain [33]. However, it is still unclear and requires further elucidation whether galanin behaves as a proper functional endogenous ligand for GPR151 in CNS structures [34] and the precise role of the GPR151 receptor system.
The calcitonin system is made up of two secretin GPCRs, CACRL (calcitonin receptor-like) and CALCR (calcitonin receptor), regulated by receptor-activity modulating proteins (RAMPs) [34,35,36], and several endogenous ligands comprising calcitonin (CT) adrenomedullin/intermedin (AM2/IMD), adrenomedullin (AM), alpha- and beta-calcitonin gene-related peptides (α-CGRP and a (β-CGRP) and amylin (AMY) [37,38]. The calcitonergic system is vastly distributed and assists many functions like, for example, hormone secretion, pain control, blood calcium regulation, food intake modulation, maintenance of vascular tone, angiogenesis, or immunoregulation [39,40,41].
Two neurotensin receptors, NTSR1 and NTSR2, bind two principal endogenous peptides, neurotensin (NT) and neuromedin N. The neurotensin system [42] influences energy balance, pain control, stress, gastrointestinal motility and secretion, and brain reward mechanisms [43,44,45,46].
Two families of peptides, apelin [47] and elabela (ELA, an acronym meaning "epibole late because endoderm late") [48,49], activate the apelin receptor (APLNR). This system distributes in several tissues (placenta, heart, lungs, brain) and influences basic physiological mechanisms affecting cardiovascular function, inflammation events, insulin secretion, or fluid homeostasis [47,50,51].
A family of endogenous ligands, including corticotropin-releasing hormone (CRH), urocortins, and two receptor types, CRH1 and CRH2, build up the corticotropin-releasing hormone system. A broad and scarce overlapping distribution of these peptides and their distinct receptor affinities determine a complex collection of actions modulating stress and neuroendocrine responses [52,53,54,55].
3. Peptidergic GPCR Structural Features
Recent advances in determining GPCR three-dimensional structures and alternative conformations revealed orthosteric binding sites' geometry and the functional significance of insurmountable antagonism, activation/inactivation states, or allosteric regulation [20,56,57,58]. The detailed understanding of the setting up of signaling complexes at the plasma membrane and in intracellular endosomes provides new insights for understanding the role of peptidergic GPCR function and cell dysfunction. Still, challenging questions defy basic research to understand how and to what extent peptidergic GPCR function influences biochemical pathways responsible for cancer development, cell survival, or cell resistance to different therapy interventions. Some peptidergic GPCR systems have been the focus of research focused on mechanisms responsible for the appearance of various pathologies, including cancer, and the design of selective pharmacological therapies [14,18,59,60,61].
Where available, some essential features about structure-activity and delineation of orthosteric and allosteric sites in some peptidergic GPCRs are described next. Figure 2 shows representative structures of peptidergic GPCR objectives of this report taken from the Protein Data Bank repository [62].
Structural studies of three opioid receptors, MOPR, DOPR, and KOPR [20,64,65], have revealed details connected with their relationship with effector proteins, such as protein effectors, agonists, and antagonists. This work will not detail the ligand binding interactions of all peptidergic GPCR mentioned. However, a few structural examples intend to illustrate the importance of defining orthosteric and allosteric grooves in designing and developing drugs to abrogate or modulate their activities. For example, the cryo-electron microscopy (EM) structure of MOPR bound to agonist fentanyl reveals its position within the orthosteric site (PDB ID 8EFL) [64], between helices 2, 3, 5, 6, and 7 of the transmembrane regions. The phenylethyl structure of fentanyl directs towards a "minor pocket" defined by residues of transmembrane 2 (TM2) and 3 (TM3), whereas the propionyl moiety establishes hydrophobic contacts with residues of TM5 and TM6. Fentanyl, like many other MOPR agonists, interacts with Asp149 in TM3 (Figure 3, A).
Interestingly, when comparing the interaction of morphine, an agonist, and naloxone, an antagonist of the MOPR agonist, it revealed that these drugs with similar chemical structure (but different bond torsion profiles) interact with MOPR in a similar groove where they accommodate distinctly [66] explaining their different outcomes when binding to the receptor. One of the main features of their diverse coupling lies in the differential dynamic recognition of residues Ile322 and Y326 (mouse MOPR) by the drugs. The mutation I322A (shortening of the aliphatic chain of Ile) attenuated morphine activity and converted naloxone into a full agonist [66].
The analysis of the structure of the NK-1R bound to the selective antagonist aprepitant unmasked the characteristics of its binding pocket compared with the binding site of the natural ligand substance P. The Met11 residue in the N-terminal region of the undecapeptide poses deep into the structure of the receptor, and the carboxy-terminal region interacts with the extracellular domains defined by extracellular domain 2 (ECL2) and the outer part of transmembrane helices I, VI and VII [58]. Aprepitant occupies a site defined mainly by the lateral chain of some amino acids. Figure 3 (B) compares the binding grooves of SP and aprepitant. The morpholine central ring of aprepitant lies between residues F268 in helix VI and Q165 in helix IV, and residues N109, P112, and I113 provide hydrophobic contacts in the surface of helix III. The positioning of F264, F268, and W261 in helix VI towards the trifluoro methyl groups of the aromatic ring fixes the positioning of W261 and determines the inactivation state of NK-1R induced by the antagonist (Figure 3, inset in B) [58,67].
The binding site of neuropeptide Y (NPY1-36) to NPY-1R is tightened mainly through weak interactions of residues of the C-terminal region with amino acids situated deeply within the core of the protein [68]. The A panel of Figure 4 represents the position of amino acid residues in the agonist and the receptor and indicates their interactions with dashed-colored lines. Briefly, NPY Tyr36 interacts through a hydrogen bond with Q219 in transmembrane domain V (TM V) and through hydrophobic engagement with I124 in TM III. Arg33 connects with N283 (TM VI) through a hydrogen bond and with F286 (TM VI) and F302 (TM VII) through π-cation interactions. Arginine35 sets electrostatic bonding with D287 (TM VI) and van der Waals forces with F173 (TM IV). Residues Thr32 and Gln34 establish non-polar interactions with Y100 (TM II).
Cryo-EM structure analysis of galanin receptor 1 (GALR1) bound to N-terminal truncated galanin (GAL1-16) showed that the peptide interacts with the receptor uniquely, with the N-terminus adopting an alpha helix conformation. The C-terminus would adopt a more disorganized loop random structure. Galanin sits on the extracellular moiety of the transmembrane helices of the receptor with an almost horizontal positioning [69]. The receptor hosts the ligand peptide in a cavity delimited by TM VI and VII and extracellular loops ECL2 and ECL3 (the main weak bonding is illustrated in Figure 4 (B).
Cryo-EM maps of the complex formed by Gs, the human calcitonin (CAL) receptor (CLR), the receptor-activity modifying protein (RAMP1), and calcitonin gene-related peptide (CGRP) have unmasked relevant features that explain the function of this peptidergic system [70]. RAMP1 extensively contacts CLR both in the extracellular domain and the transmembrane region. The carboxy-terminal region of CGRP (residues Phe37, Ala36, Lys35, and Ser34) contacts with residues F83 and P85 in the extracellular part of RAMP1 (Figure 5, A), whereas the N-terminal region of the peptide buries deeply into the transmembrane region of CLR arranging contacts with CLR residues (Figure 5, B). Residues I298 and L302 (in TM V) and M223 and Y227 (in TM III) delimit a hydrophobic bottom of the binding cavity. Positions in ECL1 and ECL2 are essential for arranging the protein receptor as an active conformer [70,71].
Crystallographic and X-ray diffraction analysis of rat neurotensin (NTS) 1 receptor (NTS1R) bound to the C-terminal sequence of NTS (amino acids 8 to 13, RRPYIL), capable of activating the receptor offers structural details worth considering. The peptide places itself into the core of the receptor protein almost perpendicularly to the plasma membrane [72]. The ligand binding hole is delimited by amino acid residues in ECL2, TM III, TM VI, and TM VII (Figure 6, A). Inverse agonist SR48692 (2-[[1-(7-chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)pyrazole-3 carbonyl]amino]adamantane-2-carboxylic acid) shares the same binding site as NTS8-13 but establish some distinct interactions. For example, in contrast with the NTS positioning, the inverse agonists hydrogen bond with Y351 (in TM VII) instead of Y146 (in ECL2). On the other hand, the antagonists, through their adamantyl group, share the same hydrophobic surface contacted by the carboxy-terminal end of NTS8-13 in the receptor core, although this surface appears more extended [73]. Recent studies emphasize the importance of recognizing the different conformers adopted by the receptor and its molecular dynamics when hosting different ligands to attain efficacious drug design [74].
The structure of AMG3054 (the apelin17 mimetic peptide), bound to the apelin receptor (APLNR), was resolved with X-ray diffraction analysis, giving details concerning the coupling coordinates of the peptide within the receptor [75]. The C-terminal end of the peptide deepens into the transmembrane structure of APLNR, whereas the N-terminus extends to the outer surface. A cavity delimited by hydrophobic amino acids provides hydrophobic interactions with the peptide: L173, Y185, and Y182 in ECL2, W85 in TM II, and F291 and Y299 in TM VII (Figure 6, B). Recently, cryo-EM analysis of APLNR bound to different ligands revealed fundamental structural features explaining biased signaling (Gs protein versus β-arrestin activity) with relevant implications in drug design [56].
Early studies confirmed that the peptide ligand CRF, adopting an alpha-helix conformation, binds primarily to the extracellular part of the CRF1R, a class B secretin GPCR, and also provided relevant details concerning the interaction of the C-terminal amidated residue of the peptide into the structure of the receptor [76]. Recent structural analysis employing X-ray free-electron laser methodology applied to a monoclinic crystal of CRF1R, bound to antagonist BMK-I-152 ([8-(4-bromo-2,6-dimethoxyphenyl)-2,7-dimethylpyrazolo[1,5-α][1,3,5]triazin-4-yl]-N, N-bis-(2-methoxyethyl) amine and docking simulations solved the structure of the complex [77]. The antagonist is accommodated in a cavity delimited by weak interactions with CRF1R residues in TM II, III, V, VI, and VII (Figure 7). The inset depicts the Gaussian volume representation of BMK-I-152.
4. Peptidergic GPCR Signaling Circuitries and Cancer
Once bound to their respective GPCRs, agonist peptides induce versatile and adaptable intracellular signaling cascades, leading to cell metabolism control and switching on or off transcriptional programs. Peptidergic GPCRs may signal through the alpha or beta-gamma subunits of different heterotrimeric G proteins and β-arrestins, as well as other ancillary proteins, for example, regulators of G-protein signaling (GRS) or receptor kinases [78], generating adaptable signaling pathways from the plasma membrane or from intracellular compartments that share intracellular effectors and mutually regulate with other signals, peptidergic or not, processed by the cell. Intracellular effectors regulate intracellular messengers (cAMP, DAG, Ca2+), modulating biochemical events encharged of short-term metabolic effects or long-term events related to gene expression (Figure 8, A and B) [16,33,79,80,81,82].
Peptidergic GPCR systems may fail in several ways, affecting harmonic cell functioning should compensatory mechanisms override. Among the causes of malfunction, we could consider abnormal overexpression, overstimulation by ligand overproduction, protein mutations, aberrant dimerization or oligomerization, truncation, and altered internalization. When the receptor does not adequately govern cellular responses associated with cell growth, migration, inflammatory outcomes, oxidative stress, increased angiogenesis, apoptosis inhibition, or mitochondrial dysfunction, it may provoke the appearance of cancer (Figure 8, right panel) [61,83,84,85].
It's important to note that not all peptidergic GPCR systems function alike or regulate cell activity in similar ways. Therefore, it is crucial to understand the specific mechanisms governed by these systems and the implications of their altered function concerning the onset and perpetuation of cancer [7,86].
Recent experimental evidence associating peptidergic GPCR systems with cancer is summarized below to underlie the importance of these receptors as possible targets in cancer therapy through strategic drug design.
4.1. The Opioid System
Opioid receptors are broadly distributed in the nervous system and peripheral tissues. Their involvement in cancer has been reported for different types of neoplasia [87]. In colorectal cancer, for example, the epithelium is struck by opioid-induced changes in the microbiota populations and altered inflammation, angiogenesis, and apoptosis mechanisms [88]. Delta opioid receptors (DOPR) appeared highly expressed in human and murine breast cancer tissues, and their stimulation-induced metastasis via the JAK1/2 signaling pathway was abolished by the administration of a DOPR antagonist [89]. The opioid drug agonist tramadol (2-[(dimethylamino)methyl]-1-(3-methoxyphenyl)cyclohexanol) activated mu-opioid receptors (MOPR) and exerted an antitumor effect in pancreatic ductal carcinoma cell lines by reducing their invasive and proliferative profile through decreasing proteins controlling the cell cycle [90].
Notwithstanding some controversy concerning whether opioids stimulate or suppress cancer development and durability, further studies should clarify their precise role in cell proliferation and migration, angiogenesis, and immunity responses in several tumors to adequately apply agonists and antagonists in potential specific treatments [91]. Also, deciphering the participation of opioids in cancer is very important since they may sustain cancer when frequently used as analgesics, for example, in neoplasia-associated pain [92,93].
4.2. The Neurokinin System
Extensive basic research performed in different experimental models, from cell cultures to in vivo procedures, has reported the involvement of neurokinin receptors, especially the NK-1R type, in various cancers [67,94,95,96,97,98,99].
In vitro and in vivo studies reported the antiproliferative activity of an NK-1R antagonist, aprepitant (a morpholine derivative), combined with 5-fluorouracil in colorectal cancer cells. The drugs displayed a synergistic effect, which upregulated genes implicated in apoptosis (Bax, p53) [100]. The undecapeptide substance P, through NK-1R, induced oxidative stress and promoted cancer progression in prostate cancer lines PC3 and LNCaP by increasing reactive oxygen species (ROS) and decreasing oxidoreductase activities. The antagonist aprepitant reversed the observed effects [101]. The NK-1R system appeared overstimulated in intrahepatic cholangiocarcinoma cells, and aprepitant showed an antiproliferative profile by activating apoptosis and autophagia via ROS increase and JNK signaling activation [102]. L760,033 (a phenyl piperidine derivative), an NK-1R antagonist, increased apoptosis in SiHa cells (a human cervical cancer line) through silencing ERK signaling and the controlling of anti-apoptotic Bcl-2 and pro-apoptotic BAX proteins [103]. The synergistic action of aprepitant and 5-aminolevulinic acid abolished cell viability and migration of glioblastoma multiforme [104].
4.3. The NPY System
The malfunction of the NPY GPCR system has been associated with more than 20 types of cancer [105].
An immunohistochemistry study showed high expression of NPY peptide and the NPY receptors 1, 2, and 5 in cells from primary prostate cancer and bone metastasis, compared with healthy tissue, suggesting the implication of this peptidergic system from the first phases of the tumor progress to advanced metastatic stages of the disease [106]. Hypoxia-induced stimulation of the NPY5R system in Erwin sarcoma cells caused mitotic alterations, leading to metastasis and colonization in other tissues [107]. NPY5R has also been implicated in the capacity of neuroblastoma cells to migrate and invade other tissues through cytoskeleton modifications mediated by RhoA [108]. In a study centered on the analysis of genes altered in colon cancer, an ensemble of 10 genes, including the one encoding NPY, acted as an altered network associated with the early and advanced stages of the disease [109]. Antagonizing NPY1R and NPY5R in breast cancer cell lines MDA-MB.231 and MCF7 in low oxygen partial pressure blocked NPY-dependent cell proliferation and migration [110]. In hypoxia conditions, the sensitivity of breast cancer cells to NPY stimulation may be due to the modulation of 5' promoter regions of the genes NPY1R and NPY5R by the hypoxia-inducible factor, HIF [111]. Applying NPY2R antagonists in human colorectal adenocarcinoma cells (HT29) reduced angiogenesis and tumor growth. ERK/MAPK was the main signaling pathway involved in the process [112].
