
Submitted:
23 September 2024
Posted:
24 September 2024
You are already at the latest version
Abstract
Gene therapy is a medical approach capable of correcting genetic and epigenetic alterations in pathological conditions, including cancer. Different strategies have been pursued for cancer gene therapy, aiming to restore the function of tumor suppressor genes or inhibit oncogenes. These comprise the use of CRISPR/Cas9 gene editing technology and therapeutic oligonucleotides, such as microRNAs, anti-microRNAs, small interfering RNAs, DNAzymes, and epigenetic modifiers. However, despite great potential, their broad clinical use is hindered due to the lack of safe and efficient systems for targeted delivery to cancer cells. To this purpose, aptamers could represent an ideal tool. They are short single-stranded oligonucleotides that bind receptors selectively present or overexpressed on cancer cell membranes, often undergoing cell-specific internalization. Thanks to this last feature, numerous internalizing aptamers have been investigated as targeting moiety to deliver therapeutic oligonucleotides, viral vectors, and nanoparticle-based systems in preclinical studies. Here, we summarize the state-of-the-art of aptamer applications in cancer gene therapy, providing key examples and discussing their advantages, limitations, and clinical perspectives.
Keywords:
aptamer
; gene therapy
; cancer
; delivery
; aptamer-based conjugate
; aptamer-based functionalization
1. Introduction
Tumorigenesis and tumor progression are intricately linked to genetic mutations and epigenetic modifications, which cause loss-of-function of tumor suppressor genes and aberrant activation of oncogenes [1]. Despite significant progress of precision medicine in targeting oncoproteins, currently available therapeutics remain ineffective in most patients, supporting the urgency in developing novel effective anti-tumor approaches [2,3,4]. In this regard, gene therapy is changing the treatment paradigm by targeting tumor cells at the DNA or RNA level, the source of their malignancy [5]. The first definition of gene therapy dates from the 1970s and has been revised numerous times, driven by the technical innovations made available by biotechnologies. Gene therapy is nowadays defined as a set of treatments that consists of inserting functional genes, correcting mutations using gene editor systems, such as CRISPR/Cas9, or regulating gene expression through small oligonucleotides, including microRNAs (miRNAs or miRs), anti-microRNAs (antimiRs), small interfering RNAs (siRNAs), DNAzymes, and epigenetic modifiers [6]. However, these nucleic acid-based therapeutics show higher molecular weight and polarity with respect to traditional small molecules, requiring a delivery system to efficiently overcome the barrier represented by cell membranes [7,8].
Viruses, physiologically predisposed to transduce exogenous genomic material through cell infection, are the primary investigated delivery modalities, leading to significant clinical results [9]. The approval of Gendicine® in 2003 in China represents a historical milestone, being the first authorized cancer gene therapy worldwide. This viral-based gene therapy consists of TP53-expressing recombinant human-type five adenoviruses (rAd-p53), successfully used for about twenty years to restore p53 function in head and neck squamous cell carcinoma [10]. Nowadays, four additional viral-based therapies (Imlygic®, Rexin-G®, Oncorine®, and Adstiladring®) have been approved by the Food and Drug Administration (FDA) and the European Medicine Agency (EMA) for cancer treatment [11]. Nevertheless, viral vectors have also shown relevant setbacks, especially regarding specificity and safety. A severe immune reaction caused the death of a patient during a clinical trial, and a correlation between retrovirus-based gene therapy and leukemia was found in four patients, evoking concerns about the immunogenicity and specificity of viral-based gene therapies [12]. Nanoparticles (NPs) can represent an alternative delivery strategy with respect to viruses [13], in particular after the successful use of lipid nanoparticles (LNPs) for the delivery of Spike mRNAs in COVID-19 vaccination [14]. However, similarly to viral vectors, NPs are not intrinsically specific for tumor targeting and provide a low delivery efficacy [15]. Therefore, improvements in current technologies or the development of alternatives for targeted gene therapy in cancer are required.
In this regard, aptamers could represent ideal tools. They are small single-stranded DNAs or RNAs capable of folding into peculiar tertiary structures and binding with high affinity and specificity to membrane oncoproteins [16,17]. In addition to the potential activity as antagonists or agonists, aptamers, following the binding to their specific target, can be internalized via receptor-mediated endocytosis, supporting their use as trojan horses for specific delivery of therapeutics in cancer cells [18,19]. Thanks to this feature, aptamers have been investigated as targeting moiety in novel gene therapy systems, being considered particularly promising to overcome current limitations of viruses and NPs.
This review provides key examples of aptamer applications for cancer gene therapy, discussing their advantages, limitations, and clinical perspectives. We briefly introduce the main historical steps in aptamer research, starting from the first description and applications, and then present an extensive collection of preclinical findings demonstrating the suitability of aptamer-based gene therapy.
2. Aptamers: Discovery and First Applications
The discovery of aptamers dated to 1990 when C. Tuerk and L. Gold described the “Systematic Evolution of Ligands by EXponential Enrichment” (SELEX), an innovative in vitro methodology to identify high-affinity RNA ligands for ideally any target of interest [20,21]. These RNA ligands, as well as those with a DNA backbone, were named “aptamers” later. Since the first description, variants of the standard SELEX have been developed (a detailed update has been recently reported by Chao Zhu and colleagues) [22]. In general, SELEX procedures consist of three distinct steps (selection): 1) incubation of a high-complexity DNA or RNA-modified aptamer pool (constituted by approximately 1015 aptamers, each characterized by a unique sequence) with a target of interest, such as single proteins, whole cells, or tissues; 2) partitioning of the aptamers bound to the target from those unbound; 3) amplification of the collected bound aptamers through PCR or RT-PCR depending on the DNA or RNA-based backbone, respectively. SELEX could also include an additional preliminary step (counter-selection) in which the aptamer pool is incubated with non-target proteins/cells/tissues aimed at eliminating aptamers with a non-specific binding activity. Counter-selection and selection are reiterated for multiple rounds (typically 8-12), gradually modifying experimental conditions, such as the ratio between the concentrations of the aptamer pool and the target, the number of counter-selection steps, binding temperature, or incubation time. This process progressively increases stringency, only allowing the binding and collection of high-affinity aptamers, while low-affinity and non-specific binders are gradually reduced. Therefore, at the end of the procedure, the high-affinity aptamers are more abundant in the final pool with respect to the initial, and their nucleotide sequences are affordably identifiable with sequencing tools [23].
Among the isolated aptamers, many recognize the extracellular domain of membrane receptors and can prevent the binding of endogenous ligands or induce a conformational change that impedes the activation of intracellular pathways, thus promoting an antagonistic action [24]. For example, the aptamer E07, which recognizes the human wild-type Epidermal growth factor receptor (EGFR) and the deleted mutant EGFRvIII, can compete with the endogenous ligand EGF, inhibiting receptor autophosphorylation and blocking the proliferation of EGFR-positive cancer cells [25]. Although fewer cases have been reported, aptamers may also promote the activation of their target, providing an agonistic function. A dimeric RNA structure constituted by two M12-23 aptamers belongs to this class [26]. M12-23 efficiently binds the murine tumor necrosis factor receptor superfamily member (4-1BB), a crucial costimulatory receptor for the survival and expansion of activated T cells. In detail, this aptameric dimeric structure was used to induce an anti-tumor immune response via the activation of CD8+ T cells and the release of IFNγ, leading to tumor regression in mouse cancer models. The two aptamers described above are only two examples of antagonist and agonist aptamers, but many others with similar activities have been identified, preclinically characterized, and proposed as valuable alternatives to monoclonal antibodies (mAbs), with which aptamers share the mode of action [27]. Even if aptamers are less investigated and developed with respect to mAbs, they show relevant advantages [28]. Aptamers are not immunogenic, can be easily modified with various chemical groups, show high stability to temperature and pH changes, lower costs of production, and no batch-to-batch variations. These distinctive features could be decisive in choosing aptamers instead of mAbs for clinical use [29].
Macugen (Pegaptanib) was the first FDA-approved aptamer in 2004, a 2’-Fluoropyrimidine (2’F-Py) modified RNA that binds vascular endothelial growth factor (VEGF) and showed efficacy for the treatment of neovascular age-related macular degeneration (AMD) [30]. Despite its efficacy, Macugen was discontinued for commercialized reasons because of superior novel treatments. Nevertheless, this was only the first proof of aptamer clinical translatability. Recently, the FDA authorized a novel 2’F-Py modified RNA aptamer inhibiting the complement factor C5 (Izervay) for geographic atrophy (GA) or advanced dry AMD [31], while many other aptamers are currently evaluated in clinical trials [32].
3. Aptamer-Guided Approaches for Delivering Cancer Gene Therapy
Although aptamers have been historically proposed as anti-cancer therapeutics due to their capacity to inhibit or stimulate membrane receptors, the discovery that aptamers can be selectively internalized once bound to their specific target on the cell surface, has extended the boundary of their application, paving the way for their use as selective delivery carriers [33,34]. In this respect, an outstanding upgrade in the area was provided by the development of Cell-Internalizing SELEX Technology, a modified SELEX protocol designed for the identification of internalizing aptamers [35,36]. Pushed by this innovation, numerous aptamers capable of internalizing in different cancer cell types have been identified and investigated for the targeted delivery of small molecules [37]. On the same line, an emerging research field is working on using aptamers to deliver oligonucleotides or to functionalize viral vectors and NPs for cancer gene therapy applications.
3.1. Aptamer-Based Conjugates for miRNA and AntimiR Delivery
MiRNAs are endogenous non-coding RNAs, typically of 20-22 nucleotides (nt) in length, that repress gene expression at the post-transcriptional level [38]. They originate from longer harpin-bearing RNAs that are processed by different cellular RNases, including the cytosolic RNase III Dicer, which lastly produces mature miRNA duplexes, each constituted by a guide and a passenger strand [39]. After the removal of the passenger strands, the guide strands are loaded into the RNA-induced silencing complex (RISC), and their nucleotide sequences are exploited to recognize targeted mRNAs based on base pairing [40]. Generally, the recognition guided by miRNAs leads to mRNA decay or translational inhibition in animals. Since miRNAs play crucial roles in different cellular processes, their dysregulation has been observed in diseases, including cancer [41]. Therefore, restoring physiological expression levels with exogenous miRNAs or miR-sequestering oligonucleotides (antimiRs) has been considered a promising therapeutic strategy. However, achieving their efficient and specific delivery to tumor cells remains a significant challenge for clinical translation [42,43]. One promising strategy to address this issue is their conjugation with internalizing aptamers, which is currently particularly affordable thanks to the progress of nucleic acid chemistry [44]. Moreover, different modified nucleotides can be introduced in miRNAs and antimiRs, as well as in aptamers, improving the pharmacokinetics and pharmacodynamics of conjugates for clinical uses [45]. In this paragraph, we describe key studies showing aptamer-mediated delivery of miRNAs and antimiRs in cancer models, underlining the conjugation strategies and the potentiality of such an approach (Supplementary Table 1).
Our group first explored the possibility of using aptamers for miRNA delivery. In detail, the GL21.T aptamer was conjugated with let-7g miRNA and tested in non-small cell lung cancer (NSCLC) models [46]. We used GL21.T since it showed specific internalization once bound to the tyrosine kinase receptor Axl, its membrane target, remarkably over-expressed in NSCLC [47]. The choice of let-7g was instead based on its reported anti-tumor role in NSCLC [48]. The conjugate was developed by fusing the aptamer and the let-7g passenger sequences in a single RNA, which was then annealed with the guide miRNA strand, thus generating a “two-blocks” conjugate (Figure 1a). To increase the resistance to serum nuclease, 2′F-Py modifications, already reported in the GL21.T aptamer, were also introduced in both miRNA strands. In this way, an aptamer conjugated to a let-7g mimic (GL21.T-let7g), compatible with in vivo studies, was developed. GL21.T-let7g allowed the selective delivery of the let-7g mimic to Axl-positive NSCLC A549 cells in vitro, its functional processing, and the silencing of its targeted genes. It additionally preserved the aptamer capacity to inhibit Axl, thus combining the biological effects of GL21.T and the let-7g mimic [47]. Promising in vivo results were also observed since the tumor growth was significantly reduced after repeated intravenous injections in xenograft mouse models. In accordance with these findings, Iaboni et al. developed an analogous GL21.T-based “two-blocks” conjugate to restore the expression of miR-212, implicated in the resistance to TNF–related apoptosis-inducing ligand (TRAIL)–mediated cell death [49,50]. The authors demonstrated that GL21.T efficiently delivered miR-212 in A549 cells, repressing the expression of the anti-apoptotic protein PED/PEA-15, a well-known target of miR-212, and inducing the re-sensitization to TRAIL treatment in NSCLC.
In addition to the “two-blocks” strategy, an alternative method of conjugation between GL21.T and let-7g was also investigated successfully [46]. GL21.T and let-7g passenger were first extended with complementary and antiparallel sequences at 3’-ends (also named sticky). The conjugate was then obtained by annealing the 2’F-Py-modified guide and passenger strands to constitute the miRNA mimic, which was further annealed with the aptamer thanks to the sticky ends. Acting in this way, the conjugate was formed by three oligonucleotide components (“three-blocks”) (Figure 1b). The “three-blocks” conjugate still recognized Axl and was efficient in let-7g delivery in vitro, revealing that the sticky extension did not negatively affect the function of both aptameric and miRNA moieties. This strategy has the advantage of distancing the two moieties through the sticky sequences, arguably promoting more efficient miRNA processing. Further GL21.T-based “three-blocks” conjugates were later developed, corroborating the broad applicability of GL21.T as a carrier of different onco-suppressive miRNAs in NSCLC, such miR-16, miR-34c, and miR-137 [46,51,52]. Despite a predominant validation in NSCLC models, GL21.T was also exploited for miRNA delivery in other Axl-positive cancers, such as breast cancer (BC) and melanoma [53]. The repeated intravenous injection of a “three-blocks” conjugate consisting of GL21.T and a miR148b mimic reduced the formation of circulating tumor cells and, consequently, the number of metastases related to the growth of BC or melanoma xenotransplants in mice. These findings indicated that GL21.T can efficiently deliver therapeutic miRNAs in both in vitro and in vivo Axl-positive cells, supporting its potentiality for clinical applications.
