
Submitted:
24 September 2024
Posted:
24 September 2024
You are already at the latest version
Abstract
Background: Slash pine (Pinus elliottii) is an important timber species in southern China, having been introduced from North America over a century ago. Recently, dozens of improved slash pine varieties have been authorized. However, the absence of efficient molecular markers hinders the identification of these improved varieties. In this study, we aimed to construct a set of DNA fingerprinting based on SNP markers for improved slash pine varieties and provide a technical procedure for their identification. Results: The genotypes of 29 improved slash pine varieties were captured using 51K liquid-phase probes developed by our team, resulting in the genotyping of 560,567 SNPs through next-generation sequencing. A total of 3502 SNPs were retained after screening for minor allele frequency, missing rate, heterozygosity rate, and other parameters. Subsequently, a random forest model was used to reduce the number of SNPs to 50. The identification rate of improved varieties with these 50 SNPs achieved 98.64%. We concluded that these SNPs effectively distinguish the improved varieties, which enabled the construction of DNA fingerprints for the improved slash pine varieties. Conclusions: This study provides a technical procedure for identifying improved slash pine varieties based on the Random Forest model and constructs a DNA fingerprinting of the 29 slash pine varieties using 50 SNPs.
Keywords:
DNA fingerprinting
; Improved variety identification
; liquid-phased probes
; Pinus elliottii
1. Introduction
Slash pine(Pinus elliottii) has been cultivated in China for over a hundred years since its introduction [1]. It is characterized by rapid growth, wide adaptability, and high oleoresin yield, making it a significant timber [1,2] and resin-producing [3] species in southern China. Improved varieties are crucial in forest tree genetics and breeding. The large planting area creates a high demand for seedlings of better types of slash pine. Recently, dozens of the improved Slash pine varieties(grafted clones) have been selected and authorized by the variety commission in China. Effective identification of improved varieties is key to tree improvement and breeding. Constructing a DNA fingerprint to highlight the molecular specificity of improved varieties is very necessary.
DNA fingerprinting is a technical tool that enables to efficiently identify plant and animal lineages [4]. Genotyping wild plant species and their cultivated relatives is now often accomplished through the use of plant DNA fingerprinting techniques. Molecular markers are crucial for constructing the DNA fingerprint. They are renowned for their high sensitivity and specificity, reproducibility, high -throughput capability, and co-dominant inheritance [5]. They allow the detection of genetic variation at the DNA sequence level, which makes it possible to identify single nucleotide polymorphisms(SNPs)[6]. DNA-based markers can be differentiated into two types [7], the first is non-PCR-based(RFLP)[8,9] and the second is PCR-based markers(RAPD, AFLP, SSR, SNP, etc.)[10,11,12,13]. Nowadays, using SNP-based probe arrays for genotyping enables the rapid acquisition of a large number of molecular markers. This is an ideal and efficient method that has been widely applied in plants [14]. SNPs offer several advantages, including their abundance, biallelic nature, relatively low mutation rate, even distribution across the genome, and relative ease of detection.[15,16,17]. SNP arrays are flexible because researchers can design specific chips based on their research needs and select particular genetic markers for analysis [14,18,19,20]. This technology of developing a SNP array has been widely applied in forestry, especially in coniferous tree species [18,21,22].
The paper of the development of 51 K liquid-phased probe array for loblolly and slash pines has been published in May 2024[23].It is designed based on the published SNPs and probes of loblolly and slash pines, novel slash pine-specific long-read transcript, and SNPs obtained from a double digest Restriction-Site Associated DNA sequencing(ddRADseq). Furthermore, targeted capture sequencing was performed using 51K liquid-phased probes on three pine species of loblolly, slash, and Caribbean pine. The probe capture had an efficiency of 68.26% on average. Principal component analysis(PCA), phylogenetic analysis, and genetic structure analysis of SNPs genotyped for the three pine species demonstrated the transferability of the SNP array for the three pines. The development of the 51K liquid-phase SNP array represents a significant breakthrough, enabling efficient and cost-effective genotyping of pine species. This technology is of great importance for identifying pine varieties, analyzing genetic diversity, and advancing germplasm research.
As another application for the 51K liquid-phased probes, this study conducted genotyping on slash pine samples, aiming to develop a technical process for identifying improved slash pine varieties. Additionally, the study sought to screen molecular markers that represent improved varieties and construct a DNA fingerprinting for them to efficiently manage the genetic resources(Supplementary Figure S1).
2. Materials and Methods
2.1. Materials and DNA Extraction
All materials were planted in Changle Forest Farm in Zhejiang Province, Hangzhou city.
We selected two set of slash pines(Figure 1): one set for constructing the DNA fingerprinting and the other set for validating the accuracy of the fingerprinting, which we also refer to as the validation population. The first set includes improved slash pine varieties, comprising 29 slash pine varieties officially recognized by the variety committee. These samples, each derived from distinct slash pine clones, were cultivated in a seed orchard. They are distinguished by their superior traits, such as fast growth speed, excellent timber characteristics, and higher turpentine yield. We utilized these 29 samples to develop a DNA fingerprinting for improved slash pine varieties. The second set of samples, referred to as the validation population, is designed to evaluate the accuracy of variety identification using the constructed DNA fingerprinting. For this validation population, we selected a progeny test forest, which includes 369 individuals. Notably, 77 of these individuals come from the 7 improved varieties family used in the first set, indicating that the validation population includes some of the known improved families(Supplementary Table 1,2).
Collect young tender needles from two sets of the slash pines for use as experimental samples. The needle was refrigerated in ice bags before DNA was extracted. M5 HiPer Universal DNA Mini Kit(Mei5 Biotechnology, Beijing) was used to extract the DNA from the sample needles. The genome target regions were captured by 51K liquid-phased probes designed by our lab for subsequently next-generating sequencing. The specific experimental steps are as follows: (1) The DNA was segmented and the end of the DNA fragment was repaired by Pre-PCR to amplify the library; (2) The probe hybridizes with the target region;(3) Hybridization probes were captured by magnetic beads using streptomycin affinity labeling;(4) The enriched target fragment was eluted and amplified by Post-PCR after capture; (5) Second generation sequencing was performed on the target region.
2.2. Data Quality Control and Genotyping
The captured DNA fragments were sequenced by Illumina Xten high-throughput Sequencing platform. The raw data were stored in FASTQ format. The adapters and low-quality data were filtered by FastQC software [24] to obtain clean reads. The steps of data filtering are as follows:(1) Remove reads with adapters;(2) Remove reads with an N content exceeding 10%;(3) Remove reads where more than 50% of bases have a quality score below 10. Subsequently, single nucleotide polymorphism(SNP) detection is implemented using the GATK software toolkit(https://gatk.broadinstitute.org/hc/en-us). According to the localization results of clean Reads in the reference genome, samtools was used to remove duplications(Mark Duplication), and GATK was used to perform local realignment and base recalibration. The reference genome we used is the reference genome of Loblolly pine and long-read transcripts specific to Slash pine. To ensure the accuracy of SNP detection, GATK was used to detect and filter single nucleotide polymorphisms, and the final SNP loci were obtained. The mutation results are displayed and saved in vcf files.