4.4. The Galanin System
The galanin peptide is highly expressed in neuroendocrine and non-neuroendocrine tumors [86,113,114].
Galanin immunohistochemical analysis of human colorectal adenocarcinoma samples revealed a higher expression of the peptide in lower stages of the disease (I-III, TMN tumor grading) than in advanced stage IV. On the other hand, a transcriptome study in the same samples showed an upregulation of specific genes concerned with the modulation of cell cycle and cell division, transcriptional control, and immunity regulation in the late stages of the disease compared with early stages[115]. This report, thus, correlates galanin downregulation with advanced colorectal adenocarcinoma stages. Immunohistochemistry studies of GAL3R in colorectal adenocarcinoma samples indicated that the high immunoreactivity signal for GAL3R correlated with a favorable clinical prognosis and extended patient survival [116]. In human glioma cell lines U251 and T986, galanin activating the GAL1R exhibited an antiproliferative profile [117].
On the other hand, secreted galanin by head and neck squamous carcinoma cells favored tumor progression [118]. A specific agonist (M89b, a truncated galanin analog) of GAL2R showed an antitumoral effect of patient xenografts of pancreatic ductal adenocarcinoma in mice [119]. Additionally, to the presence and role of the galanergic systems in different tumors, their occurrence in macrophages and other immune cells infiltrating tumors represents an essential feature of galanin as a participant in the neoplasia microenvironment regulation [86,114,120].
The effects of galanin on GAL receptors appear complex and inconspicuous and may result in proliferative or antiproliferative outcomes depending on the tumor type and the receptor ligand-triggered. Therefore, further exploration of their participation in neoplasias is needed [113].
4.5. The Calcitonin System
Several receptors and peptides build up the calcitonin signaling crossroads. The implication of this peptidergic complex in cancer is supported by in vitro and in vivo studies in different tumors [71,121,122,123].
Determination of high calcitonin levels is an excellent marker of medullary thyroid cancer; however, it is not a pathognomonic mark [124,125,126]. CGRP associated with altered dendritic development in a single cell model of medullary thyroid cancer. This association exhibited a close relationship with activated adenylyl cyclase activity, leading to augmented Kruppel-like factor 2 and consequent impairment of T-cell tumor infiltration [127]. Calcitonin receptor was found augmented in glioblastoma cells in stage T0; however, its participation in carcinogenesis mechanisms in these tumors remains undefined [128]. The upregulated presence of CGRP and its receptor CALCRL in tumor samples from patients with colorectal cancer provided novel diagnosis and prognosis tools concerning tumor stage definition. The overactivation of this system and mechanisms triggered to impulse tumor appearance require better clarification [123]. CALCR appeared as a critical regulator of cell proliferation, stemness, apoptosis, and expression of SOX2 and Oct4 in acute myeloid leukemia cells. It also has a prominent involvement in resistance mechanisms to chemotherapy [129]. Secretion of CGRP from afferent neurons added a microenvironmental tumor element influencing oral cancer growth, pain, and inflammation, making the receptor a putative target for therapy intervention [130]. A mechanistic relationship between KRAS mutation and adrenomedullin in prostatic ductal carcinoma has resulted in a disarray of the alternative complement pathway by altering the secretion of complement factor B [131]. The different types of receptors (CLR associated with varying proteins of RAMP 1, 2, and 3) impact the development of several tumors [132].
4.6. The Neurotensin System
Sound experimental evidence in patient samples and cell cultures sustains the connection of neurotensin with different neoplasias, from breast cancer to prostate, lung, brain, colon, or pancreas tumors [133,134,135,136,137,138].
By activating NTS1R and NTS2R, neurotensin augmented the appearance of neuroendocrine differentiation in animal models and cell lines related to castration-resistant prostate cancer. The NTS1R antagonist SR48692 also delayed the transdifferentiating process associated with cancer progression [139]. Some neurotensin analogs tested in a colorectal cancer cell line HCT116 diminished cell survival and reduced the percentage of cancer stem cells (CD133 positive) [140]. By triggering the NTS1R/Akt signaling route, neuron-released neurotensin promoted cell invasion and propagation in pancreatic cancer (ductal carcinoma) cell lines. NTS1R knockdown and applying GDC0941, a PI3K inhibitor, counteracted the observed effect [141]. Functional interaction between NTS2R and TRKB favored chronic lymphocytic leukemia B cell viability. The use of a peptide able to disrupt the interaction disrupted Src signaling and apoptosis mechanisms, making the protein-protein contact a sound target for therapy [142].
4.7. The Apelin System
Non-comparable methodological approaches and procedures have made it challenging to use bloodstream apelin as a marker and predictor of different neoplasias (lung, breast, prostate, ovary, brain, and more) [143,144,145]. Hence, there is an urge to standardize methods and apply them to specific tumors to ascertain the actual value of the circulating peptide in diagnosis and prognosis procedures.
The apelin/APLNR system has been shown to exhibit an antineoplastic effect in colon26 (a murine colon adenocarcinoma cell line) by increasing immunological mechanisms against the tumor cells. The agonist ligand [Pyr1]Apelin-13 administration induced CD4+ and CD8+ T cell accumulation [146]. On the other hand, the effect of apelin in immune macrophage responses to tumoral progression was analyzed in a co-culture of mouse macrophage cell line RAW264.7 with neck carcinoma cell line SCCLMT1 with suppressed (RNA interference) apelin expression. Apelin was found in this experimental model to promote the accumulation of M2 macrophages, thus facilitating tumor expansion by interfering with the microenvironment controlling tumor behavior [147]. By activating the APLNR, apelin augmented cell proliferation, migration, invasiveness, and increased glycolysis rate in cervical cancer cells (human samples and cell lines) via the PI3K/AKT signaling pathway [148]. Apelin has also been associated with liver fibrosis and hepatocellular carcinoma; moreover, the impact of this peptide on the gastrointestinal microbiota and its influence on cancer pathologies await deeper analysis and clarification [149].
One may suppose that the proliferative or antiproliferative effects observed for the apelin system may be related to experimental procedures (methodological bias) or specific characteristics of tumor types and tumor microenvironmental milieu (physio-pathological importance). In consequence, further research from this perspective should be encouraged.
4.8. The CRF System
CRF may directly influence colon cancer, as is the case for CRF-induced cell proliferation through Janus kinase activation and activation, via NF-κB, of VEGF (vascular endothelial factor) with subsequent augmented angiogenesis [150]. Indirectly, CRF and urocortins intervene in cancer progression by participating in stress and inflammation mechanisms [151]. In a murine breast cancer model, tumor-associated anxiety was partially responsible for tumor advancement. Interestingly, CRF neurons forming a circuit between the central amygdala and the lateral paragigantocellular nucleus were relevant in modulating anxiety and related tumor progression [152]. Also, CRF neurons of the paraventricular nucleus of the hypothalamus seem to contribute to tumor progression by altering the equilibrium of immune control of tumors [153]. These findings suggest an interesting salient central brain control over tumors developed in other tissues.
In human bladder cancer cell lines, CRF augmented cell migration capacity and favored metastasis in a murine-implanted xenografted tissue (originated from cancer cells). The effect was mechanistically associated with the activation, through ERK, of supervillin, an actin-binding protein involved in cell proliferation processes [154]. The participation of CRF and related peptides in cancer has also been indirectly associated with their increased expression in malignant tissues. For example, immunohistochemical analysis of CRF urocortin and CRFRs in human vulvar samples (from control to premalignant and malignant specimens) established a clear correlation between the expression patterns and malignity [155]. Also, immunolocalization analysis of CRF and its receptors, CRF1R, and CRF2R in human pancreatic cancer samples unveiled a positive correlation between augmented CRF1R signal and poor prognosis [156].
This report concerns a few peptidergic GPCR systems and their relationship with molecular mechanisms sustaining cancer. However, the list is far from complete. Other systems are also implicated. For example, glucagon-like peptide-1 (GLP-1) receptor agonists, besides their application in diabetes and obesity therapies, behave as antitumoral agents against hepatocellular carcinoma[157]. Analogs of cyclic peptide somatostatin were reported to be effective in treating neuroendocrine tumors [158]. In human-derived colorectal cancer cell lines (T84, Caco-2, HCT116), the nonapeptide bradykinin increased interleukin 6, leading to tumor cell invasion and migration [159]. Gastrin-releasing peptide, cholecystokinin, endothelins, and Tat-NTS/Tat-Cx43266-283 are associated with glioma tumors through different mechanisms [160].
5. Peptidergic GPCRs as Targets for Cancer Treatment
It is relevant to place peptidergic GPCR systems in a position of consideration and inclusion in basic and clinical studies that aim to treat different cancers by advancing structure-activity analysis, exploring molecular mechanisms, reviewing possible drug candidates, and underlying the relevance of setting appropriate drug design strategies. Since agonist and antagonist activity may regulate receptor activation and inactivation, a few representative structures that bind and impact structure conformation to the receptors under study in this review are presented.
Noteworthy, the obtention of ligands with the capacity of abrogating or stimulating peptide GPCR activity, directly related to the mechanism of cancer development and progression, would be beneficial since complex downstream signaling networks may be disconnected and complex biochemical cascades may be silenced from their initial stages. On the other hand, careful and reliable with comparable methodological procedures studies should previously establish if silencing, aborting the signaling, or promoting the signal does not interfere with physiological mechanisms in healthy cells. Hence, there is an insistence on properly unraveling the signaling circuitries for every system and in specific types of tumors.
Therapeutic strategies not only pertain to specific receptor ligands modulating orthosteric and allosteric sites and influencing dynamic receptor conformations but also include strategies aiming at obtaining safe, stable, durable, and specific drugs and compound envoys targeting the altered peptidergic system in tumor cells without affecting healthy cells. It may also include a bivalent drug designed to aim for two or more molecular objectives of the same signaling pathway or a different one. Additionally, these drugs may complement other treatments (radiotherapy, surgery, classical chemotherapy) tailored for specific cancers, tumor stages, and individuals. In practice, a canopy of pharmaceutical approaches may serve the purposes mentioned above, for example, peptides with anticancer cargo, antibodies directed against specific receptors, antibodies conjugated to drugs, or specific signaling proteins, or silencing of receptors and other proteinaceous targets with siRNA technology [161,162].
Table 1 summarizes peptidergic GPCR antagonists and agonists that, based on current research, offer promising opportunities for treating tumor development, extension, metastasis, or resistance to tumor therapy. The purpose is to underlie the importance of continuing to test and deepen knowledge in preclinical, experimental models, and clinical trials.
6. Conclusion and Future Directions
Peptidergic GPCRs have gained positions for their consideration as druggable targets for cancer management and treatment. Also, peptides, their GPCR, transducer, and effector intracellular proteins associated may serve as reliable biomarkers for diagnosing and prognosis of specific types of tumors and their stages [185].
Procedures of organic synthesis by using reference structures (for example, pyrazole rings) [186] and other synthetic strategies aimed at exploring the orthosteric and allosteric GPCR sites better may generate new compounds with possible applications in adapting, modulating, and mending GPCR dysfunction. The routes for the obtention of receptor cavity-flexible and safe, though specific, non-toxic, and potent compounds targeting and modulating conformation receptor states with functional outcomes and biased signaling routes may result in rewarding the efforts [187]. Therefore, a three-dimensional analysis that unravels receptor-drug interfaces' structural and docking characteristics is paramount.
Also of consideration is that some drugs are already marketed for other purposes, and therefore, setting clinical trials to define their therapeutical potential in cancer may result in a lower burden [188,189]. The case applies, for example, to aprepitant, an NK-1R antagonist used in clinical practice to annihilate vomit and nausea induced by chemotherapy or surgery [169]. Also, CGRP antagonists, generically called “gepants” (rimegepant or atogepant), are currently used to treat migraine [190]. Another example is a CRF1R antagonist, MK-I-152, tested for treating major depression [77].
The task is challenging, time-consuming, resource-consuming, and effort-consuming. Still, it may yield encouraging results in the context of new approaches in cancer therapy, including the repurposing of drugs already in use in different nosological entities.