Besides GL21.T, other aptamers were conjugated with miRNAs through the described strategies and validated as suitable carriers in cancer models. Especially notable is the anti-KIT DNA aptamer, successfully used to deliver miR-26a through a “two-blocks” strategy in which the two moieties were spaced with a 3-carbon chain linker (C3) [54,55]. In addition, TEG cholesterol was added at the 3’end of the passenger to improve pharmacokinetics since it delayed the blood clearance of oligonucleotides. The authors first observed that the restoration of miR-26a in KIT-expressing BC tumors promoted a synergistic effect with fluorouracil (5-FU) or Carboplatin treatments, increasing the suppression of tumor growth. Interestingly, the delivery of miR-26a was also observed in KIT expressing hematopoietic stem/progenitor (HSPCs), providing protection towards the toxicity commonly observed after administering 5-FU and Carboplatin. These data are particularly significant since they support the idea that aptamer specificity and the multifaceted miRNA roles in different tissues could be simultaneously exploited to improve therapeutic outcomes. The anti-nucleolin AS1411 aptamer, previously validated as an internalizing 2’F-Py modified aptamer, was similarly evaluated in the context of conjugate for miRNA delivery [56,57]. The primary relevance of the studies is that the authors developed a variant of the “two-blocks” conjugate, performing a post-synthesis conjugation of the aptamer and passenger moieties. In particular, they conjugated AS1411 with a mimic of let-7d miRNA via a heterobifunctional crosslinker that contained NHS-ester and maleimide reactive groups at opposite ends of a polyethylene glycol spacer (SM(PEG)2) (Figure 1c). The “two-blocks” variant could represent a valid alternative when the synthesis of a long aptamer and passenger fused sequence cannot be pursued, for example, when the aptamer length could require a synthesis of 80-90 nt oligonucleotides with evident issues related to the yield and costs. Nevertheless, additional steps of purification are required with this strategy. The authors also compared the new “two-blocks” variant with one obtained with the sticky-end extension. The SM(PEG)2-based conjugate showed higher serum stability, promoted increased intracellular let-7d levels, and, consequently, a more potent antiproliferative effect in MKN-45 gastric cancer cells. Reported data are limited to in vitro experiments, which are not sufficient to conclude the superiority of SM(PEG)2-based “two-blocks” conjugates. Nevertheless, this study suggests that an optimization of the conjugation strategy, as well of both aptamer and miRNA chemistry, could remarkably contribute to the clinical translatability of these therapeutics, and more efforts should be pursued to define the strategies to develop the best-in-class conjugate.
Unlike the miRNAs discussed so far, which exert an onco-suppressor role and need to be restored with mimics, miRNAs can also be highly expressed in tumor cells, contributing to tumorigenesis and tumor progression. For this reason, modified RNA sequences were rationally designed to be complementary to oncogenic miRNAs (commonly named antimiRs) and used to prevent their recognition in the RISC complex [58]. Our group provides the first description of aptamer applicability for selective antimiR delivery in cancer [59]. We conjugated the GL21.T aptamer with a single-strand 2’F-Py modified antimiR-222 (GL21.T-antimiR222) via a sticky-based approach and tested the “three-blocks” conjugate in glioblastoma (GBM) mouse models. The intravenous injection of GL21.T-antimiR222 provided efficient miR-222 targeting and tumor growth impairment. Nevertheless, since tumors can be characterized by concomitant dysregulation of different miRNAs, approaches for delivering more therapeutic miRNA mimics or antimiR could be necessary. In this regard, GL21.T was also conjugated with an extended 2’F-Py modified RNA sequence containing the antimiR222 and the antimiR10b in series, each separated by a C3 spacer [59]. Both antimiRs, once internalized via Axl-mediated GL21.T endocytosis, inhibited their proper oncogenic miRNA target, demonstrating the possibility to co-target different miRNAs through a single aptamer carrier. Moreover, given the overexpression of both Axl and Platelet-derived growth factor receptor (PDGFR)β in glioma-stem-like cells (GSCs), two distinct 2’F-Py modified aptamers, the GL21.T and the anti-PDGFRβ Gint.4.T aptamer, were also exploited to deliver a miR-137 mimic and an antimiR10b simultaneously [60]. The co-treatment of GSCs with the two conjugates promoted specific miRNA and antimiR uptake and a synergistic functional activity on GSC expansion.
Besides the conjugation strategies described so far, the advancements in the nanotechnology field led to the development of novel alternative approaches, such as using a three-way junction (3WJ) motif derived from the bacteriophage phi29 to assemble oligonucleotide-based nanostructures [61]. In detail, Shu et al. applied the 3-WJ motif as a scaffold to construct a self-assembling nanostructure containing multiple elements: an anti-EGFR aptamer as a carrier, antimiR-21, and an Alexa-647 molecule [62]. This all-in-one platform showed a favorable pharmacokinetic profile, tumor targeting, and delivery of antimir-21 in orthotopic triple-negative breast cancer (TNBC) models after repeated intravenous injections. Although the conjugation modality is more complicated with respect to the previously described conjugates, and a higher molecular weight could reduce tumor accessibility, the presented structure could provide better tumor retention and potentially be used for multi-targeting and diagnostic purposes, paving the way for desired theranostic applications [63].
3.2. Aptamer-Based Conjugates for siRNA Delivery
Small interfering RNAs (siRNAs) are a class of synthetic double-stranded RNAs, ranging between 20-28 base pairs, that repress gene expression by exploiting the same machinery previously described for miRNAs [64]. Different from these last, which rarely display a perfect base pairing with target mRNAs, siRNA guide strands are rationally designed to be entirely complementary to a disease-related mRNA, guiding its recognition, cleavage, and degradation. Since siRNAs can be theoretically designed toward any mRNA of interest, their potentiality for therapeutic applications is huge and has been largely investigated in recent decades. Nevertheless, similarly to miRNAs, their clinical development is hindered by the lack of efficient systems to deliver them specifically to tumor cells. Only five siRNA-based therapeutics have been approved so far (Patisiran, Givosiran, Inclisiran, Lumasiran, and Vutrisiran), but nobody for cancer indications [65,66]. Indeed, both Patisiran, consisting of chemically modified siRNAs encapsulated in LNPs, and the remaining four, based on conjugation with N-acetylgalactosamine (GalNAc) sugars, only allow efficient siRNA delivery to the liver. Therefore, developing systems for delivering siRNAs to extra-liver tissues, including tumor cells, remains an urgent medical need. Considering the features of internalizing aptamers discussed above and the analogies between siRNAs and miRNAs, conjugations of aptamers and siRNAs have been similarly pursued for cancer treatment (Supplementary Table 2) [67].
The first preclinical study reporting the suitability of aptamers for siRNA delivery was in 2006 when McNamara and colleagues used A10, an anti-prostate-specific membrane antigen (PSMA) internalizing aptamer, to deliver anti-Polo-like kinase 1 (PLK1) siRNAs in prostate cancer cells, negatively affecting cell mitosis [68]. Similarly to what was described for miRNAs, they designed a “two-blocks” conjugate, fusing the A10 2’F-Py modified RNA aptamer with the PLK1 passenger siRNA strand and annealing it with the corresponding complementary guide strand. The A10/anti-PLK1 siRNA conjugate was selectively internalized in PSMA-expressing cells, resulting in RNase III Dicer processing and downregulation of PLK1 in vitro. In addition, the authors reported that the conjugate could successfully inhibit the growth of PSMA-expressing xenograft tumors when injected intratumorally. However, this first proof only demonstrated the functionality of the conjugate when it reached the tumor area but did not demonstrate suitable pharmacokinetics for different administration routes, which are strikingly required for clinical applications. In a subsequent publication, the same research group investigated different modalities to optimize the conjugate, inserting various structural and chemical changes [69]. First, A10, originally constituted of 71 nucleotides, was reduced to 39 nucleotides (A10-3.2). The shorter A10-3.2 aptamer preserved the binding to PSMA, allowing a cheaper and more affordable chemical synthesis. Then, the authors focused on the siRNA moiety, conjugating different siRNA configurations to A10-3.2 and testing them to evaluate the effect on Dicer-mediated processing and stronger gene repression. Noteworthy, similarly to A10-3.2, the siRNA passenger strands were modified with 2’F-Py in all the developed conjugates, while the guide sequences were unmodified. In detail, they observed that conjugates harboring siRNA configurations with a -U-U- overhang at the passenger 3’end (also named OVH), a wobble base pair at the 5’ end of the guide thanks to a mismatch mutation between the strands (wobble), an inversion between the passenger and guide strands (swap), or an engineering stem-loop structure in which siRNA stem was continuous to the aptamer loop (loop), resulted more efficient in PLK1 gene repression with respect to the anti-PLK1 siRNA previously tested. In addition, Dassie et al. also studied the effect of including a 20kDa PEG tail (pegylation) at 5’ of the passenger strand to delay the clearance of an A10/anti-PLK1 siRNA conjugate (swap configuration) and, consequently, the efficacy [69]. They showed a remarkable increase in the conjugate’s blood half-life with respect to the non-pegylated conjugate, from less than 30 minutes to more than 30 hours, ameliorating the overall survival of daily intraperitoneal injected tumor-bearing mice. Reported data left a trail for the subsequent research since it demonstrated that a remarkable improvement of the first conjugate could be achieved by working on constituting moieties. Indeed, following their findings, such as more efficient configurations, OVH and swap, were used by an independent group that successfully evaluated the delivery of anti-STAT-3 siRNAs through an anti-BAFF aptamer in the context of B-cell malignancies in vitro [70].
PSMA and BAFF are specifically expressed in prostate or B-cell malignancies, respectively, limiting the exploitation of their internalization to these cancers. Therefore, delivering siRNAs using aptamers able to recognize targets more broadly expressed in different tumor types was pursued. For example, the epithelial cell adhesion molecule (EpCAM) was extensively explored since it is upregulated in stem cells and epithelial cancers, such as breast, colorectal, prostate, and pancreatic cancers. In addition, it is internalized upon ligand binding, supporting its targeting with aptamer-based conjugates for delivery applications [71]. Wang et al. conjugated an anti-EpCAM aptamer to a siRNA able to repress the expression of survivin, which was overexpressed and involved in doxorubicin resistance. The authors used a subline established from MCF-7 BC cells after long-term drug exposure (MCF-7/Adr) [72,73]. This subline also contained an enriched subpopulation of EpCAM-positive cells, which displayed relevant multi-potent differentiation and self-renewal capacities, properties typically associated with cancer stem cells. The developed conjugate was obtained with the “two-blocks” strategy by annealing the unmodified guide strand with the fused aptamer and passenger 2’F-Py modified RNA sequence. The conjugate also contained an A-A spacer between the fused elements and a 3’-U-U- siRNA overhang (OVH configuration). Experiments in MCF-7/Adr cells demonstrated efficient siRNA delivery, survivin repression, and doxorubicin sensitivity restoration. However, the authors highlighted the absence of in vivo effects in xenograft models after intravenous injection. In agreement with the findings reported by Dassie and colleagues, a short tumor accumulation was observed, which lasted less than 4 hours, suggesting the necessity to introduce the pegylation to increase serum retention. Accordingly, thanks to this optimization, a significant improvement in the overall survival of the tumor-bearing mice was achieved when they were co-treated with pegylated conjugates and doxorubicin. Nevertheless, in the same year, Gilboa-Geffen and colleagues reported the successful delivery of an anti-PLK1 siRNA through the same anti-EpCAM aptamer without pegylation [74]. Despite using the same aptamer, numerous variables are present between the two studies that could explain the different results. Indeed, the conjugate reported by Gilboa-Geffen and colleagues differed for i) the spacer between the anti-EpCAM aptamer and the siRNA, which was a U-U-U linker instead of a -A-A-; ii) the presence of dTdT overhangs at 3’ends of both siRNA strands for the amelioration of in vivo stability instead of -U-U- in the passenger. In addition, Gilboa-Geffen et al. administered the conjugate subcutaneously rather than intravenously and tested the anti-EpCAM aptamer-based conjugate in different BC cells.
Although the “two-blocks” strategy was mainly pursued in the wake of initial promising findings, “three-blocks” conjugates for siRNA delivery were also developed. For example, the anti-PDGFRβ Gint4.T aptamer was conjugated with a STAT3 siRNA through a stick-based approach and tested in different PDGFRβ-expressing GBM cells [75]. The RNA aptamer and siRNA moieties were modified with 2’F-Py, while the sticky-end sequences contained both 2’F-Py and 2’-oxygen-methyl purines (2’OMe-Pu). In addition, the aptameric and siRNA moieties were spaced from the sticky ends using C3 linkers, which were introduced to avoid interferences in the aptamer folding and siRNA processing. The Gint4.T-STAT3 conjugate led to specific and efficient repression of STAT3 expression in vitro and impaired tumor growth in GBM xenograft models upon intraperitoneal administration. Interestingly, this was observed in the absence of pegylation. Promising results using the same conjugate were also reported in patient-derived GSCs, demonstrating the suppression of GSC phenotype and propagation in vitro [76]. Moreover, the efficacy of the Gint4.T-STAT3 has been recently confirmed in a different context, besides GBM [77]. It has been shown that Gint4.T-STAT3 effectively delivered STAT3 siRNA in NSCLC cells and cancer-associated fibroblasts (CAFs), which are pivotal in the NSCLC microenvironment for promoting tumor progression. In particular, the conjugate altered the CAF phenotype and rendered epithelial cells irresponsive to CAF-pro-tumoral functions.