2.3. Core SNP Selection
The raw genotyped SNPs were screened in the following criteria: SNP loci in the Raw data were selected, and the screening criteria were as follows (Table 1): (1) biallelic sites;(2) minor allele frequency(MAF) > 0.3;(3) miss rate=0;(4) conform to the Hardy-Weinberg equilibrium; (5) linkage disequilibrium(LD) value > 0.2;(6) PIC > 0.35.(7) heterozygosity rate < 0.25[7,8,25,26,27].
2.4. Simplification of SNP Markers Based on Random Forest Model
To improve the efficiency of variety identification, it is crucial to optimize the core SNP loci. The goal is to maximize the number of identifiable individuals while minimizing the number of loci used. We employed the Random Forest algorithm to evaluate the contribution of each SNP to individual identification, using the Gini index to rank SNPs by their importance. The SNPs were ranked in descending order based on the Mean Decrease Gini index, which measures each SNP’s contribution to distinguishing individuals. We selected the 50 SNPs with the highest Gini indices for further validation.(Supplementary Table 3).
To assess the effectiveness of the selection and the genetic diversity of the candidate SNPs, we constructed phylogenetic trees for 29 improved slash pine using both the core SNPs and candidate SNPs. The phylogenetic tree was constructed using the neighbor-joining method and completed in MEGA software.
2.5. Phylogenetic Analysis and Genetic Diversity Analysis of Validation Population
The samples in the validation population underwent the same steps in sections 2.1 and 2.2 to acquire their genotype. The SNPs retained after quality control were initially filtered using the following criteria: minor allele frequency(MAF) < 0.01, missing rate > 0.8. The resulting SNPs were used for validation of population genetic structure and phylogenetic analysis. We calculate the genetic parament by the Shell script.
MEGA 11.0 software was used to construct a Neighbor-Joining phylogenetic tree. Principal component analysis(PCA) was conducted using PLINK software(version 1.07)[26] to assess population stratification and genetic relationships among the individuals. PCA was executed using the command `--noweb` in PLINK, which generates eigenvalues and eigenvectors representing the major axes of genetic variation. The top two principal components were retained for subsequent analysis. The R package ggplot2 is used for visualizing population genetic structure and PCA results.
2.6. Validation of Candidate SNPs Based on Random Forest Model and Construction of DNA Fingerprinting
To verify whether the 50 SNPs we selected have good identification accuracy of improved slash pine varieties, we trained a random forest model using the 50 SNPs from 29 superior varieties. Subsequently, we included the 50 SNP information from 370 samples in the validation population in the model for training. The model will use these 50 SNPs to predict whether the samples in the validation population are improved varieties. In fact, the samples in the validation population can be classified into two distinct categories: certified improved varieties and unimproved varieties (Supplementary Table 2). The predicted classification results from the model will be compared with the actual classifications. We used the “Random Forest” package in R for this analysis and assessed the model’s accuracy by examining the confusion matrix and error rate. Finally, we achieved satisfactory validation results, and we constructed the DNA fingerprinting of improved slash pine varieties using candidate SNPs(Supplementary Table 3).The Materials and Methods should be described with sufficient details to allow others to replicate and build on the published results. Please note that the publication of your manuscript implicates that you must make all materials, data, computer code, and protocols associated with the publication available to readers. Please disclose at the submission stage any restrictions on the availability of materials or information. New methods and protocols should be described in detail while well-established methods can be briefly described and appropriately cited.
3. Results
3.1. Screening and Simplification of Core SNPs
A total of 5,60,567 SNP loci were genotyped for 29 improved varieties of slash pine by target sequencing of 51 K liquid-phased probes. The genotyping rate was 94.61%. A total of 183,849 SNPs with a minor allele frequency(MAF) < 0.01 and a miss rate > 0.8 were retained as the primary dataset for this experiment.
Based on the MAF, miss rate, linked disequilibrium(LD) value, heterozygosity, polymorphism information content(PIC), and other indicators of SNPs, a rigorous screening process (Table 1) was formulated to screen high-quality core SNPs. A total of 3502 SNPs were finally screened. The MAF of 3502 SNPs ranged from 0.241 to 0.276 with an average of 0.258(Figure 2a). The PIC values ranged from 0.299 to 0.320, with an average value of 0.3093(Figure 2b). The heterozygosity values ranged from 0.2992 to 0.3197, with an average value of 0.3093 values ranged from 0.366 to 0.399, with an average value of 0.382(Figure 2c). The results suggest that the 3502 core SNPs have a high degree of polymorphism and are ideal for DNA fingerprinting of improved varieties of slash pine.
We constructed neighbor-joining phylogenetic trees for 29 improved varieties of slash pine using 50 candidate SNPs and 3502 core SNPs, respectively. The results showed that when we calculated phylogenetic trees using 50 SNPs and 3502 SNPs, respectively, the genetic clustering of the 29 improved varieties was largely consistent with the original clustering, with only a few individuals showing different classifications.
3.2. Population Structure of Validation Population
We selected a progeny test population as the validation cohort, consisting of 371 samples. We performed a preliminary analysis of its genetic structure and diversity, dividing it into three subpopulations(Figure 2a,b). The fixation index(Fst) and gene flow index(Nm) indicate that there is only moderate genetic differentiation and low gene flow among the three subpopulations(Table 2). Compared to the first set of samples, this population exhibits lower genetic diversity. The average MAF value of all SNP markers in the validation population is 0.129(Figure 3c), the average PIC value is 0.156(Figure 3d), and the average Het value is 0.187(Figure 3e). These genetic parameters suggest that the population has relatively low genetic diversity. Due to the small genetic differences among subpopulations, this population is more suitable for testing the accuracy of the 50 SNPs in identifying superior varieties.
3.3. Effectiveness of Identifying Improved Varieties with Candidate SNPs
We selected a progeny test population as the validation cohort, consisting of 369 samples. We performed a preliminary analysis of its genetic structure and diversity, dividing it into three subpopulations(Figure 2a,b). The fixation index(Fst) and gene flow index(Nm) indicate that there is only moderate genetic differentiation and low gene flow among the three subpopulations(Table 2). Compared to the first set of samples, this population exhibits lower genetic diversity. The average MAF value of all SNP markers in the validation population is 0.129(Figure 3c), the average PIC value is 0.156(Figure 3d), and the average Het value is 0.187(Figure 3e). These genetic parameters suggest that the population has relatively low genetic diversity. Due to the small genetic differences among subpopulations, this population is more suitable for testing the accuracy of the 50 SNPs in identifying superior varieties.
3.4. Population Structure of Validation Population
To validate whether the 50 SNPs we selected have good identification accuracy, we trained a random forest model using the 50 SNPs from 29 improved varieties. Subsequently, we included the 50 SNP from the validation population in the model for training.The random forest algorithm generated a confusion matrix to display the prediction results(Figure 4a). The results showed that only 5 individuals in the validation population were incorrectly predicted. Sample numbers 117-3, 121-5 and 126-2 which come from improved families and are authorized improved varieties, were predicted as unimproved individuals in our model. Sample numbers 154-1 and 16-3 which were authorized unimproved varieties, were predicted as improved individuals(Supplementary Table 3). The error rate plot(Figure 4b) demonstrates the performance of the Random Forest model as the number of trees increases. The overall OOB error rate (black line) decreases initially and stabilizes as more trees are added, indicating improved model accuracy. The ‘Unimproved variety’ class(green line), shows a lower and more stable error rate compared to the ‘Improved variety’ class(red line). This result indicates that our model performs more consistently when detecting unimproved individuals.