Author Contributions
Conceptualization, design, bibliography analysis, draft preparation, writing and review, F.D.R. and R.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors gratefully acknowledge support from Programa XIII para Grupos GIR (Grupo BMD, Bases moleculares del desarrollo) of the University of Salamanca (Spain).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Golemis, E.A.; Scheet, P.; Beck, T.N.; Scolnick, E.M.; Hunter, D.J.; Hawk, E.; Hopkins, N. Molecular mechanisms of the preventable causes of cancer in the United States. Genes Dev 2018, 32, 868–902. [Google Scholar] [CrossRef] [PubMed]
- Wahida, A.; Buschhorn, L.; Fröhling, S.; Jost, P.J.; Schneeweiss, A.; Lichter, P.; Kurzrock, R. The coming decade in precision oncology: six riddles. Nat Rev Cancer 2023, 23, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.C.; Zaki, T.A. Changing epidemiology of colorectal cancer - birth cohort effects and emerging risk factors. Nat Rev Gastroenterol Hepatol 2024, 21, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Obeagu, E.I.; Obeagu, G.U. Breast cancer: A review of risk factors and diagnosis. Medicine (Baltimore) 2024, 103, e36905. [Google Scholar] [CrossRef] [PubMed]
- Marino, P.; Mininni, M.; Deiana, G.; Marino, G.; Divella, R.; Bochicchio, I.; Giuliano, A.; Lapadula, S.; Lettini, A.R.; Sanseverino, F. Healthy Lifestyle and Cancer Risk: Modifiable Risk Factors to Prevent Cancer. Nutrients 2024, 16, 800. [Google Scholar] [CrossRef]
- Montégut, L.; López-Otín, C.; Kroemer, G. Aging and cancer. Mol Cancer 2024, 23, 106–z. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.K.; Kim, S. An Insight into GPCR and G-Proteins as Cancer Drivers. Cells 2021, 10, 3288. [Google Scholar] [CrossRef] [PubMed]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res 2018, 46, D440–D446. [Google Scholar] [CrossRef]
- Wu, F.; Song, G.; de Graaf, C.; Stevens, R.C. Structure and Function of Peptide-Binding G Protein-Coupled Receptors. J Mol Biol 2017, 429, 2726–2745. [Google Scholar] [CrossRef]
- Kroeze, W.K.; Sassano, M.F.; Huang, X.; Lansu, K.; McCorvy, J.D.; Giguère, P.M.; Sciaky, N.; Roth, B.L. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat Struct Mol Biol 2015, 22, 362–369. [Google Scholar] [CrossRef]
- Majumdar, S.; Chiu, Y.; Pickett, J.E.; Roth, B.L. Illuminating the understudied GPCR-ome. Drug Discov Today 2024, 29, 103848. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, T.; Lu, X.; Lan, X.; Chen, Z.; Lu, S. G protein-coupled receptors (GPCRs): advances in structures, mechanisms, and drug discovery. Signal Transduct Target Ther 2024, 9, 88. [Google Scholar] [CrossRef]
- Wacker, D.; Stevens, R.C.; Roth, B.L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427. [Google Scholar] [CrossRef]
- Foster, S.R.; Hauser, A.S.; Vedel, L.; Strachan, R.T.; Huang, X.; Gavin, A.C.; Shah, S.D.; Nayak, A.P.; Haugaard-Kedström, L.M.; Penn, R.B.; Roth, B.L.; Bräuner-Osborne, H.; Gloriam, D.E. Discovery of Human Signaling Systems: Pairing Peptides to G Protein-Coupled Receptors. Cell 2019, 179, 895–908.e21. [Google Scholar] [CrossRef]
- Beets, I.; Zels, S.; Vandewyer, E.; Demeulemeester, J.; Caers, J.; Baytemur, E.; Courtney, A.; Golinelli, L. ; Hasakioğulları, İ; Schafer, W.R.; Vértes, P.E.; Mirabeau, O.; Schoofs, L. System-wide mapping of peptide-GPCR interactions in C. elegans. Cell Rep 2023, 42, 113058. [Google Scholar] [CrossRef]
- Rodriguez, F.D.; Covenas, R. Association of Neurokinin-1 Receptor Signaling Pathways with Cancer. Curr Med Chem 2023. [Google Scholar] [CrossRef]
- Jékely, G.; Melzer, S.; Beets, I.; Kadow, I.C.G.; Koene, J.; Haddad, S.; Holden-Dye, L. The long and the short of it - a perspective on peptidergic regulation of circuits and behaviour. J Exp Biol 2018, 221, jeb166710. [Google Scholar] [CrossRef]
- Davenport, A.P.; Scully, C.C.G.; de Graaf, C.; Brown, A.J.H.; Maguire, J.J. Advances in therapeutic peptides targeting G protein-coupled receptors. Nat Rev Drug Discov 2020, 19, 389–413. [Google Scholar] [CrossRef] [PubMed]
- Abrimian, A.; Kraft, T.; Pan, Y. Endogenous Opioid Peptides and Alternatively Spliced Mu Opioid Receptor Seven Transmembrane Carboxyl-Terminal Variants. Int J Mol Sci 2021, 22, 3779. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhuang, Y.; DiBerto, J.F.; Zhou, X.E.; Schmitz, G.P.; Yuan, Q.; Jain, M.K.; Liu, W.; Melcher, K.; Jiang, Y.; Roth, B.L.; Xu, H.E. Structures of the entire human opioid receptor family. Cell 2023, 186, 413–427.e17. [Google Scholar] [CrossRef]
- Bodnar, R.J. Endogenous opiates and behavior: 2023. Peptides 2024, 179, 171268. [Google Scholar] [CrossRef] [PubMed]
- Pennefather, J.N.; Lecci, A.; Candenas, M.L.; Patak, E.; Pinto, F.M.; Maggi, C.A. Tachykinins and tachykinin receptors: a growing family. Life Sci 2004, 74, 1445–1463. [Google Scholar] [CrossRef]
- Satake, H.; Kawada, T. Overview of the primary structure, tissue-distribution, and functions of tachykinins and their receptors. Curr Drug Targets 2006, 7, 963–974. [Google Scholar] [CrossRef]
- Steinhoff, M.S.; von Mentzer, B.; Geppetti, P.; Pothoulakis, C.; Bunnett, N.W. Tachykinins and their receptors: contributions to physiological control and the mechanisms of disease. Physiol Rev 2014, 94, 265–301. [Google Scholar] [CrossRef] [PubMed]
- Holzer, P.; Reichmann, F.; Farzi, A. Neuropeptide Y, peptide YY and pancreatic polypeptide in the gut-brain axis. Neuropeptides 2012, 46, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Dumont, Y.; Bastianetto, S.; Duranton, A.; Breton, L.; Quirion, R. Immunohistochemical distribution of neuropeptide Y, peptide YY, pancreatic polypeptide-like immunoreactivity and their receptors in the epidermal skin of healthy women. Peptides 2015, 70, 7–16. [Google Scholar] [CrossRef]
- Bale, R.; Doshi, G. Cross talk about the role of Neuropeptide Y in CNS disorders and diseases. Neuropeptides 2023, 102, 102388. [Google Scholar] [CrossRef]
- Robinson, J.K.; Bartfai, T.; Langel, U. Galanin/GALP receptors and CNS homeostatic processes. CNS Neurol Disord Drug Targets 2006, 5, 327–334. [Google Scholar] [CrossRef]
- Lang, R.; Gundlach, A.L.; Holmes, F.E.; Hobson, S.A.; Wynick, D.; Hökfelt, T.; Kofler, B. Physiology, signaling, and pharmacology of galanin peptides and receptors: three decades of emerging diversity. Pharmacol Rev 2015, 67, 118–175. [Google Scholar] [CrossRef]
- Brzozowska, M.; Całka, J. Review: Occurrence and Distribution of Galanin in the Physiological and Inflammatory States in the Mammalian Gastrointestinal Tract. Front Immunol 2021, 11, 602070. [Google Scholar] [CrossRef]
- Ignatov, A.; Hermans-Borgmeyer, I.; Schaller, H.C. Cloning and characterization of a novel G-protein-coupled receptor with homology to galanin receptors. Neuropharmacology 2004, 46, 1114–1120. [Google Scholar] [CrossRef]
- Bielczyk-Maczynska, E.; Zhao, M.; Zushin, P.H.; Schnurr, T.M.; Kim, H.; Li, J.; Nallagatla, P.; Sangwung, P.; Park, C.Y.; Cornn, C.; Stahl, A.; Svensson, K.J.; Knowles, J.W. G protein-coupled receptor 151 regulates glucose metabolism and hepatic gluconeogenesis. Nat Commun 2022, 13, 7408–9. [Google Scholar] [CrossRef]
- Jiang, H.; Galtes, D.; Wang, J.; Rockman, H.A. G protein-coupled receptor signaling: transducers and effectors. Am J Physiol Cell Physiol 2022, 323, C731–C748. [Google Scholar] [CrossRef]
- Holmes, F.E.; Kerr, N.; Chen, Y.; Vanderplank, P.; McArdle, C.A.; Wynick, D. Targeted disruption of the orphan receptor Gpr151 does not alter pain-related behaviour despite a strong induction in dorsal root ganglion expression in a model of neuropathic pain. Mol Cell Neurosci 2017, 78, 35–40. [Google Scholar] [CrossRef]
- Barwell, J.; Gingell, J.J.; Watkins, H.A.; Archbold, J.K.; Poyner, D.R.; Hay, D.L. Calcitonin and calcitonin receptor-like receptors: common themes with family B GPCRs? Br J Pharmacol 2012, 166, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Pioszak, A.A.; Hay, D.L. RAMPs as allosteric modulators of the calcitonin and calcitonin-like class B G protein-coupled receptors. Adv Pharmacol 2020, 88, 115–141. [Google Scholar] [CrossRef] [PubMed]
- Amara, S.G.; Jonas, V.; Rosenfeld, M.G.; Ong, E.S.; Evans, R.M. Alternative RNA processing in calcitonin gene expression generates mRNAs encoding different polypeptide products. Nature 1982, 298, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Wimalawansa, S.J. Amylin, calcitonin gene-related peptide, calcitonin, and adrenomedullin: a peptide superfamily. Crit Rev Neurobiol 1997, 11, 167–239. [Google Scholar] [CrossRef] [PubMed]
- Kita, T.; Kitamura, K. Translational studies of adrenomedullin and related peptides regarding cardiovascular diseases. Hypertens Res 2022, 45, 389–400. [Google Scholar] [CrossRef]
- Russo, A.F.; Hay, D.L. CGRP physiology, pharmacology, and therapeutic targets: migraine and beyond. Physiol Rev 2023, 103, 1565–1644. [Google Scholar] [CrossRef]
- Spoto, S.; Basili, S.; Cangemi, R.; Yuste, J.R.; Lucena, F.; Romiti, G.F.; Raparelli, V.; Argemi, J.; D'Avanzo, G.; Locorriere, L.; Masini, F.; Calarco, R.; Testorio, G.; Spiezia, S.; Ciccozzi, M.; Angeletti, S. A Focus on the Pathophysiology of Adrenomedullin Expression: Endothelitis and Organ Damage in Severe Viral and Bacterial Infections. Cells 2024, 13, 892. [Google Scholar] [CrossRef]
- Vincent, J.P.; Mazella, J.; Kitabgi, P. Neurotensin, and neurotensin receptors. Trends Pharmacol Sci 1999, 20, 302–309. [Google Scholar] [CrossRef]
- Gereau, G.B.; Garrison, S.D.; McElligott, Z.A. Neurotensin and energy balance. J Neurochem 2023, 166, 189–200. [Google Scholar] [CrossRef]
- Torruella-Suárez, M.L.; McElligott, Z.A. Neurotensin in reward processes. Neuropharmacology 2020, 167, 108005. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.D.; Sanchez, M.L.; Covenas, R. Neurotensin, and Alcohol Use Disorders: Towards a Pharmacological Treatment. Int J Mol Sci 2023, 24, 8656. [Google Scholar] [CrossRef] [PubMed]
- Kyriatzis, G.; Khrestchatisky, M.; Ferhat, L.; Chatzaki, E.A. Neurotensin and Neurotensin Receptors in Stress-related Disorders: Pathophysiology & Novel Drug Targets. Curr Neuropharmacol 2024, 22, 916–934. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, M.N.; Stoyanov, D.S.; Pavlov, S.P.; Tonchev, A.B. Distribution, Function, and Expression of the Apelinergic System in the Healthy and Diseased Mammalian Brain. Genes (Basel) 2022, 13, 2172. [Google Scholar] [CrossRef]
- Read, C.; Nyimanu, D.; Williams, T.L.; Huggins, D.J.; Sulentic, P.; Macrae, R.G.C.; Yang, P.; Glen, R.C.; Maguire, J.J.; Davenport, A.P. International Union of Basic and Clinical Pharmacology. CVII. Structure and Pharmacology of the Apelin Receptor with a Recommendation that Elabela/Toddler Is a Second Endogenous Peptide Ligand. Pharmacol Rev 2019, 71, 467–502. [Google Scholar] [CrossRef]
- Sharma, M.; Prabhavalkar, K.S.; Bhatt, L.K. Elabela Peptide: An Emerging Target in Therapeutics. Curr Drug Targets 2022, 23, 1304–1318. [Google Scholar] [CrossRef] [PubMed]
- Dagamajalu, S.; Rex, D.A.B.; Suchitha, G.P.; Rai, A.B.; Rainey, J.K.; Prasad, T.S.K. The network map of Elabela signaling pathway in physiological and pathological conditions. J Cell Commun Signal 2022, 16, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, L.; Li, P.; Zheng, Y.; Yang, Y.; Ji, S. Apelin/APJ system in inflammation. Int Immunopharmacol 2022, 109, 108822. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Kageyama, K.; Sakihara, S.; Nigawara, T. Physiological roles of urocortins, human homologs of fish urotensin I, and their receptors. Peptides 2004, 25, 1689–1701. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K. Distribution of urocortins and corticotropin-releasing factor receptors in the cardiovascular system. Int J Endocrinol 2012, 2012, 395284. [Google Scholar] [CrossRef]
- Chrousos, G.P.; Zoumakis, E. Milestones in CRH Research. Curr Mol Pharmacol 2017, 10, 259–263. [Google Scholar] [CrossRef]