Even if aptamers show relevant advantages with respect to mAbs, these latter present two fragment-antigen-binding regions (Fab), which allows the simultaneous recognition of two antigens and increases mAb avidity toward targets, a property not pursuable using the aptamer-based conjugates described so far [78]. To achieve mAb-like bivalence and evaluate it in the context of siRNA delivery, Yan Liu et al. designed an innovative oligonucleotide-based structure using two A10-3.2 aptamers for each conjugate [79]. In addition, they included two different swap siRNAs between the two aptameric moieties for repressing EGFR and survivin. Therefore, the authors developed a mAb mimicking conjugate constituted by a PSMA aptamer, a survivin siRNA, an EGFR siRNA, and a further PSMA aptamer (PSEP). In detail, the structure was obtained through the annealing of three 2’F-Py modified RNA sequences, the first one composed by the fused sequences of the PSMA aptamer and the guide strand of the survivin siRNA; the second one composed by the PSMA aptamer and the passengers of both EGFR and survivin siRNAs in-series; the last sequence contains the guide strand of the EGFR sRNA. The aptamer and siRNA sequences were separated through an -A-A while the two siRNAs were through a -U-U-U-U bridge. The two spacers were introduced to distance the functional moieties and to confer flexibility to the structure. Notably, the authors demonstrated that the bivalent PSEP structure was more efficiently internalized and provided higher cytotoxicity compared to a control monovalent conjugate, PSEM, containing only one A10-3.2 aptamer and a non-functional second aptamer moiety, specific for the small Malachite Green organic dye. These findings suggest that bivalent conjugates could be more effective than monovalents. Nevertheless, although in vivo experiments were performed and daily intraperitoneally injections of PSEP successfully reduced the tumor burden in prostate cancer xenograft models, no comparison between bivalent conjugates and traditional “two-blocks” or “three blocks” was reported, preventing any statement regarding their superiority and supporting the necessity of additional studies. In line with this innovative bivalent aptamer-based conjugate, further similar structures, one based on two anti-HER2 aptamers (HEH), and another even based on two different aptameric moieties, an anti-HER2 and anti-HER3 aptamer (H2EH3), were developed for delivery siRNAs in BC, confirming the relevance of this approach [80,81]. Indeed, despite the absence of comparisons as previously underlined, the HEH and H2EH3 showed in vivo activity since intravenous or intraperitoneal repeated injections significantly impaired tumor growth.
3.3. Aptamer-Based Conjugated for DNAzyme Delivery
DNAzymes are synthetic catalytic ssDNAs selected from fully randomized libraries. They represent an evolution of naturally occurring ssRNA ribozymes (ranging between 40-100 nt), which fold in tertiary structures and catalyze chemical reactions [82,83]. One of the most studied ribozyme-mediated catalysis is the RNA cleavage, typical of the hammerhead and hairpin ribozymes, which were considered promising for potential therapeutic purposes [84]. Different from ribonucleases, which display a non-specific mechanism of RNA cleavage, these ribozymes only target specific RNAs [85]. Indeed, unfolded anti-sense sequences, which commonly flank the central catalytic core of the ribozymes, allow the recognition of complementary RNAs only, positioning the cleavage site adjacent to the catalytic core and promoting its cutting. Although efficiency in gene repression at the post-transcriptional level was observed in preclinical studies, no hammerhead or hairpin-like ribozymes were approved in clinics due to failures of clinal endpoints [86]. The reason could be found in their pharmacokinetics since ribozymes are more unstable to nucleases than siRNAs, which have been successfully translated to the clinics. In particular, the siRNA backbone can be easily modified to improve serum stability, for example with 2’F-Py or 2’OMe-Pu modifications, without affecting Dicer-mediated recognition and biological function. On the contrary, the introduction of modifications in ribozymes can severely inhibit their cleavage activity [87]. DNAzymes have promised to overcome this limitation and substitute ribozymes in clinics since they comprise a more nuclease-resistant DNA backbone. Nevertheless, their specific and efficient delivery to cancer cells still represents an issue for DNAzymes clinical proposals [86]. Considering this limitation, aptamers have also been recently explored as delivery carriers of DNAzymes.
An anti-HER2 DNA aptamer (42 nt) was fused to a DNAzyme (41 nt) able to repress the expression of GLUT-1. The obtained DNA sequence (83 nt) could be efficiently assembled in a metal-nucleic acid framework (MNF) with uniform morphology thanks to coordinated bonds promoted by bivalent calcium cations (H-GDz/Ca), a crucial functional element for the catalytic activity of the DNAzyme [88]. The use of a more extended DNA sequence promoted the MNF assembly. The anti-HER2 aptamer in the MNF increased its uptake in HER2-expressing gastric cells (NCI-N87 and SNU216), inducing the DNAzyme internalization and silencing of GLUT1. Noteworthy, results indicated that the aptamer-loaded MNF was also more internalized with respect to the traditional aptamer-DNAzyme conjugate (H-GDz + Ca2+), which was not previously assembled in Ca2+-coordinated MNF. This is arguably consistent with the higher avidity expected for a MNF functionalized with numerous aptamer moieties for each nanostructure. However, the control MNF (MH-GDz/Ca), functionalized with a mutated version of the anti-HER2 aptamer, was highly internalized, even higher than those provided by H-GDz/Ca, suggesting relevant non-specific uptake of MNF and indicating that more specificity could be achieved with traditional conjugates. Nevertheless, as pursued by the authors, the MNF nanostructure showed the advantage of delivering several therapeutic molecules simultaneously, even proteins, as successfully validated for the RAD-51 inhibitor IRF-1. Indeed, the intravenous injection of a MRF simultaneously loaded with the anti-GLUT1 DNAzyme and IRF-1 (IRF/H-GDz/Ca) resulted in higher tumor reduction with respect to the single treatment-loaded MRFs. In line with this study, another MRF, also named Metal-DNA nanocomplexes, was developed using the anti-nucleolin AS1411 aptamer, which was fused with a DNAzyme repressing the ATG5 mRNA, a crucial player of protective cellular autophagy [89]. The complex was constituted in the presence of Mn2+, a cofactor of the DNAzyme catalytic core and an inducer of hydroxyl radicals when internalized. This MRF is a promising example of multitargeting since the aptamer-mediated internalization in nucleolin-expressing cells led to intracellular Mn2+ accumulation, which led to cell damage prompted by hydroxyl radicals, an approach also named Chemo-dynamic therapy (CDT). At the same time, the DNAzyme repressed the expression of ATG5, preventing the activation of autophagy, which is used by tumor cells to repair damage. The authors showed that the AS1411-loaded MRFs (DACs-Mn) were selectively internalized in 4T1 cells, characterized by high nucleolin expression levels. Weaker in vitro internalization was instead observed in low-expressing nucleolin HEK-293 cells and when MRFs were assembled in the absence of AS1411 (DCs-Mn). The higher internalization of DACs-Mn promoted the rise of more late apoptotic cells (about 41%) with respect to DCs-Mn (11%) and ACs-Mn (13%), which only contained AS1411 without the DNAzyme. In vitro data were then confirmed in vivo, where tumor growth was impaired by the intravenous injection of DACs-Mn compared to ACs-Mn. Nevertheless, a study limitation should be highlighted since no characterization of DCs-Mn and ACs-Mn was reported. Based on findings from Yan et al., shorter DNA sequences could affect MRF assembly, suggesting that a deep characterization of control MRFs should be required [88]. Moreover, an innovative aspect of the study is represented by the system used to produce the fused AS1411 and DNAzyme sequences, which were biotechnologically synthesized using a rolling circle amplification (RCA) starting from a circular DNA template. This strategy allowed the synthesis of a long DNA sequence, avoiding issues of yield and costs and potentially showing great potential for developing novel DNA-based aptameric conjugates.
Overall, reported data suggest that aptamer-loaded MRFs could be promising for delivering higher amounts of DNAzymes in targeted tumor cells and achieving multitargeting with respect to traditional conjugates. Nevertheless, being a more recent strategy, further preclinical studies are required to corroborate their feasibility and potential translatability.
3.4. Aptamer-Coated Viral Vectors
As described in the introduction, viral vectors have been the primary strategy proposed for gene therapy. However, viral infection strictly depends on receptor-dependent tropism, a property that determines which cells can be successfully infected by viruses [90]. This represents one of the most limiting factors of viral-based gene therapy in cancer, as tumor cells could not express sufficient receptors to promote efficient infections.
For example, the coxsackie and adenovirus receptor (CAR), crucial in the adenoviral tropism toward hepatocytes, cardiomyocytes, myoblasts, epithelial and endothelial cells, display an oncosuppressor role in different cancers and can be downregulated in malignant cells, such as in glioma. This supports the necessity of engineering adenoviruses to achieve a CAR-independent infection and an improvement in gene therapy efficiency [91,92]. Thanks to their capacity to bind membrane cancer biomarkers, aptamers have been investigated as an emerging approach to modify natural viral tropisms artificially. Chen H. and colleagues functionalized a genetically modified Ad5, the most used adenoviral-vector serotype in humans, with the anti-nucleolin AS1411 or the anti-tenascin-C GBI-10 aptamers [93]. In detail, they introduced the biotin acceptor peptide (BAP) into the hexon, the main representative protein of the adenoviral capsid, which was consequently biotinylated during the assembly of viral particles in HEK293 host cells (Figure 2a). Then, they coated the viral surface using streptavidin as a bridge, which allowed the binding between the biotinylated AS1411 or GBI-10 aptamers with the viral biotin-bearing surface. The transduction efficiency of the aptamer-coated viruses was then tested by monitoring the expression and function of luciferase transgene, observing a 4-5 fold increase with respect to the control viral particles in a cell line naturally not infected by the Ad5 serotype.
Beyond Ad5, other commonly used types of viruses are the adeno-associated (AAV), a class of non-pathological viruses characterized by a broad tropism toward different cells thanks to their recognition of glycans, glycoconjugates, or sialic acid [94]. However, despite the broad tropism in tissues such as the liver, retina, central nervous system, kidneys, pancreas, skeletal muscles, and lungs, no cancer uses of AAVs have been achieved so far due to low cancer cell infection [11]. To improve their infection capabilities, AAV2 viral vectors, naturally able to recognize heparin sulfate proteoglycan (HSPG), were first coated with DNA-based dendrimers, a branched nano-polymers with a well-defined structure which was used as an anchorage for aptamer coating (Figure 2b). The dendrimers were covalently attached to the lysine residues of the viral capsid using a dithiobis (succinimidyl propionate) (DSP) cross-linker that allowed the constitution of -S-S bonds [95]. Then, the dendrimers were hybridized with anti-PTK7 DNA aptamers (sgc8) via a sticky-based approach since both aptamers and dendrimers were extended with base-pairing sequences. In this way, viral particles were functionalized with dendrimers that harbored four sgc8 aptamers each, generating multivalent aptamer-coated AAV2 vectors. The authors demonstrated that the sgc8 functionalized viruses efficiently infected HSPG-negative but PTK7-positive CEM cells, as shown by the exogenous expression of the green fluorescent protein (GFP) report transgene.
The two discussed findings demonstrated the possibility of extending the tropism of viruses using aptamer-based tools in vitro. However, it’s crucial to note that the engineered Ad5 and AAV2 maintained their natural tropism that would still allow the infection of such healthy tissues in vivo. This is a significant limit associated with safety concerns of viral-based therapeutics, a challenge that the field should address to ensure the safety and efficacy of these treatments. Puzzo and colleagues recently attempted a different strategy to optimize viral vectors. They developed an AS1411-coated AAV starting from a strain previously obtained in their laboratory that was deprived of the HSPG binding sites (AAV-DJR/A). In this way, the natural tropism of the progenitor virus was impaired [96]. The AS1411 coating was obtained using an innovative approach. In particular, AAV-DJR/A was further engineered using host cells capable of introducing unnatural amino acids, the azido-lysine amino acids, into the translated capsid proteins (Figure 2c). The novel AAV, named Nε-AAV, incorporated azido amino acid insertions, which allowed the conjugation of dibenzocyclooctyne (DBCO) harboring AS1411 aptamers to the capsid via a click chemistry reaction. AS1411 coated Nε-AAV infection led to an enhanced luciferase signal in MCF-7 BC cells compared to unconjugated vectors, indicating a higher infection capability promoted by the AS1411 aptamer coating in vitro. However, subcutaneous injection of AS1411 coated Nε-AAVs revealed unexpected in vivo results since lower bioluminescence in HELA subcutaneous tumors was observed with respect to uncoated Nε-AAV. Nevertheless, although less efficient than the progenitor virus, AS1411 coated Nε-AAV also displayed no detectable liver infection, suggesting that the aptamer coating could have a role in reducing liver targeting.
Overall, these findings indicate that viral vectors can be chemically conjugated with aptamers through different approaches, but their evaluation is still preliminary since biological validations were only performed using reported genes, such as luciferase and GFP. Novel investigations are mandatory to unveil whether this new approach can deliver sufficient transgenes and therapeutic effects in cancer cells without infection in healthy tissues. Nevertheless, these promising preclinical studies support the concept that aptamer functionalization could help to achieve the goal of highly specific viruses for cancer gene therapy.