The feature importance plot(Figure 4c) reveals that the top contributing SNPs in classification. SNPs are ranked by their mean decrease in Gini index, with higher values indicating greater importance in reducing classification error. As results, the validation accuracy of our selected candidate SNPs in the validation population reached 98.64% and we identified several SNP markers that made significant contributions to the classification of the samples(Figure 4c).
The result of PCA(Figure 5) illustrates the clustering of all samples based on 50 SNPs, highlighting a clear distinction between improved and unimproved varieties. While most individuals can be successfully distinguished, a small subset of individuals from the validation population were difficult to distinguish, which represented by red triangles (improved varieties) and blue circles (unimproved varieties). The clustering for unimproved varieties is particularly effective, which aligns with the lowest error rate observed for unimproved individuals in the Random Forest model(Figure 4b). These results suggest that while the Random Forest-based approach is useful for identifying improved varieties, it lacks the precision needed for accurate classification within the improved variety group.
Ultimately, we constructed DNA fingerprinting using these 50 SNPs for the 29 recognized improved varieties(Figure 6) and 77 improved individuals in the validation population(Supplementary Figure S3).
4. Discussion
4.1. An Application of the 51K Liquid-Phased Probes for Slash Pine
Slash pine has been widely cultivated in southern China, and has become an important species for afforestation since its introduction [30,31]. Fingerprinting provides core resources for the selection and application of marker-assisted breeding, thereby accelerating the breeding process [32]. The lack of complete genome information on slash pine [9,33,34] prompted us to develop a 51K liquid phase probe for rapid genotyping of individuals. This probe has been successfully applied in the breeding programs of slash, loblolly, and Caribbean pines [23]. In this study, we selected 29 improved varieties of slash pines to construct a DNA fingerprint. A total of 560,567 SNP loci were genotyped for these 29 improved varieties using target sequencing of the probe, achieving a genotyping rate of 94.61%. Ultimately, we constructed a DNA fingerprint for improved slash pine varieties with the candidate SNPs. However, due to the absence of a complete genome, we could only map the SNPs to scaffolds rather than directly to chromosomes. Although these candidate SNPs are already distributed across different scaffolds or genes, we cannot ascertain their even distribution across the 12 chromosomes of pine. This limitation may affect the precision of SNP selection and the accuracy of DNA fingerprinting predictions.
4.2. The Construction Methods of DNA Fingerprinting for the Improved Varieties of Slash Pines
Molecular markers are crucial for constructing DNA fingerprinting profiles. SSR markers are currently the most widely used molecular markers [35]. Numerous studies have examined the genetic diversity of forest tree populations and constructed fingerprint databases for improved varieties using SSR markers, such as Populus spp.[36], Xanthoceras sorbifolia [37], Sophora japonica [38] and Phellodendron amurense [39]. However, SSR markers are disadvantaged by their time-consuming nature and limited quantity, rendering them unsuitable for large-genome species such as pine. In recent years, the advancement of sequencing technology has facilitated the widespread use of SNP-based DNA fingerprinting for genotype identification and population genetic diversity analysis in crops and horticultural plants [4,40,41]. In forestry, economic crops like oil tea(Camellia oleifera)[42] and tea(Camellia sinensis)[43] have utilized SNP markers to construct DNA fingerprints, although their application remains limited.
In research related to agricultural crops and forestry, the random forest algorithm is widely utilized for plant classification [44,45] and variety identificatio [46,47,48,49]. Furthermore, when integrated with remote sensing imagery, this algorithm proves valuable for detecting plant diseases [45] and predicting yields [50,51]. The random forest algorithm holds considerable potential, offering the ability to enhance the efficiency and accuracy of target identification.
Slash pine has been planted in China for nearly a century, but the management of elite variety resources is still lacking. This study establishes a DNA fingerprinting and technical procedure for identifying improved slash pine varieties, which helps protect the intellectual property of improved varieties. Initially, we screened 3502 core SNPs representing 29 improved varieties by controlling for SNP polymorphism parameters. Subsequently, employing a random forest algorithm, we reduced the SNP count to 50 and validated their identification rate in a mixed population of improved and unimproved varieties, achieving a validation rate of 98.64%.We obtained results comparable to the previously calculated average Het, MAF, and PIC using the 51 K liquid-phase probes, indicating that the 50 candidate SNPs selected in this study are informative and highly representative. As more improved varieties receive national approval and authorization, we will continually expand the sample size in the fingerprint database to build a more accurate and efficient identification platform for improved slash pine varieties. Additionally, further optimization of model parameters and feature engineering will be essential to enhance identification accuracy.
5. Conclusions
In this study, we employed a 51K liquid-phase probe to detect SNPs in 29 improved slash pine varieties. Through a rigorous filtering process and random forest algorithm, we identified 50 SNPs for constructing a DNA fingerprint map. These SNPs were validated in a population consist of improved varieties and unimproved varieties, achieving an impressive 98.64% accuracy in variety identification. Evaluations of heterozygosity, polymorphism information content, and minor allele frequency using these 50 SNPs further confirmed their reliability. Our research has established a precise and reliable DNA fingerprint, poised to significantly enhance the rapid screening of superior slash pine germplasm in future breeding programs.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: The technical route of this study; Table S1: Basic information on improved varieties of 29 slash pines; Table S2: Basic Imformation of Samples in Validation Population; Table S3: Basic Imforation of 50 SNPs; Table S4: Random Forest Predicted vs. Actual Classification Results for Each Sample; Figure S2: DNA fingerprinting of 78 individuals in validation population improved slash pine varieties.
Author Contributions
L.Q.: Conceptualization, Supervision, Project administration, Funding acquisition W.Y.: Investigation, Resources, Data Curation, Methodology, Software, Validation, Formal analysis, Writing - Original Draft, Visualization D.S.: Writing - Review & Editing D. X.: Investigation, Resources, Methodology H.Q.: Investigation, Resources. All authors commented on the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Biological Breeding-Major Projects (Grant No. 2023ZD0405902 and 2023ZD04058)and the National Key Research and Development Program of the Ministry of Science and Technology of the People’s Republic of China(2022YFD2200023-2).
Data Availability Statement
The data have been requested for a Bioproject in the NCBI database(National Center for Biotechnology Information(nih.gov)), and specific information on the biological samples has been submitted with the accession number PRJNA1073171. BioSample submission: SUB14206957. The datasets presented in this study can be found in online repositories. The direct web link can be found below: https://zenodo.org/records/10615722.