- Vasconcelos, I.; von Hafe, M.; Adão, R.; Leite-Moreira, A.; Brás-Silva, C. Corticotropin-releasing hormone and obesity: From fetal life to adulthood. Obes Rev 2024, 25, e13763. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ji, S.; Zhang, W.; Zhang, J.; Cai, C.; Hu, R.; Zang, S.; Miao, L.; Xu, H.; Chen, L.; Yang, Z.; Guo, J.; Qin, J.; Shen, D.; Liang, P.; Zhang, Y.; Zhang, Y. Structure-based design of non-hypertrophic apelin receptor modulator. Cell 2024, 187, 1460–1475.e20. [Google Scholar] [CrossRef]
- Schöppe, J.; Ehrenmann, J.; Klenk, C.; Rucktooa, P.; Schütz, M.; Doré, A.S.; Plückthun, A. Crystal structures of the human neurokinin one receptor in complex with clinically used antagonists. Nat Commun 2019, 10, 17–8. [Google Scholar] [CrossRef] [PubMed]
- Thom, C.; Ehrenmann, J.; Vacca, S.; Waltenspühl, Y.; Schöppe, J.; Medalia, O.; Plückthun, A. Structures of neurokinin one receptor in complex with G(q) and G(s) proteins reveal substance P binding mode and unique activation features. Sci Adv 2021, 7, eabk2872. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Basith, S.; Choi, S. Recent Advances in Structure-Based Drug Design Targeting Class A G Protein-Coupled Receptors Utilizing Crystal Structures and Computational Simulations. J Med Chem 2018, 61, 1–46. [Google Scholar] [CrossRef]
- Eiden, L.E.; Goosens, K.A.; Jacobson, K.A.; Leggio, L.; Zhang, L. Peptide-Liganded G Protein-Coupled Receptors as Neurotherapeutics. ACS Pharmacol Transl Sci 2020, 3, 190–202. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: current applications and future directions. Signal Transduct Target Ther 2022, 7, 48. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res 2021, 49, W431–W437. [Google Scholar] [CrossRef]
- Zhuang, Y.; Wang, Y.; He, B.; He, X.; Zhou, X.E.; Guo, S.; Rao, Q.; Yang, J.; Liu, J.; Zhou, Q.; Wang, X.; Liu, M.; Liu, W.; Jiang, X.; Yang, D.; Jiang, H.; Shen, J.; Melcher, K.; Chen, H.; Jiang, Y.; Cheng, X.; Wang, M.; Xie, X.; Xu, H.E. Molecular recognition of morphine and fentanyl by the human μ-opioid receptor. Cell 2022, 185, 4361–4375.e19. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, M.; Zhang, X.; Guo, C.; Lei, Y.; Wang, W.; Fan, Y.; Cao, P.; Li, C.; Wang, R.; Li, X.; Yu, Y.; Yang, X. Dynamic recognition of naloxone, morphine and endomorphin1 in the same pocket of µ-opioid receptors. Front Mol Biosci 2022, 9, 925404. [Google Scholar] [CrossRef]
- Rodríguez, F.D.; Coveñas, R. The Neurokinin-1 Receptor: Structure Dynamics and Signaling. Receptors 2022, 1, 71. [Google Scholar] [CrossRef]
- Park, C.; Kim, J.; Ko, S.; Choi, Y.K.; Jeong, H.; Woo, H.; Kang, H.; Bang, I.; Kim, S.A.; Yoon, T.; Seok, C.; Im, W.; Choi, H. Structural basis of neuropeptide Y signaling through Y1 receptor. Nat Commun 2022, 13, 853–6. [Google Scholar] [CrossRef]
- Duan, J.; Shen, D.; Zhao, T.; Guo, S.; He, X.; Yin, W.; Xu, P.; Ji, Y.; Chen, L.; Liu, J.; Zhang, H.; Liu, Q.; Shi, Y.; Cheng, X.; Jiang, H.; Eric Xu, H.; Zhang, Y.; Xie, X.; Jiang, Y. Molecular basis for allosteric agonism and G protein subtype selectivity of galanin receptors. Nat Commun 2022, 13, 1364–3. [Google Scholar] [CrossRef]
- Liang, Y.; Khoshouei, M.; Deganutti, G.; Glukhova, A.; Koole, C.; Peat, T.S.; Radjainia, M.; Plitzko, J.M.; Baumeister, W.; Miller, L.J.; Hay, D.L.; Christopoulos, A.; Reynolds, C.A.; Wootten, D.; Sexton, P.M. Cryo-EM structure of the active, G(s)-protein complexed, human CGRP receptor. Nature 2018, 561, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.L.; Rodríguez, F.D.; Coveñas, R. Peptidergic Systems and Cancer: Focus on Tachykinin and Calcitonin/Calcitonin Gene-Related Peptide Families. Cancers (Basel) 2023, 15, 1694. [Google Scholar] [CrossRef] [PubMed]
- White, J.F.; Noinaj, N.; Shibata, Y.; Love, J.; Kloss, B.; Xu, F.; Gvozdenovic-Jeremic, J.; Shah, P.; Shiloach, J.; Tate, C.G.; Grisshammer, R. Structure of the agonist-bound neurotensin receptor. Nature 2012, 490, 508–513. [Google Scholar] [CrossRef]
- Deluigi, M.; Klipp, A.; Klenk, C.; Merklinger, L.; Eberle, S.A.; Morstein, L.; Heine, P.; Mittal, P.R.E.; Ernst, P.; Kamenecka, T.M.; He, Y.; Vacca, S.; Egloff, P.; Honegger, A.; Plückthun, A. Complexes of the neurotensin receptor 1 with small-molecule ligands reveal structural determinants of full, partial, and inverse agonism. Sci Adv 2021, 7, eabe5504. [Google Scholar] [CrossRef] [PubMed]
- Bumbak, F.; Bower, J.B.; Zemmer, S.C.; Inoue, A.; Pons, M.; Paniagua, J.C.; Yan, F.; Ford, J.; Wu, H.; Robson, S.A.; Bathgate, R.A.D.; Scott, D.J.; Gooley, P.R.; Ziarek, J.J. Stabilization of pre-existing neurotensin receptor conformational states by β-arrestin-1 and the biased allosteric modulator ML314. Nat Commun 2023, 14, 3328–8. [Google Scholar] [CrossRef]
- Ma, Y.; Yue, Y.; Ma, Y.; Zhang, Q.; Zhou, Q.; Song, Y.; Shen, Y.; Li, X.; Ma, X.; Li, C.; Hanson, M.A.; Han, G.W.; Sickmier, E.A.; Swaminath, G.; Zhao, S.; Stevens, R.C.; Hu, L.A.; Zhong, W.; Zhang, M.; Xu, F. Structural Basis for Apelin Control of the Human Apelin Receptor. Structure 2017, 25, 858–866.e4. [Google Scholar] [CrossRef]
- Pioszak, A.A.; Parker, N.R.; Suino-Powell, K.; Xu, H.E. Molecular recognition of corticotropin-releasing factor by its G-protein-coupled receptor CRFR1. J Biol Chem 2008, 283, 32900–32912. [Google Scholar] [CrossRef]
- Kim, H.; Lim, T.; Ha, G.E.; Lee, J.; Kim, J.; Chang, N.; Kim, S.H.; Kim, K.H.; Lee, J.; Cho, Y.; Kim, B.W.; Abrahamsson, A.; Kim, S.H.; Kim, H.; Park, S.; Lee, S.J.; Park, J.; Cheong, E.; Kim, B.M.; Cho, H. Structure-based drug discovery of a corticotropin-releasing hormone receptor one antagonist using an X-ray free-electron laser. Exp Mol Med 2023, 55, 2039–2050. [Google Scholar] [CrossRef]
- Nürnberg, B.; Beer-Hammer, S.; Reisinger, E.; Leiss, V. Non-canonical G protein signaling. Pharmacol Ther 2024, 255, 108589. [Google Scholar] [CrossRef] [PubMed]
- Crilly, S.E.; Puthenveedu, M.A. Compartmentalized GPCR Signaling from Intracellular Membranes. J Membr Biol 2021, 254, 259–271. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. GPCR-dependent and -independent arrestin signaling. Trends Pharmacol Sci 2024, 45, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Flores-Espinoza, E.; Thomsen, A.R.B. Beneath the surface: endosomal GPCR signaling. Trends Biochem Sci 2024, 49, 520–531. [Google Scholar] [CrossRef]
- Klauer, M.J.; Willette, B.K.A.; Tsvetanova, N.G. Functional diversification of cell signaling by GPCR localization. J Biol Chem 2024, 300, 105668. [Google Scholar] [CrossRef] [PubMed]
- Schoos, A.; Gabriel, C.; Knab, V.M.; Fux, D.A. Activation of HIF-1α by δ-Opioid Receptors Induces COX-2 Expression in Breast Cancer Cells and Leads to Paracrine Activation of Vascular Endothelial Cells. J Pharmacol Exp Ther 2019, 370, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Faraji, N.; Arab, S.S.; Doustmohammadi, A.; Daly, N.L.; Khosroushahi, A.Y. ApInAPDB: a database of apoptosis-inducing anticancer peptides. Sci Rep 2022, 12, 21341–6. [Google Scholar] [CrossRef] [PubMed]
- Ghaly, G.; Tallima, H.; Dabbish, E.; Badr ElDin, N.; Abd El-Rahman, M.K.; Ibrahim, M.A.A.; Shoeib, T. Anti-Cancer Peptides: Status and Future Prospects. Molecules 2023, 28, 1148. [Google Scholar] [CrossRef]
- Moll, G.N. Agonists of galanin subtype 2 receptor may prevent pancreatic cancer, and agonists of angiotensin II type 2 receptor may prevent colorectal cancer. Eur J Pharmacol 2024, 978, 176772. [Google Scholar] [CrossRef]
- Sánchez, M.L.; Rodríguez, F.D.; Coveñas, R. Involvement of the Opioid Peptide Family in Cancer Progression. Biomedicines 2023, 11, 1993. [Google Scholar] [CrossRef]
- Szczepaniak, A.; Fichna, J.; Zielińska, M. Opioids in Cancer Development, Progression and Metastasis: Focus on Colorectal Cancer. Curr Treat Options Oncol 2020, 21, 6–1. [Google Scholar] [CrossRef] [PubMed]
- Tripolt, S.; Neubauer, H.A.; Knab, V.M.; Elmer, D.P.; Aberger, F.; Moriggl, R.; Fux, D.A. Opioids drive breast cancer metastasis through the δ-opioid receptor and oncogenic STAT3. Neoplasia 2021, 23, 270–279. [Google Scholar] [CrossRef]
- Kuramochi, T.; Sano, M.; Kajiwara, I.; Oshima, Y.; Itaya, T.; Kim, J.; Ichimaru, Y.; Kitajima, O.; Masamune, A.; Ijichi, H.; Suzuki, T. Effects of tramadol via a µ-opioid receptor on pancreatic ductal adenocarcinoma in vitro and in vivo. Reg Anesth Pain Med 2024, 49, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Carli, M.; Donnini, S.; Pellegrini, C.; Coppi, E.; Bocci, G. Opioid receptors beyond pain control: The role in cancer pathology and the debated importance of their pharmacological modulation. Pharmacol Res 2020, 159, 104938. [Google Scholar] [CrossRef] [PubMed]
- Sah, D.; Shoffel-Havakuk, H.; Tsur, N.; Uhelski, M.L.; Gottumukkala, V.; Cata, J.P. Opioids and Cancer: Current Understanding and Clinical Considerations. Curr Oncol 2024, 31, 3086–3098. [Google Scholar] [CrossRef]
- Bhoir, S.; Uhelski, M.; Guerra-Londono, J.J.; Cata, J.P. The Role of Opioid Receptors in Cancer. Adv Biol (Weinh) 2023, 7, e2300102. [Google Scholar] [CrossRef]
- Robinson, P.; Coveñas, R.; Muñoz, M. Combination Therapy of Chemotherapy or Radiotherapy and the Neurokinin-1 Receptor Antagonist Aprepitant: A New Antitumor Strategy? Curr Med Chem 2023, 30, 1798–1812. [Google Scholar] [CrossRef]
- Coveñas, R.; Muñoz, M. Involvement of the Substance P/Neurokinin-1 Receptor System in Cancer. Cancers (Basel) 2022, 14, 3539. [Google Scholar] [CrossRef]
- Isorna, I.; González-Moles, M. Á; Muñoz, M., Ed.; Esteban, F. Substance P and Neurokinin-1 Receptor System in Thyroid Cancer: Potential Targets for New Molecular Therapies. J Clin Med 2023, 12, 6409. [Google Scholar] [CrossRef]
- Martín-García, D.; Téllez, T.; Redondo, M.; García-Aranda, M. The Use of SP/Neurokinin-1 as a Therapeutic Target in Colon and Rectal Cancer. Curr Med Chem 2023. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.T.; Thaggikuppe Krishnamurthy, P.; Magham, S.V. Harnessing the synergistic potential of NK1R antagonists and selective COX-2 inhibitors for simultaneous targeting of TNBC cells and cancer stem cells. J Drug Target 2024, 32, 258–269. [Google Scholar] [CrossRef]
- Rezaei, S.; Javid, H.; Iranpour, S.; Darban, R.A.; Hashemy, S.I. Unveiling the Promising Role of Substance P/Neurokinin 1 Receptor in Cancer Cell Proliferation and Cell Cycle Regulation in Human Malignancies. Curr Med Chem 2024. [Google Scholar] [CrossRef]
- Alalikhan, A.; Ebrahimi, S.; Aliee, A.; Mirzavi, F.; Hashemy, S.I. The combined anti-tumor effects of 5-fluorouracil and neurokinin receptor inhibitor, aprepitant, against colorectal cancer: In vitro and in vivo study. Med Oncol 2024, 41, 70–w. [Google Scholar] [CrossRef] [PubMed]
- Zarei Shandiz, S.; Assaran Darban, R.; Javid, H.; Ghahremanloo, A.; Hashemy, S.I. The effect of SP/NK1R on expression and activity of glutaredoxin and thioredoxin proteins in prostate cancer cells. Naunyn Schmiedebergs Arch Pharmacol 2024, 397, 5875–5882. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cao, X.; Wang, Y.; Wu, X.; Zhou, P.; Miao, L.; Deng, X. Neurokinin-1 receptor antagonist aprepitant regulates autophagy and apoptosis via ROS/JNK in intrahepatic cholangiocarcinoma. Liver Int 2024, 44, 1651–1667. [Google Scholar] [CrossRef]
- Guan, L.; Yuan, S.; Ma, J.; Liu, H.; Huang, L.; Zhang, F. Neurokinin-1 receptor is highly expressed in cervical cancer and its antagonist induces cervical cancer cell apoptosis. Eur J Histochem 2023, 67, 3570. [Google Scholar] [CrossRef]
- Ebrahimi, S.; Mirzavi, F.; Hashemy, S.I.; Khaleghi Ghadiri, M.; Stummer, W.; Gorji, A. The in vitro anti-cancer synergy of neurokinin-1 receptor antagonist, aprepitant, and 5-aminolevulinic acid in glioblastoma. Biofactors 2023, 49, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.L.; Rodríguez, F.D.; Coveñas, R. Neuropeptide Y Peptide Family and Cancer: Antitumor Therapeutic Strategies. Int J Mol Sci 2023, 24, 9962. [Google Scholar] [CrossRef] [PubMed]
- Sigorski, D.; Wesołowski, W.; Gruszecka, A.; Gulczyński, J.; Zieliński, P.; Misiukiewicz, S.; Kitlińska, J.; Iżycka-Świeszewska, E. Neuropeptide Y and its receptors in prostate cancer: associations with cancer invasiveness and perineural spread. J Cancer Res Clin Oncol 2023, 149, 5803–5822. [Google Scholar] [CrossRef]