3.5. Aptamer-Functionalized NPs
NPs are promising alternatives to viral vectors because of the better safety profiles, the more straightforward large-scale productions, and the broad possibility of customizing [97]. In addition, NPs can be designed to allow controlled-release formulations and can be more easily loaded with multiple therapeutics. However, these non-viral carriers, initially developed for the safe delivery of traditional chemotherapies, have shown low delivery efficiency in cancer, as demonstrated by the modest clinical results obtained [98]. These results mainly depend on the lack of specific targeting, which is not intrinsically endowed in the standard nanomaterials used to assemble NPs. For this reason, preclinical studies have attempted the optimization of NPs through functionalization with targeting ligands, including aptamers, to promote cell-specific and efficient cellular uptake. Since the literature on this topic is extensive and has grown remarkably in the recent decade, we are going to focus on the preclinical findings in which already clinical approved NP types were functionalized with the aptamer that reached the more advanced stage in cancer clinical trials, the anti-nucleolin aptamer AS1411 [99,100]. Thus, examples of AS1411-functionalized liposomes, metallic-based, and polyplexes are provided [101].
Among clinically approved NPs in cancer therapy, about 60% are lipid-based [102]. It is not surprising that evaluations of potential benefits associated with aptamer-based functionalization were extensively made in this context [103,104]. For example, Yu et al. developed cationic liposomes using Dioleoylphosphatidylethanolamine (DOPE), Sphingomyelin (SM), didecyldimethylammonium bromide (DDAB), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[(polyethyleneglycol)-2000] DSPE-PEG2000, and DSPE-PEG2000 maleimide (DSPE-PEG2000-MAL) (Figure 3a) [105]. The maleimide group in this last component was introduced to allow the functionalization of liposomes with AS1411 via a post-insertion method. The authors loaded the hydrophobic layer of the liposomes with Paclitaxel (PTX) and promoted the absorption of anti-PLK1 siRNAs onto the external liposomal surface. The developed AS1411/Lipo-PTX-siPLK1 successfully delivered PTX and siPLK1 to MCF-7 BC cells, synergistically affecting cell proliferation. In vitro data were then corroborated in mouse models where AS1411/Lipo-PTX-siPLK1 impaired tumor growth and extended the survival of tumor-bearing mice, demonstrating that aptamer-based functionalization could significantly improve liposome performances. Promising results were also reported exploiting aptamers in the context of metallic-based nanoparticles, used in about 6% of approved NP-based pharmaceuticals [102]. An interesting example is the gold(Au)-based nanoparticles described by Deng et al., simultaneously functionalized through the constitution of -S-S bonds with anti-nucleolin AS1411 aptamers and antimiRs targeting miR-221 (Figure 3b) [106]. Notably, the authors also functionalized the Au surfaces with nuclear localization signal (NLS) peptides since they reported that most miR-221 and nucleolin resided and functioned in the nucleus. Thus, an Au-based nanoparticle contemporary functionalized with AS1411 aptamers, antimiR-221 oligonucleotides, and NLS peptides was developed (NPsN-AS1411/a221). The functionalization with AS1411 significantly improved the effect of the NPs, providing a synergistic inhibitory effect with antimiR-221 on the nucleolin/miR-221/NFκB/DNMT1 signaling both in vitro and in vivo. These data are particularly relevant since they confirm the optimizing role of aptamer and the potentiality of NPs for multitargeting approaches. However, no evaluation of the effective contribution of NLS can be instead obtained from the study since the authors did not compare NPs in the presence or absence of NLS. Besides liposomes and Au-based, although not similarly developed for clinical applications, polyplexes were also functionalized with aptamers [107]. Aliabadi and colleagues have recently developed a copolymer-based polyplex constituted of Poly(lactic acid)121-b-Poly(N-3-aminopropyl)methacrylamide)103 (PLA121-b-PAPMA103) (Figure 3c) [108]. Thanks to its polycation nature, this polyplex self-assembled in a spherical micelle that absorbed plasmids encoding anti-survivin shRNAs and AS1411 aptamers. The study demonstrated that AS1411-based functionalization improved the in vivo effect of the polyplexes, reducing the tumor masses and increasing the survival rate in mouse models. This article is interesting not only for developing an innovative nanomaterial and its functionalization with AS1411 but also for attempting to deliver a plasmid, an option not reported for aptamer-based conjugates. In general, these three exemplary studies underline the benefit of aptamer functionalization in improving the performance of NPs independently from the different nanomaterials used to assemble them. These promising data are not only limited to AS1411, but several other aptamers have been investigated for similar purposes, demonstrating flexibility in what aptamer can be conjugated to NPs, paving the way for efficient delivery of therapeutic transgenes, RNAs, or oligonucleotides in different cancer types [109,110].
In addition to the above discussion regarding NP-based delivery systems of therapeutic oligonucleotides or plasmids, a particular mention should be made to innovative findings associated with CRISPR/Cas9 technology, the most powerful gene editing technique available nowadays [111]. Although it seems promising for cancer applications, concerns about off-target genetic alterations in healthy tissues are holding its use in vivo, and the development of customized delivery vehicles is considered a crucial medical need. With this aim, a single adenoviral vector for the expression of the entire CRISPR/Cas9 machinery was developed, but the previously described limits associated with viral vectors support their further improvements before clinical proposals [112]. Aptamer coating of this viral vector could be pursued, but no references are reported in the literature to our knowledge. In this scenario, exploiting NPs and their aptamer-functionalized upgrade could represent an ideal tool [113,114]. Indeed, the contemporary presence of a single guide RNA (sgRNA) and the Cas9 nuclease is necessary for the technology functionality, requiring the delivery of the pre-constituted macro-complex or the delivery of a DNA plasmid for its post-delivery cellular expression, both characterized by large size and not easily compatible with aptamer-based conjugates. Liu and colleagues opted to develop a protein-based NP, representing 38% of already approved NPs in clinics [102,115]. In detail, they complexed a CDK11 silencing CRISPR/Cas9 expressing plasmid with protamine, a small polycationic natural peptide, constituting the core of the polymeric NP (Figure 3d). Then, the central core was coated with AS1411-incorporated Carboxymethyl chitosan (CMC), which was absorbed thanks to its negative charges via electrostatic interactions. Lastly, the NP was completed with KALA, a positive-charged peptide. KALA and AS1411 were introduced to improve the cellular internalization of the gene delivery system. In line with this in silico design, KALA and AS1411 remarkably increased the internalization in MCF-7 BC cells. As expected by higher internalization of the plasmid, CDK11 expression was silenced due to CRISPR/Cas9 nuclear function. Although no in vivo validation was reported, this study highlighted preliminary information about the feasibility of aptamer-functionalization of NPs to deliver CRISPR/Cas9 machinery in cancer cells selectively. This result was recently achieved with a different AS1411-functionalized NP [116]. In this case, Khademi et al. incorporated a plasmid encoding the entire CRISPR/Cas9 machinery to impair the expression of FOXM1 into a chitosan-based core (CS). The core was then coated with hyaluronic acid (HA) and AS1411 (AS) for targeting delivery (Figure 3e). Apt-HA-CS-CRISPR/Cas9 successfully delivered CRISPR/Cas9 into nucleolin-expressing cells (MCF-7, SK-MES-1, HeLa) but not into those that were negative (HEK293). Furthermore, data were confirmed in vivo, where Apt-HA-CS-CRISPR/Cas9 promoted efficient tumor inhibition with no observable distribution in other organs.
In general, these study collection, in addition to further preclinical findings not only limited to the AS1411 aptamer, strongly suggest that aptamer functionalization could be decisive in achieving selective delivery of CRISPR/Cas9 technology.
4. Aptamers as CRISPR/Cas9 Regulators and Epigenetic Modifiers
Although the most investigated application of aptamers in gene therapy is in the context of delivery, recent findings suggest that aptamers could have interesting roles in intracellular compartments, thus opening new avenues in the field. Remaining in the context of CRISPR/Cas9, aptamers have been recently used as allosteric sites to modulate the gene editing machinery. The sequence encoding for the anti-theophylline RNA aptamer was first introduced in various positions of an sgRNA [117]. Kundert and colleagues demonstrated that such anti-theophylline aptamer-included sgRNAs could show relevant conformational changes because of theophylline binding, enabling the assembly or disassembly of CRISPR/Cas9 machinery on DNA and, consequently, modulation of its function. These ligand-activated or ligand-deactivated sgRNAs were named ligRNAs, which permitted rapid (within minutes) and reversible CRISPR/Cas9 regulation depending on theophylline concentration in E. coli. However, despite being an innovative approach, the originated liRNAs were not active in mammalian cells, underlining the necessity of optimizations. In this perspective, Yan Liu and colleagues exploited the crystal structure of the Cas9/sgRNA complex, identifying novel positions for the introduction of the anti-theophylline aptamer [118]. They observed two positions in which the presence of the anti-theophylline aptamer could remarkably impair the function of the sgRNA, even if the authors underlined that a total inhibition was not achieved. However, the addition of theophylline remarkably increased the formation of the ternary complexes constituted of a sgRNA, which harbored the guiding RNA sequence for the ASCL1 gene, Cas9, and targeted DNA in electrophoretic shift assays, supporting validation in living cells. A CRISPR-mediated gene activation (CRISPRa) assay was then performed in HEK293 cells, which stably expressed Cas9-VP64-p65-Rta (VPR). In line with the previous in vitro assay, the authors observed that the incubation of cells with theophylline positively controlled the activation of the ASCL1 gene, demonstrating that the novel engineered anti-theophylline aptamer bearing sgRNAs could also modulate the CRISR/Cas9 recognition of its targeted DNA in a human cellular context. Noteworthy, since the introduction of the aptameric allosteric site was rationally included in positions unrelated to the guiding RNA sequence, at least theoretically, these sgRNAs could be modified to recognize other genes of interest.
Besides their potential involvement as additional constituents and regulators of CRISPR/Cas9 machinery, aptamers can also play a role in epigenetics. Since epigenetic aberrations, including DNA methylation and histone acetylation, have been commonly found in cancer, developing molecules able to restore normal epigenetic profiles in tumor cells has attracted interest [119]. Wang et al. first reported the selection of DNA aptamers able to target the DNA maintenance methyltransferase DNMT1 [120]. The isolated anti-DNMT1 aptamer (Apt. #9) used at the sub-micromolar scale could compete with the hemi-methylated DNA duplex, the original substrate of DNMT1, promoting an effective enzymatic inhibition thanks to the presence in its structure of a stem-loop. Apt. #9 displayed selectivity for DNMT1, since no binding activity toward DNMT3A and DNMT3B was observed. Intriguingly, the authors reported a significative internalization of Apt. #9 by simple incubation with DNMT1 high-expressing HeLa cells, also showing colocalization with DNMT1 inside the nuclei. This phenomenon also determined a more than 40% reduction in DNA methylation when Apt. #9 was incubated with cells at 3µM final concentration. However, the effect provided by Apt. #9 only caused a faint reduction of HeLa cell viability (about 20%). Further validations in different cancer cells could reveal if the modest result was related to the tumor model or the selected aptamer. Indeed, Apt. #9 showed fast degradation in the culture medium (11 hours) that, in association with the affinity in the high nM range (770nM), could suggest a temporary modification of the cellular epigenetic status and a faint biological effect. A further step in using aptamer as a modifier of the DNA methylation status was recently made. Starting from a class of RNAs with an inherent capability of inhibiting DNMT1 (DNMT1-interacting RNAs, also shortly named DiRs), our group applied a novel doped SELEX approach to produce 2’-Fluro pyrimidine-modified RNA aptamers that maintained the capacity of neutralizing DNMT1 but showed higher affinity and stability for potential therapeutic applications [121,122]. The procedure led to the identification of an aptameric DiRs (aptaDiRs), Ce-49 sh that showed nM affinity for DNMT1 (<100nM), no significative interaction with DNMT3A and DNMT3B, and longer stability in human serum (half-life longer than 48 h). Regarding functionality, Ce-49 sh exhibited efficient deregulation of DNA methylation in K562 leukemic cells and promising inhibition of tumor growth in vivo. Targeting epigenetics with aptamers is not limited to DNA methylation since DNA aptamers were also used to neutralize Histone acetyltransferase 1 (HAT1). Indeed, a novel DNA aptamer (apHAT610) that efficiently targeted HAT1 in lung cancer cells has been recently reported by Klett-Mingo and colleagues [123]. Therefore, although aptamer-mediated targeting of epigenetic modifications has been preliminarily evaluated only, further studies could reveal potentiality for cancer therapies.
5. Discussion
Although gene therapy has emerged as an outstanding perspective direction in the field of cancer therapy, its clinical application remains limited due to the low delivery efficiency and important concerns about the onset of off-target effects. As discussed in this review, aptamers have been extensively investigated as targeting moiety in novel gene therapy systems to allow a more efficient and selective delivery. They have been directly conjugated to therapeutic oligonucleotides (miRNAs, antimiRs, siRNAs, and DNAzymes) or used to functionalize the surface of therapeutic oligonucleotide-loaded NPs. Aptamer-based conjugates were the first to be developed, but it should be noted that the trend on this subject has slowed down in the last few years, shifting greater attention to combining aptamers with NPs. This could be partly due to the increasing interest in multi-targeting approaches that can be more easily achieved by exploring the multi-loading capability of NPs. Regarding the use of aptamers to coat viral surfaces, despite interesting results, data are still preliminary, and additional studies are required.