Acknowledgments
We thank the Changle Forest Farm and the China Shengshidai(Beijing) Biotech Co., Ltd. for their technical assistance in DNA capture sequencing based on 51K liquid-phased probes.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| SNP: | single nucleotide polymorphism; |
| SSR: | simple sequence repeats; |
| AFLP: | amplified fragment length polymorphism; |
| RAPD: | random amplified polymorphic DNA; |
| RFLP: | restriction fragment length polymorphism; |
| MAS: | molecular marker-assisted selection; |
| MAF: | minor allele frequency; |
| PIC: | polymorphism information content; |
| LD: | linked disequilibrium; |
| hdw: | Hardy-Weinberg; |
| Het: | The expected heterozygosity |
References
- Xianyin, D.; Xueyu, T. a. O.; Shu, D.; Qifu, L.; Jingmin, J. Estimation of Wood Basic Density in a Pinus Elliottii Stand Using Pilodyn and Resistograph Measurements. JOURNAL OF NANJING FORESTRY UNIVERSITY 2020, 44, 142. [Google Scholar] [CrossRef]
- Zhao, D.; Bullock, B.P.; Montes, C.R.; Wang, M.; Greene, D.; Sutter, L. Loblolly Pine Outperforms Slash Pine in the Southeastern United States – A Long-Term Experimental Comparison Study. Forest Ecology and Management 2019, 450, 117532. [Google Scholar] [CrossRef]
- Ding, X.; Li, Y.; Zhang, Y.; Diao, S.; Luan, Q.; Jiang, J. Genetic Analysis and Elite Tree Selection of the Main Resin Components of Slash Pine. Front. Plant Sci. 2023, 14. [Google Scholar] [CrossRef]
- Nybom, H.; Weising, K.; Rotter, B. DNA Fingerprinting in Botany: Past, Present, Future. Investig Genet 2014, 5, 1. [Google Scholar] [CrossRef]
- Wang, X.-R.; Szmidt, A.E. Molecular Markers in Population Genetics of Forest Trees. Scandinavian Journal of Forest Research 2001, 16, 199–220. [Google Scholar] [CrossRef]
- Nadeem, M.A.; Nawaz, M.A.; Shahid, M.Q.; Doğan, Y.; Comertpay, G.; Yıldız, M.; Hatipoğlu, R.; Ahmad, F.; Alsaleh, A.; Labhane, N.; et al. DNA Molecular Markers in Plant Breeding: Current Status and Recent Advancements in Genomic Selection and Genome Editing. Biotechnology & Biotechnological Equipment 2018, 32, 261–285. [Google Scholar] [CrossRef]
- Kumar, P.; Gupta, V.K.; Misra, A.K.; Modi, D.R.; Pandey, B.K. Potential of Molecular Markers in Plant Biotechnology. Plant Omics 2020, 2, 141–162. [Google Scholar] [CrossRef]
- Tsumura, Y.; Yoshimura, K.; Tomaru, N.; Ohba, K. Molecular Phytogeny of Conifers Using RFLP Analysis of PCR-Amplified Specific Chloroplast Genes. Theoret. Appl. Genetics 1995, 91, 1222–1236. [Google Scholar] [CrossRef] [PubMed]
- De La Torre, A.R.; Birol, I.; Bousquet, J.; Ingvarsson, P.K.; Jansson, S.; Jones, S.J.M.; Keeling, C.I.; MacKay, J.; Nilsson, O.; Ritland, K.; et al. Insights into Conifer Giga-Genomes. Plant Physiology 2014, 166, 1724–1732. [Google Scholar] [CrossRef]
- Nesbitt, K.A.; Potts, B.M.; Vaillancourt, R.E.; West, A.K.; Reid, J.B. Partitioning and Distribution of RAPD Variation in a Forest Tree Species, Eucalyptus Globulus (Myrtaceae). Heredity 1995, 74, 628–637. [Google Scholar] [CrossRef]
- DeVerno, L.L.; Mosseler, A. Genetic Variation in Red Pine (Pinus Resinosa) Revealed by RAPD and RAPD-RFLP Analysis. Can. J. For. Res. 1997, 27, 1316–1320. [Google Scholar] [CrossRef]
- Rawat, A.; Barthwal, S.; Ginwal, H.S. Comparative Assessment of SSR, ISSR and AFLP Markers for Characterization of Selected Genotypes of Himalayan Chir Pine (Pinus Roxburghii Sarg.) Based on Resin Yield. Silvae Genetica 2014, 63, 94–108. [Google Scholar] [CrossRef]
- Ganea, S.; Ranade, S.S.; Hall, D.; Abrahamsson, S.; García-Gil, M.R. Development and Transferability of Two Multiplexes nSSR in Scots Pine (Pinus Sylvestris L.). J. For. Res. 2015, 26, 361–368. [Google Scholar] [CrossRef]
- Guo, Z.; Yang, Q.; Huang, F.; Zheng, H.; Sang, Z.; Xu, Y.; Zhang, C.; Wu, K.; Tao, J.; Prasanna, B.M.; et al. Development of High-Resolution Multiple-SNP Arrays for Genetic Analyses and Molecular Breeding through Genotyping by Target Sequencing and Liquid Chip. Plant Communications 2021, 2, 100230. [Google Scholar] [CrossRef] [PubMed]
- Batley, J.; Edwards, D. SNP Applications in Plants. In Association Mapping in Plants; Oraguzie, N.C., Rikkerink, E.H.A., Gardiner, S.E., De Silva, H.N., Eds.; Springer New York: New York, NY, 2007; pp. 95–102. ISBN 978-0-387-35844-4. [Google Scholar]
- Mammadov, J.; Aggarwal, R.; Buyyarapu, R.; Kumpatla, S. SNP Markers and Their Impact on Plant Breeding. International Journal of Plant Genomics 2012, 2012, 728398. [Google Scholar] [CrossRef]
- Jehan, T.; Lakhanpaul, S. Single Nucleotide Polymorphism (SNP)–Methods and Applications in Plant Genetics: A Review. IJBT Vol.5(4) [October 2006] 2006.