- Lu, C.; Mahajan, A.; Hong, S.; Galli, S.; Zhu, S.; Tilan, J.U.; Abualsaud, N.; Adnani, M.; Chung, S.; Elmansy, N.; Rodgers, J.; Rodriguez, O.; Albanese, C.; Wang, H.; Regan, M.; Zgonc, V.; Blancato, J.; Krawczyk, E.; Gallicano, G.I.; Girgis, M.; Cheema, A.; Iżycka-Świeszewska, E.; Cavalli, L.R.; Pack, S.D.; Kitlinska, J. Hypoxia-activated neuropeptide Y/Y5 receptor/RhoA pathway triggers chromosomal instability and bone metastasis in Ewing sarcoma. Nat Commun 2022, 13, 2323–x. [Google Scholar] [CrossRef]
- Abualsaud, N.; Caprio, L.; Galli, S.; Krawczyk, E.; Alamri, L.; Zhu, S.; Gallicano, G.I.; Kitlinska, J. Neuropeptide Y/Y5 Receptor Pathway Stimulates Neuroblastoma Cell Motility Through RhoA Activation. Front Cell Dev Biol 2021, 8, 627090. [Google Scholar] [CrossRef]
- Chen, B.; Chakrobortty, N.; Saha, A.K.; Shang, X. Identifying colon cancer stage related genes and their cellular pathways. Front Genet 2023, 14, 1120185. [Google Scholar] [CrossRef]
- Pascetta, S.A.; Kirsh, S.M.; Cameron, M.; Uniacke, J. Pharmacological inhibition of neuropeptide Y receptors Y1 and Y5 reduces hypoxic breast cancer migration, proliferation, and signaling. BMC Cancer 2023, 23, 494–1. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, P.J.; Pascetta, S.A.; Kirsh, S.M.; Al-Khazraji, B.K.; Uniacke, J. Expression of hypoxia inducible factor-dependent neuropeptide Y receptors Y1 and Y5 sensitizes hypoxic cells to NPY stimulation. J Biol Chem 2022, 298, 101645. [Google Scholar] [CrossRef] [PubMed]
- Chakroborty, D.; Goswami, S.; Fan, H.; Frankel, W.L.; Basu, S.; Sarkar, C. Neuropeptide Y, a paracrine factor secreted by cancer cells, is an independent regulator of angiogenesis in colon cancer. Br J Cancer 2022, 127, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Rauch, I.; Kofler, B. The galanin system in cancer. Exp Suppl 2010, 102, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.L.; Coveñas, R. The Galaninergic System: A Target for Cancer Treatment. Cancers (Basel) 2022, 14, 3755. [Google Scholar] [CrossRef]
- Talaat, I.M.; Yakout, N.M.; Soliman, A.S.A.; Venkatachalam, T.; Vinod, A.; Eldohaji, L.; Nair, V.; Hareedy, A.; Kandil, A.; Abdel-Rahman, W.M.; Hamoudi, R.; Saber-Ayad, M. Evaluation of Galanin Expression in Colorectal Cancer: An Immunohistochemical and Transcriptomic Study. Front Oncol 2022, 12, 877147. [Google Scholar] [CrossRef]
- Kiezun, J.; Godlewski, J.; Krazinski, B.E.; Kozielec, Z.; Kmiec, Z. Galanin Receptors (GalR1, GalR2, and GalR3) Expression in Colorectal Cancer Tissue and Correlations to the Overall Survival and Poor Prognosis of CRC Patients. Int J Mol Sci 2022, 23, 3735. [Google Scholar] [CrossRef]
- Mei, Z.; Yang, Y.; Li, Y.; Yang, F.; Li, J.; Xing, N.; Xu, Z.D. Galanin suppresses proliferation of human U251 and T98G glioma cells via its subtype 1 receptor. Biol Chem 2017, 398, 1127–1139. [Google Scholar] [CrossRef]
- de Medeiros, M.C.; Liu, M.; Banerjee, R.; Bellile, E.; D'Silva, N.J.; Rossa, C.J. Galanin mediates tumor-induced immunosuppression in head and neck squamous cell carcinoma. Cell Oncol (Dordr) 2022, 45, 241–256. [Google Scholar] [CrossRef]
- Namsolleck, P.; Kofler, B.; Moll, G.N. Galanin 2 Receptor: A Novel Target for a Subset of Pancreatic Ductal Adenocarcinoma. Int J Mol Sci 2023, 24, 10193. [Google Scholar] [CrossRef] [PubMed]
- Falkenstetter, S.; Leitner, J.; Brunner, S.M.; Rieder, T.N.; Kofler, B.; Weis, S. Galanin System in Human Glioma and Pituitary Adenoma. Front Endocrinol (Lausanne) 2020, 11, 155. [Google Scholar] [CrossRef] [PubMed]
- Wende, B.; Beyer, A.L.; Ruhnke, N.; Kaemmerer, D.; Sänger, J.; Schulz, S.; Lupp, A. Expression of the Calcitonin Receptor-like Receptor (CALCRL) in Normal and Neoplastic Tissues. Int J Mol Sci 2023, 24, 3960. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Nishibata, M.; Maruyama, T.; Sunami, S.; Isono, K.; Kawamata, T. Activation of Transient Receptor Potential Vanilloid 1 Is Involved in Both Pain and Tumor Growth in a Mouse Model of Cancer Pain. Neuroscience 2024, 538, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Șerban, R.; Stepan, M.; Florescu, D.N.; Boldeanu, M.; Florescu, M.; Șerbănescu, M.; Ionescu, M.; Streba, L.; Drăgoescu, N.; Christopher, P.; Obleagă, V.; Constantin, C.; Vere, C.C. Expression of Calcitonin Gene-Related Peptide and Calcitonin Receptor-like Receptor in Colorectal Adenocarcinoma. Int J Mol Sci 2024, 25, 4461. [Google Scholar] [CrossRef]
- Toledo, S.P.A.; Lourenço, D.M.J.; Santos, M.A.; Tavares, M.R.; Toledo, R.A.; Correia-Deur, J.E.d.M. Hypercalcitoninemia is not pathognomonic of medullary thyroid carcinoma. Clinics (Sao Paulo) 2009, 64, 699–706. [Google Scholar] [CrossRef]
- Master, S.R.; Mathias, P.M.; Burns, B. Medullary Thyroid Cancer. In StatPearlsStatPearls Publishing LLC: Treasure Island (FL), 2024;
- Fugazzola, L. Medullary thyroid cancer - An update. Best Pract Res Clin Endocrinol Metab 2023, 37, 101655. [Google Scholar] [CrossRef]
- Hou, Y.; Lin, B.; Xu, T.; Jiang, J.; Luo, S.; Chen, W.; Chen, X.; Wang, Y.; Liao, G.; Wang, J.; Zhang, J.; Li, X.; Xiang, X.; Xie, Y.; Wang, J.; Peng, S.; Lv, W.; Liu, Y.; Xiao, H. The neurotransmitter calcitonin gene-related peptide shapes an immunosuppressive microenvironment in medullary thyroid cancer. Nat Commun 2024, 15, 5555–7. [Google Scholar] [CrossRef]
- Alberti, G.; Sánchez-López, C.M.; Marcilla, A.; Barone, R.; Caruso Bavisotto, C.; Graziano, F.; Conway de Macario, E.; Macario, A.J.L.; Bucchieri, F.; Cappello, F.; Campanella, C.; Rappa, F. Hsp70 and Calcitonin Receptor Protein in Extracellular Vesicles from Glioblastoma Multiforme: Biomarkers with Putative Roles in Carcinogenesis and Potential for Differentiating Tumor Types. Int J Mol Sci 2024, 25, 3415. [Google Scholar] [CrossRef]
- Tang, S.; Zhu, H.; Sheng, L.; Mu, Q.; Wang, Y.; Xu, K.; Zhou, M.; Xu, Z.; Wu, A.; Ouyang, G. CALCRL knockdown suppresses cancer stemness and chemoresistance in acute myeloid leukemia with FLT3-ITD and DNM3TA-R882 double mutations. Drug Dev Res 2024, 85, e22137. [Google Scholar] [CrossRef]
- Tu, N.H.; Inoue, K.; Lewis, P.K.; Khan, A.; Hwang, J.H.; Chokshi, V.; Dabovic, B.B.; Selvaraj, S.; Bhattacharya, A.; Dubeykovskaya, Z.; Pinkerton, N.M.; Bunnett, N.W.; Loomis, C.A.; Albertson, D.G.; Schmidt, B.L. Calcitonin Related Polypeptide Alpha Mediates Oral Cancer Pain. Cells 2023, 12, 1675. [Google Scholar] [CrossRef]
- Lee, M.J.; Cho, J.; Bae, S.; Jung, H.S.; Kang, C.M.; Kim, S.H.; Choi, H.J.; Lee, C.; Kim, H.; Jo, D.; Paik, Y. Inhibition of the Alternative Complement Pathway May Cause Secretion of Factor B, Enabling an Early Detection of Pancreatic Cancer. J Proteome Res 2024, 23, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Jailani, A.B.A.; Bigos, K.J.A.; Avgoustou, P.; Egan, J.L.; Hathway, R.A.; Skerry, T.M.; Richards, G.O. Targeting the adrenomedullin-2 receptor for the discovery and development of novel anti-cancer agents. Expert Opin Drug Discov 2022, 17, 839–848. [Google Scholar] [CrossRef]
- Ouyang, Q.; Zhou, J.; Yang, W.; Cui, H.; Xu, M.; Yi, L. Oncogenic role of neurotensin and neurotensin receptors in various cancers. Clin Exp Pharmacol Physiol 2017, 44, 841–846. [Google Scholar] [CrossRef]
- Qiu, S.; Pellino, G.; Fiorentino, F.; Rasheed, S.; Darzi, A.; Tekkis, P.; Kontovounisios, C. A Review of the Role of Neurotensin and Its Receptors in Colorectal Cancer. Gastroenterol Res Pract 2017, 2017, 6456257. [Google Scholar] [CrossRef]
- Nikolaou, S.; Qiu, S.; Fiorentino, F.; Simillis, C.; Rasheed, S.; Tekkis, P.; Kontovounisios, C. The role of Neurotensin and its receptors in non-gastrointestinal cancers: a review. Cell Commun Signal 2020, 18, 68–y. [Google Scholar] [CrossRef] [PubMed]
- Christou, N.; Blondy, S.; David, V.; Verdier, M.; Lalloué, F.; Jauberteau, M.; Mathonnet, M.; Perraud, A. Neurotensin pathway in digestive cancers and clinical applications: an overview. Cell Death Dis 2020, 11, 1027–8. [Google Scholar] [CrossRef]
- Moody, T.W.; Ramos-Alvarez, I.; Jensen, R.T. Adding of neurotensin to non-small cell lung cancer cells increases tyrosine phosphorylation of HER3. Peptides 2022, 156, 170858. [Google Scholar] [CrossRef]
- Sánchez, M.L.; Coveñas, R. The Neurotensinergic System: A Target for Cancer Treatment. Curr Med Chem 2022, 29, 3231–3260. [Google Scholar] [CrossRef]
- Zhu, S.; Tian, H.; Niu, X.; Wang, J.; Li, X.; Jiang, N.; Wen, S.; Chen, X.; Ren, S.; Xu, C.; Chang, C.; Flores-Morales, A.; Shang, Z.; Sun, Y.; Niu, Y. Neurotensin and its receptors mediate neuroendocrine transdifferentiation in prostate cancer. Oncogene 2019, 38, 4875–4884. [Google Scholar] [CrossRef]
- Szaryńska, M.; Olejniczak-Kęder, A.; Podpłońska, K.; Prahl, A.; Iłowska, E. Bradykinin and Neurotensin Analogues as Potential Compounds in Colon Cancer Therapy. Int J Mol Sci 2023, 24, 9644. [Google Scholar] [CrossRef]
- Hung, Y.; Wang, H.; Hsu, S.; Wang, L.; Tsai, Y.; Su, Y.; Hung, W.; Chen, L. Neuron-derived neurotensin promotes pancreatic cancer invasiveness and gemcitabine resistance via the NTSR1/Akt pathway. Am J Cancer Res 2024, 14, 448–466. [Google Scholar] [CrossRef] [PubMed]
- Ikhlef, L.; Yassine, M.; Chandouri, B.; Rivière, L.; Naves, T.; Dmytruk, N.; Gachard, N.; Jauberteau, M.; Gallet, P. Targeting the NTSR2/TrkB oncogenic pathway in chronic lymphocytic leukemia. Sci Rep 2024, 14, 6084–5. [Google Scholar] [CrossRef] [PubMed]
- Masoumi, J.; Jafarzadeh, A.; Khorramdelazad, H.; Abbasloui, M.; Abdolalizadeh, J.; Jamali, N. Role of Apelin/APJ axis in cancer development and progression. Adv Med Sci 2020, 65, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Grinstead, C.; Yoon, S. Apelin, a Circulating Biomarker in Cancer Evaluation: A Systematic Review. Cancers (Basel) 2022, 14, 4656. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Z.; Zhao, Q.; Chen, L. Roles of apelin/APJ system in cancer: Biomarker, predictor, and emerging therapeutic target. J Cell Physiol 2022, 237, 3734–3751. [Google Scholar] [CrossRef]
- Hu, L.; Hayashi, Y.; Kidoya, H.; Takakura, N. Endothelial cell-derived Apelin inhibits tumor growth by altering immune cell localization. Sci Rep 2021, 11, 14047–5. [Google Scholar] [CrossRef]
- Çelik, F.S.; Güneş, C.E.; Yavuz, E.; Kurar, E. Apelin triggers macrophage polarization to M2 type in head and neck cancer. Immunobiology 2023, 228, 152353. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, B.; Zhang, W.; Zhang, T.; Liu, Q.; Jiao, X.; Ye, J.; Hao, Y.; Gao, Q.; Ma, G.; Hao, C.; Cui, B. APLN promotes the proliferation, migration, and glycolysis of cervical cancer through the PI3K/AKT/mTOR pathway. Arch Biochem Biophys 2024, 755, 109983. [Google Scholar] [CrossRef]
- Effenberger, M.; Grander, C.; Hausmann, B.; Enrich, B.; Pjevac, P.; Zoller, H.; Tilg, H. Apelin and the gut microbiome: Potential interaction in human MASLD. Dig Liver Dis 2024, 56, 932–940. [Google Scholar] [CrossRef]
- Fang, X.; Hong, Y.; Dai, L.; Qian, Y.; Zhu, C.; Wu, B.; Li, S. CRH promotes human colon cancer cell proliferation via IL-6/JAK2/STAT3 signaling pathway and VEGF-induced tumor angiogenesis. Mol Carcinog 2017, 56, 2434–2445. [Google Scholar] [CrossRef]
- Zhu, C.; Li, S. Role of CRH in colitis and colitis-associated cancer: a combinative result of central and peripheral effects? Front Endocrinol (Lausanne) 2024, 15, 1363748. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Wen, H.; Dai, L.; Lou, Y.; Wang, Z.; Yi, Y.; Yan, X.; Wu, Y.; Sun, W.; Chen, P.; Yang, S.; Qi, X.; Zhang, Y.; Wu, G. A brain-tumor neural circuit controls breast cancer progression in mice. J Clin Invest 2023, 133, e167725. [Google Scholar] [CrossRef]
- Yoshida, S.; Hamada, Y.; Narita, M.; Sato, D.; Tanaka, K.; Mori, T.; Tezuka, H.; Suda, Y.; Tamura, H.; Aoki, K.; Kuzumaki, N.; Narita, M. Elucidation of the mechanisms underlying tumor aggravation by the activation of stress-related neurons in the paraventricular nucleus of the hypothalamus. Mol Brain 2023, 16, 18–0. [Google Scholar] [CrossRef] [PubMed]
- Mao, R.; Zhou, F.; Hong, Y.; Li, Y.; Zhu, C.; Jin, L.; Li, S. CRH upregulates supervillin through ERK and AKT pathways to promote bladder cancer cell migration. Cell Biol Int 2024. [Google Scholar] [CrossRef]