Even if findings regarding aptamer-based conjugates and aptamer-functionalized NPs are promising and could support the next steps toward clinical translation, the lack of comparative studies has presumably prevented their further development, representing the main limitation of this research field. Indeed, numerous research groups have developed and characterized functional aptamer-based conjugates, which differ for the aptamer and the therapeutic oligonucleotide used, the conjugation strategy applied, the spacing between the two moieties, and the chemical modifications introduced. The authors rarely describe the process that supported the design of the presented conjugate, and no references regarding negative results before the definition of the final conjugate are commonly discussed. Since no retrospective comparison between conjugates in different publications can be consistently made, information about which could be the best conjugate type or if such conjugates perform better in specific tumoral contexts should be mandatory. An analogous statement can be reported for NPs, developed using several nanomaterials, loaded with different therapeutics, and functionalized with aptamers applying various methods. Therefore, since the efficacy of these aptamer-tailored delivery systems depends on multiple variables, including stability in biological fluids, biodistribution, elimination, aptamer binding, expression level of recognized receptors, rate of internalization, percentage of escape from the endosomes, and pharmacological effect once in the cytosol or nucleus, it seems evident that each modification could remarkably have an impact on each of indicated processes, strongly suggesting more deeply investigations concerning these aspects. Moreover, besides comparing systems belonging to the same class, comparisons between aptamer-based conjugates or functionalized NPs are analogously necessary. For example, a therapeutic siRNA could be delivered using the same aptamer in the context of an aptamer-based conjugate or a functionalized NP. However, despite the aptamer in common, the two delivery systems are remarkably different in pharmacokinetics and pharmacodynamics, and the lack of comparisons between them could make challenging the role of pharmaceutical companies, which should select the best aptamer-guided gene therapy approach and invest remarkable funds in more advanced preclinical studies and potentially clinical trials. In this scenario, while recognizing the difficulties of achieving this, more collaborations between academic groups should be pursued, allowing the creation of multidisciplinary teams able to perform crucial comparative experiments. At the same time, since these studies are costly for academia, the involvement of private stakeholders could be decisive, and more funds from pharmaceutical companies should be invested in these early development stages. This cooperation could also help to validate these systems in more advanced in vivo models, such as orthotopic and patient-derived xenografts, which are mainly missing and necessary to move forward with a technology that could be a promising player for future generations of gene therapies.
Despite recent and less investigated, it is also worth mentioning the contribution that aptamers could make in delivering or even modulating CRISPR/Cas9 gene editing technology, supporting medical approaches still hindered today. Moreover, although the leading utility of aptamers seems to be in the context of delivery, they can be endowed with their own nuclear function as epigenetic modifiers. This is a particularly interesting field in which aptamers can emerge as innovative tools in the next decade with potential advantages over conventional epigenetic therapies in specificity and safety.
6. Conclusions
Aptamers have unique and diversified features that make them ideal for numerous applications in gene therapies. Further efforts are necessary to complete their validations, but reported findings strongly support additional studies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Table 1: Aptamer-based miRNA and antimiR conjugates; Supplementary Table 2: Aptamer-based siRNA conjugates.
Author Contributions
Conceptualization, supervision, and funding acquisition, S.C. and C.L.E.; writing—original draft preparation, G.C. (Gabriele Coppola), F.C., G.C. (Giuseppe Ciccone), M.L.I., A.D.V.; writing—review and editing A.D.V., S.C. and C.L.E. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chakravarthi, B.V.S.K.; Nepal, S.; Varambally, S. Genomic and Epigenomic Alterations in Cancer. Am J Pathol 2016, 186, 1724–1735. [Google Scholar] [CrossRef] [PubMed]
- The Lancet 20 Years of Precision Medicine in Oncology. The Lancet 2021, 397, 1781. [CrossRef] [PubMed]
- The Global Challenge of Cancer. Nat Cancer 2020, 1, 1–2. [CrossRef] [PubMed]
- Haier, J.; Schaefers, J. Economic Perspective of Cancer Care and Its Consequences for Vulnerable Groups. Cancers (Basel) 2022, 14, 3158. [Google Scholar] [CrossRef]
- Bauer, G.; Anderson, J.S. History of Gene Therapy. In; 2014; pp. 9–15.
- Landhuis, E. The Definition of Gene Therapy Has Changed. Nature 2021. [Google Scholar] [CrossRef]
- Dowdy, S.F. Overcoming Cellular Barriers for RNA Therapeutics. Nat Biotechnol 2017, 35, 222–229. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in Oligonucleotide Drug Delivery. Nat Rev Drug Discov 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Zhao, Z.; Anselmo, A.C.; Mitragotri, S. Viral Vector-based Gene Therapies in the Clinic. Bioeng Transl Med 2022, 7. [Google Scholar] [CrossRef]
- Qi, L.; Li, G.; Li, P.; Wang, H.; Fang, X.; He, T.; Li, J. Twenty Years of Gendicine® RAd-P53 Cancer Gene Therapy: The First-in-Class Human Cancer Gene Therapy in the Era of Personalized Oncology. Genes Dis 2024, 11, 101155. [Google Scholar] [CrossRef]
- Wang, J.-H.; Gessler, D.J.; Zhan, W.; Gallagher, T.L.; Gao, G. Adeno-Associated Virus as a Delivery Vector for Gene Therapy of Human Diseases. Signal Transduct Target Ther 2024, 9, 78. [Google Scholar] [CrossRef]
- Anguela, X.M.; High, K.A. Entering the Modern Era of Gene Therapy. Annu Rev Med 2019, 70, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Sayour, E.J.; Boczkowski, D.; Mitchell, D.A.; Nair, S.K. Cancer MRNA Vaccines: Clinical Advances and Future Opportunities. Nat Rev Clin Oncol 2024. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. MRNA Vaccines for Infectious Diseases: Principles, Delivery and Clinical Translation. Nat Rev Drug Discov 2021, 20, 817–838. [Google Scholar] [CrossRef] [PubMed]
- Mirón-Barroso, S.; Domènech, E.B.; Trigueros, S. Nanotechnology-Based Strategies to Overcome Current Barriers in Gene Delivery. Int J Mol Sci 2021, 22, 8537. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.; Zhang, X. Aptamers as Versatile Ligands for Biomedical and Pharmaceutical Applications. Int J Nanomedicine 2020, Volume 15, 1059–1071. [Google Scholar] [CrossRef]
- Bauer, M.; Strom, M.; Hammond, D.S.; Shigdar, S. Anything You Can Do, I Can Do Better: Can Aptamers Replace Antibodies in Clinical Diagnostic Applications? Molecules 2019, 24, 4377. [Google Scholar] [CrossRef]
- Porciani, D.; Cardwell, L.N.; Tawiah, K.D.; Alam, K.K.; Lange, M.J.; Daniels, M.A.; Burke, D.H. Modular Cell-Internalizing Aptamer Nanostructure Enables Targeted Delivery of Large Functional RNAs in Cancer Cell Lines. Nat Commun 2018, 9, 2283. [Google Scholar] [CrossRef]
- Esposito, C.L.; Catuogno, S.; Condorelli, G.; Ungaro, P.; De Franciscis, V. Aptamer Chimeras for Therapeutic Delivery: The Challenging Perspectives. Genes (Basel) 2018, 9, 529. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic Evolution of Ligands by Exponential Enrichment: RNA Ligands to Bacteriophage T4 DNA Polymerase. Science (1979) 1990, 249, 505–510. [Google Scholar] [CrossRef]
- Ellington, A.D.; Szostak, J.W. In Vitro Selection of RNA Molecules That Bind Specific Ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef]
- Zhu, C.; Feng, Z.; Qin, H.; Chen, L.; Yan, M.; Li, L.; Qu, F. Recent Progress of SELEX Methods for Screening Nucleic Acid Aptamers. Talanta 2024, 266, 124998. [Google Scholar] [CrossRef] [PubMed]
- Catuogno, S.; Esposito, C.L. Aptamer Cell-Based Selection: Overview and Advances. Biomedicines 2017, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Mayer, G.; Wulffen, B. The Chemical Biology of Aptamers: Synthesis and Applications. In The Chemical Biology of Nucleic Acids; Wiley, 2010; pp. 377–400. [Google Scholar]
- Li, N.; Nguyen, H.H.; Byrom, M.; Ellington, A.D. Inhibition of Cell Proliferation by an Anti-EGFR Aptamer. PLoS One 2011, 6, e20299. [Google Scholar] [CrossRef]
- McNamara, J.O.; Kolonias, D.; Pastor, F.; Mittler, R.S.; Chen, L.; Giangrande, P.H.; Sullenger, B.; Gilboa, E. Multivalent 4-1BB Binding Aptamers Costimulate CD8+ T Cells and Inhibit Tumor Growth in Mice. Journal of Clinical Investigation 2008, 118, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J. Aptamers as Targeted Therapeutics: Current Potential and Challenges. Nat Rev Drug Discov 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.H.; Elsherbiny, M.E.; Emara, M. Updates on Aptamer Research. Int J Mol Sci 2019, 20, 2511. [Google Scholar] [CrossRef]
- Esawi, E.; Nsairat, H.; Mahmoud, I.S.; Lafi, Z.; Al-Kadash, A.; Al-Ragheb, B.A.; Ismail, S.I.; Alhaer, W. Clinical Use and Future Perspective of Aptamers. In Aptamers Engineered Nanocarriers for Cancer Therapy; Elsevier, 2023; pp. 481–520. [Google Scholar]
- Gragoudas, E.S.; Adamis, A.P.; Cunningham, E.T.; Feinsod, M.; Guyer, D.R. Pegaptanib for Neovascular Age-Related Macular Degeneration. New England Journal of Medicine 2004, 351, 2805–2816. [Google Scholar] [CrossRef]
- Mullard, A. FDA Approves Second RNA Aptamer. Nat Rev Drug Discov 2023, 22, 774–774. [Google Scholar] [CrossRef]
- Xiao, X.; Li, H.; Zhao, L.; Zhang, Y.; Liu, Z. Oligonucleotide Aptamers: Recent Advances in Their Screening, Molecular Conformation and Therapeutic Applications. Biomedicine & Pharmacotherapy 2021, 143, 112232. [Google Scholar] [CrossRef]
- Wan, L.-Y.; Yuan, W.-F.; Ai, W.-B.; Ai, Y.-W.; Wang, J.-J.; Chu, L.-Y.; Zhang, Y.-Q.; Wu, J.-F. An Exploration of Aptamer Internalization Mechanisms and Their Applications in Drug Delivery. Expert Opin Drug Deliv 2019, 16, 207–218. [Google Scholar] [CrossRef]
- Yoon, S.; Rossi, J.J. Aptamers: Uptake Mechanisms and Intracellular Applications. Adv Drug Deliv Rev 2018, 134, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Thiel, W.H.; Thiel, K.W.; Flenker, K.S.; Bair, T.; Dupuy, A.J.; McNamara, J.O.; Miller, F.J.; Giangrande, P.H. Cell-Internalization SELEX: Method for Identifying Cell-Internalizing RNA Aptamers for Delivering SiRNAs to Target Cells. In; 2015; pp. 187–199.
- Thiel, K.W.; Hernandez, L.I.; Dassie, J.P.; Thiel, W.H.; Liu, X.; Stockdale, K.R.; Rothman, A.M.; Hernandez, F.J.; McNamara, J.O.; Giangrande, P.H. Delivery of Chemo-Sensitizing SiRNAs to HER2+-Breast Cancer Cells Using RNA Aptamers. Nucleic Acids Res 2012, 40, 6319–6337. [Google Scholar] [CrossRef] [PubMed]
- Gamboa, J.; Lourenço, P.; Cruz, C.; Gallardo, E. Aptamers for the Delivery of Plant-Based Compounds: A Review. Pharmaceutics 2024, 16, 541. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Shang, R.; Lee, S.; Senavirathne, G.; Lai, E.C. MicroRNAs in Action: Biogenesis, Function and Regulation. Nat Rev Genet 2023, 24, 816–833. [Google Scholar] [CrossRef]
- Iwakawa, H.; Tomari, Y. Life of RISC: Formation, Action, and Degradation of RNA-Induced Silencing Complex. Mol Cell 2022, 82, 30–43. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The Role of MicroRNAs in Human Cancer. Signal Transduct Target Ther 2016, 1, 15004. [Google Scholar] [CrossRef]
- Menon, A.; Abd-Aziz, N.; Khalid, K.; Poh, C.L.; Naidu, R. MiRNA: A Promising Therapeutic Target in Cancer. Int J Mol Sci 2022, 23, 11502. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, D.-Y.; Huang, L. In Vivo Delivery of MiRNAs for Cancer Therapy: Challenges and Strategies. Adv Drug Deliv Rev 2015, 81, 128–141. [Google Scholar] [CrossRef]
- Odeh, F.; Nsairat, H.; Alshaer, W.; Ismail, M.A.; Esawi, E.; Qaqish, B.; Bawab, A. Al; Ismail, S.I. Aptamers Chemistry: Chemical Modifications and Conjugation Strategies. Molecules 2019, 25, 3. [Google Scholar] [CrossRef]
- Esposito, C.L.; Catuogno, S.; de Franciscis, V. Aptamer-MiRNA Conjugates for Cancer Cell-Targeted Delivery. In; 2016; pp. 197–208.