- Pavy, N.; Gagnon, F.; Rigault, P.; Blais, S.; Deschênes, A.; Boyle, B.; Pelgas, B.; Deslauriers, M.; Clément, S.; Lavigne, P.; et al. Development of High-Density SNP Genotyping Arrays for White Spruce (Picea Glauca) and Transferability to Subtropical and Nordic Congeners. Molecular Ecology Resources 2013, 13, 324–336. [Google Scholar] [CrossRef]
- Ganal, M.W.; Wieseke, R.; Luerssen, H.; Durstewitz, G.; Graner, E.-M.; Plieske, J.; Polley, A. High-Throughput SNP Profiling of Genetic Resources in Crop Plants Using Genotyping Arrays. In Genomics of Plant Genetic Resources: Volume 1. Managing, sequencing and mining genetic resources; Tuberosa, R., Graner, A., Frison, E., Eds.; Springer Netherlands: Dordrecht, 2014; pp. 113–130. ISBN 978-94-007-7572-5. [Google Scholar]
- Montanari, S.; Deng, C.; Koot, E.; Bassil, N.V.; Zurn, J.D.; Morrison-Whittle, P.; Worthington, M.L.; Aryal, R.; Ashrafi, H.; Pradelles, J.; et al. A Multiplexed Plant–Animal SNP Array for Selective Breeding and Species Conservation Applications. G3 Genes|Genomes|Genetics 2023, 13, jkad170. [Google Scholar] [CrossRef]
- Jackson, C.; Christie, N.; Reynolds, S.M.; Marais, G.C.; Tii-kuzu, Y.; Caballero, M.; Kampman, T.; Visser, E.A.; Naidoo, S.; Kain, D.; et al. A Genome-Wide SNP Genotyping Resource for Tropical Pine Tree Species. Molecular Ecology Resources 2022, 22, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Graham, N.; Telfer, E.; Frickey, T.; Slavov, G.; Ismael, A.; Klápště, J.; Dungey, H. Development and Validation of a 36K SNP Array for Radiata Pine (Pinus Radiata D.Don). Forests 2022, 13, 176. [Google Scholar] [CrossRef]
- Diao, S.; Ding, X.; Luan, Q.; Chen, Z.-Q.; Wu, H.X.; Li, X.; Zhang, Y.; Sun, J.; Wu, Y.; Zou, L.-H.; et al. Development of 51 K Liquid-Phased Probe Array for Loblolly and Slash Pines and Its Application to GWAS of Slash Pine Breeding Population. Industrial Crops and Products 2024, 216, 118777. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A Tool for Multi-Genome Mapping and Quality Control. F1000Res 2018, 7, 1338. [Google Scholar] [CrossRef]
- Carvalho, A.; Matos, M.; Lima-Brito, J.; Guedes-Pinto, H.; Benito, C. DNA Fingerprint of F1 Interspecific Hybrids from the Triticeae Tribe Using ISSRs. Euphytica 2005, 143, 93–99. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. The American Journal of Human Genetics 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Boban, S.; Maurya, S.; Jha, Z. DNA Fingerprinting: An Overview on Genetic Diversity Studies in the Botanical Taxa of Indian Bamboo. Genet Resour Crop Evol 2022, 69, 469–498. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luan, Q.; Jiang, J.; Li, Y. Prediction and Utilization of Malondialdehyde in Exotic Pine Under Drought Stress Using Near-Infrared Spectroscopy. Front. Plant Sci. 2021, 12. [Google Scholar] [CrossRef]
- Lai, M.; Dong, L.; Su, R.; Zhang, L.; Jia, T.; Chen, T.; Yi, M. Needle Functional Features in Contrasting Yield Phenotypes of Slash Pine at Three Locations in Southern China. Industrial Crops and Products 2023, 206, 117613. [Google Scholar] [CrossRef]
- Jiang, G.-L. Molecular Marker-Assisted Breeding: A Plant Breeder’s Review. In Advances in Plant Breeding Strategies: Breeding, Biotechnology and Molecular Tools; Al-Khayri, J.M., Jain, S.M., Johnson, D.V., Eds.; Springer International Publishing: Cham, 2015; pp. 431–472. ISBN 978-3-319-22521-0. [Google Scholar]
- Prunier, J.; Verta, J.-P.; MacKay, J.J. Conifer Genomics and Adaptation: At the Crossroads of Genetic Diversity and Genome Function. New Phytologist 2016, 209, 44–62. [Google Scholar] [CrossRef]
- Rodríguez, S.M.; Ordás, R.J.; Alvarez, J.M. Conifer Biotechnology: An Overview. Forests 2022, 13, 1061. [Google Scholar] [CrossRef]
- Singh, N.; Choudhury, D.R.; Singh, A.K.; Kumar, S.; Srinivasan, K.; Tyagi, R.K.; Singh, N.K.; Singh, R. Comparison of SSR and SNP Markers in Estimation of Genetic Diversity and Population Structure of Indian Rice Varieties. PLOS ONE 2013, 8, e84136. [Google Scholar] [CrossRef]
- Liu, C. SSR Fingerprinting of Poplar Clones and Construction of Core Germplasm Bank. Master’s Thesis, Central South University of forestry and technology, 2022.
- Shen, Z. Evaluation and Selection of Xanthoceras Sorbifolia Germplasm Resources Based on Morphological Characteristics and SSR Molecular Markers. PhD Thesis, Beijing Forestry University, 2017.
- Lu, Y. Genetic Diversity Evaluation and Molecular Identification of Clones of Sophora Japonica. PhD Thesis, Chinese Academy of Forestry, 2019.
- Zhang, L. Genetic Variation Analysis of the plus Tree Population of Pinus Lumenensis and Construction of Core Germplasm. PhD Thesis, Beijing Forestry University, 2020.
- Wang, Y.; Lv, H.; Xiang, X.; Yang, A.; Feng, Q.; Dai, P.; Li, Y.; Jiang, X.; Liu, G.; Zhang, X. Construction of a SNP Fingerprinting Database and Population Genetic Analysis of Cigar Tobacco Germplasm Resources in China. Front. Plant Sci. 2021, 12. [Google Scholar] [CrossRef]
- Yang, Y.; Lyu, M.; Liu, J.; Wu, J.; Wang, Q.; Xie, T.; Li, H.; Chen, R.; Sun, D.; Yang, Y.; et al. Construction of an SNP Fingerprinting Database and Population Genetic Analysis of 329 Cauliflower Cultivars. BMC Plant Biol 2022, 22, 522. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Wang, K.; Yao, X.; Ren, H. Construction of Molecular ID Cards for Main Camellia Oleifera Germplasm Resources Based on Transcriptome SNPs. China Agricultural Science 2023, 56, 217–235. [Google Scholar]
- Fan, X.; Yu, W.; Cai, C.; Lin, Y.; Wang, Z.; Fang, W.; Zhang, J.; Ye, N. Construction of Molecular ID Card for Tea Variety Resources Using SNP Markers. China Agricultural Science 2021, 54, 1751–1772. [Google Scholar]
- Zhang, L.; Liu, Z.; Ren, T.; Liu, D.; Ma, Z.; Tong, L.; Zhang, C.; Zhou, T.; Zhang, X.; Li, S. Identification of Seed Maize Fields With High Spatial Resolution and Multiple Spectral Remote Sensing Using Random Forest Classifier. Remote Sensing 2020, 12, 362. [Google Scholar] [CrossRef]
- Pankaja, K.; Suma, V. Plant Leaf Recognition and Classification Based on the Whale Optimization Algorithm (WOA) and Random Forest (RF). J. Inst. Eng. India Ser. B 2020, 101, 597–607. [Google Scholar] [CrossRef]
- Qiu; Shen, B.; Li, T.; Guo, J.; Wang, J.; Sun L.; Chen X.; Hu S. Fragrant Osmanthus Varieties Identification Method Based on Random Forest Algorithm and SRAP Molecular Markers. Forestry Science 2018, 54, 32–45.
- Zhang, M.; He, Z.; Ma, J.; Sang, Z.; Zhu, Z.; Zhang, D.; Ma, L.; Chen, F. Genetic Relationship Analysis and Molecular Identification of Magnolia japonica Varieties Based on SSR and SRAP Markers. Journal of Beijing Forestry University 2019, 41, 69–80. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang Establishment of Alfalfa Forage Variety Database Based on Terahertz Technology and Discussion on Color Mechanism. Master Thesis, 10.27643/d.cnki.gsybu.2021.001585, 2021.
- Guo, M.; Gu, W.; Xie, X.; Mao, Y. ; Chen; Xiong Variety Identification Method Based on Single Nucleotide Polymorphism Screening. Computer Application 2024, 44, 369–373. [Google Scholar]
- Everingham, Y.; Sexton, J.; Skocaj, D.; Inman-Bamber, G. Accurate Prediction of Sugarcane Yield Using a Random Forest Algorithm. Agron. Sustain. Dev. 2016, 36, 27. [Google Scholar] [CrossRef]
- Prasad, N.R.; Patel, N.R.; Danodia, A. Crop Yield Prediction in Cotton for Regional Level Using Random Forest Approach. Spat. Inf. Res. 2021, 29, 195–206. [Google Scholar] [CrossRef]
Figure 1.
The relationship of two set of slash pine. There are 33 families in validation population, and 7 of them were the progeny of 7 improved varieties which included in the 29 improved varieties.
Figure 1.