- Dimas, A.; Goussia, A.; Papoudou-Bai, A.; Politi, A.; Paschopoulos, M.; Navrozoglou, I.; Makrigiannakis, A.; Vrekoussis, T. The expression of corticotropin-releasing hormone family peptides in premalignant and malignant vulvar lesions. Clin Transl Oncol 2024, 26, 260–268. [Google Scholar] [CrossRef]
- Sato, N.; Motoi, F.; Tajiki, H.; Kawaguchi, K.; Ohtsuka, H.; Takadate, T.; Nakagawa, K.; Takagi, K.; Suzuki, T.; Katayose, Y.; Fukudo, S.; Unno, M. Expression of Corticotropin-Releasing Hormone and Its Receptors May Be Associated With Survival Rate in Pancreatic Cancer. Gastro Hep Adv 2022, 2, 147–155. [Google Scholar] [CrossRef]
- Arvanitakis, K.; Koufakis, T.; Kotsa, K.; Germanidis, G. How Far beyond Diabetes Can the Benefits of Glucagon-like Peptide-1 Receptor Agonists Go? A Review of the Evidence on Their Effects on Hepatocellular Carcinoma. Cancers (Basel) 2022, 14, 4651. [Google Scholar] [CrossRef] [PubMed]
- Faggiano, A. Long-acting somatostatin analogs and well differentiated neuroendocrine tumors: a 20-year-old story. J Endocrinol Invest 2024, 47, 35–46. [Google Scholar] [CrossRef]
- Wang, G.; Ye, Y.; Zhang, X.; Song, J. Bradykinin stimulates IL-6 production and cell invasion in colorectal cancer cells. Oncol Rep 2014, 32, 1709–1714. [Google Scholar] [CrossRef]
- Sánchez, M.L.; Mangas, A.; Coveñas, R. Glioma and Peptidergic Systems: Oncogenic and Anticancer Peptides. Int J Mol Sci 2024, 25, 7990. [Google Scholar] [CrossRef]
- Rodríguez, F.D.; Coveñas, R. Antitumor Strategies Targeting Peptidergic Systems. Encyclopedia 2024, 4, 487. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, H.; Tan, L.; Siu, K.T.H.; Guan, X. Exploring treatment options in cancer: Tumor treatment strategies. Signal Transduct Target Ther 2024, 9, 175. [Google Scholar] [CrossRef]
- Li, Z.; You, Y.; Griffin, N.; Feng, J.; Shan, F. Low-dose naltrexone (LDN): A promising treatment in immune-related diseases and cancer therapy. Int Immunopharmacol 2018, 61, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Yan, L.; Shan, F.; Wang, X.; Qu, N.; Handley, M.K.; Ma, M. Low-dose naltrexone plays antineoplastic role in cervical cancer progression through suppressing PI3K/AKT/mTOR pathway. Transl Oncol 2021, 14, 101028. [Google Scholar] [CrossRef]
- Qu, N.; Meng, Y.; Handley, M.K.; Wang, C.; Shan, F. Preclinical and clinical studies into the bioactivity of low-dose naltrexone (LDN) for oncotherapy. Int Immunopharmacol 2021, 96, 107714. [Google Scholar] [CrossRef]
- Vijayakumar, J.; Haddad, T.; Gupta, K.; Sauers, J.; Yee, D. An open label phase II study of safety and clinical activity of naltrexone for treatment of hormone refractory metastatic breast cancer. Invest New Drugs 2023, 41, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Ciwun, M.; Tankiewicz-Kwedlo, A.; Pawlak, D. Low-Dose Naltrexone as an Adjuvant in Combined Anticancer Therapy. Cancers (Basel) 2024, 16, 1240. [Google Scholar] [CrossRef]
- Akbari, S.; Assaran Darban, R.; Javid, H.; Esparham, A.; Hashemy, S.I. The anti-tumoral role of Hesperidin and Aprepitant on prostate cancer cells through redox modifications. Naunyn Schmiedebergs Arch Pharmacol 2023, 396, 3559–3567. [Google Scholar] [CrossRef]
- Coveñas, R.; Rodríguez, F.D.; Robinson, P.; Muñoz, M. The Repurposing of Non-Peptide Neurokinin-1 Receptor Antagonists as Antitumor Drugs: An Urgent Challenge for Aprepitant. Int J Mol Sci 2023, 24, 15936. [Google Scholar] [CrossRef]
- Nishibe-Toyosato, S.; Ando, Y.; Torii, Y.; Ichikawa, R.; Owaki, A.; Miyamura, H.; Nishio, E.; Matsuda, H.; Tsujii-Fujii, N.; Shimato-Isobe, A.; Mukaiji, K.; Ito, K.; Hayashi, T.; Fujii, T.; Yamada, S. Comparing Injection Site Reactions of Aprepitant and Fosaprepitant in Gynecologic Cancer Chemotherapy. In Vivo 2024, 38, 2374–2382. [Google Scholar] [CrossRef]
- Valdivia, V.; Recio, R.; Lerena, P.; Pozo, E.; Serrano, R.; Calero, R.; Pintado, C.; Leal, M.P.; Moreno-Rodríguez, N.; Organero, J. Á; Khiar, N.; Fernández, I. Biological evaluation of carbohydrate-based aprepitant analogs for neuroblastoma treatment. Eur J Med Chem 2024, 264, 116021. [Google Scholar] [CrossRef]
- Beirith, I.; Renz, B.W.; Mudusetti, S.; Ring, N.S.; Kolorz, J.; Koch, D.; Bazhin, A.V.; Berger, M.; Wang, J.; Angele, M.K.; D'Haese, J.G.; Guba, M.O.; Niess, H.; Andrassy, J.; Werner, J.; Ilmer, M. Identification of the Neurokinin-1 Receptor as Targetable Stratification Factor for Drug Repurposing in Pancreatic Cancer. Cancers (Basel) 2021, 13, 2703. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Lee, M.; Gao, Y.; Bu, P.; Coarfa, C.; Miles, B.; Sreekumar, A.; Creighton, C.J.; Ayala, G. Neuropeptide Y nerve paracrine regulation of prostate cancer oncogenesis and therapy resistance. Prostate 2021, 81, 58–71. [Google Scholar] [CrossRef]
- Kuipers, A.; Balaskó, M.; Pétervári, E.; Koller, A.; Brunner, S.M.; Moll, G.N.; Kofler, B. Intranasal Delivery of a Methyllanthionine-Stabilized Galanin Receptor-2-Selective Agonist Reduces Acute Food Intake. Neurotherapeutics 2021, 18, 2737–2752. [Google Scholar] [CrossRef] [PubMed]
- Deigin, V.; Linkova, N.; Vinogradova, J.; Vinogradov, D.; Polyakova, V.; Medvedev, D.; Krasichkov, A.; Volpina, O. The First Reciprocal Activities of Chiral Peptide Pharmaceuticals: Thymogen and Thymodepressin, as Examples. Int J Mol Sci 2024, 25, 5042. [Google Scholar] [CrossRef] [PubMed]
- Koller, A.; Rid, R.; Beyreis, M.; Bianchini, R.; Holub, B.S.; Lang, A.; Sternberg, F.; Brodowicz, B.; Velickovic, O.; Jakab, M.; Kerschbaum, H.; Önder, K.; Kofler, B. In vitro toxicity of the galanin receptor 3 antagonist SNAP 37889. Neuropeptides 2016, 56, 83–88. [Google Scholar] [CrossRef]
- Avgoustou, P.; Jailani, A.B.A.; Zirimwabagabo, J.; Tozer, M.J.; Gibson, K.R.; Glossop, P.A.; Mills, J.E.J.; Porter, R.A.; Blaney, P.; Bungay, P.J.; Wang, N.; Shaw, A.P.; Bigos, K.J.A.; Holmes, J.L.; Warrington, J.I.; Skerry, T.M.; Harrity, J.P.A.; Richards, G.O. Discovery of a First-in-Class Potent Small Molecule Antagonist against the Adrenomedullin-2 Receptor. ACS Pharmacol Transl Sci 2020, 3, 706–719. [Google Scholar] [CrossRef]
- Zirimwabagabo, J.; Jailani, A.B.A.; Avgoustou, P.; Tozer, M.J.; Gibson, K.R.; Glossop, P.A.; Mills, J.E.J.; Porter, R.A.; Blaney, P.; Wang, N.; Skerry, T.M.; Richards, G.O.; Harrity, J.P.A. Discovery of a First-In-Class Small Molecule Antagonist against the Adrenomedullin-2 Receptor: Structure-Activity Relationships and Optimization. J Med Chem 2021, 64, 3299–3319. [Google Scholar] [CrossRef] [PubMed]
- Gluexam, T.; Grandits, A.M.; Schlerka, A.; Nguyen, C.H.; Etzler, J.; Finkes, T.; Fuchs, M.; Scheid, C.; Heller, G.; Hackl, H.; Harrer, N.; Sill, H.; Koller, E.; Stoiber, D.; Sommergruber, W.; Wieser, R. CGRP Signaling via CALCRL Increases Chemotherapy Resistance and Stem Cell Properties in Acute Myeloid Leukemia. Int J Mol Sci 2019, 20, 5826. [Google Scholar] [CrossRef]
- Zhu, W.; Sheng, D.; Shao, Y.; Zhang, Q.; Peng, Y. Neuronal calcitonin gene-related peptide promotes prostate tumor growth in the bone microenvironment. Peptides 2021, 135, 170423. [Google Scholar] [CrossRef]
- Suekane, A.; Ichikawa, T.; Saito, Y.; Nakahata, S.; Morishita, K. The CGRP Receptor Antagonist MK0974 Induces EVI1(high) AML Cell Apoptosis by Disrupting ERK Signaling. Anticancer Res 2022, 42, 4743–4752. [Google Scholar] [CrossRef]
- Liu, J.; Agopiantz, M.; Poupon, J.; Wu, Z.; Just, P.; Borghese, B.; Ségal-Bendirdjian, E.; Gauchotte, G.; Gompel, A.; Forgez, P. Neurotensin Receptor 1 Antagonist SR48692 Improves Response to Carboplatin by Enhancing Apoptosis and Inhibiting Drug Efflux in Ovarian Cancer. Clin Cancer Res 2017, 23, 6516–6528. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.; Ehrlich, L.; Venter, J.; O'Brien, A.; White, T.; Zhou, T.; Dang, T.; Meng, F.; Invernizzi, P.; Bernuzzi, F.; Alpini, G.; Lairmore, T.C.; Glaser, S. Inhibition of the apelin/apelin receptor axis decreases cholangiocarcinoma growth. Cancer Lett 2017, 386, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Ying, H.; Yu, Z.; Chang, L.; Chen, Z.; Chen, J.; Chang, S.; Qiu, Y.; Lin, X. Apelin Receptor Can Act as a Specific Marker and Promising Therapeutic Target for Infantile Hemangioma. J Invest Dermatol 2023, 143, 566–577.e12. [Google Scholar] [CrossRef]
- Savage, S.R.; Yi, X.; Lei, J.T.; Wen, B.; Zhao, H.; Liao, Y.; Jaehnig, E.J.; Somes, L.K.; Shafer, P.W.; Lee, T.D.; Fu, Z.; Dou, Y.; Shi, Z.; Gao, D.; Hoyos, V.; Gao, Q.; Zhang, B. Pan-cancer proteogenomics expands the landscape of therapeutic targets. Cell 2024, 187, 4389–4407.e15. [Google Scholar] [CrossRef]
- Lusardi, M.; Signorello, M.G.; Russo, E.; Caviglia, D.; Ponassi, M.; Iervasi, E.; Rosano, C.; Brullo, C.; Spallarossa, A. Structure-Activity Relationship Studies on Highly Functionalized Pyrazole Hydrazones and Amides as Antiproliferative and Antioxidant Agents. Int J Mol Sci 2024, 25, 4607. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Ashrafizadeh, M.; Aref, A.R.; Sethi, G.; Ertas, Y.N. Peptide-functionalized, -assembled and -loaded nanoparticles in cancer therapy. Drug Discov Today 2024, 29, 103981. [Google Scholar] [CrossRef]
- Suresh, V.; Bardhan, M.; Dave, T.; Shamim, M.A.; Suresh, D.; Satish, P.; Dhakal, B.; Bhonsale, A.; Roy, P.; Padhi, B.K.; Monteith, T. Zavegepant for Acute Treatment of Migraine: A Systematic Review and Meta-analysis of Randomized Controlled Trials. Clin Neuropharmacol 2024, 47, 72–81. [Google Scholar] [CrossRef]
- Jerra, V.S.; Ramachandran, B.; Shareef, S.; Carrillo-Bermejo, A.; Sundararaj, R.; Venkatesan, S. Molecular docking aided machine learning for the identification of potential VEGFR inhibitors against renal cell carcinoma. Med Oncol 2024, 41, 198–0. [Google Scholar] [CrossRef]
- Caronna, E.; Alpuente, A.; Torres-Ferrus, M.; Pozo-Rosich, P. CGRP monoclonal antibodies and CGRP receptor antagonists (Gepants) in migraine prevention. Handb Clin Neurol 2024, 199, 107–124. [Google Scholar] [CrossRef]
Figure 1.
Natural peptide ligands for eight representative GPCRs. The lines indicate the binding affinity (in different degrees) of peptides for the receptor type. The figure is based on data on pairing peptides and GPCRs reported by Foster et al. [14]. Abbreviations: AM2/IMD, Adrenomedullin/Intermedin; AMY, amylin; APLNR, apelin receptor; CALCR, calcitonin receptor; CALCRL, Calcitonin receptor-like; α-CGRP, Alpha-calcitonin gene-related peptide; CRHR, Corticotrophin releasing hormone receptor; CT, Calcitonin; DOPR, delta-opioid receptor; ELA, Elabela; GALR, Galanin receptor; GPR151, G protein-coupled receptor 151 KOPR, kappa opioid receptor; Leu-Enk, Leucine-enkephalin; Met-Enk, Methionine-enkephalin; MOPR, Mu opioid receptor; NKA, Neurokinin A; NKB, Neurokinin B; NKR, Neurokinin receptor; NPYR, neuropeptide Y receptor; NTSR, Neurotensin receptor; PP, pancreatic peptide; PYY, Peptide YY; PrRP, Prolactin-releasing peptide; RFRP, R(Arg) F(Phe)-amide-related peptide SF11, [N-(4-ethoxyphenyl)-4-(hydroxydiphenylmethyl)-1-piperidinecarbothioamide]; SP, substance P; UCN, Urocortin.
Figure 1.
Natural peptide ligands for eight representative GPCRs. The lines indicate the binding affinity (in different degrees) of peptides for the receptor type. The figure is based on data on pairing peptides and GPCRs reported by Foster et al. [14]. Abbreviations: AM2/IMD, Adrenomedullin/Intermedin; AMY, amylin; APLNR, apelin receptor; CALCR, calcitonin receptor; CALCRL, Calcitonin receptor-like; α-CGRP, Alpha-calcitonin gene-related peptide; CRHR, Corticotrophin releasing hormone receptor; CT, Calcitonin; DOPR, delta-opioid receptor; ELA, Elabela; GALR, Galanin receptor; GPR151, G protein-coupled receptor 151 KOPR, kappa opioid receptor; Leu-Enk, Leucine-enkephalin; Met-Enk, Methionine-enkephalin; MOPR, Mu opioid receptor; NKA, Neurokinin A; NKB, Neurokinin B; NKR, Neurokinin receptor; NPYR, neuropeptide Y receptor; NTSR, Neurotensin receptor; PP, pancreatic peptide; PYY, Peptide YY; PrRP, Prolactin-releasing peptide; RFRP, R(Arg) F(Phe)-amide-related peptide SF11, [N-(4-ethoxyphenyl)-4-(hydroxydiphenylmethyl)-1-piperidinecarbothioamide]; SP, substance P; UCN, Urocortin.
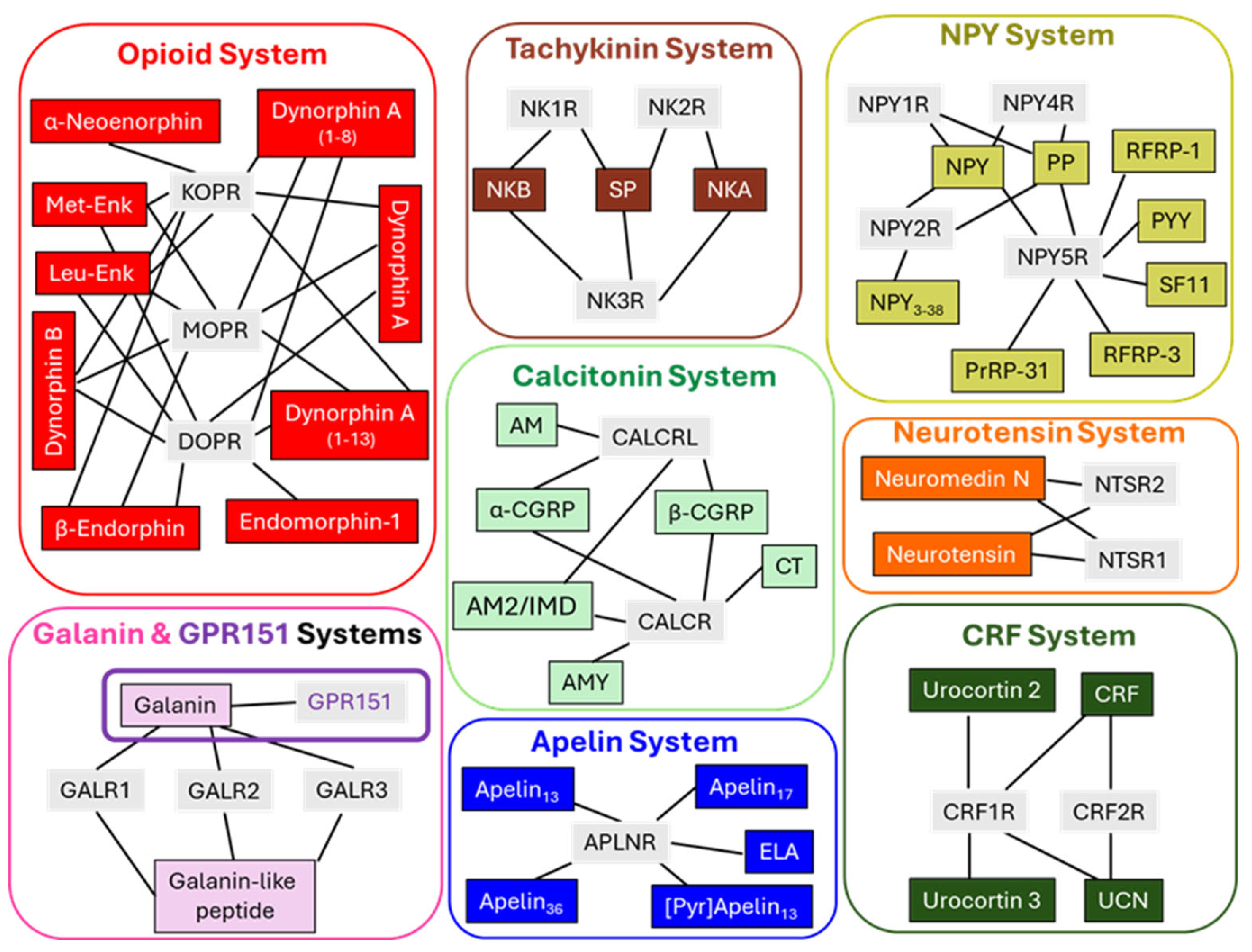
Figure 2.