- Esposito, C.L.; Cerchia, L.; Catuogno, S.; De Vita, G.; Dassie, J.P.; Santamaria, G.; Swiderski, P.; Condorelli, G.; Giangrande, P.H.; de Franciscis, V. Multifunctional Aptamer-MiRNA Conjugates for Targeted Cancer Therapy. Mol Ther 2014, 22, 1151–1163. [Google Scholar] [CrossRef] [PubMed]
- Cerchia, L.; Esposito, C.L.; Camorani, S.; Rienzo, A.; Stasio, L.; Insabato, L.; Affuso, A.; de Franciscis, V. Targeting Axl With an High-Affinity Inhibitory Aptamer. Molecular Therapy 2012, 20, 2291–2303. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.S.; Erkeland, S.J.; Pester, R.E.; Chen, C.Y.; Ebert, M.S.; Sharp, P.A.; Jacks, T. Suppression of Non-Small Cell Lung Tumor Development by the Let-7 MicroRNA Family. Proceedings of the National Academy of Sciences 2008, 105, 3903–3908. [Google Scholar] [CrossRef] [PubMed]
- Iaboni, M.; Russo, V.; Fontanella, R.; Roscigno, G.; Fiore, D.; Donnarumma, E.; Esposito, C.L.; Quintavalle, C.; Giangrande, P.H.; de Franciscis, V.; et al. Aptamer-MiRNA-212 Conjugate Sensitizes NSCLC Cells to TRAIL. Mol Ther Nucleic Acids 2016, 5, e289. [Google Scholar] [CrossRef]
- Chen, W.; Song, J.; Bian, H.; Yang, X.; Xie, X.; Zhu, Q.; Qin, C.; Qi, J. The Functions and Targets of MiR-212 as a Potential Biomarker of Cancer Diagnosis and Therapy. J Cell Mol Med 2020, 24, 2392–2401. [Google Scholar] [CrossRef]
- Russo, V.; Paciocco, A.; Affinito, A.; Roscigno, G.; Fiore, D.; Palma, F.; Galasso, M.; Volinia, S.; Fiorelli, A.; Esposito, C.L.; et al. Aptamer-MiR-34c Conjugate Affects Cell Proliferation of Non-Small-Cell Lung Cancer Cells. Mol Ther Nucleic Acids 2018, 13, 334–346. [Google Scholar] [CrossRef]
- Nuzzo, S.; Catuogno, S.; Capuozzo, M.; Fiorelli, A.; Swiderski, P.; Boccella, S.; de Nigris, F.; Esposito, C.L. Axl-Targeted Delivery of the Oncosuppressor MiR-137 in Non-Small-Cell Lung Cancer. Mol Ther Nucleic Acids 2019, 17, 256–263. [Google Scholar] [CrossRef]
- Quirico, L.; Orso, F.; Esposito, C.L.; Bertone, S.; Coppo, R.; Conti, L.; Catuogno, S.; Cavallo, F.; de Franciscis, V.; Taverna, D. Axl-148b Chimeric Aptamers Inhibit Breast Cancer and Melanoma Progression. Int J Biol Sci 2020, 16, 1238–1251. [Google Scholar] [CrossRef]
- Zhao, N.; Pei, S.-N.; Qi, J.; Zeng, Z.; Iyer, S.P.; Lin, P.; Tung, C.-H.; Zu, Y. Oligonucleotide Aptamer-Drug Conjugates for Targeted Therapy of Acute Myeloid Leukemia. Biomaterials 2015, 67, 42–51. [Google Scholar] [CrossRef]
- Tanno, T.; Zhang, P.; Lazarski, C.A.; Liu, Y.; Zheng, P. An Aptamer-Based Targeted Delivery of MiR-26a Protects Mice against Chemotherapy Toxicity While Suppressing Tumor Growth. Blood Adv 2017, 1, 1107–1119. [Google Scholar] [CrossRef]
- Ramezanpour, M.; Daei, P.; Tabarzad, M.; Khanaki, K.; Elmi, A.; Barati, M. Preliminary Study on the Effect of Nucleolin Specific Aptamer–MiRNA Let-7d Chimera on Janus Kinase-2 Expression Level and Activity in Gastric Cancer (MKN-45) Cells. Mol Biol Rep 2019, 46, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Daei, P.; Ramezanpour, M.; Khanaki, K.; Tabarzad, M.; Nikokar, I.; Hedayati Ch, M.; Elmi, A. Aptamer-Based Targeted Delivery of MiRNA Let-7d to Gastric Cancer Cells as a Novel Anti-Tumor Therapeutic Agent. Iran J Pharm Res 2018, 17, 1537–1549. [Google Scholar] [PubMed]
- Crooke, S.T.; Baker, B.F.; Crooke, R.M.; Liang, X. Antisense Technology: An Overview and Prospectus. Nat Rev Drug Discov 2021, 20, 427–453. [Google Scholar] [CrossRef] [PubMed]
- Catuogno, S.; Rienzo, A.; Di Vito, A.; Esposito, C.L.; de Franciscis, V. Selective Delivery of Therapeutic Single Strand AntimiRs by Aptamer-Based Conjugates. Journal of Controlled Release 2015, 210, 147–159. [Google Scholar] [CrossRef]
- Esposito, C.L.; Nuzzo, S.; Kumar, S.A.; Rienzo, A.; Lawrence, C.L.; Pallini, R.; Shaw, L.; Alder, J.E.; Ricci-Vitiani, L.; Catuogno, S.; et al. A Combined MicroRNA-Based Targeted Therapeutic Approach to Eradicate Glioblastoma Stem-like Cells. Journal of Controlled Release 2016, 238, 43–57. [Google Scholar] [CrossRef]
- Fang, Y.; Shu, D.; Xiao, F.; Guo, P.; Qin, P.Z. Modular Assembly of Chimeric Phi29 Packaging RNAs That Support DNA Packaging. Biochem Biophys Res Commun 2008, 372, 589–594. [Google Scholar] [CrossRef]
- Shu, D.; Li, H.; Shu, Y.; Xiong, G.; Carson, W.E.; Haque, F.; Xu, R.; Guo, P. Systemic Delivery of Anti-MiRNA for Suppression of Triple Negative Breast Cancer Utilizing RNA Nanotechnology. ACS Nano 2015, 9, 9731–9740. [Google Scholar] [CrossRef]
- Arnold, C. Theranostics Could Be Big Business in Precision Oncology. Nat Med 2022, 28, 606–608. [Google Scholar] [CrossRef]
- Dana, H.; Chalbatani, G.M.; Mahmoodzadeh, H.; Karimloo, R.; Rezaiean, O.; Moradzadeh, A.; Mehmandoost, N.; Moazzen, F.; Mazraeh, A.; Marmari, V.; et al. Molecular Mechanisms and Biological Functions of SiRNA. Int J Biomed Sci 2017, 13, 48–57. [Google Scholar] [CrossRef]
- Zhang, M.M.; Bahal, R.; Rasmussen, T.P.; Manautou, J.E.; Zhong, X. The Growth of SiRNA-Based Therapeutics: Updated Clinical Studies. Biochem Pharmacol 2021, 189, 114432. [Google Scholar] [CrossRef]
- Friedrich, M.; Aigner, A. Therapeutic SiRNA: State-of-the-Art and Future Perspectives. BioDrugs 2022, 36, 549–571. [Google Scholar] [CrossRef] [PubMed]
- Ali Zaidi, S.S.; Fatima, F.; Ali Zaidi, S.A.; Zhou, D.; Deng, W.; Liu, S. Engineering SiRNA Therapeutics: Challenges and Strategies. J Nanobiotechnology 2023, 21, 381. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.O.; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell Type–Specific Delivery of SiRNAs with Aptamer-SiRNA Chimeras. Nat Biotechnol 2006, 24, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Dassie, J.P.; Liu, X.; Thomas, G.S.; Whitaker, R.M.; Thiel, K.W.; Stockdale, K.R.; Meyerholz, D.K.; McCaffrey, A.P.; McNamara, J.O.; Giangrande, P.H. Systemic Administration of Optimized Aptamer-SiRNA Chimeras Promotes Regression of PSMA-Expressing Tumors. Nat Biotechnol 2009, 27, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tiemann, K.; Chomchan, P.; Alluin, J.; Swiderski, P.; Burnett, J.; Zhang, X.; Forman, S.; Chen, R.; Rossi, J. Dual Functional BAFF Receptor Aptamers Inhibit Ligand-Induced Proliferation and Deliver SiRNAs to NHL Cells. Nucleic Acids Res 2013, 41, 4266–4283. [Google Scholar] [CrossRef]
- Mohtar, M.; Syafruddin, S.; Nasir, S.; Low, T.Y. Revisiting the Roles of Pro-Metastatic EpCAM in Cancer. Biomolecules 2020, 10, 255. [Google Scholar] [CrossRef]
- Wang, T.; Gantier, M.P.; Xiang, D.; Bean, A.G.; Bruce, M.; Zhou, S.-F.; Khasraw, M.; Ward, A.; Wang, L.; Wei, M.Q.; et al. EpCAM Aptamer-Mediated Survivin Silencing Sensitized Cancer Stem Cells to Doxorubicin in a Breast Cancer Model. Theranostics 2015, 5, 1456–1472. [Google Scholar] [CrossRef]
- Shigdar, S.; Lin, J.; Yu, Y.; Pastuovic, M.; Wei, M.; Duan, W. RNA Aptamer against a Cancer Stem Cell Marker Epithelial Cell Adhesion Molecule. Cancer Sci 2011, 102, 991–998. [Google Scholar] [CrossRef]
- Gilboa-Geffen, A.; Hamar, P.; Le, M.T.N.; Wheeler, L.A.; Trifonova, R.; Petrocca, F.; Wittrup, A.; Lieberman, J. Gene Knockdown by EpCAM Aptamer–SiRNA Chimeras Suppresses Epithelial Breast Cancers and Their Tumor-Initiating Cells. Mol Cancer Ther 2015, 14, 2279–2291. [Google Scholar] [CrossRef]
- Esposito, C.L.; Nuzzo, S.; Catuogno, S.; Romano, S.; de Nigris, F.; de Franciscis, V. STAT3 Gene Silencing by Aptamer-SiRNA Chimera as Selective Therapeutic for Glioblastoma. Mol Ther Nucleic Acids 2018, 10, 398–411. [Google Scholar] [CrossRef]
- Esposito, C.L.; Nuzzo, S.; Ibba, M.L.; Ricci-Vitiani, L.; Pallini, R.; Condorelli, G.; Catuogno, S.; de Franciscis, V. Combined Targeting of Glioblastoma Stem-Like Cells by Neutralizing RNA-Bio-Drugs for STAT3. Cancers (Basel) 2020, 12, 1434. [Google Scholar] [CrossRef] [PubMed]
- Ibba, M.L.; Ciccone, G.; Rotoli, D.; Coppola, G.; Fiorelli, A.; Catuogno, S.; Esposito, C.L. STAT3 Silencing by an Aptamer-Based Strategy Hampers the Crosstalk between NSCLC Cells and Cancer-Associated Fibroblasts. Mol Ther Nucleic Acids 2023, 32, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Oostindie, S.C.; Lazar, G.A.; Schuurman, J.; Parren, P.W.H.I. Avidity in Antibody Effector Functions and Biotherapeutic Drug Design. Nat Rev Drug Discov 2022, 21, 715–735. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Yu, X.; Liu, H.; Wu, D.; She, J.-X. Co-Targeting EGFR and Survivin with a Bivalent Aptamer-Dual SiRNA Chimera Effectively Suppresses Prostate Cancer. Sci Rep 2016, 6, 30346. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Maihle, N.J.; Yu, X.; Tang, S.-C.; Liu, H.Y. Synergistic Targeting HER2 and EGFR with Bivalent Aptamer-SiRNA Chimera Efficiently Inhibits HER2-Positive Tumor Growth. Mol Pharm 2018, 15, 4801–4813. [Google Scholar] [CrossRef]
- Yu, X.; Ghamande, S.; Liu, H.; Xue, L.; Zhao, S.; Tan, W.; Zhao, L.; Tang, S.-C.; Wu, D.; Korkaya, H.; et al. Targeting EGFR/HER2/HER3 with a Three-in-One Aptamer-SiRNA Chimera Confers Superior Activity against HER2+ Breast Cancer. Mol Ther Nucleic Acids 2018, 10, 317–330. [Google Scholar] [CrossRef]
- Walter, N.G.; Engelke, D.R. Ribozymes: Catalytic RNAs That Cut Things, Make Things, and Do Odd and Useful Jobs. Biologist (London) 2002, 49, 199–203. [Google Scholar]
- Goleij, P.; Babamohamadi, M.; Rezaee, A.; Sanaye, P.M.; Tabari, M.A.K.; Sadreddini, S.; Arefnezhad, R.; Motedayyen, H. Types of RNA Therapeutics. In; 2024; pp. 41–63.
- Reza, Md.S.; Mim, F.; Quader, M.R.; Khan, M.J.R.; Hossain, Md.S.; Uddin, K.R.; Akter, S.; Rahman, S.; Roy, S.; Rumman, Md.A. The Possibility of Nucleic Acids to Act as Anti-Viral Therapeutic Agents—A Review. Open J Med Microbiol 2021, 11, 198–248. [Google Scholar] [CrossRef]
- Khan, A.U. Ribozyme: A Clinical Tool. Clinica Chimica Acta 2006, 367, 20–27. [Google Scholar] [CrossRef]
- Feng, R.; Patil, S.; Zhao, X.; Miao, Z.; Qian, A. RNA Therapeutics - Research and Clinical Advancements. Front Mol Biosci 2021, 8. [Google Scholar] [CrossRef]
- Sioud, M. Ribozymes and SiRNAs: From Structure to Preclinical Applications. In RNA Towards Medicine; Springer-Verlag: Berlin/Heidelberg; pp. 223–242.