The relationship of two set of slash pine. There are 33 families in validation population, and 7 of them were the progeny of 7 improved varieties which included in the 29 improved varieties.
Figure 2.
(a-c) Genetic parameter information of 3502 SNPs including MAF, PIC, and heterozygosity rate, respectively. (d-e) Phylogenetic tree of 29 improved varieties of slash pine constructed using 3502 SNPs and 50 SNPs, respectively.
Figure 2.
(a-c) Genetic parameter information of 3502 SNPs including MAF, PIC, and heterozygosity rate, respectively. (d-e) Phylogenetic tree of 29 improved varieties of slash pine constructed using 3502 SNPs and 50 SNPs, respectively.
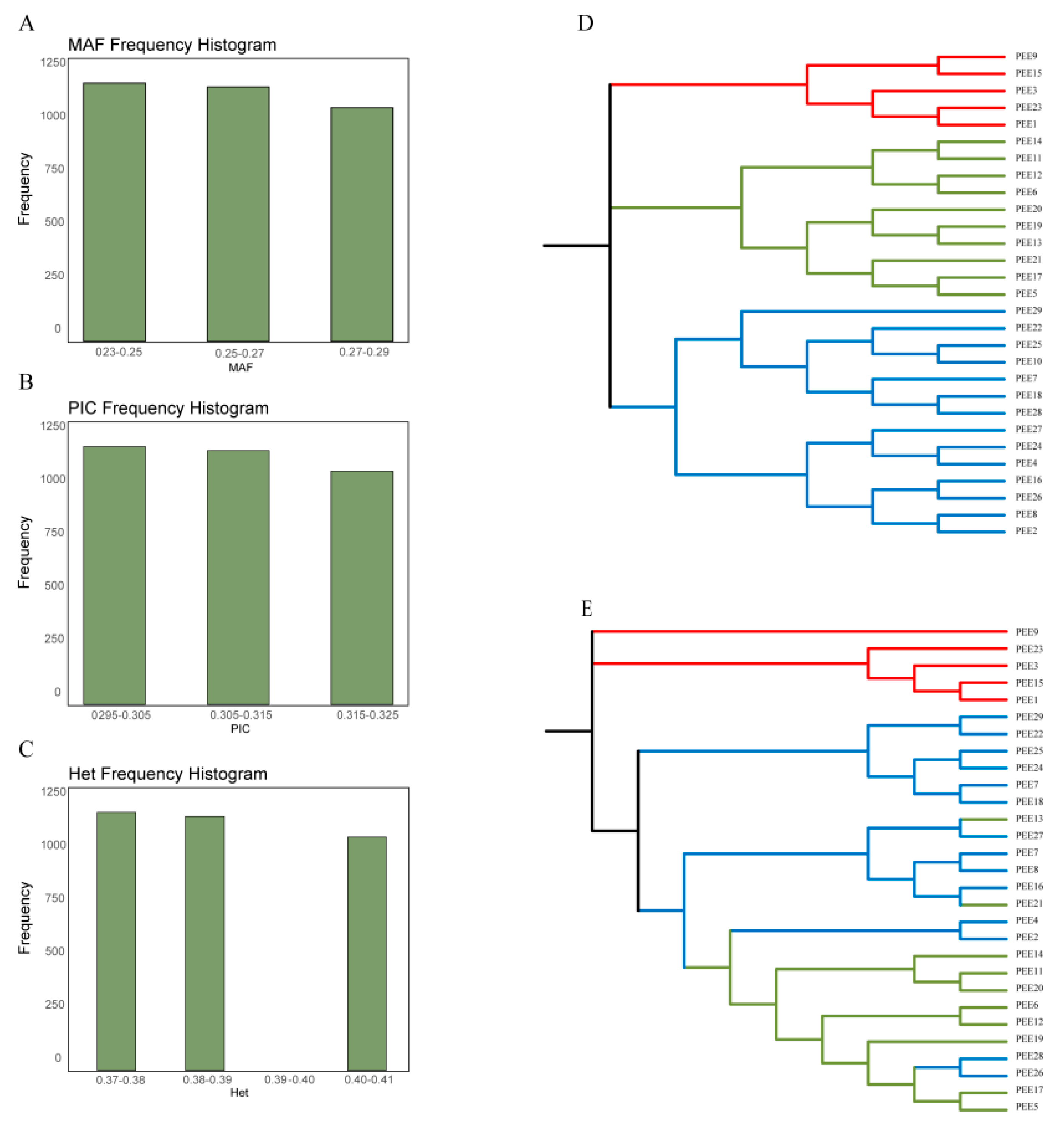
Figure 3.
Phylogenetic and population genetic analyses of validation population. (a) The phylogenetic tree of samples in validation population. (b) Principal component analysis, the colors of the circles were consistent with the phylogenetic tree. (c-e) Histogram of genetic diversity parameters of the validation population.
Figure 3.
Phylogenetic and population genetic analyses of validation population. (a) The phylogenetic tree of samples in validation population. (b) Principal component analysis, the colors of the circles were consistent with the phylogenetic tree. (c-e) Histogram of genetic diversity parameters of the validation population.
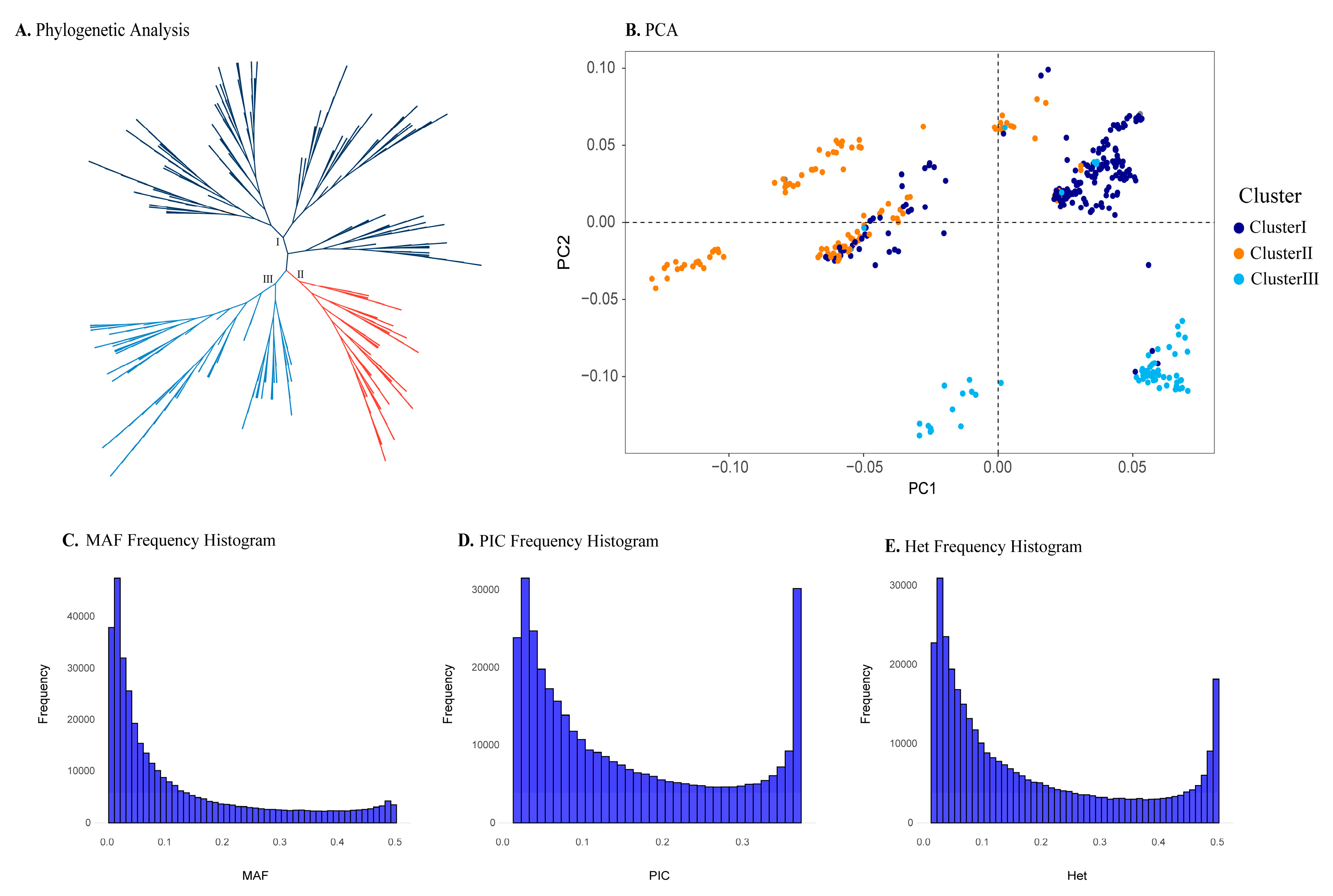
Figure 4.