Three-dimensional structures representing peptidergic GPCRs determined by X-ray crystallography or cryo-electron microscopy. All structures shown were obtained from the protein data bank [62] and drawn with the Molstar online free software [63]. Each figure depicts the receptor abbreviated name and the PDB identity code (four characters). Abbreviations: APLNR, apelin receptor; CLR-RAMP1, calcitonin-like receptor—receptor activity modifying protein 1; CRF1R, corticotropin releasing factor receptor 1; GALR1, galanin receptor 1; MOP, mu-opioid receptor; NK-1R, neurokinin 1 receptor; NPY-1R, neuropeptide receptor 1; NTS1R, neurotensin receptor 1.
Figure 2.
Three-dimensional structures representing peptidergic GPCRs determined by X-ray crystallography or cryo-electron microscopy. All structures shown were obtained from the protein data bank [62] and drawn with the Molstar online free software [63]. Each figure depicts the receptor abbreviated name and the PDB identity code (four characters). Abbreviations: APLNR, apelin receptor; CLR-RAMP1, calcitonin-like receptor—receptor activity modifying protein 1; CRF1R, corticotropin releasing factor receptor 1; GALR1, galanin receptor 1; MOP, mu-opioid receptor; NK-1R, neurokinin 1 receptor; NPY-1R, neuropeptide receptor 1; NTS1R, neurotensin receptor 1.
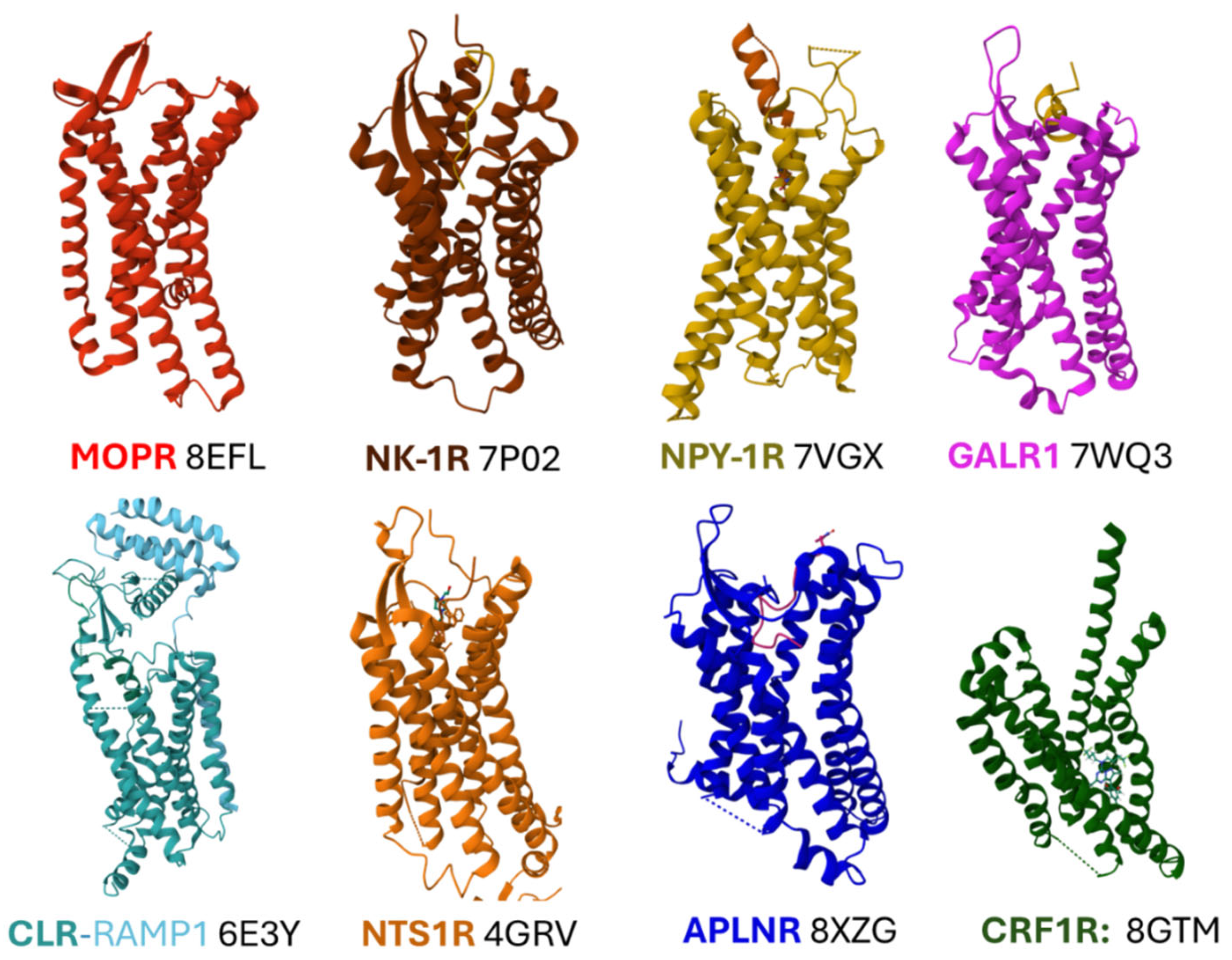
Figure 3.
A depicts the binding site of the opioid agonist fentanyl to MOPR. Transmembrane domains (TM) are indicated, as well as the amino acid residues with a one-letter code and the number in superscript indicating their position in the primary sequence of the receptor protein. The color code of the residues denotes the TM where the residues belong. A so-called minor pocket lodging the aromatic ring, as defined by TM2 and TM3, appears to be highlighted in yellow. The inset in A presents the two-dimensional structure of fentanyl. B shows the binding pockets of substance P (SP) and aprepitant within the NK-1R. ECL2 denotes the extracellular domain 2. The inset in B contains the two-dimensional structure of aprepitant with the positioning of amino acids that delimit the binding cavity. The three-dimensional representations were from the protein data bank [62] sketched with the Molstar online free software [63]. The two-dimensional chemical structures were drawn using KingDraw, a free chemical structure editor (version 3.0 for Windows).
Figure 3.
A depicts the binding site of the opioid agonist fentanyl to MOPR. Transmembrane domains (TM) are indicated, as well as the amino acid residues with a one-letter code and the number in superscript indicating their position in the primary sequence of the receptor protein. The color code of the residues denotes the TM where the residues belong. A so-called minor pocket lodging the aromatic ring, as defined by TM2 and TM3, appears to be highlighted in yellow. The inset in A presents the two-dimensional structure of fentanyl. B shows the binding pockets of substance P (SP) and aprepitant within the NK-1R. ECL2 denotes the extracellular domain 2. The inset in B contains the two-dimensional structure of aprepitant with the positioning of amino acids that delimit the binding cavity. The three-dimensional representations were from the protein data bank [62] sketched with the Molstar online free software [63]. The two-dimensional chemical structures were drawn using KingDraw, a free chemical structure editor (version 3.0 for Windows).
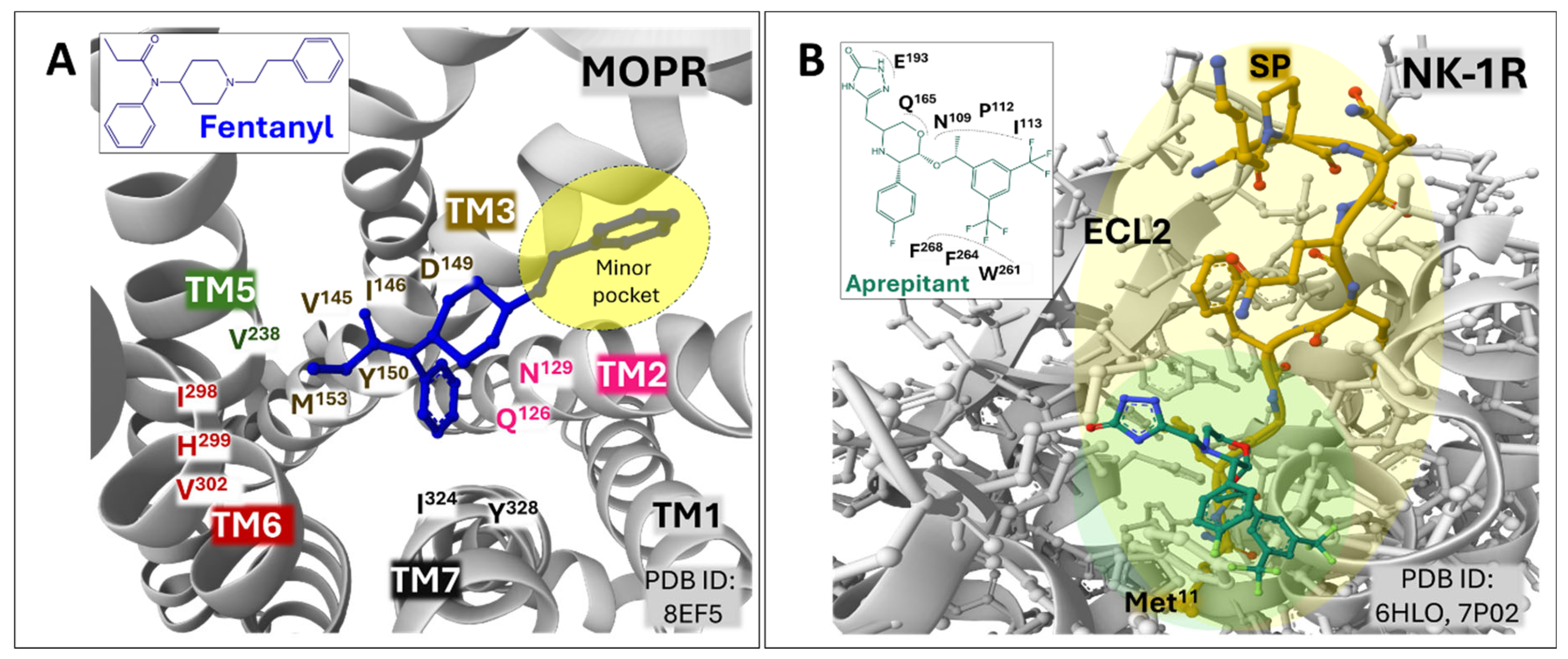
Figure 4.
Panel A depicts the accommodation of the C-terminal region of NPY to the NPY-1R (see texts for details). Panel B represents the N-terminal end (residues 1 to 16) of galanin (GAL), with the network of weak interactions defining the binding site in the GALR1. The B inset represents the galanin receptor site delimited by extracellular loops 2 and 3 (ECL2, ECL3) and transmembrane domains (TM) II, VI, and VII. Dashed colored lines in both panels represent weak interactions between the ligands and their respective receptors. The amino acid residues were drawn with a one-letter code for the receptor proteins and a three-letter code for the peptide ligands. The superscript number denotes their position in the primary sequence of the receptor or the peptide ligand. The three-dimensional representations were acquired from the protein data bank [62] sketched with the Molstar online free software [63].
Figure 4.
Panel A depicts the accommodation of the C-terminal region of NPY to the NPY-1R (see texts for details). Panel B represents the N-terminal end (residues 1 to 16) of galanin (GAL), with the network of weak interactions defining the binding site in the GALR1. The B inset represents the galanin receptor site delimited by extracellular loops 2 and 3 (ECL2, ECL3) and transmembrane domains (TM) II, VI, and VII. Dashed colored lines in both panels represent weak interactions between the ligands and their respective receptors. The amino acid residues were drawn with a one-letter code for the receptor proteins and a three-letter code for the peptide ligands. The superscript number denotes their position in the primary sequence of the receptor or the peptide ligand. The three-dimensional representations were acquired from the protein data bank [62] sketched with the Molstar online free software [63].
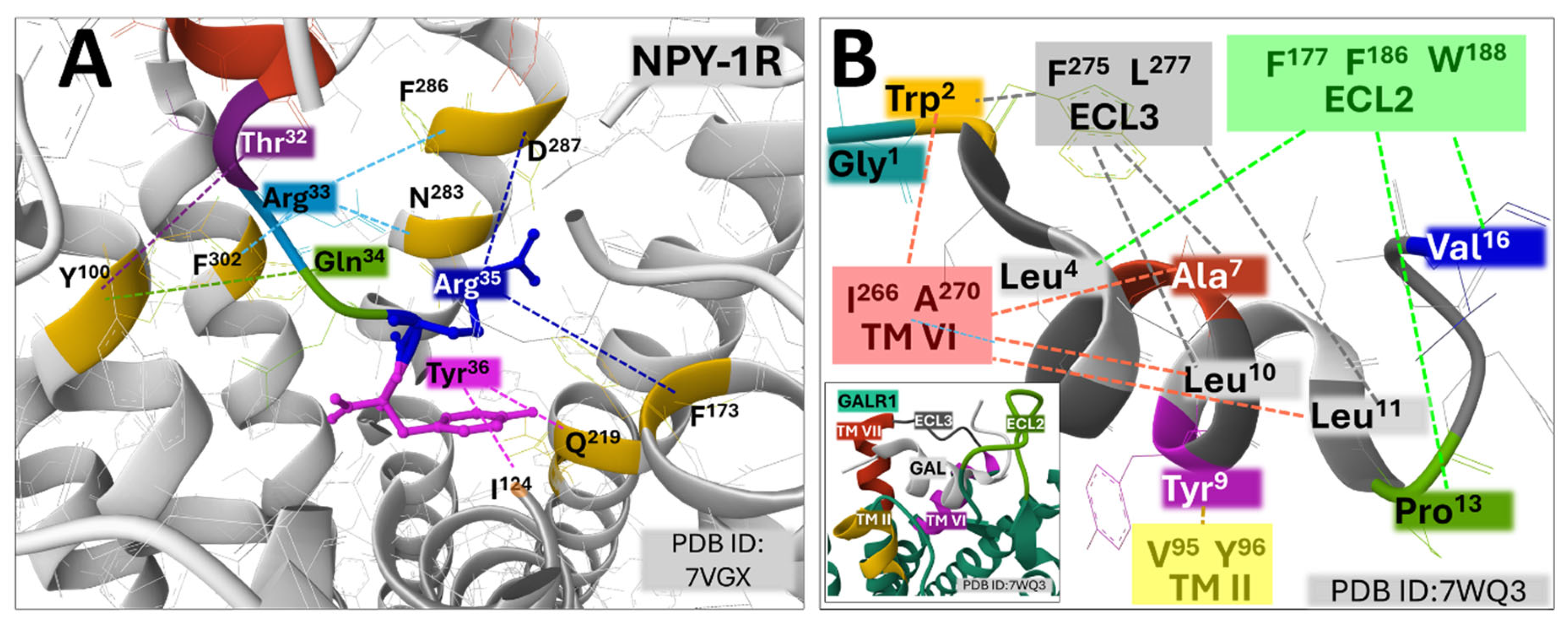
Figure 5.
Panel A represents the contacts between the C-terminal region of CGRP and RAMP1 (see texts for details). Panel B depicts the N-terminal end of CGRP with the circuitry of weak interactions defining the binding site in CLR. Some residues in extracellular loops 2 and 3 (ECL2, ECL3) and transmembrane domains (TM.) V marking out the peptide binding site are highlighted. The amino acid residues were drawn with a one-letter code for the receptor proteins and a three-letter code for the peptide ligand. The superscript number denotes their position in the primary sequence of the receptor or the peptide ligand. The three-dimensional representations were acquired from the protein data bank [62] drawn with the Molstar online free software [63].
Figure 5.