- Yan, J.; Bhadane, R.; Ran, M.; Ma, X.; Li, Y.; Zheng, D.; Salo-Ahen, O.M.H.; Zhang, H. Development of Aptamer-DNAzyme Based Metal-Nucleic Acid Frameworks for Gastric Cancer Therapy. Nat Commun 2024, 15, 3684. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Liu, X.; Luo, L.; Chen, J.; Zhou, X.; Chen, G.; Huang, X.; Yu, L.; Chen, Q.; Yang, Y.; et al. Metal-DNA Nanocomplexes Enhance Chemo-dynamic Therapy by Inhibiting Autophagy-Mediated Resistance. Angewandte Chemie International Edition 2023, 62. [Google Scholar] [CrossRef] [PubMed]
- Nomaguchi, M.; Fujita, M.; Miyazaki, Y.; Adachi, A. Viral Tropism. Front Microbiol 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Sumerel, L.A.; Belousova, N.; Lyons, G.R.; Carey, D.E.; Krasnykh, V.; Douglas, J.T. The Coxsackievirus and Adenovirus Receptor Acts as a Tumour Suppressor in Malignant Glioma Cells. Br J Cancer 2003, 88, 1411–1416. [Google Scholar] [CrossRef]
- Tessarollo, N.G.; Domingues, A.C.M.; Antunes, F.; Luz, J.C. dos S. da; Rodrigues, O.A.; Cerqueira, O.L.D.; Strauss, B.E. Nonreplicating Adenoviral Vectors: Improving Tropism and Delivery of Cancer Gene Therapy. Cancers (Basel) 2021, 13, 1863. [Google Scholar] [CrossRef]
- Chen, H.; Zheng, X.; Di, B.; Wang, D.; Zhang, Y.; Xia, H.; Mao, Q. Aptamer Modification Improves the Adenoviral Transduction of Malignant Glioma Cells. J Biotechnol 2013, 168, 362–366. [Google Scholar] [CrossRef]
- Dhungel, B.P.; Bailey, C.G.; Rasko, J.E.J. Journey to the Center of the Cell: Tracing the Path of AAV Transduction. Trends Mol Med 2021, 27, 172–184. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, L.; Cui, C.; Cansiz, S.; Liang, H.; Wu, C.; Teng, I.-T.; Chen, W.; Liu, Y.; Hou, W.; et al. Enhanced Targeted Gene Transduction: AAV2 Vectors Conjugated to Multiple Aptamers via Reducible Disulfide Linkages. J Am Chem Soc 2018, 140, 2–5. [Google Scholar] [CrossRef]
- Puzzo, F.; Zhang, C.; Powell Gray, B.; Zhang, F.; Sullenger, B.A.; Kay, M.A. Aptamer-Programmable Adeno-Associated Viral Vectors as a Novel Platform for Cell-Specific Gene Transfer. Mol Ther Nucleic Acids 2023, 31, 383–397. [Google Scholar] [CrossRef]
- Dogbey, D.M.; Torres, V.E.S.; Fajemisin, E.; Mpondo, L.; Ngwenya, T.; Akinrinmade, O.A.; Perriman, A.W.; Barth, S. Technological Advances in the Use of Viral and Non-Viral Vectors for Delivering Genetic and Non-Genetic Cargos for Cancer Therapy. Drug Deliv Transl Res 2023, 13, 2719–2738. [Google Scholar] [CrossRef]
- Germain, M.; Caputo, F.; Metcalfe, S.; Tosi, G.; Spring, K.; Åslund, A.K.O.; Pottier, A.; Schiffelers, R.; Ceccaldi, A.; Schmid, R. Delivering the Power of Nanomedicine to Patients Today. Journal of Controlled Release 2020, 326, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Bambury, R.M.; Van Allen, E.M.; Drabkin, H.A.; Lara, P.N.; Harzstark, A.L.; Wagle, N.; Figlin, R.A.; Smith, G.W.; Garraway, L.A.; et al. A Phase II Trial of AS1411 (a Novel Nucleolin-Targeted DNA Aptamer) in Metastatic Renal Cell Carcinoma. Invest New Drugs 2014, 32, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Van den Avont, A.; Sharma-Walia, N. Anti-Nucleolin Aptamer AS1411: An Advancing Therapeutic. Front Mol Biosci 2023, 10. [Google Scholar] [CrossRef] [PubMed]
- Mendes, B.B.; Conniot, J.; Avital, A.; Yao, D.; Jiang, X.; Zhou, X.; Sharf-Pauker, N.; Xiao, Y.; Adir, O.; Liang, H.; et al. Nanodelivery of Nucleic Acids. Nature Reviews Methods Primers 2022, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, F.; Caruana, P.; De la Fuente, N.; Español, P.; Gámez, M.; Balart, J.; Llurba, E.; Rovira, R.; Ruiz, R.; Martín-Lorente, C.; et al. Nano-Based Approved Pharmaceuticals for Cancer Treatment: Present and Future Challenges. Biomolecules 2022, 12, 784. [Google Scholar] [CrossRef] [PubMed]
- Luiz, M.T.; Dutra, J.A.P.; Tofani, L.B.; de Araújo, J.T.C.; Di Filippo, L.D.; Marchetti, J.M.; Chorilli, M. Targeted Liposomes: A Nonviral Gene Delivery System for Cancer Therapy. Pharmaceutics 2022, 14, 821. [Google Scholar] [CrossRef]
- Wong, K.-Y.; Wong, M.-S.; Liu, J. Aptamer-Functionalized Liposomes for Drug Delivery. Biomed J 2024, 47, 100685. [Google Scholar] [CrossRef]
- Yu, S.; Bi, X.; Yang, L.; Wu, S.; Yu, Y.; Jiang, B.; Zhang, A.; Lan, K.; Duan, S. Co-Delivery of Paclitaxel and PLK1-Targeted SiRNA Using Aptamer-Functionalized Cationic Liposome for Synergistic Anti-Breast Cancer Effects In Vivo. J Biomed Nanotechnol 2019, 15, 1135–1148. [Google Scholar] [CrossRef]
- Deng, R.; Shen, N.; Yang, Y.; Yu, H.; Xu, S.; Yang, Y.-W.; Liu, S.; Meguellati, K.; Yan, F. Targeting Epigenetic Pathway with Gold Nanoparticles for Acute Myeloid Leukemia Therapy. Biomaterials 2018, 167, 80–90. [Google Scholar] [CrossRef]
- Ayatollahi, S.; Salmasi, Z.; Hashemi, M.; Askarian, S.; Oskuee, R.K.; Abnous, K.; Ramezani, M. Aptamer-Targeted Delivery of Bcl-XL ShRNA Using Alkyl Modified PAMAM Dendrimers into Lung Cancer Cells. Int J Biochem Cell Biol 2017, 92, 210–217. [Google Scholar] [CrossRef]
- Aliabadi, A.; Vakili-Azghandi, M.; Abnous, K.; Taghdisi, S.M.; Ramezani, M.; Alibolandi, M. Nucleolin-Targeted Cationic Nanoparticle for Delivery of Survivin ShRNA against Colorectal Cancer in Vitro and in Vivo. Eur Polym J 2024, 208, 112872. [Google Scholar] [CrossRef]
- Fu, Z.; Xiang, J. Aptamer-Functionalized Nanoparticles in Targeted Delivery and Cancer Therapy. Int J Mol Sci 2020, 21, 9123. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; He, Z.; Ge, W.; Zhao, Z. Application of Aptamer Functionalized Nanomaterials in Targeting Therapeutics of Typical Tumors. Front Bioeng Biotechnol 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, Y.; Qi, H.; Cui, W.; Zhang, L.; Fu, X.; He, X.; Liu, M.; Li, P.; Yu, T. CRISPR/Cas9 Therapeutics: Progress and Prospects. Signal Transduct Target Ther 2023, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Ehrke-Schulz, E.; Schiwon, M.; Leitner, T.; Dávid, S.; Bergmann, T.; Liu, J.; Ehrhardt, A. CRISPR/Cas9 Delivery with One Single Adenoviral Vector Devoid of All Viral Genes. Sci Rep 2017, 7, 17113. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Ouyang, K.; Xu, X.; Xu, L.; Wen, C.; Zhou, X.; Qin, Z.; Xu, Z.; Sun, W.; Liang, Y. Nanoparticle Delivery of CRISPR/Cas9 for Genome Editing. Front Genet 2021, 12. [Google Scholar] [CrossRef]
- Yip, B. Recent Advances in CRISPR/Cas9 Delivery Strategies. Biomolecules 2020, 10, 839. [Google Scholar] [CrossRef]
- Liu, B.-Y.; He, X.-Y.; Zhuo, R.-X.; Cheng, S.-X. Tumor Targeted Genome Editing Mediated by a Multi-Functional Gene Vector for Regulating Cell Behaviors. Journal of Controlled Release 2018, 291, 90–98. [Google Scholar] [CrossRef]
- Khademi, Z. , Ramezani, M., Alibolandi, M., Zirak, M. R., Salmasi, Z., Abnous, K., & Taghdisi, S. M.. A novel dual-targeting delivery system for specific delivery of CRISPR/Cas9 using hyaluronic acid, chitosan and AS1411. Carbohydrate Polymers 2022, 292, 119691. [Google Scholar] [CrossRef]
- Kundert, K.; Lucas, J.E.; Watters, K.E.; Fellmann, C.; Ng, A.H.; Heineike, B.M.; Fitzsimmons, C.M.; Oakes, B.L.; Qu, J.; Prasad, N.; et al. Controlling CRISPR-Cas9 with Ligand-Activated and Ligand-Deactivated SgRNAs. Nat Commun 2019, 10, 2127. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Lin, J.; Xu, L. Theophylline-Induced Synergic Activation of Guide RNA to Control CRISPR/Cas9 Function. Chemical Communications 2021, 57, 5418–5421. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Kim, M.-Y. Cancer Epigenetics: Past, Present and Future. Semin Cancer Biol 2022, 83, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lee, J.Y.; Gao, L.; Yin, J.; Duan, Y.; Jimenez, L.A.; Adkins, G.B.; Ren, W.; Li, L.; Fang, J.; et al. A DNA Aptamer for Binding and Inhibition of DNA Methyltransferase 1. Nucleic Acids Res 2019. [Google Scholar] [CrossRef] [PubMed]
- Di Ruscio, A.; Ebralidze, A.K.; Benoukraf, T.; Amabile, G.; Goff, L.A.; Terragni, J.; Figueroa, M.E.; De Figueiredo Pontes, L.L.; Alberich-Jorda, M.; Zhang, P.; et al. DNMT1-Interacting RNAs Block Gene-Specific DNA Methylation. Nature 2013, 503, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Esposito, C.L.; Autiero, I.; Sandomenico, A.; Li, H.; Bassal, M.A.; Ibba, M.L.; Wang, D.; Rinaldi, L.; Ummarino, S.; Gaggi, G.; et al. Targeted Systematic Evolution of an RNA Platform Neutralizing DNMT1 Function and Controlling DNA Methylation. Nat Commun 2023, 14, 99. [Google Scholar] [CrossRef]
- Klett-Mingo, J.I.; Pinto-Díez, C.; Cambronero-Plaza, J.; Carrión-Marchante, R.; Barragán-Usero, M.; Pérez-Morgado, M.I.; Rodríguez-Martín, E.; Toledo-Lobo, M.V.; González, V.M.; Martín, M.E. Potential Therapeutic Use of Aptamers against HAT1 in Lung Cancer. Cancers (Basel) 2022, 15, 227. [Google Scholar] [CrossRef]
Figure 1.
Overview of aptamer-based delivery strategies. (a) Two-blocks strategy: the aptamer is elongated with the passenger strand of the therapeutic RNA, the guide is then annealed through base complementarity; (b) Three-blocks strategy: the passenger and the guide strands of the therapeutic RNA are paired up through an annealing reaction, further, “sticky-ends” were added to the 3’ extremity of both aptamer and passenger strand, and the two moieties were annealed; (c) Crosslinker strategy: the aptamer and the therapeutic RNA are synthetized with activable groups, which react with a crosslinker, allowing covalent conjugation.
Figure 1.
Overview of aptamer-based delivery strategies. (a) Two-blocks strategy: the aptamer is elongated with the passenger strand of the therapeutic RNA, the guide is then annealed through base complementarity; (b) Three-blocks strategy: the passenger and the guide strands of the therapeutic RNA are paired up through an annealing reaction, further, “sticky-ends” were added to the 3’ extremity of both aptamer and passenger strand, and the two moieties were annealed; (c) Crosslinker strategy: the aptamer and the therapeutic RNA are synthetized with activable groups, which react with a crosslinker, allowing covalent conjugation.