Performance evaluation of the Random Forest model. (a) Confusion Matrix Heatmap: Displays the classification results of the Random Forest model, with predicted versus actual labels for “Improved variety” and “Unimproved variety” classes. (b) Random Forest Error Rates: Shows the out-of-bag (OOB) error rates across different numbers of trees in the Random Forest, with the black line representing the overall error, the green line showing the error for “Unimproved variety”, and the red line showing the error for “Improved variety”. (c) Feature Importance: A bar plot illustrating the importance of different features in the Random Forest model, based on the mean decrease in Gini index, indicating their contribution to the accuracy of the model’s predictions.
Figure 4.
Performance evaluation of the Random Forest model. (a) Confusion Matrix Heatmap: Displays the classification results of the Random Forest model, with predicted versus actual labels for “Improved variety” and “Unimproved variety” classes. (b) Random Forest Error Rates: Shows the out-of-bag (OOB) error rates across different numbers of trees in the Random Forest, with the black line representing the overall error, the green line showing the error for “Unimproved variety”, and the red line showing the error for “Improved variety”. (c) Feature Importance: A bar plot illustrating the importance of different features in the Random Forest model, based on the mean decrease in Gini index, indicating their contribution to the accuracy of the model’s predictions.
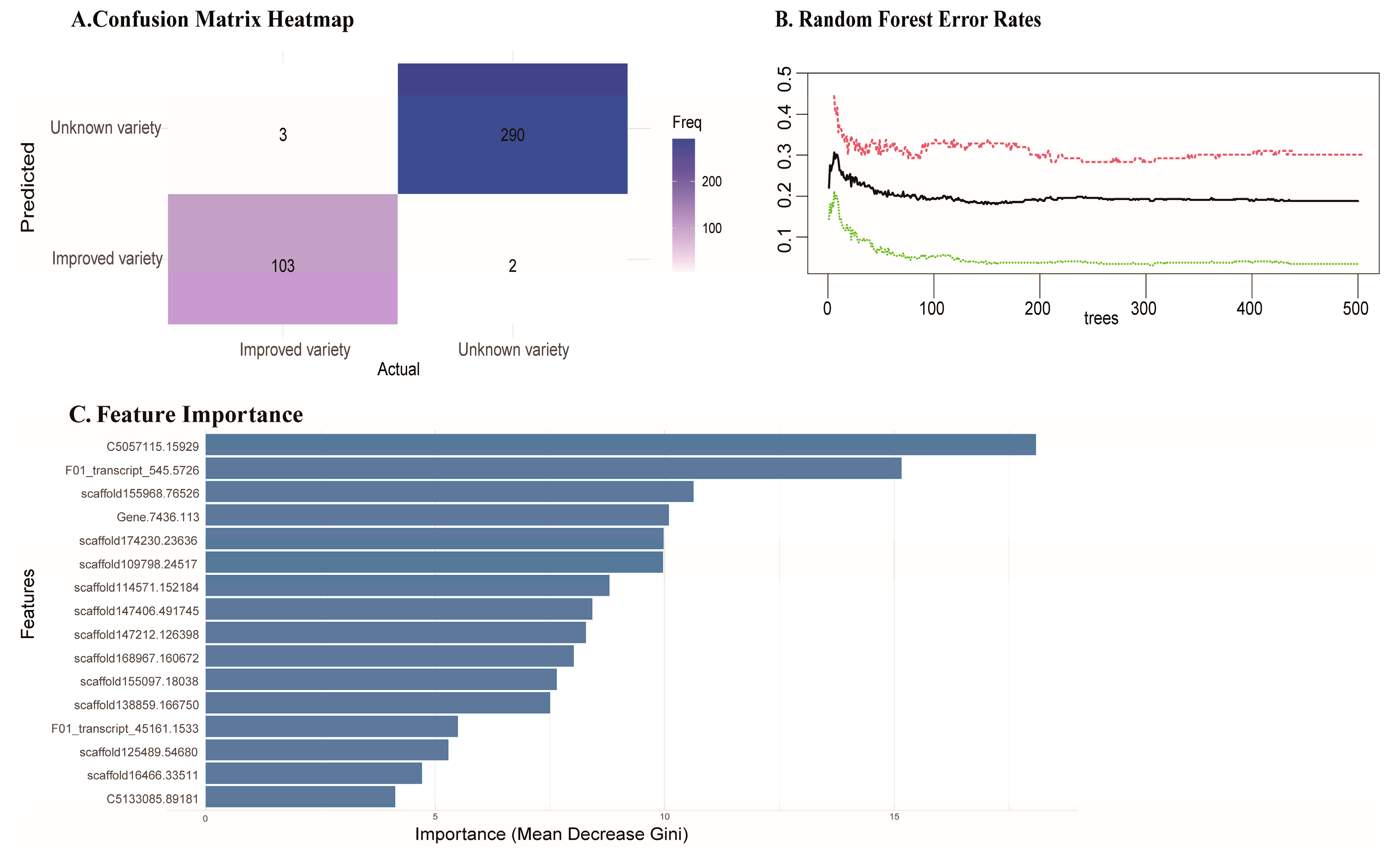
Figure 5.
Principal component analysis of 29 improved slash pine varieties and the 369 individuals in the validation population. The orange triangle, the red triangle, and the blue circle represent 29 improved slash pine varieties, improved slash pine varieties in validation population, and unimproved varieties in validation population, respectively.
Figure 5.
Principal component analysis of 29 improved slash pine varieties and the 369 individuals in the validation population. The orange triangle, the red triangle, and the blue circle represent 29 improved slash pine varieties, improved slash pine varieties in validation population, and unimproved varieties in validation population, respectively.