Panel A represents the contacts between the C-terminal region of CGRP and RAMP1 (see texts for details). Panel B depicts the N-terminal end of CGRP with the circuitry of weak interactions defining the binding site in CLR. Some residues in extracellular loops 2 and 3 (ECL2, ECL3) and transmembrane domains (TM.) V marking out the peptide binding site are highlighted. The amino acid residues were drawn with a one-letter code for the receptor proteins and a three-letter code for the peptide ligand. The superscript number denotes their position in the primary sequence of the receptor or the peptide ligand. The three-dimensional representations were acquired from the protein data bank [62] drawn with the Molstar online free software [63].
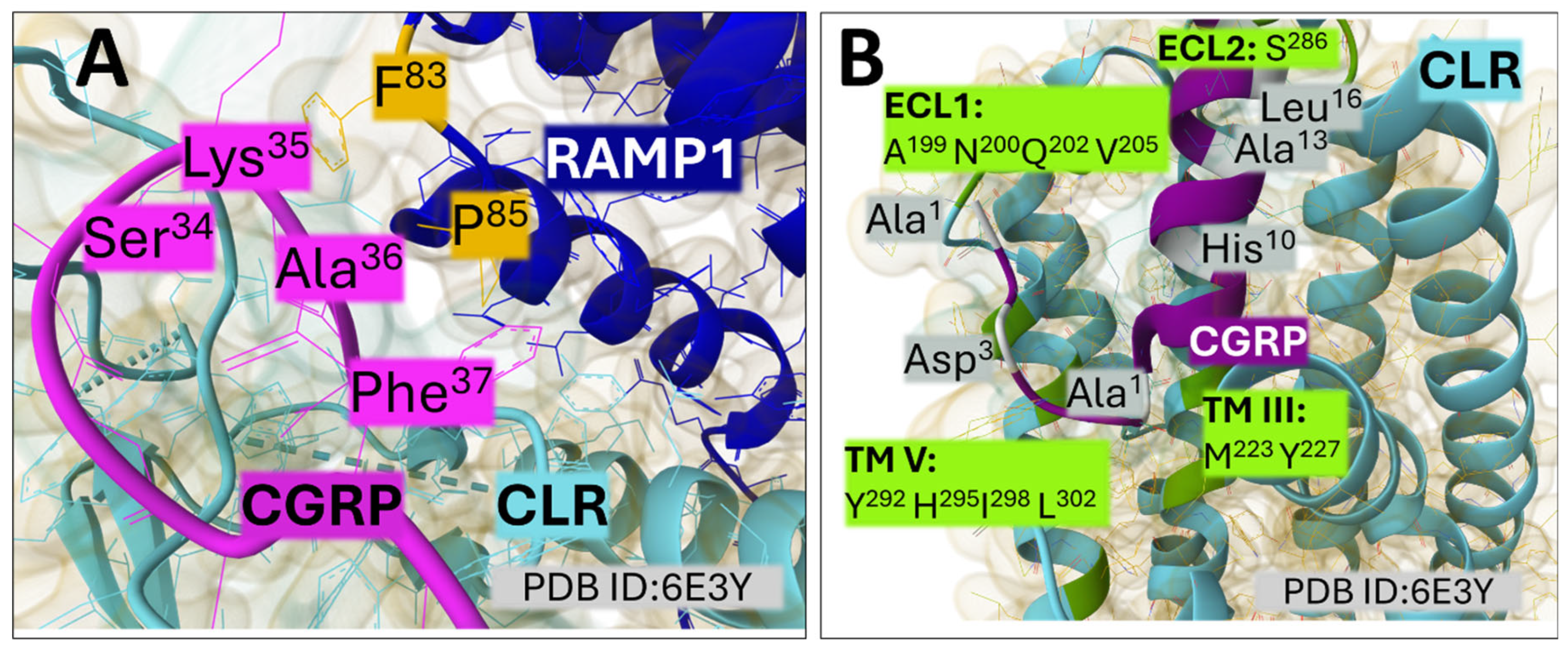
Figure 6.
Panel A highlights the primary contacts between the C-terminal region of NTS8-13 and NTS1R (PDB ID: 4GRV). The inset in A illustrates the structure of inverse agonist SR48692 (PDB ID: 6ZIN) (refer to text for details). Panel B depicts the binding site of the apelin17 mimetic peptide, AMG3054 (PDB ID: 5VBL), in the apelin receptor (APLNR). The amino acid residues were drawn with a one-letter code for the receptor proteins and a three-letter code for the peptide ligands. The superscript number denotes their position in the primary sequence of the receptor or the peptide ligand. Transmembrane domains (TM) and extracellular loops (ECL) are indicated. The three-dimensional representations were acquired from the protein data bank [62] drawn with the Molstar online free software [63].
Figure 6.
Panel A highlights the primary contacts between the C-terminal region of NTS8-13 and NTS1R (PDB ID: 4GRV). The inset in A illustrates the structure of inverse agonist SR48692 (PDB ID: 6ZIN) (refer to text for details). Panel B depicts the binding site of the apelin17 mimetic peptide, AMG3054 (PDB ID: 5VBL), in the apelin receptor (APLNR). The amino acid residues were drawn with a one-letter code for the receptor proteins and a three-letter code for the peptide ligands. The superscript number denotes their position in the primary sequence of the receptor or the peptide ligand. Transmembrane domains (TM) and extracellular loops (ECL) are indicated. The three-dimensional representations were acquired from the protein data bank [62] drawn with the Molstar online free software [63].

Figure 7.
The allosteric antagonist of BMK-I-152 binds to the core of CRF1R (PDB ID: 8GTM), depicting the surface contacts of the receptor with the antagonist. The site has three regions: the upper, defined by residues in TM II, III, VI, and VII; the middle, by residues in TM III, V, and VI; and the bottom, by residues in TM III, V, and VII. The inset illustrates the Gaussian volume representation of BMK-I-152 (PDB ID: 8GTM). The amino acid residues were drawn with a one-letter code. The superscript number denotes their position in the primary sequence of the receptor and the background color of the TM to which they belong. The structures were acquired from the protein data bank [62] and drawn with the Molstar online free software [63].
Figure 7.
The allosteric antagonist of BMK-I-152 binds to the core of CRF1R (PDB ID: 8GTM), depicting the surface contacts of the receptor with the antagonist. The site has three regions: the upper, defined by residues in TM II, III, VI, and VII; the middle, by residues in TM III, V, and VI; and the bottom, by residues in TM III, V, and VII. The inset illustrates the Gaussian volume representation of BMK-I-152 (PDB ID: 8GTM). The amino acid residues were drawn with a one-letter code. The superscript number denotes their position in the primary sequence of the receptor and the background color of the TM to which they belong. The structures were acquired from the protein data bank [62] and drawn with the Molstar online free software [63].
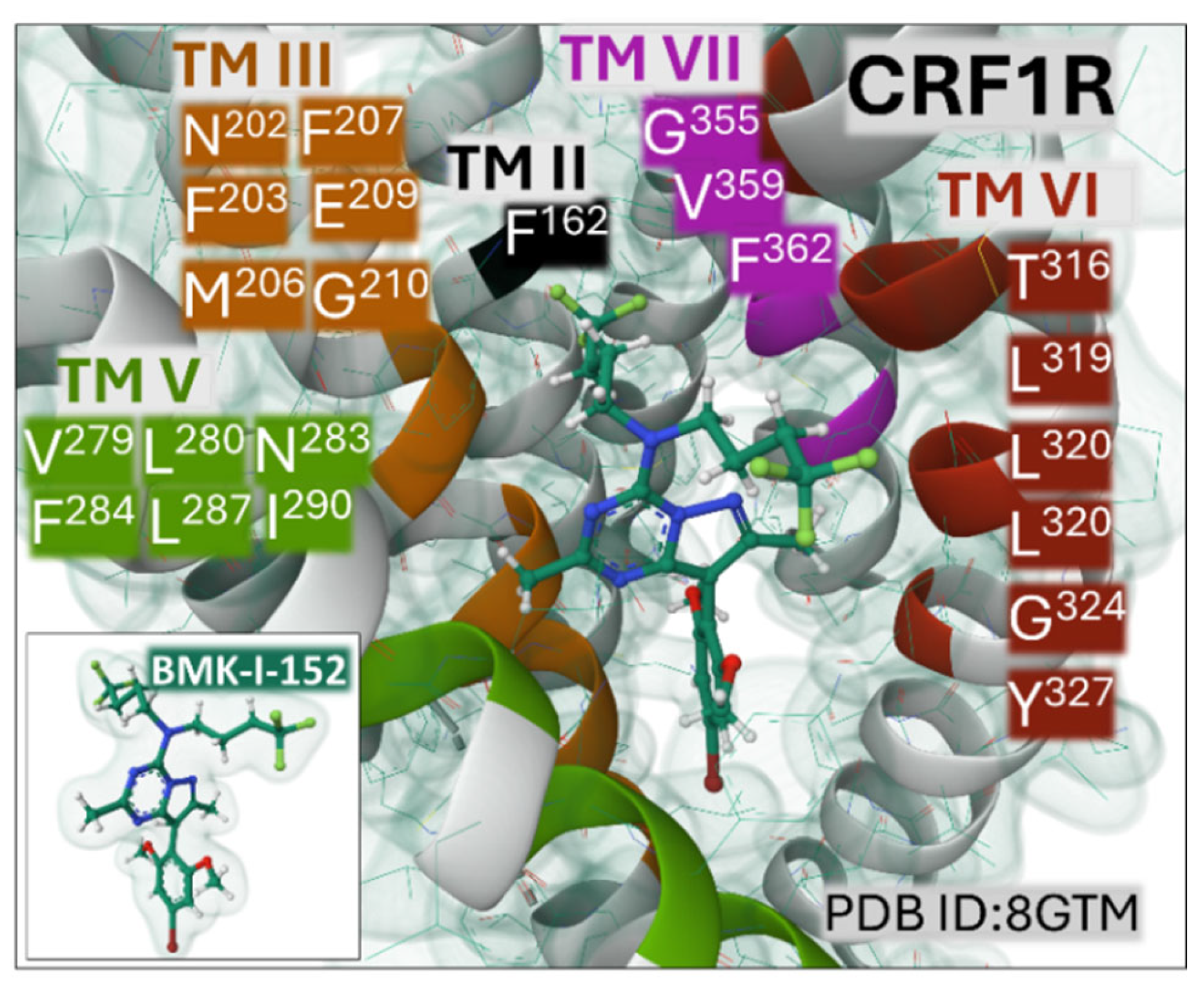
Figure 8.
Schematic representation of signaling pathways triggered by peptides binding to their respective receptors (A). B indicates how GPCRs activated by peptides may signal from the plasma membrane or, once internalized in endosomes, from signalosomes, determining complex and modulated intracellular signals that affect cell metabolic processes and transcriptional programs. Alteration of GPCR homeostasis through uncontrolled overexpression, down-regulation, truncation, dimerization, or overstimulation leads to changes inducing transitory or sustained cellular events, leading to aberrant cell growth, cell migration, inhibition of apoptosis, or mitochondrial dysfunction compromising energy metabolism and adaptative cell responses, ending eventually in abnormal cell division and growth, among other disturbances. The three-dimensional representation of GPCR was drawn from the protein data bank (PDB ID: 8XZG) [62] with the Molstar online free software [63]. Abbreviations: AC, adenylyl cyclase; AKT, Ak strain transforming, a protein kinase B; β-arr, beta-arrestin; cAMP, cyclic 3´-5´ adenosine monophosphate; ERK, extracellular receptor kinase; NF-kB, nuclear factor kappa B; PI3K, phosphatidyl inositol 3-kinase; PLC, phospholipase C; PKA, protein kinase A; PKC, protein kinase C; RAS, rat sarcoma, ; ROCK, Rho-associated coiled-coil kinase.
Figure 8.
Schematic representation of signaling pathways triggered by peptides binding to their respective receptors (A). B indicates how GPCRs activated by peptides may signal from the plasma membrane or, once internalized in endosomes, from signalosomes, determining complex and modulated intracellular signals that affect cell metabolic processes and transcriptional programs. Alteration of GPCR homeostasis through uncontrolled overexpression, down-regulation, truncation, dimerization, or overstimulation leads to changes inducing transitory or sustained cellular events, leading to aberrant cell growth, cell migration, inhibition of apoptosis, or mitochondrial dysfunction compromising energy metabolism and adaptative cell responses, ending eventually in abnormal cell division and growth, among other disturbances. The three-dimensional representation of GPCR was drawn from the protein data bank (PDB ID: 8XZG) [62] with the Molstar online free software [63]. Abbreviations: AC, adenylyl cyclase; AKT, Ak strain transforming, a protein kinase B; β-arr, beta-arrestin; cAMP, cyclic 3´-5´ adenosine monophosphate; ERK, extracellular receptor kinase; NF-kB, nuclear factor kappa B; PI3K, phosphatidyl inositol 3-kinase; PLC, phospholipase C; PKA, protein kinase A; PKC, protein kinase C; RAS, rat sarcoma, ; ROCK, Rho-associated coiled-coil kinase.


Table 1.
Representative compounds with antineoplastic properties when tested in different types of tumors in experimental preclinical models and clinical trials. Chemical structures were drawn with KingDraw free chemical structure editor software (version 3.0 for Windows).
Table 1.
Representative compounds with antineoplastic properties when tested in different types of tumors in experimental preclinical models and clinical trials. Chemical structures were drawn with KingDraw free chemical structure editor software (version 3.0 for Windows).
| Antitumoral compounds | Receptor | Tumor | References |
Naltrexone (antagonist)
|
OPR | Metastatic breast cancer (phase II study) Gastrointestinal cancer Cervical cancer |
[163,164,165,166,167] |
Aprepitant (antagonist)
|
NK-1R | Gall bladder cancer Colorectal cancer Gynecologic cancer Cholangiocarcinoma Neuroblastoma Prostate cancer Thyroid cancer Pancreatic cancer |
[96,102,168,169,170,171,172] |
BIP3226 (antagonist) L152,804 (antagonist) 
|
NPY1R (BIBP 3226), NPY5R (L152,804)- |
Breast cancer Colon cancer Prostate cancer |
[105,110,112,173] |
| M89b, a galanin analogue (agonist) Amino acid sequence: pEWNLNAAGYLLATHACG (pE stands for pyro-Glu) |
GAL2R | Pancreatic ductal adenocarcinoma Colorectal cancer |
[114,119,174,175] |
SNAP 37889 (antagonist)
|
GAL3R | Promyelocytic leukemia cells | [176] |
Indolinone Compound 25 (antagonist)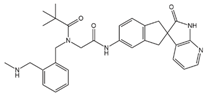
|
CLR-RAMP3 (AM2R) |
Human pancreatic cancer cell lines In vivo models of breast cancer |
[132,177,178] |
Olcegepant (antagonist)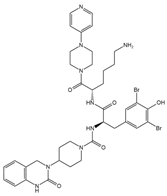 Telcagepant or MK0974) (antagonist) 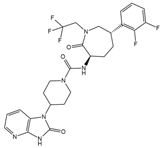
|
CLR-RAMP1 | Prostate cancer Acute myeloid leukemia |
[179,180,181] |
SR 48692 (antagonist)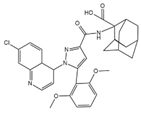
|
NTS1R | Ovarian cancer | [182] |
ML 221 (antagonist)
|
APLNR | Cholangiocarcinoma Breast cancer Infantile hemangioma |
[143,183,184] |
BMK-I-152 (antagonist)
|
CRFR1 | Not tested in cancer therapy* Tested for major depression |
[77] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