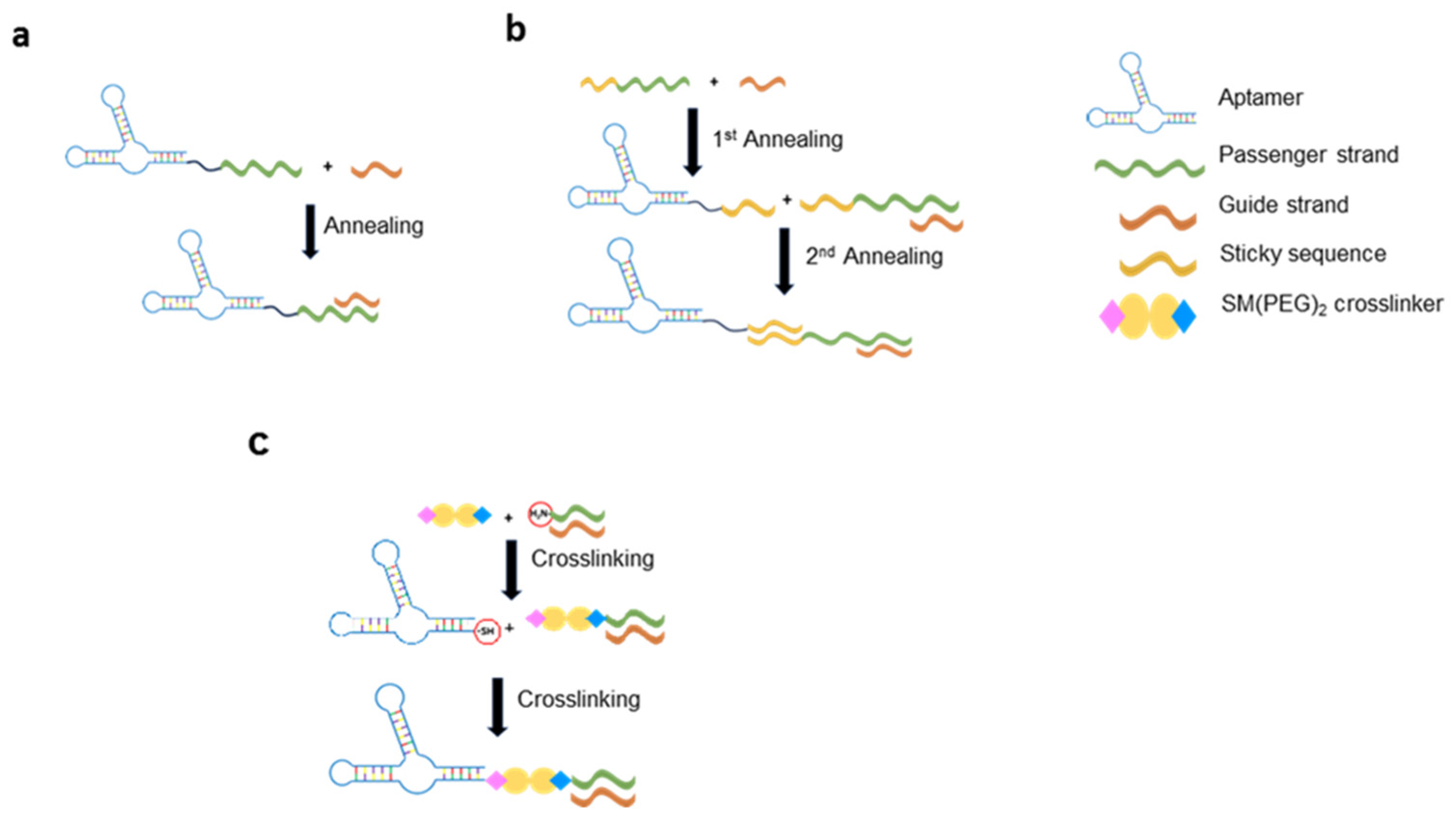
Figure 2.
Aptamer-coated viral vectors approaches for targeted delivery. (a) Adenovectors coated with aptamers, through the binding between biotinylated aptamers and the Biotin Acceptor Peptide (BAP) introduced in the capsid proteins of the virus, via a streptavidin bridge; (b) AAV covalently attached with a DNA dendrimer-aptamer complex through S-S bonding with capsid proteins; (c) AAV coated with aptamers thanks to the introduction of the azido-unnatural peptide in the capsid sequence which is able to react with dibenzocyclooctyne (DBCO) bound to the aptamers.
Figure 2.
Aptamer-coated viral vectors approaches for targeted delivery. (a) Adenovectors coated with aptamers, through the binding between biotinylated aptamers and the Biotin Acceptor Peptide (BAP) introduced in the capsid proteins of the virus, via a streptavidin bridge; (b) AAV covalently attached with a DNA dendrimer-aptamer complex through S-S bonding with capsid proteins; (c) AAV coated with aptamers thanks to the introduction of the azido-unnatural peptide in the capsid sequence which is able to react with dibenzocyclooctyne (DBCO) bound to the aptamers.
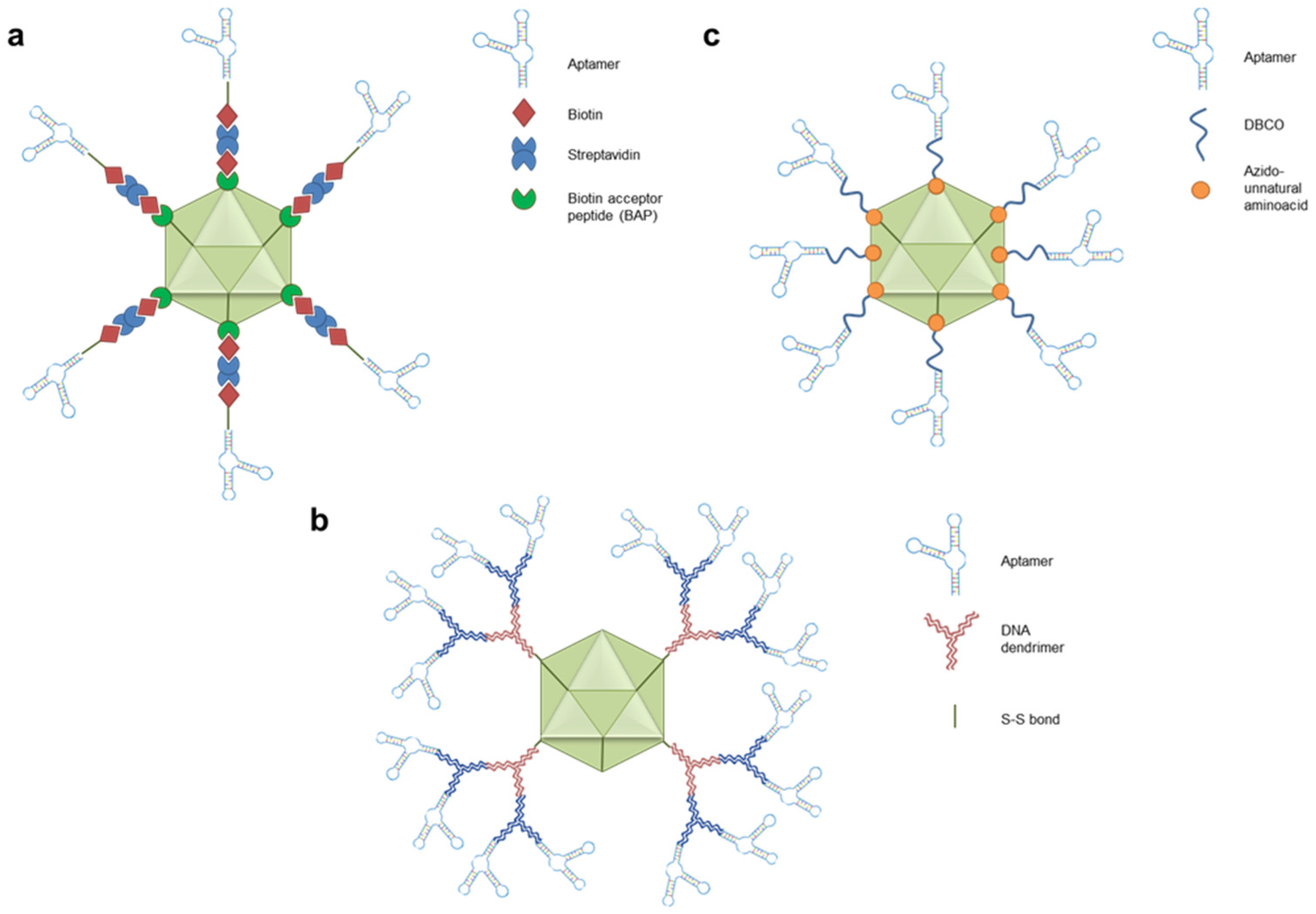
Figure 3.
Examples of AS1411-based functionalization of the most investigated NPs. (a) Cationic liposomes functionalized with AS1411 aptamer, loaded with paclitaxel (PTX) in its hydrophobic lipid component and the anti-PLK1 siRNA (siPLK1) on its hydrophilic surface; (b) Gold(Au)-based multitargeting nanoparticles simultaneously coated with AS1411 aptamer, a nuclear localization signal (NLS) peptide and anti-miR-221; (c) Copolymer-based polyplex constituted of self-assembled Poly(lactic acid)121 -b-Poly(N-3-aminopropyl)methacrylamide)103 (PLA121 -b-PAPMA103 ) in spherical micelles, electrostatically decorated with AS1411 aptamer and packed with plasmids encoding anti-survivin shRNAs; (d) Protein-based nanoparticles composed of a core containing a CDK11 silencing CRISPR/Cas9 expressing plasmid complexed with protamine sulfate, coated with AS1411-incorporated carboxymethyl chitosan (CMC) via electrostatic interactions, further functionalized with the positively charged KALA peptide for enhanced cellular internalization; (e) Chitosan-based nanoparticles with a chitosan core (CS) incorporating the entire CRISPR/Cas9 machinery plasmid encoding FOXM1, coated with hyaluronic acid (HA) and AS1411 aptamer for targeted delivery.
Figure 3.
Examples of AS1411-based functionalization of the most investigated NPs. (a) Cationic liposomes functionalized with AS1411 aptamer, loaded with paclitaxel (PTX) in its hydrophobic lipid component and the anti-PLK1 siRNA (siPLK1) on its hydrophilic surface; (b) Gold(Au)-based multitargeting nanoparticles simultaneously coated with AS1411 aptamer, a nuclear localization signal (NLS) peptide and anti-miR-221; (c) Copolymer-based polyplex constituted of self-assembled Poly(lactic acid)121 -b-Poly(N-3-aminopropyl)methacrylamide)103 (PLA121 -b-PAPMA103 ) in spherical micelles, electrostatically decorated with AS1411 aptamer and packed with plasmids encoding anti-survivin shRNAs; (d) Protein-based nanoparticles composed of a core containing a CDK11 silencing CRISPR/Cas9 expressing plasmid complexed with protamine sulfate, coated with AS1411-incorporated carboxymethyl chitosan (CMC) via electrostatic interactions, further functionalized with the positively charged KALA peptide for enhanced cellular internalization; (e) Chitosan-based nanoparticles with a chitosan core (CS) incorporating the entire CRISPR/Cas9 machinery plasmid encoding FOXM1, coated with hyaluronic acid (HA) and AS1411 aptamer for targeted delivery.
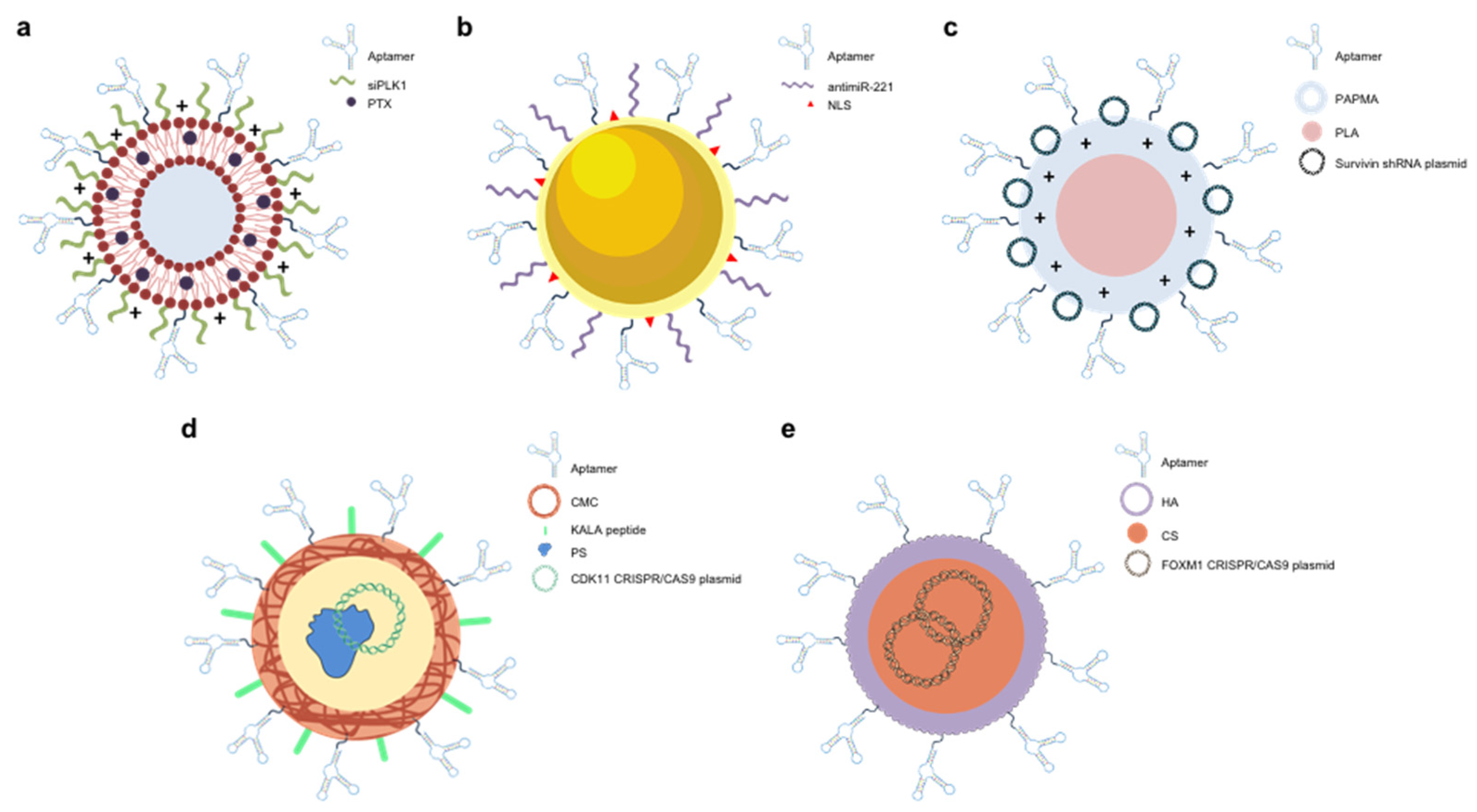
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