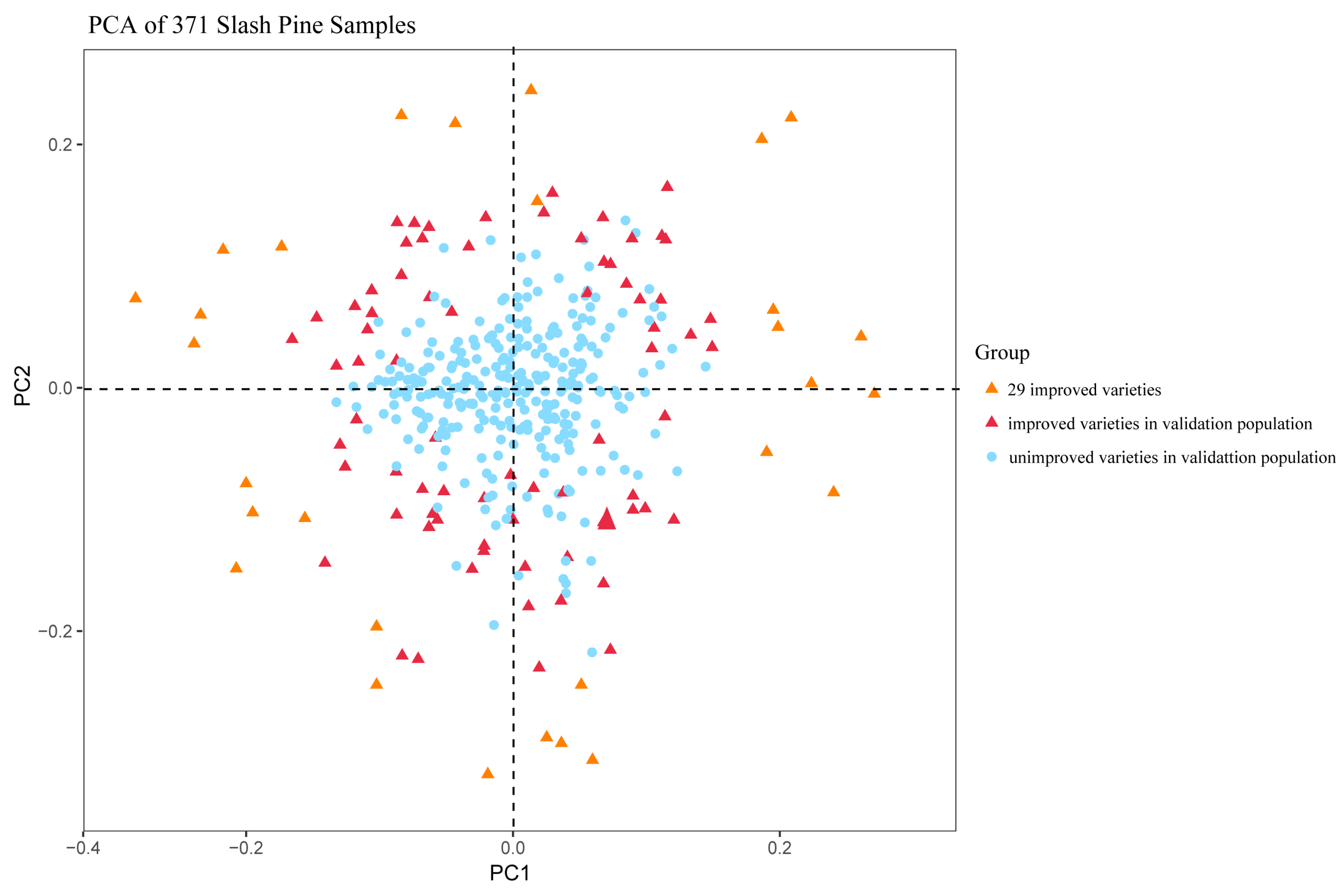
Figure 6.
DNA fingerprinting of 29 improved slash pine varieties. Each line represents one improved slash pine variety, and each column represents one SNP locus. Blue, yellow, and orange represent 0/0, 0/1, and 1/1, respectively.
Figure 6.
DNA fingerprinting of 29 improved slash pine varieties. Each line represents one improved slash pine variety, and each column represents one SNP locus. Blue, yellow, and orange represent 0/0, 0/1, and 1/1, respectively.
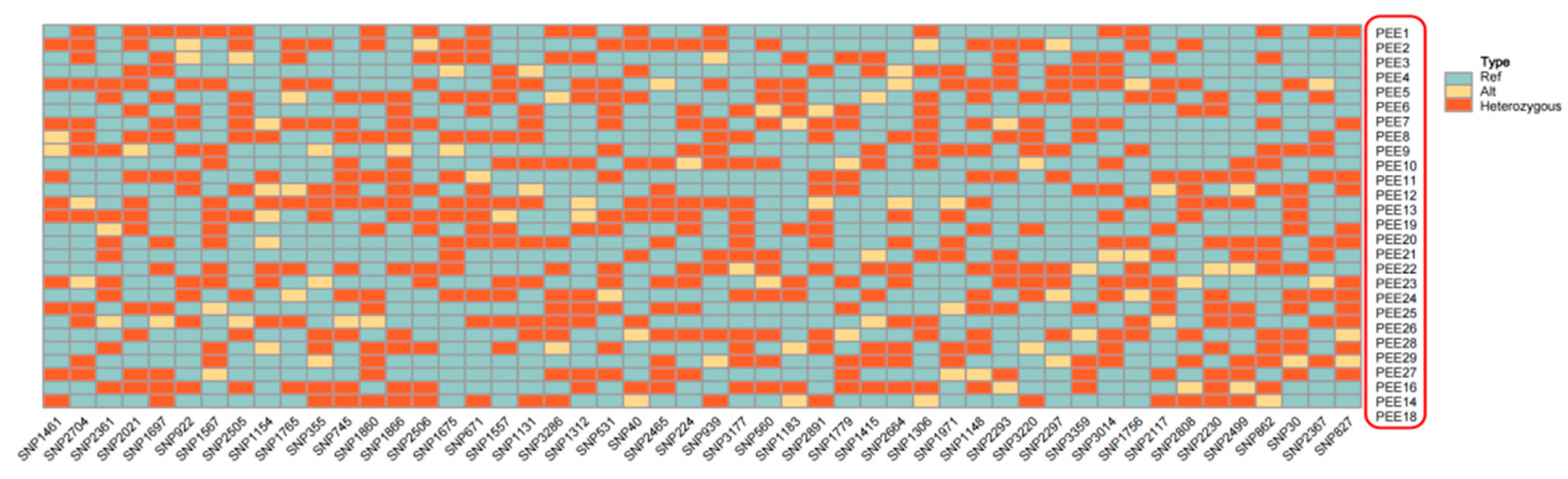

Table 1.
The setting parameters for screening core SNPs.
| Screening Step | The number of remaining SNPs |
|---|---|
| Raw data | 1,83,849 |
| MAF>0.1, miss rate=0 | 64,193 |
| hdw>0.01 | 51,413 |
| ld>0.2 | 30,394 |
| PIC>0.35, | 12,841 |
| Het<0.4 | 3,502 |
Table 2.
The details of Fst(below diagonal) and Nm(above diagonal) among subpopulations.
| PopⅠ | PopⅡ | PopⅢ | |
|---|---|---|---|
| Pop1Ⅰ | 0.022 | 0.033 | |
| PopⅡ | 0.099 | 0.032 | |
| PopⅢ | 0.154 | 0.15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



